
Submitted:
13 January 2026
Posted:
14 January 2026
You are already at the latest version
Abstract
Pancreatic cancer remains a highly lethal malignancy, with standard therapies offering limited benefits in advanced stages; thus, novel strategies that exploit specific cancer cell vulnerabilities are urgently needed. Building on our previous findings that nanosecond pulsed electric fields (nsPEF) combined with cold atmospheric plasma (CAP) produce enhanced cytotoxicity, this study investigates the mo-lecular mechanisms underlying this synergy. Pan02 pancreatic cancer cells were subjected to nsPEF, CAP, or a combination of both. We assessed cell viability, reactive oxygen species (ROS) production, and mitochondrial integrity using metabolic assays, flow cytometry, and fluorescence microscopy. Apoptotic markers were evaluated via Western blotting and caspase activity assays. Combined nsPEF–CAP treatment significantly outperformed either modality alone in inducing cell death. Mechanistically, dual treatment triggered a surge in intracellular ROS, particularly mitochondrial superoxide, indicating severe oxidative stress. Distinct mitochondrial responses were observed: nsPEF reduced mitochondrial membrane potential (ΔΨm), whereas CAP alone caused a slight elevation. Notably, while CAP induced apoptosis (evidenced by increased cleaved caspase-3 and caspase-3/7 activity), lethal nsPEF (100 pulses) caused cell death without triggering apoptotic signaling. However, mild nsPEF (20 pulses) significantly potentiated CAP-induced apoptosis. These findings suggest that nsPEF sensitizes cells to CAP treat-ment by amplifying oxidative stress and mitochondrial dysfunction. This synergistic combination repre-sents a promising therapeutic approach for targeting pancreatic cancer cells resistant to conventional therapies.
Keywords:
pancreatic cancer
1. Introduction
Despite decades of research, pancreatic cancer remains characterized by exceptional lethality and a profound intrinsic resistance to conventional chemotherapeutic and immunotherapeutic interventions [1,2,3]. This poor prognosis is largely attributed to the tumor’s dense stromal barrier, early metastatic dissemination, and limited response to chemotherapy [4] and radiation [5]. Survival statistics remain persistently low, with an overall 5-year survival rate of approximately 12% across all stages [6]. For patients diagnosed with distant metastatic disease, the prognosis is particularly dismal, with a 5-year survival rate of only ~2.9% [7]. Therefore, novel therapeutic strategies are urgently needed to overcome these barriers and improve patient outcomes.
Nanosecond pulsed electric field (nsPEF), also known as nano-pulse stimulation, has emerged as a promising non-thermal physical modality for cancer ablation [8,9,10]. nsPEF employs ultra-short, high-voltage pulses [11,12,13] to induce nanoporation in the plasma membrane and affect intracellular structures, including the mitochondria, endoplasmic reticulum, and nucleus [14,15,16,17]. These interactions disrupt calcium signaling [11,18,19,20], and mitochondrial homeostasis [11], while generating reactive oxygen species (ROS) ultimately triggering both apoptotic and non-apoptotic cell death pathways. Beyond direct ablation, nsPEF can sensitize tumors to drug delivery, gene transfer, and immune activation [21,22]. While efficacy has been demonstrated in melanoma [23], breast [24], pancreatic [21] and hepatocellular carcinoma [8,25], recent studies highlight its potential to suppress tumor growth and reduce multi-drug resistance in pancreatic cancer models [26,27].
However, the therapeutic efficacy of nsPEF is currently constrained by tumor volume. At diagnosis, human pancreatic tumors typically exceed 2 cm, presenting a challenge for effective local nsPEF treatment. Preclinical studies indicate an inverse relationship between tumor size and nsPEF efficacy: while small tumors (4–7 mm) exhibit high regression rates, significantly reduced tumor control is observed in larger tumors (8–11 mm) treated with identical parameters [21]. This suggests that nsPEF monotherapy may be insufficient for durable control of clinically relevant pancreatic masses, highlighting the need for combinatorial strategies that enhance efficacy without escalating treatment intensity.
Cold atmospheric plasma (CAP) offers a complementary approach. CAP is a non-thermal plasma generated at atmospheric pressure [28] comprising a dynamic mixture of reactive oxygen and nitrogen species (ROS/RNS), UV photons, and charged particles [29,30,31]. The synergistic interplay of long-lived (e.g., H₂O₂, NO₂⁻) and short-lived species (e.g., •OH, ¹O₂) [32,33,34] allows CAP to selectively inactivate cancer cells by exploiting their altered redox homeostasis [29]. CAP induces diverse cell death pathways, including apoptosis, ferroptosis [35], and pyroptosis [36], and has demonstrated potential in stimulating anti-tumor immunity [13,37,38,39]. Importantly, CAP-based technology has been developed for early clinical trials, where it was used to clear post-surgical tumor residuals [40]. Importantly, despite these advantages, CAP monotherapy faces significant limitations, primarily regarding tissue penetration. Therapeutic effects of plasma are typically restricted to superficial cell layers (3–5 cell layers) [41,42] making complete eradication of deep-seated or non-superficial lesions difficult [43,44]. Furthermore, the complexity of in vivo mechanisms and the lack of standardized "plasma dosing" complicates its independent clinical application [45,46].
The combination of nsPEF and CAP presents a compelling theoretical advantage: nsPEF acts as an intracellular stressor that permeabilizes membranes, potentially facilitating the entry of CAP-generated reactive species [31,47]. Building on prior work demonstrating the synergistic cytotoxicity of this combination in pancreatic cancer [31] the specific molecular mechanisms driving this effect remain undefined. While both modalities induce ROS, the extent to which nsPEF modulates intracellular and mitochondrial ROS to sensitize cells to CAP is unknown. To address this gap, this study investigates ROS generation, mitochondrial oxidative stress, and apoptotic signaling in pancreatic cancer cells treated with combined nsPEF and CAP. By specifically examining mild to sublethal nsPEF conditions, we aim to elucidate how nanosecond pulses alter cellular susceptibility to CAP-induced oxidative damage.
2. Materials and Methods
2.1. Cell Culture
Pan02 murine pancreatic adenocarcinoma cells were originally provided by the Division of Cancer Treatment and Diagnosis (DCTD, NCI) and have been maintained in RPMI 1640 medium (ATCC 30-2001) supplemented with 10% fetal bovine serum and antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin, Atlanta Biologicals). Frozen stocks (passage 4–6) were thawed for expansion, and cells with passage numbers 10 to 20 were used for all described experiments. Cells were tested periodically to ensure no mycoplasma contamination occurred.
2.2. In Vitro nsPEF and CAP Treatment
For nsPEF treatment, Pan02 cells (5 × 10⁶ cells/mL, 100 µL) in a 0.1 cm gap electroporation cuvette were exposed to 5 kV, 60 ns pulses at 1 Hz, with various pulse numbers, 20, 30, or 100. To obtain a total volume of 300 µL per condition, three cuvettes were combined.
For cold atmospheric plasma (CAP) treatment, cells were seeded in 24-well plates (300 µL/well; 5 × 10⁶ cells/mL). Exposure was delivered using a nanosecond pulsed atmospheric pressure plasma jet (ns-APPJ; 200 ns, 9 kV, 2 kHz) as previously described [31]. Helium was used as the working gas at a fixed flow rate of 355 sccm. The high-voltage electrode nozzle tip was positioned at a fixed gap distance of 5 mm from the liquid surface. The CAP dosage was defined by exposure durations of 2, 3, 5, and 10 minutes (min).
For combination treatments, a 5-minute interval was maintained between modalities. Two sequences were evaluated: (1) CAP (3 min) followed by nsPEF (20 pulses), and (2) nsPEF (20 pulses) followed by CAP (3 min). Untreated cells served as controls. A helium-gas-only control was not included based on prior validation showing negligible effects on Pan02 viability [31]. All conditions were tested in triplicate and on three separate dates.
2.3. Viability Determined by Metabolic Activity Assay
Following treatment, cell concentration was normalized to 1 × 10⁶ cells/mL. Suspensions (10 µL) were transferred to clear, flat-bottom 96-well plates containing 90 µL of complete medium and incubated for 18 h at 37 °C in a 5% CO₂ humidified atmosphere. Cell viability was assessed using the WST-1 reagent (Roche Applied Science). WST-1 (10 µL) was added to each well, followed by a 2-hour (h) incubation. Absorbance was measured at 450 nm (reference wavelength: 630 nm) using a MultiSkan MCC/340 microplate reader (Fisher Scientific). Relative viability was calculated using the following formula: Treated sample (OD450-OD630)/control (OD450-OD630) × 100%. Pan02 cells without treatment but otherwise handled identically were used as the control.
For ROS scavenging experiments, cells were pre-incubated with 10 mM sodium pyruvate (Gibco) for 30 min at 37 °C prior to nsPEF, CAP, or combination treatments.
2.4. Oxidative Stress and Mitochondrial Membrane Potential (ΔΨm) Analysis
Following the treatment, cells were incubated for 2 h at 37 °C. During the final 30 min, cells were stained with the following fluorescent probes: MitoSOX™ Red (5 µM; Invitrogen) for mitochondrial superoxide, carboxy-H₂DCFDA (5 µM; Invitrogen) for general intracellular ROS, and TMRE (100 nM; Invitrogen) for ΔΨm. Hoechst 33342 (Invitrogen) was used for nuclear counterstaining. Fluorescence was analyzed via flow cytometry using the following excitation/emission settings: MitoSOX (~510/580 nm), H₂DCFDA (485/535 nm), and TMRE (~549/575 nm). Fluorescence microscopy was performed under identical staining conditions to visualize mitochondrial superoxide distribution.
2.5. Caspase 3/7 Activity
Caspase activation was evaluated in the following groups: untreated control, nsPEF alone (30 or 100 pulses), CAP alone (5 or 10 min), and combination treatments (CAP 3 min + nsPEF 20 pulses; or nsPEF 20 pulses + CAP 3 min). Post-treatment, cells were plated (3 × 10⁵ cells/mL) in 6-well plates and incubated for 24 h. Caspase-3/7 activity was quantified using CellEvent™ Caspase-3/7 Green Detection Reagent (Invitrogen). The reagent (500 nM final concentration) was added to samples and incubated for 25 min at 37 °C in the dark. Green fluorescence (Ex/Em: 511/533 nm) was quantified by flow cytometry.
2.6. Western Blot Assay
Cells were harvested at indicated time points (2, 6, or 24 h) following treatment with nsPEF, CAP, combination treatment, or Staurosporine (STS; positive control). Lysates were prepared in RIPA buffer, and total protein was quantified via BCA assay. Proteins were resolved on 10% SDS–PAGE gels (160 V, 60 min) and transferred to PVDF membranes (60 V, 90 min). Membranes were blocked in Tris-buffered saline (TBS) for 30 min at room temperature and probed overnight at 4 °C with primary antibodies against cleaved caspase-3 and GAPDH (Cell Signaling Technology). Bands were detected using goat anti-rabbit IRDye 680 secondary antibodies (LI-COR Biosciences) and visualized using a Bio-Rad ChemiDoc imaging system.
2.7. Statistical Analysis
Data are presented as mean ± standard deviation (SD) of at least three independent biological replicates performed on separate days to ensure reliability. Statistical significance was determined using one-way or two-way ANOVA, with p<0.05 considered significant. Flow cytometry data were processed using FlowJo software. All statistical analyses and graphing were performed using GraphPad Prism 10.
3. Results
3.1. Combined nsPEF–CAP Treatment Markedly Reduces Pancreatic Cancer Cell Viability
First, to confirm our previous finding of synergy between nsPEF and CAP [31], we treated Pan02 pancreatic cancer cells using mild to modest parameters. nsPEF parameters were set to pulse duration 60 ns, 50 kV/cm, 1 Hz, and 20 pulses. CAP was generated using an atmospheric pressure plasma jet (ns-APPJ) with nanosecond high-voltage pulses (200 ns, 9 kV, 2 kHz) for a 3-minute exposure. Cells were treated with either modality alone or in combination sequences (CAP→nsPEF or nsPEF→CAP) as shown in Figure 1. Cell viability, assessed 24 h post-treatment using the WST-1 assay, revealed that nsPEF and CAP monotherapies reduced viability by 7.3% and 49.3%, respectively, relative to untreated controls. In contrast, the combined CAP→nsPEF and nsPEF→CAP treatments resulted in near-complete eradication of metabolic activity, causing 97.4% and 97.3% cell death, respectively. This profound cytotoxicity substantially exceeds the theoretical additive effect of the individual treatments, confirming a strong synergistic interaction that severely compromises cell survival [31]. Given that, both nsPEF and CAP are known inducers of ROS [29,48,49], we next investigated the relationship between ROS generation, and this enhanced cell death.
3.2. 2-nsPEF–CAP Treatments Elevate Mitochondrial Superoxide and Cytoplasmic ROS Production
Flow cytometry revealed distinct patterns of mitochondrial superoxide production across treatment groups. While both nsPEF and CAP monotherapies increased MitoSOX fluorescence compared to controls, CAP induced higher levels than nsPEF (Figure 2A). Notably, the highest mitochondrial superoxide levels were detected in the combination groups, particularly in the nsPEF→CAP sequence, which showed a marked amplification beyond either single treatment. This indicates that combining nsPEF and CAP intensifies mitochondrial oxidative stress more effectively than either modality alone.
To visually confirm these findings, we evaluated mitochondrial ROS using fluorescence microscopy. Control and single-treatment groups exhibited weak to moderate MitoSOX staining (Figure 2B), consistent with flow cytometry data (Figure 2A). In contrast, both combination treatments produced bright, widespread mitochondrial ROS signals, reinforcing that dual exposure drives robust superoxide accumulation at the mitochondrial level.
Cytoplasmic ROS levels were primarily influenced by nsPEF. nsPEF monotherapy triggered a strong increase in H₂DCFDA fluorescence, whereas CAP produced only a moderate elevation. In the combination groups, nsPEF→CAP resulted in higher cytoplasmic ROS levels than either single treatment; however, the magnitude of this increase did not exceed the sum of the individual effects. The CAP→nsPEF sequence did not enhance cytoplasmic ROS beyond the nsPEF baseline (Figure 2C). Overall, no synergistic generation of general cytoplasmic ROS was observed, despite the combination treatments resulting in >97% cell death compared to only 7% for nsPEF alone. The discrepancy between cytoplasmic ROS levels and cell viability suggests distinct mechanisms are at play and will be addressed in the discussion. Representative flow cytometry plots (Figure 2D) confirmed these trends: nsPEF shifted the population markedly to higher ROS levels, while CAP produced a smaller shift, and the combination showed a further but non-additive increase [50,51,52,53].
3.3. ROS Scavenging Rescues Cells from Dual Treatment-Induced Death
To determine whether ROS are the key drivers of cytotoxicity, we evaluated cell viability in the presence of the antioxidant sodium pyruvate. Cells were pre-incubated with sodium pyruvate (10 mM) for 30 min prior to treatment, and viability was quantified 24 h later (Figure 3). In the absence of antioxidant, nsPEF alone maintained 79.8% viable, while CAP reduced viability to 68.5%. As observed previously, the combined CAP→nsPEF treatment caused a profound loss of metabolic activity, dropping viability to 18.6%. Importantly, sodium pyruvate alone did not increase viability in untreated controls. However, in the presence of sodium pyruvate, the cytotoxic effects were completely reversed. Interestingly, both nsPEF and CAP single treatments in the presence of the scavenger resulted in viability exceeding 100% (nsPEF+SP: 134.8%; CAP+SP: 134.6%), suggesting compensatory metabolic stimulation under these conditions. Crucially, sodium pyruvate rescued the combination group, restoring viability from 18.6% to 78.8%. These findings demonstrate that ROS generation is an essential driver of cell death following both single and combined exposures. The strong protective effect of sodium pyruvate highlights oxidative stress as the central contributor to the synergistic cytotoxicity.
3.4. nsPEF Induces Mitochondrial Depolarization Whereas CAP Hyperpolarizes Mitochondria
To investigate mitochondrial contributions to cell death, we measured the mitochondrial membrane potential (ΔΨm) using TMRE at 2 h post-treatment and compared these early changes to 24 h viability data. nsPEF alone induced a significant decrease in ΔΨm, reducing TMRE intensity to approximately two-thirds of the control baseline (Figure 4A). However, despite this depolarization, cell viability remained largely unaffected (only 7.3% reduction, Figure 1), suggesting that this level of ΔΨm loss is likely mild, transient, or reversible, and not independently lethal.
In contrast, CAP treatment 3 min, which caused ~50% cell death (Figure1), resulted in a modest increase (hyperpolarization) in ΔΨm compared to controls (Figure 4A&B). This indicates a lack of direct correlation between depolarization and CAP-induced cell death. Combination treatments (nsPEF→CAP and CAP→nsPEF) resulted in near-complete cell death (>97%), yet the observed ΔΨm changes were either similar to nsPEF alone (for nsPEF→CAP) or showed only moderate reduction (for CAP→nsPEF) (Figure 4A&B). Therefore, the dramatic enhancement of cytotoxicity in combination treatments cannot be attributed primarily to gross alterations in ΔΨm. While ΔΨm changes can correlate with cell death under some conditions [15], in this system, early ΔΨm shifts were not predictive of long-term survival.
3.5. Enhanced Apoptosis Likely Contributes to Synergistic Cytotoxicity of nsPEF-CAP Combination Treatment
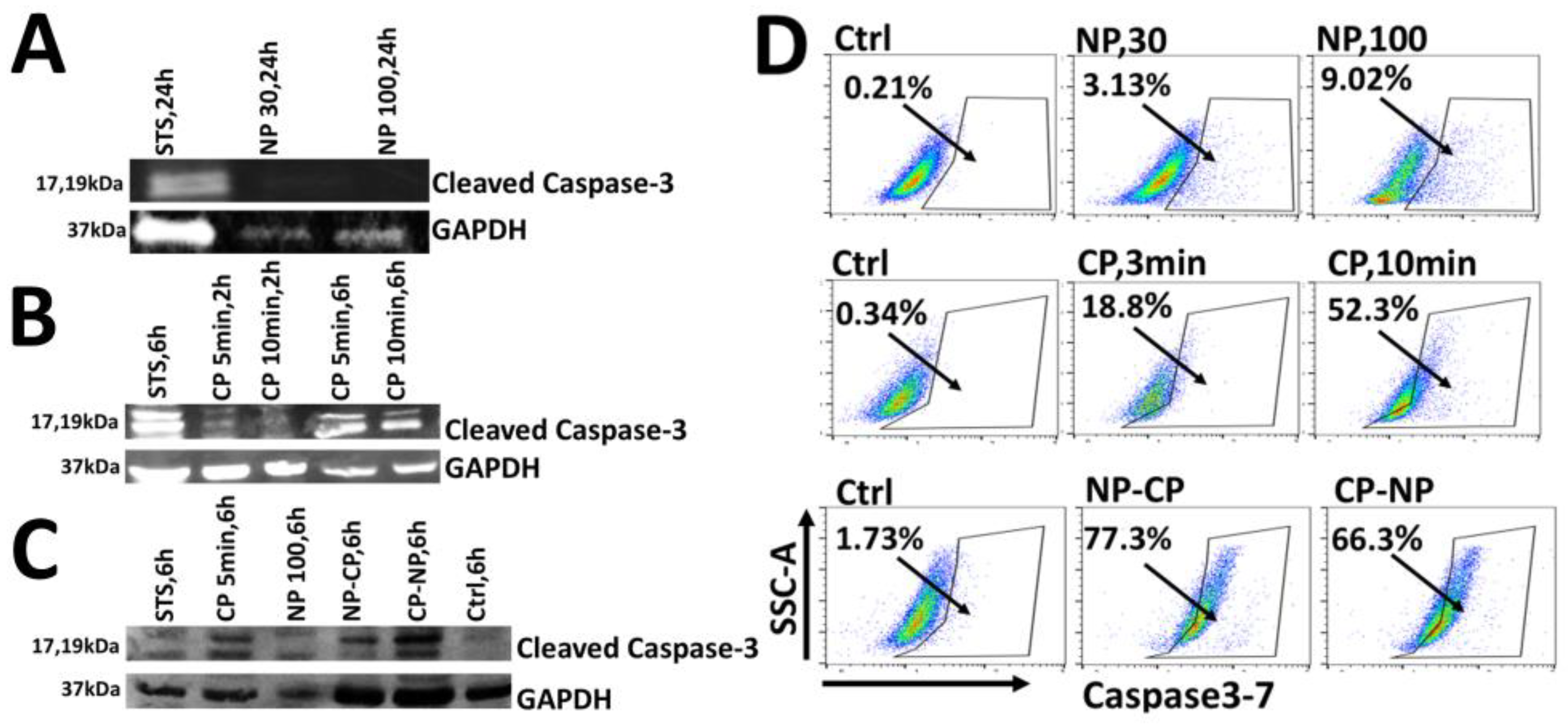
To determine the mode of cell death, we first investigated whether nsPEF induces apoptosis. Pan02 cells were treated with sublethal or lethal doses of nsPEF (30 or 100 pulses), resulting in 20% or > 80% cell death, respectively [31]. As shown in (Figure 5A), cleaved caspase-3 was detected only in the positive control (staurosporine); no cleavage was observed in nsPEF-treated cells. This demonstrates that, under the parameters tested, nsPEF induces non-apoptotic cell death in Pan02 cells.
Next, we assessed CAP-induced apoptosis. Cells treated with CAP (5 or 10 min) showed clear, time-dependent caspase-3 cleavage (17 kDa and 19 kDa fragments), with stronger activation observed at 6 h post-treatment compared to 2 h (Figure 5B). This confirms that CAP robustly activates apoptotic pathways at the protein level.
Having established that CAP engages apoptotic signaling while nsPEF does not, we examined the combination. As shown in (Figure 5C), both sequential treatments significantly enhanced caspase-3 cleavage compared to either modality alone. To quantify this, we employed a functional caspase-3/7 activity assay. As shown in (Figure 5D), nsPEF alone induced minimal caspase activity (3.13% positive cells for 30 pulses; 9.02% for 100 pulses). In contrast, CAP treatment markedly enhanced apoptosis in a dose-dependent manner (18.8% positive for CAP 3 min; 52.3% positive for CAP 10 min). Notably, the combination of mild nsPEF (20 pulses) followed by CAP (3 min) resulted in the strongest caspase-3/7 activation (77.3% positive cells), while CAP followed by nsPEF yielded 66.3% positive. These results suggest that although nsPEF does not inherently trigger apoptosis, it effectively primes cells to undergo CAP-induced apoptosis. Collectively, these data demonstrate that nsPEF functions as a sensitizer, synergistically amplifying the caspase-dependent apoptotic cascade initiated by CAP.
4. Discussion
In this study, we demonstrate that nanosecond pulsed electric fields (nsPEF) and cold atmospheric plasma (CAP) act synergistically to induce Pan02 pancreatic cancer cell death, with the combined treatment yielding substantially greater cytotoxicity than either modality alone. Our findings suggest that this synergy arises from nsPEF sensitizing cells, via ROS amplification, to CAP-induced apoptosis. Mechanistically, we show that while CAP is the primary driver of ROS-mediated apoptosis, nsPEF functions as a "primer" that escalates this process. Notably, lethal nsPEF alone failed to activate apoptotic signaling, instead inducing non-apoptotic cell death. Furthermore, we observed a distinct divergence in mitochondrial responses: changes in mitochondrial membrane potential (ΔΨm) did not correlate with cell death levels across treatments, challenging the assumption that early depolarization is a prerequisite for cytotoxicity in this context [54]. A common paradigm posits that extracellular CAP-generated ROS penetrate the cell or stimulate endogenous ROS production, subsequently activating downstream death pathways [55,56]. Indeed, recent reports have linked CAP-induced ROS to alternative death mechanisms, such as GSDME-dependent pyroptosis [36]. Beyond ROS, CAP alters membrane permeability and ion flux, particularly calcium homeostasis [57,58]. Mild increases in intracellular Ca²⁺ can stimulate mitochondrial dehydrogenases in the TCA cycle and transiently enhance electron transport chain (ETC) activity, resulting in elevated ΔΨm [39]. Such hyperpolarization likely reflects an early stress-adaptation response, where cells temporarily boost bioenergetic output to counteract oxidative stress. Consistent with this "biphasic" model, we observed that ΔΨm remained intact or even hyperpolarized at 2 h post-treatment, despite robust ROS accumulation. However, by 6 h, prominent caspase-3 activation occurred, indicating that sustained oxidative stress eventually overwhelmed this adaptive capacity, triggering mitochondrial dysfunction and apoptosis. The complete rescue of cell viability by antioxidants confirms that ROS are the indispensable mediators of this CAP-induced toxicity [38,39,59,60].
In stark contrast, nsPEF alone produced significant early ROS and disrupted ΔΨm but failed to activate caspase-3, even under conditions causing lethal (>90%) cytotoxicity [31]. This absence of caspase cleavage indicates that nsPEF predominantly triggers non-apoptotic cell death in Pan02 cells [12,16,61], aligning with observations by Pakhomova et al. regarding delayed or absent apoptotic features in certain cell lines. Collectively, these data suggest that classical apoptosis contributes marginally to nsPEF monotherapy efficacy. Crucially, however, mild nsPEF, which was non-lethal on its own, markedly enhanced CAP-mediated apoptosis in the combination setting. This supports a model where nsPEF functions as a priming stimulus, increasing cellular susceptibility to the ROS overload delivered by CAP. Our results also reveal an intriguing divergence in mitochondrial behavior. nsPEF caused the greatest early drop in ΔΨm [15,61], whereas CAP induced slight hyperpolarization early on, despite being the stronger apoptotic inducer later [62,63]. This mismatch suggests: (1) early ΔΨm depolarization is neither the initial cause of CAP-induced apoptosis nor the primary driver of synergistic cell death; and (2) severe ΔΨm loss in CAP-treated cells likely occurs as a secondary consequence of downstream caspase execution [64]. While previous studies in melanoma cells have linked nsPEF-induced depolarization directly to cell death , the dissociation observed here suggests that in Pan02 cells, the nsPEF-induced potential loss is likely transient or reversible [15] and not the sole executioner mechanism.
Although we clearly demonstrate that combination treatment amplifies ROS and cytotoxicity, several specific interactions warrant further study. The sources of ROS differ fundamentally: CAP delivers extracellular species (e.g., H₂O₂, NOx) entering via diffusion or peroxiporins [13,48,65], while nsPEF induces intracellular ROS via mitochondrial and calcium-dependent pathways [49,71,72]. The synergy likely stems from nsPEF-induced nanoporation facilitating the influx of CAP-derived species, while simultaneous calcium mobilization compounds mitochondrial stress. Prior studies indicate that such sustained oxidative stress can activate intrinsic apoptotic cascades [11,25,73], providing a plausible framework for the enhanced caspase-3/7 activity we observed.
Integrating these observations, we propose a synergistic model where CAP and nsPEF operate via complementary, temporally distinct mechanisms converging on catastrophic oxidative stress. CAP provides a rich source of extracellular RONS which enter cells, potentially facilitated by nsPEF-induced nanopores [66,67] while nsPEF simultaneously triggers endogenous ROS production and calcium influx. This "double hit" overwhelms antioxidant defenses and the endoplasmic reticulum [49,68], driving the cell past a survivable threshold. Notably, the lack of caspase activation by lethal nsPEF alone confirms its role is not to initiate apoptosis directly, but to create a pro-oxidant state that amplifies the efficacy of CAP, which acts as the apoptotic executioner[19,23,27,69].
5. Limitations
Several limitations of this study should be acknowledged. First, the precise signaling intermediates connecting the ROS burst to caspase activation remain to be fully mapped. Whether apoptosis proceeds predominantly via intrinsic (mitochondrial) or extrinsic (receptor) pathways or involves other modalities like ferroptosis or pyroptosis, requires further investigation. Second, this study was conducted exclusively in vitro using a murine pancreatic cancer cell line. In vivo tumors possess dense stroma, complex vasculature, and immune components that influence therapeutic response. Future studies should validate these findings in 3D culture systems and immuno-competent animal models to assess the translational potential of this combinatorial approach.
6. Conclusion
The present study demonstrates that nanosecond pulsed electric fields (nsPEF) function as a potent sensitizer for cold atmospheric plasma (CAP)-induced apoptosis in pancreatic cancer cells. This synergistic cytotoxicity is mediated by amplified mitochondrial ROS accumulation and enhanced activation of the apoptotic cascade. Distinctly, while nsPEF alone induces non-apoptotic death and transient mitochondrial depolarization, it effectively primes cells to undergo ROS-dependent apoptosis when combined with CAP. These findings support the further development of nsPEF–CAP combination therapy as a novel, apoptosis-targeted strategy for treating refractory pancreatic cancer.
Funding
This work was supported by funding from the ODU Multidisciplinary Biomedical Research Seed Funding (MBRSF) grant to S. Guo and funding from the U.S. Air Force Office of Scientific Research under Grant FA9550-17-1-0257 to C. Jiang.
Acknowledgments
The authors thank Dr. Stephen Beebe for his support and helpful discussions, and Dr. Yu Jing for her assistance with flow cytometry and fluorescence microscopy. Bayan Hassan Alharbi for assistance with Western blot setup and moral support. Brittany Ruedlinger for scientific advice and general navigating through graduate life.
Conflicts of Interest
The authors declare the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Silva, H.M. and R.C.A. Rosa, Pancreatic cancer in the era of precision medicine: challenges, advances, and the future of therapeutic strategies. Journal of the Egyptian National Cancer Institute, 2025. 37(1): p. 64. [CrossRef]
- Zhu, H., et al., Pancreatic cancer: challenges and opportunities. BMC medicine, 2018. 16(1): p. 214. [CrossRef]
- Tesfaye, A.A., et al., The evolution into personalized therapies in pancreatic ductal adenocarcinoma: challenges and opportunities. Expert review of anticancer therapy, 2018. 18(2): p. 131–148. [CrossRef]
- Long, J., et al., Overcoming drug resistance in pancreatic cancer. Expert opinion on therapeutic targets, 2011. 15(7): p. 817–828. [CrossRef]
- Miller, C.S., et al., Feasibility and safety of endoscopic ultrasound-guided diffusing alpha emitter radiation therapy for advanced pancreatic cancer: Preliminary data. Endoscopy International Open, 2024. 12(10): p. E1085–E1091. [CrossRef]
- Rahib, L., T. Coffin, and B. Kenner, Factors Driving Pancreatic Cancer Survival Rates. Pancreas, 2024: p. 10.1097. [CrossRef]
- Sohal, D.P., et al., Metastatic pancreatic cancer: ASCO guideline update. Journal of Clinical Oncology, 2020. 38(27): p. 3217–3230. [CrossRef]
- Nuccitelli, R. and A. McDaniel, Nano-Pulse Stimulation Therapy in Oncology. Bioelectricity, 2024. 6(2): p. 72–79. [CrossRef]
- McDaniel, A., et al., Nano-pulse stimulation™ therapy (NPS™) is superior to cryoablation in clearing murine melanoma tumors. Frontiers in Oncology, 2023. 12: p. 948472. [CrossRef]
- Pastori, C., et al., Neoadjuvant chemo-immunotherapy is improved with a novel pulsed electric field technology in an immune-cold murine model. Plos one, 2024. 19(3): p. e0299499. [CrossRef]
- Beebe, S.J., Mechanisms of Nanosecond Pulsed Electric Field (NsPEF)-induced cell death in cells and tumors. Journal of Nanomedicine Research, 2015. 2(1). [CrossRef]
- Nuccitelli, R., et al., Nanosecond pulsed electric fields cause melanomas to self-destruct. Biochemical and biophysical research communications, 2006. 343(2): p. 351–360. [CrossRef]
- Guo, S., et al., The role of reactive oxygen species in the immunity induced by nano-pulse stimulation. Scientific reports, 2021. 11(1): p. 23745. [CrossRef]
- Yao, C., et al., Simulation and experimental study on the responses of subcellular structures in tumor cells induced by 5 ns pulsed electric fields. Applied Sciences, 2023. 13(14): p. 8142. [CrossRef]
- Beebe, S.J., et al., Transient features in nanosecond pulsed electric fields differentially modulate mitochondria and viability. PLoS One, 2012. 7(12): p. e51349. [CrossRef]
- Beebe, S.J., N.M. Sain, and W. Ren, Induction of cell death mechanisms and apoptosis by nanosecond pulsed electric fields (nsPEFs). Cells, 2013. 2(1): p. 136–162. [CrossRef]
- Schoenbach, K.H., S.J. Beebe, and E.S. Buescher, Intracellular effect of ultrashort electrical pulses. Bioelectromagnetics: Journal of the Bioelectromagnetics Society, The Society for Physical Regulation in Biology and Medicine, The European Bioelectromagnetics Association, 2001. 22(6): p. 440–448.
- Zaklit, J., et al., 2-ns Electrostimulation of Ca2+ influx into chromaffin cells: Rapid modulation by field reversal. Biophysical journal, 2021. 120(3): p. 556–567. [CrossRef]
- Semenov, I., S. Xiao, and A.G. Pakhomov, Primary pathways of intracellular Ca2+ mobilization by nanosecond pulsed electric field. Biochimica et Biophysica Acta (BBA)-Biomembranes, 2013. 1828(3): p. 981–989. [CrossRef]
- Craviso, G.L., et al., Nanosecond electric pulses: a novel stimulus for triggering Ca2+ influx into chromaffin cells via voltage-gated Ca2+ channels. Cellular and molecular neurobiology, 2010. 30(8): p. 1259–1265. [CrossRef]
- Guo, S., et al., Nano-pulse stimulation for the treatment of pancreatic cancer and the changes in immune profile. Cancers, 2018. 10(7): p. 217. [CrossRef]
- Zhao, J., et al., Corrigendum: Antitumor effect and immune response of nanosecond pulsed electric fields in pancreatic cancer. Frontiers in Oncology, 2022. 12: p. 1052763. [CrossRef]
- Rossi, A., et al., Mechanisms and immunogenicity of nsPEF-induced cell death in B16F10 melanoma tumors. Scientific Reports, 2019. 9(1): p. 431. [CrossRef]
- Xu, Z., et al., Nanosecond pulsed electric field induces an antitumor effect in triple-negative breast cancer via CXCL9 axis dependence in mice. Cancers, 2023. 15(7): p. 2076. [CrossRef]
- Nuccitelli, R., et al., Nanoelectroablation of murine tumors triggers a CD8-dependent inhibition of secondary tumor growth. PLoS one, 2015. 10(7): p. e0134364. [CrossRef]
- Liang, Y.-y., et al., Anti-tumor effects of nanosecond pulsed electric fields in a murine model of pancreatic cancer. Bioelectrochemistry, 2025. 161: p. 108803. [CrossRef]
- Szlasa, W., et al., Nanosecond pulsed electric field suppresses growth and reduces multi-drug resistance effect in pancreatic cancer. Scientific Reports, 2023. 13(1): p. 351. [CrossRef]
- Yan, D., et al., Multi-modal biological destruction by cold atmospheric plasma: capability and mechanism. Biomedicines, 2021. 9(9): p. 1259. [CrossRef]
- Jiang, C., et al., Modulation of ROS in nanosecond-pulsed plasma-activated media for dosage-dependent cancer cell inactivation in vitro. Physics of Plasmas, 2020. 27(11). [CrossRef]
- Jiang, C., et al., Synergistic effects of an atmospheric-pressure plasma jet and pulsed electric field on cells and skin. IEEE Transactions on Plasma Science, 2021. 49(11): p. 3317–3324. [CrossRef]
- Oshin, E.A., et al., Synergistic effects of nanosecond pulsed plasma and electric field on inactivation of pancreatic cancer cells in vitro. Scientific reports, 2024. 14(1): p. 885. [CrossRef]
- Hamza, I.A., et al., Cold atmospheric plasma: a sustainable approach to inactivating viruses, bacteria, and protozoa with remediation of organic pollutants in river water and wastewater. Environmental Science and Pollution Research, 2023. 30(54): p. 116214–116226. [CrossRef]
- Fridman, G., et al., Applied plasma medicine. Plasma processes and polymers, 2008. 5(6): p. 503–533. [CrossRef]
- Zhai, S.-y., M.G. Kong, and Y.-m. Xia, Cold atmospheric plasma ameliorates skin diseases involving reactive oxygen/nitrogen species-mediated functions. Frontiers in Immunology, 2022. 13: p. 868386. [CrossRef]
- Jo, A., et al., Plasma-activated medium induces ferroptosis by depleting FSP1 in human lung cancer cells. Cell death & disease, 2022. 13(3): p. 212.
- Yang, X., et al., Cold atmospheric plasma induces GSDME-dependent pyroptotic signaling pathway via ROS generation in tumor cells. Cell death & disease, 2020. 11(4): p. 295.
- Kniazeva, V., et al., Adjuvant composite cold atmospheric plasma therapy increases antitumoral effect of doxorubicin hydrochloride. Frontiers in Oncology, 2023. 13: p. 1171042. [CrossRef]
- Semmler, M.L., et al., Molecular mechanisms of the efficacy of cold atmospheric pressure plasma (CAP) in cancer treatment. Cancers, 2020. 12(2): p. 269. [CrossRef]
- Min, T., et al., Therapeutic effects of cold atmospheric plasma on solid tumor. Frontiers in Medicine, 2022. 9: p. 884887. [CrossRef]
- Canady, J., et al., The first cold atmospheric plasma phase I clinical trial for the treatment of advanced solid tumors: a novel treatment arm for cancer. Cancers, 2023. 15(14): p. 3688. [CrossRef]
- Chen, Z., et al., Cold atmospheric plasma discharged in water and its potential use in cancer therapy. Journal of Physics D: Applied Physics, 2016. 50(1): p. 015208. [CrossRef]
- Partecke, L.I., et al., Tissue tolerable plasma (TTP) induces apoptosis in pancreatic cancer cells in vitro and in vivo. BMC cancer, 2012. 12(1): p. 473. [CrossRef]
- Honnorat, B., Application of cold plasma in oncology, multidisciplinary experiments, physical, chemical and biological modeling. 2018, Sorbonne Université.
- Yan, D., J. Sherman, and M. Keidar, Cold atmospheric plasma, a novel promising anti-cancer treatment modality. Oncotarget 8: 15977–15995. 2017. [CrossRef]
- Pouvesle, J.-M., et al. Plasma/target interactions in biomedical applications of cold atmospheric pressure plasmas. in PSE 2018. 2018.
- Metelmann, P., et al., Clinical studies applying physical plasma in head and neck cancer-key points and study design. Int. J. Clin. Res. Trials, 2016. 1: p. 103. [CrossRef]
- Wolff, C.M., J.F. Kolb, and S. Bekeschus, Combined in vitro toxicity and immunogenicity of cold plasma and pulsed electric fields. Biomedicines, 2022. 10(12): p. 3084. [CrossRef]
- Pakhomova, O.N., et al., Oxidative effects of nanosecond pulsed electric field exposure in cells and cell-free media. Archives of biochemistry and biophysics, 2012. 527(1): p. 55–64. [CrossRef]
- Nuccitelli, R., et al., Nanosecond pulsed electric field stimulation of reactive oxygen species in human pancreatic cancer cells is Ca2+-dependent. Biochemical and biophysical research communications, 2013. 435(4): p. 580–585. [CrossRef]
- Kim, J., J. Kim, and J.-S. Bae, ROS homeostasis and metabolism: a critical liaison for cancer therapy. Experimental & molecular medicine, 2016. 48(11): p. e269–e269.
- Noh, J., et al., Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nature communications, 2015. 6(1): p. 6907. [CrossRef]
- Pourahmad, J., A. Salimi, and E. Seydi, Role of oxygen free radicals in cancer development and treatment, in Free radicals and diseases. 2016, IntechOpen.
- Policastro, L.L., et al., The tumor microenvironment: characterization, redox considerations, and novel approaches for reactive oxygen species-targeted gene therapy. Antioxidants & redox signaling, 2013. 19(8): p. 854–895.
- Xu, D., et al., In situ OH generation from O2− and H2O2 plays a critical role in plasma-induced cell death. PloS one, 2015. 10(6): p. e0128205. [CrossRef]
- Kim, S.J. and T. Chung, Cold atmospheric plasma jet-generated RONS and their selective effects on normal and carcinoma cells. Scientific reports, 2016. 6(1): p. 20332. [CrossRef]
- Yun, J.H., et al., Non-thermal atmospheric pressure plasma induces selective cancer cell apoptosis by modulating redox homeostasis. Cell Communication and Signaling, 2024. 22(1): p. 452. [CrossRef]
- Schneider, C., et al., Cold atmospheric plasma causes a calcium influx in melanoma cells triggering CAP-induced senescence. Scientific reports, 2018. 8(1): p. 10048. [CrossRef]
- Iuchi, K., et al., Cold atmospheric nitrogen plasma induces metal-initiated cell death by cell membrane rupture and mitochondrial perturbation. Cell Biochemistry and Function, 2023. 41(6): p. 687–695. [CrossRef]
- Yan, D., J.H. Sherman, and M. Keidar, Cold atmospheric plasma, a novel promising anti-cancer treatment modality. Oncotarget, 2016. 8(9): p. 15977. [CrossRef]
- Bauer, G., et al., Dynamics of singlet oxygen-triggered, RONS-based apoptosis induction after treatment of tumor cells with cold atmospheric plasma or plasma-activated medium. Scientific reports, 2019. 9(1): p. 13931. [CrossRef]
- Gianulis, E.C., et al., Electroporation of mammalian cells by nanosecond electric field oscillations and its inhibition by the electric field reversal. Scientific reports, 2015. 5(1): p. 13818. [CrossRef]
- Dezest, M., et al., Mechanistic insights into the impact of Cold Atmospheric Pressure Plasma on human epithelial cell lines. Scientific reports, 2017. 7(1): p. 41163. [CrossRef]
- Murakami, T., Numerical modelling of the effects of cold atmospheric plasma on mitochondrial redox homeostasis and energy metabolism. Scientific reports, 2019. 9(1): p. 17138. [CrossRef]
- Ricci, J.-E., et al., Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell, 2004. 117(6): p. 773–786. [CrossRef]
- Asadipour, K., et al., Data Supporting Nanosecond Pulsed Electric Fields Modulate Electron Transport in in the Plasma Membrane and Mitochondria. Available at SSRN 4489182.
- Vernier, P.T., et al., Nanopore formation and phosphatidylserine externalization in a phospholipid bilayer at high transmembrane potential. Journal of the American Chemical Society, 2006. 128(19): p. 6288–6289. [CrossRef]
- Vernier, P.T., Y. Sun, and M.A. Gundersen, Nanoelectropulse-driven membrane perturbation and small molecule permeabilization. BMC cell biology, 2006. 7(1): p. 37. [CrossRef]
- Asadipour, K., et al., Nanosecond pulsed electric fields (nsPEFs) modulate electron transport in the plasma membrane and the mitochondria. Bioelectrochemistry, 2024. 155: p. 108568. [CrossRef]
- Semenov, I., S. Xiao, and A. Pakhomov, Non-Ligand Mobilization of Intracellular Free Calcium by Nanosecond Pulsed Electric Field (NsPEF). Biophysical Journal, 2013. 104(2): p. 617a. [CrossRef]
Figure 1.
Effect of combined nanosecond pulsed electric field and cold atmospheric plasma treatment on Pan02 cell viability. Pan02 cells were subjected to various treatments: untreated control (Ctrl), nanosecond pulsed electric field ( NP) alone (60 ns, 50 kV/cm, 1 Hz, 20 pulses), cold atmospheric plasma alone 3 min plasma treatment time (CP), sequential application of CP followed by NP (CP-NP), or sequential application of NP followed by CP (NP-CP). Cell viability was assessed 24 hours (h) post-treatment using a WST-1 metabolic activity assays. Data is presented as mean ± SD.*: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (One way ANOVA).
Figure 1.
Effect of combined nanosecond pulsed electric field and cold atmospheric plasma treatment on Pan02 cell viability. Pan02 cells were subjected to various treatments: untreated control (Ctrl), nanosecond pulsed electric field ( NP) alone (60 ns, 50 kV/cm, 1 Hz, 20 pulses), cold atmospheric plasma alone 3 min plasma treatment time (CP), sequential application of CP followed by NP (CP-NP), or sequential application of NP followed by CP (NP-CP). Cell viability was assessed 24 hours (h) post-treatment using a WST-1 metabolic activity assays. Data is presented as mean ± SD.*: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (One way ANOVA).
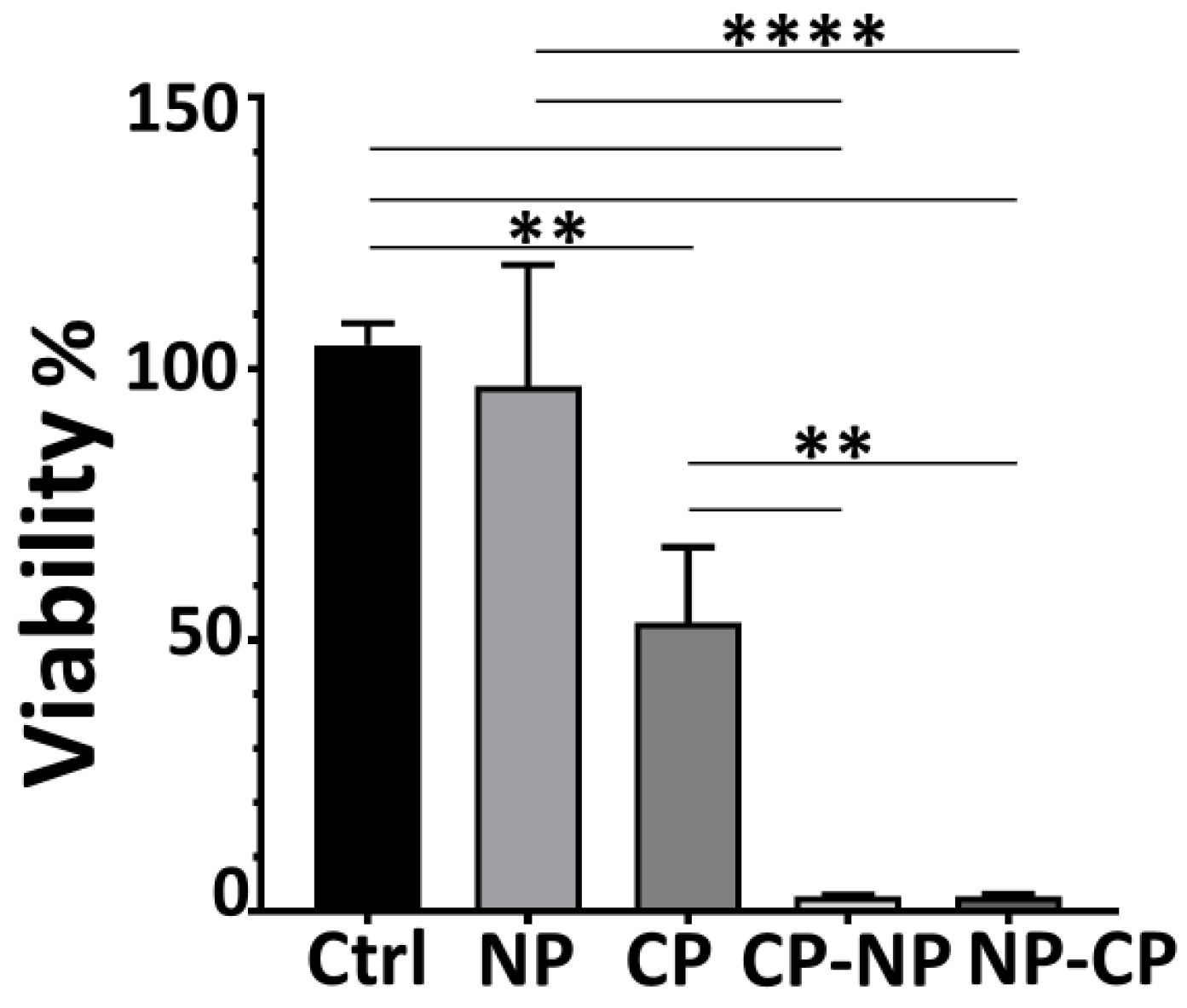
Figure 2.
Effect of combined nanosecond pulsed electric field and cold atmospheric plasma treatment on reactive oxygen species generation in Pan02 cells. Pan02 cells were treated with nanosecond pulsed electric field (NP, 60 ns, 50 kV/cm, 1 Hz, 20 pulses) alone, cold atmospheric plasma (CP, 3 min plasma treatment time) alone, sequential application of NP followed by CP (NP-CP), or sequential application of CP followed by NP (CP-NP). The control group (Ctrl) represents untreated cells maintained in media only. All assessments were performed at 2-h post-treatment. (A) Percentage of positive cells for mitochondrial superoxide (Mito-O2*-). (B) Representative flow plots and fluorescent microscopic images using MitoSOX (5 µM) staining (red) and DAPI nuclear staining (blue). (C) Percentage of positive cells for cytoplasmic ROS. (D) Representative flow plots using H2DCFDA staining. Data is presented as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (One way ANOVA).
Figure 2.
Effect of combined nanosecond pulsed electric field and cold atmospheric plasma treatment on reactive oxygen species generation in Pan02 cells. Pan02 cells were treated with nanosecond pulsed electric field (NP, 60 ns, 50 kV/cm, 1 Hz, 20 pulses) alone, cold atmospheric plasma (CP, 3 min plasma treatment time) alone, sequential application of NP followed by CP (NP-CP), or sequential application of CP followed by NP (CP-NP). The control group (Ctrl) represents untreated cells maintained in media only. All assessments were performed at 2-h post-treatment. (A) Percentage of positive cells for mitochondrial superoxide (Mito-O2*-). (B) Representative flow plots and fluorescent microscopic images using MitoSOX (5 µM) staining (red) and DAPI nuclear staining (blue). (C) Percentage of positive cells for cytoplasmic ROS. (D) Representative flow plots using H2DCFDA staining. Data is presented as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (One way ANOVA).
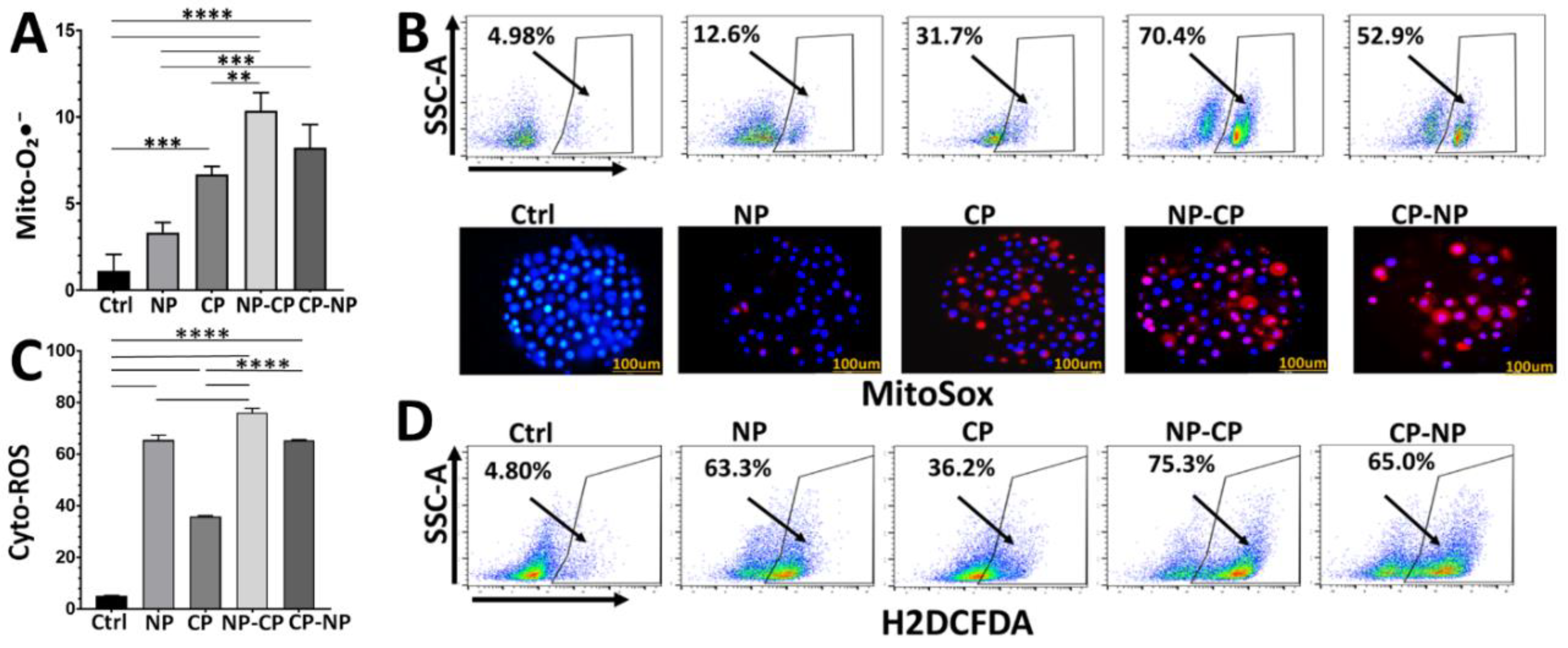
Figure 3.
Protection of Pan02 cells from ROS-induced death by sodium pyruvate. Pan02 cells were subjected to various treatments: nanosecond pulsed electric field (NP, 60 ns, 50 kV/cm, 1 Hz, 20 pulses) alone, cold atmospheric plasma (CP, 3 min plasma treatment time) alone, or sequential application of CP followed by NP (CP-NP). Untreated cells served as controls (Ctrl). Prior to treatment, cells were pre-incubated with sodium pyruvate (SP) or without (No SP). Cell viability was assessed 24 h post-treatment using a WST-1 metabolic activity assay, with results expressed as a percentage of untreated control cells (Ctrl). Data is presented as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (Two-way ANOVA).
Figure 3.
Protection of Pan02 cells from ROS-induced death by sodium pyruvate. Pan02 cells were subjected to various treatments: nanosecond pulsed electric field (NP, 60 ns, 50 kV/cm, 1 Hz, 20 pulses) alone, cold atmospheric plasma (CP, 3 min plasma treatment time) alone, or sequential application of CP followed by NP (CP-NP). Untreated cells served as controls (Ctrl). Prior to treatment, cells were pre-incubated with sodium pyruvate (SP) or without (No SP). Cell viability was assessed 24 h post-treatment using a WST-1 metabolic activity assay, with results expressed as a percentage of untreated control cells (Ctrl). Data is presented as mean ± SD. *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001 (Two-way ANOVA).
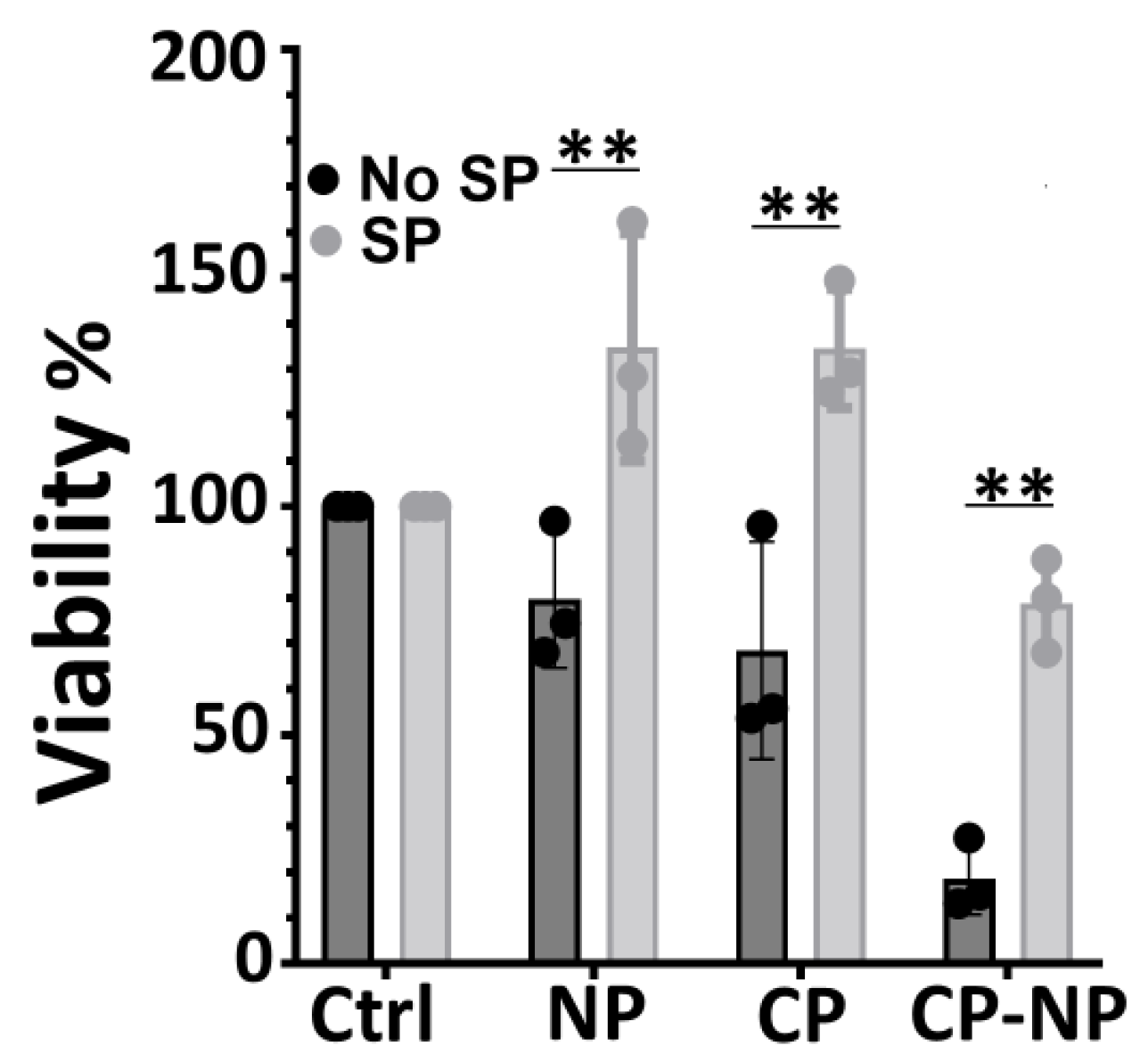
Figure 4.
Distinct impact of nanosecond pulsed electric field and cold atmospheric plasma on mitochondrial membrane potential ΔΨm) in Pan02 cells. Pan02 cells were treated with media control (Ctrl), nanosecond pulsed electric field (NP, 60 ns, 50 kV/cm, 1 Hz, 20 pulses) alone, cold atmospheric plasma (CP, 3 min plasma treatment time) alone, sequential application of NP followed by CP (NP-CP), or sequential application of CP followed by NP (CP-NP). ΔΨm was assessed at 2 h post-treatment using the TMRE fluorescent probe. (A) Bar graph shows ΔΨm, indicated by TMRE fluorescence intensity. (B) Representative flow cytometric plots illustrating ΔΨm shift. Data is presented as mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 (One way ANOVA).
Figure 4.
Distinct impact of nanosecond pulsed electric field and cold atmospheric plasma on mitochondrial membrane potential ΔΨm) in Pan02 cells. Pan02 cells were treated with media control (Ctrl), nanosecond pulsed electric field (NP, 60 ns, 50 kV/cm, 1 Hz, 20 pulses) alone, cold atmospheric plasma (CP, 3 min plasma treatment time) alone, sequential application of NP followed by CP (NP-CP), or sequential application of CP followed by NP (CP-NP). ΔΨm was assessed at 2 h post-treatment using the TMRE fluorescent probe. (A) Bar graph shows ΔΨm, indicated by TMRE fluorescence intensity. (B) Representative flow cytometric plots illustrating ΔΨm shift. Data is presented as mean ± SD. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 (One way ANOVA).
Figure 5.
The analysis of caspase-3 cleavage and caspase-3/7 activity in Pan02 cells following nanosecond pulsed electric field and cold atmospheric plasma treatments. Pan02 cells were subjected to various treatments with nanosecond pulsed electric field (NP; 60 ns, 50 kV/cm, 1 Hz) and/or cold atmospheric plasma (CP). Staurosporine (STS) served as a positive control for apoptosis induction). Representative image of Western blot for cleaved caspase-3 expression in Pan02 cells treated with NP 30 pulses or 100 pulses at 24-h (A); CP treatment 5- or 10-min at 2 or 6-h (B); and NP 100 pulses, CP-5 min, NP 20 pulses followed by CP 3-min (NP-), and CP 3-min followed by NP 20 pulses (CP- NP) at 6-h (C). (D) Representative flow cytometric plots illustrating caspase 3/7 activity in Pan02 cells 24 h post-treatment. Cells were untreated, or treated with nsPEF (NP, 30 pulses; NP, 100 pulses), CAP (CP, 3-min; CP, 10-min), or sequential combined treatments (NP-CP: NP 20 pulses followed by CAP 3-min; CP-NP: CAP 3-min followed by NP 20 pulses).
Figure 5.
The analysis of caspase-3 cleavage and caspase-3/7 activity in Pan02 cells following nanosecond pulsed electric field and cold atmospheric plasma treatments. Pan02 cells were subjected to various treatments with nanosecond pulsed electric field (NP; 60 ns, 50 kV/cm, 1 Hz) and/or cold atmospheric plasma (CP). Staurosporine (STS) served as a positive control for apoptosis induction). Representative image of Western blot for cleaved caspase-3 expression in Pan02 cells treated with NP 30 pulses or 100 pulses at 24-h (A); CP treatment 5- or 10-min at 2 or 6-h (B); and NP 100 pulses, CP-5 min, NP 20 pulses followed by CP 3-min (NP-), and CP 3-min followed by NP 20 pulses (CP- NP) at 6-h (C). (D) Representative flow cytometric plots illustrating caspase 3/7 activity in Pan02 cells 24 h post-treatment. Cells were untreated, or treated with nsPEF (NP, 30 pulses; NP, 100 pulses), CAP (CP, 3-min; CP, 10-min), or sequential combined treatments (NP-CP: NP 20 pulses followed by CAP 3-min; CP-NP: CAP 3-min followed by NP 20 pulses).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



