
Submitted:
09 January 2026
Posted:
12 January 2026
You are already at the latest version
Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder in which gut-brain interactions, enteric alpha-synuclein (αS) pathology, and neuroimmune signals contribute to nigrostriatal degeneration. Alongside the frequent occurrence of early gastrointestinal dysfunction, findings from enteric αS pathology and epidemiological studies support the existence of a gut-first subtype of PD. This article provides an integrated review of current evidence linking gastrointestinal and brain αS pathology, gut barrier dysfunction, inflammation, and microbial imbalance in PD. We discuss how these processes may interact through the microbiota-gut-brain axis to drive neurodegeneration, and explore emerging microbiome-based therapeutic strategies including fecal microbiota transplantation, rifaximin, and probiotic approaches. We also point out future research directions, including improved enteric αS imaging and longitudinal microbiome studies starting before PD diagnosis.
Keywords:
alpha-synuclein
; Parkinson's disease
; gut-brain axis
; gut
; microbiome
1. Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease (AD) (Kalia & Lang, 2015). PD affects 1% to 2% of individuals older than 60, and its prevalence rises to 3.5% at age 85 to 89 years (Rizek et al., 2016). The prevalence of PD has increased substantially from approximately 3.15 million cases in 1990 to 11.77 million cases in 2021 (Li et al., 2025). Projections indicate that the number of people living with PD worldwide could rise to nearly 25 million by 2050 (Su et al., 2025).
PD pathology is primarily characterized by the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), most notably in the ventrolateral region whose neurons connect to the dorsal putamen of the striatum (Kalia & Lang, 2015). Despite clinical heterogeneity in motor symptoms, the defining motor symptoms of PD include bradykinesia, rigidity, and resting tremor (Obeso et al., 2017). Non-motor symptoms, including constipation, hyposmia, rapid eye movement sleep behavior disorder (RBD), and depression frequently emerge before the onset of motor symptoms, representing the premotor stage of PD (Tolosa et al., 2007). Mutations in the SNCA gene encoding α-synuclein (αS) can cause familial forms of PD (Polymeropoulos et al., 1997). Together with the finding that αS aggregates into Lewy bodies, the pathological hallmark of sporadic PD (Spillantini et al., 1998), this strongly implicates αS in PD pathogenesis and progression. Currently available PD medications act solely as symptomatic treatments and fail to alter disease progression, whereas promising disease-modifying treatments designed to delay pathogenesis are currently still under clinical development (McFarthing et al., 2024).
αS is a 140-amino-acid protein that is highly abundant in the nervous system (Goedert, 2001), accounting for around 1% of cytosolic protein (Stefanis, 2011). It is also very abundant, for unclear reasons, in erythrocytes and platelets (Barbour et al., 2008). αS is enriched at presynaptic terminals, where it contributes to the regulation of synaptic vesicle pools (Murphy et al., 2000). Upon binding to phospholipid membranes, αS shifts from a largely unstructured conformation to a partially folded state with amphipathic α-helical conformation (Nuscher et al., 2004). Soluble oligomeric forms of αS, rather than fibrillar species, are thought to be chiefly responsible for toxicity, and membrane interactions have been reported to influence its misfolding and aggregation (Bendor et al., 2013). Consistent with the importance of membrane binding, familial PD mutations in αS can abolish its interaction with presynaptic vesicles (Jensen, 1998). Importantly, αS pathology in PD is not restricted to the brain but is also observed in the gastrointestinal (GI) tract (Lebouvier et al., 2008; Beach et al., 2010; Annerino et al., 2012; Stokholm et al., 2016; Ohlsson & Englund, 2019; Tanei et al., 2020; Emmi et al., 2023). αS is expressed in intestinal enteroendocrine cells (Chandra et al., 2017) and certain enteric neurons (Paillusson, 2013). This observation brings the enteric nervous system (ENS) into focus as a potential site of early αS misfolding and disease initiation.
The ENS, often termed the ‘second brain’, regulates GI function autonomously but remains in continuous communication with the central nervous system (CNS) (Rao & Gershon, 2016; Gershon & Margolis, 2021), via the vagus nerve, transmitting extensive sensory information from the gut to the brain (Rao & Gershon, 2016). This bidirectional communication underlies the gut-brain axis, through which peripheral changes may influence central pathology. The anatomical and functional properties of the ENS and its vagal connections are the basis for models in which PD begins in the gut and ascends to the brain, as formalized in the Braak staging hypothesis. According to this framework, misfolded αS may plausibly propagate retrogradely along vagal pathways from the GI tract to the brainstem (Braak et al., 2003; Braak et al., 2002; Braak et al., 2005). This proposed gut-to-brain route is supported by experimental GI-seeding models showing vagus-dependent spread of αS pathology from the ENS to the brain (Pan-Montojo et al., 2012; Holmqvist et al., 2014; Uemura et al., 2018; Kim et al., 2019), and by epidemiological vagotomy cohorts in which full truncal vagotomy is associated with reduced subsequent PD risk (Svensson et al., 2015; Liu et al., 2017).
In this review, we synthesize evidence for gut-first mechanisms in PD by first outlining anatomical and neuropathological support for gut-initiated disease, then examining barrier and immune pathways that link intestinal pathology to nigrostriatal degeneration, and finally summarize microbiome alterations and microbiota-targeted interventions. We also highlight the emerging research direction of enteric αS imaging and propose longitudinal microbiome cohort studies to clarify causality and guide future therapies.
2. Gut-Brain Framework: ENS, Braak Hypothesis and αS Spread
This section outlines the gut-brain framework in PD, beginning with the anatomy and physiology of the ENS and then linking the Braak hypothesis to experimental and clinicopathological evidence that misfolded αS can propagate in a prion-like fashion from gut to brain along autonomic circuits.
2.1. The ENS and the Gut-Brain Axis
Although capable of independent integrative activity, the ENS transmits extensive sensory information to the brain, primarily through the vagus nerve, and contains more than 100 million neurons using nearly every neurotransmitter found in the CNS; approximately 90% of vagal fibers are afferent, indicating that the brain is predominantly receiving signals from the gut rather than sending them (Rao & Gershon, 2016). Within the GI wall, the ENS is organized into two major plexuses: the myenteric (Auerbach) plexus and the submucosal (Meissner) plexus (Gershon & Margolis, 2021), the former located between the circular and longitudinal muscle layers and coordinating GI motility, and the latter situated beneath the mucosa and containing secretomotor and vasomotor neurons that regulate epithelial secretion and mucosal blood flow (Fleming et al., 2020). The ENS comprises intrinsic primary afferent neurons, interneurons, and motor neurons that sense the intestinal environment and coordinate motility and secretion (Gershon & Margolis, 2021; Fleming et al., 2020). The gut-brain axis represents a dynamic, bidirectional neuroendocrine system in which information is transmitted by neuronal signaling, predominantly vagal afferents, endocrine messengers such as gut hormones, and immune mediators including cytokines (Westfall, 2017), creating an integrated communication framework linking the GI tract to CNS function (Gershon & Margolis, 2021). Within this network, the gut transmits information to the brain through spinal and vagal visceral afferent pathways, while the brain sends sympathetic and parasympathetic efferent inputs to the gut (Gershon & Margolis, 2021). The intestinal microbiome acts as an active third participant in this bidirectional signaling (Gershon & Margolis, 2021), with microbial and enteroendocrine signals integrated by the ENS within the submucosal and myenteric plexuses, rather than through direct microbial contact with vagal fibers, before being relayed centrally (Dicks, 2022).
2.2. The Braak Hypothesis
Idiopathic PD was proposed by Heiko Braak and colleagues to follow a stereotyped six-stage progression of Lewy neurites and Lewy bodies, beginning in specific lower brainstem and olfactory regions and ascending through autonomic and brainstem nuclei to limbic and widespread neocortical territories (Braak et al., 2003; Braak et al., 2002). Within this scheme, early pathology is suggested to appear in the dorsal motor nucleus of the vagus nerve (DMV) together with anterior olfactory structures, then to ascend through interconnected brainstem and midbrain nuclei, including the substantia nigra pars compacta, before reaching limbic and association cortices (Braak et al., 2002). Because these regions are linked by well-defined fiber pathways, Braak and colleagues proposed that pathology propagates via retrograde axonal and transneuronal transport along preexisting circuits, identifying the DMV as the most plausible intracerebral starting point rather than the anterior olfactory system (Braak et al., 2003). Extending this framework, they articulated a gut-origin hypothesis in which a pathogen crossing the GI mucosa enters the enteric nervous system and reaches the brain via unmyelinated preganglionic vagal fibers, projecting to the DMV and from which pathology can spread to selectively vulnerable brainstem, midbrain, and cortical targets (Braak et al., 2003). The stomach was highlighted as a likely portal of entry because of its extensive vagal innervation, thin single-cell epithelial barrier, and prolonged exposure to luminal contents (Braak et al., 2003; Braak et al., 2005). Supporting this model, an autopsy study demonstrated αS immunoreactive inclusions in neurons of the submucosal Meissner plexus and myenteric Auerbach plexus of the stomach, with affected axons extending into the gastric mucosa near fundic glands (Braak et al., 2005). These altered enteric neurons provide an anatomical link between the gut and the brain and suggest a plausible site where αS misfolding could begin and then ascend to the CNS via vagal retrograde transport (Braak et al., 2005; Braak et al., 2003).
2.3. Experimental Evidence for Prion-Like αS Propagation
In vitro, aggregated αS drives nucleation-dependent fibrillogenesis, bypassing the lag phase in a dose-dependent manner and recruiting both wild-type (WT) and mutant αS monomer, consistent with a self-propagating seeding mechanism (Wood et al., 1999). In transgenic mice overexpressing human A53T αS (M83 line), intracerebral inoculation of diseased brain homogenate or synthetic preformed fibrils led to neuronal uptake, seeded recruitment of endogenous αS, and propagation of pathology along anatomically connected CNS pathways (Luk et al., 2012a). The induced lesions resembled Lewy bodies and Lewy neurites, spread bilaterally along white matter tracts, and were associated with relatively uniform incubation periods and reduced survival, with phosphorylated, insoluble host αS aggregates indicating templated conversion (Luk et al., 2012a). In WT mice, intrastriatal injection of preformed αS fibrils induced Lewy body-like and Lewy neurite-like inclusions in anatomically connected regions that progressed to the substantia nigra pars compacta, where dopaminergic neurons developed inclusions and underwent progressive loss, accompanied by reduced striatal dopamine and motor impairment, demonstrating that transmission of pathologic αS is sufficient to initiate key pathological and behavioral features of PD-like neurodegeneration (Luk et al., 2012b). Across mouse, rat and nonhuman primate models, intracerebral exposure to patient-derived aggregates or recombinant fibrils similarly seed endogenous αS and produces progressive pathology with neurodegenerative and behavioral consequences constrained by anatomical connectivity and strain properties (Peelaerts et al., 2015; Recasens et al., 2014; Watts et al., 2013; Masuda-Suzukake et al., 2013; Paumier et al., 2015). Collectively, these studies demonstrate that pathological αS can behave in a prion-like manner, with seed-dependent templating, connectivity-constrained propagation that can lead to Lewy-type pathology, neurodegeneration, and motor deficits across species.
Human transplantation data provide convergent support: postmortem analyses of PD patients who received fetal dopaminergic grafts reveal Lewy body-type, αS-positive inclusions within grafted neurons 11 to 16 years after transplantation (Li et al., 2008), consistent with transfer and accumulation of host αS in transplanted cells and with prion-like models of disease spread (Li et al., 2008; Kordower et al., 2008).
Within this prion-like framework, gut-focused models test whether αS pathology can originate in the ENS and ascend to the CNS along autonomic pathways. In mice, gut-administered rotenone led to αS accumulation that advanced from ENS to CNS along vagal and sympathetic routes, and hemivagotomy or sympathectomy blocked CNS progression (Pan-Montojo et al., 2012), supporting an ENS-to-CNS trajectory aligned with Braak staging. In rats, injection of PD patient brain lysate or recombinant αS into the intestinal wall resulted in human αS appearing in vagal fibers within days and in cholinergic neurons of the DMV shortly thereafter, providing direct evidence that peripherally introduced αS can propagate from gut to brain along the vagus nerve (Holmqvist et al., 2014).
Gastric αS preformed fibril (PFF) models define the subsequent pattern of spread. In mice, PFF injection into the muscularis of the pyloric stomach and upper duodenum, near the myenteric plexus, induced pathological αS locally in the gut, and by 1 month, in the DMV; Over subsequent months, aggregates extended to additional brainstem and limbic regions with substantia nigra pars compacta involvement, dopaminergic neuron loss, reduced striatal dopamine, and motor deficits. Truncal vagotomy prevented brain spread of αS pathology, preserved nigrostriatal markers, and normalized motor performance, indicating that the vagus nerve is required for gut-to-brain transmission in this model (Kim et al., 2019). In WT mice, gastric-wall PFF inoculation induces phosphorylated, Lewy body-like αS aggregates in the DMV that were eliminated by prior cervical vagotomy; over longer follow-up, DMV involvement diminished while phosphorylated αS persisted in the myenteric plexus, consistent with transmission from ENS to brainstem along vagal pathways (Uemura et al., 2018). Other gut PFF models indicate that age and lysosomal vulnerability constrain gut-to-brain propagation after duodenal seeding (Challis et al., 2020), while αS expression and transgenic overexpression facilitate DMV pathology but not caudo-rostral spread (Uemura et al., 2019).
Vagotomy cohorts provide complementary human evidence for a vagus-dependent route. In Denmark, full truncal vagotomy was associated with lower subsequent PD risk compared with both superselective gastric vagotomy and matched population controls, with the strongest protective signal emerging more than twenty years after surgery (Svensson et al., 2015). In Sweden, a matched-cohort analysis of 9,430 vagotomized patients and 377,200 reference individuals found no overall association between vagotomy and PD risk. However, when analyses were limited to diagnoses made more than five years after surgery, patients who had undergone truncal vagotomy showed a lower risk of developing PD, whereas selective and highly selective vagotomies were not associated with decreased risk (Liu et al., 2017). Beyond five years of follow-up, truncal vagotomy was also associated with a lower PD risk than selective vagotomy (Liu et al., 2017). The consistent pattern, with risk reduction confined to full truncal vagotomy, supports a vagus-dependent route from the GI tract to the brain (Svensson et al., 2015; Liu et al., 2017) and is consistent with experimental evidence that gut-initiated, vagus-mediated αS delivery reaches the DMV (Pan-Montojo et al., 2012; Holmqvist et al., 2014; Kim et al., 2019; Uemura et al., 2018).
Neuropathological studies show that αS pathology frequently involves peripheral autonomic and enteric structures in patterns compatible with an enteric-vagus-CNS axis (Beach et al., 2010; Tanei et al., 2020; Emmi et al., 2023). In a large autopsy series, PD and dementia with Lewy bodies exhibited widespread phosphorylated αS pathology in spinal cord and peripheral nervous system, with normal elderly controls lacking lesions; within the GI tract, immunoreactivity was common, and the spinal cord, paraspinal sympathetic ganglia, vagus nerve, and ENS were frequently involved (Beach et al., 2010). In a community-based autopsy series, esophageal ENS αS pathology was absent in controls, present in roughly one third of preclinical or prodromal Lewy body disease, and increased with stage, correlating with autonomic dysfunction (Tanei et al., 2020). Quantitative analyses show that enteric αS-positive neuritic pathology is present in PD and absent in controls, with mixed findings regarding myenteric neuronal loss, but consistently indicating substantial ENS involvement aligned with vagal territories (Annerino et al., 2012; Ohlsson & Englund, 2019). Early and advanced PD patients’ duodenal biopsies show increased mucosal T lymphocytes expressing the cluster of differentiation 3 marker and B lymphocytes expressing cluster of differentiation 20, higher expression of human leukocyte antigen-DR on antigen-presenting cells, and hypertrophic enteric glial cells positive for glial fibrillary acidic protein compared with controls, indicating a chronic inflammatory response with enteric gliosis in the proximal small intestine (Campagnolo et al., 2024). These immune and glial changes co-occurred with a significantly greater area occupied by aggregated, thread-like αS deposits along mucosal nerve fibers in PD, a pattern not detected in any control samples, supporting a disease-specific inflammatory enteric αS pathology that may contribute to ongoing αS misfolding and aggregation in the enteric nervous system (Campagnolo et al., 2024). Biopsy studies extend these observations into the prodromal phase. Colonic biopsy series have identified phosphorylated αS within the submucosal plexus of PD patients but not in controls (Lebouvier et al., 2008), with approximately 72% of patients positive in one cohort (Lebouvier et al., 2010). In archival GI biopsies from individuals who later developed PD, phosphorylated αS immunoreactivity was detected in enteric neural structures in 56% of prodromal cases (Stokholm et al., 2016). In this cohort, prodromal samples were obtained on average 7 years before PD diagnosis, with sampling times ranging from about 20 years to a few months before diagnosis, demonstrating that enteric phosphorylated αS can be present in the gut up to two decades before clinical onset (Stokholm et al., 2016).
Molecular and anatomical studies suggest that the ENS provides a distinctive substrate for initiating αS pathology that can access vagal pathways. Intact-cell and tissue cross-linking studies show that αS assembly state is context-dependent, with lysis-sensitive tetramers and oligomers detectable in neurons and non-neural cells (Dettmer et al., 2013). By contrast, enteric neurons and human intestinal tissue show predominantly monomeric αS under similar cross-linking conditions (Corbillé et al., 2016), suggesting that local triggers in the gut may be required to generate seeds competent for retrograde spread. In the proximal GI tract, a defined subset of myenteric neurons and vagal preganglionic efferent fibers express αS (Phillips et al., 2008). Vagal fibers form ring-like contacts onto myenteric neurons and subdiaphragmatic vagotomy markedly reduces αS-positive neurites in the myenteric plexus, indicating that much of the αS-immunoreactive fiber network is vagal in origin (Phillips et al., 2008).
3. Future Directions in Enteric αS Imaging
Despite the findings introduced above, αS immunoreactivity was also detected in gastric and colonic mucosa from individuals without a PD diagnosis at the time of biopsy (Chung et al., 2015; Shin et al., 2017; Ruffmann et al., 2018; Visanji et al., 2015). These observations might suggest that αS aggregation could begin in the ENS years before clinical onset, consistent with gut-first models of PD pathogenesis. This hypothesis needs to be validated in future research, which could include the development of specific in vivo imaging biomarkers of aggregated αS in the ENS. Aggregated αS and Lewy-type inclusion bodies cannot currently be assessed in living humans using SPECT or PET radiotracers because available candidate ligands show suboptimal binding and physicochemical properties, including insufficient affinity and selectivity for αS over other β-sheet aggregates, non-ideal lipophilicity and brain pharmacokinetics, and problematic radiometabolites (Alzghool et al., 2022). The predominantly intracellular localization and low abundance of αS aggregates further complicate their detection with current tracers (Alzghool et al., 2022). Although several αS PET tracers are in preclinical or early clinical studies, none has yet been approved for routine clinical imaging (Guo et al., 2025). Enteric immunohistochemistry indicates that coarse Lewy body-like aggregates within myenteric ganglia are characteristic of PD, whereas finer synaptic or cytoplasmic αS staining appear in both groups, making intraganglionic αS aggregates the most disease-relevant tissue target (Aldecoa et al., 2015). Given the large amounts of αS and αS-like proteins from dietary, microbial and endogenous sources within the intestinal lumen and epithelial layers, any enteric imaging tracer must distinguish intraganglionic aggregated αS from luminal or epithelial signals within the neuro-epithelial-glial interface (Lerner, 2021). Recent ligands represent promising advances: DABTA-based 18F tracers, notably [18F]d8, show subnanomolar affinity for αS fibrils, more than 200-fold selectivity over amyloid-β and tau in vitro, and in preclinical mouse studies show no detectable brain radiometabolites at early time points (Uzuegbunam et al., 2022). More recently, ligands such as 18F-C05-05 and 18F-SPAL-T-06 visualize αS pathology in human brain tissue and show favorable safety profiles with prominent hepatobiliary and intestinal uptake (Goto et al., 2025; Guo et al., 2025). These next-generation tracers mark substantial progress toward ante-mortem αS imaging.
4. Barriers, Immunity, and Neurodegeneration Along the Microbiota-Gut-Brain Axis
This section examines how gut barrier disruption, lipopolysaccharide (LPS) exposure and blood-brain barrier (BBB) compromise link intestinal dysbiosis to central neuroinflammation, and how microglial activation, oxidative stress and mitochondrial dysfunction amplify αS toxicity to drive selective nigrostriatal degeneration.
4.1. Intestinal Barrier Dysfunction, Lipopolysaccharide Signaling, and Blood-Brain Barrier Disruption
In newly diagnosed, untreated PD, colonic permeability to sucralose is approximately doubled compared with controls and is positively correlated with mucosal Escherichia coli (E. coli), nitrotyrosine, and enteric αS, while plasma LPS-binding protein is reduced, consistent with increased exposure to Gram-negative bacteria (Forsyth et al., 2011). Convergent work suggests that gut dysbiosis and increased intestinal permeability expose the ENS to bacterial products, promoting αS misfolding and accumulation that could seed pathology spreading to the CNS (Mulak, 2015; Rietdijk et al., 2017; Kelly et al., 2013; Olanow et al., 2014; Lubomski et al., 2019). At the systemic level, LPS from Gram-negative bacteria induces pro-inflammatory cytokines, downregulates tight-junction proteins and compromises the BBB (Goyal et al., 2020; Perez-Pardo et al., 2018; Perez Pardo, 2017). Increased intestinal permeability and enrichment of LPS-producing bacteria in PD may augment LPS exposure, which activates enteric neurons and glial cells, drives production of pro-inflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α), and sustains systemic and gut inflammation along the microbiota-gut-brain axis (Baizabal-Carvallo & Alonso-Juarez, 2020). In PD-relevant gut-first models, oral administration of the Enterobacteriaceae member Proteus mirabilis increases fecal and serum LPS, reduces expression of the tight-junction protein occludin, and upregulates TNF-α and Toll-like receptor 4 (TLR4) in the distal colon, resulting in LPS-dependent epithelial barrier disruption and colonic inflammation that mechanistically links dysbiosis to increased gut permeability (Choi et al., 2018). Bacterial LPS impairs colonic barrier function by reducing tight-junction proteins such as occludin and promoting TNF-α release through TLR4-mediated activation of lamina propria macrophages and T helper 1 cell differentiation, thereby sustaining mucosal inflammation (Choi et al., 2018). In metabolic stress models, leaky gut with elevated circulating LPS enhances TLR4 signaling, BBB disruption and brain cytokine levels and is ameliorated by depleting intestinal Gram-negative bacteria (Kurita et al., 2020), whereas psychological stress models demonstrate dysbiosis with coordinated loss of tight-junction proteins at gut and brain barriers (Geng et al., 2020), supporting a gut-to-circulation-to-brain LPS pathway in which intestinal leakiness and dysbiosis amplify neuroinflammation via TLR4 and BBB compromise.
4.2. Microglial Activation and αS-LPS Crosstalk
Microglia are immune cells of the CNS and shift from homeostatic surveillance to reactive states that become neurotoxic as PD progresses (Ho, 2019). Systemic and gut-derived LPS activates microglia, elevating pro-inflammatory cytokines such as TNF-α, interleukin 6 and IL-1β (Brown, 2019; Bodea et al., 2014; Fitzgerald et al., 2019; Baizabal-Carvallo & Alonso-Juarez, 2020). Extracellular misfolded αS activates microglia through Toll-like receptor 2, leading to the release of cytokines, reactive oxygen species (ROS) and nitric oxide (George & Brundin, 2015), while non-aggregated extracellular αS variants modulate cytokine release, ROS and microglial phagocytosis (Roodveldt et al., 2010). αS-driven microglial activation is reduced in TLR4-deficient microglia, highlighting TLR4 as a key upstream regulator (Fellner et al., 2012). In vivo, αS fibrils rapidly trigger a major histocompatibility complex II response in substantia nigra and recruit peripheral monocytes, macrophages and lymphocytes (Harms et al., 2017). Positron emission tomography imaging using the 18-kDa translocator protein shows sustained microglial activation in PD that correlates with motor severity (Politis, 2012), and postmortem studies reveal persistent microgliosis in the substantia nigra (Croisier et al., 2005; Joers et al., 2016). Together with evidence for peripheral immune cell infiltration and broader neuroimmune engagement (Whitton, 2007; Joers et al., 2016; Williams et al., 2020), these findings indicate that chronic microglial activation and peripheral immune engagement are key features of PD-related neuroinflammation.
Biophysical and cell-based studies show that gut-derived LPS binds α-helical intermediates of αS to form lipid-protein complexes that stabilize nucleating species and accelerate fibril formation by shortening the lag phase and half-time of aggregation (Bhattacharyya, 2019). Conceptual models of the microbiota-gut-brain axis further propose that microbiota-driven increases in intestinal and BBB permeability allow LPS and other microbiota-derived metabolites to access the CNS, contribute to the formation of fibrillar αS pathogenic species that propagate via the vagus nerve, and template additional αS molecules to convert into β-sheet-rich aggregates (Fitzgerald et al., 2019). LPS-induced activation of inducible nitric oxide synthase in microglia and macrophages increases nitric oxide and peroxynitrite production, leading to nitration of αS, formation of oligomeric αS species, and dopaminergic neuron injury (Choi et al., 2010; Shavali et al., 2006). Inflammasome activation by bacterial LPS converts procaspase-1 into its active form, caspase-1, which cleaves αS at aspartic acid 121 to produce truncated fragments that are highly prone to aggregate, enriched in detergent-insoluble inclusions, and neurotoxic (Wang et al., 2016; Baizabal-Carvallo & Alonso-Juarez, 2020). In a complementary pathway, neuronal co-expression of αS and iNOS was shown to increase nitric oxide production, promote αS nitration, and accelerate the formation of larger aggregated αS species resembling those observed in PD (Stone et al., 2012). Collectively, these data support the view that LPS-driven inflammatory signaling promotes iNOS-dependent nitrosative stress and caspase-1-mediated truncation of αS, producing modified αS species that are especially prone to aggregation and may contribute to dopaminergic neurodegeneration.
4.3. Oxidative Stress, Mitochondrial Dysfunction and Selective Nigral Vulnerability
Once αS pathology and microglial activation are established in the CNS, oxidative stress and mitochondrial dysfunction can act as amplifiers of dopaminergic neurodegeneration, as shown in neuronal and dopaminergic cell models, where αS overexpression induces mitochondrial abnormalities, elevates ROS, and promotes inclusion-like aggregates (Hsu et al., 2000; Hu et al., 2019; Chong et al., 2017). In the substantia nigra pars compacta, calcium entry through L-type channels elevates mitochondrial oxidant stress, and the formation of αS Lewy body-like inclusions further increases oxidant stress in susceptible compartments (Dryanovski et al., 2013). Converging evidence from αS preformed fibril models, complex I inhibition and human PD tissue shows that aggregated αS can compromise mitochondrial respiration (Tapias et al., 2017; Ding et al., 2018; Malpartida et al., 2020) and is oxidized and nitrated in vivo (Tapias et al., 2017; Musgrove et al., 2019). Epidemiological links to complex I-inhibiting pesticides (Ascherio & Schwarzschild, 2016) and genetic links to mitochondrial quality-control gene defects (Kalinderi et al., 2016) support an oxidative-mitochondrial axis as an amplifier of αS-related neurodegeneration (Gao et al., 2011).
5. Gut Microbiome and Microbial Metabolites in PD
This section reviews alterations in gut microbiome composition in PD patients, considers methodological and clinical confounders, and then focuses on specific microbial metabolites and taxa, particularly short-chain fatty acid (SCFA) producers and mucin-degrading species, as mechanistic mediators connecting dysbiosis to barrier damage, immune dysregulation and enteric αS aggregation.
5.1. Gut Microbiome Composition in PD
The gut microbiota is a dense microbial ecosystem of approximately 1013–1014 microorganisms (Flint, 2012), contains millions of genes (Qin et al., 2010) and has widespread effects on host physiology, including the regulation of barrier, immune and neural functions (Spielman et al., 2018; Fülling et al., 2019; Flint, 2012). Studies in germ-free and antibiotic-treated mice show that absence or disruption of microbiota impairs hippocampal neurogenesis, alters CNS signaling (Fülling et al., 2019; Möhle et al., 2016), and increases BBB permeability (Braniste et al., 2014; Lin et al., 2021). Recolonization or SCFA supplementation restores tight-junction proteins such as occludin and claudin-5 (Braniste et al., 2014) and ZO-1 (Spielman et al., 2018), implicating microbiota-derived SCFAs as key barrier-stabilizing mediators.
The intestinal barrier and the BBB therefore act as dual gatekeepers that regulate entry of microbe-derived molecules and immune signals into the circulation and brain, shaping gut-brain communication in health and neurodegenerative disease (Pellegrini et al., 2022; Welcome, 2019; Wang et al., 2020; Martens et al., 2018). In PD, a pro-inflammatory shift in the intestinal microbiota, with increased LPS exposure and “leaky gut” (Mulak, 2015; Keshavarzian et al., 2015), is proposed to drive αS misfolding and central neuroinflammation along the brain-gut-microbiota axis (Mulak, 2015; Lin et al., 2021). Accordingly, dysbiosis is now a major focus in PD, and multiple case-control cohorts have profiled the gut microbiome in patients versus controls (Table 1).
Across most cohorts, PD has been associated with reduced relative abundances of SCFA-producing taxa, including Prevotellaceae, Faecalibacterium and Lachnospiraceae, as well as genera such as Blautia, Coprococcus and Roseburia, compared with neurologically healthy controls (Scheperjans et al., 2015; Keshavarzian et al., 2015; Unger et al., 2016; Hill-Burns et al., 2017; Li et al., 2017; Petrov et al., 2017; Bedarf et al., 2017; Lin et al., 2018; Li et al., 2019; Pietrucci et al., 2019; Cirstea et al., 2020; Cosma-Grigorov et al., 2020; Vascellari et al., 2020; Aho et al., 2021). Declines in Lachnospiraceae and Prevotellaceae are among the most frequently reported PD-associated microbiome changes (Chen et al., 2021). These taxa are major producers of SCFAs which help maintain intestinal barrier integrity and GI motility, suggesting that their depletion may weaken the barrier and increase exposure of enteric neurons to microbial products that drive excessive αS production within the ENS (Chen et al., 2021). Conversely, increased relative abundances of Akkermansia and, in multiple cohorts, Enterobacteriaceae, Lactobacillaceae, Bifidobacterium, Desulfovibrionaceae and Christensenellaceae have been reported in PD (Unger et al., 2016; Hill-Burns et al., 2017; Li et al., 2017; Petrov et al., 2017; Bedarf et al., 2017; Lin et al., 2018; Li et al., 2019; Pietrucci et al., 2019; Cirstea et al., 2020; Vascellari et al., 2020; Zhang et al., 2020; Qian et al., 2020; Zhang et al., 2022).
Within this pattern of PD-associated enrichment of facultative anaerobic Enterobacteriaceae whose abundance in the gut correlates with motor dysfunction, a specific nitrate-respiration-dependent metabolic pathway has been identified by which these bacteria may initiate intestinal αS aggregation (Ortiz de Ora et al., 2024). In anaerobic culture, nitrate-respiring E. coli K-12 reduced nitrate to nitrite, creating an oxidizing redox potential that shifted labile iron from ferrous iron (Fe2+) to ferric iron (Fe3+) (Ortiz de Ora et al., 2024). This oxidation creates an environment in which dopamine is converted to dopamine-derived quinones that promote aggregation of monomeric αS (Ortiz de Ora et al., 2024).
Under fermentative conditions, or when E. coli K-12 lacked nitrate respiration or nitrate reduction was inhibited with sodium tungstate, αS aggregation was noticeably reduced, demonstrating that bacterial nitrate reduction and nitrite production are crucial for this aggregation cascade (Ortiz de Ora et al., 2024). In enteroendocrine STC-1 cells, which natively express αS and line the intestinal tract, exposure to extracellular nitrite but not nitrate produced concentration-dependent increases in intracellular αS aggregates, supporting a model in which Enterobacteriaceae nitrate respiration and luminal nitrite production may create oxidizing conditions that promote αS aggregation within intestinal epithelial cells, supporting a bacterial metabolic mechanism that could initiate gut αS pathology in PD (Ortiz de Ora et al., 2024).
In dopaminergic-differentiated human neuroblastoma SH-SY5Y neurons, exposure to microbiota-derived molecules showed that rhamnolipid and LPS increase αS messenger RNA and intracellular protein levels with minimal effects on viability across the tested concentration range, whereas curli CsgA and phenol-soluble modulin α1 cause little or no change in αS expression (Ioghen et al., 2024), indicating that specific bacterial amphiphiles can directly upregulate αS in dopaminergic neurons as a potential early molecular step toward aggregation. Curli proteins are extracellular β-sheet-rich amyloid fibers produced by Enterobacteriaceae such as E. coli, with CsgA as the major structural subunit whose polymerization is nucleated by CsgB (Chapman et al., 2002), and they share structural and biophysical properties with human pathological amyloids implicated in neurodegenerative disease (Zhang et al., 2023; Mehra et al., 2019). Curli fibrils from E. coli have been found to enhance αS fibrillation and promote the propagation of αS aggregates within the brain (Lundmark et al., 2005). In vivo, oral exposure to E. coli producing curli fibers increases αS deposition in enteric neurons and enhances aggregated, proteinase K-resistant αS in brain regions, together with microgliosis, astrogliosis and elevated brain cytokines, findings consistent with curli–αS cross-seeding and neuroinflammation (Chen et al., 2016). Moreover, Curli CsgA homologs encoded by human gut Enterobacteriaceae form functional amyloid fibers and differentially accelerate αS fibril formation in vitro, with certain gut-derived CsgA variants more potently promoting αS aggregation in a manner consistent with cross-seeding by specific CsgA-αS complexes (Bhoite et al., 2022).
However, the reported microbiome differences in PD may be strongly influenced by clinical and treatment factors, particularly medication exposure. For example, Scheperjans et al. (2014) reported that, beyond the effect of PD diagnosis, Catechol-O-methyltransferase (COMT) inhibitor use accounted for additional variance in the abundances of fecal Lactobacillaceae and Clostridiales Incertae Sedis IV (Scheperjans et al., 2014), and Hill-Burns et al. (2017) showed that COMT inhibitors have an independent gut-microbiome signature in PD patients (Hill-Burns et al., 2017). In a separate cohort, entacapone use was negatively correlated with the abundance of Faecalibacterium prausnitzii and with both absolute and relative fecal butyrate concentrations, a key SCFA, changes that overlap with PD-associated losses of butyrate-producing taxa (Unger et al., 2016). More broadly, antibiotics and other commonly used drugs substantially reshape gut communities and metabolites, indicating that observed PD-microbiome associations may partly reflect treatment rather than disease alone (Ianiro et al., 2016). These medication effects, together with cross-sectional designs, dietary and regional differences and heterogeneous sequencing and analytic pipelines, preclude firm inferences about whether microbiome changes precede PD or arise secondarily from prodromal symptoms, therapy or disease progression.
5.2. Future Directions: Longitudinal Microbiome Cohorts
To address this causality gap, we propose a prospective, multi-continent longitudinal study enrolling individuals before PD onset and following them through the prodromal window into incident disease. Recruitment would deliberately enrich for at-risk groups spanning genetic susceptibility and prodromal clinical markers, including individuals with idiopathic RBD and carriers of PD-associated variants such as GBA or LRRK2 (Kalia & Lang, 2015), alongside an age- and sex-matched comparator cohort from the same source population. The protocol should standardize biospecimen collection across sites, obtain repeated stool samples for strain-resolved shotgun metagenomics plus metabolomics, and capture time-varying covariates with harmonized medication and dietary histories. Sampling anchored to preclinical baseline, prodromal and post-diagnosis intervals, and analyzed with time-to-event models and mediation-based target trial emulation, would distinguish taxa and microbial functions that consistently precede PD onset from those that shift with prodromal features, treatment or disease stage.
5.3. Short Chain Fatty Acids in Microbiota-Gut-Brain Mechanisms
Many of the taxa consistently depleted in PD, including Lachnospiraceae, Roseburia spp. and Faecalibacterium prausnitzii (Scheperjans et al., 2014; Keshavarzian et al., 2015; Unger et al., 2016; Hill-Burns et al., 2017; Li et al., 2017; Petrov et al., 2017; Bedarf et al., 2017; Lin et al., 2018; Li et al., 2019; Pietrucci et al., 2019; Cirstea et al., 2020; Cosma-Grigorov et al., 2020; Vascellari et al., 2020; Aho et al., 2021), are prominent producers of SCFAs, particularly butyrate and, under appropriate substrates, propionate and acetate (Fernández et al., 2016; Flint et al., 2014). Prevotella enrichment is associated with increased propionate production (Chen et al., 2017). The observed loss of these commensals in PD cohorts therefore supports a model of reduced colonic SCFA production, with downstream effects on epithelial barrier integrity and broader immune-neural interactions that align with the gut-first and dysbiosis frameworks outlined above. SCFAs, especially butyrate, enhance intestinal epithelial barrier function by promoting tight-junction assembly (Peng et al., 2009), reducing paracellular permeability and reversing tight-junction lesions (Plöger et al., 2012; Bischoff et al., 2014), and their deficiency is linked to impaired barrier integrity (Bischoff et al., 2014). PD-associated dysbiosis includes altered fecal SCFA profiles together with increased intestinal permeability and endotoxin exposure (Kujawska & Jodynis-Liebert, 2018; Chen & Lin, 2022), supporting a “leaky gut” state in which weakening of the epithelial barrier may permit luminal microbial products and metabolites to reach the intestinal mucosa and systemic circulation. In gut-brain axis models, SCFA dysregulation and barrier dysfunction are proposed to facilitate entry of intestinal αS into the ENS and its retrograde propagation along vagal pathways to the brainstem, linking dysbiosis-driven leaky gut to caudo-rostral spread of Lewy pathology (Kujawska & Jodynis-Liebert, 2018; Schaeffer et al., 2020; Chen & Lin, 2022). In transgenic rats that overexpress human αS, overexpression is associated with age-dependent gut dysbiosis, changes in microbial metabolites including SCFAs and increased succinate, age-related increases in intestinal permeability, and both intestinal and systemic inflammation (Singh et al., 2023). With ageing, human αS also accumulates in phosphorylated and aggregated forms in the colonic myenteric plexus (Singh et al., 2023). Short-term treatment with a broad-spectrum antibiotic cocktail eliminates fecal SCFAs, lowers succinate concentrations, and reduces αS levels in the olfactory bulb without altering enteric αS (Singh et al., 2023). Together, these findings implicate microbiota-derived metabolites as modulators of αS pathology along the microbiota–gut–brain axis (Singh et al., 2023). Similar mechanisms extend to the BBB. In germ-free mice, absence of microbiota delays BBB maturation, increases permeability and reduces tight-junction proteins, whereas colonization with microbiota or SCFA-producing strains, or butyrate treatment, restores barrier integrity (Al-Asmakh & Hedin, 2015). SCFAs such as butyrate and propionate also increase tight-junction expression and reduce LPS-induced BBB permeability in vitro (Caspani & Swann, 2019). In neonatal antibiotic-induced dysbiosis, reduced fecal SCFAs, disrupted intestinal physiology and abnormal prefrontal myelination and behavior are partly corrected by oral tributyrin, linking microbiota-derived butyrate to both gut barrier function and CNS structure (Keogh et al., 2021). These barrier stabilizing actions provide a direct mechanistic counterpoint to the LPS-driven gut and BBB disruption described earlier.
Beyond structural effects, SCFAs act as signaling molecules via G-protein-coupled receptors and histone deacetylase inhibition (Koh et al., 2016; Li et al., 2018), exerting context-dependent pro- and anti-inflammatory effects on Nuclear Factor kappa-light-chain-enhancer of activated B cells and Mitogen-Activated Protein Kinase signaling, cytokine production, endothelial activation and leukocyte recruitment (Li et al., 2018). Reduced SCFA levels may therefore contribute to heightened peripheral inflammatory tone and altered central immune states, including microglial reactivity (Erny et al., 2015; Heiss & Olofsson, 2019). At physiological plasma concentrations, SCFA mixtures protect human dopaminergic-like SH-SY5Y neurons from oxidative stress via G protein-coupled receptor 43-dependent pathways that limit mitochondrial injury and apoptosis (Saikachain et al., 2023), and sodium butyrate shows neuroprotective effects on dopaminergic neurons in rodent models, linking microbial metabolites to neuronal resilience in PD-relevant circuits (Wu et al., 2008; Kidd & Schneider, 2010). Early-life antibiotic-induced dysbiosis in mice produces lasting increases in gut permeability, cortical myelination abnormalities and cognitive and anxiety-like deficits that are reversed by oral butyrate, further implicating SCFA signaling in gut-brain coupling (Keogh et al., 2021). SCFAs, particularly butyrate, modulate enteric neuronal activity and colonic contractility (Canani, 2011; Unger et al., 2016), and reduced fecal SCFA levels in PD have been linked to impaired GI colonic motility, a feature evident in both prodromal and established PD (Unger et al., 2016).
5.4. Mucin-Degrading Taxa and αS Aggregation
Conversely, PD-associated enrichment of mucin-degrading taxa such as Akkermansia muciniphila, a specialist that uses mucin as its sole carbon and nitrogen source, may promote early mucosal αS pathology (Derrien et al., 2004; Hirayama et al., 2023). Increased Akkermansia, together with reduced butyrate-producing taxa such as Faecalibacterium and Roseburia, may lead to thinning of the intestinal mucosal layer and increased epithelial permeability, allowing inflammatory substances such as LPS, enteric E. coli and even pesticides and herbicides to cross the mucosal barrier, reach the intestinal neural plexus and expose normally expressed αS in enteroendocrine cells and ENS neurons to these stressors, thereby promoting formation of insoluble αS fibrils that can ascend the vagus nerve to the dorsal motor nucleus (Hirayama et al., 2023). Conditioned medium from A. muciniphila, particularly under mucin-deprived conditions, induces endoplasmic-reticulum calcium release, mitochondrial calcium overload, reactive oxygen species generation and αS overexpression, phosphorylation and aggregation in enteroendocrine cells in vitro, and oral A. muciniphila increases αS aggregation in enteroendocrine cells in aged mice (Neto et al., 2022). Taken together, depletion of SCFA-producing commensals and expansion of mucin specialists provides a coherent mechanistic link from PD-associated dysbiosis to barrier weakness, immune dysregulation and early, gut-initiated αS aggregation within the ENS. The proposed interactions between gut dysbiosis, barrier dysfunction, enteric αS aggregation and vagus-mediated propagation of αS to the brain are summarized in Figure 1.
6. Therapeutic Implications Along the Microbiota-Gut-Brain Axis
This section explores microbiota-targeted strategies that seek to modify PD pathogenesis along the microbiota-gut-brain axis, including fecal microbiota transplantation (FMT), rifaximin, and probiotic, prebiotic and synbiotic approaches, and highlights how these interventions may reshape microbial communities, restore barrier integrity and modulate neuroimmune pathways with the potential for disease modification.
6.1. Fecal Microbiota Transplantation
If gut dysbiosis, barrier failure and altered SCFA signaling contribute to PD pathogenesis, deliberately reshaping the intestinal microbiota becomes an attractive candidate for disease modification. Accordingly, fecal microbiota transplantation (FMT) has been proposed as a disease-modifying strategy in PD because gut dysbiosis, barrier dysfunction and microbiota-driven inflammation appear to contribute to nigrostriatal degeneration along the microbiota-gut-brain axis (Lorente-Picón & Laguna, 2021), and because experimental work suggests that correcting dysbiosis via FMT can modulate these pathways (Zhao et al., 2021; Eslami et al., 2025). FMT delivers processed stool from carefully screened healthy donors to the recipient GI tract via capsules, tubes or colonoscopy to restore a more balanced, diverse microbial community (Vongsavath et al., 2023).
In MPTP-induced and chronic rotenone-induced PD mouse models, FMT from healthy donor mice reversed toxin-induced dysbiosis and improved motor function (Sun et al., 2018; Zhao et al., 2021). Specifically, in the MPTP model, FMT increased striatal dopamine and serotonin and reduced microglial and astrocytic activation in the substantia nigra (Sun et al., 2018). In the chronic rotenone model, FMT alleviated chronic inflammation, preserved intestinal and BBB integrity, and suppressed LPS-TLR4-mediated inflammatory signaling along the microbiota-gut-brain axis (Zhao et al., 2021; Eslami et al., 2025).
Randomized controlled trials (RCTs) in mild to moderate PD have tested FMT as an adjunct to dopaminergic therapy using oral capsules, nasoenteric delivery or colonoscopy (Cheng et al., 2023; DuPont et al., 2023; Bruggeman et al., 2024; Scheperjans et al., 2024; Wang et al., 2025). A large capsule study reported greater 12-week improvements than placebo in total Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) and several non-motor and GI outcomes (Cheng et al., 2023). The GUT-PARFECT trial found a modest but significant advantage of donor over autologous nasojejunal FMT for off-state motor scores over 12 months (Bruggeman et al., 2024). A colonoscopic RCT from China reported that donor FMT improved motor symptoms, constipation and mood versus placebo in mild to moderate PD (Wang et al., 2025), whereas other small trials suggest that FMT is well tolerated and may improve constipation and quality of life but that objective motor changes in capsule and colonoscopic studies often parallel placebo or wane over time (Cheng et al., 2023; DuPont et al., 2023). However, a large multicentre double-blind colonoscopic trial found no superiority of donor FMT over placebo on global MDS-UPDRS I–III at six months (Scheperjans et al., 2024). Together, these data suggest that correcting dysbiosis can normalize microbial metabolites (Eslami et al., 2025), strengthen barrier function (Sun et al., 2018; Zhao et al., 2021), and attenuate innate immune pathways that couple the gut to nigrostriatal degeneration (Zhao et al., 2021; Lorente-Picón & Laguna, 2021; Eslami et al., 2025). Non-placebo-controlled studies, including an open-label preliminary trial and a prospective single-arm series of PD patients with constipation, generally report improvements in motor, non-motor and bowel symptoms after FMT (Kuai et al., 2021; Xue et al., 2020; Huang et al., 2019). In addition, a single case report described marked relief of refractory constipation and transient motor improvement in a PD patient following FMT (Huang et al., 2019). Where gut microbiota were profiled, these studies described shifts toward increased diversity and changes in taxa interpreted as beneficial (Kuai et al., 2021; Huang et al., 2019), although the absence of rigorous controls limits inference. Across these studies and broader FMT experience, adverse events are usually mild, transient GI complaints in capsule, nasoenteric and colonoscopic protocols (Cheng et al., 2023; DuPont et al., 2023; Bruggeman et al., 2024; Scheperjans et al., 2024; Wang et al., 2025), and serious procedure- or microbiota-related complications appear rare when rigorous donor screening and monitoring are used (Marcella et al., 2020).
Mechanistically, clinical and preclinical work indicates that FMT increases microbial diversity and shifts community structure toward healthier-donor profiles, often enriching SCFA-producing taxa (Kuai et al., 2021; Cheng et al., 2023; Eslami et al., 2025). In toxin-based PD models it has been shown to strengthen epithelial and blood-brain barriers and attenuate inflammatory signaling in gut and brain (Zhao et al., 2021; Sun et al., 2018), consistent with broader microbiota-gut-brain axis frameworks proposed by other researchers (Lorente-Picón & Laguna, 2021; Eslami et al., 2025). However, metagenomic analyses show that donor strain engraftment is highly variable and depends on donor-recipient compatibility, arguing for microbiome-informed precision strategies such as rational donor selection or defined microbial consortia rather than a universal stool product (Li et al., 2016; Chen et al., 2024). Animal studies consistently show restoration of dopaminergic signalling (Sun et al., 2018; Zhao et al., 2021), attenuation of neuroinflammation (Sun et al., 2018; Zhao et al., 2021; Sampson et al., 2016) and improved motor behavior (Sun et al., 2018; Zhao et al., 2021; Sampson et al., 2016), whereas human trials are small, short and mixed, with one large colonoscopic study negative on global motor outcomes (Scheperjans et al., 2024) and others showing modest-to-moderate benefits in specific motor, non-motor and GI domains (Cheng et al., 2023; DuPont et al., 2023; Bruggeman et al., 2024). Large, multicentre, placebo-controlled trials with deep microbiome and metabolomic profiling and harmonized endpoints are needed before FMT can be considered a reliable disease-modifying therapy.
6.2. Antibiotic Modulation: Rifaximin
Another strategy to target the gut ecosystem is the use of non-absorbable antibiotics such as rifaximin, a gut-selective agent that modulates the microbiota-host interface. Rifaximin is widely used for small intestinal bacterial overgrowth (Barboza et al., 2015; Rao and Bhagatwala, 2019; Sroka et al., 2022), and achieves high intraluminal concentrations with limited systemic exposure (Ponziani et al., 2017). Beyond its antimicrobial activity, rifaximin activates intestinal pregnane X receptor, which inhibits nuclear factor kappa B-dependent production of pro-inflammatory cytokines such as IL-1β and TNF-α, and it promotes the growth of beneficial genera including Bifidobacterium, Faecalibacterium prausnitzii and Lactobacillus without major disruption of overall community composition (Ponziani et al., 2017). In a small open-label study in probable Alzheimer’s disease, rifaximin treatment was associated with reduced serum neurofilament light levels (Suhocki et al., 2022). In a chronic stress model, rifaximin preserved microbial richness, restored key SCFA-producing taxa and normalized brain butyrate levels (Li et al., 2021), while in MitoPark mice it reshaped the gut microbiota, lowered circulating pro-inflammatory cytokines, preserved BBB integrity and improved motor and recognition memory performance (Hong et al., 2022). In PD, rifaximin-based eradication of small intestinal bacterial overgrowth improved motor fluctuations, reducing daily “off” time and delayed-on episodes without altering levodopa pharmacokinetics (Fasano et al., 2013).
6.3. Probiotic, Prebiotic and Synbiotic Approaches
Probiotic, prebiotic and synbiotic strategies aim to correct gut dysbiosis and support a healthier gut microbial ecosystem (Hill et al., 2014; Fijan, 2014; Slavin, 2013). A meta-analysis indicates that oral probiotic supplementation can improve motor and several non-motor symptoms (Park et al., 2023), consistent with evidence that these interventions can enhance SCFA production, particularly butyrate (Markowiak-Kopeć & Śliżewska, 2020), and may foster an anti-inflammatory, barrier-supporting and psychobiotic profile via the microbiota-gut-brain axis (Dinan et al., 2013; Long-Smith et al., 2019).
Clinically, small PD trials with synbiotics and psychobiotics report convergent symptomatic signals. In one study, a synbiotic combining Lactobacillus strains with inulin administered alongside levodopa/carbidopa improved MDS-UPDRS Parts I-III scores and reduced inflammatory and oxidative stress markers while increasing brain-derived neurotrophic factor levels (Ramadan et al., 2025); Adjunctive Lactiplantibacillus plantarum PS128 supplementation significantly improved off- and on-state UPDRS-III motor scores, reduced daily off time, and enhanced quality of life in patients with PD in an open-label, single-arm, baseline-controlled pilot study (Lu et al., 2021). Preclinical work with PS128 in toxin-based PD models shows neuroprotection of dopaminergic neurons, reduced neuroinflammation and prevention of pathogen-associated microbial shifts, implicating both central and gut-mediated mechanisms (Liao et al., 2020; Lee et al., 2023). Overall, these clinical findings suggest that microbiota-targeted strategies can beneficially influence gut composition, SCFA production, barrier function and neuroimmune pathways with downstream effects on motor and selected non-motor outcomes. However, larger and randomized controlled trials are needed before microbiome-targeted interventions can be regarded as disease-modifying in PD.
7. Conclusions
This review integrates neuropathological, experimental, epidemiological and clinical evidence to argue that PD is not solely a brain-limited synucleinopathy but, in many cases, a gut-brain disorder. Braak staging, gut-first propagation models and vagotomy cohorts collectively support a scenario in which misfolded αS can arise within the ENS, ascend along vagal pathways to the DMV and then spread through interconnected brainstem, limbic and cortical networks. Enteric αS pathology in prodromal and clinically manifest PD, together with the molecular and anatomical specialization of the ENS, further supports the plausibility of a gut-origin trajectory. Building on this framework, this article highlights a hypothesis for how intestinal dysbiosis may promote early enteric αS aggregation, elaborated as follows:
Depletion of SCFA-producing commensals and increase in pro-inflammatory, mucin-degrading, and LPS-producing taxa weaken the intestinal barrier, which increases LPS translocation. Parallel effects on the BBB facilitate passage of harmful microbial products and inflammatory mediators into the circulation and CNS. In addition, experimental studies of LPS, nitrate-respiring Enterobacteriaceae that generate nitrite-driven oxidative conditions, and bacterial amyloid curli indicate that specific microbial products can directly promote nitrosative modification, cross-seeding and aggregation of αS in enteroendocrine cells and enteric neurons. In the gut and brain, LPS-TLR4 signaling, microglial and enteric glial activation, iNOS-dependent nitrosative stress and inflammasome-driven caspase-1 cleavage may create an environment that favors nitration, truncation and aggregation of αS. These modified αS species are especially prone to form fibrillar, Lewy body-like assemblies and to propagate along the vagus nerve to the CNS. Furthermore, oxidative stress and mitochondrial dysfunction may act as downstream amplifiers of αS toxicity, contributing to the selective vulnerability of nigrostriatal dopaminergic neurons.
A distinctive contribution of this article is linking these mechanistic pathways to specific microbiome-targeted strategies. Human microbiome studies and preclinical models are synthesized to show how changing the gut environment through FMT, rifaximin and probiotic, prebiotic and synbiotic interventions, can alter microbial metabolites, strengthen barrier integrity and improve motor and selected non-motor outcomes. At the same time, current trials remain heterogeneous, highlighting the need for the development of specific enteric aggregated/misfolded αS imaging tools, longitudinal multi-continent cohorts beginning before PD diagnosis, and rigorously controlled and mechanistically informed intervention studies with microbiome-specific donor and strain selection. Addressing these gaps will be essential to move from correlative gut-brain associations toward disease-modifying therapies that rationally manipulate the intestinal microbiome to alter the course of PD.
References
- Kalia, L. V.; Lang, A. E. Parkinson’s disease. The Lancet 2015, 386(9996), 896–912. [Google Scholar] [CrossRef] [PubMed]
- Rizek, P.; Kumar, N.; Jog, M. S. An update on the diagnosis and treatment of Parkinson disease. Canadian Medical Association Journal 2016, 188(16), 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ye, X.; Huang, Z.; Ye, L.; Chen, C. Global burden of Parkinson’s disease from 1990 to 2021: a population-based study. BMJ Open 2025, 15(4), e095610. [Google Scholar] [CrossRef]
- Su, D.; Cui, Y.; He, C.; Yin, P.; Bai, R.; Zhu, J.; Lam, J. S. T.; Zhang, J.; Yan, R.; Zheng, X.; Wu, J.; Zhao, D.; Wang, A.; Zhou, M.; Feng, T. Projections for prevalence of Parkinson’s disease and its driving factors in 195 countries and territories to 2050: modelling study of Global Burden of Disease Study 2021. BMJ 2025, 388, e080952. [Google Scholar] [CrossRef]
- Obeso, J. A.; Stamelou, M.; Goetz, C. G.; Poewe, W.; Lang, A. E.; Weintraub, D.; Burn, D.; Halliday, G. M.; Bezard, E.; Przedborski, S.; Lehericy, S.; Brooks, D. J.; Rothwell, J. C.; Hallett, M.; DeLong; Marras, C.; Tanner, C. M.; Ross, G. W.; Langston, J. W.; Klein, C.; Bonifati, V.; Jankovic, J.; Lozano, A. M.; Deuschl, G.; Bergman, H.; Tolosa, E.; Rodriguez-Violante, M.; Fahn, S.; Postuma, R. B.; Berg, D.; Marek, K.; Standaert, D. G.; Surmeier, D. J.; Olanow, C. W.; Kordower, J. H.; Calabresi, P.; Schapira, A. H. V.; Stoessl, A. J. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Movement Disorders 2017, 32(9), 1264–1310. [Google Scholar] [CrossRef]
- Tolosa, E.; Compta, Y.; Gaig, C. The premotor phase of Parkinson’s disease. Parkinsonism & Related Disorders 2007, 13, S2–S7. [Google Scholar] [CrossRef]
- Polymeropoulos, M. H.; Lavedan, C.; Leroy, E.; Ide, S. E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; Stenroos, E. S.; Chandrasekharappa, S.; Athanassiadou, A.; Papapetropoulos, T.; Johnson, W. G.; Lazzarini, A. M.; Duvoisin, R. C.; Di Iorio, G.; Golbe, L. I.; Nussbaum, R. L. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science 1997, 276(5321), 2045–2047. [Google Scholar] [CrossRef]
- Spillantini, M. G.; Crowther, R. A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proceedings of the National Academy of Sciences 1998, 95(11), 6469–6473. [Google Scholar] [CrossRef]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Pitzer, K.; Fiske, B.; Navangul, A.; Beissert, K.; Pilcicka, A.; Fuest, R.; Wyse, R. K.; Stott, S. R. W. Parkinson’s Disease Drug therapies in the clinical Trial pipeline: 2024 update. Journal of Parkinson’s Disease 2024, 14(5), 899–912. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nature Reviews. Neuroscience 2001, 2(7), 492–501. [Google Scholar] [CrossRef]
- Stefanis, L. -Synuclein in Parkinson’s disease. Cold Spring Harbor Perspectives in Medicine 2012, 2(2), a009399. [Google Scholar] [CrossRef] [PubMed]
- Barbour, R.; Kling, K.; Anderson, J. P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J. M.; Soriano, F.; Seubert, P.; Chilcote, T. J. Red blood cells are the major source of Alpha-Synuclein in blood. Neurodegenerative Diseases 2008, 5(2), 55–59. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D. D.; Rueter, S. M.; Trojanowski, J. Q.; Lee, V. M. -y. Synucleins are developmentally expressed, and A-Synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. Journal of Neuroscience 2000, 20(9), 3214–3220. [Google Scholar] [CrossRef] [PubMed]
- Nuscher, B.; Kamp, F.; Mehnert, T.; Odoy, S.; Haass, C.; Kahle, P. J.; Beyer, K. A-Synuclein has a high affinity for packing defects in a bilayer membrane. Journal of Biological Chemistry 2004, 279(21), 21966–21975. [Google Scholar] [CrossRef]
- Bendor, J. T.; Logan, T. P.; Edwards, R. H. The function of A-Synuclein. Neuron 2013, 79(6), 1044–1066. [Google Scholar] [CrossRef]
- Jensen, P. H.; Nielsen, M. S.; Jakes, R.; Dotti, C. G.; Goedert, M. Binding of A-Synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. Journal of Biological Chemistry 1998, 273(41), 26292–26294. [Google Scholar] [CrossRef]
- Rao, M.; Gershon, M. D. The bowel and beyond: the enteric nervous system in neurological disorders. Nature Reviews Gastroenterology & Hepatology 2016, 13(9), 517–528. [Google Scholar] [CrossRef]
- Gershon, M. D.; Margolis, K. G. The gut, its microbiome, and the brain: connections and communications. Journal of Clinical Investigation 2021, 131(18). [Google Scholar] [CrossRef]
- Fleming, M. A.; Ehsan, L.; Moore, S. R.; Levin, D. E. The enteric nervous system and its emerging role as a therapeutic target. Gastroenterology Research and Practice 2020, 2020, 1–13. [Google Scholar] [CrossRef]
- Westfall, S.; Lomis, N.; Kahouli, I.; Dia, S. Y.; Singh, S. P.; Prakash, S. Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cellular and Molecular Life Sciences 2017, 74(20), 3769–3787. [Google Scholar] [CrossRef]
- Dicks, L. M. T. Gut bacteria and neurotransmitters. Microorganisms 2022, 10(9), 1838. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W. P.; Del Tredici, K. Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. Journal of Neural Transmission 2003, 110(5), 517–536. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R. A. I.; Steur, E. N. H. J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging 2002, 24(2), 197–211. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; De Vos, R. A. I.; Bohl, J.; Del Tredici, K. Gastric α-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neuroscience Letters 2005, 396(1), 67–72. [Google Scholar] [CrossRef]
- Wood, S. J.; Wypych, J.; Steavenson, S.; Louis, J.-C.; Citron, M.; Biere, A. L. A-Synuclein fibrillogenesis is nucleation-dependent. Journal of Biological Chemistry 1999, 274(28), 19509–19512. [Google Scholar] [CrossRef]
- Luk, K. C.; Kehm, V. M.; Zhang, B.; O’Brien, P.; Trojanowski, J. Q.; Lee, V. M. Y. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. The Journal of Experimental Medicine 2012a, 209(5), 975–986. [Google Scholar] [CrossRef]
- Luk, K. C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J. Q.; Lee, V. M. -y. Pathological A-Synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012b, 338(6109), 949–953. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van Der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van Den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522(7556), 340–344. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.; Blesa, J.; Parent, A.; Perier, C.; Fariñas, I.; Obeso, J. A.; Bezard, E.; Vila, M. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Annals of Neurology 2014, 75(3), 351–362. [Google Scholar] [CrossRef]
- Watts, J. C.; Giles, K.; Oehler, A.; Middleton, L.; Dexter, D. T.; Gentleman, S. M.; DeArmond, S. J.; Prusiner, S. B. Transmission of multiple system atrophy prions to transgenic mice. Proceedings of the National Academy of Sciences 2013, 110(48), 19555–19560. [Google Scholar] [CrossRef] [PubMed]
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D. M. A.; Hasegawa, M. Prion-like spreading of pathological α-synuclein in brain. Brain 2013, 136(4), 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Paumier, K. L.; Luk, K. C.; Manfredsson, F. P.; Kanaan, N. M.; Lipton, J. W.; Collier, T. J.; Steece-Collier, K.; Kemp, C. J.; Celano, S.; Schulz, E.; Sandoval, I. M.; Fleming, S.; Dirr, E.; Polinski, N. K.; Trojanowski, J. Q.; Lee, V. M.; Sortwell, C. E. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiology of Disease 2015, 82, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Englund, E.; Holton, J. L.; Soulet, D.; Hagell, P.; Lees, A. J.; Lashley, T.; Quinn, N. P.; Rehncrona, S.; Björklund, A.; Widner, H.; Revesz, T.; Lindvall, O.; Brundin, P. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nature Medicine 2008, 14(5), 501–503. [Google Scholar] [CrossRef]
- Kordower, J. H.; Chu, Y.; Hauser, R. A.; Freeman, T. B.; Olanow, C. W. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nature Medicine 2008, 14(5), 504–506. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G. A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.-M.; Rodrigo-Angulo, M.; Gille, G.; Funk, R. H. W.; Reichmann, H. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Scientific Reports 2012, 2(1), 898. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathologica 2014, 128(6), 805–820. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S. S.; Lee, S.; Lee, J. H.; Kim, W. R.; Kook, M.; Foss, C. A.; Shen, C.; Lee, H.; Kulkarni, S.; Pasricha, P. J.; Lee, G.; Pomper, M. G.; Dawson, V. L.; Dawson, T. M.; Ko, H. S. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103(4), 627–641.e7. [Google Scholar] [CrossRef]
- Uemura, N.; Yagi, H.; Uemura, M. T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Molecular Neurodegeneration 2018, 13(1), 21. [Google Scholar] [CrossRef]
- Challis, C.; Hori, A.; Sampson, T. R.; Yoo, B. B.; Challis, R. C.; Hamilton, A. M.; Mazmanian, S. K.; Volpicelli-Daley, L. A.; Gradinaru, V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nature Neuroscience 2020, 23(3), 327–336. [Google Scholar] [CrossRef]
- Uemura, N.; Yagi, H.; Uemura, M. T.; Yamakado, H.; Takahashi, R. Limited spread of pathology within the brainstem of α-synuclein BAC transgenic mice inoculated with preformed fibrils into the gastrointestinal tract. Neuroscience Letters 2019, 716, 134651. [Google Scholar] [CrossRef]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R. W.; Djurhuus, J. C.; Pedersen, L.; Borghammer, P.; Sørensen, H. T. Vagotomy and subsequent risk of Parkinson’s disease. Annals of Neurology 2015, 78(4), 522–529. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, F.; Pedersen, N. L.; Tillander, A.; Ludvigsson, J. F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease. Neurology 2017, 88(21), 1996–2002. [Google Scholar] [CrossRef]
- Beach, T. G.; Adler, C. H.; Sue, L. I.; Vedders, L.; Lue, L.; White, C. L., III; Akiyama, H.; Caviness, J. N.; Shill, H. A.; Sabbagh, M. N.; Walker, D. G. Multi-organ distribution of phosphorylated α-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathologica 2010, 119(6), 689–702. [Google Scholar] [CrossRef] [PubMed]
- Tanei, Z.-I.; Saito, Y.; Ito, S.; Matsubara, T.; Motoda, A.; Yamazaki, M.; Sakashita, Y.; Kawakami, I.; Ikemura, M.; Tanaka, S.; Sengoku, R.; Arai, T.; Murayama, S. Lewy pathology of the esophagus correlates with the progression of Lewy body disease: a Japanese cohort study of autopsy cases. Acta Neuropathologica 2020, 141(1), 25–37. [Google Scholar] [CrossRef]
- Annerino, D. M.; Arshad, S.; Taylor, G. M.; Adler, C. H.; Beach, T. G.; Greene, J. G. Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathologica 2012, 124(5), 665–680. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, B.; Englund, E. Atrophic myenteric and submucosal neurons are observed in Parkinson’s disease. Parkinson S Disease 2019, 2019, 1–5. [Google Scholar] [CrossRef]
- Lebouvier, T.; Chaumette, T.; Damier, P.; Coron, E.; Touchefeu, Y.; Vrignaud, S.; Naveilhan, P.; Galmiche, J.; Varannes, S. B. D.; Derkinderen, P.; Neunlist, M. Pathological lesions in colonic biopsies during Parkinson’s disease. Gut 2008, 57(12), 1741–1743. [Google Scholar] [CrossRef]
- Lebouvier, T.; Neunlist, M.; Varannes, S. B. D.; Coron, E.; Drouard, A.; N’Guyen, J.-M.; Chaumette, T.; Tasselli, M.; Paillusson, S.; Flamand, M.; Galmiche, J.-P.; Damier, P.; Derkinderen, P. Colonic Biopsies to Assess the Neuropathology of Parkinson’s Disease and Its Relationship with Symptoms. PLoS ONE 2010, 5(9), e12728. [Google Scholar] [CrossRef]
- Stokholm, M. G.; Danielsen, E. H.; Hamilton-Dutoit, S. J.; Borghammer, P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Annals of Neurology 2016, 79(6), 940–949. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A. J.; Luth, E. S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of A-Synuclein and Β-Synuclein in neurons and non-neural cells. Journal of Biological Chemistry 2013, 288(9), 6371–6385. [Google Scholar] [CrossRef] [PubMed]
- Corbillé, A.; Neunlist, M.; Derkinderen, P. Cross-linking for the analysis of α-synuclein in the enteric nervous system. Journal of Neurochemistry 2016, 139(5), 839–847. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R. J.; Walter, G. C.; Wilder, S. L.; Baronowsky, E. A.; Powley, T. L. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: Autonomic pathway implicated in Parkinson’s disease? Neuroscience 2008, 153(3), 733–750. [Google Scholar] [CrossRef]
- Alzghool, O. M.; Van Dongen, G.; Van De Giessen, E.; Schoonmade, L.; Beaino, W. A-Synuclein Radiotracer development and in vivo imaging: recent advancements and new perspectives. Movement Disorders 2022, 37(5), 936–948. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xiang, J.; Ye, K.; Zhang, Z. Development of Positron Emission Tomography Radiotracers for Imaging α-Synuclein Aggregates. Cells 2025, 14(12), 907. [Google Scholar] [CrossRef] [PubMed]
- Aldecoa, I.; Navarro-Otano, J.; Stefanova, N.; Sprenger, F. S.; Seppi, K.; Poewe, W.; Cuatrecasas, M.; Valldeoriola, F.; Gelpi, E.; Tolosa, E. Alpha-synuclein immunoreactivity patterns in the enteric nervous system. Neuroscience Letters 2015, 602, 145–149. [Google Scholar] [CrossRef]
- Lerner, A. The intestinal luminal sources of α-synuclein: a gastroenterologist perspective. Nutrition Reviews 2021, 80(2), 282–293. [Google Scholar] [CrossRef]
- Uzuegbunam, B. C.; Li, J.; Paslawski, W.; Weber, W.; Svenningsson, P.; Ågren, H.; Yousefi, B. H. Toward novel [18F]Fluorine-Labeled radiotracers for the imaging of A-Synuclein fibrils. Frontiers in Aging Neuroscience 2022, 14, 830704. [Google Scholar] [CrossRef]
- Goto, R.; Matsuoka, K.; Kimura, Y.; Kataoka, Y.; Oya, M.; Hirata, K.; Tagai, K.; Takahata, K.; Seki, C.; Kawamura, K.; Zhang, M.-R.; Higuchi, M.; Endo, H. Human biodistribution and radiation dosimetry of two novel α-synuclein PET tracers, 18F-SPAL-T-06 and 18F-C05-05. Scientific Reports 2025, 15(1), 8640. [Google Scholar] [CrossRef]
- Forsyth, C. B.; Shannon, K. M.; Kordower, J. H.; Voigt, R. M.; Shaikh, M.; Jaglin, J. A.; Estes, J. D.; Dodiya, H. B.; Keshavarzian, A. Increased Intestinal Permeability Correlates with Sigmoid Mucosa alpha-Synuclein Staining and Endotoxin Exposure Markers in Early Parkinson’s Disease. PLoS ONE 2011, 6(12), e28032. [Google Scholar] [CrossRef]
- Mulak, A. Brain-gut-microbiota axis in Parkinson’s disease. World Journal of Gastroenterology 2015, 21(37), 10609. [Google Scholar] [CrossRef]
- Rietdijk, C. D.; Perez-Pardo, P.; Garssen, J.; Van Wezel, R. J. A.; Kraneveld, A. D. Exploring Braak’s hypothesis of Parkinson’s disease. Frontiers in Neurology 2017, 8, 37. [Google Scholar] [CrossRef]
- Kelly, L. P.; Carvey, P. M.; Keshavarzian, A.; Shannon, K. M.; Shaikh, M.; Bakay, R. a. E.; Kordower, J. H. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Movement Disorders 2013, 29(8), 999–1009. [Google Scholar] [CrossRef]
- Olanow, C. W.; Wakeman, D. R.; Kordower, J. H. Peripheral alpha-synuclein and Parkinson’s disease. Movement Disorders 2014, 29(8), 963–966. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Kliest, T.; Dodiya, H. B.; Broersen, L. M.; Garssen, J.; Keshavarzian, A.; Kraneveld, A. D. The gut-brain axis in Parkinson’s disease: Possibilities for food-based therapies. European Journal of Pharmacology 2017, 817, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H. B.; Engen, P. A.; Forsyth, C. B.; Huschens, A. M.; Shaikh, M.; Voigt, R. M.; Naqib, A.; Green, S. J.; Kordower, J. H.; Shannon, K. M.; Garssen, J.; Kraneveld, A. D.; Keshavarzian, A. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut 2018, 68(5), 829–843. [Google Scholar] [CrossRef] [PubMed]
- Goyal, D.; Ali, S. A.; Singh, R. K. Emerging role of gut microbiota in modulation of neuroinflammation and neurodegeneration with emphasis on Alzheimer’s disease. Progress in Neuro-Psychopharmacology and Biological Psychiatry 2020, 106, 110112. [Google Scholar] [CrossRef] [PubMed]
- Kurita, N.; Yamashiro, K.; Kuroki, T.; Tanaka, R.; Urabe, T.; Ueno, Y.; Miyamoto, N.; Takanashi, M.; Shimura, H.; Inaba, T.; Yamashiro, Y.; Nomoto, K.; Matsumoto, S.; Takahashi, T.; Tsuji, H.; Asahara, T.; Hattori, N. Metabolic endotoxemia promotes neuroinflammation after focal cerebral ischemia. Journal of Cerebral Blood Flow & Metabolism 2020, 40(12), 2505–2520. [Google Scholar] [CrossRef]
- Geng, S.; Yang, L.; Cheng, F.; Zhang, Z.; Li, J.; Liu, W.; Li, Y.; Chen, Y.; Bao, Y.; Chen, L.; Fei, Z.; Li, X.; Hou, J.; Lin, Y.; Liu, Z.; Zhang, S.; Wang, H.; Zhang, Q.; Wang, H.; Wang, X.; Zhang, J. Gut microbiota are associated with psychological Stress-Induced defections in intestinal and Blood–Brain barriers. Frontiers in Microbiology 2020, 10, 3067. [Google Scholar] [CrossRef]
- Ho, M. S. Microglia in Parkinson’s disease. Advances in Experimental Medicine and Biology 2019, 1175, 335–353. [Google Scholar] [CrossRef]
- Brown, G. C. The Endotoxin Hypothesis of Neurodegeneration 2019, Vol. 16, 180. [CrossRef]
- Bodea, L. -g.; Wang, Y.; Linnartz-Gerlach, B.; Kopatz, J.; Sinkkonen, L.; Musgrove, R.; Kaoma, T.; Muller, A.; Vallar, L.; Di Monte, D. A.; Balling, R.; Neumann, H. Neurodegeneration by activation of the Microglial Complement-Phagosome pathway. Journal of Neuroscience 2014, 34(25), 8546–8556. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Brundin, P. Immunotherapy in Parkinson’s Disease: Micromanaging Alpha-Synuclein aggregation. Journal of Parkinson S Disease 2015, 5(3), 413–424. [Google Scholar] [CrossRef] [PubMed]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Fernandez-Montesinos, R.; Caro, M.; Lachaud, C. C.; Waudby, C. A.; Delgado, M.; Dobson, C. M.; Pozo, D. Glial Innate Immunity Generated by Non-Aggregated Alpha-Synuclein in Mouse: Differences between Wild-type and Parkinson’s Disease-Linked Mutants. PLoS ONE 2010, 5(10), e13481. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G. K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2012, 61(3), 349–360. [Google Scholar] [CrossRef]
- Harms, A. S.; Delic, V.; Thome, A. D.; Bryant, N.; Liu, Z.; Chandra, S.; Jurkuvenaite, A.; West, A. B. α-Synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathologica Communications 2017, 5(1), 85. [Google Scholar] [CrossRef]
- Croisier, E.; Moran, L. B.; Dexter, D. T.; Pearce, R. K.; Graeber, M. B. Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. Journal of Neuroinflammation 2005, 2(1), 14. [Google Scholar] [CrossRef]
- Williams, G. P.; Marmion, D. J.; Schonhoff, A. M.; Jurkuvenaite, A.; Won, W.-J.; Standaert, D. G.; Kordower, J. H.; Harms, A. S. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathologica 2020, 139(5), 855–874. [Google Scholar] [CrossRef]
- Whitton, P. S. Inflammation as a causative factor in the aetiology of Parkinson’s disease. British Journal of Pharmacology 2007, 150(8), 963–976. [Google Scholar] [CrossRef]
- Joers, V.; Tansey, M. G.; Mulas, G.; Carta, A. R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Progress in Neurobiology 2016, 155, 57–75. [Google Scholar] [CrossRef]
- Politis, M. Imaging of microglia in patients with neurodegenerative disorders. Frontiers in Pharmacology 2012, 3, 96. [Google Scholar] [CrossRef] [PubMed]
- Choi, J. G.; Kim, N.; Ju, I. G.; Eo, H.; Lim, S.-M.; Jang, S.-E.; Kim, D.-H.; Oh, M. S. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Scientific Reports 2018, 8(1), 1275. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-Y.; Zhang, J.; Bing, G. Aging enhances the neuroinflammatory response and α-synuclein nitration in rats. Neurobiology of Aging 2010, 31(9), 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, D.; Mohite, G. M.; Krishnamoorthy, J.; Gayen, N.; Mehra, S.; Navalkar, A.; Kotler, S. A.; Ratha, B. N.; Ghosh, A.; Kumar, R.; Garai, K.; Mandal, A. K.; Maji, S. K.; Bhunia, A. Lipopolysaccharide from Gut Microbiota Modulates α-Synuclein Aggregation and Alters Its Biological Function. ACS Chemical Neuroscience 2019, 10(5), 2229–2236. [Google Scholar] [CrossRef]
- Wang, W.; Nguyen, L. T. T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O. S.; Miller, D. W.; Datta, D.; Wu, F.; Wu, C.-X.; Landeru, A.; Wells, J. A.; Cookson, M. R.; Boxer, M. B.; Thomas, C. J.; Gai, W. P.; Ringe, D.; Petsko, G. A.; Hoang, Q. Q. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proceedings of the National Academy of Sciences 2016, 113(34), 9587–9592. [Google Scholar] [CrossRef]
- Chen, S. G.; Stribinskis, V.; Rane, M. J.; Demuth, D. R.; Gozal, E.; Roberts, A. M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; Son, F.; Jin, S.; Kerber, R.; Adame, A.; Masliah, E.; Friedland, R. P. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Scientific Reports 2016, 6(1), 34477. [Google Scholar] [CrossRef]
- Shavali, S.; Combs, C. K.; Ebadi, M. Reactive macrophages increase oxidative stress and Alpha-Synuclein nitration during death of dopaminergic neuronal cells in Co-Culture: Relevance to Parkinson’s Disease. Neurochemical Research 2006, 31(1), 85–94. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J. F.; Alonso-Juarez, M. The Link between Gut Dysbiosis and Neuroinflammation in Parkinson’s Disease. Neuroscience 2020, 432, 160–173. [Google Scholar] [CrossRef]
- Hsu, L. J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. A-Synuclein promotes mitochondrial deficit and oxidative stress. American Journal of Pathology 2000, 157(2), 401–410. [Google Scholar] [CrossRef]
- Hu, D.; Sun, X.; Liao, X.; Zhang, X.; Zarabi, S.; Schimmer, A.; Hong, Y.; Ford, C.; Luo, Y.; Qi, X. Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathologica 2019, 137(6), 939–960. [Google Scholar] [CrossRef]
- Chong, W.; Jiménez, J.; McIIvin, M.; Saito, M. A.; Kwakye, G. F. A-Synuclein enhances cadmium uptake and neurotoxicity via oxidative stress and caspase activated cell death mechanisms in a dopaminergic cell model of Parkinson’s disease. Neurotoxicity Research 2017, 32(2), 231–246. [Google Scholar] [CrossRef] [PubMed]
- Dryanovski, D. I.; Guzman, J. N.; Xie, Z.; Galteri, D. J.; Volpicelli-Daley, L. A.; Lee, V. M.-Y.; Miller, R. J.; Schumacker, P. T.; Surmeier, D. J. Calcium entry and -Synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. Journal of Neuroscience 2013, 33(24), 10154–10164. [Google Scholar] [CrossRef] [PubMed]
- Tapias, V.; Hu, X.; Luk, K. C.; Sanders, L. H.; Lee, V. M.; Greenamyre, J. T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cellular and Molecular Life Sciences 2017, 74(15), 2851–2874. [Google Scholar] [CrossRef] [PubMed]
- Malpartida, A. B.; Williamson, M.; Narendra, D. P.; Wade-Martins, R.; Ryan, B. J. Mitochondrial dysfunction and mitophagy in Parkinson’s Disease: From mechanism to therapy. Trends in Biochemical Sciences 2020, 46(4), 329–343. [Google Scholar] [CrossRef]
- Ding, H.; Xiong, Y.; Sun, J.; Chen, C.; Gao, J.; Xu, H. Asiatic acid prevents oxidative stress and apoptosis by inhibiting the translocation of A-Synuclein into mitochondria. Frontiers in Neuroscience 2018, 12, 431. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M. A. The epidemiology of Parkinson’s disease: risk factors and prevention. The Lancet Neurology 2016, 15(12), 1257–1272. [Google Scholar] [CrossRef]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: current progress and future prospects. Acta Neurologica Scandinavica 2016, 134(5), 314–326. [Google Scholar] [CrossRef]
- Gao, H.-M.; Zhang, F.; Zhou, H.; Kam, W.; Wilson, B.; Hong, J.-S. Neuroinflammation and A-Synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environmental Health Perspectives 2011, 119(6), 807–814. [Google Scholar] [CrossRef]
- Flint, H. J. The impact of nutrition on the human microbiome. Nutrition Reviews 2012, 70, S10–S13. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K. S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; Mende, D. R.; Li, J.; Xu, J.; Li, S.; Li, D.; Cao, J.; Wang, B.; Liang, H.; Zheng, H.; Xie, Y.; Tap, J.; Lepage, P.; Bertalan, M.; Batto, J.-M.; Hansen, T.; Paslier, D. L.; Linneberg, A.; Nielsen, H. B.; Pelletier, E.; Renault, P.; Sicheritz-Ponten, T.; Turner, K.; Zhu, H.; Yu, C.; Li, S.; Jian, M.; Zhou, Y.; Li, Y.; Zhang, X.; Li, S.; Qin, N.; Yang, H.; Wang, J.; Brunak, S.; Doré, J.; Guarner, F.; Kristiansen, K.; Pedersen, O.; Parkhill, J.; Weissenbach, J.; Bork, P.; Ehrlich, S. D.; Wang, J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464(7285), 59–65. [Google Scholar] [CrossRef]
- Spielman, L. J.; Gibson, D. L.; Klegeris, A. Unhealthy gut, unhealthy brain: The role of the intestinal microbiota in neurodegenerative diseases. Neurochemistry International 2018, 120, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Fülling, C.; Dinan, T. G.; Cryan, J. F. Gut Microbe to Brain Signaling: What Happens in Vagus…. Neuron 2019, 101(6), 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Möhle, L.; Mattei, D.; Heimesaat, M. M.; Bereswill, S.; Fischer, A.; Alutis, M.; French, T.; Hambardzumyan, D.; Matzinger, P.; Dunay, I. R.; Wolf, S. A. Ly6Chi Monocytes Provide a Link between Antibiotic-Induced Changes in Gut Microbiota and Adult Hippocampal Neurogenesis. Cell Reports 2016, 15(9), 1945–1956. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L. G.; Kundu, P.; Gulyás, B.; Halldin, C.; Hultenby, K.; Nilsson, H.; Hebert, H.; Volpe, B. T.; Diamond, B.; Pettersson, S. The gut microbiota influences blood-brain barrier permeability in mice. Science Translational Medicine 2014, 6(263), 263ra158. [Google Scholar] [CrossRef]
- Lin, X.; Liu, Y.; Ma, L.; Ma, X.; Shen, L.; Ma, X.; Chen, Z.; Chen, H.; Li, D.; Su, Z.; Chen, X. Constipation induced gut microbiota dysbiosis exacerbates experimental autoimmune encephalomyelitis in C57BL/6 mice. Journal of Translational Medicine 2021, 19(1), 317. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; D’Antongiovanni, V.; Antonioli, L.; Bernardini, N.; Derkinderen, P. The intestinal barrier in disorders of the central nervous system. the Lancet. Gastroenterology & Hepatology 2022, 8(1), 66–80. [Google Scholar] [CrossRef]
- Welcome, M. O. Gut microbiota Disorder, gut epithelial and Blood–Brain barrier dysfunctions in etiopathogenesis of dementia: molecular mechanisms and signaling pathways. NeuroMolecular Medicine 2019, 21(3), 205–226. [Google Scholar] [CrossRef]
- Martens, E. C.; Neumann, M.; Desai, M. S. Interactions of commensal and pathogenic microorganisms with the intestinal mucosal barrier. Nature Reviews Microbiology 2018, 16(8), 457–470. [Google Scholar] [CrossRef]
- Wang, Y.; An, Y.; Ma, W.; Yu, H.; Lu, Y.; Zhang, X.; Wang, Y.; Liu, W.; Wang, T.; Xiao, R. 27-Hydroxycholesterol contributes to cognitive deficits in APP/PS1 transgenic mice through microbiota dysbiosis and intestinal barrier dysfunction. Journal of Neuroinflammation 2020, 17(1), 199. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S. J.; Engen, P. A.; Voigt, R. M.; Naqib, A.; Forsyth, C. B.; Mutlu, E.; Shannon, K. M. Colonic bacterial composition in Parkinson’s disease. Movement Disorders 2015, 30(10), 1351–1360. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P. a. B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; Kinnunen, E.; Murros, K.; Auvinen, P. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Movement Disorders 2014, 30(3), 350–358. [Google Scholar] [CrossRef]
- Unger, M. M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.-H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism & Related Disorders 2016, 32, 66–72. [Google Scholar] [CrossRef]
- Hill-Burns, E. M.; Debelius, J. W.; Morton, J. T.; Wissemann, W. T.; Lewis, M. R.; Wallen, Z. D.; Peddada, S. D.; Factor, S. A.; Molho, E.; Zabetian, C. P.; Knight, R.; Payami, H. Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Movement Disorders 2017, 32(5), 739–749. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Science China Life Sciences 2017, 60(11), 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V. A.; Saltykova, I. V.; Zhukova, I. A.; Alifirova, V. M.; Zhukova, N. G.; Dorofeeva, Yu. B.; Tyakht, A. V.; Kovarsky, B. A.; Alekseev, D. G.; Kostryukova, E. S.; Mironova, Yu. S.; Izhboldina, O. P.; Nikitina, M. A.; Perevozchikova, T. V.; Fait, E. A.; Babenko, V. V.; Vakhitova, M. T.; Govorun, V. M.; Sazonov, A. E. Analysis of Gut Microbiota in Patients with Parkinson’s Disease. Bulletin of Experimental Biology and Medicine 2017, 162(6), 734–737. [Google Scholar] [CrossRef]
- Bedarf, J. R.; Hildebrand, F.; Coelho, L. P.; Sunagawa, S.; Bahram, M.; Goeser, F.; Bork, P.; Wüllner, U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Medicine 2017, 9(1), 61. [Google Scholar] [CrossRef]
- Lin, A.; Zheng, W.; He, Y.; Tang, W.; Wei, X.; He, R.; Huang, W.; Su, Y.; Huang, Y.; Zhou, H.; Xie, H. Gut microbiota in patients with Parkinson’s disease in southern China. Parkinsonism & Related Disorders 2018, 53, 82–88. [Google Scholar] [CrossRef]
- Li, F.; Wang, P.; Chen, Z.; Sui, X.; Xie, X.; Zhang, J. Alteration of the fecal microbiota in North-Eastern Han Chinese population with sporadic Parkinson’s disease. Neuroscience Letters 2019, 707, 134297. [Google Scholar] [CrossRef]
- Pietrucci, D.; Cerroni, R.; Unida, V.; Farcomeni, A.; Pierantozzi, M.; Mercuri, N. B.; Biocca, S.; Stefani, A.; Desideri, A. Dysbiosis of gut microbiota in a selected population of Parkinson’s patients. Parkinsonism & Related Disorders 2019, 65, 124–130. [Google Scholar] [CrossRef]
- Cirstea, M. S.; Yu, A. C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L. H.; Mackenzie, M.; Huan, T.; Finlay, B. B.; Appel-Cresswell, S. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Movement Disorders 2020, 35(7), 1208–1217. [Google Scholar] [CrossRef]
- Cosma-Grigorov, A.; Meixner, H.; Mrochen, A.; Wirtz, S.; Winkler, J.; Marxreiter, F. Changes in gastrointestinal microbiome composition in PD: a pivotal role of covariates. Frontiers in Neurology 2020, 11, 1041. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P.; Perra, D.; Madau, V.; Sarchioto, M.; Oppo, V.; Simola, N.; Morelli, M.; Santoru, M. L.; Atzori, L.; Melis, M.; Cossu, G.; Manzin, A. Gut Microbiota and Metabolome Alterations Associated with Parkinson’s Disease. mSystems 2020, 5(5). [Google Scholar] [CrossRef] [PubMed]
- Aho, V. T. E.; Houser, M. C.; Pereira, P. a. B.; Chang, J.; Rudi, K.; Paulin, L.; Hertzberg, V.; Auvinen, P.; Tansey, M. G.; Scheperjans, F. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Molecular Neurodegeneration 2021, 16(1), 6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yue, L.; Fang, X.; Wang, G.; Li, C.; Sun, X.; Jia, X.; Yang, J.; Song, J.; Zhang, Y.; Guo, C.; Ma, G.; Sang, M.; Chen, F.; Wang, P. Altered gut microbiota in Parkinson’s disease patients/healthy spouses and its association with clinical features. Parkinsonism & Related Disorders 2020, 81, 84–88. [Google Scholar] [CrossRef]
- Qian, Y.; Yang, X.; Xu, S.; Huang, P.; Li, B.; Du, J.; He, Y.; Su, B.; Xu, L.-M.; Wang, L.; Huang, R.; Chen, S.; Xiao, Q. Gut metagenomics-derived genes as potential biomarkers of Parkinson’s disease. Brain 2020, 143(8), 2474–2489. [Google Scholar] [CrossRef]
- Zhang, K.; Paul, K. C.; Jacobs, J. P.; Chou, H.-C.; Folle, A. D.; Del Rosario, I.; Yu, Y.; Bronstein, J. M.; Keener, A. M.; Ritz, B. Parkinson’s Disease and the gut microbiome in rural California. Journal of Parkinson S Disease 2022, 12(8), 2441–2452. [Google Scholar] [CrossRef]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: between good and evil. Gut 2016, 65(11), 1906–1915. [Google Scholar] [CrossRef]
- Fernández, J.; Redondo-Blanco, S.; Gutiérrez-Del-Río, I.; Miguélez, E. M.; Villar, C. J.; Lombó, F. Colon microbiota fermentation of dietary prebiotics towards short-chain fatty acids and their roles as anti-inflammatory and antitumour agents: A review. Journal of Functional Foods 2016, 25, 511–522. [Google Scholar] [CrossRef]
- Flint, H. J.; Duncan, S. H.; Scott, K. P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proceedings of the Nutrition Society 2014, 74(1), 13–22. [Google Scholar] [CrossRef]
- Chen, T.; Long, W.; Zhang, C.; Liu, S.; Zhao, L.; Hamaker, B. R. Fiber-utilizing capacity varies in Prevotella- versus Bacteroides-dominated gut microbiota. Scientific Reports 2017, 7(1), 2594. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.-R.; Green, R. S.; Holzmanr, I. R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-Activated protein kinase in CACO-2 cell monolayers. Journal of Nutrition 2009, 139(9), 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Plöger, S.; Stumpff, F.; Penner, G. B.; Schulzke, J.; Gäbel, G.; Martens, H.; Shen, Z.; Günzel, D.; Aschenbach, J. R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Annals of the New York Academy of Sciences 2012, 1258(1), 52–59. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S. C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.-D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J. M. Intestinal permeability – a new target for disease prevention and therapy. BMC Gastroenterology 2014, 14(1), 189. [Google Scholar] [CrossRef] [PubMed]
- Al-Asmakh, M.; Hedin, L. Microbiota and the control of blood-tissue barriers. Tissue Barriers 2015, 3(3), e1039691. [Google Scholar] [CrossRef]
- Caspani, G.; Swann, J. Small talk: microbial metabolites involved in the signaling from microbiota to brain. Current Opinion in Pharmacology 2019, 48, 99–106. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-Chain fatty acids as key bacterial metabolites. Cell 2016, 165(6), 1332–1345. [Google Scholar] [CrossRef]
- Keogh, C. E.; Kim, D. H. J.; Pusceddu, M. M.; Knotts, T. A.; Rabasa, G.; Sladek, J. A.; Hsieh, M. T.; Honeycutt, M.; Brust-Mascher, I.; Barboza, M.; Gareau, M. G. Myelin as a regulator of development of the microbiota-gut-brain axis. Brain Behavior and Immunity 2020, 91, 437–450. [Google Scholar] [CrossRef]
- Erny, D.; De Angelis, A. L. H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; Schwierzeck, V.; Utermöhlen, O.; Chun, E.; Garrett, W. S.; McCoy, K. D.; Diefenbach, A.; Staeheli, P.; Stecher, B.; Amit, I.; Prinz, M. Host microbiota constantly control maturation and function of microglia in the CNS. Nature Neuroscience 2015, 18(7), 965–977. [Google Scholar] [CrossRef]
- Heiss, C. N.; Olofsson, L. E. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. Journal of Neuroendocrinology 2019, 31(5), e12684. [Google Scholar] [CrossRef]
- Saikachain, N.; Sungkaworn, T.; Muanprasat, C.; Asavapanumas, N. Neuroprotective effect of short-chain fatty acids against oxidative stress-induced SH-SY5Y injury via GPR43-dependent pathway. Journal of Neurochemistry 2023, 166(2), 201–214. [Google Scholar] [CrossRef]
- Wu, X.; Chen, P. S.; Dallas, S.; Wilson, B.; Block, M. L.; Wang, C.-C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; Chuang, D.-M.; Zhang, W.; Lu, R. B.; Hong, J.-S. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. The International Journal of Neuropsychopharmacology 2008, 11(08), 1123. [Google Scholar] [CrossRef]
- Kidd, S. K.; Schneider, J. S. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Research 2010, 1354, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Canani, R. B. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World Journal of Gastroenterology 2011, 17(12), 1519. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Vaughan, E. E.; Plugge, C. M.; De Vos, W. M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. INTERNATIONAL JOURNAL OF SYSTEMATIC AND EVOLUTIONARY MICROBIOLOGY 2004, 54(5), 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Neto, D. P. A.; Bosque, B. P.; De Godoy, J. V. P.; Rodrigues, P. V.; Meneses, D. D.; Tostes, K.; Tonoli, C. C. C.; De Carvalho, H. F.; González-Billault, C.; De Castro Fonseca, M. Akkermansia muciniphila induces mitochondrial calcium overload and α -synuclein aggregation in an enteroendocrine cell line. iScience 2022, 25(3), 103908. [Google Scholar] [CrossRef]
- Lorente-Picón, M.; Laguna, A. New avenues for Parkinson’s Disease therapeutics: Disease-Modifying Strategies based on the gut Microbiota. Biomolecules 2021, 11(3), 433. [Google Scholar] [CrossRef]
- Zhao, Z.; Ning, J.; Bao, X.-Q.; Shang, M.; Ma, J.; Li, G.; Zhang, D. Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome 2021, 9(1), 226. [Google Scholar] [CrossRef]
- Eslami, M.; Adampour, Z.; Dowlat, B. F.; Yaghmayee, S.; Tabaei, F. M.; Oksenych, V.; Naderian, R. A novel frontier in Gut–Brain axis Research: The transplantation of fecal microbiota in neurodegenerative Disorders. Biomedicines 2025, 13(4), 915. [Google Scholar] [CrossRef]
- Vongsavath, T.; Laeeq, T.; Tun, K. M.; Hong, A. S. The Potential Role of fecal microbiota transplantation in Parkinson’s Disease: A systematic literature review. Applied Microbiology 2023, 3(3), 993–1002. [Google Scholar] [CrossRef]
- Sun, M.-F.; Zhu, Y.-L.; Zhou, Z.-L.; Jia, X.-B.; Xu, Y.-D.; Yang, Q.; Cui, C.; Shen, Y.-Q. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behavior and Immunity 2018, 70, 48–60. [Google Scholar] [CrossRef]
- Sampson, T. R.; Debelius, J. W.; Thron, T.; Janssen, S.; Shastri, G. G.; Ilhan, Z. E.; Challis, C.; Schretter, C. E.; Rocha, S.; Gradinaru, V.; Chesselet, M.-F.; Keshavarzian, A.; Shannon, K. M.; Krajmalnik-Brown, R.; Wittung-Stafshede, P.; Knight, R.; Mazmanian, S. K. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167(6), 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tan, G.; Zhu, Q.; Wang, C.; Ruan, G.; Ying, S.; Qie, J.; Hu, X.; Xiao, Z.; Xu, F.; Chen, L.; Chen, M.; Pei, Y.; Zhang, H.; Tian, Y.; Chen, D.; Liu, X.; Huang, H.; Wei, Y. Efficacy of fecal microbiota transplantation in patients with Parkinson’s disease: clinical trial results from a randomized, placebo-controlled design. Gut Microbes 2023, 15(2), 2284247. [Google Scholar] [CrossRef] [PubMed]
- DuPont, H. L.; Suescun, J.; Jiang, Z.-D.; Brown, E. L.; Essigmann, H. T.; Alexander, A. S.; DuPont, A. W.; Iqbal, T.; Utay, N. S.; Newmark, M.; Schiess, M. C. Fecal microbiota transplantation in Parkinson’s disease—A randomized repeat-dose, placebo-controlled clinical pilot study. Frontiers in Neurology 2023, 14, 1104759. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, A.; Vandendriessche, C.; Hamerlinck, H.; De Looze, D.; Tate, D. J.; Vuylsteke, M.; De Commer, L.; Devolder, L.; Raes, J.; Verhasselt, B.; Laukens, D.; Vandenbroucke, R. E.; Santens, P. Safety and efficacy of faecal microbiota transplantation in patients with mild to moderate Parkinson’s disease (GUT-PARFECT): a double-blind, placebo-controlled, randomised, phase 2 trial. EClinicalMedicine 2024, 71, 102563. [Google Scholar] [CrossRef]
- Scheperjans, F.; Levo, R.; Bosch, B.; Lääperi, M.; Pereira, P. a. B.; Smolander, O.-P.; Aho, V. T. E.; Vetkas, N.; Toivio, L.; Kainulainen, V.; Fedorova, T. D.; Lahtinen, P.; Ortiz, R.; Kaasinen, V.; Satokari, R.; Arkkila, P. Fecal microbiota transplantation for treatment of Parkinson disease. JAMA Neurology 2024, 81(9), 925–938. [Google Scholar] [CrossRef]
- Wang, J.; Xue, L.; Zhang, M.; Shen, P.; Zhao, W.; Tong, Q.; Wu, S.; Dai, W.; Yang, X.; Wang, H. Colonoscopic fecal microbiota transplantation for Mild-to-Moderate Parkinson’s Disease: A randomized controlled trial. Brain Behavior and Immunity 2025, 130, 106086. [Google Scholar] [CrossRef]
- Kuai, X.-Y.; Yao, X.-H.; Xu, L.-J.; Zhou, Y.-Q.; Zhang, L.-P.; Liu, Y.; Pei, S.-F.; Zhou, C.-L. Evaluation of fecal microbiota transplantation in Parkinson’s disease patients with constipation. Microbial Cell Factories 2021, 20(1), 98. [Google Scholar] [CrossRef]
- Xue, L.-J.; Yang, X.-Z.; Tong, Q.; Shen, P.; Ma, S.-J.; Wu, S.-N.; Zheng, J.-L.; Wang, H.-G. Fecal microbiota transplantation therapy for Parkinson’s disease. Medicine 2020, 99(35), e22035. [Google Scholar] [CrossRef]
- Huang, H.; Xu, H.; Luo, Q.; He, J.; Li, M.; Chen, H.; Tang, W.; Nie, Y.; Zhou, Y. Fecal microbiota transplantation to treat Parkinson’s disease with constipation. Medicine 2019, 98(26), e16163. [Google Scholar] [CrossRef]
- Marcella, C.; Cui, B.; Kelly, C. R.; Ianiro, G.; Cammarota, G.; Zhang, F. Systematic review: the global incidence of faecal microbiota transplantation-related adverse events from 2000 to 2020. Alimentary Pharmacology & Therapeutics 2020, 53(1), 33–42. [Google Scholar] [CrossRef]
- Li, S. S.; Zhu, A.; Benes, V.; Costea, P. I.; Hercog, R.; Hildebrand, F.; Huerta-Cepas, J.; Nieuwdorp, M.; Salojärvi, J.; Voigt, A. Y.; Zeller, G.; Sunagawa, S.; De Vos, W. M.; Bork, P. Durable coexistence of donor and recipient strains after fecal microbiota transplantation. Science 2016, 352(6285), 586–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wu, C.; Xu, J.; Ye, C.; Chen, X.; Tian, H.; Zong, N.; Zhang, S.; Li, L.; Gao, Y.; Zhao, D.; Lv, X.; Yang, Q.; Wang, L.; Cui, J.; Lin, Z.; Lu, J.; Yang, R.; Yin, F.; Qin, N.; Li, N.; Xu, Q.; Qin, H. Donor-recipient intermicrobial interactions impact transfer of subspecies and fecal microbiota transplantation outcome. Cell Host & Microbe 2024, 32(3), 349–365.e4. [Google Scholar] [CrossRef] [PubMed]
- Barboza, J. L.; Okun, M. S.; Moshiree, B. The treatment of gastroparesis, constipation and small intestinal bacterial overgrowth syndrome in patients with Parkinson’s disease. Expert Opinion on Pharmacotherapy 2015, 16(16), 2449–2464. [Google Scholar] [CrossRef] [PubMed]
- Rao, S. S. C.; Bhagatwala, J. Small intestinal bacterial overgrowth: clinical features and therapeutic management. Clinical and Translational Gastroenterology 2019, 10(10), e00078. [Google Scholar] [CrossRef]
- Sroka, N.; Rydzewska-Rosołowska, A.; Kakareko, K.; Rosołowski, M.; Głowińska, I.; Hryszko, T. Show Me What You Have Inside—The Complex Interplay between SIBO and Multiple Medical Conditions—A Systematic Review. Nutrients 2022, 15(1), 90. [Google Scholar] [CrossRef]
- Ponziani, F. R.; Zocco, M. A.; D’Aversa, F.; Pompili, M.; Gasbarrini, A. Eubiotic properties of rifaximin: Disruption of the traditional concepts in gut microbiota modulation. World Journal of Gastroenterology 2017, 23(25), 4491. [Google Scholar] [CrossRef]
- Li, H.; Xiang, Y.; Zhu, Z.; Wang, W.; Jiang, Z.; Zhao, M.; Cheng, S.; Pan, F.; Liu, D.; Ho, R. C. M.; Ho, C. S. H. Rifaximin-mediated gut microbiota regulation modulates the function of microglia and protects against CUMS-induced depression-like behaviors in adolescent rat. Journal of Neuroinflammation 2021, 18(1), 254. [Google Scholar] [CrossRef]
- Hong, C.-T.; Chan, L.; Chen, K.-Y.; Lee, H.-H.; Huang, L.-K.; Yang, Y.-C. S. H.; Liu, Y.-R.; Hu, C.-J. Rifaximin modifies gut microbiota and attenuates inflammation in Parkinson’s Disease: Preclinical and clinical studies. Cells 2022, 11(21), 3468. [Google Scholar] [CrossRef]
- Suhocki, P. V.; Ronald, J. S.; Diehl, A. M. E.; Murdoch, D. M.; Doraiswamy, P. M. Probing gut-brain links in Alzheimer’s disease with rifaximin. Alzheimer S & Dementia Translational Research & Clinical Interventions 2022, 8(1), e12225. [Google Scholar] [CrossRef]
- Fasano, A.; Bove, F.; Gabrielli, M.; Petracca, M.; Zocco, M. A.; Ragazzoni, E.; Barbaro, F.; Piano, C.; Fortuna, S.; Tortora, A.; Di Giacopo, R.; Campanale, M.; Gigante, G.; Lauritano, E. C.; Navarra, P.; Marconi, S.; Gasbarrini, A.; Bentivoglio, A. R. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Movement Disorders 2013, 28(9), 1241–1249. [Google Scholar] [CrossRef]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G. R.; Merenstein, D. J.; Pot, B.; Morelli, L.; Canani, R. B.; Flint, H. J.; Salminen, S.; Calder, P. C.; Sanders, M. E. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nature Reviews Gastroenterology & Hepatology 2014, 11(8), 506–514. [Google Scholar] [CrossRef] [PubMed]
- Fijan, S. Microorganisms with Claimed Probiotic Properties: An Overview of Recent Literature. International Journal of Environmental Research and Public Health 2014, 11(5), 4745–4767. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J. Fiber and Prebiotics: Mechanisms and health benefits. Nutrients 2013, 5(4), 1417–1435. [Google Scholar] [CrossRef] [PubMed]
- Park, J. M.; Lee, S. C.; Ham, C.; Kim, Y. W. Effect of probiotic supplementation on gastrointestinal motility, inflammation, motor, non-motor symptoms and mental health in Parkinson’s disease: a meta-analysis of randomized controlled trials. Gut Pathogens 2023, 15(1), 9. [Google Scholar] [CrossRef]
- Markowiak-Kopeć, P.; Śliżewska, K. The effect of probiotics on the production of Short-Chain fatty acids by human intestinal microbiome. Nutrients 2020, 12(4), 1107. [Google Scholar] [CrossRef]
- Dinan, T. G.; Stanton, C.; Cryan, J. F. Psychobiotics: a novel class of psychotropic. Biological Psychiatry 2013, 74(10), 720–726. [Google Scholar] [CrossRef]
- Long-Smith, C.; O’Riordan, K. J.; Clarke, G.; Stanton, C.; Dinan, T. G.; Cryan, J. F. Microbiota-Gut-Brain Axis: new therapeutic opportunities. The Annual Review of Pharmacology and Toxicology 2019, 60(1), 477–502. [Google Scholar] [CrossRef]
- Ramadan, M. E.; Mostafa, T. M.; Ghali, A. A.; El-Afify, D. R. Randomized controlled trial evaluating synbiotic supplementation as an adjuvant therapy in the treatment of Parkinson’s disease. Inflammopharmacology 2025, 33(7), 3897–3908. [Google Scholar] [CrossRef]
- Lu, C.-S.; Chang, H.-C.; Weng, Y.-H.; Chen, C.-C.; Kuo, Y.-S.; Tsai, Y.-C. The Add-On Effect of Lactobacillus plantarum PS128 in Patients With Parkinson’s Disease: A Pilot Study. Frontiers in Nutrition 2021, 8, 650053. [Google Scholar] [CrossRef]
- Liao, J.-F.; Cheng, Y.-F.; You, S.-T.; Kuo, W.-C.; Huang, C.-W.; Chiou, J.-J.; Hsu, C.-C.; Hsieh-Li, H.-M.; Wang, S.; Tsai, Y.-C. Lactobacillus plantarum PS128 alleviates neurodegenerative progression in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse models of Parkinson’s disease. Brain Behavior and Immunity 2020, 90, 26–46. [Google Scholar] [CrossRef]
- Lee, Y. Z.; Cheng, S.-H.; Chang, M.-Y.; Lin, Y.-F.; Wu, C.-C.; Tsai, Y.-C. Neuroprotective Effects of Lactobacillus plantarum PS128 in a Mouse Model of Parkinson’s Disease: The Role of Gut Microbiota and MicroRNAs. International Journal of Molecular Sciences 2023, 24(7), 6794. [Google Scholar] [CrossRef]
- Emmi, A.; Sandre, M.; Russo, F. P.; Tombesi, G.; Garrì, F.; Campagnolo, M.; Carecchio, M.; Biundo, R.; Spolverato, G.; Macchi, V.; Savarino, E.; Farinati, F.; Parchi, P.; Porzionato, A.; Bubacco, L.; De Caro, R.; Kovacs, G. G.; Antonini, A. Duodenal alpha-Synuclein pathology and enteric gliosis in advanced Parkinson’s disease. Movement Disorders 2023, 38(5), 885–894. [Google Scholar] [CrossRef]
- Chung, S. J.; Kim, J.; Lee, H. J.; Ryu, H.; Kim, K.; Lee, J. H.; Jung, K. W.; Kim, M. J.; Kim, M.; Kim, Y. J.; Yun, S.; Lee, J.; Hong, S.; Myung, S. Alpha-synuclein in gastric and colonic mucosa in Parkinson’s disease: Limited role as a biomarker. Movement Disorders 2016, 31(2), 241–249. [Google Scholar] [CrossRef]
- Shin, C.; Park, S.-H.; Yun, J. Y.; Shin, J. H.; Yang, H.-K.; Lee, H.-J.; Kong, S.-H.; Suh, Y.-S.; Shen, G.; Kim, Y.; Kim, H.-J.; Jeon, B. Fundamental limit of alpha-synuclein pathology in gastrointestinal biopsy as a pathologic biomarker of Parkinson’s disease: Comparison with surgical specimens. Parkinsonism & Related Disorders 2017, 44, 73–78. [Google Scholar] [CrossRef]
- Ruffmann, C.; Bengoa-Vergniory, N.; Poggiolini, I.; Ritchie, D.; Hu, M. T.; Alegre-Abarrategui, J.; Parkkinen, L. Detection of alpha-synuclein conformational variants from gastro-intestinal biopsy tissue as a potential biomarker for Parkinson’s disease. Neuropathology and Applied Neurobiology 2018, 44(7), 722–736. [Google Scholar] [CrossRef]
- Visanji, N. P.; Marras, C.; Kern, D. S.; Dakheel, A. A.; Gao, A.; Liu, L. W. C.; Lang, A. E.; Hazrati, L.-N. Colonic mucosal α-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 2015, 84(6), 609–616. [Google Scholar] [CrossRef]
- Lubomski, M.; Tan, A. H.; Lim, S.-Y.; Holmes, A. J.; Davis, R. L.; Sue, C. M. Parkinson’s disease and the gastrointestinal microbiome. Journal of Neurology 2020, 267(9), 2507–2523. [Google Scholar] [CrossRef]
- Musgrove, R. E.; Helwig, M.; Bae, E.-J.; Aboutalebi, H.; Lee, S.-J.; Ulusoy, A.; Di Monte, D. A. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular α-synuclein transfer. Journal of Clinical Investigation 2019, 129(9), 3738–3753. [Google Scholar] [CrossRef]
- Li, M.; Van Esch, B. C. A. M.; Wagenaar, G. T. M.; Garssen, J.; Folkerts, G.; Henricks, P. A. J. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. European Journal of Pharmacology 2018, 831, 52–59. [Google Scholar] [CrossRef]
- Campagnolo, M.; Weis, L.; Sandre, M.; Tushevski, A.; Russo, F. P.; Savarino, E.; Carecchio, M.; Stocco, E.; Macchi, V.; Caro, R.; Parchi, P.; Bubacco, L.; Porzionato, A.; Antonini, A.; Emmi, A. Immune Landscape of the Enteric Nervous System Differentiates Parkinson’s Disease Patients from Controls: The PADUA-CESNE Cohort. Neurobiology of Disease 2024, 196, 106609. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.; Alonso-Juarez, M. The Link between Gut Dysbiosis and Neuroinflammation in Parkinson’s Disease. Neuroscience 2020, 432, 160–173. [Google Scholar] [CrossRef]
- Fitzgerald, E.; Murphy, S.; Martinson, H. Alpha-Synuclein Pathology and the Role of the Microbiota in Parkinson’s Disease. Frontiers in Neuroscience 2019, 13, 369. [Google Scholar] [CrossRef]
- Stone, D. K.; Kiyota, T.; Mosley, R.; Gendelman, H. A Model of Nitric Oxide Induced α-Synuclein Misfolding in Parkinson’s Disease. Neuroscience Letters 2012, 523, 167–171. [Google Scholar] [CrossRef]
- Chen, Z.-J.; Liang, C.-Y.; Yang, L.-Q.; Ren, S.; Xia, Y.; Cui, L.; Li, X.-F.; Gao, B. Association of Parkinson’s Disease With Microbes and Microbiological Therapy. Frontiers in Cellular and Infection Microbiology 2021, 11, 619354. [Google Scholar] [CrossRef]
- Ortiz de Ora, L.; Uyeda, K. S.; Bess, E. Discovery of a Gut Bacterial Metabolic Pathway that Drives α-Synuclein Aggregation. ACS Chemical Biology 2024, 19, 1323–1335. [Google Scholar] [CrossRef]
- Ioghen, O.; Gaina, G.; Lambrescu, I.; Manole, E.; Pop, S.; Niculescu, T. M.; Mosoia, O.; Ceafalan, L.; Popescu, B. Bacterial Products Initiation of Alpha-Synuclein Pathology: An In Vitro Study. Scientific Reports 2024, 14, 81020. [Google Scholar] [CrossRef]
- Chapman, M.; Robinson, L. S.; Pinkner, J.; Roth, R.; Heuser, J.; Hammar, M.; Normark, S.; Hultgren, S. Role of Escherichia coli Curli Operons in Directing Amyloid Fiber Formation. Science 2002, 295, 851–855. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, B.; Guo, J. Parkinson’s Disease and Gut Microbiota: From Clinical to Mechanistic and Therapeutic Studies. Translational Neurodegeneration 2023, 12, 57. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S. α-Synuclein Misfolding and Aggregation: Implications in Parkinson’s Disease Pathogenesis. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Lundmark, K.; Westermark, G.; Olsén, A.; Westermark, P. Protein Fibrils in Nature Can Enhance Amyloid Protein A Amyloidosis in Mice: Cross-Seeding as a Disease Mechanism. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 6098–6102. [Google Scholar] [CrossRef]
- Bhoite, S. S.; Han, Y.; Ruotolo, B.; Chapman, M. Mechanistic Insights into Accelerated α-Synuclein Aggregation Mediated by Human Microbiome-Associated Functional Amyloids. Journal of Biological Chemistry 2022, 298, 102088. [Google Scholar] [CrossRef]
- Kujawska, M.; Jodynis-Liebert, J. What Is the Evidence that Parkinson’s Disease Is a Prion Disorder, Which Originates in the Gut? International Journal of Molecular Sciences 2018, 19, 3573. [Google Scholar] [CrossRef]
- Chen, S.-J.; Lin, C.-H. Gut Microenvironmental Changes as a Potential Trigger in Parkinson’s Disease through the Gut–Brain Axis. Journal of Biomedical Science 2022, 29, 57. [Google Scholar] [CrossRef]
- Schaeffer, E.; Kluge, A.; Böttner, M.; Zunke, F.; Cossais, F.; Berg, D.; Arnold, P. Alpha Synuclein Connects the Gut–Brain Axis in Parkinson’s Disease Patients – A View on Clinical Aspects, Cellular Pathology and Analytical Methodology. Frontiers in Cell and Developmental Biology 2020, 8, 573696. [Google Scholar] [CrossRef]
- Singh, Y.; Trautwein, C.; Romani, J.; Salker, M.; Neckel, P.; Fraccaroli, I.; Abeditashi, M.; Woerner, N.; Admard, J.; Dhariwal, A.; Dueholm, M.; Schäfer, K.; Lang, F.; Otzen, D.; Lashuel, H.; Riess, O.; Casadei, N. Overexpression of Human Alpha-Synuclein Leads to Dysregulated Microbiome/Metabolites with Ageing in a Rat Model of Parkinson Disease. Molecular Neurodegeneration 2023, 18, 57. [Google Scholar] [CrossRef]
- Hirayama, M.; Nishiwaki, H.; Hamaguchi, T.; Ohno, K. Gastrointestinal Disorders in Parkinson’s Disease and Other Lewy Body Diseases. NPJ Parkinson’s Disease 2023, 9, 57. [Google Scholar] [CrossRef]
- Ponziani, F.; Zocco, M.; D’Aversa, F.; Pompili, M.; Gasbarrini, A. Eubiotic Properties of Rifaximin: Disruption of the Traditional Concepts in Gut Microbiota Modulation. World Journal of Gastroenterology 2017, 23, 4491–4499. [Google Scholar] [CrossRef]
- Chandra, R.; Hiniker, A.; Kuo, Y.; Nussbaum, R.; Liddle, R. α-Synuclein in Gut Endocrine Cells and Its Implications for Parkinson’s Disease. JCI Insight 2017, 2(12), e92295. [Google Scholar] [CrossRef]
- Paillusson, S.; Clairembault, T.; Biraud, M.; Neunlist, M.; Derkinderen, P. Activity-Dependent Secretion of Alpha-Synuclein by Enteric Neurons. Journal of Neurochemistry 2013, 125(4), 512–517. [Google Scholar] [CrossRef]
Figure 1.
A schematic depiction illustrating the proposed mechanisms linking the gut and brain in PD. On the left, aggregated αS accumulates in the ENS and propagates retrogradely along the vagus nerve toward the DMV and brain. In the middle panel, a healthy gut microbiome rich in SCFA-producing commensals supports the mucus layer, maintains intact epithelial tight junctions, and preserves barrier integrity. In the right panel, PD-associated gut dysbiosis with reduced SCFA-producing bacteria and increased LPS-producing and mucin-degrading taxa disrupts tight junctions, increases intestinal permeability, activates dendritic cells and macrophages via TLR4, and drives the release of pro-inflammatory cytokines, creating an environment that favors enteric αS aggregation and its vagus-mediated spread to the CNS. Some image components in this figure were taken from NIAID Visual & Medical Arts (https://bioart.niaid.nih.gov).
Figure 1.
A schematic depiction illustrating the proposed mechanisms linking the gut and brain in PD. On the left, aggregated αS accumulates in the ENS and propagates retrogradely along the vagus nerve toward the DMV and brain. In the middle panel, a healthy gut microbiome rich in SCFA-producing commensals supports the mucus layer, maintains intact epithelial tight junctions, and preserves barrier integrity. In the right panel, PD-associated gut dysbiosis with reduced SCFA-producing bacteria and increased LPS-producing and mucin-degrading taxa disrupts tight junctions, increases intestinal permeability, activates dendritic cells and macrophages via TLR4, and drives the release of pro-inflammatory cytokines, creating an environment that favors enteric αS aggregation and its vagus-mediated spread to the CNS. Some image components in this figure were taken from NIAID Visual & Medical Arts (https://bioart.niaid.nih.gov).
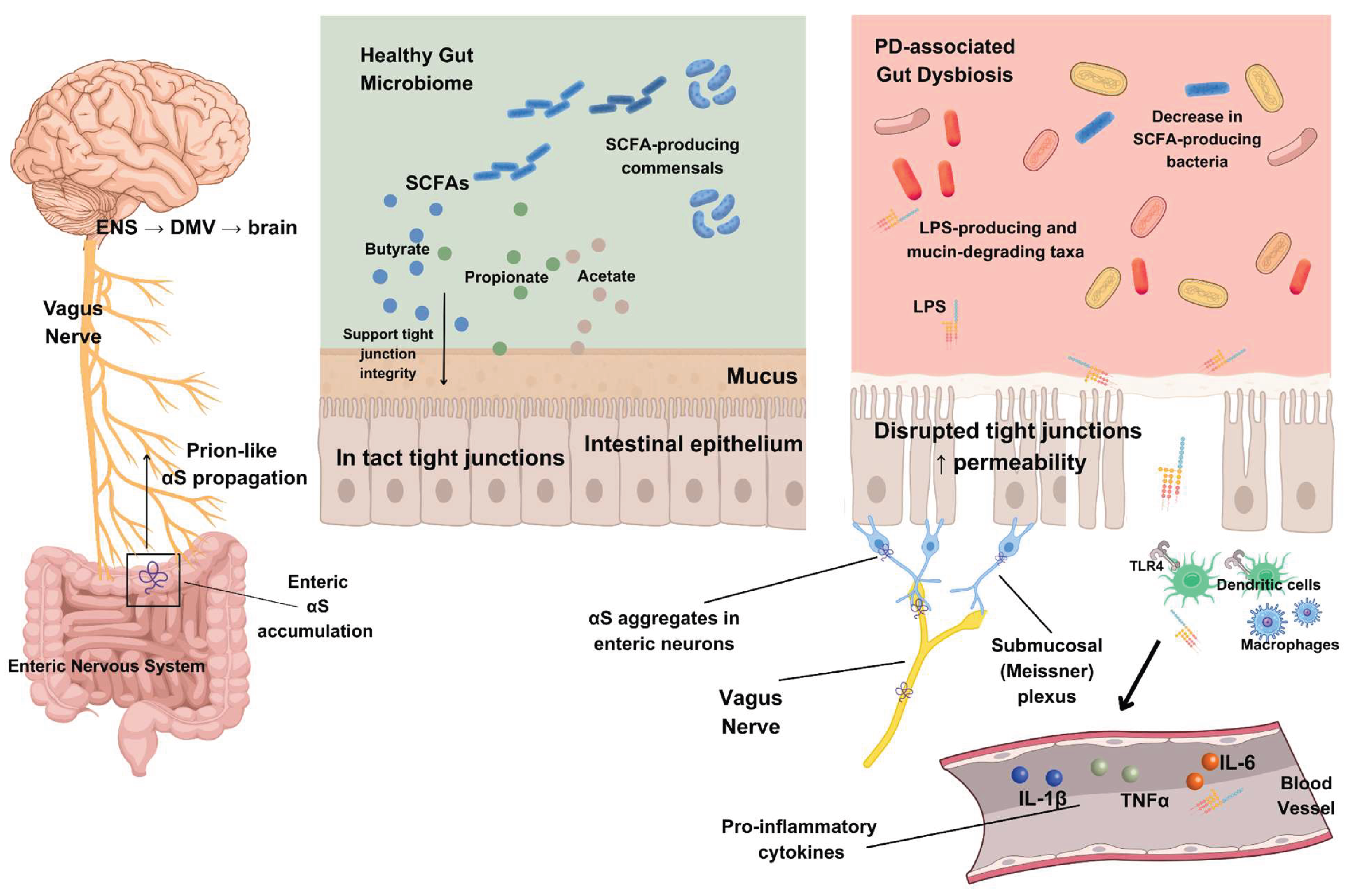

Table 1.
Summary of studies investigating alterations in gut microbiome composition in Parkinson’s disease (PD) cohorts compared to healthy controls (HC). Direction of change: ↑ = higher in PD vs HC (significant); ↓ = lower in PD vs HC (significant); ↔ = measured, not significant; NM = not measured / not reported.
Table 1.
Summary of studies investigating alterations in gut microbiome composition in Parkinson’s disease (PD) cohorts compared to healthy controls (HC). Direction of change: ↑ = higher in PD vs HC (significant); ↓ = lower in PD vs HC (significant); ↔ = measured, not significant; NM = not measured / not reported.
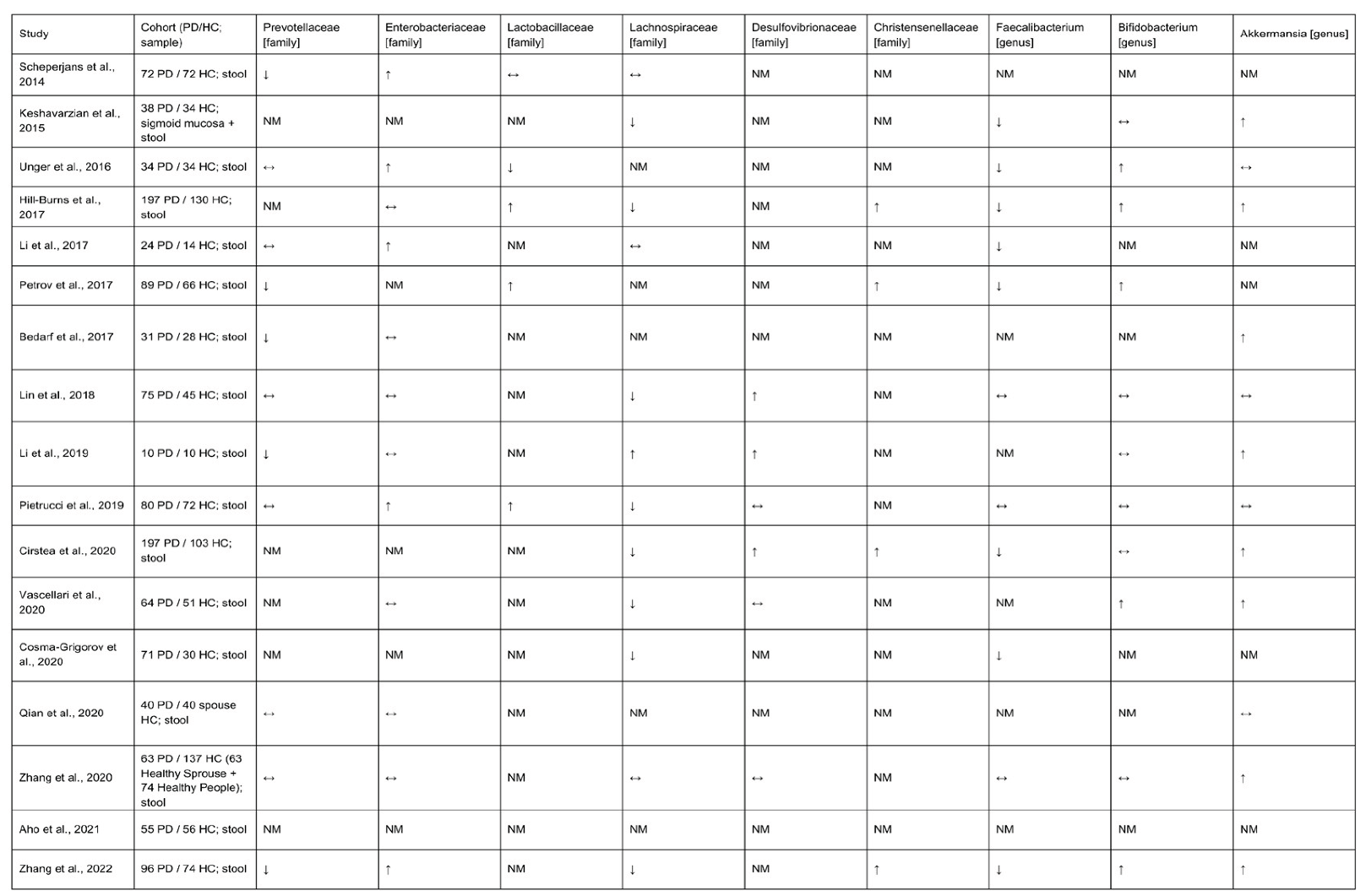 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



