
Submitted:
09 January 2026
Posted:
12 January 2026
You are already at the latest version
Abstract
Sulfur compounds are extremely toxic and highly corrosive (e.g. mercaptans and hy-drogen sulfide) and are commonly found in natural gas streams and can be damaging even if only minute amounts are present in natural gas streams because it can affect the quality of fuels and cause failure of downstream equipment. Many metal oxides have been used as adsorbent/catalyst for the removal of sulfur compounds from natural gas; however, they vary greatly in how well they can remove sulfur compounds, and the underlying mechanisms of these processes are still not fully understood. Therefore, the purpose of this study was to examine the adsorption/removal performance of many metal oxides on halloysite support at the same conditions to identify the relationship between the electronic properties (specifically bandgap energy) and breakthrough time (a measure of removal/adsorption efficiency). The experimental results indicate large differences in the adsorption performance of the studied oxides and some commercial metal oxides had lower than expected adsorption performance. Conversely, all the studied oxides with the lowest bandgap energies showed higher sulfur compound (e.g. ethyl mercaptan) uptake and longer breakthrough times indicating that the electronic properties of the oxides are important in determining the strength of interaction be-tween the sulfur compounds and the metal oxide. The experimental results from this study will provide understanding of why certain metal oxides may not perform as good as others during natural gas desulfurization and assist in developing a systematic method for selecting adsorbents/catalysts that will improve the overall natural gas desulfurization process. Furthermore, incorporating palladium oxides into the base catalyst formulation achieved a maximum breakthrough time of 630 minutes at 25°C 200 psi, and 36 mL/min. These findings provide critical insights for developing catalysts that integrate metal oxides to enhance adsorption efficiency while reducing hazardous byproducts during sulfur compounds (e.g. mercaptans and hydrogen sulfide) removal from natural gas.
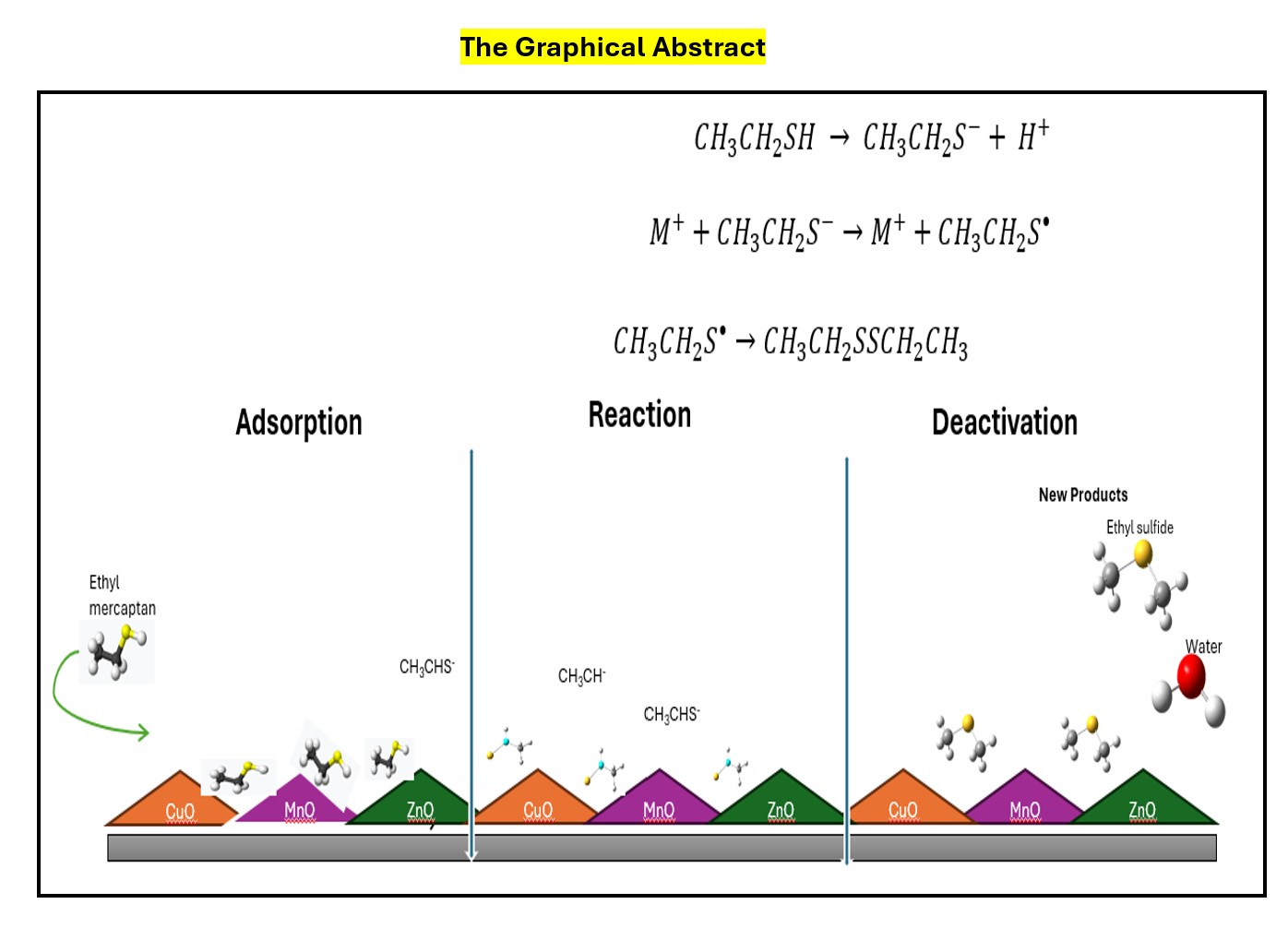
Keywords:
metal oxides
; transition-metals
; bandgap
; breakthrough time
; mercaptan
; adsorption
1. Introduction
Sulfur compounds (e.g. ethanethiol, CH₃CH₂SH) are colorless gas with a characteristic odor resembling rotten or cooked cabbage. They are common volatile organic compound found in natural gas, petroleum gas, and water gas [1]. Ethyl mercaptan has an exceptionally low odor detection threshold, around 0.002 ppm, but is highly toxic at relatively high concentrations. It can poison catalysts used in methanol and ammonia synthesis. For instance, just 4 mg of sulfur per gram of Fe-Cu-K catalyst can reduce the catalyst's activity in the Fischer-Tropsch process by approximately 50%. Consequently, it is crucial to completely remove sulfur compounds (e.g. mercaptans) from supply gas [2].
Beyond economic implications, sulfur compounds also pose environmental risks. In the troposphere and stratosphere, they undergo a series of oxidation steps that ultimately form sulfur dioxide (SO₂) and, subsequently, sulfuric acid (H₂SO₄). Once formed, sulfuric acid contributes to sulfate aerosols, which are well known to modify atmospheric radiative balance by scattering incoming solar radiation and enhancing cloud formation. These aerosols are therefore linked to regional climate forcing and acid deposition [3,4]. This effect arises after oxidation, not during the oxidation reaction itself but because of the sulfate aerosol particles produced. [5]. Over recent decades, substantial efforts have focused on the effective removal of sulfur compounds (e.g. mercaptans and hydrogen sulfide) from gas streams due to its significant impact in industrial catalysis and atmospheric chemistry [6,7,8,9,10].
Various methods have been employed to remove mercaptans from gas streams, including adsorption [11], photocatalytic oxidation [12], catalytic incineration [13], thermal decomposition (CH₃CH₂SH → Hydrocarbons + H₂S) [14], and biological degradation [15,16]. Each of these methods has unique advantages and limitations, with varying degrees of cost-effectiveness. Among these, catalytic oxidation is regarded as a highly effective approach for the complete removal of mercaptans from natural gas due to its high desulfurization efficiency, mild operating conditions, and low cost. This method also produces minimal byproducts, making it a cost-effective and environmentally favorable option [17,18].
Activated carbons are widely recognized for their outstanding capacity to remove certain acidic gases due to their porosity, large internal surface area, and surface chemistry. The use of activated carbon to eliminate mercaptans is well documented in literature [19,20,21,22]. Previous studies discovered that at room temperature, mercaptans adsorbed on activated carbon and quickly oxidize to diethyl disulfide, which stays highly adsorbed on the carbon surface [23,24]. Pore size, pore volume and surface area all have a significant impact on activated carbon adsorption capacity. Chemical and thermal treatments, as well as impregnation methods, can be applied to modify the functional groups of the carbon surface [25]. Furthermore, alkali metals (e.g., K, Na), alkaline earth metals (e.g., Mg, Ca), transition metal oxides (e.g., CuO, ZnO, NiO), and rare earth metal oxides (e.g., CeO₂, La₂O₃) have been demonstrated to be outstanding active components [26].
The sulfur compounds (e.g. ethyl mercaptan) first anchor onto the alkaline sites of the catalyst surface and then migrate by surface diffusion toward the nearby redox-active centers, where it undergoes oxidative transformation into the corresponding products [27]. Choosing an appropriate catalyst is essential for efficient removal of mercaptan. Most studies utilize two types of catalysts for treating mercaptans (e.g. CH₃CH₂SH): supported and unsupported catalysts. Supported catalysts are further classified based on the type of support material used. Oxide supports can be either non-metallic, such as activated carbon [28], or metallic, such as Al₂O₃ and TiO₂ [29]. Unsupported catalysts typically involve layered double hydroxides (LDHs) and mixed metal oxides (MMOs) [30]. Heating LDHs to a specific temperature result in MMO catalysts, which are favored for their high dispersion, large specific surface area, and synergistic interactions with different metal oxides [31,32]. The application of solid alkaline-based material catalysts has proven successful for the catalytic oxidation of sulfur-based VOCs over a variety of operational ranges [33]. Catalyst performance is significantly affected by metal composition since the selection of metal components influences how individual oxides will interact with LDH precursors and among themselves [33]. For example, transition metals like Ni and Fe have demonstrated the ability to enhance electron transport for the oxidation of sulfur-based species; this may improve both activation and adsorption of the target molecule [34]. As such, NiCuFe-LDH was synthesized through a coprecipitation route with subsequent thermal treatment at temperature ranges that have been reported to favor oxide formation while maintaining redox function [31,32,33,34].
In addition, a number of metal-oxide catalysts were prepared using an impregnation method and tested for sulfur compounds (e.g. mercaptans, hydrogen sulfur) removal. This research investigates how various metal oxides affect breakthrough behavior (measure of adsorption/removal efficiency) and specifically seeks to obtain longer breakthrough times using the same operational parameters. Structure-performance relationships were established between the catalysts and their characterization using FTIR, EDS and BET surface analysis. The data provided practical knowledge about the treatment of sulfur compounds like mercaptans and a basis for choosing and designing metal-oxide catalysts that have improved adsorption properties.
1.1. The Transition Metal Oxides
Transition metal oxides (TMOs) are being employed as catalysts due to their high levels of structural durability and high degree of chemical versatility that allows them to operate well within difficult reaction conditions. The growing use of TMOs is also a result of their ability to function well in electrochemical systems and electronic materials. More importantly, however, TMOs are becoming increasingly popular in the field of gas phase catalysis, where both structural durability and resistance to deactivation are essential. Recent experimental and mechanistic studies of reactions occurring between ethyl-mercaptan and transition metal oxide surfaces have provided considerable insight into how these reactions occur at the gas/solid interfaces of TMOs and the role that both the electronic structure of the surface and metal-sulfur interactions play [35].
Catalytic activity of TMOs can be influenced by changes in both composition and surface chemistry, which will in turn influence the amount of acidity/basicity/redox activity available on the surface [36]. The availability of acidity/basicity/redox activity on the surface will determine how sulfur containing species will initially adsorb onto the surface, be activated, and then transformed on the surface. Lewis’s acid-base interactions assist in the initial adsorption of sulfur containing species onto the surface, whereas redox active sites facilitate the electron transfer during subsequent reaction steps [37]. TMO support also provides contributions to the overall catalytic performance of TMO based catalysts by stabilizing intermediate products formed during the reaction and facilitating an exchange of oxygen with the environment which ultimately influences the overall reaction pathway and long-term stability of the catalyst. Therefore, careful management of surface chemistry is critical to achieving optimal TMO based catalysts for heterogeneous sulfur removal applications. Advances in surface science have enabled detailed investigations of TMO surface structures, compositions, and electrical properties. These insights are highly relevant to the present work because the same surface characteristics, particularly defect density, metal–support interactions, and nanoscale electronic effects governs how CH₃CH₂SH anchors, migrates, and ultimately oxidized on the catalysts [38].
TMOs are used as photocatalysts and electrocatalysts and are active in various chemical reactions, such as oxidation, selective oxidation, reduction, dehydrogenation, the water-gas shift reaction (WGS), and CO₂ hydrogenation. For example, FeO/Pt and CuO/Ag catalysts show high reactivity for aerobic oxidation of benzyl alcohol, with the metal-oxide interface acting as the active site. Mixed-valent manganese oxides exhibit redox versatility and can form structures like layered, spinel, perovskite, and bixbyite, with oxidation states ranging from 2+ to 4+ [39]. Studies have also developed MnO₂@FeOOH catalysts for formaldehyde removal in indoor air, where reactive oxygen species (ROS) generated on the catalyst surface facilitate oxidation. Superoxide anions are instrumental in this oxidation process, as shown by ROS quenching experiments in both aqueous and gaseous phases [40].
Applying an ultrathin MnO₂ coating on FeOOH reduces electron transfer resistance and increases surface oxygen vacancies, which aids in ROS production [41]. Lithium-promoted mesoporous manganese oxides, used for mild oxidation of allyl ethers, also exhibit enhanced surface activity due to lithium’s influence on radical pathways [42]. Other examples include Al₂O₃-supported vanadia (VOx) and MoO₃-Fe₂O₃ catalysts, which are effective in oxidative dehydrogenation (ODH) of alkanes such as ethane and propane to octane [43,44,45,46]. CeO₂-x/CoO₁-x/Co interfaces have shown promising WGS activity, with CeO₂-x modulating Co oxidation states, reducing Co poisoning, and enhancing H₂ production [47]. A Ni-NiOx-Y₂O₃ catalyst has also been shown to outperform pure Ni catalysts in WGS reactions, delivering high reactivity at moderate temperatures.
In electrochemical applications, molybdenum-based oxides are preferred for their versatile crystal structures and adjustable Mo oxidation states. Defect Engineering is an effective method for increasing catalytic performance. Defects are formed through the introduction of oxygen vacancies, which decrease the barrier to electron flow and increase the movement of ions through the lattice of the oxide. A good example of this defect engineered catalyst is mesoporous MoOx that was synthesized using an inverse micelle route, which has shown an increase in the activity and selectivity in electrophilic aromatic substitution involving benzyl alcohol to methyl-diphenylmethane [48]. Oxygen vacancies have a major role in maintaining and creating active sites when rapid charge redistribution occurs and repeated reaction cycles occur allowing for sustained catalytic activity [49].
1.2. Selection of TMOs for the Catalyst Preparation
Based on their properties and characteristics, researchers have focused on band-gap size, chemical reactivity and cost as key factors linked to effective desulfurization [50]. This study compared the reactivity of ethyl mercaptan on various metal oxides, including ZnO, Cu₂O, MnO, and NiO supported on halloysite, and established the following reactivity order: Al₂O₃ < NiO < ZnO < Cu₂O < MnO. Although alumina exhibited the roughest surface, it showed the lowest reactivity toward sulfur, indicating that surface defect sites alone could not account for the observed trend. In heterogeneous catalysis, the band gap: the energy difference between the valence band (filled states) and the conduction band (empty states), governs the ease with which electron transfer occurs between the adsorbate and the catalyst surface. Materials with narrower band gaps generally facilitate stronger S-M interactions because electrons can more readily participate in bond formation or redox steps. Thus, differences in metal-oxide band gaps provide a more consistent explanation for the observed reactivity sequence than surface morphology alone.
The activity of these metal oxides is further influenced by factors such as porosity, limited surface area, low resistance to sintering at elevated temperatures, and attrition. Metal oxides with narrower band gaps, such as PdO (BG 0.8 eV), CuO (BG 2.1 eV), ZnO (BG 3.3 eV), and CeO2 (BG 3.2 eV), demonstrate higher reactivity toward mercaptans, as illustrated in Figure 1. Narrower band gaps result in less stable valence bands, leading to stronger bonding interactions between the oxide and HS or S species due to the increased reactivity of a less stable oxide valence band [51].
In comparison, the metallic Cu bands provide better energy alignment with the frontier orbitals of sulfur than alumina bands [52]. Cu particles supported on alumina experience electronic perturbations; however, these perturbations are insufficient to fully bridge the energy differences observed when compared to metallic copper.
The influence of adsorption energy on the partially occupied orbitals in HS and S (denoted as ECB) and the top of the valence band energy of the metal oxide (EVB) was analyzed using perturbation theory [56,57]. This relationship is expressed by the proportionality Q∝EVB−ECB. Insights into the interactions between adsorbates and metal surfaces have led to key descriptors for binding and catalysis, significantly impacting the design of metal catalysts. The experimental results indicate that oxides with lower band-gap energies exhibit enhanced reactivity toward sulfur-containing molecules, suggesting that electronic structure plays a controlling role in adsorption behavior.
2. Results and Discussion
The observed trends were placed into context with the previously conducted research on metal-oxide desulfurization and it closely align with the metal-retention challenges identified in our samples prepared by filtration methods, which have been found to result in the same type of metal retention issues [58,59] as those reported here. Filtration methods have also been shown to result in the loss of active metals and non-uniform deposition on the support, which is expected to limit the amount of adsorption sites available and accelerate degradation of performance over time when exposed to sulfur compounds [60]. Therefore, the significance of the data collected in this study was due to the demonstration that metal retention during the sample preparation process significantly impacts both adsorption efficiency (i.e., break-through behavior) and operational stability. These findings indicate a need to develop new sample preparation techniques that do not include filtration induced losses, which will be investigated in further detail in future work.
2.1. The Analysis of Breakthrough Time
2.1.1. Single Metal on the Halloysite Support
The breakthrough behavior of ethyl mercaptan over halloysite-supported single-metal oxides catalyst are shown in Figure 2, demonstrating the differences in adsorption performance (measure of removal efficiency) among the various catalysts tested.
The zinc oxide reached breakthrough after 210 minutes of testing, demonstrating a useful interaction with sulfur compounds. The breakthrough observed with zinc oxide is consistent with the electronic characteristics of ZnO; the relatively low band gap of ZnO provides an opportunity for charge transfer between the adsorbed sulfur species and the surface oxygen sites on the ZnO, thus providing a means of sustaining chemisorption and extending the time required for saturation to occur.
Copper oxide showed a similar but better response than zinc oxide, reaching breakthrough time after 215 minutes of testing. Copper oxide is characterized by its ability to rapidly redistribute electrons on the surface due to the relatively small band gap of CuO; these characteristics favor the polarizing of the Cu-S bond and facilitate the activation of thiol groups on the copper surface and sustain adsorption; this resulted in a slight increase in breakthrough time relative to ZnO [39].
Of the single-metal oxides tested, manganese oxide showed the greatest response to sulfur compounds, with a breakthrough delayed to 240 minutes. Although MnO does not possess the smallest band gap among the metal oxides tested, it is believed that the redox active Mn²⁺/Mn³⁺ chemistry of MnO contributes to the continued interaction with sulfur-containing molecules. The redox flexibility exhibited by MnO limits the early site saturation and results in a higher overall uptake. Additionally, the increased ionic radius of Mn²⁺ relative to Zn²⁺ and Cu²⁺ creates a greater degree of adaptability in the coordination of sulfur containing molecules on the halloysite surface, which would also serve to enhance the stability of sulfur adsorption [61].
Unlike the other metal oxides, nickel oxide behaved differently, with a rapid breakthrough time at approximately 32 minutes of testing. The rapid breakthrough observed with nickel oxide demonstrates an early saturation of the Ni²⁺ sites on the support (halloysite) with sulfur compounds and as such, suggests that there was limited effective interaction between the nickel sites and ethyl mercaptan under the test conditions used. NiO has a comparatively wider bandgap and less advantageous band-edge positions, restricting surface electron mobility and limiting the strength of Ni-S interactions. These electronic constraints reduce the availability of reactive surface sites and hinder sulfur chemisorption, leading to early breakthroughs. Similar behavior has been reported for sulfur compounds, where wide-band-gap oxides exhibited weak electron exchange and poor adsorption strength [62]. Reduced chemisorption has also been observed with non-sulfur molecules such as CO and NO on insulating oxides, further confirming that limited electronic flexibility suppresses surface reactivity [63].
2.1.2. Multiple Metals on the Halloysite Support
The single-metal results in Figure 2 show that although ZnO, CuO, and MnO individually demonstrated moderate to strong sulfur (e.g. mercaptans, H2S) adsorption, no single metal provided an optimal balance of bandgap energy, redox flexibility, and sulfur-binding strength compared to previous work [17]. Each oxide exhibited specific strengths such as MnO’s long breakthrough time or CuO’s strong charge-transfer capability but also clear limitations, including NiO’s poor performance and the restricted adsorption capacity of the other single-metal systems. These findings indicated that sulfur compounds adsorption is controlled by multiple simultaneous electronic and structural factors that no single oxide can fully satisfy. This motivated the transition to multi-metal formulations, where combining complementary metals could enhance oxygen-vacancy density, broaden redox behavior, and strengthen sulfur-metal interactions, ultimately enabling superior breakthrough performance beyond what single-metal catalysts could achieve.
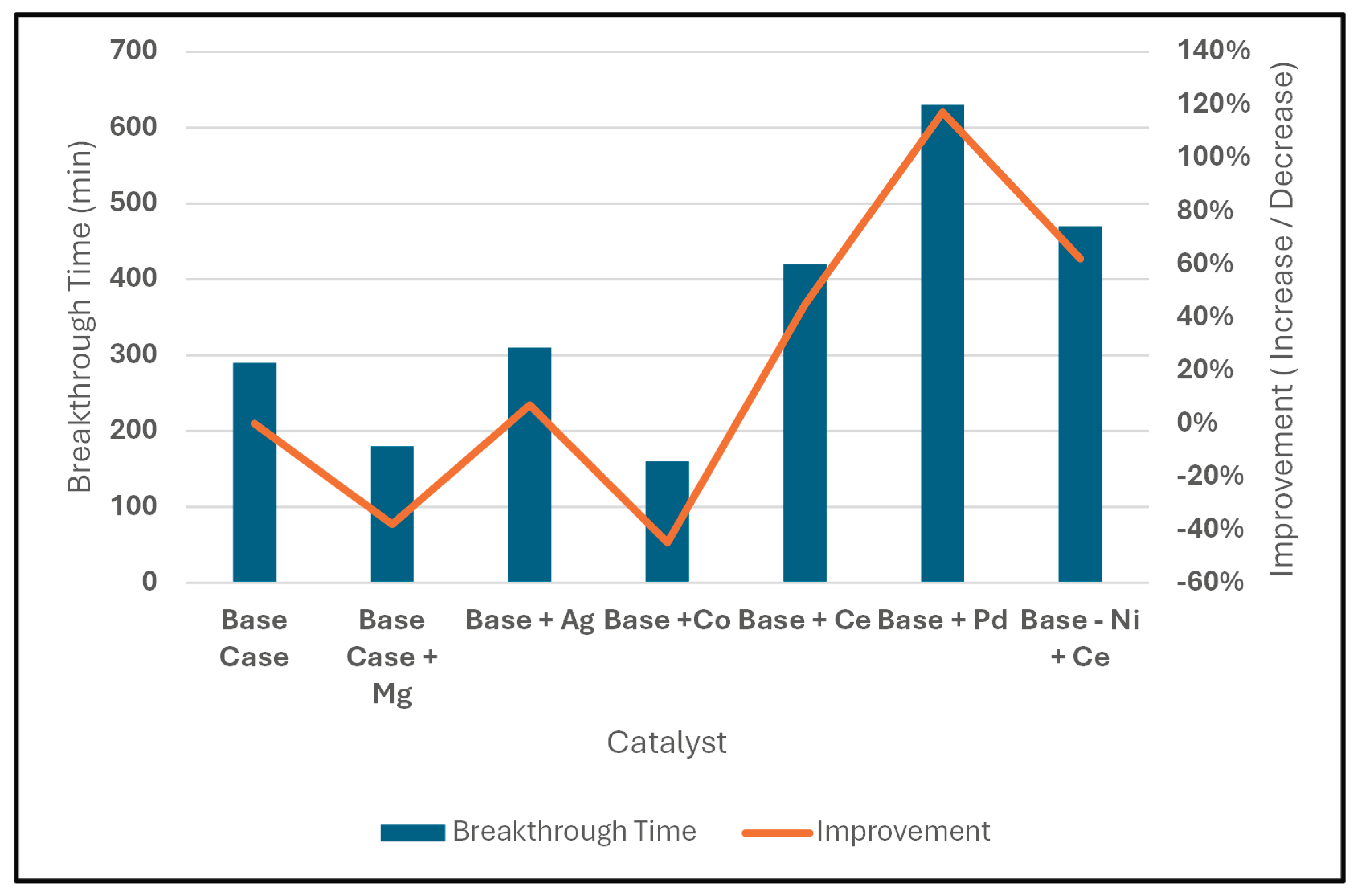
Figure 3 compares the breakthrough performance of sulfur compounds across the base catalyst system (Zn-Cu-Mn-Ni on halloysite) and its modified formulations containing (one at a time) Mg, Ag, Co, Ce, Pd, and Ce as a replacement for Ni. The base catalyst provides 290 minutes of removal of sulfur compounds at 10 ppm before the breakthrough time, when tested under conditions of 200 psi and a gas flow rate of 36 mL/min at 25°C. The observed performance of the catalyst was due to the additive effects of all four included metal oxides as opposed to one metal oxide being dominant. The combination of band gap energy differences, and redox activity, results in surface oxygen vacancy formation, and charge transfer through the surface that allows sulfur to be retained on the surface for a greater amount of time. Therefore, the increased break-through time (BT) of the base catalyst demonstrates that it has more surface interactions than other single-metal catalysts and is less likely to undergo sulfur poisoning during catalytic operations.
The addition of magnesium to the base catalyst resulted in a decrease to approximately 180 minutes of BT, or a loss in performance of 50%. Although MgO can stabilize nickel and enhance oxygen mobility in some catalyst systems [64,65], its addition in this context diluted the concentration of redox-active transition-metal sites necessary for strong sulfur bonding. Consequently, Mg acted more as a structural promoter than a functional sulfur-binding species, lowering the overall electron-transfer capacity and shortening the adsorption lifespan.
Silver-modified catalyst exhibited only a slight improvement relative to the base system, with breakthrough occurring at 310 min. Despite its electronic similarity to Cu, Ag rapidly forms multilayer Ag₂S films at room temperature, passivating the surface and blocking further sulfur chemisorption [66,67]. The limited enhancement is therefore attributed to fast silver sulfide formation, which reduces the number of accessible metal sites and restricts sulfur uptake. This deactivation mechanism aligns with established behavior of Ag in sulfur environments [68].
The cobalt-modified catalyst also demonstrated reduced performance, showing breakthrough at 160 min. While Co₃O₄ normally promotes thiol oxidation due to its mixed Co²⁺/Co³⁺ redox centers and spinel structure, EDS analysis (Figure 5) revealed that most cobalt was lost during filtration likely due to acetic-acid-induced multilayer formation that prevented uniform deposition. As a result, the intended redox enhancement was not realized, and the catalyst instead exhibited diminished sulfur adsorption relative to the baseline formulation.
Cerium was shown to be an effective promoter of sulfur adsorption, with a break-through time of about 420 minutes compared to 300 minutes for the base catalyst. The increased adsorption is attributed to the redox properties of ceria and the ability of reversible Ce³⁺/Ce⁴⁺ site changes and oxygen vacancy formation to form stronger interactions with surface bound sulfur. Although the increase in adsorption capacity due to the addition of ceria was significant, it was less than would have been expected based on prior studies of redox active materials [69,70]. Prior studies showed a higher affinity of transition metals to bind sulfur than was observed with ceria. The difference may be due to interactions between ceria and the silica portion of the halloysite support. These interactions can result in the formation of amorphous cerium silicates, which are known to decrease the number of surface ceria sites (Ce³⁺) available for sulfur adsorption as well as the amount of oxygen vacancies present in the surface [71,72].
Addition of palladium resulted in the largest increase in adsorption capacity of any additive tested. Palladium increased the breakthrough time to just under 630 minutes or almost double the base catalyst. The large increase in adsorption capacity due to palladium is consistent with the narrow band gap of PdO and the ability of palladium to form strong bonds with sulfur. During adsorption, the Pd-O bond breaks and a Pd-S bond forms. The energy required for the breaking of the Pd-O bond is reflected in a shift in the Pd core level binding energy. The incorporation of palladium into a metal oxide matrix has been previously demonstrated to produce cooperative effects between the palladium and other metal oxides in the matrix. In particular, the electronic states of the surrounding metal oxides (ZnO, CuO, NiO, and MnO) allow for charge redistribution at the palladium/sulfur interface. Cooperative effects such as these have been shown to enhance sulfur activation greater than what palladium oxide alone could accomplish [73,74]. Therefore, the combination of strong S-Pd bonds and cooperative effects within the composite matrix results in long term adsorption, resulting in the largest increase in adsorption capacity of any additive tested.
Lastly, replacing nickel in the base composition with ceria increased the break-through time to approximately 570 minutes, producing a very large increase in adsorption capacity. NiO can occupy interstitial vacancies and limit the formation of active oxygen-deficient sites, so its removal increases the availability of CeO₂ redox centers and the density of surface defects. The enhanced breakthrough performance suggests that CeO₂ is a more effective sulfur-adsorbing component than NiO under these conditions, and that nickel may have hindered vacancy formation within the base catalyst structure.
Overall, the multi-metal comparison clearly demonstrates that sulfur compound adsorption/removal performance is governed by a combination of bandgap size, redox flexibility, oxygen vacancy concentration, and metal-sulfur binding strength. Pd and Ce produced the most substantial improvements, while Mg, Ag, and Co provided little or negative contributions. These results highlight the importance of electronic structure and surface defect engineering in designing high-efficiency sulfur adsorption catalysts.
FTIR Analysis and Catalytic Implications of Multi-Metal Catalyst
Figure 4 presents the FTIR spectra of the base catalyst (Zn-Cu-Mn-Ni) and the modified formulations incorporating Pd, Mg, Co, Ce, Ag, and Ce (without Ni). All spectra exhibit the characteristic vibrational features of halloysite-supported metal oxides, with distinct variations reflecting the electronic structure and surface chemistry introduced by each incorporated metal species. These differences directly influence sulfur adsorption by modifying surface hydroxyl density, Lewis’s acidity, oxygen-vacancy concentration, and metal-sulfur binding strength.
Across all catalysts, broad absorption in the 3600-3000 cm⁻¹ region corresponds to O-H stretching of surface hydroxyl groups and interlayer water. Differences in intensity among samples indicate varying degrees of surface hydration and hydroxyl density, which are known to influence thiol adsorption through hydrogen bonding and proton transfer pathways. The weak features around 1630-1650 cm⁻¹ are assigned to H-O-H bending, confirming physiosorbed water common to aluminosilicate supports. The 1000-1100 cm⁻¹ region contains the strong Si-O-Si and Si-O-Al stretching modes of halloysite, while additional shoulders at 900-950 cm⁻¹ indicate metal-oxygen interactions due to metal-O-Al or metal-O-Si linkages. Peaks observed between 650-800 cm⁻¹ correspond to Al-O-Si bending and lattice vibrations of incorporated metal oxides.
The base formulation exhibits hydroxyl bands of consistent intensity and a moderate, uniform shift in the Si-O-Si vibration, indicating that the metal oxides are evenly dispersed on the halloysite surface without any single species dominating the surface chemistry. The subtle peak near 2360-2400 cm⁻¹ suggests weak surface carbonate species, which can interact with thiols. The presence of multiple transition-metal-oxygen vibrations in the 550-750 cm⁻¹ range reflects redox-active metal sites that support sulfur chemisorption and oxidative transformations.
Pd incorporation causes a noticeable reduction in the O-H band intensity and a stronger, sharper feature near 950-1000 cm⁻¹, indicative of increased metal-oxygen bonding and higher surface electron density. Palladium-oxygen vibrational influences in the 650-700 cm⁻¹ region signal successful PdO incorporation, enhancing Lewis’s acidity and enabling strong Pd-S chemisorption. These structural features are consistent with the FTIR evidence of fewer hydroxyls but more electron-rich Pd sites, explaining this catalyst’s superior sulfur breakthrough time (630 min).
The Mg-containing catalyst shows a broadened O-H region and increased transmittance around 3700-3500 cm⁻¹, indicating stronger Mg-OH formation. The enhanced hydroxyl character increases basicity but reduces the density of redox-active transition-metal sites. The relatively unchanged Si-O-Si region and weak metal-O bands suggest that Mg dilutes active sites, aligning with its poorer breakthrough performance (180 min)
Co-modified samples show new or intensified bands around 660-690 cm⁻¹, corresponding to Co-O vibrations characteristic of Co₃O₄ spinel structures. However, the overall spectrum exhibits lower shifts in the Si-O-Si region, consistent with low Co loading. Limited incorporation reduces oxygen-vacancy generation and redox activity, explaining its reduced breakthrough time (160 min). Minimal changes in hydroxyl regions further support weak Co-support interaction.
Ce incorporation produces clear broadening and shifts near 1050-1100 cm⁻¹, reflecting Ce-O-Si interactions and partial cerium-silicate formation. A subtle shoulder around 800-850 cm⁻¹ corresponds to Ce-O vibrational modes. The enhanced O-H intensity suggests increased surface polarizability and oxygen-vacancy formation associated with Ce³⁺/Ce⁴⁺ redox cycling.
The spectral characteristics of this study correlate with the increased retention of sulfur (e.g. C2H5SH) of the catalyst containing Ce, which achieved breakthrough after approximately 420 minutes; this can be attributed to the formation of Ce-O-S bonds on its surface. On the other hand, the Ag-modified sample had very little decrease in intensity of the O-H band and very minor changes in the 900-1100 cm⁻¹ area; this suggests poor interaction between Ag-species and halloysite support. Also, since there are no notable Ag-O bands present, it can be inferred that the Ag-species were not well-dispersed as Ag₂O domains instead of being well-anchored to the surface. This data when compared with well-documented tendencies of silver forming Ag₂S rapidly [66,67,68], agrees with minimal performance improvements observed with this catalyst, which reached breakthrough after approximately 310 minutes due to rapid sulfide passivation. More pronounced spectral changes were observed when Ni was removed from the formula and replaced by Ce. The addition of Ce caused more obvious changes in both the hydroxyl region and the Si-O-Al framework vibration, indicating Ce has a greater impact on the surface chemistry and metal-support interaction of this catalyst system. The O-H band becomes more intense and slightly shifted, indicating increased hydrogen-bonding and oxygen-vacancy density. Ce-O features at 760-850 cm⁻¹ become more pronounced than in the Ce+ base sample, reflecting higher Ce incorporation.
EDS Analysis and Catalytic Implications of Multi-Metal Halloysite Catalyst
The EDS data (Figure 5) demonstrates the presence of each of the four metals used as part of the catalyst preparation process and shows the relative proportion of these metals present on the halloysite surface post-synthesis, thus confirming retention of each of the metals through the entire preparation process. The EDS spectrum obtained from the base catalyst shows the typical signals for the metals present (Mn, Ni, Cu, and Zn), as well as the Al-Si signals resulting from the halloysite support. Signals from each of the four metals can be seen clearly in the EDS data acquired from the halloysite surface, indicating the metals have been incorporated into the halloysite support because of the synthesis method. The intensity of the metal signal suggests there are no significant variations in the amount of any of the metals, suggesting they are uniformly distributed on the surface of the halloysite support. Thus, simultaneous involvement of multiple metal sites in sulfur adsorption will likely occur; this should eliminate dominance of adsorption by any one metal. This does not imply the absence of synergy; rather, the catalyst performance reflects the combined effect of multiple metals whose interactions create a collective, moderate breakthrough response, instead of one metal dominating the surface chemistry.
When magnesium was added to this system, the EDS plot changes noticeably. The typical halloysite peaks remain, but Mg itself barely appears. This is not surprising given how soluble magnesium precursors are and how weakly Mg binds to the aluminosilicate surface. During filtration, much of the Mg is simply washed away, leaving the catalyst surface practically unchanged from the base. The poorer adsorption performance of this sample is consistent with what the EDS is showing, there was very little Mg present to influence catalytic behavior.
Silver tells a similar story, but for different chemical reasons. The base catalyst with Ag shows only small Ag peaks in the EDS spectrum. Silver ions readily form stable aqueous complexes such as [Ag(AcO)₂] ⁻ or Ag-acetate species, and without tight control of pH and ionic strength, much of the Ag remains in solution rather than anchoring to the halloysite surface [75]. Halloysite surfaces exhibit predominantly positive charges under mildly acidic conditions, offering limited electrostatic attraction for Ag⁺ complexes and promoting their loss during filtration [76]. Even the Ag that remains is known to sulfate or sulfurize rapidly upon exposure to thiols, forming passivating Ag₂S layers [77]. Thus, the weak EDS signal and the limited improvement in breakthrough time are consistent: a significant fraction of Ag was removed during filtration, and the fraction retained becomes chemically deactivated by sulfur species.
Cobalt behaves even more drastically. In the base-with-Co spectrum, the Co peaks are nearly undetectable, indicating that most of the cobalt precursor was washed away during filtration. Cobalt acetate remains strongly solvated in aqueous media and undergoes limited hydrolysis or ligand exchange, preventing the formation of surface-bound Co-OH or Co-O-Si linkages on halloysite [78]. In addition, Co²⁺ interacts weakly with aluminosilicate surfaces because its hydration shell is comparatively stable and resists dehydration at the interface, reducing its ability to bind to the clay surface [79]. As a result, the final catalyst contains almost no cobalt, and its adsorption behavior resembles that of the base material but slightly weaker, since the overall distribution of active metal sites becomes less uniform.
Cerium, however, is retained very differently, in the base with Ce sample, the Ce peaks stand out clearly. Cerium has a natural tendency to bind strongly to oxygen-rich surfaces, and Ce precursors do not wash out easily. The strong EDS signal suggests a meaningful presence of Ce on the surface, and this aligns with the longer breakthrough observed experimentally. The Ce³⁺/Ce⁴⁺ redox pair and the oxygen vacancies associated with ceria are well-known to interact beneficially with sulfur compounds, which helps explain the improved sulfur adsorption.
Palladium is also strongly retained, the base with Pd spectrum contains sharp, clean Pd peaks, which indicate that Pd remains attached to the surface after filtration. Palladium precursors hydrolyze and anchor well to aluminosilicates, and Pd itself shows strong affinity for sulfur-containing molecules. It is not surprising that this catalyst shows the longest breakthrough time among all samples; the EDS confirms that Pd is present in enough quantity to influence surface chemistry significantly.
The final sample, Base (without Ni) + Ce, shows what happens when Ni is removed from the initial composition. The Ce peaks become stronger compared to the previous Ce-containing sample. With Ni absence, Ce has more available surface sites to occupy, and the EDS reflects this shift clearly. The sulfur adsorption tests demonstrated that this formulation was competitive with the Pd containing catalyst. The reason for this competitive nature is believed to be due to an increased number of cerium species on the surface. It appears that the cerium species occupy positions that would have been occupied by nickel species, thereby increasing the surface area of oxygen vacancy-related positions; allowing for an increase in the amount of ethyl mercaptan that can interact and be taken up by the catalyst. Overall, the data generated from the EDS studies demonstrate that it is not only important to select metals to be used as catalysts but also to retain them throughout the synthesis and filtration processes. Metals such as Pd and Ce appear to be well retained onto the halloysite while other metals tend to be lost. These differences in retention appear to influence the adsorption behaviors exhibited by the catalysts.
Others, such as Mg, Ag, and Co, are only present in small amounts after filtration, so their influence on sulfur removal is limited. The EDS patterns therefore match well with the adsorption trends and provide a direct explanation for the differences in catalytic behavior across the modified samples.
2.2. The Analysis of Surface Morphology of the Prepared Catalyst
The findings show that doping causes notable structural alterations as shown in Table 2. All additional metals had an increase in the base (Zn, Cu, Mn, Ni) catalyst's surface area (36.253 m²/g), with the addition of magnesium exhibiting the largest surface area (50.9865 m²/g). To a much lesser degree, other metals such as Ag, Co, Ce, and Ce-Ni also increase surface area. Out of all the modified catalyst, the Pd-doped catalyst exhibits the least amount of surface area growth (43.2712 m²/g). The relatively large pore volume of the Mg-containing sample (0.2544 cm³/g), followed by the Ag- and Co-modified catalysts, indicates that these metals contribute most strongly to the overall porosity development of the material. Out of all the doped samples, the Pd-modified catalyst had the smallest pore volume (0.2225 cm³/g). Doping causes the pores to shrink; the base catalyst has the biggest pores size (238.8216 Å). The lowest pore size is caused by Ce doping (188.7434 Å), which is followed by Ce-Ni (190.4318 Å) and Co (192.4566 Å). In contrast to other doped samples, the Pd-modified catalyst maintains a larger pore size (205.6373 Å). Overall, the findings imply that doping alters the base catalyst's textural characteristics. Ce result is the smallest pore size, indicating a more compact structure, whereas Mg doping is the most successful at increasing surface area and pore volume.
Figure 6.
Micromeritics ASAP 2020.


Table 1.
Surface area, Pore volume and Pore size of the modified catalyst prepared.
| Catalyst | Surface Area (m2/g) | Pore Volume (cm3/g) | Pore Size (Å) |
|---|---|---|---|
| Base | 36.253 | 0.2165 | 238.8216 |
| Base + Mg | 50.9865 | 0.2544 | 194.1288 |
| Base + Ag | 48.3956 | 0.2420 | 199.9837 |
| Base + Co | 48.6031 | 0.2339 | 192.4566 |
| Base + Ce | 47.5022 | 0.2241 | 188.7434 |
| Base + Pd | 43.2712 | 0.2225 | 205.6373 |
| Base + Ce -Ni | 48.5709 | 0.2312 | 190.4318 |
2.3. Performance Summary
After establishing how individual metal oxides influence the performance of the base catalyst in sulfur compounds removal from natural gas, the next step is to determine which metals most effectively enhance breakthrough behavior under the defined operating conditions of flow rate, pressure, and temperature. Each modified catalyst was tested in duplicate, and the measurements showed minimal variation, indicating high reproducibility and low standard deviation in the data.
Table 2 shows that the base catalyst having Zn, Cu, Mn and Ni as active metals supported on alumina-silicates has good (albeit limited) sulfur removal capabilities; the breakthrough occurred at 290 minutes and had a sulfur capacity of approximately 3967 mg S g⁻¹. However, adding Palladium to the base formulation leads to a marked change in performance; the breakthrough time increased to 630 minutes, and the sulfur capacity increased to approximately 8618 mg S g⁻¹ which suggests that the longer breakthrough time was due to an actual increase in sulfur sorption rather than a change in reaction kinetics. A similar but smaller trend occurs when Nickel is removed and Cerium is added; in this case the breakthrough time was extended to 570 minutes, and the sulfur capacity is 7798 mg S g⁻¹. On the other hand, catalysts which have been modified with either Magnesium or Cobalt produce both shorter breakthrough times and lower sulfur capacities than the base catalyst which suggests that these modifications decrease the number of available adsorption sites rather than increasing them. Overall, the high correlation between the breakthrough time and the sulfur capacity for all samples indicates that the performance of the catalysts in this system is primarily driven by how much sulfur the catalyst surface can retain before becoming saturated.
Table 2.
The Performance Summary of all the Experimental runs.
| Metal Oxide | Testing Conditions | Breakthrough | Improvement | Sulphur Capacity (q) | ||
|---|---|---|---|---|---|---|
| Flowrate(mL/min) | Pressure (psi) | Temp (OC) | (mins) | (%) | mg S/g | |
| Base Case | 36 | 200 | 25 | 290 | 0% | 3967.2 |
| Base Case + Mg | 36 | 200 | 25 | 180 | -38% | 2462.2 |
| Base Case + Ag | 36 | 200 | 25 | 310 | 7% | 4240.8 |
| Base Case + Co | 36 | 200 | 25 | 160 | -45% | 2188.8 |
| Base Case + Ce | 36 | 200 | 25 | 420 | 45% | 5745.6 |
| Base Case + Pd | 36 | 200 | 25 | 630 | 117% | 8618.4 |
| Base Case - Ni +Ce | 36 | 200 | 25 | 570 | 62% | 7797.6 |
Figure 7.
Performance Summary.
The rate of sulfur adsorption on modified metal oxide catalyst surfaces is significantly faster than on pure oxides. In metal/oxide systems, the supported metal provides numerous electronic states that facilitate efficient bonding interactions with sulfur-containing molecules. Sulfur attaches to the metal oxide through two primary mechanisms: (1) charge transfer from the metal to vacant sulfur orbitals (σ-back-donation) and (2) electron transfer from sulfur orbitals into the unoccupied 4sp states of the substrate (σ- and π-donation). These strong electron-donor and electron-acceptor interactions are reflected in the catalyst’s activity, as measured by breakthrough time (measure of removal/adsorption efficiency. When the oxide (support) has a large band gap (Al₂O₃), the sulfur-containing molecules interact primarily with the metal species deposited on the support, rather than with the support itself, because the wide band gap limits the support’s ability to participate in electron transfer during adsorption [80].
3. Materials and Methods
3.1. Materials
The chemicals used in the experiments include zinc acetate dihydrate (97%), nickel (II) acetate tetrahydrate (99%), copper (II) nitrate trihydrate (99%), manganese acetate tetrahydrate (98%), palladium acetate, silver acetate, magnesium acetate tetrahydrate, cerium (III) acetate and cobalt (II) acetate. Additional reagents include sodium hydroxide, acetic acid (99.7%), and 200-proof ethanol.
Catalyst Preparation
All chemicals were of reagent-grade quality and used without further purification. The catalyst preparation steps are as follows: to achieve a particle size of 0.4 mm, halloysite pure was crushed and sieved using a 40-mesh screen and 3g was weigh. Then, 120 ml of 99.5% ethanol was added to the halloysite pure (solution 2) and subject to ultrasonication. Subsequently, the following compounds one at a time or combine base on the catalyst under preparation: manganese acetate tetrahydrate (Mn(CH₃COO)₂·4H₂O), zinc acetate dihydrate (Zn(CH₃COO)₂·2H₂O), nickel(II) acetate tetrahydrate (Ni(CH₃COO)₂·4H₂O), copper(II) nitrate trihydrate (Cu(NO₃)₂·3H₂O), and zinc acetate dihydrate (Zn(CH₃COO)₂·2H₂O) were dissolved in 240 ml of 99.5% ethanol to create Solution 1. The metal composition in this solution could be adjusted based on their mass percentage ratio (w/w). The metal solution and the halloysite solution were mixed, to adjust the pH of the resulting mixture to 8 with 1M NaOH solution and then subject to further ultrasonication, as shown in Figure 7.
The precursor CuO-ZnO-NiO-MgO-MnO/Halloysite pure was then prepared by refluxing the mixture for three hours at 85°C. The remaining ethanol was removed through filtration using filter paper as indicated in Figure 7. The retained product was allowed to air dry for 8-12 hours. When the solid sample was ultimately extracted, it was calcined for 2.5 hours at 300 °C in a furnace and then cooled to undergo performance testing in ethyl-mercaptan adsorption. The metals selection was based on their properties (example bandgap, reactivity) and cost. The metals with smaller/ moderate bandgap and cost effective were selected and later modified with other metals.
3.2. Methods
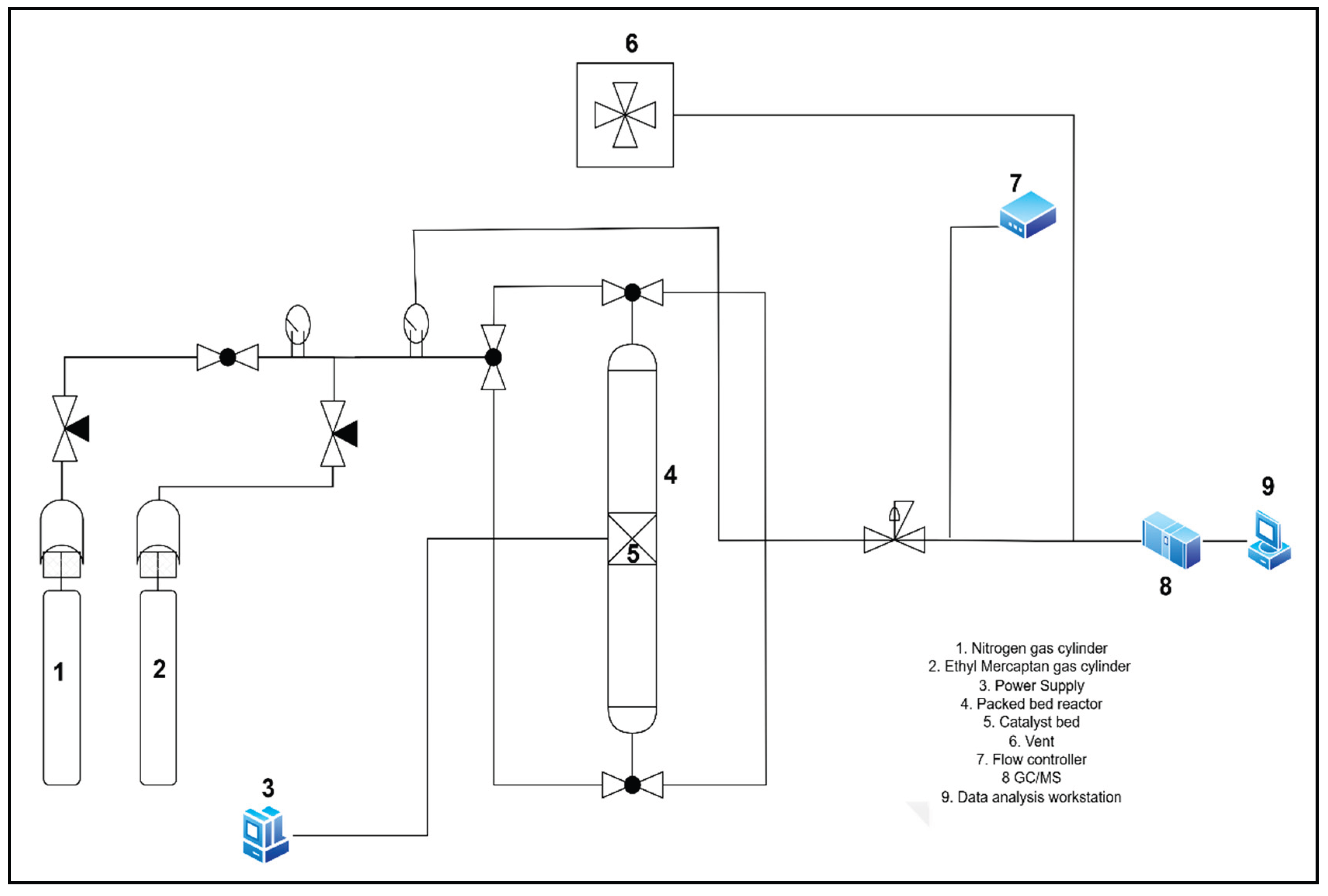
The catalytic activity test was carried out in a stainless-steel tube packed bed reactor with dimensions of 310 mm in length and an inner diameter of 6.35 mm as shown in Figure 8. In each experimental run, 0.5 g of catalyst was loaded in the reactor with the help of glass wool to keep it in place. After loading, the reactor bed was pressured at 60 psi with a nitrogen gas flow rate of 36 mL/min. Once the operational pressure was attained, the system was leak tested using Snoop TM and then left for 30 minutes to see if there were any minor leaks that Snoop TM did not detect by measuring the pressure drop on the pressure gauge. This enabled for leak testing of all connections, from the gas tank to the downstream mass flow controller. Once the system was found to be leak-free, to start the ethyl mercaptan removal process, the feed was changed from N2 to a gas mixture with 200 ppm ethyl mercaptan in methane. The gas combination was introduced axially into the packed bed reactor at 250C, 200psi and 36 mL/min. Once the flow was stabilized at the proper pressure, the GC-MS procedure began immediately. Breakthrough data was collected once breakthrough (10ppm) attained. The sulfur capacity of each experiment at breakthrough (q), used to evaluate and compare the results across experimental runs, was calculated using the following formula.
Ci is the initial concentration of CH3CH2SH, Cf is the concentration of the outlet gas, including CH3CH2SH, CH3CH2SSCH3CH2, CH3CH2SCH3CH2, in ppm, Q denotes the volumetric flow rate of the model gas, measured in mL/min, t corresponds to the breakthrough time, measured in minutes, M indicates the mass of the loaded catalyst and f is a correction factor that accounts for deviations from ideal plug-flow behaviour, including gas compressibility, pressure effects, and sulphur speciation in the outlet stream, in this study, was taken as unity under steady-state conditions
4. Conclusions
- Metals with smaller bandgaps exhibited superior sulfur adsorption due to enhanced electron transfer and stronger surface sulfur interactions. Narrow bandgap oxides provided more active sites and faster charge mobility, leading to longer breakthrough times and better adsorption performance. In contrast, wider bandgap oxides limited charge exchange, resulting in weaker adsorption and reduced desulfurization efficiency.
- The Mg-modified catalyst is good for high dispersion and adsorption-dependent reactions since it has the largest surface area and pore volume. The smallest pores are seen in Ce-modified catalysts (Ce and Ce-Ni), which may increase catalytic stability and selectivity. With its moderate surface area and lowest pore volume, the Pd-modified catalyst exhibits a denser structure that may be advantageous for oxidation or hydrogenation processes. All things considered, added metals greatly improve texture and increase catalyst efficiency according to reaction needs.
- The breakthrough study demonstrates the influence of metal oxides and band gaps on the adsorption capabilities of the base catalyst. Composed primarily of aluminum silicate, zinc, copper, manganese, and nickel. The base catalyst achieved a breakthrough time of 290 minutes, reducing sulfur compounds concentration to a minimum of 10 ppm.
- The results indicate that optimal catalytic performance is attained when the base catalyst was modified with palladium oxide, extending the breakthrough time from 290 to 630 minutes and an improvement of nearly 117%. Addition of palladium oxide to the base catalyst showed the highest performance improvement, followed by the substitution of nickel oxide by cerium oxide. The superior performance of the modified catalyst is likely due to its chemical composition and metal band gap, which significantly enhances its catalytic activity in sulfur compounds adsorption (removal from natural gas stream) capacity.
Author Contributions
Conceptualization, R.H. and M.E.Z.; methodology, R.H., M.E.Z., and S.A.; formal analysis, R.H., S.A., and W.E.H.; data curation, S.A and W.E.H.; writing-original draft preparation, S.A. and O.O.; writing-review and editing, R.H., M.E.Z., S.A., D.F.,T.K., A.G., and E.R.; project administration, R.H, M.E.Z, and W.E.H.; funding acquisition, R.H and M.E.Z. All authors have read and agreed to the published version of the manuscript
Funding
This research was supported by the Guangdong Basic and Applied Basic Research Foundation (no. 2021A1515012342) U.S.–Israel Fossil Energy Center (FEC19) administered by the BIRD Foundation and funded by the Israeli Energy Ministry and the U.S. Department of Energy.
Data Availability Statement
All data have been provided in the manuscript.
Acknowledgments
The authors acknowledge the financial support of the U.S. – Israel Fossil Energy Center (FEC19) administered by the BIRD Foundation and funded by the Israeli Energy Ministry and the U.S. Department of Energy for this work.
Conflicts of Interest
All Authors declare that there is no conflict of interest regarding the manuscript.
Future Work
In reference to the FTIR and EDS, most of the metals were lost through filtration and hence other alternative preparation method will be considered to retain all the metals in our future work. Most of the metals expected performance were not achieved and we suspect that the added acetic acid formed layers with the metals and depreciates its performance. Future work will consider comparison of “with and without” acetic acid. This can be concluded with their performance and scanning electron microscopic (SEM) images.
References
- Zhao, S.; Yi, H.; Tang, X.; Gao, F.; Zhang, B.; Wang, Z.; Zuo, Y. Methyl mercaptan removal from gas streams using metal-modified activated carbon. Journal of Cleaner Production 2015, 87, 856–861. [Google Scholar] [CrossRef]
- Guo, C.; Zhu, J.; He, J.; Hu, L.; Zhang, P.; Li, D. Catalytic oxidation/photocatalytic degradation of ethyl mercaptan on α-MnO2@ H4Nb6O17-NS nanocomposite. Vacuum 2020, 182, 109718. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric chemistry and physics: from air pollution to climate change; John Wiley & Sons, 2016. [Google Scholar]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the upper and lower atmosphere: theory, experiments, and applications; Elsevier, 1999. [Google Scholar]
- Bauer, S.E.; Mishchenko, M.I.; Lacis, A.A.; Zhang, S.; Perlwitz, J.; Metzger, S.M. Do sulfate and nitrate coatings on mineral dust have important effects on radiative properties and climate modeling? Journal of Geophysical Research: Atmospheres 2007, 112(D6). [Google Scholar] [CrossRef]
- Bashkova, S.; Bagreev, A.; Bandosz, T.J. Activated Carbons as Adsorbents of Methyl Mercaptan. In Fuel Chemistry Deivienv Preprints; 2002. [Google Scholar]
- Ito, E.; Van Veen, J.R. On novel processes for removing sulphur from refinery streams. Catalysis today 2006, 116(4), 446–460. [Google Scholar] [CrossRef]
- Agbroko, O.W.; Piler, K.; Benson, T.J. A comprehensive review of H2S scavenger technologies from oil and gas streams. ChemBioEng Reviews 2017, 4(6), 339–359. [Google Scholar] [CrossRef]
- Mortaheb, H.R.; Ghaemmaghami, F.; Mokhtarani, B. A review on removal of sulfur components from gasoline by pervaporation. Chemical Engineering Research and Design 2012, 90(3), 409–432. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Hu, L.; He, J.; 10. Adsorption and separation of ethyl mercaptan from methane gas on HNb3O8 nanosheets. Industrial & Engineering Chemistry Research 2021, 60(23), 8504–8515. [Google Scholar] [CrossRef]
- Ryzhikov, A.; Hulea, V.; Tichit, D.; Leroi, C.; Anglerot, D.; Coq, B.; Trens, P. Methyl mercaptan and carbonyl sulfide traces removal through adsorption and catalysis on zeolites and layered double hydroxides. Applied Catalysis A: General 2011, 397(1-2), 218–224. [Google Scholar] [CrossRef]
- Jie, H.E.; ZHAO, J.B.; LAN, Y.X. Adsorption and photocatalytic oxidation of dimethyl sulfide and ethyl mercaptan over layered K1-2xMnxTiNbO5 and K1-2xNixTiNbO5. Journal of Fuel Chemistry and Technology 2009, 37(4), 485–488. [Google Scholar]
- Chu, H.; Wu, L.W. The catalytic incineration of ethyl mercaptan over a MnO/Fe2O3 catalyst. Journal of Environmental Science & Health Part A 1998, 33(6), 1119–1148. [Google Scholar]
- Sehon, A.H.; deB. Darwent, B. The thermal decomposition of mercaptans. Journal of the American Chemical Society 1954, 76(19), 4806–4810. [Google Scholar] [CrossRef]
- Van den Bosch, M.; Sang, Å.O. Urban natural environments as nature-based solutions for improved public health–A systematic review of reviews. Environmental research 2017, 158, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Sedighi, M.; Zamir, S. M.; Vahabzadeh, F. Cometabolic degradation of ethyl mercaptan by phenol-utilizing Ralstonia eutropha in suspended growth and gas-recycling trickle-bed reactor. Journal of environmental management 2016, 165, 53–61. [Google Scholar] [CrossRef]
- Zuniga, G.M. Developing Catalysts for the Removal of Methyl Mercaptan from Natural Gas; University of Louisiana at Lafayette, 2023. [Google Scholar]
- Shingan, B.; Timung, S.; Jain, S.; Singh, V.P. Technological horizons in natural gas processing: A comprehensive review of recent developments. Separation Science and Technology 2024, 59(10-14), 1216–1240. [Google Scholar] [CrossRef]
- Bandosz, T.J. Activated carbon surfaces in environmental remediation; Elsevier, 2006. [Google Scholar]
- Lyu, Y.; Liu, X.; Liu, W.; Tian, Y.; Qin, Z. Adsorption/oxidation of ethyl mercaptan on Fe-N-modified active carbon catalyst. Chemical Engineering Journal 2020, 393, 124680. [Google Scholar] [CrossRef]
- Jana, S.; Sarkar, U. Alkaline functionalization of granular activated carbon for the removal of volatile organo sulphur compounds (VOSCs) generated in sewage treatment plants. Journal of Environmental Chemical Engineering 2018, 6(2), 3510–3519. [Google Scholar] [CrossRef]
- Jana, S.; Sarkar, U. Alkaline functionalization of granular activated carbon for the removal of volatile organo sulphur compounds (VOSCs) generated in sewage treatment plants. Journal of Environmental Chemical Engineering 2018, 6(2), 3510–3519. [Google Scholar] [CrossRef]
- Bagreev, A.; Katikaneni, S.; Parab, S.; Bandosz, T.J. Desulfurization of digester gas: prediction of activated carbon bed performance at low concentrations of hydrogen sulfide. Catalysis Today 2005, 99(3-4), 329–337. [Google Scholar] [CrossRef]
- Cui, H.; Turn, S.Q.; Reese, M.A. Removal of sulfur compounds from utility pipelined synthetic natural gas using modified activated carbons. Catalysis Today 2009, 139(4), 274–279. [Google Scholar] [CrossRef]
- Hsi, H.C.; Rood, M.J.; Rostam-Abadi, M.; Chang, Y.M. Effects of sulfur, nitric acid, and thermal treatments on the properties and mercury adsorption of activated carbons from bituminous coals. Aerosol and Air Quality Research 2013, 13(2), 730–738. [Google Scholar] [CrossRef]
- Conti-Ramsden, M. G.; Asghar, H. M. A.; Hussain, S. N.; Roberts, E. P. L.; Brown, N. W. Removal of mercaptans from a gas stream using continuous adsorption-regeneration. Water science and technology 2012, 66(9), 1849–1855. [Google Scholar] [CrossRef]
- Lyu, Y.; Liu, X.; Liu, W.; Tian, Y.; Qin, Z. Adsorption/oxidation of ethyl mercaptan on Fe-N-modified active carbon catalyst. Chemical Engineering Journal 2020, 393, 124680. [Google Scholar] [CrossRef]
- Wen, Y.; Zhao, S.; Yi, H.; Gao, F.; Yu, Q.; Liu, J.; Tang, T.; Tang, X. Efficient catalytic oxidation of methyl mercaptan to sulfur dioxide with NiCuFe mixed metal oxides. Environmental Technology & Innovation 2022, 26, 102252. [Google Scholar]
- Ma, N.; Yi, F.; Zhu, J. Characterization of aroma-active compounds and perceptual interaction between esters and sulfur compounds in Xi baijiu. European Food Research and Technology 2020, 246, 2517–2535. [Google Scholar] [CrossRef]
- Sun, Z.; Liao, T.; Dou, Y.; Hwang, S.M.; Park, M.S.; Jiang, L.; Kim, J.H.; Dou, S.X. Generalized self-assembly of scalable two-dimensional transition metal oxide nanosheets. Nature communications 2014, 5(1), 3813. [Google Scholar] [CrossRef]
- Hu, L.; Zhu, J.; He, J. Adsorption and conversion of ethyl mercaptan on the surface of Ag-Mn/NS-HNbMoO6. Journal of Chemical Technology & Biotechnology 2022, 97(9), 2496–2510. [Google Scholar]
- Dong, W.; Xu, C.; Zhao, W.; Xin, M.; Xiang, Y.; Fu, Y.; Cao, X.; Yuan, H.; Ye, W.; Qiu, L.; Xu, G. Insight into the poisoning effects and mechanisms of CH3SH, CH3CH2SH, CH3SCH3, and CH3CH2SCH2CH3 on Pt/C catalysts in fuel cell HOR. International Journal of Hydrogen Energy 2023, 48(81), 31745–31757. [Google Scholar] [CrossRef]
- Yi, Y.; Liu, H.; Chu, B.; Qin, Z.; Dong, L.; He, H.; Bin, L. Catalytic removal NO by CO over LaNi0. 5M0. 5O3 (M= Co, Mn, Cu) perovskite oxide catalysts: tune surface chemical composition to improve N2 selectivity. Chemical Engineering Journal 2019, 369, 511–521. [Google Scholar] [CrossRef]
- Xue, T.; Wang, Y.; Yang, L.; Li, Z.; Gao, Y.; Wang, Q. Development of quinary layered double hydroxide-derived high-entropy oxides for toluene catalytic removal. Catalysts 2023, 13(1), 119. [Google Scholar] [CrossRef]
- Sahoo, S.; Wickramathilaka, K. Y.; Njeri, E.; Silva, D.; Suib, S. L. A review on transition metal oxides in catalysis. Frontiers in Chemistry 2024, 12, 1374878. [Google Scholar] [CrossRef]
- Baral, S. C.; Maneesha, P.; Sen, S.; Sen, S.; Sen, S. An introduction to metal oxides. In Optical Properties of Metal Oxide Nanostructures; Springer Nature Singapore: Singapore, 2023; pp. 1–34. [Google Scholar]
- Wu, X.; Xu, L.; Chen, M.; Lv, C.; Wen, X.; Cui, Y.; Hu, X. Recent progresses in the design and fabrication of highly efficient Ni-based catalysts with advanced catalytic activity and enhanced anti-coke performance toward CO2 reforming of methane. Frontiers in Chemistry 2020, 8, 581923. [Google Scholar] [CrossRef]
- Beniya, A.; Higashi, S. Towards dense single-atom catalysts for future automotive applications. Nature Catalysis 2019, 2(7), 590–602. [Google Scholar] [CrossRef]
- Abrashev, M. V.; Chernev, P.; Kubella, P.; Mohammadi, M. R.; Pasquini, C.; Dau, H.; Zaharieva, I. Origin of the heat-induced improvement of catalytic activity and stability of MnO x electrocatalysts for water oxidation. Journal of Materials Chemistry A 2019, 7(28), 17022–17036. [Google Scholar] [CrossRef]
- VS, M. M.; Jose, S.; Varghese, A. Harnessing transition metal oxide-carbon heterostructures: Pioneering electrocatalysts for energy systems and other applications. Journal of Energy Storage 2024, 99, 113171. [Google Scholar]
- Wang, C.; Han, Z.; Zou, X.; Liu, H.; Wang, H.; Shu, D.; Suib, S. L. Ultrathin MnO2-coated FeOOH catalyst for indoor formaldehyde oxidation at ambient temperature: new insight into surface reactive oxygen species and in-field testing in an air cleaner. Environmental Science & Technology 2022, 56(15), 10963–10976. [Google Scholar]
- Datta, P.; Roy, D.; Jain, D.; Kumar, S.; Sil, S.; Bhunia, A.; Mandal, S. K. Uncovering the On-Pathway Reaction Intermediates for Metal-Free Atom Transfer Radical Addition to Olefins through Photogenerated Phenalenyl Radical Anion. ACS Catalysis 2024, 14(5), 3420–3433. [Google Scholar] [CrossRef]
- Gao, Y.; Li, F. Natural Gas Conversion to Olefins via Chemical Looping. In Direct Natural Gas Conversion to Value-Added Chemicals; CRC Press, 2020; pp. 71–94. [Google Scholar]
- Gao, Y. Ethylene Production with Active Lattice Oxygen Under a Cyclic Redox Scheme; North Carolina State University, 2019. [Google Scholar]
- Zhou, H.; Wang, L.; Xiao, F. S. Zeolites Containing Heteroatoms/Metal Nanoparticles for Catalytic Conversion of Light Alkanes. Micro-Mesoporous Metallosilicates: Synthesis, Characterization, and Catalytic Applications 2024, 423–446. [Google Scholar]
- Zhao, J., Liu, Z., Wang, D., Zhao, L., Shi, D., He, Z., ... & Valtchev, V. Atomically Dispersed Zinc Sites Embedded in a Layered Silicalite-2 Zeolite with Enhanced Stability for Propane Dehydrogenation. Available at SSRN 4975063.
- Ping, L.; Zhang, Y.; Wang, B.; Fan, M.; Ling, L.; Zhang, R. Unraveling the surface state evolution of IrO2 in ethane chemical looping oxidative dehydrogenation. ACS Catalysis 2023, 13(2), 1381–1399. [Google Scholar] [CrossRef]
- Fu, X. P.; Wu, C. P.; Wang, W. W.; Jin, Z.; Liu, J. C.; Ma, C.; Jia, C. J. Boosting reactivity of water-gas shift reaction by synergistic function over CeO2-x/CoO1-x/Co dual interfacial structures. Nature Communications 2023, 14(1), 6851. [Google Scholar] [CrossRef]
- Zhang, L. Heterogeneous Catalyst Design and Synthesis with Controlled and Well-defined Strategies; The University of Wisconsin-Madison, 2020. [Google Scholar]
- Rodriguez, J. A.; Chaturvedi, S.; Kuhn, M.; Hrbek, J. Reaction of H2S and S2 with metal/oxide surfaces: band-gap size and chemical reactivity. The Journal of Physical Chemistry B 1998, 102(28), 5511–5519. [Google Scholar] [CrossRef]
- Sun, S. Recent advances in hybrid Cu 2 O-based heterogeneous nanostructures. Nanoscale 2015, 7(25), 10850–10882. [Google Scholar] [CrossRef]
- Chaturvedi, S.; Rodriguez, J. A.; Brito, J. L. Characterization of pure and sulfided NiMoO 4 catalysts using synchrotron-based X-ray absorption spectroscopy (XAS) and temperature-programmed reduction (TPR). Catalysis letters 1998, 51, 85–93. [Google Scholar] [CrossRef]
- Ping, Y. Solvation effects on the band edge positions of photocatalyst surfaces. In Phys. Chem. Chem. Phys.; RSC Publishing, 2015; Volume 17, pp. 7075–7084. [Google Scholar]
- Chen, S. “Prediction of semiconductor band edge positions in aqueous environments from first-principles” 2012, arXiv:1203.1970v1.
- Abdullah, E. A. Band edge positions as a key parameter to a systematic classification of semiconductor photocatalysts. European Journal of Chemistry 2019, 10(1), 82–94. [Google Scholar] [CrossRef]
- Kessler, A.; Hedberg, J.; Blomberg, E.; Odnevall, I. Reactive oxygen species formed by metal and metal oxide nanoparticles in physiological media—a review of reactions of importance to nanotoxicity and proposal for categorization. Nanomaterials 2022, 12(11), 1922. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Chaturvedi, S.; Kuhn, M.; Hrbek, J. Reaction of H2S and S2 with metal/oxide surfaces: band-gap size and chemical reactivity. The Journal of Physical Chemistry B 1998, 102(28), 5511–5519. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y. Influence of preparation route on metal dispersion and sulfur adsorption efficiency of mixed metal oxides. Applied Catalysis B: Environmental 2018, 232, 145–156. [Google Scholar]
- Tawfik, A. M.; Shaban, M. Enhanced desulfurization performance of transition-metal-oxide catalysts: Role of preparation method on metal retention and surface chemistry. Journal of Environmental Chemical Engineering 2021, 9(5), 105–112. [Google Scholar]
- Zhang, L.; Wang, J.; Liu, Q. Metal leaching and dispersion challenges in supported catalysts prepared via filtration: Impacts on structural stability and reactivity. Catalysis Today 2019, 327, 194–203. [Google Scholar]
- Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and oxides. Acta Crystallographica Section A 1976, 32(5), 751–767. [Google Scholar] [CrossRef]
- Li, C.; Yu, J.; Wang, X.; Li, Z. Adsorption and activation of thiol and sulfide molecules on metal oxide surfaces: Influence of band structure and surface electron density. Applied Catalysis B: Environmental 2017, 203, 1–10. [Google Scholar]
- Campbell, C. T. Energetics of supported metal nanoparticles: Impact of size and support electronic properties on adsorption and reactivity. Journal of Catalysis 2013, 308, 380–390. [Google Scholar]
- Bartholomew, C. H.; Farrauto, R. J. Fundamentals of Industrial Catalytic Processes, 2nd ed.; Wiley, 2011. [Google Scholar]
- Chen, A.; Li, S.; Zhou, R.; Wang, X. Enhanced oxygen mobility and nickel stabilization in MgO-modified Ni catalysts for oxidation and reforming reactions. Applied Catalysis B: Environmental 2018, 224, 218–228. [Google Scholar]
- Szczepaniak, W.; Stojek, Z. Kinetics of silver sulfide film formation on silver in sulfide-containing environments. Corrosion Science 1997, 39(4–5), 889–908. [Google Scholar]
- Briand, L. E.; Mosqueda, H.; Henderson, M. A. The interaction of sulfur- and thiol-containing molecules with silver surfaces: Formation and growth of Ag₂S layers. Surface Science 2002, 515, 33–48. [Google Scholar]
- Hultquist, G.; Leger, P.; Seo, M. Sulphidation of silver and the formation of passivating Ag₂S layers. Corrosion Science 1991, 32(9), 1041–1053. [Google Scholar]
- Topsøe, H.; Nielsen, R. M. Activation of sulfur compounds on nickel-based catalysts: Fundamentals and performance expectations. Journal of Catalysis 1996, 161, 1–10. [Google Scholar]
- Oyama, S. T. Sulfur-resistant catalysts: Structure and reactivity of nickel and transition-metal sulfides. Catalysis Reviews 2003, 45(2), 231–287. [Google Scholar]
- Yao, H. C.; Yu Yao, Y. F. Ceria in catalysis: Formation of cerium–silicate phases and their impact on redox properties. Journal of Catalysis 1997, 172, 367–373. [Google Scholar]
- Li, G.; Smith, R. L.; Inomata, H. Effects of silica–ceria interactions on oxygen-vacancy formation and redox behavior in CeO₂–SiO₂ mixed oxides. Chemistry of Materials 2008, 20, 859–867. [Google Scholar]
- Bera, P.; Ray, B. Synergistic interactions in Pd-modified mixed metal oxides: Influence of support band structure on redox and adsorption behavior. Journal of Catalysis 2006, 244, 1–10. [Google Scholar]
- Rodriguez, J. A.; Goodman, D. W.; Bauschlicher, C. W. Electronic and structural effects in Pd-based oxide systems: Charge transfer, band-gap modification, and enhanced adsorption. Surface Science Reports 2000, 35, 1–81. [Google Scholar]
- Harris, W. R.; Raymond, K. N. Silver(I) complexation in aqueous solution: Stability and ligand-exchange behavior. Inorganic Chemistry 2005, 44, 1180–1187. [Google Scholar]
- Joussein, E.; et al. Halloysite clay minerals: Structure, properties and uses. Clay Minerals 2005, 40, 383–426. [Google Scholar] [CrossRef]
- Briand, L. E.; Mosqueda, H.; Henderson, M. A. The interaction of sulfur- and thiol-containing molecules with silver surfaces: Formation and growth of Ag₂S layers. Surface Science 2002, 515, 33–48. [Google Scholar]
- Rulíšek, L.; Havlas, Z. Aqueous solvation and hydrolysis of metal acetates: A theoretical study. Journal of the American Chemical Society 2000, 122, 10428–10439. [Google Scholar] [CrossRef]
- Sposito, G. The Chemistry of Soils, 2nd ed.; Oxford University Press, 2008. [Google Scholar]
- Campbell, C. T. Ultrathin metal films and supported metal catalysts: Bonding, energetics, and interactions with oxide supports. Surface Science Reports 1997, 27, 1–111. [Google Scholar] [CrossRef]
- Yi, D.; Huang, H.; Meng, X.; Shi, L. Desulfurization of liquid hydrocarbon streams via adsorption reactions by silver-modified bentonite. Industrial & Engineering Chemistry Research 2013, 52(18), 6112–6118. [Google Scholar] [CrossRef]
- Chen, J.; Wu, X.; Selloni, A. Electronic structure and bonding properties of cobalt oxide in the spinel structure. Physical Review B—Condensed Matter and Materials Physics 2011, 83(24), 245204. [Google Scholar] [CrossRef]
- Wöhrle, D.; Buck, T.; Hündorf, U.; Schulz-Ekloff, G.; Andreev, A. Phthalocyanines on mineral carriers, 4. Low-molecular-weight and polymeric phthalocyanines on SiO2, γ-Al2O3 and active charcoal as catalysts for the oxidation of 2-mercaptoethanol. Die Makromolekulare Chemie: Macromolecular Chemistry and Physics 1989, 190(5), 961–974. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, Y.; Chen, L.; Yang, L.; Zhou, L.; Luo, X.; Dai, W. Promoting the catalytic activity and SO2 resistance of CeO2 by Ti-doping for low-temperature NH3-SCR: Increasing surface activity and constructing Ce3+ sites. Chemical Engineering Journal 2023, 473, 145272. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Rodrıguez-Reinoso, F.; Sepúlveda-Escribano, A. Improved metal-support interaction in Pt/CeO2/SiO2 catalysts after zinc addition. Journal of Catalysis 2002, 210(1), 127–136. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, Y.; Chen, L.; Yang, L.; Zhou, L.; Luo, X.; Dai, W. Promoting the catalytic activity and SO2 resistance of CeO2 by Ti-doping for low-temperature NH3-SCR: Increasing surface activity and constructing Ce3+ sites. Chemical Engineering Journal 2023, 473, 145272. [Google Scholar] [CrossRef]
- Silvestre-Albero, J.; Rodrıguez-Reinoso, F.; Sepúlveda-Escribano, A. Improved metal-support interaction in Pt/CeO2/SiO2 catalysts after zinc addition. Journal of Catalysis 2002, 210(1), 127–136. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Chaturvedi, S.; Jirsak, T. The bonding of sulfur to Pd surfaces: photoemission and molecular–orbital studies. Chemical physics letters 1998, 296(3-4), 421–428. [Google Scholar] [CrossRef]
- Georgiadis, A. G.; Charisiou, N. D.; Goula, M. A. Removal of hydrogen sulfide from various industrial gases: A review of the most promising adsorbing materials. Catalysts 2020, 10(5), 521. [Google Scholar] [CrossRef]
- Bauer, S.E.; Mishchenko, M.I.; Lacis, A.A.; Zhang, S.; Perlwitz, J.; Metzger, S.M. Do sulfate and nitrate coatings on mineral dust have important effects on radiative properties and climate modeling? Journal of Geophysical Research: Atmospheres 2007, 112(D6). [Google Scholar] [CrossRef]
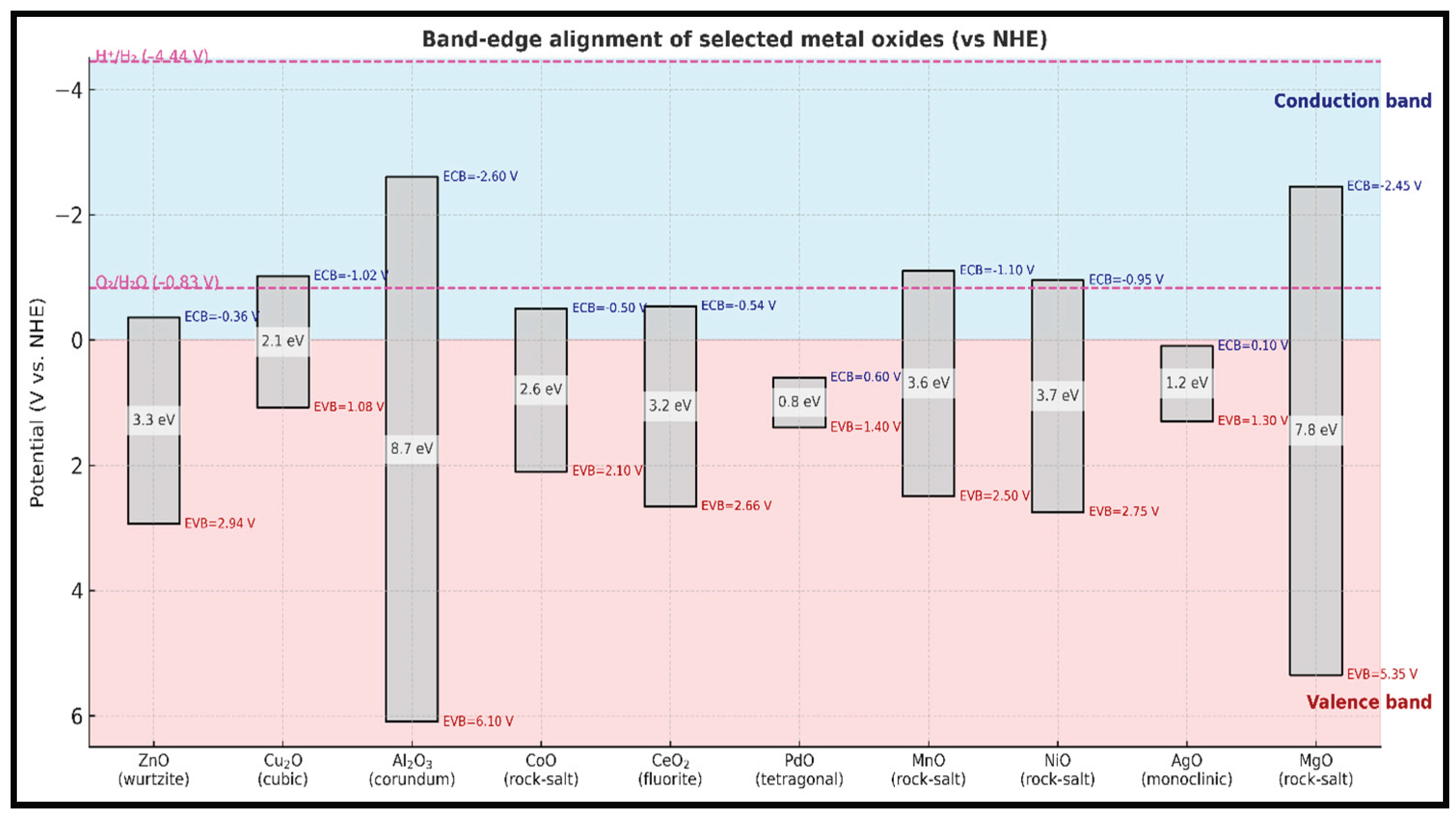
Figure 2.
Single metal on halloysite support catalyst performance.
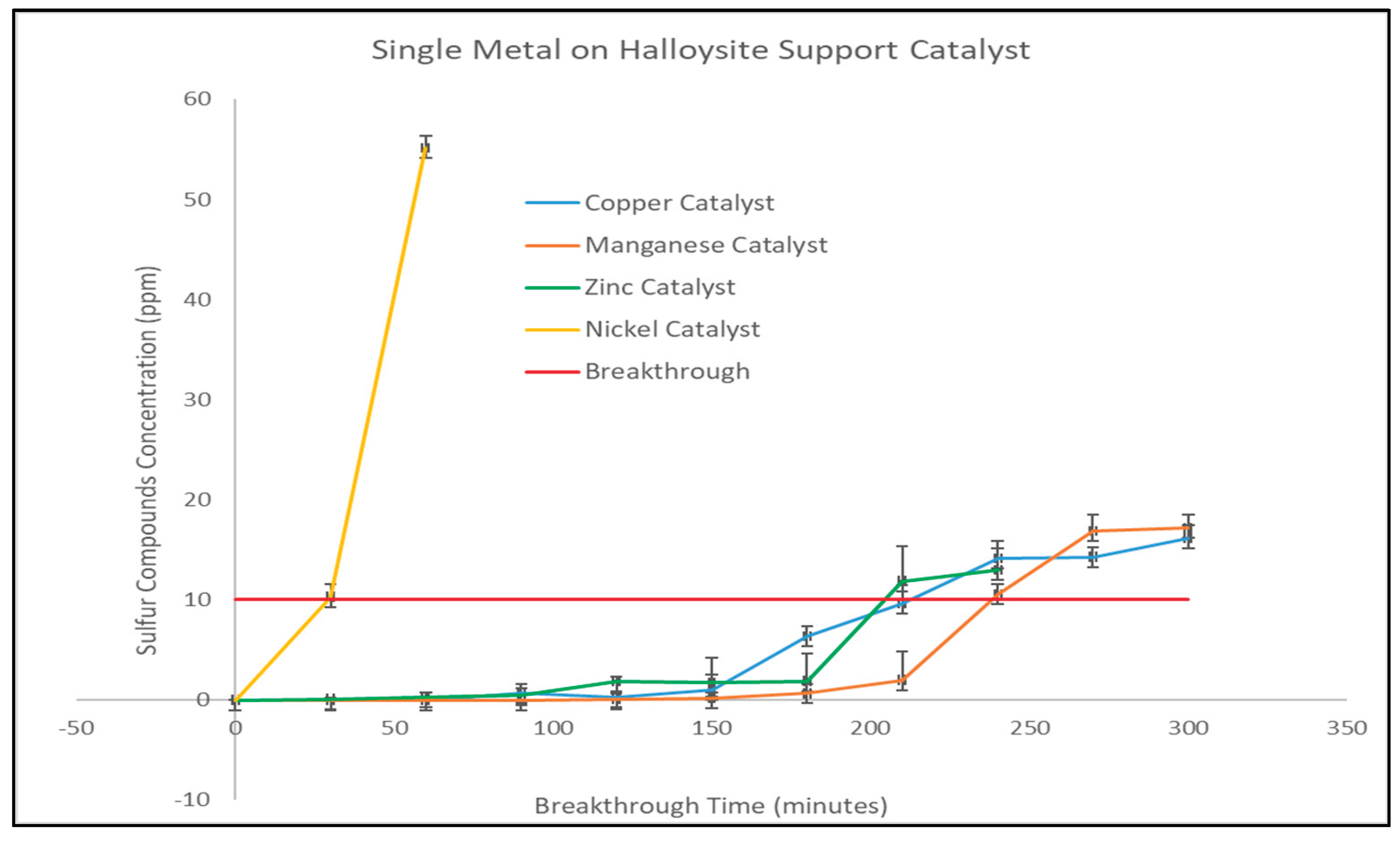
Figure 3.
Multiple metals adsorption performance.
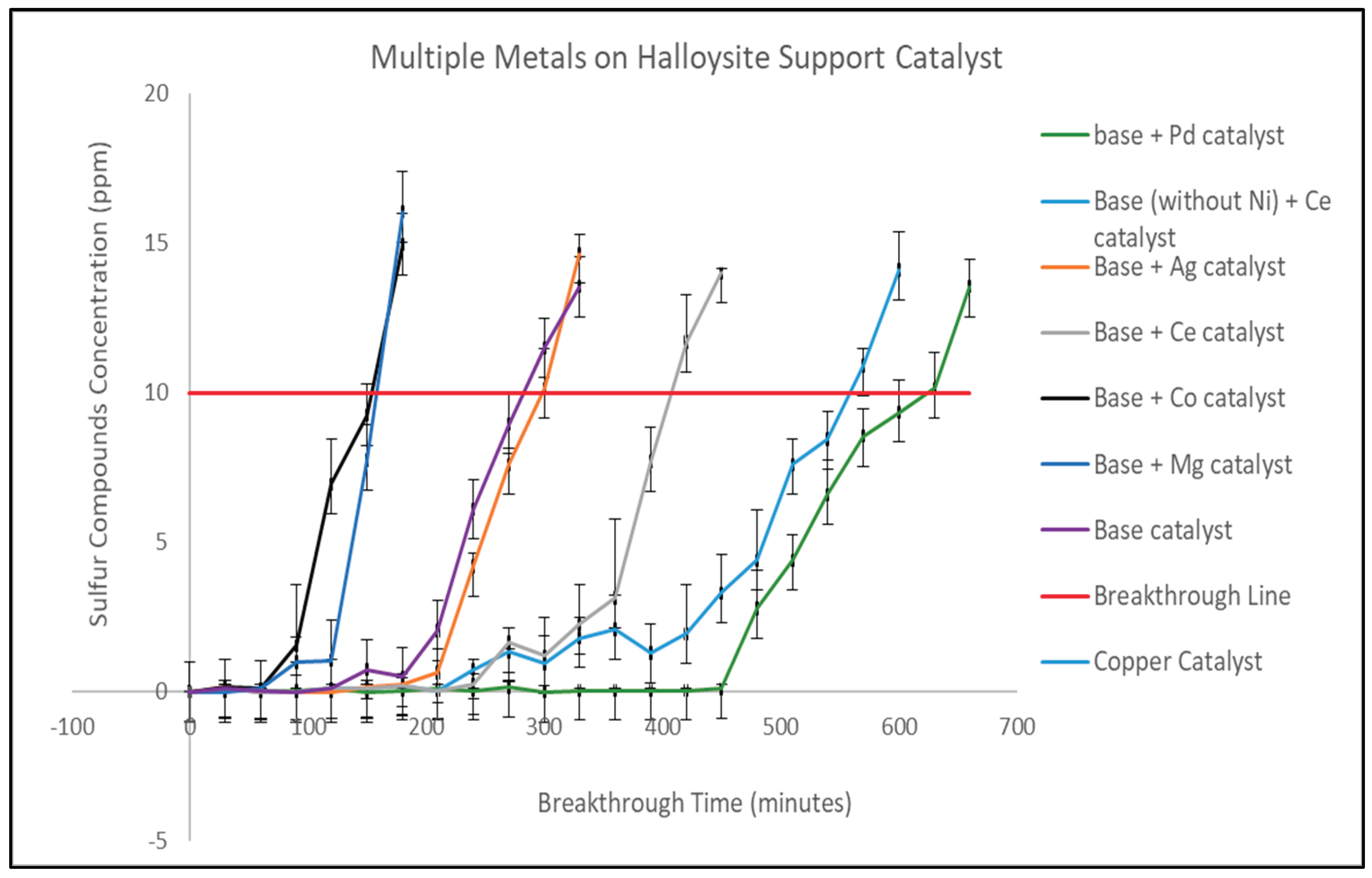
Figure 4.
FTIR Spectrum of the Multi-metal Catalyst.
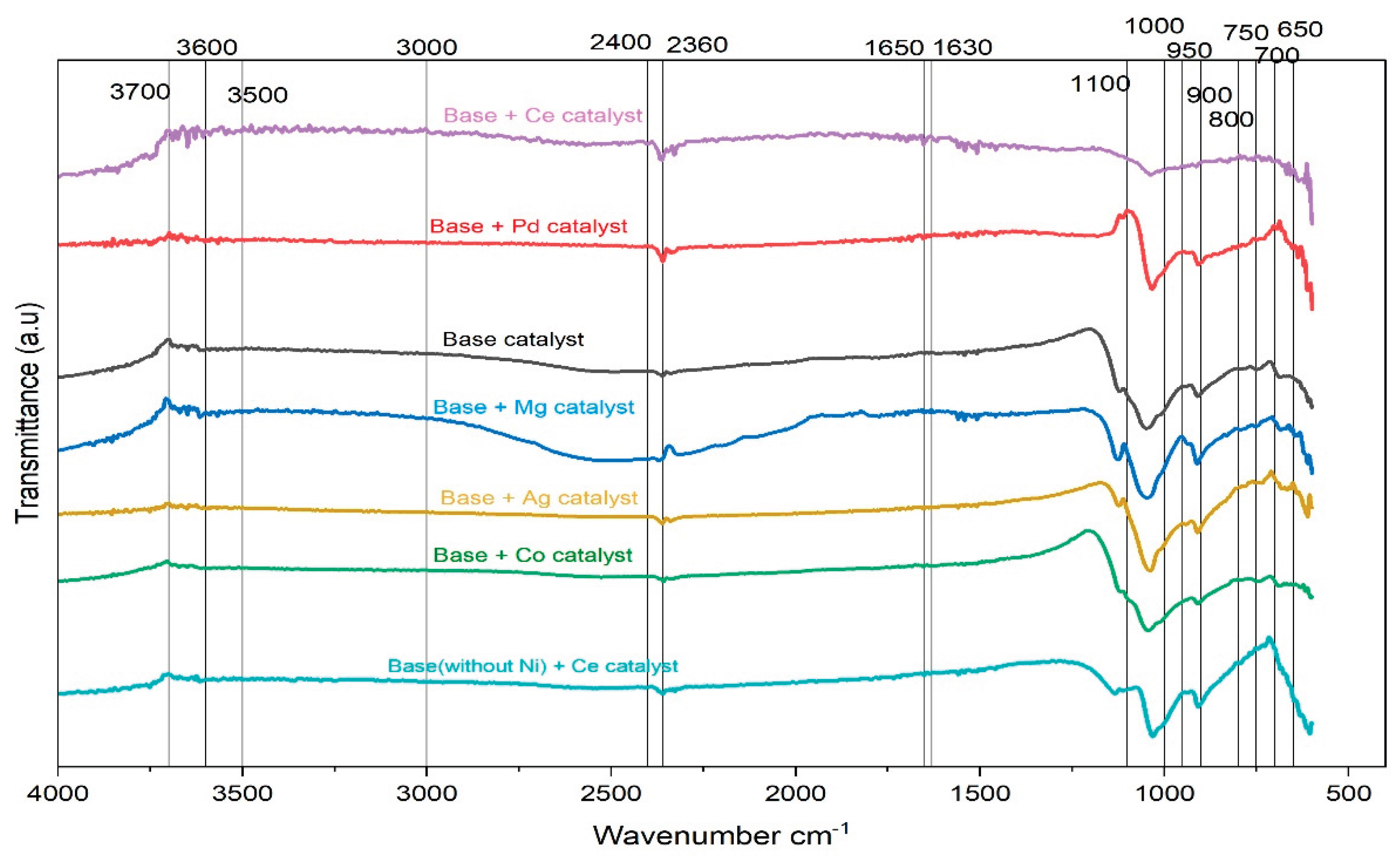
Figure 5.
EDS Spectrum of the Multi-metal Catalyst.
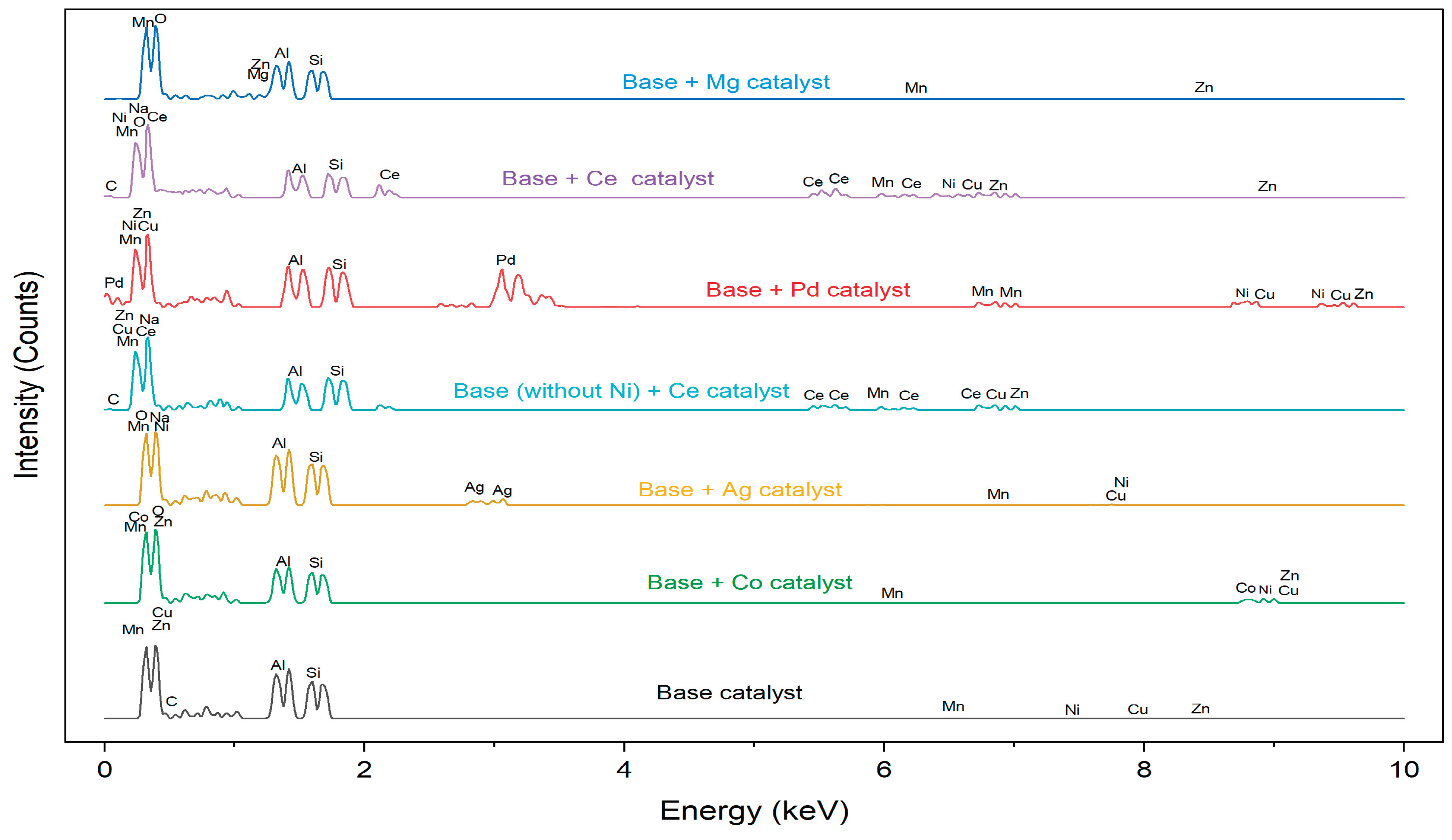
Figure 7.
Catalyst preparation process.
Figure 8.
The Experimental (Catalyst Testing) Set-up.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



