Submitted:
08 January 2026
Posted:
09 January 2026
You are already at the latest version
Abstract
For decades, biologists have been struggling to determine the structure of proteins efficiently. They were experimenting to find a method to accurately predict protein structures from their amino acid sequence. As a result of this research, Google DeepMind was able to introduce a Deep Learning approach to predict protein structures. It is AlphaFold2. AlphaFold2 could grab the attention of biologists because of its high impact on human beings. Also, the Nobel Prize for chemistry in 2024 was offered for this breakthrough invention. However, even though AlphaFold2’s exceptional accuracy, it has some limitations. This comprehensive review provides a systematic analysis of the main limitations of the AlphaFold 2 framework. The AlphaFold2 struggles to predict multiple conformations for the same sequence, the effects of point mutations, and antigen-antibody interactions. And AlphaFold2 fails to predict protein-DNA and protein-RNA complexes, nucleic acid structure, ligand and ion binding, post-translational modifications, and membrane plane for transmembrane domains. By systematically reviewing these limitations, we can use this review as a roadmap for future research to improve the legendary AlphaFold2 framework for the well-being of human beings.
Keywords:
structural biology
; computational biology AlphaFold
; protein structure prediction
; deep learning
; Evoformer
; multiple sequence alignment (MSA)
1. Introduction
In the 21st century, many breakthrough innovations and developments can be seen in the field of Artificial Intelligence due to the development of infrastructure and data availability. One of the major disciplines that is being rapidly developed is Deep Learning. The foundation of Deep Learning is Artificial Neural Networks that are inspired by human neurons. Deep learning allows computational models that are composed of multiple processing layers to learn representations of data with multiple levels of abstraction. Deep Learning can be used in various tasks like speech recognition, visual object recognition, object detection, and many other domains, such as drug discovery and genomics [1]. A milestone was achieved in 2012 by a deep convolutional neural network(CNN) named AlexNet, by updating records in the ImageNet competition [2]. This achievement proved that Deep Learning can also be used in complex computer vision tasks. This pace of development in deep learning was continued in the domain of Natural Language Processing (NLP) with the invention of the Transformer architecture [3]. Deep learning approaches have been able to achieve exceptional accuracy in many fields. The ability of deep learning for solving complex problems and deciphering complex patterns in nature inspired the scientist to apply this sophisticated technique to one of biology’s most formidable challenges: the protein folding problem.
Proteins are the building blocks of life, and every cell in the human body contains protein. The basic structure of a protein is an amino acid sequence. In nature, proteins exist as three-dimensional structures. Identifying these 3D structures is very significant for biological experiments. Because 3D structure is directly linked to its functionality, and knowing the structure allows scientists to understand its biological role. This knowledge is essential for designing new drugs and proteins, predicting how other molecules will interact with the protein, and understanding and treating diseases [4,5]. But identifying the 3D structure of a protein was a formidable challenge for decades. This challenge was defined as “Protein Folding Problem.” [6]. Scientists used experimental methods like X-ray crystallography and cryo-electron microscopy to discover the 3D structure of proteins. But these methods were very time-consuming and costly. Therefore, with the development of computer science, scientists tried to solve this problem using computational methods.
But many computational solutions did not achieve considerable accuracy. As the first reliable and successful solution was Google DeepMind’s AlphaFold2 [6]. Demonstrating unprecedented accuracy in the 14th Critical Assessment of protein Structure Prediction (CASP14). This deep learning-based system introduced a solution to this scientific challenge. This solution was recognized with the 2024 Nobel Prize in Chemistry. Before AlphaFold 2, determining protein structures was a time-consuming and resource-intensive process. Over more than 60 years, scientists worldwide have discovered around 150,000 protein structures using experimental methods. In contrast, within one and a half years, AlphaFold 2 predicted the structures of over 200 million proteins [6] — covering nearly every protein known to exist in nature.
However, AlphaFold2 has its own limitations. The single, static structural models predicted by AlphaFold2 are unable to fully capture the dynamic and context-dependent nature of protein function [4,6]. Also, it struggles to predict Multiple conformations for the same sequence, the effects of point mutations, and Antigen-antibody interactions. Furthermore, it can’t predict protein-DNA and protein-RNA complexes, nucleic acid structure, ligand and ion binding, post-translational modifications, and membrane plane for transmembrane domains. As the scientific community heavily depends on AlphaFold2's predictions for hypothesis design and experimental design, a critical and systematic understanding of its limitations is essential to prevent misinterpretation and to guide the next wave of innovation in the field.
This review provides a comprehensive analysis of the importance of the AlphaFold architecture, primary architectural features, the limitations of the AlphaFold2 framework, and approaches to overcome these limitations. We first present a concise overview of the model's core components, with a main focus on AlphaFold2, including its reliance on multiple sequence alignments. We then categorize and critically evaluate its key limitations, including its inability to model protein dynamics and conformational ensembles, its insensitivity to the structural effects of point mutations and post-translational modifications, and its architectural blind spots for intrinsically disordered proteins. Finally, we discuss emerging solutions, including the recent release of AlphaFold3, and outline the remaining challenges that define the future frontiers of computational structural biology.
“The rest of the paper is organized as follows: Section 2reviews the evolution of AlphaFold Architecture. Section 3describes the core limitations and challenges of the AlphaFold2 Framework. Section 4details the emerging solutions and the future. Section 5presents the conclusion.”
2. The Evolution of AlphaFold Architecture
2.1. The Predecessor: AlphaFold
The first version of AlphaFold, hereafter referred to as AlphaFold1, was introduced by DeepMind in 2018. In 2018, AlphaFold1 competed in CASP 13(Critical Assessment of protein Structure Prediction) [6]. In this competition, AlphaFold1 was able to predict previously unknown protein structures with state-of-the-art performance by outperforming all other competing applications, reaching a summed z-score of 52.8 versus 36.6 from the second place and, combining FM and FM/ TBM categories, achieved 68.3 z-score versus 48.2 .Even without using a template, AlphaFold1 also scored well in the TBM category [5].
AlphaFold1’s architecture was fundamentally a two-step process. The main component of AlphaFold1 is a convolutional neural network that was trained on the PDB (Protein Data Bank). The second major component is the gradient descent procedure that runs at inference time. To predict the structure of a protein, AlphaFold1 follows this two-step process. At the first step, it predicts the pairwise distances(distogram) between amino acid residues using a CNN. Second, this predicted distogram was used to construct a potential of mean force, which essentially served as a score function. An optimization technique, gradient descent, was then utilized to generate a 3D protein structure by adjusting its backbone torsion angles to satisfy the distance constraints defined by this potential. Even AlphaFold1 showed a state-of-the-art accuracy, it was unable to achieve the benchmark accuracy of 90%. Also, the separation of prediction and structure generation is another core limitation. These limitations motivated the DeepMind team to redesign the AlphaFold1 architecture to achieve benchmark accuracy of 90% [6].
2.2. The Breakthrough: The End-to-End Architecture of AlphaFold2
AlphaFold2 is considered one of the greatest contributions of Artificial Intelligence to the scientific field and one of the most significant scientific breakthroughs in the 21st century [7]. The release of AlphaFold2 marked a paradigm shift in protein structure prediction. AlphaFold2 adopts an architecture that is completely different from previous Deep Learning models, including AlphaFold1 [7,8]. While AlphaFold1 depends on a two-step process (predicting a distogram using a CNNs followed by a separate gradient descent optimization), AlphaFold2 introduced an end-to-end deep learning architecture that can predict 3D structure of proteins from a given amino acid sequence [6]. This state-of-the-art framework is heavily based on the attention mechanism-based Transformer architecture used in NLP [3]. AlphaFold2 competed at CASP14 in 2020, and AlphaFold2 predicted protein structures with an exceptionally high accuracy than other competing applications, demonstrating a root-mean-square deviation (RMSD) among prediction and experimental backbone structures of 0.8Å versus the 2.8Å from the next best performing application [5]. Check the Figure 1.
The AlphaFold2 architecture operates as a unified pipeline consisting of three integrated modules. They are Input Module, Evoformer module and Structure Module (check Figure 2) [6,7]. Unlike traditional Deep Learning approaches that process one input at a time, AlphaFold2 processes evolutionary and geometric data of the provided amino acid sequence simultaneously. The pipeline begins with two database searches. One is a genetic database search to construct a Multiple Sequence Alignment (MSA). The other one is structural database search for identify the template structures of similar amino acid sequences. Then these inputs are passed through the Evoformer module. Finally, the structure module, which achieves the transition from the abstract representation of protein structure to 3D atom coordinates of the target protein [7].
A key distinguishing architectural feature of AlphaFold2 is parallel data representation. The AlphaFold2 model maintains two parallel data streams. They are MSA representation and Pair representation. The input module takes the amino acid sequence and finds its homologs in sequence databases, and constructs MSA by aligning the input sequence and its homolog sequences. AF2 also checks whether any of the homologs has a 3D structure available in protein structure databases, and constructs a pairwise distance matrix between amino acids. Then AF2 generates MSA representation and pair representation [6,7]. Here homolog is a protein with a shared evolutionary origin, that they evolved from a ancestor [9]. This same ancestry strongly implies they fold into similar 3D structures and perform related functions, even if their sequences differ [10].
The heart of the AlphaFold2 architecture is the Evoformer (check Figure 3), a custom transformer-based module that contains 48 blocks [5,6]. Unlike conventional transformers used for Natural Language Processing, the Evoformer is designed to exchange information between the MSA representation (evolutionary) and Pairwise representation (geometric). The MSA information can be reinterpreted as the pairwise information is improved, and in a similar way, the pairwise information can be further improved as the MSA information is reinterpreted [7]. Within each block of evoformer, the model employs novel attention mechanisms, such as Triangular Self-Attention. This mechanism refines the pair representation by not violating the triangle inequality rule on the distance matrix. This ensures that the predicted distances between residues are geometrically consistent.
In the Evoformer module, AlphaFold2 takes two inputs from the input module and passes them through a deep neural network. Each block in 48 blocks take two inputs: an MSA representation and a pair representation. The outputs from each Evoformer block are an updated MSA representation and an updated pair representation (check Figure 3). Each Evoformer block contains two pathways and two communication channels. The first pathway works on the MSA. It computes attention over a large matrix of protein symbols. The MSA attention is factorized in row-wise gated self-attention and column-wise gated self-attention components. The row-wise gated self-attention allows the network to identify which pairs of amino acids are more related and combines the information from the input pair representation with MSA. The column-wise gated self-attention allows the network to determine which sequences are more informative. The second pathway works on the pair representation. The main feature of this pathway is that attention is organized around the triangles of residues, which is based on the triangle inequality theorem. As shown in Figure 3, the first two steps are triangular multiplicative updates, which are based on non-attention method. The second two steps are triangular self-attention. They update the pair representation in the Evoformer block.
The final module of the pipeline, the structure module, is responsible for generating the 3D structure(check figure 4) [6,7]. Instead of predicting a static distance map (as in AlphaFold1 [8]), this module predicts the rotation and translation of each amino acid residue. To achieve this, DeepMind introduced Invariant Point Attention (IPA), a specialized attention mechanism that ensures the prediction respects the laws of Euclidean geometry [6,7]. This allows the network to construct the protein backbone and side chains directly, predicting the specific torsion angles for every atom.To gain maximum accuracy, AlphaFold2 utilizes a process known as “recycling”. The output 3D structure from the Structure module are fed back into the network as input for three iterations. This allows the model to progressively refine the output structure [6,7].
Multiple reasons contribute to the outstanding success of AlphaFold2. Unarguably, the most critical factor is the delicate algorithms used, such as attention mechanisms-based transformers [6,7]. In AlphaFold2, several types of attention mechanisms are used, with each one focusing on a specific learning task. The training method is also a cause of the success of AlphaFold2. The researchers have utilized the concept of self-distillation for the training phase [12]. The25% of the training data comes from the known structures in PDB(Protein Data Bank), while 75% of the data was from the new self-distillation data set. Other approaches that may contribute to the success of AlphaFold2 are the recycling approach and the end-to-end framework.
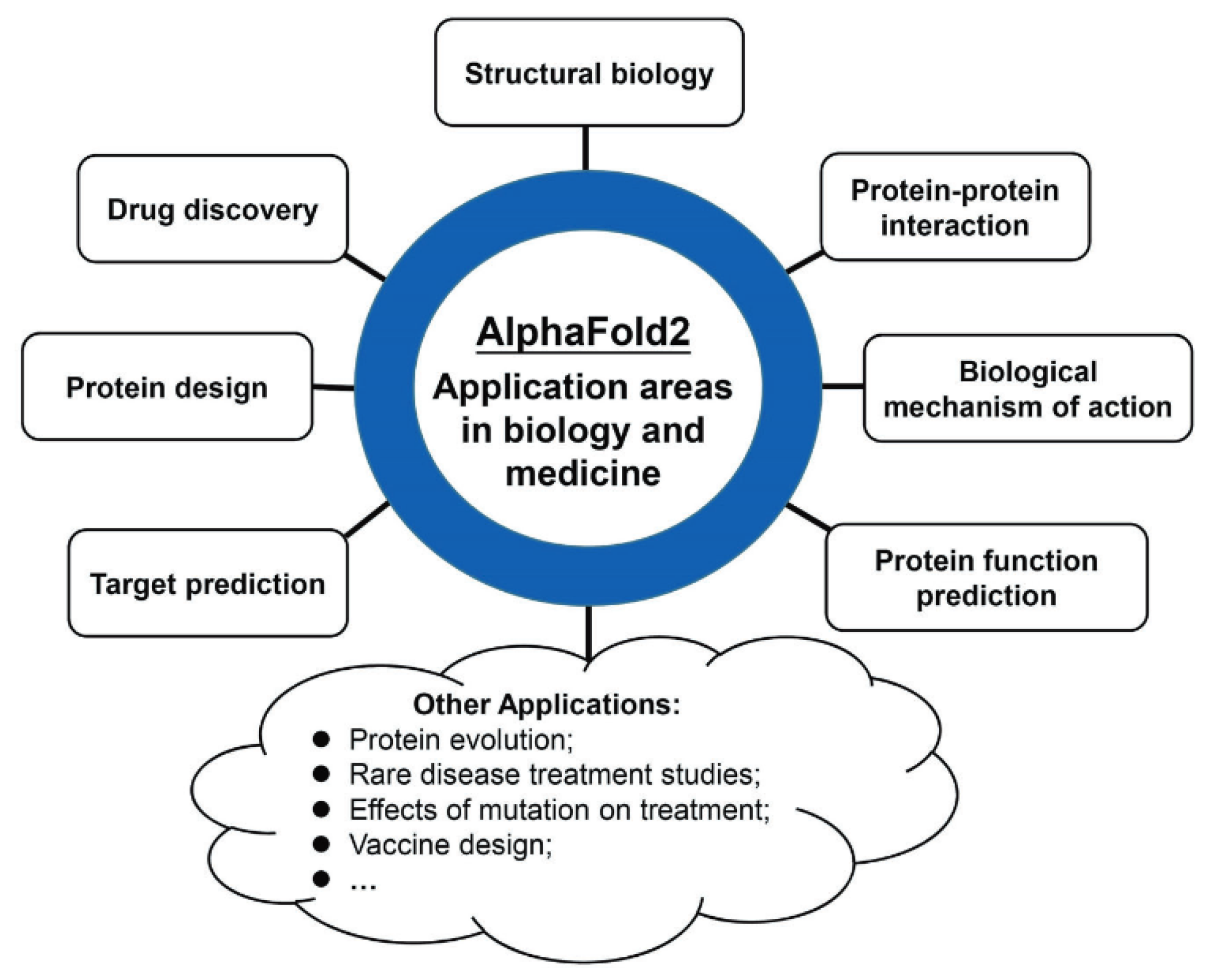
The outstanding accuracy of AlphaFold2 has revolutionized multiple disciplines in biology and medicine. The applications of AlphaFold2 can be categorized into 8 categories: structural biology, drug discovery, protein design, target prediction, protein function prediction, protein-protein interaction, biological mechanism of action, and others (check Figure 5) [7]. In structural biology, AlphaFold2 predictions are now used to assist experimental methods, such as solving the ‘phasing problem’ in X-ray crystallography and fitting atomic models into Cryo-EM density maps. Also, AlphaFold2 has significantly improved drug discovery. Furthermore, the architecture has been modified to protein design, enabling the engineering of new enzymes and therapeutics. By predicting protein structures of 200 million proteins, AlphaFold2 has fundamentally accelerated research in the fields of biology and medicine.
3. Core Limitations And Unsolved Challenges Of The Static Model
While AlphaFold2 has been recognized as the most reliable answer to the protein folding problem, recent research suggests that it has successfully predicted “static folds” rather than solving the complex reality of protein structures [13]. AlphaFold2 was originally trained on single protein chains, so it excels at predicting their structures. But there are some aspects of the structures that AlphaFold2 can not predict accurately. Also, another common disadvantage of AlphaFold2 is that it requires a lot of computational resources to run the model locally (about 3TB of disk space and a powerful NVIDIA GPU with GBs of memory). Another cause that affects to accuracy of AlphaFold2 is not utilizing energy functions that seek to identify native-like protein conformations [13]. Crucially, AlphaFold2 does not simply replicate known protein structures. Recent research has proved that AlphaFold2 can predict structures never previously seen by the model or seen in PDB [14,15,16].
3.1. The Static Structure Problem: Modeling Conformational Dynamics
Protein dynamics is a very important area in structural biology [17,18,19,20,21]. The protein structure predicted by AlphaFold2 is a static state structure. However, proteins are naturally very dynamic with multiple states. Many significant pathological and physical proteins have multiple subtle conformational changes. And these proteins show ever-changing spatial configurations. But AlphaFold2 usually gives a single optimal solution, which is hard to cover the conformational diversity of proteins. Although this does not mean that it is not possible to predict the dynamic nature of proteins with AF2 [7,13]. According to some recent research, AlphaFold2 can still be used for some predictions of protein dynamics.
3.2. Challenges in Predicting Biomolecular Interactions
While AlphaFold2 has achieved a state-of-the-art accuracy for monomeric polypeptide chains, it faces significant difficulties when predicting interactions within complex biological environments. The standard version of AlphaFold2 model was trained on amino acid sequences, creating a “blind spot” for essential biomolecules such as DNA, RNA, lipid membranes, and small-molecule ligands [7] This is a significant disadvantage for drug discovery, as the model can not predict the location of metal ions, cofactors, or post-translational modifications(PTMs), which are usually required for protein function [5]. The AlphaFold2 model also exhibits difficulties with environmental context. In transmembrane proteins like ChRmine, AlphaFold2 fails to account for the physical constraints of the lipid bilayer. leading to predictions where transmembrane domains are distorted or non-physiologically placed [13].
3.3. Insensitivity to Point Mutations
AlphaFold2 is not sensitive to point mutations that change a single residue, due to the alteration in the DNA sequence. Point mutations can be frequently seen in proteins, especially in pathological states. Understanding the effects of missense mutations of protein structures will help to understand their biological mechanisms. Even though AlphaFold2 predicts the static structure of proteins very well, it performs poorly in predicting the effect of missense mutations of proteins’ 3D structures.
Furthermore, predicting structures of proteins with post-translational modifications, orphan proteins, and artificially designed proteins remains a significant challenge for AlphaFold2.
4. Future Directions and Outlook
Despite AlphaFold2’s excellent accuracy, understanding its core limitations inspires us to modify its architecture and training strategies to overcome these limitations in future versions of AlphaFold architecture. The next phase of computational biology is focused on modeling the dynamic, interactive behavior of proteins.
4.1. From Static Snapshots to Dynamic Ensembles
The "protein folding problem" was never just about finding a single set of coordinates; it was about understanding how a protein moves through an energy landscape. As discussed in Section 3, AlphaFold2 is fundamentally limited by its training on static crystal structures, forcing it to output a single "ground state." The future lies in Generative AI, specifically diffusion models—the same technology used by image generators like Midjourney. Unlike the approach of AlphaFold2, which aims for the single "best" answer, stochastic diffusion models can sample the full Boltzmann distribution of a protein. This shift is critical. It will allow researchers to generate not just one structure, but a movie of structures, capturing the transition states of enzymes and the opening-and-closing mechanisms of ion channels that AlphaFold2 currently misses [22].
4.2. The Challenge of "Structural Hallucination"
The shift toward generative models introduces a new risk that was less prevalent in the multiple sequence alignment (MSA)-based era of AlphaFold2: hallucination. Because newer architectures like AlphaFold 3 and RFdiffusion rely less on evolutionary history and more on learned physical probabilities, they possess the capacity to invent plausible-looking structures that do not exist in nature. A model might generate a protein with perfect bond angles and helical geometry that, in reality, would fail to fold or aggregate in a test tube.[23] As these models become central to de novo protein design, the field faces an urgent need for robust verification metrics. We must develop "guardrail" algorithms that can distinguish between a novel, stable protein design and a convincing deep-learning hallucination.
4.3. Unified Biomolecular Modeling for Drug Discovery
Perhaps the most immediate frontier is the expansion of the "biological context." Proteins rarely function in the isolation of a pure peptide chain; they operate in a crowded environment, interacting with DNA, RNA, ions, and small molecules. The release of AlphaFold 3 in 2024 marked the first major step toward solving this by replacing the Evoformer with the Pairformer architecture, allowing the system to process general chemical tokens rather than just amino acids [24]. This evolution is set to revolutionize drug discovery. Instead of predicting a protein pocket and then using separate physics simulators to dock a drug (a process prone to error), future models will predict the protein-drug complex in a single inference step. This capability to model "induced fit"—where the drug and protein change shape to accommodate each other—will likely become the standard for pharmaceutical research in the coming decade.
4.4. The Synergy of AI and Experimentation
Ultimately, the relationship between AI and wet-lab experiments is shifting from competition to integration. AI will not replace experimental structural biology; it will guide it. We are already seeing the emergence of "Active Learning" loops, where an AI model predicts a structure, a robot in the lab synthesizes it, and the physical results are immediately fed back to fine-tune the model [25]. Furthermore, deep learning is becoming indispensable for interpreting noisy experimental data, such as resolving blurry Cryo-EM density maps or assigning complex NMR spectra. The future represents a hybrid workflow where experimental data provides the ground truth, and AI provides the speed and interpolation necessary to map the entire proteome.
5. Conclusions
The evolution of AlphaFold represents a milestone of structural biology. By transitioning from CNN based architecture of AlphaFold1 to the attention-based Evoformer architecture of AlphaFold2, DeepMind achieved near experimental-level accuracy. The integration of deep learning concepts with fundamental knowledge of chemistry, biology and mathematics for designing this architecture is really brilliant. Also, their training strategies, like self-distillation, contributed to its exceptional accuracy. This top-notch architecture’s contribution for structural biology was recognized with the Nobel Prize in chemistry in 2024. However, as this review has highlighted, a static structure is not able to fully represent the real world behavior of a protein. Furthermore, limitations like the bias toward rigid crystal structures, the insensitivity to point mutations, and the inability to natively model complex biochemical environments reveal that the protein folding problem is not fully resolved. To resolve these limitations, experts in this field are focusing on generative diffusion models rather than predictive models. These diffusion models are capable of sampling dynamic conformational ensembles. As a result of this, Alpha fold3 was introduced. Ultimately, the AlphaFold will not replace the experimental methods of structural biology, but rather establishing a powerful synergy where deep learning serves as the primary engine for hypothesis generation. The continued evolution of the AlphaFold architecture promises to drive transformative advances in drug discovery and our understanding of life at the molecular level.
References
- Y. LeCun, Y. Bengio, and G. Hinton, “Deep learning,”. Nature 2015, vol. 521(no. 7553), 436–444. [CrossRef]
- A. Krizhevsky, I. Sutskever, and G. E. Hinton, “ImageNet classification with deep convolutional neural networks,”. Commun. ACM 2017, vol. 60(no. 6), 84–90. [CrossRef]
- Vaswani et al., “Attention Is All You Need,” Aug. 02, 2023, arXiv:1706.03762. 2023. [CrossRef]
- R. Nussinov, M. Zhang, Y. Liu, and H. Jang, “AlphaFold, Artificial Intelligence (AI), and Allostery,”. J. Phys. Chem. B 2022, vol. 126(no. 34), 6372–6383. [CrossRef]
- L. M. F. Bertoline, A. N. Lima, J. E. Krieger, and S. K. Teixeira, “Before and after AlphaFold2: An overview of protein structure prediction,”. Front. Bioinforma. 2023, vol. 3, 1120370. [CrossRef]
- J. Jumper et al., “Highly accurate protein structure prediction with AlphaFold,”. Nature 2021, vol. 596(no. 7873), 583–589. [CrossRef] [PubMed]
- Z. Yang, X. Zeng, Y. Zhao, and R. Chen, “AlphaFold2 and its applications in the fields of biology and medicine,”. Signal Transduct. Target. Ther. 2023, vol. 8(no. 1), 115. [CrossRef] [PubMed]
- A. W. Senior et al., “Improved protein structure prediction using potentials from deep learning,”. Nature 2020, vol. 577(no. 7792), 706–710. [CrossRef]
- W. M. Fitch, “Distinguishing Homologous from Analogous Proteins,”. Syst. Zool. 1970, vol. 19(no. 2), 99. [CrossRef]
- Chothia and A. M. Lesk, “The relation between the divergence of sequence and structure in proteins.,”. EMBO J. 1986, vol. 5(no. 4), 823–826. [CrossRef]
- Q. Xie, M.-T. Luong, E. Hovy, and Q. V. Le, “Self-training with Noisy Student improves ImageNet classification,”. arXiv 2019. [CrossRef]
- V. Agarwal and A. C. McShan, “The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins,”. Nat. Chem. Biol. 2024, vol. 20(no. 8), 950–959. [CrossRef]
- N. Bordin et al., “AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms,”. Commun. Biol. 2023, vol. 6(no. 1), 160. [CrossRef]
- Barrio-Hernandez et al., “Clustering predicted structures at the scale of the known protein universe,”. Nature 2023, vol. 622(no. 7983), 637–645. [CrossRef] [PubMed]
- Durairaj et al., “Uncovering new families and folds in the natural protein universe,”. Nature 2023, vol. 622(no. 7983), 646–653. [CrossRef] [PubMed]
- M. Dobson, “Protein folding and misfolding,”. Nature 2003, vol. 426(no. 6968), 884–890. [CrossRef] [PubMed]
- V. Daggett and A. R. Fersht, “Is there a unifying mechanism for protein folding?,”. Trends Biochem. Sci. 2003, vol. 28(no. 1), 18–25. [CrossRef]
- S. Glazer, R. J. Radmer, and R. B. Altman, “Improving Structure-Based Function Prediction Using Molecular Dynamics,”. Structure 2009, vol. 17(no. 7), 919–929. [CrossRef]
- M. C. Childers and V. Daggett, “Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles,”. J. Phys. Chem. B 2018, vol. 122(no. 26), 6673–6689. [CrossRef]
- Y. I. Yang, Q. Shao, J. Zhang, L. Yang, and Y. Q. Gao, “Enhanced sampling in molecular dynamics,”. J. Chem. Phys. 2019, vol. 151(no. 7), 070902. [CrossRef]
- H. K. Wayment-Steele et al., “Predicting multiple conformations via sequence clustering and AlphaFold2,”. Nature 2024, vol. 625(no. 7996), 832–839. [CrossRef]
- L. Watson et al., “De novo design of protein structure and function with RFdiffusion,”. Nature 2023, vol. 620(no. 7976), 1089–1100. [CrossRef]
- J. Abramson et al., “Accurate structure prediction of biomolecular interactions with AlphaFold 3,”. Nature 2024, vol. 630(no. 8016), 493–500. [CrossRef]
- T. C. Terwilliger et al., “AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination,”. Nat. Methods 2024, vol. 21(no. 1), 110–116. [CrossRef]
Figure 1.
The performance of AlphaFold on the CASP14 dataset, (n = 87 protein domains) relative to the top15 entries (out of 146 entries) (Adapted from [6]).
Figure 1.
The performance of AlphaFold on the CASP14 dataset, (n = 87 protein domains) relative to the top15 entries (out of 146 entries) (Adapted from [6]).
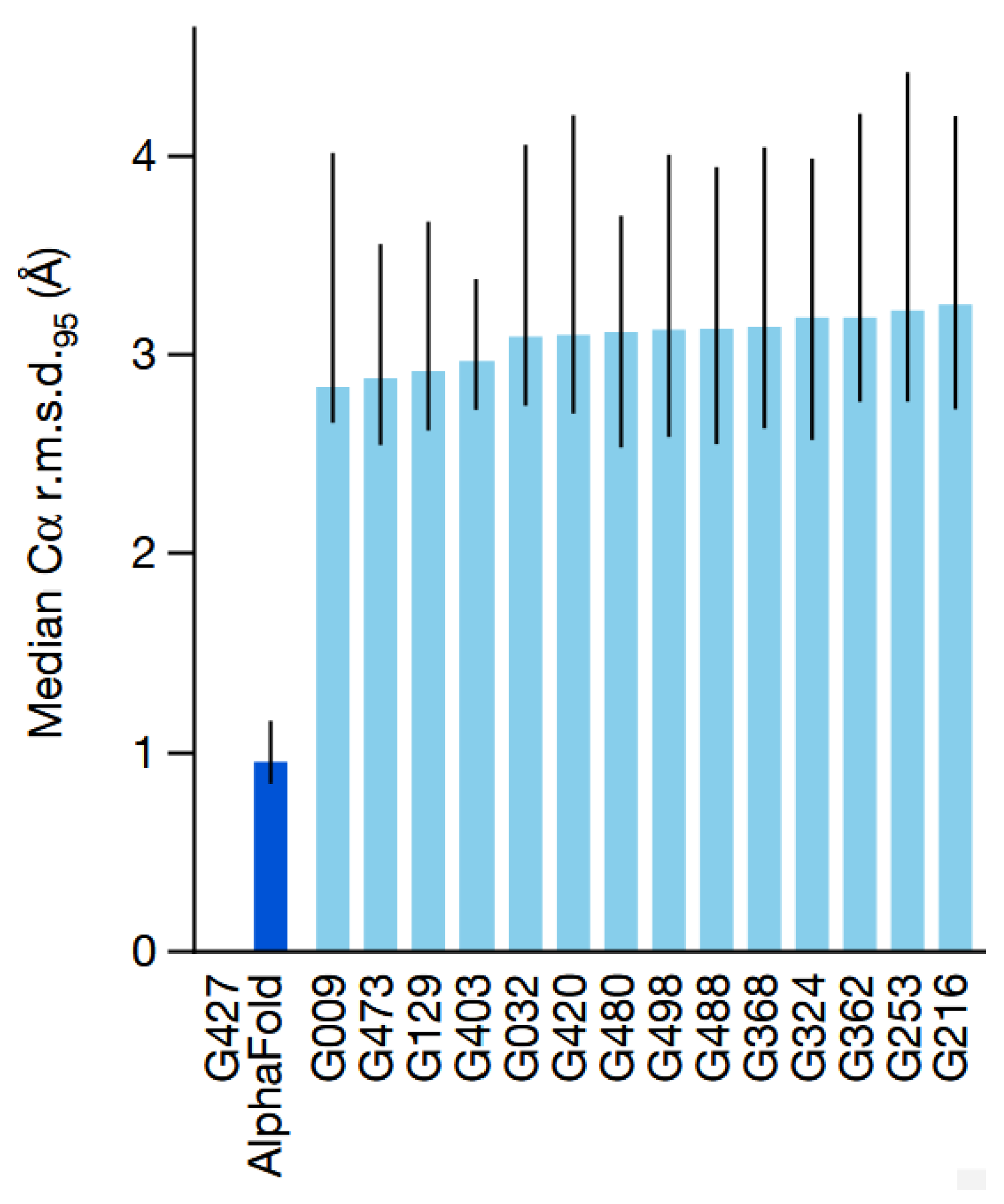
Figure 2.
AlphaFold2 architectural diagram. Arrows show the information flow among the various components described in this paper. Array shapes are shown in parentheses with s, number of sequences r, number of residues c, number of channels. (Adapted from [6]).
Figure 2.
AlphaFold2 architectural diagram. Arrows show the information flow among the various components described in this paper. Array shapes are shown in parentheses with s, number of sequences r, number of residues c, number of channels. (Adapted from [6]).
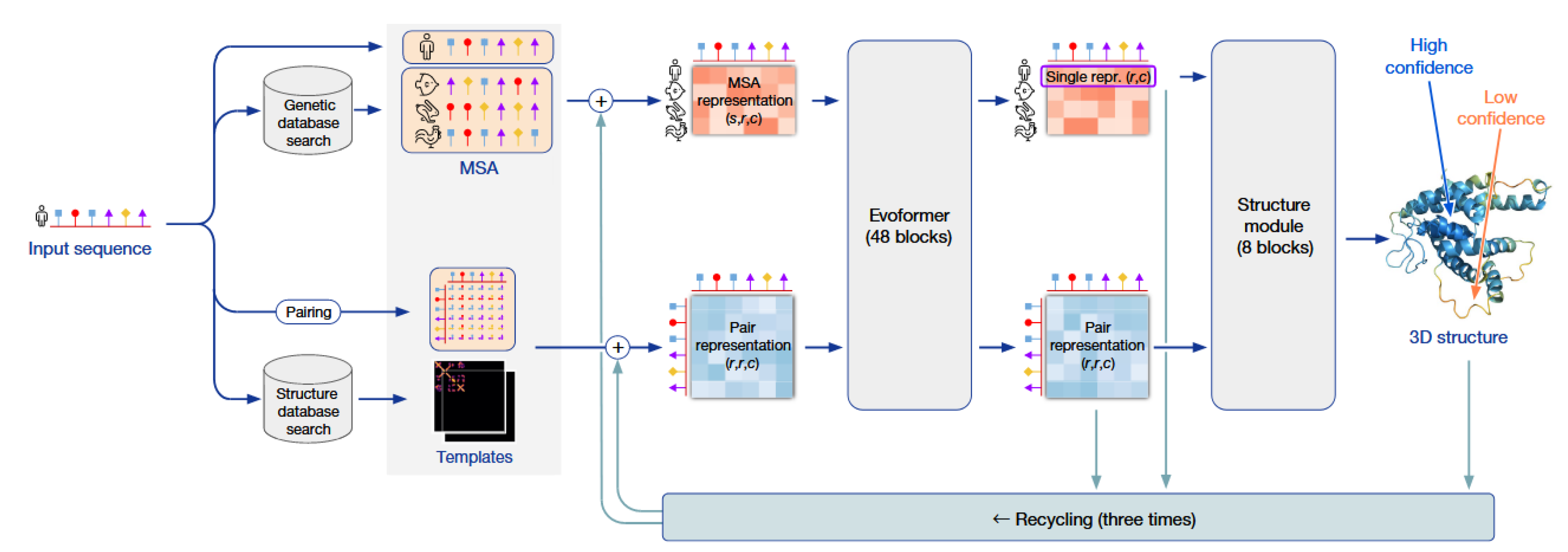
Figure 3.
Components of a block in Evoformer. Evoformer contains 48 blocks with weights not shared. The MSA representation and the pair representation are renewed through each block. (Adapted from [7]).
Figure 3.
Components of a block in Evoformer. Evoformer contains 48 blocks with weights not shared. The MSA representation and the pair representation are renewed through each block. (Adapted from [7]).
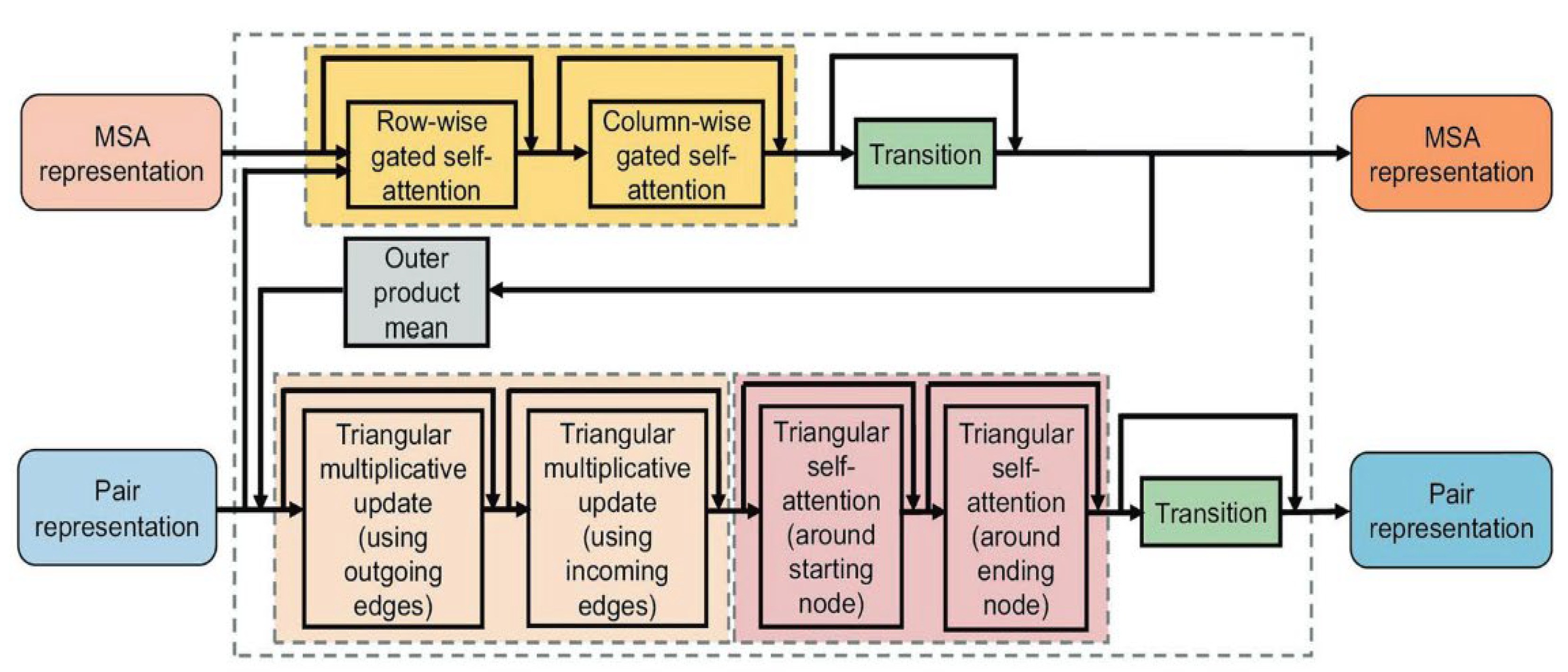
Figure 4.
Components of a block in the structure module. Structure module contains 8 blocks with shared weights. Single representation and backbone frames are updated through each block of the structure module. (Adapted from [7]).
Figure 4.
Components of a block in the structure module. Structure module contains 8 blocks with shared weights. Single representation and backbone frames are updated through each block of the structure module. (Adapted from [7]).
Figure 5.
Application areas of AF2 in the fields of biology and medicine. (Adapted from [7]).
Figure 5.
Application areas of AF2 in the fields of biology and medicine. (Adapted from [7]).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



