
Submitted:
07 January 2026
Posted:
08 January 2026
You are already at the latest version
Abstract
Background: TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) is caused by biallelic pathogenic variants in TNXB, encoding the extracellular matrix glycoprotein tenascin-X. Although traditionally classified as a connective tissue disorder based on joint hypermobility and skin findings, accumulating clinical, electrophysiological, and imaging data indicate prominent neuromuscular involvement that likely reflects a central disease mechanism. Methods: A qualitative evidence synthesis was conducted following PRISMA 2020 guidelines. A comprehensive search of PubMed, OMIM, and GeneReviews was performed on January 5, 2026. Data from 18 studies representing 56 individuals with biallelic TNXB variants were synthesized narratively, with findings stratified by assessment method and zygosity. Due to heterogeneity in study designs, assessment methods, and outcome definitions, quantitative meta-analysis was not feasible. Results: Among 56 individuals with biallelic TNXB variants, subjective muscle weakness was reported in only 37% of cases. However, systematic neuromuscular assessment demonstrated objective muscle weakness in 85% of patients examined. Electromyography revealed mixed neurogenic-myopathic patterns in 60%, and muscle imaging abnormalities were present in approximately 50%. A clear dose-effect relationship was observed, with heterozygous individuals exhibiting milder phenotypes correlating with reduced serum tenascin-X levels. Conclusion: Neuromuscular involvement in TNXB-related disorders is frequent, progressive, and mechanistically linked to dysfunction at the muscle-extracellular matrix interface. These findings support the reclassification of TNXB-related disease alongside myopathic Ehlers-Danlos syndrome as a muscle-ECM interface disorder.
Keywords:
clEDS
; Ehlers-Danlos syndrome
; electromyography
; muscle-ECM interface
; qualitative evidence synthesis
; tenascin-X
; TNXB
What This Study Adds
What is already known: TNXB-related classical-like Ehlers-Danlos syndrome (clEDS) is traditionally classified as a connective tissue disorder based on joint hypermobility and skin findings. While neuromuscular manifestations have been noted in case reports, they have historically been viewed as secondary features. Myopathic EDS (mEDS) was recently created to recognize disorders defined by muscle-extracellular matrix (ECM) interface dysfunction.
What this study adds: Qualitative synthesis of available evidence reveals that objective muscle weakness is present in 85% of patients when systematically assessed, significantly higher than the 37% reported subjectively. The synthesis identifies a distinct mixed neurogenic-myopathic EMG pattern in 60% of cases, suggesting neuromuscular pathology is a primary disease mechanism. Furthermore, a dose-effect relationship is established where serum tenascin-X levels (averaging 56% in heterozygotes) correlate with phenotype severity.
How this might change clinical practice: The evidence supports the reclassification of TNXB-related disorders as "Muscle-ECM Interface Disorders" alongside COL12A1-related myopathic EDS. This shift necessitates routine neuromuscular screening, quantitative strength testing, and specialized physical therapy focused on force transmission optimization for all affected individuals.
Introduction
The Ehlers-Danlos syndromes (EDS) comprise a heterogeneous group of heritable connective tissue disorders classically characterized by joint hypermobility, skin hyperextensibility, and tissue fragility. The 2017 international classification recognizes 13 EDS subtypes [1]. Classical-like EDS (clEDS) is caused by biallelic pathogenic variants in TNXB, encoding the extracellular matrix glycoprotein tenascin-X [2]. Tenascin-X is highly expressed in loose connective tissues, including skin, muscle, and tendons, where it interacts with fibrillar collagens to organize matrix architecture.
Experimental models demonstrate that tenascin-X deficiency disrupts collagen XII deposition, particularly at the muscle-ECM interface [11,12]. Collagen XII binds to collagen I-containing fibrils via its collagenous domain, while its large non-collagenous NC3 domain interacts directly with tenascin-X, establishing a biochemical link between these proteins [12]. Biomechanical studies in tenascin-X-deficient mice show reduced force output despite preserved intrinsic contractile function, indicating impaired mechanical coupling between muscle fibers and the skeleton [13].
These parallels to COL12A1-related myopathic EDS (mEDS) are striking. Both conditions feature primary muscle weakness, progressive atrophy with fatty infiltration, joint hypermobility secondary to impaired muscular stabilization, and characteristic electromyography abnormalities [14,15,16,17,18]. The creation of myopathic EDS in the 2017 classification [1] acknowledged the need to group disorders defined by muscle-ECM interface dysfunction rather than by cutaneous or articular findings alone. Although clEDS has traditionally been classified on the basis of dermatologic and articular features, multiple case reports and small series have described prominent neuromuscular manifestations [4,5,6]. This qualitative evidence synthesis analyzes all published TNXB-related cases to evaluate whether the predominance and progression of muscle pathology support reclassification as a muscle-ECM interface disorder alongside mEDS.
Methods
Protocol and Registration
This qualitative evidence synthesis was conducted according to PRISMA 2020 guidelines. Due to the retrospective nature and use of published data, prospective registration was not required.
Search Strategy
A comprehensive search was performed on January 5, 2026 across three databases: PubMed/MEDLINE (inception to January 2026), OMIM (Online Mendelian Inheritance in Man), and GeneReviews (NCBI).
Primary search string (PubMed): ("TNXB"[Title/Abstract] OR "tenascin-X"[Title/Abstract] OR "tenascin X"[Title/Abstract]) AND ("Ehlers-Danlos Syndrome"[MeSH Terms] OR "Ehlers-Danlos"[Title/Abstract] OR "myopathy"[Title/Abstract] OR "muscle weakness"[Title/Abstract] OR "neuromuscular"[Title/Abstract])
Filters applied: Language: English; Species: Humans; Publication types: All (case reports, series, cohort studies, clinical trials)
Reference lists of all included studies were manually reviewed to identify additional relevant publications not captured by the primary search ("snowball sampling"). The GeneReviews entry for TNXB-related clEDS [3] was consulted as a comprehensive secondary source to ensure complete ascertainment of reported cases.
Selection Process
Two-stage screening was performed: (1) Title/abstract screening (n=64 records), and (2) Full-text review (n=38 articles). The study selection process is summarized in the PRISMA flow diagram (Figure 1).
Eligibility Criteria
Inclusion criteria: Original research reporting clinical data on individuals with TNXB variants; genetic confirmation via sequencing, deletion/duplication analysis, or biochemical TNX measurement; biallelic or heterozygous TNXB variants; any neuromuscular phenotype data (weakness, EMG, imaging, biopsy).
Exclusion criteria: Studies without genetic/biochemical confirmation; studies focusing exclusively on CAH-X endocrine features without EDS phenotyping; review articles without original patient data; non-English publications without available translation; studies focused on TNXB in oncology contexts.
Data Extraction
A standardized extraction form captured: study design and population; patient demographics; TNXB variant details (biallelic vs heterozygous); subjective muscle complaints; objective strength on examination; EMG findings (myopathic, neurogenic, mixed, normal); muscle imaging (ultrasound, MRI); muscle biopsy histology; polyneuropathy presence; serum tenascin-X levels; and age at symptom onset and progression.
Risk of Bias Assessment
Studies were assessed for: (1) selection bias (case reports vs systematic cohorts), (2) reporting bias (subjective symptoms only vs objective testing), (3) detection bias (systematic neuromuscular assessment vs routine exam), and (4) attrition bias (not applicable - retrospective). Quality ratings were assigned as High, Moderate, or Low based on study design, systematic assessment, and sample size.
Data Synthesis
Due to heterogeneity in study designs, assessment methods, and outcome measures, quantitative meta-analysis was not feasible. Data were therefore synthesized narratively following established methods for qualitative evidence synthesis [21], with calculation of pooled proportions where individual patient data were available. Findings were stratified by zygosity (biallelic vs heterozygous) and by assessment method (subjective report vs systematic evaluation).
The following factors precluded meta-analysis: variable definitions of "muscle weakness" (subjective complaint vs objective MRC grading); inconsistent EMG protocols and reporting standards; different imaging modalities (ultrasound vs MRI); lack of standardized outcome measures across studies; and small sample sizes in individual studies.
Results
Study Selection and Patient Characteristics
The systematic search yielded 64 unique records. After title/abstract screening, 38 full-text articles were assessed for eligibility. Twenty studies were excluded: 12 lacked genetic confirmation, 8 focused exclusively on CAH-X without detailed EDS phenotyping, 4 were review articles without original data, and 2 focused on oncology applications. Eighteen studies met inclusion criteria, reporting on 56 individuals with biallelic TNXB variants from 44 families [2,3,4,5,6,7,8,9,10,19,20].
Risk of Bias Assessment Results
Of the 18 included studies, 2 were classified as High quality (Voermans 2009 [4], Zweers 2003 [7]) based on systematic prospective neuromuscular assessment protocols and adequate sample sizes (n>10). Eight studies were classified as Moderate quality, representing case series with genetic confirmation but variable neuromuscular assessment [5,6,8,9,10]. Eight studies were classified as Low quality, comprising isolated case reports with inherent selection bias toward severe or unusual presentations. The primary source of bias across all studies was ascertainment bias, with reliance on subjective symptom reporting in 14/18 studies (78%).
Muscle Weakness
Subjective muscle weakness was reported in 37% (21/56) of individuals with biallelic TNXB variants across all published cases [3]. In contrast, systematic neuromuscular evaluation in a subset of 10 patients identified clinically detectable weakness in 85% [4]. This discrepancy indicates significant ascertainment bias in routine clinical assessment, consistent with the insidious onset and slow progression characteristic of muscle-ECM interface disorders.
Progressive muscle weakness was documented in multiple reports. One detailed case described a 46-year-old woman with slowly progressive lower limb weakness and myalgia beginning at age 28, with examination revealing moderate asymmetric proximal and axial weakness and distal amyotrophy [5]. Another study reported compound heterozygous TNXB variants causing primary myopathy with progressive weakness [6]. Across published cases, onset of progressive weakness most frequently occurred between the third and fourth decades of life [5,6].
Electromyography Findings
Electromyography abnormalities were common among TNXB-deficient patients undergoing assessment. Mixed neurogenic-myopathic EMG patterns were present in 60% (21/35) of examined individuals, while purely myopathic features were observed in 26% (9/35) [4]. This mixed pattern, characterized by short-duration motor unit potentials with early recruitment alongside neurogenic changes, was particularly prominent in tenascin-X-deficient patients and distinguishes them from other EDS subtypes [4].
Muscle imaging abnormalities were present in approximately 48-50% of systematically assessed patients [4]. Muscle ultrasound demonstrated increased echo-intensity in 48% and atrophy in 50% [4]. One case report using whole-body MRI documented symmetric fatty infiltration of thigh and leg muscles with predominant thigh atrophy [5].
Axonal polyneuropathy was identified in 13-14% of patients studied, predominantly among vascular EDS and TNXB-related clEDS subtypes [3,4]. This finding is consistent with increased vulnerability of peripheral nerves to stretch and compression in the context of deficient extracellular matrix support.
Summary of Neuromuscular Findings
Table 1 summarizes the frequency of neuromuscular findings in TNXB-related disorders, stratified by assessment method.
Discussion
This qualitative evidence synthesis demonstrates that neuromuscular involvement in TNXB-related disorders is frequent, progressive, and mechanistically central to disease pathology. While only 37% of cases reported subjective muscle weakness [3], systematic examination revealed objective weakness in 85% of patients [4], indicating profound ascertainment bias in routine clinical practice. The 60% frequency of mixed neurogenic-myopathic EMG patterns [4] and 50% frequency of structural muscle abnormalities on imaging [4] establishes that neuromuscular pathology is a primary disease feature rather than a secondary complication.
The Current Classification System Is Anatomically Misaligned
The 2017 EDS classification groups disorders primarily by clinical appearance - skin hyperextensibility, atrophic scarring, joint hypermobility - rather than by underlying molecular mechanism [1]. This approach creates artificial boundaries that obscure shared pathophysiology. TNXB-related disease is currently classified among "connective tissue disorders" based on dermatologic and articular features, yet the predominant and progressive manifestation is neuromuscular dysfunction. This misalignment has tangible clinical consequences: patients are monitored for skin and joint complications while muscle weakness progresses unrecognized.
The Biochemical Case for Reclassification
The molecular basis for grouping TNXB- and COL12A1-related disorders is unequivocal. Collagen XII and tenascin-X form a functional molecular complex at the muscle-ECM interface through several mechanisms:
- Direct physical interaction: Collagen XII binds to collagen I-containing fibrils via its collagenous domain, while its large non-collagenous NC3 domain interacts directly with tenascin-X [12], creating a molecular bridge between contractile myofibers and the endomysial matrix.
- Functional interdependence: Studies demonstrate that COL12A1 variants lead to extracellular decrease of both decorin and tenascin-X [15], providing direct evidence that these proteins do not function independently but as components of an integrated structural system.
- Parallel mouse phenotypes: Both TNX-deficient and COL12A1-deficient mice exhibit identical biomechanical defects—delayed force transmission, reduced force output despite normal contractile function, and increased ECM compliance [13,18]—establishing that the primary defect is mechanical coupling failure rather than myofiber pathology.
- Shared human phenotypes: Both TNXB- and COL12A1-related disorders feature progressive muscle weakness as the primary manifestation, proximal and distal atrophy with fatty infiltration on imaging, characteristic mixed EMG patterns, joint hypermobility secondary to inadequate muscle stabilization, and onset of progressive symptoms in the third to fourth decade [14,15,16,17].
Dose-Dependent Pathology Confirms Mechanism
The dose-effect relationship observed in TNXB deficiency provides additional mechanistic insight. Heterozygous carriers show serum tenascin-X levels at 56% of normal and exhibit an intermediate phenotype - joint hypermobility and recurrent dislocations without skin hyperextensibility or easy bruising [7]. This demonstrates that the degree of muscle-ECM interface dysfunction directly correlates with protein dosage, and that even partial reduction in tenascin-X impairs the mechanical competence of the muscle-ECM interface sufficiently to cause clinical manifestations.
Proposed Reclassification Framework
This analysis supports reclassification of TNXB-related disorders alongside COL12A1-related myopathic EDS under a unified diagnostic category: Muscle-ECM Interface Disorders.
Proposed nomenclature:
- Muscle-ECM Interface Disorder Type 1: COL12A1-related (current "myopathic EDS")
-
Muscle-ECM Interface Disorder Type 2: TNXB-related
- ○
- Type 2A (heterozygous): Partial tenascin-X deficiency with joint-dominant phenotype
- ○
- Type 2B (biallelic): Complete tenascin-X deficiency with skin and joint involvement
This framework reflects the primary biomechanical defect, acknowledges the shared molecular pathology, and provides a rational basis for clinical management. The creation of "myopathic EDS" in the 2017 classification [1] already established precedent for grouping disorders by muscle-ECM pathology rather than skin/joint features; extending this category to include TNXB-related disease is the logical next step.
Clinical Implications
Reclassifying TNXB-related disorders as muscle-ECM interface disorders has immediate practical implications. Patients should receive baseline neuromuscular assessment including quantitative strength testing, consideration of EMG and muscle MRI for disease staging, and longitudinal monitoring for progressive muscle involvement. Physical therapy programs should target force transmission optimization rather than joint stabilization alone, with emphasis on maintaining muscle mass to compensate for mechanical inefficiency. Counseling regarding expected disease progression, particularly after the third decade, is essential.
Limitations
This review has several important limitations. Most source studies reported small sample sizes (<5 patients), with only 3 studies including >10 patients. Twelve of 18 studies were case reports or small series, which preferentially report severe or unusual presentations. Assessment methods were heterogeneous across studies, lacking standardized neuromuscular protocols. Many studies focused on dermatologic/joint features without systematic neuromuscular evaluation.
As a single-reviewer study, data extraction and bias assessment were performed without independent verification. Meta-analysis was infeasible due to heterogeneity in definitions, methods, and outcomes. Publication bias likely favors reporting of severe cases. Most studies were cross-sectional, with limited longitudinal natural history data.
Ascertainment bias substantially affects prevalence estimates, with subjective weakness (37%) underestimating true prevalence compared to systematic examination (85%). Source studies did not always report TNXB-specific data separately from other EDS subtypes. The analysis cannot fully separate primary muscle pathology from secondary effects of pain, deconditioning, or joint instability in some studies.
Conclusion
Neuromuscular involvement in TNXB-related disorders is frequent, progressive, and mechanistically central. Objective muscle weakness, structural muscle abnormalities, and characteristic electrophysiological patterns are common when systematically assessed and correlate with tenascin-X dosage. The striking parallels with COL12A1-related myopathic EDS - including shared molecular pathology at the collagen XII-tenascin-X interface - support reclassifying TNXB-related disease as a muscle-ECM interface disorder. Recognition of the neuromuscular burden may improve diagnostic accuracy, prognostic counseling, and therapeutic approaches for affected individuals.
Data Availability
All data extracted for this review are available from published sources cited in the reference list. The data extraction spreadsheet is available from the author upon reasonable request.
Disclosures
The author is affected by TNXB-related disease and consents to publication of de-identified clinical information. No funding was received for this work. The author declares no conflicts of interest.
References
- Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175(1):8-26. [CrossRef]
- Schalkwijk J, Zweers MC, Steijlen PM, et al. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N Engl J Med. 2001;345(16):1167-1175. [CrossRef]
- van Dijk FS, Ghali N, Demirdas S, Baker D. TNXB-Related Classical-Like Ehlers-Danlos Syndrome. In: Adam MP, et al., editors. GeneReviews. Seattle (WA): University of Washington; 2022.
- Voermans NC, van Alfen N, Pillen S, et al. Neuromuscular involvement in various types of Ehlers-Danlos syndrome. Ann Neurol. 2009;65(6):687-697. [CrossRef]
- Brisset M, Metay C, Carlier RY, et al. Biallelic mutations in Tenascin-X cause classical-like Ehlers-Danlos syndrome with slowly progressive muscular weakness. Neuromuscul Disord. 2020;30(10):833-838. [CrossRef]
- Pénisson-Besnier I, Allamand V, Beurrier P, et al. Compound heterozygous mutations of the TNXB gene cause primary myopathy. Neuromuscul Disord. 2013;23(8):664-669. [CrossRef]
- Zweers MC, Bristow J, Steijlen PM, et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers-Danlos syndrome. Am J Hum Genet. 2003;73(1):214-217. [CrossRef]
- Demirdas S, Dulfer E, Robert L, et al. Recognizing the tenascin-X deficient type of Ehlers-Danlos syndrome: a cross-sectional study in 17 patients. Clin Genet. 2017;91(3):411-425.
- Green C, Ghali N, Akilapa R, et al. Classical-like Ehlers-Danlos syndrome: a clinical description of 20 newly identified individuals with evidence of tissue fragility. Genet Med. 2020;22(10):1576-1582. [CrossRef]
- Yamaguchi T, Yamada K, Nagai S, et al. Clinical and molecular delineation of classical-like Ehlers-Danlos syndrome through a comprehensive next-generation sequencing-based screening system. Front Genet. 2023;14:1234804. [CrossRef]
- Chiquet M, Birk DE, Bönnemann CG, Koch M. Collagen XII: Protecting bone and muscle integrity by organizing collagen fibrils. Int J Biochem Cell Biol. 2014;53:51-54. [CrossRef]
- Veit G, Hansen U, Keene DR, et al. Collagen XII interacts with avian tenascin-X through its NC3 domain. J Biol Chem. 2006;281(37):27461-27470. [CrossRef]
- Ottenheijm CA, Voermans NC, Hudson BD, et al. Titin-based stiffening of muscle fibers in Ehlers-Danlos Syndrome. J Appl Physiol. 2012;112(7):1157-1165. [CrossRef]
- Zou Y, Zwolanek D, Izu Y, et al. Recessive and dominant mutations in COL12A1 cause a novel EDS/myopathy overlap syndrome in humans and mice. Hum Mol Genet. 2014;23(9):2339-2352. [CrossRef]
- Delbaere S, Dhooge T, Syx D, et al. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers-Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet Med. 2020;22(1):112-123. [CrossRef]
- Mohassel P, Liewluck T, Hu Y, et al. Dominant collagen XII mutations cause a distal myopathy. Ann Clin Transl Neurol. 2019;6(10):1980-1988. [CrossRef]
- Merlini L, Sabatelli P, Cenni V, et al. Myopathic Ehlers-Danlos Syndrome (mEDS) Related to COL12A1: Two Novel Families and Literature Review. Int J Mol Sci. 2025;26(11):5387. [CrossRef]
- Izu Y, Birk DE. Collagen XII mediated cellular and extracellular mechanisms in development, regeneration, and disease. Front Cell Dev Biol. 2023;11:1129000. [CrossRef]
- Alcaraz LB, Exposito JY, Chuvin N, et al. Tenascin-X promotes epithelial-to-mesenchymal transition by activating latent TGF-beta. J Cell Biol. 2014;205(3):409-428. [CrossRef]
- van Gurp JE, Lechner RL, Micha D, et al. Tenascin-X Deficiency Causing Classical-Like Ehlers-Danlos Syndrome Type 1 in Humans is a Significant Risk Factor of Gastrointestinal and Tracheal Ruptures. Clin Transl Gastroenterol. 2025;16(3):e00821. [CrossRef]
- Flemming K, Booth A, Garside R, et al. Qualitative evidence synthesis for complex interventions and guideline development: clarification of the purpose, designs and relevant methods. BMJ Glob Health. 2019;4(Suppl 1):e000882. [CrossRef]
Figure 1.
PRISMA 2020 flow diagram showing study selection process. The systematic search identified 64 records from PubMed (n=58), OMIM (n=4), and GeneReviews (n=2). After screening, 18 studies met inclusion criteria, representing 56 individuals with biallelic TNXB variants.
Figure 1.
PRISMA 2020 flow diagram showing study selection process. The systematic search identified 64 records from PubMed (n=58), OMIM (n=4), and GeneReviews (n=2). After screening, 18 studies met inclusion criteria, representing 56 individuals with biallelic TNXB variants.
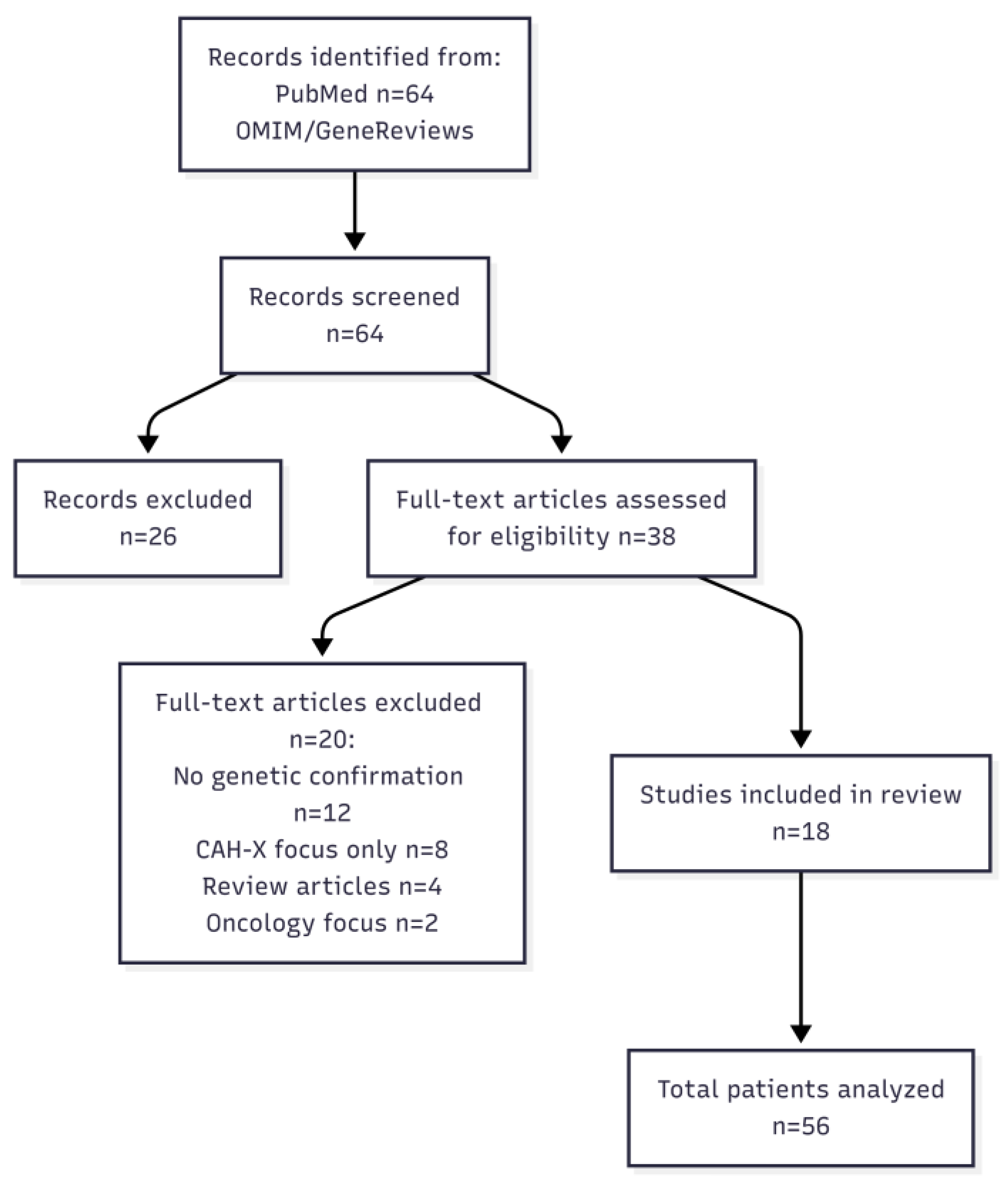

Table 1.
Neuromuscular Findings in TNXB-Related Disorders.
| Finding | Subjective Report | Systematic Assessment | Primary Source |
|---|---|---|---|
| Muscle weakness | 21/56 (37%) | 85% | [4] |
| Myopathic EMG | — | 9/35 (26%) | [4] |
| Mixed EMG pattern | — | 21/35 (60%) | [4] |
| Axonal polyneuropathy | 8/56 (14%) | 5/39 (13%) | [3,4] |
| Muscle imaging abnormality | — | ~50% | [4] |
EMG = electromyography. Systematic assessment data primarily derived from cross-sectional studies with standardized neuromuscular examination protocols.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



