
Submitted:
01 January 2026
Posted:
08 January 2026
You are already at the latest version
Abstract
Recombinogenic DNA damage can initiate chromosomal rearrangements that can alter gene expression or accelerate cancer progression in higher eukaryotes. Thus, there is a critical need to identify genes that suppress chromosomal rearrangements and environmental exposures that promote genetic instability. Cell cycle checkpoints modulate the cell cycle so that DNA repair occurs before the replication or segregation of damaged chromosomes. Saccharomyces cerevisiae (budding yeast) RAD9 was the first cell cycle checkpoint gene identified, which initiated intensive research studies into the mechanisms of checkpoint activation and the phenotypes of checkpoint mutants. The budding yeast Rad9 protein serves as both an adaptor and scaffold that facilitates downstream effector activation to orchestrate a DNA damage response at multiple stages of the cell cycle, which facilitate double-strand break (DSB) repair by sister chromatid recombination. However, the role of RAD9 in homologous recombination and in suppressing gross chromosomal rearrangements (GCRs) is not completely understood. In this review we discuss how RAD9 can promote genome instability resulting from aberrant DNA replication intermediates, while suppressing DSB-associated rearrangements. We also discuss possible mechanisms accounting for the synergistic increase in genomic instability in double mutants defective in both RAD9 and recombinational repair. We emphasize that while there is an overlap between checkpoint and recombinational repair pathways, RAD9 and checkpoint pathways can function independently to suppress chromosomal instability. These studies thus elucidate checkpoint mechanisms that control homologous recombination between repeated sequences.
Keywords:
homologous recombination
; Genome instability
; DNA damage
; cell-cycle checkpoint
; budding yeast
1. Introduction
Biological organisms have finely tuned DNA repair pathways to maintain genetic integrity. Such mechanisms repair a multitude of DNA lesions, including base pair damage, bulky DNA adducts, DNA cross-links and single and double-strand breaks (DSBs). However, cell cycle progression in the presence of unrepaired DNA lesions, can lead to replication fork collapse and the persistence of chromosomal fragments and genotoxic lesions, which generate mutations and chromosomal rearrangements in subsequent cell cycles. Hartwell and Weinert [1] coined cell cycle checkpoints as “control mechanisms that enforce dependency in the cell cycle”. RAD9 [2] was the first identified cell-cycle checkpoint gene. Cell-cycle checkpoint genes arrest or delay the cell cycle so that repair of DNA is completed before DNA lesions are replicated or segregated to the next cell cycle. This is particularly important in DSB repair, where aberrant repair of DSBs can reshape the genome via recombination between repeated sequences [3], while the persistence of a single DSB can confer lethality [4]
Cell cycle checkpoints include those that function at G1/S, intra S phase and at G2/M. Components of these checkpoints are well-conserved from yeast to mammalian cells. In general, protein sensors recognize DNA damage, which, in turn, activate apical kinases that signal to effector kinases, amplifying the DNA damage signal (for review, see [5,6]). These kinases, in turn, activate factors that arrest the cell cycle, modulate DNA repair, and control the transcriptional response to DNA damage [7,8]. While both apical and effector kinases can directly and indirectly promote their own self-regulation (for review, see [6,9]), phosphatases can directly remove phosphates from activated targets, and once DNA repair is completed, the absence of the DNA damage switches off activation signal (for review see, [10]).
Checkpoint activation is initiated by diverse DNA structures. While single-stranded DNA (ssDNA) is a general signal for checkpoint activation in Saccharomyces cerevisiae (budding yeast), DNA damage signals include DSBs [10,11,12], stalled replication forks [13], unresolved Holliday structures [14], ssDNA at telomeres [15,16], fragile DNA sites [17]. and unresolved DNA replication intermediates, such as 5’ single-strand flaps [18], trapped topoisomerase-DNA structures [19,20]. However, developmentally programmed DSBs, such and those generated by Spol11 in meiosis [21] and programmed HO endonuclease-induced breaks during mating-type switching, do not generate a checkpoint response [22].These observations indicate that activation of the checkpoint response is tightly controlled and partially dependent on the efficiency of repairing DNA lesions, such as DNA DSBs.
The redundancy of DSB-mediated repair pathways in eukaryotic organisms confers resistance to ionizing radiation and radiomimetic chemicals. These pathways include non-homologous end joining (NHEJ), microhomology-mediated end joining (MMEJ), and homologous recombination (HR) (for review, see [23]). HR pathways for DSB repair (Figure 1) include gap repair mechanisms, single-strand annealing (SSA), and break-induced replication (BIR) [23]. Common steps in HR pathways are the resection of the DSBs to reveal ssDNA, formation of a recombination intermediate involving heteroduplex DNA, and resolution of the heteroduplex [23]. The choice of which pathway is used in DSB repair depends on the cell cycle phase and ploidy; for example, NHEJ facilitates DSB repair in G1 haploid cells since sister chromatids and homologs are absent [4]. Considering the abundance of repeated sequences in yeast, DSBs could initiate diverse genome rearrangements [3], including deletions, duplications, and translocations (Figure 2).
The higher radiation resistance of G2 diploid cells, compared to G1 diploid cells [24,25] and the significant role of HR in DSB repair, supports the assertion that sister chromatids are the preferred substrates for recombinational repair [25]. Considering that sister chromatids are transient in the cell cycle, checkpoint control of DSB repair by has been intensively studied [26]. The central players in this pathway in budding yeast are the PI-3K like kinase encoding genes TEL1 and MEC1 [27], which are the orthologs of the ataxia telangiectasia mutated (ATM) and ATM-related gene (ATR), respectively [28]. DSB signaling involves the rapid recruitment of the Mre11/Sae2 complex and subsequent resection, which leads to ssDNA bound to single-strand binding protein (RPA) (Figure 3). Sae2 activation, in turn, is controlled by the Cdk2 so that resection occurs at the G2 stage of the cell cycle [29]. The ssDNA is then a substrate for binding by the Mec1-Ddc2 complex, which is facilitated by the 9-1-1 complex. Mec1 also phosphorylates DNA repair proteins, including Sgs1 [30], Rad51 [31], Rad55 [32], and Exo1[30] that facilitate sister chromatid recombination. The Mec1 kinase phosphorylates Rad53 [33], the CHK2 ortholog, and Chk1 [34]. Rad9 serves to bind Rad53 and facilitates Mec1’s phosphorylation of Rad53, which then triggers Rad53 autophosphorylaton and its release from Rad9 [35,36]. Rad53, in turn, phosphorylates effector proteins, whose functions are to inhibit late replication firing [13] , upregulate deoxynucleotide levels [37] and facilitate formation of Rad51 filaments [32]. Cell cycle arrest is achieved by Mec1-dependent activation of Chk1, which in turn phosphorylates Pds1 (securin), which blocks the degradation of cohesin by anaphase promoting complex (APCCdc20) [38]. Rad53 (Chk2) inhibits Cdc20-Pds1 interaction [39] and phosphorylates Dun1, which in turn, inhibits mitotic exit by activating the Bfa1-Bub2 complex [40]. Additional roles of the checkpoint response are to inhibit de novo telomere addition [41], promote the mobility of broken chromatids [42,43], and inhibit asymmetric resection [44], which could promote genome rearrangements [45]. Thus, checkpoints serve to both arrest the cell cycle and facilitate DNA repair.
An intermediate substrate in both checkpoint signaling and recombinational repair is ssDNA bound to single-strand binding protein [46,47]. ssDNA generated by DSB resection is a substrate for Rad51 filament formation, which is facilitated by Rad52, Rad55, and Rad57 (for review see [48]). Rad51-coated DNA then catalyzes DNA strand invasion and with the assistance of Rad54 generate Holliday intermediates. The Sgs1 helicase can abort recombination intermediates [49], while additional proteins, such as Yen1, can resolve recombination intermediates to yield crossover events [50]. DSB processing thus produces intermediates which could bind either MR repair proteins or checkpoint proteins. Rad9 controls resection through its interaction with the Mrell complex [51], thus affecting both DSB-repair mechanisms and checkpoint signaling.
While the checkpoint response and RAD50 group genes are required for efficient DSB gap repair and ionizing radiation resistance, individual genes within the RAD50 group are not required at all stages of DNA damage-associated or spontaneous HR. Differential requirements can be due to multiple sources of spontaneous DNA lesions, including DNA replication errors, transcription, and base excision repair. While all HR events require RAD52 [48], RAD51 mutations only modestly affect spontaneous SCR [52] and spontaneous unequal sister chromatid recombination events (uSCR) are RAD51-independent [53]; additional pathways have been identified that participate in uSCR [54]. On the other hand, heteroallelic recombination is RAD51-dependent [48]. Rad50-mediated resection delays mating-type switching [55] but is not required to initiate X ray-associated crossovers [56]. Other studies have also shown multiple pathways for both SSA and BIR events [57,58]. RAD9 functionality at all stages of the cell cycle [59] thus underscores the importance of understanding checkpoint function in diverse HR events and a better understanding of the Rad9 protein.
2. Rad9 Protein and Function
The yeast RAD9 gene encodes a protein of 1309 amino acids and is an ortholog of the human 53BP1 and the Schizosaccharomyces pombe crb2+ genes [60,61], and shows similarity the human BRCA1 gene [61]. RAD9 mediates DNA damage-activated checkpoints at G1 [63]), S [18,64], and G2/M stages of the cell cycle [65]. An artificial intelligence (AI)-generated alpha fold structure [59] of the Rad9 protein is shown in Figure 3. This structure indicates both predicted and disordered domains; namely, with high confidence shown for the predicted structures of the tudor domains. The relevant structure includes five major features; these include the Chk1-activating domain (CAD), the Mec1/Tel1-phosphorylated SQ/TQ cluster domain (SCD), the Rad53 binding domain, the hydrophobic tandem tudor domain, and the BRCT domain [61]. Regulation is achieved by 98 phosphorylation sites mediated by Mec1, Rad53, Cdc28 -Clb2, and ubiquitylation on position K1139 [68].
Rad9 protein binds to methylated histone H3 on nucleosomes in undamaged cells via its tandem tudor domain [69], and Rad9’s tandem BRCT domain enhances its concentration at DSBs by interacting with phosphorylated Histone H2A (S119). Rad9 is further recruited to DNA damage sites by the Dpb11 (TOPBP1), which interacts with the 9-1-1 complex that is loaded onto ssDNA-dsDNA junctions [70]. This, in turn, facilitates Rad9 phosphorylation by Mec1, which induces Rad9 multimerization via its BRCT domain and enables Rad9 to recruit Rad53 [71]; thus, Rad9 serves as an adaptor so that Mec1 can phosphorylate Rad53 [72]. The oligomerized Rad9 can also serve as a scaffold for the aggregation of multiple Rad53 molecules, which facilitates Rad53 trans autophosphorylation[73] leading to the subsequent release of Rad53. Rad9 thus controls one branch of the checkpoint pathway, the other of which is controlled by Mrc1, whose role in checkpoint activation is restricted to DNA replication forks. Rad9 can also function as an adaptor for Mec1-mediated Chk1 activation [74]. These observations illustrate that Rad9 serves multiple functions in checkpoint activation at distinct stages of the cell cycle.
As a consequence of G2/M checkpoint inactivation, irradiated rad9 cells form microcolonies on agar plates. Such microcolonies accumulate inviable cells due to mis-segregation and loss of chromosomal fragments [75]. Similar phenotypes are also evident after exposure to select chemical DNA damaging agents, including methyl methanesulfonate (MMS), cisplatin, and topoisomerase inhibitors; spontaneous chromosome loss is also observed [76]. Thus, in the presence of either spontaneous or environmentally-induced DNA damage, chromosomal fragments or aberrant DNA replication structures could initiate aberrant recombination events that could lead to higher frequencies of chromosomal rearrangements resulting from homologous (HR) or NHEJ.
RAD9 facilitates the completion of DNA replication initiated during S phase. First, it facilitates chromosomal DNA replication when there are large distances between origins, a function which does not require recombination [77]. Second, it backups MRC1’s function to extend the length of checkpoint signaling during replication stress [78]. Third, it is required for recombination-mediated resolution of aberrant structures that are formed in DNA replication. For example, rad9 exhibits synthetic lethality with rad27[79], a mutant defective in the processing of 5’ flaps generated during lagging strand synthesis [80]. The hyper-recombination of rad27 and the synthetic lethality with rad27 and rad52 [81] suggest that HR is required to resolve such aberrant structures that would otherwise confer lethality.
RAD9’s function in controlling HR between sister chromatids and homologs, and ectopic recombination between repeated sequences is not fully understood. Depending on the assay to measure HR, rad9 mutants may exhibit enhanced recombination, decrease recombination, or no effect. In addition, particular phenotypes may depend on whether the assay measures spontaneous or DNA damage-associated recombination or whether the assay is performed in haploid or diploid cells. Our effort to elucidate these apparently contradictory phenotypes will include: 1) summarizing different recombination assays that exhibit rad9 phenotypes, 2) describing interactions with RAD9 and recombinational repair pathways, and 3) comparing phenotypes of rad9 mutants with mutants in other checkpoint genes. Finally, we will present possible mechanisms that may elucidate these phenotypes and future experiments.
3. Hr Phenotypes of Rad9 Mutants
The RAD9 requirement for HR and NHEJ events is shown in Table 1. The arrangement of repeated sequences that generate different chromosomal rearrangements is shown in Figure 2. Factors that may affect rates of HR include the size and orientation of the repeats, the distance between repeats, and whether the repeats are identical or divergent. Additional factors in Rad+ strains also include ploidy and whether both MATa and MATa are expressed.
3.a. RAD9 Requirement for SCR
Recombination between sister chromatids proceeds by multiple mechanisms, including template-switching and DSB-initiated HR. While RAD9 is not required for spontaneous sister chromatid exchange, RAD9 is required for DSB-associated sister chromatid recombination as demonstrated in assays to measure either equal or unequal sister chromatid recombination [82,83]. The assertion that X ray-associated recombination requires a G2/M checkpoint is supported by observations that rad9’s X-ray sensitivity is suppressed when cells are irradiated after pretreating with the microtubule inhibitor nocodazole. Both X-ray and HO-induced DSBs do not stimulate as many uSCR events in rad9 mutants as they do in wild type [82,83] have shown that RAD9 is also required for DSB-initiated equal SCR in a pathway that involves stabilization of cohesin; cohesin is absolutely required for repair of DSBs in yeast [84] and participates in the DNA damage response [85]. This observation is further supported by observations that one downstream effector of Mec1 activation, Rad53, which contributes to the cohesin stabilization and maintenance of cell cycle arrest, is required for X-ray associated SCR [86]. While Chk1 is also a downstream effector of Mec1 activation, chk1 mutants are not X-ray sensitive [87], nor do they exhibit a clear defect in sister chromatid cohesion [88] or X-ray associated uSCR [86]. These results suggest that Rad53 plays a major role in the RAD9-mediated checkpoint pathway that facilitates DSB-associated SCR.
rad9 mutants exhibit fewer DNA damage-associated uSCRs after exposure to selective chemical agents [25,82,89], particularly those that indirectly generate DSBs, such as methyl methane sulfonate (MMS) and camptothecin, a topoisomerase inhibitor. However, rad9 mutants exhibit only a minor decrease in UV-associated uSCR [82] and no decrease in the 4-nitroquinoline 1-oxide (4-NQO)-associated uSCR events [89]; 4NQO is a UV-mimetic agent. Since UV promotes RAD5-dependent template switch events [90] , and both UV and 4-NQO-associated uSCR require RAD5 [91], these observations suggest that RAD9 is not required for template switching.
3.b. rDNA Repeat Instability and CNV
While RAD9 promotes DSB-associated SCRs, it suppresses rDNA repeat instability that results from insufficient DNA replication firing due to non-functional origin recognition complex (Orc) proteins, as present at the restrictive temperature in orc2-1 mutants [92]. RAD9 confers lethality in orc2-1 diploid mutants at the restrictive temperatures. However, the viable colonies obtained in rad9 orc2-1 diploid mutant contain reduced numbers of rDNA repeat units. The authors suggest that because rDNA contains many Orc2 binding sites, limited amounts of Orc2 protein at the restrictive temperature are insufficient to initiate replication from other chromosomal origins, and that rDNA may be particularly vulnerable to recombinogenic lesions [68]. Such recombinogenic lesions could promote uSCR, unequal homolog recombination, and intrachromatidal recombination. Reduced rDNA copy number thus allows the initiation of DNA replication at other chromosomal origins and thus confers viability.
Copy number variation (CNV) has also been measured in checkpoint mutants containing multiple juxtaposed CUP1 repeats (Table 2). In these haploid strains, nicotinamide-mediated suppression of the histone deacetylase H3K56ac induces CUP1 transcriptional induction initiating a replication fork impediment and CNV contraction. CNV contraction is RAD52-dependent and proceeds through HR. Interestingly, MRC1, not RAD9 suppresses CNV [93]. In addition, rad27 mutants exhibit enhanced CNV, suggesting aberrant replication intermediates are leading to recombinogenic lesions. Considering that the double mus81 yen1 mutant defective in Holiday junction resolution does not exhibit CNV, an attractive mechanism is that CUP1 transcriptional induction induces replication fork cleavage in S phase initiating a replication restart mechanism. Thus, RAD9 can suppress instability at the rDNA locus while facilitating instability at CUP1 repeats [93].
3.c. RAD9 Is Not Required for SSA Unless There Are Significant Mismatches Between Annealing Sequences
RAD9 is not required for the completion of SSA when an enzymatic-induced DSB initiates recombination between two non-tandem repeats. These assays included one strain that contained tandem his3 fragments, where an HO endonuclease cut site (HOcs) was inserted in one his3 fragment, or an HOcs was inserted between ura3 repeats [94]. However, RAD9 is required for intrachromatidal recombination between homeologous repeats that contain 3% divergent DNA [85] in which an HOcs was inserted between the 200bp ura3 repeat units. The RAD9-dependence of SSA is suppressed by nocodazole, suggesting that the critical factor for completing recombinational repair between divergent repeats is maintenance in G2 [95]. The authors suggest that SSA between heterologous repeats requires mismatch repair proteins, which require an extended G2. While RAD9 is not explicitly required for SSA, other checkpoint proteins, such as Mec1, do affect SSA by phosphorylating Slx4 [96] and affecting Rad1/Rad10 cleavage on non-homologous 3’ tails [97]. Thus, while checkpoint proteins regulate SSA, the RAD9 function has not been explicitly defined.
3.d. RAD9 Affects the Outcome of DSB-induced Homolog Recombination
The overall conclusion of RAD9-dependence of homolog recombination is that RAD9 is not required for spontaneous recombination but does alter the type of DSB-induced recombination event. Several strain constructs used to measure rates of spontaneous homolog recombination include those that 1) simply measure gene conversion events between two heteroalleles and 2) those that measure cross-overs between two heteroalleles of two or more gene. In the first type of construct, RAD9 is not required for spontaneous homolog recombination between heteroalleles in either diploids or in a haploid disomic from chromosomes VII [98,99,100]. In the second strain construct, CanR and Thr+ recombinants were selected in a diploid that was heterozygous with wild type at both CAN1 and HOM3. The authors observed a two-fold increase in rates of spontaneous homolog recombination [101]. Thus, RAD9 has a minimal effect on recombination between homologs.
However, RAD9 does alter the types of recombination events that are induced by an I-Sce1 restriction endonuclease when the endonuclease recognition site is placed in one copy of an ade2 gene in a diploid strain containing two ade2 heteroalleles flanked by different markers [102]. In this strain construction, gene conversion events, cross-overs, and break-induced replication (BIR) events could be identified by scoring the presence of the ade2 allele and whether the flanking marker was present. Based on these studies, the authors demonstrated that the rad9 mutant exhibited a higher percentage of short-track gene conversion and a reduced frequency of break induced replication and cross-over events. By Chip analysis, they also showed that Rad9 limits the Sgs1 and Mph1 helicase binding, suggesting that Rad9 facilitates the recombinogenic repair of DSBs by stable annealing of the recipient and donor strands during recombination [102].
3.e. RAD9 Suppresses Ectopic Recombination That Generates Translocations
RAD9 suppresses translocations generated by HR between his3 repeated sequences located at centromere-linked loci on non-homologous chromosomes II and IV. Compared to the wild-type diploid, the rate of spontaneous homology-directed translocations in homozygous rad9 diploid mutants increases by seven-fold [82,100]. A modest but significant increase is also observed in rad9 haploids, compared to the wild type [82]. The hyper-Rec phenotype of the rad9 mutant is further enhanced if cells are exposed to radiation; an approximately thousand-fold stimulation was observed after rad9 diploid mutants are exposed to 15.6 krads [82]. The radiation-associated recombination is suppressed by pre-arresting cells with the microtubule inhibitor nocodazole before UV or X ray exposure. These data suggest that cell cycle delay is sufficient to suppress DNA damage-associated translocations.
Many homology-mediated translocations in rad9 mutants may result from recombinogenic acentric and centromere-containing chromosomal fragments that are inherited in repeated cells cycles (Figure 4). Consistent with this proposal, many radiation-associated translocations are non-reciprocal events, also referred to as half-crossovers (HCs). Although BIR can theoretically generate these HCs, BIR has not been observed to transverse centromeric DNA [103]. Instead, it is likely that multiple rearrangements generate radiation-associated His+ recombinants, especially those where the His+ phenotype is unstable. This interpretation is supported by independent observations that DSBs can efficiently remodel the genome [3] and that chromosomal instability in rad9 mutants can be initiated at telomeric sites [99], which would subsequently trigger recombination at other loci.
The higher frequency of DNA damage-associated homology-directed translocations in rad9 mutants depends on the DNA damaging agent [89]. While DNA damaging agents that directly or indirectly generate DSBs s, such hydrogen peroxide, camptothecin, bleomycin, phleomycin, stimulate more homology-directed translocations in rad9 mutants, other DNA damaging agents do not. For example, rad9 diploid cells that are exposed to N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) or the UV-mimetic 4-NQO do not exhibit enhanced recombination compared to exposed wild-type cells. These studies suggest that many of these DNA damaging agents trigger recombination between ectopic repeats by mechanisms other than formation of DSBs.
An increase in homology-directed translocations in rad9 diploids has also been observed in independent studies [104]. In a strain construction to measure loss of heterozygosity (LOH) on chromosome III, where the URA3 gene is positioned on one chromosome III homolog, rad9 mutants exhibit a higher frequency of 5-fluoroorotic acid resistant (Ura-) isolates containing an accompanying chromosomal rearrangement [104]. While the increase in the frequency of spontaneous translocations was a modest threefold, the increase observed in diploid orc1-4 rad9 mutants was 42-fold. Many of these cross-over events occurred at Ty1 sequences. While the rad9 mutants did not exhibit an increase in LOH due to gene conversion, LOH events due to cross-over events increased three-fold in the rad9 diploid and 14-fold in the orc1-4 rad9 diploid mutant, compared to wild type. These data thus support the notion that rad9 mutants exhibit more ectopic recombination due to cross-over events.
3.f. RAD9 and RAD50 Group Genes Are Separate Pathways For Suppressing Ectopic Events in Diploid Strains
Models for DSB-initiated checkpoint signaling and HR repair of DSBs suggest that there is cross-talk between the two pathways. The Mre11/Rad50/Xrs2 complex, which facilitates DSB recission, is required in both DSBs-mediated checkpoint signaling and in DSB repair. Because Rad9 is required for Mec1-mediated activation of RAD53, the model would suggest recombinational repair and RAD9-mediated checkpoint pathways participate in the same pathway for suppressing homology-directed translocations [105]. Indeed, in haploid mutants RAD9 is epistatic to recombinational repair [51,106] in conferring ionizing radiation resistance. While RAD9 suppresses frequencies of spontaneous homology-directed translocations by seven-fold, diploid mutants defective in RAD9 either RAD51, RAD55, and RAD57 exhibit a synergistic (57-78-fold) increase in the frequencies of spontaneous ectopic recombination (Table 3). In addition, diploid mutants defective in RAD9 in and either MRE11 or XRS2 exhibit synergistic (57-fold) increases in spontaneous homology-directed translocations. These studies suggest that RAD9 and RAD50 group genes that participate in independent pathways for suppressing spontaneous HR between repeated sequences in diploid strains. The lesions that initiate these events, however, are unknown.
One possible interpretation of the synergistic increase in spontaneous translocation in rad9 rad51 double mutants is that the ectopic recombination between the repeated sequences is mediated by SSA. Mutations in Rad9 confer more ssDNA while RAD51 inhibits single-strand annealing SSA [107,108,109]. Since Rad9 inhibits resection of DSBs and Rad51 inhibits SSA, an increase in ssDNA and lack of inhibition to reanneal these sequences may increase recombination events. Similar interactions would also apply to the interaction of rad9 and the other rad mutants. An alternative explanations is that more DNA lesions accumulate in rad51 mutants, and these lesions are tolerated in rad9 mutants. At present, we cannot distinguish the two and it is possible that both mechanisms are important in promoting chromosomal rearrangements that occur by single-strand annealing.
3.g. RAD9 and SGS1 Suppress HR Between Divergent Genes and Ty1 Elements
Schmidt and Kolodner [110] observed that SGS1 suppresses the formation of gross genome rearrangements (GCRs) that occur by recombination between the CAN1 positioned on chr V and the LYP1 or ALP1 genes, positioned on chr XIV. CAN1, LYP1, and ALP1 encode basic amino acid transporters, which share more than 50% sequence identity. The haploid strains contain URA3 and CAN1 genes located on the non-essential chromosomal V arm; double selection against both URA3 and CAN1 using 5-fluororotic acid (5-FOA) and canavanine, respectively, generates drug resistant isolates containing chromosomal rearrangements. While most FOAR and CanR isolates result from NHEJ events, in sgs1 mutants, the rate of GCRs is increased 22-fold above wild type, while the sgs1 rad9 mutant exhibited a 213-fold increase above wild type. While only ~3% of these drug-resistant isolates were translocations due to HR between these sequences, no homology-directed translocations were observed in either the sgs1 or the rad9 mutants; translocations found in the single mutants are due to NHEJ or micro-homology end joining [110]. These experiments thus indicate that SGS1 and RAD9 constitute independent pathways in suppressing GCRs in haploid strains through multiple mechanisms, including homeologous recombination between sequences.
RAD9 and SGS1 were also observed to suppress DSB-initiated ectopic rearrangements that directly result from BIR. Vasan et al. [111] characterized recombinants that resulted from HCs initiated by galactose-induced DSBs at a HOcs positioned on chromosome III in a chromosome III disomic strain. Deletion of RAD9 and SGS1 conferred a higher frequency of recombinants that resulted from compromised BIR leading to cascades of genome instability [111]. One interpretation is that rad9 mutants exhibit greater mis-segregation of chromosomal fragments while Sgs1 serves as an anti-recombinogenic factor that reverses recombination intermediates generated by BIR. Additionally, Rad9 protects replication intermediates from excessive degradation [112]. Thus, aborted BIR (Figure 1) may lead to aberrant recombination events at ectopic loci. Recombination sites involving HC during compromised HC include Ty1 elements. These data thus indicate that rad9 mutants exhibit both higher frequencies of spontaneous and DSB-associated recombination between Ty1 sequences.
3.h. RAD9 Is Required for Enhanced Genetic Instability Due to HR Exhibited in mec1 Hypomorphs and Promotes Genetic Instability Resulting from DNA Replication Defects
Various mutants defective in the stabilization of DNA replication intermediates accumulate recombinogenic lesions in S phase, due to DNA replication fork collapse, failure to adequately process Okazaki fragments or resulting from lower levels of dNTPs. This is particularly true of mec1 hypomorphs, such as mec1-21 and mec1-srf mutants [113], which exhibit decreased viability when RAD52 is inactivated. The hyper-recombination of mec1-21 is suppressed by mutations in SML1, an inhibitor of ribonucleotide reductase, suggesting that elevated dNTP levels reduce the accumulation of recombinogenic substrates [114]. Unlike rad9 mutants, which do not exhibit higher rates of spontaneous uSCR and heteroallelic recombination, mec1-21 mutants exhibit hyper-recombination in spontaneous uSCR and heteroallelic recombination. RAD9 is required for the hyper-recombination phenotypes that are exhibited by mec1-21 mutants and mec1-21 rad9 double mutants exhibit similar rates of homology-directed translocations as rad9 diploids. An attractive model is that RAD9 is required to delay the cell cycle so that recombinogenic DNA damage produced by replication fork collapse or replication errors can be repaired. However, additional observations indicate that RAD9 is required to protect stalled or collapsed replication forks from excessive degradation [112], and that rad9 mutants exhibit hyper-resection leading to Mec1-mediated Sgs1 phosphorylation [115]. Thus, a combination of factors may function to ensure that recombinogenic lesions generated during DNA replication can be adequately repaired so that replication fork progression may be complete.
Phenotypes of mec1-21 rad9 mutants are partially mimicked by mec1-21 chk1 and mec1-21 pds1 mutants, while higher frequencies of homology-directed recombination observed in rad9 diploids are also exhibited by rad53 diploids [87]. For example, mutations in either PDS1 or CHK1 reduce the hyper-recombination phenotype of mec1-21, and also increase radiation sensitivity in mec1-21, as observed in mec1-21 rad9 mutants. One interpretation of these results is that mutations in either CHK1 or PDS1 confer toleration under-replicated DNA, which would otherwise be a substrate for HR proteins. These observations suggest that RAD9 functions in promoting genome instability observed in S phase checkpoints function through downstream effectors.
Besides promoting recombination in S phase checkpoint mutants, RAD9 promotes triplet GAA repeat expansion in cdc13-1 mutants [116]. These mutants exhibit telomeric single strands and such single strands may sequester Mrc1 and additional proteins required for replication fork signaling. Although slowed replication progression is not associated with GAA repeat expansion in the cdc13 strain, the RAD9-mediated checkpoint pathway genes, RAD9, RAD53, MEC1, and EXO1 are required. The authors postulate that RAD9 is required in post-replicative repair and that polymerase replication over such replicative gaps may promote the repeat expansion [116].
4. Rad9 Role in Suppressing Gcrs
RAD9 functions in suppressing gross GCRs that result from fusion of indirect repeated sequences, which does not require HR functions. This was demonstrated in a chromosome VII disomic strain where one copy of chromosome VII contains a telomeric CAN1 gene and internal short, inverted repeats. Spontaneous CanR mutants containing rearrangements generated by fusion of these inverted repeats can then be selected and screened based on sectored colony phenotype; sectors typically contain unstable dicentric and acentric chromosomes [117,118]. rad9 mutants exhibit 17-fold higher frequencies of CanR isolates containing such rearrangements. The authors suggest that inverted repeat fusions result from aberrant template switching events during S phase.
However, RAD9 and G2/M checkpoint genes have a minor role in suppressing spontaneous Chr V rearrangements, compared to S phase checkpoint mutants [119]. The GCR assay used a haploid strain containing URA3 and CAN1 genes located on the non-essential chromosomal V arm; double selection against both URA3 and CAN1 using 5-fluororotic acid (5-FOA) and canavanine, respectively, generates drug resistant isolates containing chromosomal rearrangements. rad9 mutants have slightly elevated rates (threefold) of GCRs, while the double rad9 exol mutant exhibits a slightly more elevated rate of GCR, compared to the single mutants [120]. Even when URA3 gene is placed in a position adjacent to Ty1 element, the overall rate of GCRs in rad9 mutant is only two-fold greater than that observed in wild type (Table 2). Since non-homologous end joining (NHEJ) is a major mechanism for generating GCRs, one possible explanation for the modest increase in GCRs in rad9 mutants is the rad9 deficiency in NHEJ [121,122]). Another contributing factor is that rad9 mutants exhibit chromosome loss [76], which may be lethal in haploid strains.
While RAD9 function in suppressing spontaneous double drug-resistant isolates is minor, its function in suppressing frequencies of DNA damage-associated GCRs is significant. Cells exposed to 0.07% MMS, exhibit -168-fold more GCRs compared to the spontaneous frequency in rad9 [123], while wild-type exhibits a 68-fold more GCRs, compared to the spontaneous frequency in wild type. While this fold increase may seem minor, the overall frequencies of MMS-associated GCRs is approximately 1200-fold greater than the frequency of spontaneous GCRs in wild type [123], compared to the 98-fold increase in the frequency of MMS-associated GCRs observed in wild type compared to the spontaneous frequency observed in wild type. MMS is known to significantly impede DNA replication [124], supporting observations that S phase DNA errors are the major initiating cause for GCRs.
Comparing the genetic control and DNA damage-inducibility of GCRs and HR-directed translocations reveals important similarities and differences. The major similarity is that defects in S phase checkpoint defects confer the highest increases in frequencies of either GCRs or HR-mediated rearrangements, likely due to recombinogenic structures generated during replication. Additionally, the higher rates of spontaneous GCRs and HR-directed translocations observed in the mec1 mutant can be synergistically increased by knocking-out RAD51 [125], suggesting that the checkpoint and recombinational repair pathway are independent in suppressing both NHEJ-mediated and HR-mediated rearrangements. RAD9 suppresses both DNA damage-associated GCRs or HR-directed translocations, particularly when cells are exposed to the X-ray mimetic chemical, MMS. Both frequencies of GCRs and HR-directed translocations are stimulated by diverse DNA damaging agents, ranging from simple alkylating agents to both X rays and UV.
The difference between the genetic control and DNA damage-inducibility of GCR and HR-directed translocations are also notable. DNA damage-associated but not spontaneous GCRs require yKu70 [123,126,127], a gene required for NHEJ; whereas, the highest frequencies of radiation-associated HR-directed translocations are observed in diploid and not haploid strains [128], in which NHEJ is repressed. Indeed, in yku70 haploid mutants, DNA damage-associated GCRs are abolished [123] while five to six-fold increase in X-ray associated homology-directed translocations are observed in haploid yku70 mutants [129]. While the increase in DNA damage-associated GCRs is significant, the total number of radiation-associated HR translocations is significantly higher. These observations underscore observations that DSBs are important lesions in remodeling the yeast genome even when NHEJ is abolished.
5. Similarities of Rad9 Orthologs in Promoting Genetic Stability
The budding yeast RAD9 ortholog in Schizosacharomyces pombe (fission yeast) is crb2+ and the human ortholog is 53BP1, which share structural and functional similarities. Similar to budding yeast Rad9, 53BP1 and Crb2 contain Tudor(2) and BRCT(2) domains, which enable these proteins to bind to chromatin and DSBs and provide a scaffold for additional checkpoint proteins [130].They are also recognized and phosphorylated by cyclin-dependent kinases and ATM, and promote HR-mediated repair of DSBs and cross-overs while enabling checkpoint-mediated arrest. They differ, however, in how they regulate HR and promote NHEJ.
In S. pombe, crb2 mutants are defective in DSB repair and exhibit enhanced loss of linear mini-chromosomes and LOH, likely due to BIR [131]. This was shown in haploid strains that contain a linear mini-chromosome in which a HO-endonuclease cut site is flanked by a non-tandem duplication, and SSA was measured upon induction of the DSB. In an independent study, the X-ray sensitivity of crb2 mutant deficient in Cdk-mediated phosphorylation was shown to be suppressed by mutations in topoisomerase III, suggesting that lack of crb2 leads to the accumulation of recombination intermediates [132]. These studies have suggested that S. pombe crb2+ may function at multiple stages in HR. However, since diploidy is unstable in S. pombe [133], it is difficult to ascertain to the function of crb2+ in suppressing the formation of rearrangements that would confer lethality in haploids.
While 53BP1 is a well-known marker for DSBs in mammalian cells, 53BP1 has opposing roles in promoting HR; while it limits resection and thus promotes NHEJ, it promotes DSB repair by HR at heterochromatin in G2 cells [134]. The role of limiting end resection, however, serves important in VD(J) recombination and at unprotected telomeres. Thus, both Crb2 and 53BP1 function to promote HR in particular contexts. These studies indicate evolutionary conservations of some functions of budding yeast RAD9.
6. Summary
RAD9 plays opposing roles in both promoting genetic instability and suppressing genetic instability. By arresting the cell cycle at the G2/M stage, RAD9 provides time for recombinogenic DNA damage to be adequately repaired by SCR and by SSA between sequences that have significant mismatches. On the other hand, by triggering a checkpoint response due to replication errors, RAD9 triggers downstream effectors that may promote genome instability resulting in both ectopic recombination and repeat amplifications. While RAD9’s role in controlling DNA damage-associated checkpoint activation has been well-studied, how RAD9 suppresses recombination due to spontaneous DNA damage [136] is unknown.
At the molecular level, there are two alternative mechanisms by which RAD9 influences HR: 1) RAD9 controls cell cycle arrest at particular points in the cell cycle by facilitating Rad53 and Chk1 activation, and 2) RAD9 promotes cross-overs, both in its role in controlling DNA end resection and in its role in inhibiting both Sgs1 and Mph1. How these RAD9 activities function in the context of ploidy, position of repeat units, sequence divergence, and sequence orientation has yet been fully investigated. Understanding these functions will translate into a better understanding of the role of human 53BP1 in HR and in DSB repair.
Author Contributions
Michael Fasullo contributed the information concerning checkpoint pathways and data concerning the homologous recombination phenotypes observed in rad9 mutants.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data provided in this review is provided with references attached. Unpublished data may be provided upon request.
Acknowledgments
This work was supported by National Institutes of Health (This research was supported by grants from the National Institutes of Health, R01CA70105 and R21ES015954, R15ES02685 and a grant from the Center for Advancement of Nanotechnology (CATN).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
CNV, Copy number variation
DSB, double-strand breaks
5-FOA, 5-fluororotic acid
GCR, gross chromosomal rearrangements
HC, half crossover
HOcs, HO endonuclease cut site
HR, homologous recombination
LOH, loss of heterozygosity
MMEJ, microhomology-mediated end joining
MMS, methyl methanesulfonate
NHEJ, non-homologous end-joining
4-NQO, 4-nitroquinoline 1-oxide
Orc, Origin recognition complex
SSB, single-strand breaks
SCR, sister chromatid recombination
uSCR, unequal sister chromatid recombination
References
- Hartwell, L.H.; Weinert, T.A. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989, 246, 629–634. [Google Scholar] [CrossRef]
- Prakash, L. Lack of chemically induced mutation in repair-deficient mutants of yeast. Genetics 1974, 78, 1101–1118. [Google Scholar] [CrossRef]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. U.S.A 2008, 105, 11845–11850. [Google Scholar] [CrossRef]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef]
- Cussiol, J.R.; Soares, B.L.; Oliveira, F.M.B. From yeast to humans: Understanding the biology of DNA Damage Response (DDR) kinases. Genet Mol Biol. 2019, 43, e20190071. [Google Scholar] [CrossRef]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: checkpoint and repair at 30 years. EMBO J. 2019, 16(38(18)), e101801. [Google Scholar] [CrossRef]
- Aboussekhra, A.; Vialard, J.E.; Morrison, D.E.; de la Torre-Ruiz, M.A.; Cernakova, L.; Fabre, F.; Lowndes, N. F. A novel role for the budding yeast RAD9 checkpoint gene in DNA damage-dependent transcription. The EMBO Journal 1996, 15, 3912–3922. [Google Scholar] [CrossRef] [PubMed]
- Jaehnig, E.J.; Kuo, D.; Hombauer, H.; Ideker, T.G.; Kolodner, R.D. Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 2013, 4, 174–188. [Google Scholar] [CrossRef]
- Clerici, M.; Trovesi, C.; Galbiati, A.; Lucchini, G.; Longhese, M.P. Mec1/ATR regulates the generation of single-stranded DNA that attenuates Tel1/ATM signaling at DNA ends. EMBO J. 2014, 33, 198–216. [Google Scholar] [CrossRef] [PubMed]
- Casari, E.; Tisi, R.; Longhese, M. P. Checkpoint activation and recovery: regulation of the 9-1-1 axis by the PP2A phosphatase. DNA repair 2025, 103854–103864. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niu, H.; Yu, Y.; Wang, J.; Zhu, S.; Zhou, J.; Papusha, A.; Cui, D.; Pan, X.; Kwon, Y.; Sung, P.; Ira, G. Enrichment of Cdk1-cyclins at DNA double-strand breaks stimulates Fun30 phosphorylation and DNA end resection. Nucleic Acids Res. 2016, 44, 2742–2753. [Google Scholar] [CrossRef]
- Waterman, D.P.; Haber, J.E.; Smolka, M.B. Checkpoint Responses to DNA Double-Strand Breaks. Annu Rev Biochem. 2020, 89, 103–133. [Google Scholar] [CrossRef]
- Tercero, J.A.; Diffley, J.F. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001, 412, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.; Gn, K.; Lichten, M. Unresolved Recombination Intermediates Cause a RAD9-Dependent Cell Cycle Arrest in Saccharomyces cerevisiae. Genetics 2019, 213, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Garvik, B.; Carson, M.; Hartwell, L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol. 1995, 15, 6128–6138. [Google Scholar] [CrossRef]
- Lazzaro, V.; Sapountzi, M.; Granata, A.; Pellicioli, M.; Vaze, J.E.; Haber, J.; Plevani, P.; Lydall, D.; Muzi-Falconi, M. Histone methyltransferase Dot1 and Rad9 inhibit single-stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J. 2008, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Andreadis, C.; Nikolaou, C.; Fragiadakis, G.S.; Tsiliki, G.; Alexandraki, D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014, 42, 12650–12667. [Google Scholar] [CrossRef]
- Paulovich, A.G.; Margulies, R.U.; Garvik, B.M.; Hartwell, L.H. RAD9, RAD17, and RAD24 are required for S phase regulation in Saccharomyces cerevisiae in response to DNA damage. Genetics 1997, 145, 45–62. [Google Scholar] [CrossRef]
- Levin, N.A.; Bjornsti, M.A.; Fink, G.R. A novel mutation in DNA topoisomerase I of yeast causes DNA damage and RAD9-dependent cell cycle arrest. Genetics 1993, 133, 799–814. [Google Scholar] [CrossRef]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.H.; Seiler, J.A.; Zhang, H.; Marchand, C.; Agama, K.; Nitiss, J.L.; Redon, C. Repair of topoisomerase I-mediated DNA damage. Prog Nucleic Acid Res Mol Biol. 2006, 81, 179–229. [Google Scholar]
- Usui, T.; Shinohara, A. Rad9, a 53BP1 Ortholog of Budding Yeast, Is Insensitive to Spo11-Induced Double-Strand Breaks During Meiosis. Front Cell Dev Biol. 2021, 9, 635383. [Google Scholar] [CrossRef]
- Lyndaker, A.M.; Goldfarb, T.; Alani, E. Mutants defective in Rad1-Rad10-Slx4 exhibit a unique pattern of viability during mating-type switching in Saccharomyces cerevisiae. Genetics 2008, 179, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb Perspect Biol. 2014, 6(9), a016428. [Google Scholar] [CrossRef] [PubMed]
- Brunborg, G.; Resnick, M.A.; Williamson, D.H. Cell-cycle-specific repair of DNA double strand breaks in Saccharomyces cerevisiae. Radiat Res. 1980, 82, 547–558. [Google Scholar] [CrossRef]
- Kadyk, L.C.; Hartwell, L.H. Sister chromatids are preferred over homologs as substrates for recombinational repair in Saccharomyces cerevisiae. Genetics 1992, 132, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Lustig, A.J.; Petes, T.D. Identification of yeast mutants with altered telomere structure. Proc Natl Acad Sci U S A 1986, 83, 1398–1402. [Google Scholar] [CrossRef]
- Kato, R.; Ogawa, H. An essential gene, ESR1, is required for mitotic cell growth, DNA repair, and meiotic recombination in Saccharomyces cerevisiae. Nucleic Acids Res. 1994, 22, 3104–3112. [Google Scholar] [CrossRef]
- Shiloh, Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. 2003, 3, 3155–3168. [Google Scholar] [CrossRef]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; Haber, J.E.; Foiani, M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef]
- Sanford, E. J.; Comstock, W. J.; Faça, V. M.; Vega, S. C.; Gnügge, R.; Symington, L. S.; et al. Phosphoproteomics reveals a distinctive Mec1/ATR signaling response upon DNA end hyper-resection. EMBO J. 2021, 40, e104566. [Google Scholar] [CrossRef]
- Flott, S.; Kwon, Y.; Pigli, Y.Z.; Rice, P.A.; Sung, P.; Jackson, S.P. Regulation of Rad51 function by phosphorylation. EMBO Rep. 2011, 12, 833–839. [Google Scholar] [CrossRef]
- Herzberg, K.; Bashkirov, V.I.; Rolfsmeier, M.; Haghnazari, E.; McDonald, W.H.; Anderson, S.; Bashkirova, E.V.; Yates, J.R.; Heyer, W.D. Phosphorylation of Rad55 on serines 2, 8, and 14 is required for efficient homologous recombination in the recovery of stalled replication forks. Mol Cell Biol. 2006, 26, 8396–8409. [Google Scholar] [CrossRef]
- Chen, Y.; Caldwell, J.M.; Pereira, E.; Baker, R.W.; Sanchez, Y. ATRMec1 phosphorylation-independent activation of Chk1 in vivo. J Biol Chem. 2009, 284, 182–190. [Google Scholar] [CrossRef]
- Sanchez, Y.; Desany, B.A.; Jones, W.J; Liu, Q.; Wang, B.; Elledge, S.J. Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science. 1996, 271, 357–360. [Google Scholar] [CrossRef]
- Toh, G.W.; Lowndes, N.F. Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem Soc Trans. 2003, 31, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Bachant, J.; Wang, H.; Hu, F.; Liu, D.; Tetzlaff, M.; Elledge, S.J. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 1999, 286, 1166–1171. [Google Scholar] [CrossRef]
- Zhao, X.; Chabes, A.; Domkin, V.; Thelander, L.; Rothstein, R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 2001, 20, 3544–3553. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Foster, S.S.; Petrini, J.H. Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol Cell. 2009, 33, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Tang, Z.; Yu, H.; Cohen-Fix, O. Two distinct pathways for inhibiting pds1 ubiquitination in response to DNA damage. J Biol Chem. 2003, 278, 45027–45033. [Google Scholar] [CrossRef]
- Hu, F.; Wang, Y.; Liu, D.; Li, Y.; Qin, J.; Elledge, S.J. Regulation of the Bub2/Bfa1 GAP complex by Cdc5 and cell cycle checkpoints. Cell 2001, 107, 655–665. [Google Scholar] [CrossRef]
- Makovets, S.; Blackburn, E.H. DNA damage signaling prevents deleterious telomere addition at DNA breaks. Nat Cell Biol. 2009, 11, 1383–1386. [Google Scholar] [CrossRef]
- Dion, V.; Kalck, V.; Horigome, C.; Towbin, B.D.; Gasser, S.M. Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol. 2012, 14, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Miné-Hattab, J.; Rothstein, R. Increased chromosome mobility facilitates homology search during recombination. Nat Cell Biol. 2012, 14, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Ngo, G.H.; Balakrishnan, L.; Dubarry, M.; Campbell, J.L.; Lydall, D. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res. 2014, 42, 10516–10528. [Google Scholar] [CrossRef]
- Marcomini, I.; Shimada, K.; Delgoshaie, N.; Yamamoto, I.; Seeber, A.; Cheblal, A.; Horigome, C.; Naumann, U.; Gasser, S.M. Asymmetric Processing of DNA Ends at a Double-Strand Break Leads to Unconstrained Dynamics and Ectopic Translocation. Cell Rep. 2018, 24, 2614–2628.e4. [Google Scholar] [CrossRef]
- Lazzaro, F.; Giannattasio, M.; Puddu, F.; Granata, M.; Pellicioli, A.; Plevani, P.; Muzi-Falconi, M. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair (Amst) 2009, 8, 1055–1067. [Google Scholar] [CrossRef]
- Finn, K.; Lowndes, N.F.; Grenon, M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci. 2012, 69, 1447–1473. [Google Scholar] [CrossRef] [PubMed]
- Krogh, B.O.; Symington, L.S. Recombination proteins in yeast. Annu. Rev. Genet. 2004, 38, 233–271. [Google Scholar] [CrossRef]
- Gangloff, S.; McDonald, J.P.; Bendixen, C.; Arthur, L.; Rothstein, R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol. 1994, 14, 8391–8398. [Google Scholar] [CrossRef]
- Ip, S.C.; Rass, U.; Blanco, M.G.; Flynn, H.R.; Skehel, J.M.; West, S.C. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008, 20, 357–361. [Google Scholar] [CrossRef]
- Ferrari, M.; Dibitetto, D.; De Gregorio, G.; Eapen, V.V.; Rawal, C.C.; Lazzaro, F.; Tsabar, M.; Marini, F.; Haber, J.E.; Pellicioli, A. Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet. 2015, 11, e1004928. [Google Scholar] [CrossRef]
- Claussin, C.; Porubský, D.; Spierings, D.C.; Halsema, N.; Rentas, S.; Guryev, V.; Lansdorp, P.M.; Chang, M. Genome-wide mapping of sister chromatid exchange events in single yeast cells using Strand-seq. Elife 2017, 6e30560. [Google Scholar]
- Fasullo, M.; Giallanza, P.; Dong, Z.; Cera, C.; Bennett, T. Saccharomyces cerevisiae rad51 mutants are defective in DNA damage-associated sister chromatid exchanges but exhibit increased rates of homology-directed translocations. Genetics 2001, 158, 959–972. [Google Scholar] [CrossRef]
- Dong, Z.; Fasullo, M. Multiple recombination pathways for sister chromatid exchange in Saccharomyces cerevisiae: role of RAD1 and the RAD52 epistasis group genes. Nucleic Acids Res. 2003, 31, 2576–2785. [Google Scholar] [CrossRef]
- Ivanov, E.L.; Sugawara, N.; White, C.I.; Fabre, F.; Haber, J.E. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1994, 14(5), 3414–3425. [Google Scholar] [PubMed]
- Westmoreland, J.W.; Resnick, M.A. Recombinational repair of radiation-induced double-strand breaks occurs in the absence of extensive resection. Nucleic Acids Res. 2016, 29, 695–704. [Google Scholar] [CrossRef]
- Ivanov, E.L.; Sugawara, N.; Fishman-Lobell, J.; Haber, J.E. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 1996, 142, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Prado, F.; Aguilera, A. Role of reciprocal exchange, one-ended invasion crossover and single-strand annealing on inverted and direct repeat recombination in yeast: different requirements for the RAD1, RAD10, and RAD52 genes. Genetics 1995, 139, 109–123. [Google Scholar] [CrossRef]
- Mathiasen, D.P.; Lisby, M. Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae. FEMS Microbiology Reviews 2014, 38, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Saka, Y.; Esashi, F.; Matsusaka, T.; Mochida, S.; Yanagida, M. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997, 11, 3387–3400. [Google Scholar] [CrossRef]
- Alpha-Bazin, B.; Lorphelin, A.; Nozerand, N.; Charier, G.; Marchetti, C.; Bérenguer, F.; Couprie, J.; Gilquin, B.; Zinn-Justin, S.; Quéméneur, E. Boundaries and physical characterization of a new domain shared between mammalian 53BP1 and yeast Rad9 checkpoint proteins. Protein Sci. 2005, 14, 1827–1839. [Google Scholar] [CrossRef] [PubMed]
- Nnakwe, C.C.; Altaf, M.; Côté, J.; Kron, S.J. Dissection of Rad9 BRCT domain function in the mitotic checkpoint response to telomere uncapping. DNA Repair (Amst) 2009, 8, 1452–1461. [Google Scholar] [CrossRef]
- Siede, W.; Friedberg, A.S.; Friedberg, E.C. RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc Natl Acad Sci U. S. A 1993, 90, 7985–7989. [Google Scholar] [CrossRef] [PubMed]
- Bacal, J.; Moriel-Carretero, M.; Pardo, B.; Barthe, A.; Sharma, S.; Chabes, A.; Lengronne, A.; Pasero, P. Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J. 2018, 37, (21):e99319. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niu, H.; Yu, Y.; Wang, J.; Zhu, S.; Zhou, J.; Papusha, A.; Cui, D.; Pan, X.; Kwon, Y.; Sung, P.; Ira, G. Enrichment of Cdk1-cyclins at DNA double-strand breaks stimulates Fun30 phosphorylation and DNA end resection. Nucleic Acids Res. 2016, 44, 2742–2753. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; Bridgland, A.; Meyer, C.; Kohl, S.A.A.; Ballard, A.J.; Cowie, A.; Romera-Paredes, B.; Nikolov, S.; Jain, R.; Adler, J.; Back, T.; Petersen, S.; Reiman, D.; Clancy, E.; Zielinski, M.; Steinegger, M.; Pacholska, M.; Berghammer, T.; Bodenstein, S.; Silver, D.; Vinyals, O.; Senior, A.W.; Kavukcuoglu, K.; Kohli, P.; Hassabis, D. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. Available online: https://alphafold.ebi.ac.uk/entry/P14737. [CrossRef]
- Usui, T.; Foster, S.S.; Petrini, J.H. Maintenance of the DNA-damage checkpoint requires DNA-damage-induced mediator protein oligomerization. Mol Cell. 2009, 33, 147–159. [Google Scholar] [CrossRef]
- Lancelot, N.; Charier, G.; Couprie, J.; Duband-Goulet, I.; Alpha-Bazin, B.; Quémeneur, E.; Ma, E.; Marsolier-Kergoat, M.C.; Ropars, V.; Charbonnier, J.B.; Miron, S.; Craescu, C.T.; Callebaut, I.; Gilquin, B.; Zinn-Justin, S. The checkpoint Saccharomyces cerevisiae Rad9 protein contains a tandem tudor domain that recognizes DNA. Nucleic Acids Res. 2007, 35, 5898–5912. [Google Scholar] [CrossRef]
- Pfander, B; Diffley, JF. Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J 2011, 30(24), 4897–907. [Google Scholar] [CrossRef]
- Schwartz, M.F.; Duong, J.K.; Sun, Z.; Morrow, J.S.; Pradhan, D.; Stern, D.F. Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell. 2002, 9, 1055–1065. [Google Scholar] [CrossRef]
- van den Bosch, M.; Lowndes, NF. Remodeling the Rad9 checkpoint complex: preparing Rad53 for action. Cell Cycle 2004, 3, 119–122. [Google Scholar] [CrossRef]
- Vialard, J.E.; Gilbert, C.S.; Green, C.M.; Lowndes, NF. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 1998, 17, 5679–5688. [Google Scholar] [CrossRef]
- Blankley, R.T.; Lydall, D. A domain of Rad9 specifically required for activation of Chk1 in budding yeast. J Cell Sci. 2004, 117, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T.A.; Hartwell, L.H. Cell cycle arrest of cdc mutants and specificity of the RAD9 checkpoint. Genetics 1993, 134, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.L. Spontaneous chromosome loss in Saccharomyces cerevisiae is suppressed by DNA damage checkpoint functions. Genetics 2001, 159, 1501–1509. [Google Scholar] [CrossRef]
- Theis, J.F.; Irene, C.; Dershowitz, A.; Brost, R.L.; Tobin, M.L.; di Sanzo, F.M.; Wang, J.Y.; Boone, C.; Newlon, C.S. The DNA damage response pathway contributes to the stability of chromosome III derivatives lacking efficient replicators. PLoS Genet. 2010, 6(12), e1001227. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Crabbé, L.; Pasero, P. Signaling pathways of replication stress in yeast. FEMS Yeast Res. 2017, 1(17(2)). [Google Scholar] [CrossRef]
- Vallen, E.A.; Cross, F.R. Mutations in RAD27 define a potential link between G1 cyclins and DNA replication. Mol Cell Biol. 1995, 15, 4291–4302. [Google Scholar] [CrossRef]
- Medina-Rivera, M.; Phelps, S.; Sridharan, M.; Becker, J.; Lamb, N.A.; Kumar, C.; Sutton, M.D.; Bielinsky, A.; Balakrishnan, L.; Surtees, J.A. Elevated MSH2 MSH3 expression interferes with DNA metabolism in vivo. Nucleic Acids Res. 2023, 51, 12185–12206. [Google Scholar] [CrossRef]
- Debrauwère, H.; Loeillet, S.; Lin, W.; Lopes, J.; Nicolas, A. Links between replication and recombination in Saccharomyces cerevisiae: a hypersensitive requirement for homologous recombination in the absence of Rad27 activity. Proc Natl Acad Sci U S A 2001, 98, 8263–8269. [Google Scholar] [CrossRef]
- Fasullo, M.; Bennett, T.; AhChing, P.; Koudelik, J. The Saccharomyces cerevisiae RAD9 checkpoint reduces the DNA damage-associated stimulation of directed translocations. Mol Cell Biol. 1998, 18, 1190–1200. [Google Scholar] [CrossRef]
- Conde, F.; Refolio, E; Cordón-Preciado, V; Cortés-Ledesma, F; Aragón, L; Aguilera, A; San-Segundo, PA. The Dot1 histone methyltransferase and the Rad9 checkpoint adaptor contribute to cohesin-dependent double-strand break repair by sister chromatid recombination in Saccharomyces cerevisiae. Genetics 2009, 182, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Ström, L.; Karlsson, C.; Lindroos, H.B.; Wedahl, S.; Katou, Y.; Shirahige, K.; Sjögren, C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 2007, 317, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Litwin, I.; Pilarczyk, E.; Wysocki, R. The Emerging Role of Cohesin in the DNA Damage Response. Genes (Basel) 2018, 9, 581–605. [Google Scholar] [CrossRef]
- Fasullo, M.; Dong, Z.; Sun, M.; Zeng, L. Saccharomyces cerevisiae RAD53 (CHK2) but not CHK1 is required for double-strand break-initiated SCE and DNA damage-associated SCE after exposure to X rays and chemical agents. DNA Repair (Amst). 2005, 4, 1240–1251. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Sun, M. The Saccharomyces cerevisiae checkpoint genes RAD9, CHK1 and PDS1 are required for elevated homologous recombination in a mec1 (ATR) hypomorphic mutant. Cell Cycle 2008, 7, 2418–2426. [Google Scholar] [CrossRef]
- Covo, S.; Chiou, E.; Gordenin, D.A.; Resnick, M.A. Suppression of allelic recombination and aneuploidy by cohesin is independent of Chk1 in Saccharomyces cerevisiae. PLoS One. 2014, 9(12), e113435. [Google Scholar] [CrossRef]
- Fasullo, M.; Zeng, L.; Giallanza, P. Enhanced stimulation of chromosomal translocations by radiomimetic DNA damaging agents and camptothecin in Saccharomyces cerevisiae rad9 checkpoint mutants. Mutat Res. 2004, 547, 123–132. [Google Scholar] [CrossRef]
- Zhang, H.; Lawrence, C.W. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci U S A 2005, 102, 15954–15959. [Google Scholar] [CrossRef]
- Fasullo, M; Sun, M. Both RAD5-dependent and independent pathways are involved in DNA damage-associated sister chromatid exchange in budding yeast. AIMS Genet 2017, 4, 84–102. [Google Scholar] [CrossRef]
- Ide, S.; Watanabe, K.; Watanabe, H.; Shirahige, K.; Kobayashi, T.; Maki, H. Abnormality in initiation program of DNA replication is monitored by the highly repetitive rRNA gene array on chromosome XII in budding yeast. Mol Cell Biol. 2007, 27, 568–578. [Google Scholar] [CrossRef]
- Whale, A.J.; King, M.; Hull, R.M.; Krueger, F.; Houseley, J. Stimulation of adaptive gene amplification by origin firing under replication fork constraint. Nucleic Acids Res. 2022, 50, 915–936. [Google Scholar] [CrossRef]
- DeMase, D.; Zeng, L.; Cera, C.; Fasullo, M. The Saccharomyces cerevisiae PDS1 and RAD9 checkpoint genes control different DNA double-strand break repair pathways. DNA Repair 2005, 4, 59–69. [Google Scholar] [CrossRef]
- George, C.M.; Lyndaker, A.M.; Alani, E. The DNA damage checkpoint allows recombination between divergent DNA sequences in budding yeast. DNA Repair (Amst). 2011, 10, 1086–1094. [Google Scholar] [CrossRef]
- Flott, S.; Alabert, C.; Toh, G.W.; Toth, R.; Sugawara, N.; Campbell, D.G.; Haber, J.E.; Pasero, P.; Rouse, J. Phosphorylation of Slx4 by Mec1 and Tel1 regulates the single-strand annealing mode of DNA repair in budding yeast. Mol Cell Biol. 2007, 27, 6433–6445. [Google Scholar] [CrossRef]
- Toh, G.W.; Sugawara, N.; Dong, J.; Toth, R.; Lee, S.E.; Haber, J.E.; Rouse, J. Mec1/Tel1-dependent phosphorylation of Slx4 stimulates Rad1-Rad10-dependent cleavage of non-homologous DNA tails. DNA Repair (Amst) 2010, 9, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Weinert, T. A.; Hartwell, L. H. Characterization of the RAD9 gene of Saccharomyces cerevisiae and evidence that it acts posttranslationally in cell cycle arrest after DNA damage. Mol. Cell. Biol. 1990, 10, 6554–6564. [Google Scholar]
- Beyer, T.; Weinert, T. Ontogeny of Unstable Chromosomes Generated by Telomere Error in Budding Yeast. PLoS Genet. 2016, 12(10), e1006345. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Sun, M.; Fasullo, M. Checkpoint and recombination pathways independently suppress rates of spontaneous homology-directed chromosomal translocations in budding yeast. Front Genet. 2025, 6, 1479307. [Google Scholar] [CrossRef]
- Craven, R.J.; Greenwell, P.W.; Dominska, M.; Petes, T.D. Regulation of genome stability by TEL1 and MEC1, yeast homologs of the mammalian ATM and ATR genes. Genetics 2002, 161, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Rawal, C.C.; Lodovichi, S.; Vietri, M.Y.; Pellicioli, A. Rad9/53BP1 promotes DNA repair via crossover recombination by limiting the Sgs1 and Mph1 helicases. Nat Commun. 2020, 11, 3181. [Google Scholar] [CrossRef] [PubMed]
- Morrow, D.M.; Connelly, C.; Hieter, P. Break copy" duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 1997, 147, 371–382. [Google Scholar] [CrossRef]
- Watanabe, K.; Morishita, J.; Umezu, K.; Shirahige, K.; Maki, H. Involvement of RAD9-dependent damage checkpoint control in arrest of cell cycle, induction of cell death, and chromosome instability caused by defects in origin recognition complex in Saccharomyces cerevisiae. Eukaryot. Cell 2002, 1, 200–212. [Google Scholar] [CrossRef]
- Finn, K.; Lowndes, N.F.; Grenon, M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell Mol Life Sci. 2012, 69, 1447–1473. [Google Scholar] [CrossRef]
- Game, J.C.; Williamson, M.S.; Spicakova, T.; Brown, J.M. The RAD6/BRE1 histone modification pathway in Saccharomyces confers radiation resistance through a RAD51-dependent process that is independent of RAD18. Genetics 2006, 173, 1951–1968. [Google Scholar] [CrossRef]
- Pannunzio, N.R.; Manthey, G.M.; Bailis, A. M. RAD59 and RAD1 cooperate in translocation formation by single-strand annealing in Saccharomyces cerevisiae. Current genetics 210(56), 87–100. [CrossRef] [PubMed]
- Wu, Y.; Kantake, N.; Sugiyama, T.; Kowalczykowski, S.C. Rad51 protein controls Rad52-mediated DNA annealing. J Biol Chem. 2008, 283, 14883–14892. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.N.; Pham, N.; Tsai, A.M.; Janto, N.V.; Choi, J.; Ira, G.; Haber, J.E. A Rad51-independent pathway promotes single-strand template repair in gene editing. PLoS Genet. 2020, 16(10), e1008689. [Google Scholar] [CrossRef]
- Schmidt, K.H.; Wu, J.; Kolodner, R.D. Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol Cell Biol. 2006, 26, 5406–5420. [Google Scholar] [CrossRef]
- Vasan, S.; Deem, A.; Ramakrishnan, S.; Argueso, J.L.; Malkova, A. Cascades of genetic instability resulting from compromised break-induced replication. PLoS Genet. 2014, 10(2), e1004119. [Google Scholar] [CrossRef]
- Villa, M.; Bonetti, D.; Carraro, M.; Longhese, M.P. Rad9/53BP1 protects stalled replication forks from degradation in Mec1/ATR-defective cells. EMBO Rep. 2018, 19, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Merrill, B.J.; Holm, C. A requirement for recombinational repair in Saccharomyces cerevisiae is caused by DNA replication defects of mec1 mutants. Genetics. 1999, 153, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Fasullo, M.; Tsaponina, O.; Sun, M.; Chabes, A. Elevated dNTP levels suppress hyper-recombination in Saccharomyces cerevisiae S-phase checkpoint mutants. Nucleic Acids Res. 2010, 38, 1195–1203. [Google Scholar] [CrossRef]
- Xie, B.; Sanford, E. J.; Hung, S. H.; Wagner, M.; Heyer, W. D.; Smolka, M. B. Multi-step control of homologous recombination via Mec1/ATR suppresses chromosomal rearrangements. EMBO J. 2024, 43, 3027–3043. [Google Scholar] [CrossRef]
- Spivakovsky-Gonzalez, E.; Polleys, E.J.; Masnovo, C.; Cebrian, J.; Molina-Vargas, A.M.; Freudenreich, C.H.; Mirkin, S.M. Rad9-mediated checkpoint activation is responsible for elevated expansions of GAA repeats in CST-deficient yeast. Genetics 2021, 219(2), iyab125. [Google Scholar] [CrossRef] [PubMed]
- Paek, A.L.; Kaochar, S.; Jones, H.; Elezaby, A.; Shanks, L.; Weinert, T. Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev. 2009, 23, 2861–2875. [Google Scholar] [CrossRef]
- Kaochar, S.; Shanks, L.; Weinert, T. Checkpoint genes and Exo1 regulate nearby inverted repeat fusions that form dicentric chromosomes in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 2010, 107, 21605–21610. [Google Scholar] [CrossRef]
- Myung, K.; Datta, A.; Kolodner, R.D. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell 2001, 104, 397–408. [Google Scholar] [CrossRef]
- Putnam, C.D.; Kolodner, R.D. Pathways and Mechanisms that Prevent Genome Instability in Saccharomyces cerevisiae. Genetics. 2017, 206, 1187–1225. [Google Scholar] [CrossRef]
- de la Torre-Ruiz, M.; Lowndes, N.F. The Saccharomyces cerevisiae DNA damage checkpoint is required for efficient repair of double strand breaks by non-homologous end joining. FEBS Lett. 2000, 467, 311–315. [Google Scholar] [CrossRef]
- Lewis, L.K.; Kirchner, J.M.; Resnick, M.A. Requirement for end-joining and checkpoint functions, but not RAD52-mediated recombination, after EcoRI endonuclease cleavage of Saccharomyces cerevisiae DNA. Mol Cell Biol. 1998, 18, 1891–1902. [Google Scholar] [CrossRef]
- Myung, K.; Kolodner, R.D. Induction of genome instability by DNA damage in Saccharomyces cerevisiae. DNA Repair (Amst) 2003, 2, 243–258. [Google Scholar] [CrossRef]
- Bonner, J.N.; Zhao, X. Replication-Associated Recombinational Repair: Lessons from Budding Yeast. Genes (Basel) 2016, 17, 48–68. [Google Scholar] [CrossRef] [PubMed]
- Pennaneach, V.; Kolodner, RD. Recombination and the Tel1 and Mec1 checkpoints differentially effect genome rearrangements driven by telomere dysfunction in yeast. Nat Genet. 2004, 36, 612–617. [Google Scholar] [CrossRef]
- Banerjee, S.; Smith, S.; Myung, K. Suppression of gross chromosomal rearrangements by yKu70-yKu80 heterodimer through DNA damage checkpoints. Proc Natl Acad Sci U S A. 2006, 103, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kolodner, R.D. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999, 23, 81–85. [Google Scholar] [CrossRef]
- Fasullo, M.; Dave, P. Mating type regulates the radiation-associated stimulation of reciprocal translocation events in Saccharomyces cerevisiae. Mol Gen Genet. 1994, 243, 63–70. [Google Scholar] [CrossRef]
- Fasullo, M.; St Amour, C.; Zeng, L. Enhanced stimulation of chromosomal translocations and sister chromatid exchanges by either HO-induced double-strand breaks or ionizing radiation in Saccharomyces cerevisiae yku70 mutants. Mutat Res. 2005, 578, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Du, L.L.; Nakamura, T.M; Russell, P. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 2006, 20, 1583–1596. [Google Scholar] [CrossRef]
- Blaikley, E.J.; Tinline-Purvis, H.; Kasparek, T.R.; Marguerat, S.; Sarkar, S.; Hulme, L.; Hussey, S.; Wee, B.Y.; Deegan, R.S.; Walker, C.A.; Pai, C.C.; Bähler, J.; Nakagawa, T.; Humphrey, T.C. The DNA damage checkpoint pathway promotes extensive resection and nucleotide synthesis to facilitate homologous recombination repair and genome stability in fission yeast. Nucleic Acids Res. 2014, 42, 5644–5656. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Ismail, A.; Klement, K.; Goodarzi, A.A.; Conrad, S.; Freire, R.; Shibata, A.; Lobrich, M.; Jeggo, P.A. Opposing roles for 53BP1 during homologous recombination. Nucleic Acids Res. 2013, 41, 9719–9731. [Google Scholar] [CrossRef] [PubMed]
- Park, J.M.; Pinski, D.F.; Forsburg, S.L. Diploidy confers genomic instability in Schizosaccharomyces pombe. Genetics. 2025, 230(2), iyaf078. [Google Scholar] [CrossRef] [PubMed]
- Caspari, T.; Murray, J.M.; Carr, A.M. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 2002, 16, 1195–1208. [Google Scholar] [CrossRef]
- Alvaro, D.; Lisby, M.; Rothstein, R. Genome-wide analysis of Rad52 foci reveals diverse mechanisms impacting recombination. PLoS Genet. 2007, 3(12), e228. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Double-strand break (DSB) repair pathways including sister chromatid recombinational repair (left panel), single-strand annealing (middle panel), and break-induced replication (right panel). A). The initiation of the DNA lesion, B). The position of the break, C). The resection of the break revealing 3’ overhangs, D). Homology search and annealing complementary DNA. E). DNA single-strand gaps that are filled by DNA polymerases where newly synthesized strands are shown in purple, F). Resolution, ligation of nicks, and reconstitution of the chromatid. Single strands of the 5’ -3’ polarity are shown in blue and 3’-5’ polarity is shown in yellow. The newly synthesized DNA is shown in purple, where the arrow indicates the 3’ polarity.
Figure 1.
Double-strand break (DSB) repair pathways including sister chromatid recombinational repair (left panel), single-strand annealing (middle panel), and break-induced replication (right panel). A). The initiation of the DNA lesion, B). The position of the break, C). The resection of the break revealing 3’ overhangs, D). Homology search and annealing complementary DNA. E). DNA single-strand gaps that are filled by DNA polymerases where newly synthesized strands are shown in purple, F). Resolution, ligation of nicks, and reconstitution of the chromatid. Single strands of the 5’ -3’ polarity are shown in blue and 3’-5’ polarity is shown in yellow. The newly synthesized DNA is shown in purple, where the arrow indicates the 3’ polarity.
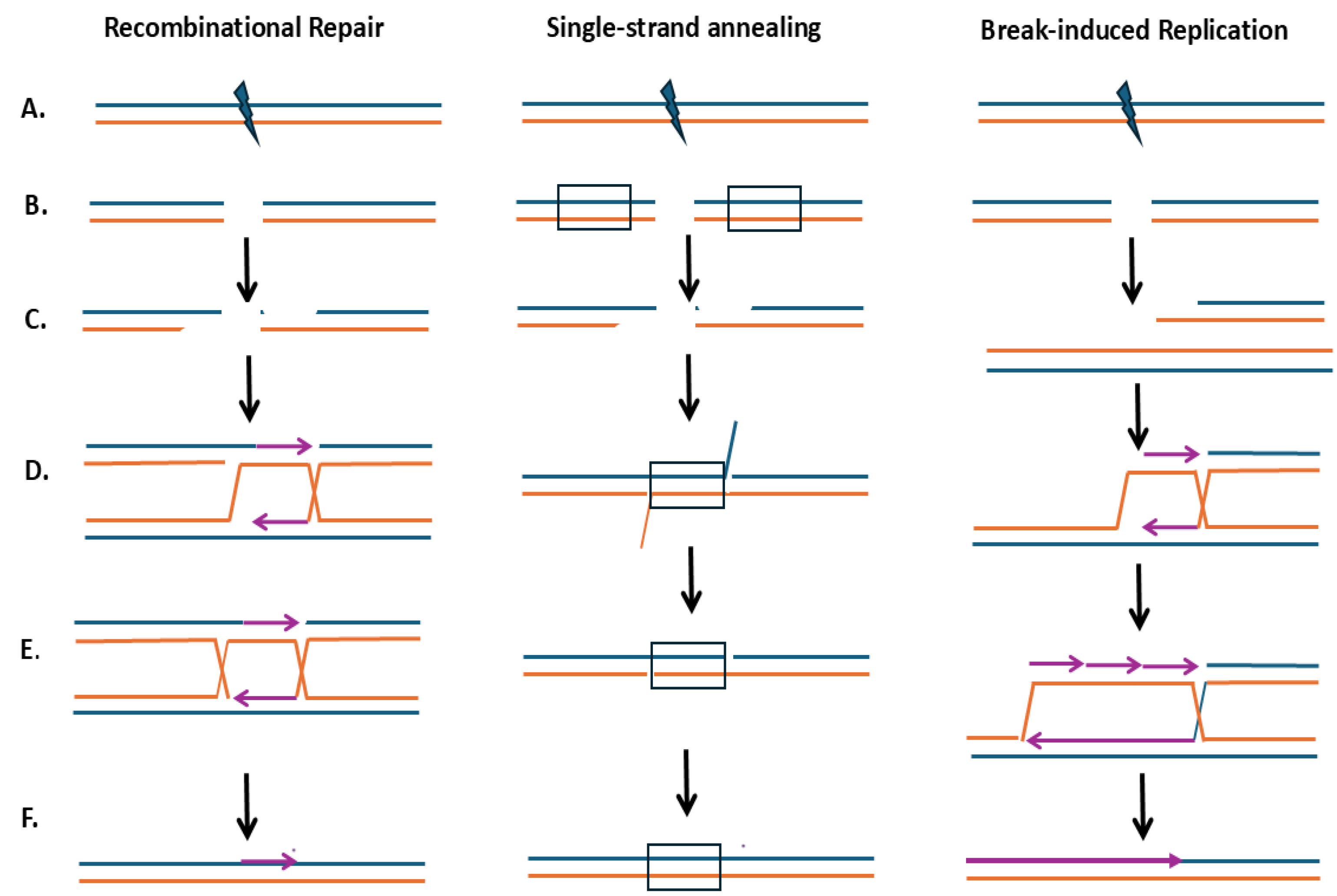
Figure 2.
Arrangements of repeated sequences aligned in direct orientations on (A) chromosome, (B) sister chromatids, (C) homologs, and (D) non-homologous chromosomes. Recombination between these repeats generates, deletion, duplication, and reciprocal recombination. The chromosome is shown as a single line representing duplex DNA. For simplicity, the left arms of the chromosomes are not shown. The oval represents the centromere, and the rectangular box represents a repeated sequence. The blue and red colors are indicative of two different non-homologous chromosomes. The URA3 is shown as an example of a gene located between repeated sequences.
Figure 2.
Arrangements of repeated sequences aligned in direct orientations on (A) chromosome, (B) sister chromatids, (C) homologs, and (D) non-homologous chromosomes. Recombination between these repeats generates, deletion, duplication, and reciprocal recombination. The chromosome is shown as a single line representing duplex DNA. For simplicity, the left arms of the chromosomes are not shown. The oval represents the centromere, and the rectangular box represents a repeated sequence. The blue and red colors are indicative of two different non-homologous chromosomes. The URA3 is shown as an example of a gene located between repeated sequences.
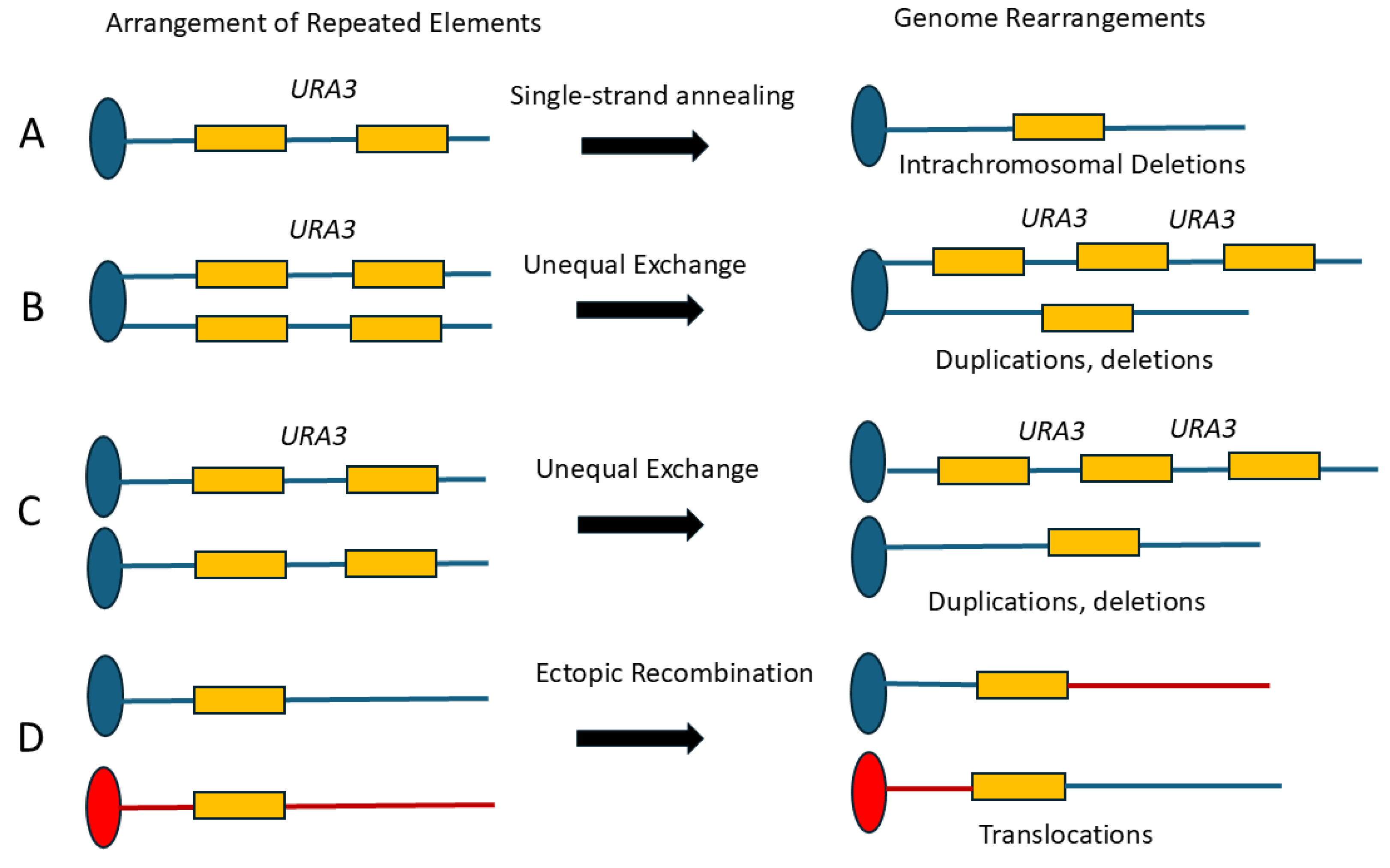
Figure 3.
Domain structures and alpha-fold predicted structure of the Rad9 protein (1309 amino acids) and Rad9-mediated checkpoint pathway. On the left are the domains within the amino acid sequence, starting from the N-terminal to the C-terminal end. Each color code represents a different domain; red, blue, orange, green and purple represent the Chk1-activating domain, the SQ domain, the Rad53 binding domain, the tudor domain, the BRCT domain, respectively. The alpha fold structure is shown on the right, where the highest confidence structures are colored green and dark brown; these two domains cover both the tudor and the BRCT domains, respectively. On the right is the Rad9-mediated checkpoint pathway. The checkpoint pathway is initiated by a double-strand break (DSB), followed by Tel1 binding, MRX recruitment of Sae2 and nucleases, and Mec1 Ddc2 recruitment. Rad9 protein concentrates at the restricted DSB by binding to modified chromatin and then serves as a scaffold to recruit Chk1 (left) and Rad53 (right). Mec1 phosphorylates Chk1 and Rad53; Rad53p catalyzes its own phosphorylation and the polyphosphorylated Rad53 is released from the Rad9 scaffold.
Figure 3.
Domain structures and alpha-fold predicted structure of the Rad9 protein (1309 amino acids) and Rad9-mediated checkpoint pathway. On the left are the domains within the amino acid sequence, starting from the N-terminal to the C-terminal end. Each color code represents a different domain; red, blue, orange, green and purple represent the Chk1-activating domain, the SQ domain, the Rad53 binding domain, the tudor domain, the BRCT domain, respectively. The alpha fold structure is shown on the right, where the highest confidence structures are colored green and dark brown; these two domains cover both the tudor and the BRCT domains, respectively. On the right is the Rad9-mediated checkpoint pathway. The checkpoint pathway is initiated by a double-strand break (DSB), followed by Tel1 binding, MRX recruitment of Sae2 and nucleases, and Mec1 Ddc2 recruitment. Rad9 protein concentrates at the restricted DSB by binding to modified chromatin and then serves as a scaffold to recruit Chk1 (left) and Rad53 (right). Mec1 phosphorylates Chk1 and Rad53; Rad53p catalyzes its own phosphorylation and the polyphosphorylated Rad53 is released from the Rad9 scaffold.
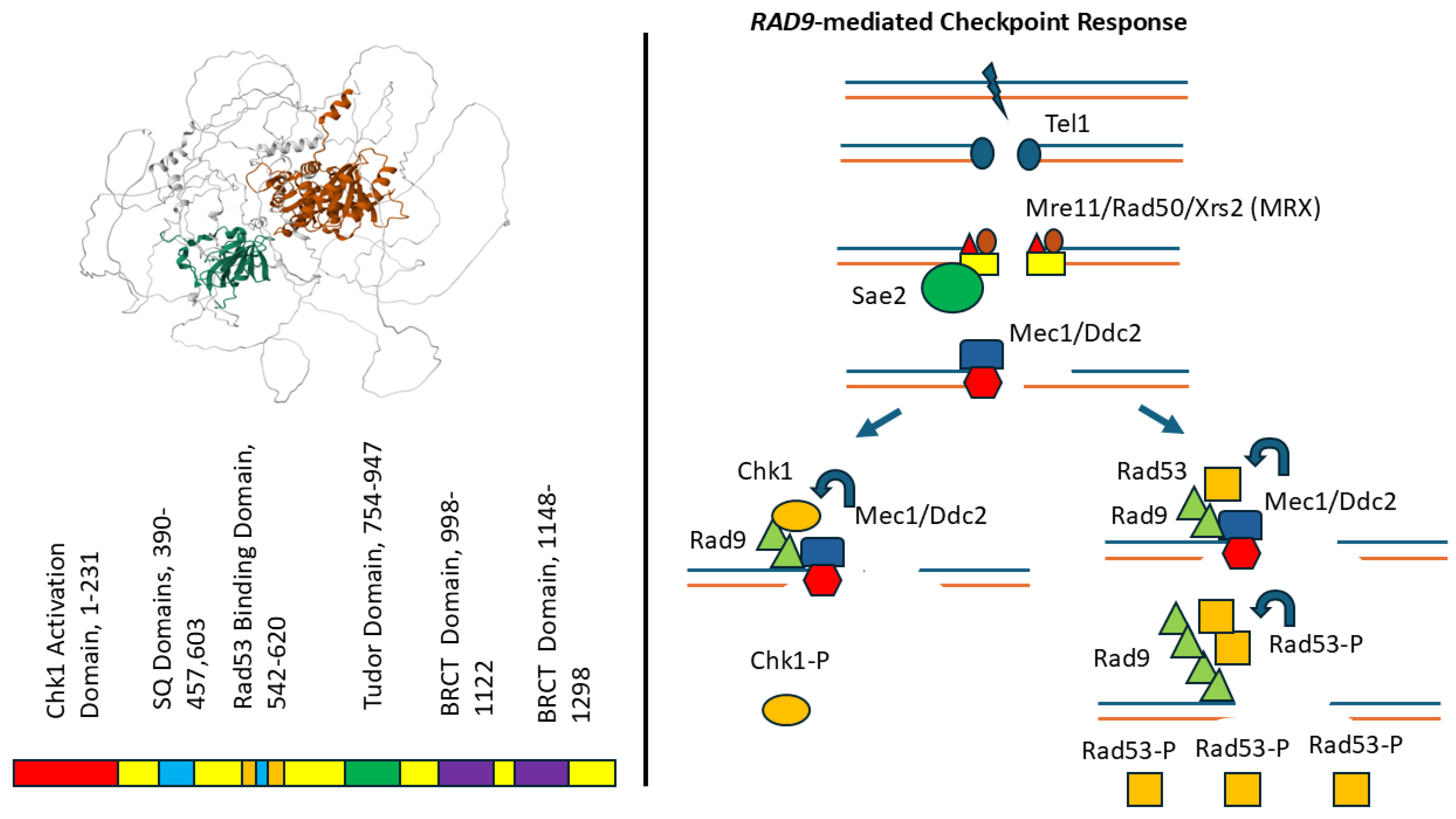
Figure 4.
RAD9 functions in suppressing and promoting homology-directed translocations. The left panel shows mechanisms by which RAD9 suppresses homology-directed translocations. RAD9 can promote G2/M arrest allowing for addition time in the cell cycle to facilitate DSB and DNA repair; otherwise, acentric chromosome fragments are segregated and HR between repeated sequences promotes translocations. In the middle panel, RAD9 promotes cross-overs by inhibiting helicase function of MPH1 and SGS1, thereby promoting SCR. In the last panel, RAD9 promotes ectopic recombination in S phase by promoting HR between single-stranded DNA present in repeated DNA sequences.
Figure 4.
RAD9 functions in suppressing and promoting homology-directed translocations. The left panel shows mechanisms by which RAD9 suppresses homology-directed translocations. RAD9 can promote G2/M arrest allowing for addition time in the cell cycle to facilitate DSB and DNA repair; otherwise, acentric chromosome fragments are segregated and HR between repeated sequences promotes translocations. In the middle panel, RAD9 promotes cross-overs by inhibiting helicase function of MPH1 and SGS1, thereby promoting SCR. In the last panel, RAD9 promotes ectopic recombination in S phase by promoting HR between single-stranded DNA present in repeated DNA sequences.

Table 1.
Recombination phenotypes of rad9 mutants.
| Genetic Assay | DNA damaging agent and assay | rad9 phenotype1 | Ploidy specificity | Reference |
|---|---|---|---|---|
| Intrachromatid recombination | ||||
| SSA between homeologous repeats | HO-induced DSBs | Decreased | Haploid | [22] |
| SSA | HO-induced DSBs | NC | Haploid | [94] |
| Deletions at the rDNA locus | Spontaneous | Increased in orc1-4 mutants | Haploid | [92] |
| Sister chromatid recombination | ||||
| uSCR | Spontaneous | NC | Haploid | [82] |
| uSCR | X rays | Decreased | Haploid | [82] |
| uSCR | MMS | Decreased | Haploid | [82] |
| Equal SCR | 1-Sce1 induced break | Decreased | Haploid | [83] |
| Homolog recombination between heteroalleles | ||||
| Homolog recombination occurring between two heteroalleles | Spontaneous | Two-fold increase | Diploid | [101] |
| Allelic recombination | Spontaneous | MC | Diploid | [100,117] |
| Gross chromosomal rearrangements (GCRs) | ||||
| GCR | Spontaneous | Increased | Haploid | [119,123,127] |
| GCR | Spontaneous | Increased | Disome for VII | [117] |
| Recombination between repeats on non-homologous chromosomes | ||||
| Directed translocations | Spontaneous | Increased | Haploid and Diploid | [82] |
| Directed translocations | Radiation (X-ray, UV) | Increased | Diploid | [82] |
| Directed translocations | Topoisomerase Inhibitors | Increased | Diploid | [89] |
| Directed translocations | MMS | Increased | Diploid | [82,89] |
| Directed translocations | 4-NQO | No change | Diploid | [89] |
| Directed translocations | HO-induced breaks | No change | Diploid | [82] |
| Chromosoe III translocations | Spontaneous | Increased in orc1-4 mutants | Diploid | [104] |
| Ectopic Gene Conversion | Spontaneous | No Change | Haploid and Diploid Strain | Fasullo (unpublished) |
| Non-homologous end-joining between cohesive ends | ||||
| Chromosome Breaks | HO-induced breaks | Decrease | Haploid | [94,122] |
| Plasmid Breaks | pRS315 digested with BamH1 | Decrease | Haploid | [121] |
1Rate or frequency in comparison to wild type, see reference for more detail.
Table 2.
Fold Stimulation of Recombination-directed and Gross Chromosomal Rearrangements Generated in rad9, checkpoint, and rad mutants.
Table 2.
Fold Stimulation of Recombination-directed and Gross Chromosomal Rearrangements Generated in rad9, checkpoint, and rad mutants.
| Genotype | RAD52-Dependent Rearrangements | Gross Chromosomal Rearrangements (GCRs) | |||
|---|---|---|---|---|---|
| his3-repeat-directed translocations1 | CUP1 Copy Number Variation(CNV)2 | yel069c::URA3 CAN13 |
yel072w::URA3 CAN4 |
CenVII ade6 ADE3/ hxk2:CAN1 cenVII ADE6 ade3e. |
|
| WT | 1 | 1 | 1 | 1 | 1 |
| rad9 | 7 | 0.8 | 6 | 1.9 | 23 |
| mrc1 | 4.1 | 2 | 19 | 4.2 | |
| rad53 sml1 | 10 | 1 | 27 | 16 | 42 |
| rad5 | 3.3 | 0.9 | 127 | 19 | 27 |
| rad27 | 2.1 | 1100 | 140 | ||
| rad52 | <0.1 | 0.08 | 126 | 0.6 | 73 |
| mec1-21 | 23 | ||||
| mec1-21 rad51 | 100 | ||||
| mec1 sml1 | 1 | 194 | 7.6 | 97 | |
| mec1 sml1 rad51 | 1271 | ||||
1Rate of spontaneous recombination in diploid wild type is 3 x 10-8, Fold increase is the (rate in mutant)/(rate in wild type). 2 CNV is measured by Southern blot by comparing the intensity of variation in wild type with that of the mutant. 3 Rate of spontaneous GCRs in haploid wild type is 3.5 x 10-10, Fold increase is the (rate in mutant)/(rate in wild type).4 Rate of GCRs in haploid wild type is 2 x 10-8, Fold increase is the (rate in mutant)/(rate in wild type). 5 Rate of GCRs is Chr VII disome is 2.3 x 10-5, Fold increase is the (rate in mutant)/(rate in wild type).
Table 3.
Synergistic effects of rad9 and DNA repair mutants on generating homology-mediated translocations.
Table 3.
Synergistic effects of rad9 and DNA repair mutants on generating homology-mediated translocations.
| Genotype of Single and Double Mutant | Recombination Assay | Ploidy | Fold Increase relative to WT1 | Fold Increase Compared to rad9 | Fold Increase Compared to single DNA metabolism mutant | References |
|---|---|---|---|---|---|---|
| rad9 | Directed translocations | Diploid | 7 | 1 | NA | [82,100] |
| rad9 rad51 | Directed translocations | Diploid | 57 |
8 | 5.3 | [100] |
| rad9 rad55 | Directed translocations | Diploid | 77 | 11 | 6.2 | [100] |
| rad9 rad57 | Directed translocations | Diploid | 78 | 11 | 5.3 | [100] |
| rad9 rad54 | Directed translocations | Diploid | 55 | 8 | 24 | [100] |
| rad9 mre11 | Directed translocations | Diploid | 57 | 8 | 2 | [100] |
| rad9 mec1 | Directed translocations | Diploid | 6 | 1 | 0.3 | [100] |
| rad9 | GCR | Haploid | 6 | 1 | NA | [110] |
| rad9 sgs1 | GCR | Haploid | 213 (3%) | 37 | 9.7 | [110] |
1Wild type strain is the corresponding Rad+ haploid or diploid form which the single or the double mutant is derived from.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



