Submitted:
05 January 2026
Posted:
06 January 2026
You are already at the latest version
Abstract
Cadmium (Cd) is a ubiquitous environmental pollutant that enters the circulation from the lungs and gastrointestinal tract. For most people, staple foods form the main route of Cd exposure. Current evidence suggests that Cd may increase the prevalence of iron deficiency and anemia in environmentally exposed people. Concerningly, intravenous iron administration to treat iron deficiency anemia has resulted in adverse bone outcomes in a higher-than-expected frequency; for which reasons remain unclear. The bone-derived hormone, fibroblast growth factor 23 (FGF23), the regulator of vitamin D and phosphate homeostasis, has been speculatively implicated, given that anemia, iron deficiency and inflammatory conditions all are known to increase FGF23 expression levels in osteoblasts. Additionally, early studies demonstrated that Cd increased FGF23 expression by osteoblast-like cells and suppressed FGF23 cleavage leading to an abrupt rise in serum FGF23, which, in turn, mediated an effect of Cd on tubular phosphate reabsorption. In this review, experimental breakthrough studies showing Cd-induced iron deficiency, and a reduction in iron absorption by Cd are summarized together with intestinal absorption of Cd, and an increment of Cd uptake and Cd body burden in those with low body iron stores. Potential contributions of Cd, anemia and iron deficiency in the context of hypophosphatemic osteomalacia development after intravenous iron supplementation, are discussed. Mechanism of Cd-induced ferroptosis in pathogenesis of osteoporosis, emphasizing heme oxygenase-1 (HO-1)/bilirubin axis and zinc deficiency are presented.
Keywords:
anemia
; bilirubin
; bone fragility
; cadmium
; fibroblast growth factor 23
; heme oxygenase-1
; iron deficiency
; osteomalacia
; zinc
1. Introduction
Iron deficiency anemia (IDA) is defined based on the levels of blood hemoglobin and the indicators of body iron content, like serum ferritin, free erythrocyte protoporphyrin and a transferrin saturation index [1,2,3,4,5]. Ferritin is an iron storage protein, as such, its concentration in the serum below 20 and 30 µg/L are indicative of body iron store depletion, and a low body iron store status, respectively [2,6]. However, because of the rising serum ferritin levels in chronic inflammatory conditions, soluble transferrin receptors (sTfR), and the sTfR–ferritin index have been employed for an accurate IDA assessment [2,6].
Based on blood hemoglobin levels below 12.0 g/dL in women and 13.0 g/dL in men, the 2021 global prevalence of anemia across all ages was 24.3% [7,8]. The predominant cause of anemia is iron deficiency, followed by hemoglobinopathies, hemolysis and chronic disease. IDA is a significant problem, especially among children (1-4 years), women of childbearing age, pregnant women [9,10,11], and people with chronic diseases, chronic kidney disease (CKD) included [6,12,13]. Moreover, IDA is a known risk factor for osteoporosis [14,15], while a high risk of bone fracture has been linked to low blood hemoglobin levels in men [16]. In a comprehensive review, Lichtler and Cowley have presented evidence connecting the prevalence of IDA and anemia with environmental exposure to cadmium (Cd), lead (Pb) and indoor and ambient air pollution, and fluoride [17].
Oral and intravenous iron supplementation have been in current use for IDA treatment and management [18,19,20]. Intravenous iron administration is used to bypass the gastrointestinal tract because of the low absorption rate of oral iron and its side effects. Iron infusion therapy increased hemoglobin concentrations more efficiently than oral iron in CKD patients, and resulted in higher hemoglobin and ferritin levels in pregnant women, compared with oral iron [10,11,12,13].
Concerningly, hypophosphatemic osteomalacia occurred in a particularly high frequency (50%) of patients who received intravenous iron therapy using ferric carboxymaltose preparation [19,20]. FGF23, a bone derived hormone, has been implicated; specifically, ferric carboxymaltose may inhibit the cleavage of FGF23, leading to an abrupt rise of active, intact FGF23 (iFGF23) [19,20]. Notably, iron deficiency per se can affect both synthesis and degradation of FGF23 from which iFGF23 and cleaved C-terminal FGF23 [(Cter)-FGF23] peptides are generated [21], and a Cter-FGF23 fragment has been linked to iron homeostasis independent of an effect of iFGF23 on renal phosphate reabsorption [22]. Moreover, other intravenous iron formulations also resulted in hypophosphatemic osteomalacia although in low frequencies (4-5%).
Hypophosphatemic osteomalacia due to chronic exposure to a high-dose Cd (>100 µg/day), experienced by itai-itai disease is now rare; however, the evidence connecting iron deficiency with environmental Cd exposure [17] together with hypophosphatemia osteomalacia in iron infusion therapy [19,20] has prompted us to the present work. Effects of Cd on intestinal absorption of iron leading to iron deficiency and are highlighted along with effects of IDA on Cd absorption and body burden of Cd. We discuss the potential role for Cd in the context of the adverse bone outcomes of iron infusion therapy. Additionally, we provide insight into effects of Cd on cellular stress response mechanisms involving heme oxygenase-1 (HO-1)/bilirubin axis. The two-hit hypothesis of Cd-induced cytotoxicity is presented.
2. Absorption and Accumulation of Cd in the Human Body
In this section, the intestinal absorption of Cd is discussed along with the use of blood Cd and urinary Cd as indicators of exposure to the metal. Effects of Cd on iron assimilation and a reduced cellular iron uptake revealed in recent long-term feeding trials are highlighted. Special emphasis is given to increased Cd absorption and Cd body burden in those with low body iron status.
2.1. Cd Exposure Route and Bone Outcome
Cd is present as a contaminant in virtually all food types, especially staple foods; inevitably, normal diets have become a common route of exposure [23,24,25]. Additional Cd exposure routes are tobacco smoke and airborne particle pollution, a concern especially among urban populations [26,27,28,29]. An exposure to a Cd dose between 10 and 15 µg/day may increase risk of osteoporosis [30,31,32].
In high doses; daily exposure to Cd at 100 µg or a life-time exposure of 1 g can cause itai-itai disease [33,34], where osteomalacia was observed in the presence of markedly reduced proximal tubular reabsorption of various filtered substances, phosphate included [35,36,37,38]. The manifestations of severe Cd poisoning were replicated using ovariectomized cynomolgus monkeys [38]. In a study using osteoblast-like cells, Kido et al. found that Cd increased expression of fibroblast growth factor 23 (FGF23), the regulator of vitamin D and phosphate homeostasis [40]. In a study by Aranami et al, a reduction in kidney tubular phosphate reabsorption in Cd-intoxicated mice was found to be mediated by FGF23 [41].
2.2. Intestinal Absorption of Cd
All living organisms cannot generate nor destroy any metal, and consequently, specialized metal transport proteins and pathways have been evolved to acquire all essential metals iron, zinc, manganese, calcium, and cobalt from exogenous sources (the diet) [42,43,44]. Even though Cd has no physiological role or nutritional value, its electronegativity and ionic radius are close to zinc, calcium, and iron; consequently, it is absorbed by the enterocytes. through the transport mechanisms and pathways for essential metals (Figure 1).
Examples of metal transport proteins responsible for absorption of Cd in an ionic form (Cd2+) are those for calcium (TRPV6), zinc (ZIP14), iron (DMT1) [45,46,47,48,49,50]. Cd in complexes with metallothionein (MT) and the plant metal binding ligand phytochelatin (PC) are absorbed through transcytosis [51] and endocytosis mediated by human neutrophil gelatinase-associated lipocalin (hNGAL) [52,53].
The rate of individual metal assimilation is regulated by the specificity and the enterocyte levels of individuals transporters expressed by enterocytes. Such regulatory modes are pivotal to prevent metal deficiency as well as metal overload. Two metal efflux transporters have been found to display an absolute specificity; ferroportin1 (FNP1) has an absolute specificity for iron (Fe2+) [54,55], whereas ZnT1 is for the extrusion of only Zn (2+) [56]. These suggests that Cd is retained within the cells due to no exit route; a long residence time of Cd in cells.
The transport protein for iron or zinc only means also that the absorption rate of Cd exceeds that of iron or zinc, given that Cd enters the enterocytes through multiple routes. At any given time, the whole blood Cd level is indicative of recent exposure because the average lifespan of erythrocytes is 120 days. The biological half-life of blood Cd ranged between 75 and 128 days [57]. The half-life of Cd in the body varied from 7.4 to 30 years; the lower the bodily burden, the longer the half-life of Cd [58,59,60].
2.3. Urinary Cd Is Indicative of Body Burden and Toxicity at the Present Time
Acquired Cd accumulates mostly within the kidney tubular cells, where its levels increase through to the age of 50 years but decline thereafter due to its release into the urine as the injured tubular cells die for any reason [61]. Thus, urinary Cd reflects its nephrotoxicity at the present time [61]. A study of environmentally exposed Chinese subjects aged 2.8 to 86.8 years (n = 1235) showed that Cd excretion levels increased with age, peaking at 50 years in non-smoking women and 60 years in non-smoking men [62].
Based on a direct relationship between urinary Cd and the accumulation of Cd in the kidney cortex, urinary Cd is an indicator of the body burden of the metal [63,64,65,66]. Using data from kidney transplant donors, urinary Cd of 0.42 μg/g cr corresponded to kidney Cd of 25 μg/g wet tissue weight [65]; urinary Cd 0.34 μg/g cr in women corresponded to kidney Cd of 17.1 μg/g, and urinary Cd of 0.23 μg/g cr in men corresponded to kidney Cd of 12.5 μg/g [66].
2.4. Iron Deficiency Induced by Cd: Breakthrough Studies
Using a long-term feeing strategy, Tokumoto et al. have shown, for the first time, an effect of Cd on iron absorption, leading to iron deficiency in Cd toxicity targets [67]. A significant reduction in hepatic iron content was observed in groups of female C57BL/6J mice given a diet containing 300 ppm Cd for 12, 15, 19 and 21 months [67]. Such a decrease in hepatic iron content was attributable to Cd-induced suppression of duodenal expression of the HCP1 and Cybrd1 genes, encoding the influx transporters for heme iron and non-heme iron, respectively. As Figure 1 presents, CYBRD1 is required to reduce Fe3+ to absorbable form, Fe2+. Interestingly, Cd did not seem to influence the duodenal expression of the iron efflux transporters [67]. The conditions may favor Cd absorption. In summary, Cd reduced duodenal absorption of both heme iron and non-heme iron, leading to a decrease in hepatic iron storage (ferritin), indicative of the low body iron store status.
In another breakthrough study where Sprague Dawley male rats were given Cd in drinking water at 0, 50, 75 mg/L CdCl2 for 1 and 6 months, Zhang et al. observed a decrease in iron content in the proximal tubular cells of the kidneys [68]. They found, like the study by Tokumoto et al. that Cd affected the duodenal expression of the genes encoding specialized transport proteins for metals. Specifically, Cd suppressed the expression of SLC11A2 gene and SLC40A1 genes, encoding divalent metal transporter1 (DMT1) and ferroportin1 (FPN1). Additionally, Cd lowered the expression levels of various metal transporters by the kidney tubular cells which included SLC11A2 (DMT1), CUBN (cubilin), LRP2 (megalin), SLC39A14 (ZIP14), and SLC39A8 (ZIP8). These data suggest that a long-term Cd exposure may induce iron insufficient state in tubular cells through decreasing the duodenal absorption of iron, systemic transport, and uptake of iron by tubular cells.
The above findings lend support to a connection between low environmental exposure to Cd and a high prevalence of iron deficiency, especially in vulnerable subpopulation groups. They also prompted us to explore potential impact of Cd on undesirable bone outcomes of intravenous iron supplementation, through inducing iron deficiency (Section 3).
3. Undesirable Bone Outcomes in Iron Infusion Therapy
Speculatively, hypophosphatemic osteomalacia was linked to the use of ferric carboxymaltose preparation. Nonetheless, such bone outcome occurred also with other iron formulations [19,20], which may indicate potential involvement of different pathogenic factors. Recent research studies show that inflammation, anemia, and dysregulated iron homeostasis (overload/deficiency) can impact FGF23 synthesis and its cleavage to iFGF23 and Cter-FGF23 [21,22]. Moreover, the Cter-FGF23 fragment may have biological role distinct from iFGF-23 [21,22].
To reveal similarities/differences in pathological features, we review reported cases of bone complications following iron infusion therapy. Also, we review a proven case of hypophosphatemic osteomalacia in an Indian jewellery male worker, exposed to a high dose of Cd in fumes [69]. His blood Cd level was 6-time higher than an occupational exposure limit of 5 µg/L [69].
3.1. Hypophosphatemic Osteomalacia
Samões et al. reported a case of a 70-year-old man who had Rendu–Osler–Weber disease and developed hypophosphatemic osteomalacia as a complication of frequent infusions of ferric carboxymaltose for more than 10 years [70]. Vilaca et al. have published a systematic review, in which the authors included 28 case reports (30 patients, aged 28-80 years) of osteomalacia developed as a complication of intravenous iron infusions [71]. Initially, all 30 individuals had IDA from gastrointestinal diseases or gynecological bleeding. These subjects underwent treatment with repeated intravenous iron infusions using various iron preparations (saccharated ferric oxide, iron polymaltose, and ferric carboxymaltose), which led to osteomalacia marked by musculoskeletal pain, fractures, and pseudofractures. Most cases had rising plasma iFGF23 levels; nonetheless, C-terminal FGF23 (Cter-FGF23) levels were also elevated in some cases, along with hypophosphatemia and increased plasma FGF23 levels. Clinical picture involved bone pain and onset of fractures. Additionally, some of the included subjects had comorbidities such as Crohn disease, or were treated with glucocorticoids [72], both of which may significantly influence bone quality [73,74].
Establishing a link between intravenous iron supplementation and the onset of osteomalacia is a challenging task. Namely, the majority or all of these individuals had IDA prior to iron infusions, and IDA itself may damage bone [75,76,77,78]. Thus, the effects of IDA and iron infusion were indistinguishable. Moreover, individuals with IDA may have had elevated blood Cd concentrations [79,80,81], which is toxic to bones [82]. It would not be possible to determine whether osteomalacia is a consequence of IDA, Cd toxicity, or a transient iron overload.
A case report on Cd-induced hypophosphatemic osteomalacia has recently been published. Namely, the authors reported a case of a man in his 40s who complained of lower-back pain. Using X-ray imaging, pseudofractures were observed [83]. His blood Cd level of 30 µg/L was sixfold above an occupational exposure limit of 5.0 µg/L. He also had hypochromic microcytic anemia and elevated cFGF23, and was ultimately diagnosed with hypophosphatemic osteomalacia due to severe Cd toxicity [83].
Comparative overview of the main characteristics of cases of the reported iron infusion–mediated osteomalacia and cases of Cd-mediated osteomalacia shows that the two conditions share all major characteristics, namely presence of IDA, increased plasma FGF23 levels, hypophosphatemia, and bone pain. Unfortunately, Cd exposure levels were not measured in the cases where iron was found “guilty” for the onset of hypophosphatemic osteomalacia; measuring of Cd exposure levels in those cases would be helpful to clarify whether Cd contributed or caused osteomalacia.
Changing therapy from intravenous to oral administration of iron brings improvement in those individuals treated with intravenous iron supplementation [71]. This improvement can be easily explained since Cd and iron share the same intestinal metal transporters and pathways (Section 2.2). DMT1, ZIP8, and ZIP14 metal transporters are expressed in the duodenum [84], and they are likely responsible for assimilation of iron, zinc, and Cd [85,86]. This further suggests that iron and Cd would compete for same metal transport proteins; consequently, less Cd would enter the circulation in response to oral iron supplementation.
Interestingly, although it was speculated that plasma FGF23 levels would drop after the cessation of intravenous iron supplementation, Klein et al. reported that FGF23 remained elevated for months after the last infusion [87]. This may indicate involvement of factors influencing FGF23 synthesis and cleavage. This should not suggest hypophosphatemic osteomalacia as alone consequence of a low-dose Cd exposure. Rather, the contributions of environmental Cd, iron deficiency for any cause, and iron infusion cannot be separated, and previous studies have considered only iron infusion as the sole cause. Our working hypothesis to explain involvement of Cd through iron deficiency is presented in Figure 2.
Osteoblasts can easily uptake circulating Cd when it is bound to transferrin [88] and transferrin receptor 1 (TfR1) is expressed on the osteoblast surface [89]. The expression of Trf1 is upregulated by IDA due to the accumulation of hypoxia-inducible factors (HIFs) [90]. Following its entry into osteoblasts, Cd promotes the secretion of iFGF23, which then reduces tubular reabsorption of phosphate, demonstrated previously [41,42]. IDA increased also the abundance of TfR1 on the cell surface [91,92], resulting in an enhanced uptake of Cd since, as discussed, Cd is bound to transferrin [92]. In effect, osteoblasts could take up and accumulate Cd for a prolonged period, and Cd promotes excessive ROS (Section 4), leading to some extent cell damage. When there is suddenly a significant amount of iron in circulation, osteoblasts uptake it as well. Results of an in vivo study demonstrated that Cd toxicity is potentiated in the presence of iron [93]. It would not be possible to ascertain whether those individuals would develop osteomalacia after iron infusion if they were minimally exposed to Cd. However, this could be shown if chelation therapy were applied before iron infusion.
3.2. Fracture Risk
Anemia and iron deficiency have been linked to risk of fractures and postoperative complications after hip fracture [94,95,96,97]. Lee et al. analyzed fracture risk in a large retrospective study that recruited over 70,000 individuals, of which 10,568 (15.1%) had anemia (not classified) [97]. They observed that both men and women with anemia had an increased risk of sustaining vertebral and femoral fractures; moreover, a negative correlation was found between hemoglobin levels and fracture risk for both sites examined. A higher fracture risk was independently associated with current smoking status in another study [97].
In a study from Sweden, an increased risk of incident osteoporosis-related fractures was observed in those who never smoked; the risk of fracture rose 58% per μg/L increase in blood Cd. This result was obtained after adjustment for age, sex, BMI, physical activity, and fiber consumption [98]. Blood Cd as little as 0.31 µg/L appeared to be sufficient to increase fracture risk by 21%, compared to blood Cd <0.15 μg/L [98]
A Swedish prospective study conducted on 1005 men (66 with anemia) who participated in the Osteoporotic Fractures in Men (MrOS) study, evaluated whether anemia (not classified) could be associated with an increased fracture risk [92,99]. The median follow-up time in that study was 10.1 years, during which 346 participants sustained fractures. The hazard ratio for any fracture type was almost doubling, even after adjusting for age and hip bone mineral density (BMD). Of note, individuals with anemia had higher circulating levels of intact fibroblast growth factor 23 (iFGF23) independent of age, erythropoietin levels, and eGFR) [99].
Similarly, Valderrábano et al. examined the association between anemia and fracture risk in 3632 aged men, 249 had anemia [100]. Their results showed that the presence of anemia increased the risk of any fracture type by 67%, while the risk for non-vertebral fractures was even slightly more pronounced [100].
A study from Norway examined whether anemia could predict non-vertebral fractures [101]. This study included 5286 individuals (2511 men and 2775 women), aged 55 to 74 years. Like the Swedish men study [100], Jørgensen and colleagues found that men with anemia had a twofold higher risk of non-vertebral fractures compared with men with desirable hemoglobin levels. However, after adjusting for various variables such as lipid profiles, forearm BMD, hand grip strength, BMI, smoking status, and creatinine levels, the significant effect of anemia on fracture risk was lost for women, but remained significant for men [101].
Teng and colleagues conducted a meta-analysis of data from seven original articles [95]. They found that the pooled relative risk for any fracture type in individuals with anemia was 1.26. After stratifying the risk by location, they reported a higher fracture risk for hip fractures than for vertebral fractures in individuals with anemia. Additionally, men had a greater fracture risk than women, although the presence of anemia led to increased fracture risk in both sexes and for both examined sites [95]. Results from another study from the US, which included 160,080 women, indicated an increased risk of spine, hip, and all-type fractures, with hip fracture risk being particularly high in individuals with anemia (hazard ratio 1.81) [102].
3.3. IDA
Most studies have shown that IDA and iron deficiency had direct effects on bone quality [76,77]. Pioneering work in the field of IDA-induced bone alterations revealed that rats fed with an iron-deficient diet had lower bone mineral density in the femur and spine and worse mechanical properties compared with rats with adequate iron intake [77].
In an in vivo study where rats were fed a low-iron diet for 5 weeks, the low-iron-intake group had lower whole-body and femur DXA scores and deteriorated lumbar microarchitecture. This deterioration was reflected in lower bone volume fraction (BV/TV), trabecular thickness (Tb.Th), and trabecular number (Tb.N), along with a higher structure model index and trabecular separation. In another study using Wistar rats fed with an iron-deficient diet, Diaz-Castro et al. observed decreasing levels of procollagen type I N-terminal propeptide, coupled with rising serum parathormone (PTH), indicative of bone resorption [76].
3.4. Other Potential Contributors
Osteomalacia due to vitamin D deficiency is characterized by altered mineralization of newly formed osteoid. Chwalba et al. divided 140 children into two groups based on whether their blood Cd levels were above or below the median (0.27 µg/L), and reported that children with blood Cd above median had 23% lower serum vitamin D levels [103]. There is some evidence that Cd could disrupt the final hydroxylation reaction of provitamin D in the kidneys [104,105].
In summary, IDA impacted unequivocally, bone quality. This means that IDA-induced adverse bone effects may also have contributed to fracture, especially in the cases where osteomalacia followed an iron infusion treatment. Namely, Vilaca et al. reported that 22 individuals sustained fractures [71]. Since IDA causes bone alteration and it is speculated that iron infusion can also damage bones, it is difficult distinguish whether the fractures occurred due to IDA, iron infusion, or a combination of these. Moreover, ID can raise the circulating iFGF23 concentrations, as confirmed in studies in vivo [106,107,108] and humans [109,110]. The serum iFGF23 concentrations inversely correlated with serum Fe levels [110]. Evidence that anemia, iron deficiency and inflammation, through HIF1α and erythropoietin, upregulate the FGF23 gene is increasingly reported.
4. Bone Toxicity Mechanism of Cd
In this section, we summarize results of empirical studies supporting epidemiological/clinical data discussed in Section 3. A two-hit hypothesis is presented to explain the molecular basis of Cd cytotoxicity involving stress response and defense against bone cell death through ferroptosis; an iron-dependent form of regulated cell death induced by lipid peroxidation.
4.1. Experimental Data
Using in vitro experiments, Cd-intoxicated osteoblasts were less viable and they secreted less alkaline phosphatase [111]. Another study provided evidence that Cd stimulated bone resorption by upregulating RANK expression [112]. Interestingly, Wan et al. demonstrated that Cd per se may affect bone mineralization [113]. Namely, they investigated effects of Cd on human bone marrow mesenchymal stem cells in vitro and obtained that Cd-exposed cells at the doses of 2.5 and 5 μM CdCl2 lacked intracellular calcification nodules in terms of quantity and volume detected by alizarin red staining [113,115]. In the second part of their research, Wan et al. showed a negative relationship between bone morphogenetic protein 4 and urinary Cd levels [114]. Bone morphogenetic protein 4 has a significant role in bone mineralization [115].
Liu et al. investigated mechanisms by which Cd induced osteoblast death [118], where they employed osteoblasts derived from Sprague-Dawley rat fetuses treated with 0, 1, 2, and 5 μM Cd. They observed changes in osteoblast nucleus morphology and the upregulation of Bax but the downregulation of Bcl-2 (which has antiapoptotic activity) [116]. In another in vitro study utilizing MC-3T3-E1 cells, exposure to CdCl2 at 0–20 μM decreased cell viability, and promoted osteoblast apoptosis, which resulted from a decreased Bcl-2 level and accumulation of Bax mRNA and protein, and interfered with osteoblast formation by decreasing the expression of RANKL [117].
In additional to induction of FGF23 expression in the osteoblast-like cells [40], a series of experimental studies have ascertained that Cd effects on osteoclasts and osteoblasts via multiple mechanisms, including RANKL-RANK axis [116,117]. Ran et al. demonstrated that SIRT1/PGC-1α/P53Lys382 signaling pathway could mediate osteoporosis due to Cd, while noting that bones from patients with osteoporosis had an elevated Cd level [118].
In summary, Cd promotes bone resorption by causing premature death (ferroptosis) of osteoblasts, while increasing the formation of osteoclasts. These findings are in line with current views on the significance of ferroptosis in normal bone health and pathogenesis of Cd-induced osteoporosis [119,120].
4.2. Hypothetical Two-Hit Mechanism of the Cytotoxiicty of Cd
Potential contribution of iron-dependent cell death (ferroptosis) in pathogenesis of osteomalacia in itai-itai disease patients was first evident from a study by Noda et al. who examined 23 autopsy cases of itai-itai disease and 18 cases of sudden death as controls [121]. Using histochemical staining and x-ray microanalysis, iron was found in approximately half of mineralization fronts, and the bone content of Cd was 4.5-time higher in patients with itai-itai disease, compared to control subjects [121]. Iron presence at mineralization fronts was also found in rats and monkeys treated with Cd [39,122].
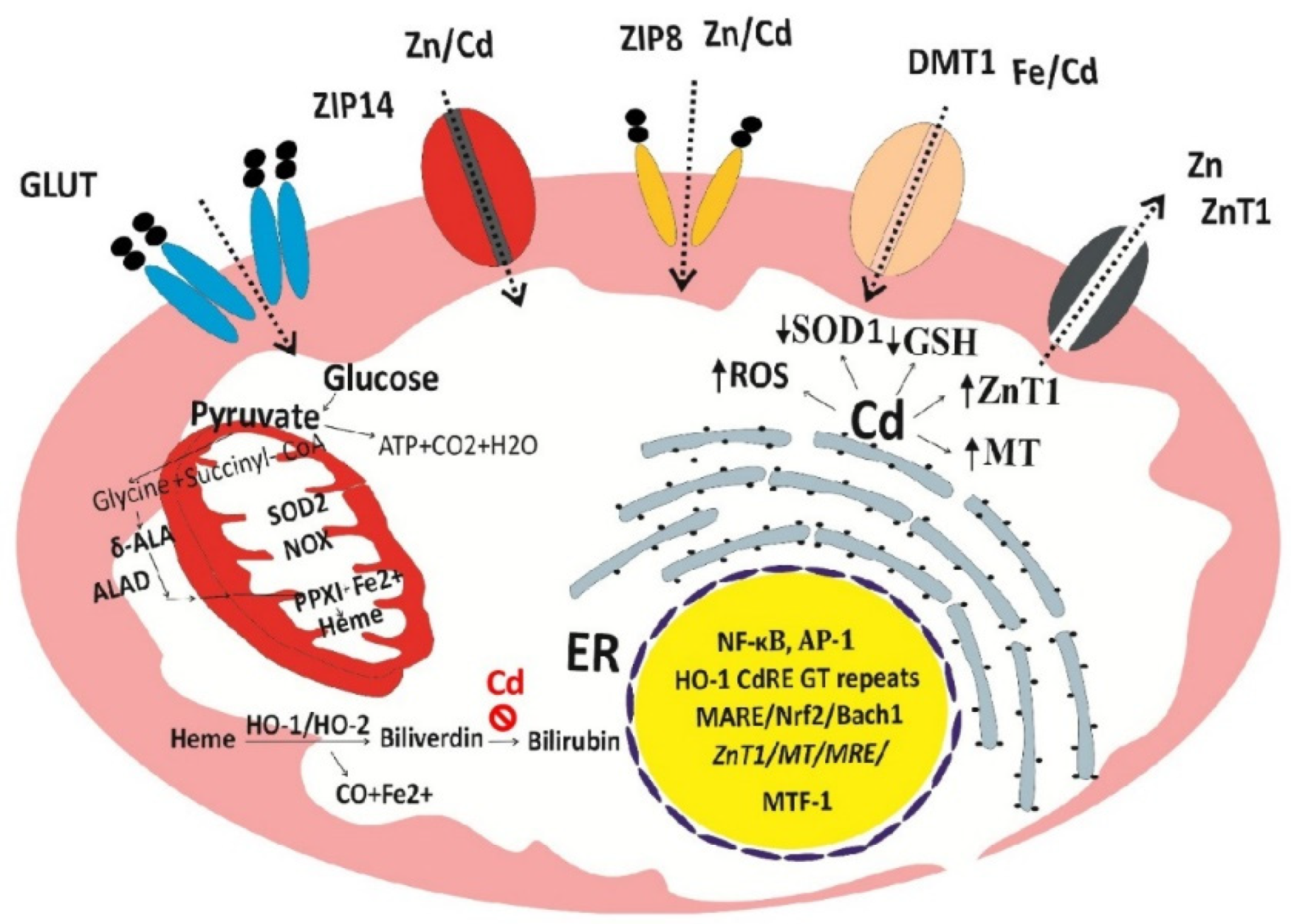
Given a unique role of heme oxygenase-1 (HO-1) in cellular stress response and defense against oxidative damage [123,124], we hypothesize that through excessive ROS and damage to mitochondria, Cd causes a release iron from heme, ultimately leading to cell death (ferroptosis) (Figure 3).
It is important to note; firstly that Cd at very low concentrations can cause a massive increase in HO-1 protein and catalytic activity because it activates the HO-1 gene through cadmium response element (CdRE) in additional to antioxidant response element (ARE) [124,125,126,127]. Secondly, the increase in HO-1 enzyme activity by Cd is not coupled with bilirubin synthesis [128,129]. In effect, Cd increases intracellular levels of Fe2+, while depriving cells of the ability to protect cells against lipid peroxidation and ferroptosis ensues. Expression of ZnT1 by Cd can lead to cellular zinc depletion because ZnT1 has an absolute specificity for zinc as is FNT1, a cell exit route for iron only. Experimental studies show that zinc reduced osteoclast activities and increased the number of osteoblasts [130,131] and that oxidative stress due to zinc deficiency promoted RANKL expression in rat bones [133].
5. Conclusions
A combined effect of iron deficiency and Cd bone toxicity may explain partially bone complications of intravenous iron supplementation therapy. The synthesis and cleavage of the bone-derived FGF23 forming intact (iFGF23) and Cter-FGF23 peptide fragments are affected by IDA and iron deficiency. The Cter-FGF23 fragment has been linked to iron homeostasis independent of iFGF23 role in renal phosphate reabsorption. In experimental studies, Cd increased FGF23 expression by osteoblast-like cells and suppressed FGF23 cleavage leading to a rise of serum FGF23, which, in turn, mediated an effect of Cd on tubular phosphate reabsorption. Moreover, Cd may interfere with vitamin D metabolism and bone mineralization.
While a rising Cd body burden might be a contributing factor, IDA may, in part, account for an increased prevalence of fractures due to Cd because IDA increases Cd absorption and accumulation in bones; bone tissues from patients with osteoporosis had higher Cd levels, compared to controls. Unfortunately, Cd exposure was not quantified in any report relating intravenous iron therapy to hypophosphatemic osteomalacia; noting, however that iron deficiency and Cd could raise plasma iFGF23 levels independently. Current evidence suggests distinct role for Cter-FGF23 fragment in the regulation of iron homeostasis, while suppressing erythropoietin synthesis in kidneys by FGF23 contributes further to anemia.
Evidence that environmental Cd increases the prevalence of iron deficiency and IDA in the general population calls for an effort to address a range of environmental exposures in future iron supplementation programs. The ability of Cd to reduce iron absorption and iron deficiency has recently been demonstrated using long-term feeding studies. Previous short-term dosing experiments failed to reveal such effects of Cd on iron absorption that can lead to iron deficiency if exposure to the metal continues; a likely scenario because Cd exposure occurs through a normal diet.
In theory, Cd can enter any cell in the body using metal transport proteins, receptors and pathways for iron and zinc (e.g., DMT1, ZIP14, ZIP8, TfR). Through CdRE and MARE/NrF2, Cd induces a massive increase in HO-1 enzyme activity resulting in a release of iron (Fe2+) from heme. However, by the mechanism remains unknown, the upregulation of HO-1 in response to Cd is not coupled with the generation of bilirubin, a potent lipid peroxidation chain breaker; consequently, extensive cellular oxidative damage and cell death (ferroptosis) may ensue.
At very low concentrations, Cd induces the expression of ZnT1, a highly specific zinc efflux transporter; consequently, zinc deficiency is inevitable because zinc is extruded from cells by ZnT1 while Cd is retained within cells. Like the bilirubin synthesis hit by Cd, cellular zinc deficiency is a universal cytotoxic mechanism of Cd.
Avoidance of foods containing high levels of Cd and smoking cessation are pivotal as is the maintenance of optimal body contents of iron and zinc. Dietary antioxidants provide a complementary preventive measure.
Author Contributions
Conceptualization, A.C., P.M. and S.S.; writing—original draft preparation, A.C. and S.S.; writing—review and editing, P.M. and S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent: Not applicable.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
This work was supported with resources from the Centre for Kidney Disease Research, Translational Research Institute, and the Department of Kidney and Transplant Services, Princess Alexandra Hospital, QLD, Australia.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lopez, A.; Cacoub, P.; Macdougall, I.C.; Peyrin-Biroulet, L. Iron deficiency anaemia. Lancet 2016, 387, 907–916. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Musallam, K.M.; Taher, A.T. Iron deficiency anaemia revisited. J. Intern. Med. 2020, 287, 153–170. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Sharma, E.; Marley, A.; Samaan, M.A.; Brookes, M.J. Iron deficiency anaemia: pathophysiology, assessment, practical management. BMJ Open Gastroenterol. 2022, 9, e000759. [Google Scholar] [CrossRef]
- Auerbach, M.; DeLoughery, T.G.; Tirnauer, J.S. Iron Deficiency in Adults: A Review. JAMA 2025, 333, 1813–1823. [Google Scholar] [CrossRef]
- Li, X.; Finberg, K.E. Iron Deficiency Anemia. Adv. Exp. Med. Biol. 2025, 1480, 163–178. [Google Scholar] [PubMed]
- Ueda, N.; Takasawa, K. Impact of Inflammation on Ferritin, Hepcidin and the Management of Iron Deficiency Anemia in Chronic Kidney Disease. Nutrients 2018, 10, 1173. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Motta, I. Anemia in Clinical Practice-Definition and Classification: Does Hemoglobin Change With Aging? Semin. Hematol. 2015, 52, 261–269. [Google Scholar] [CrossRef]
- GBD 2021 Anaemia Collaborators. Prevalence, years lived with disability, and trends in anaemia burden by severity and cause, 1990-2021: findings from the Global Burden of Disease Study 2021. Lancet Haematol. 2023, 10, e713–e734. [Google Scholar] [CrossRef]
- Safiri, S.; Kolahi, A.A.; Noori, M.; Nejadghaderi, S.A.; Karamzad, N.; Bragazzi, N.L.; Sullman, M.J.M.; Abdollahi, M.; Collins, G.S.; Kaufman, J.S.; et al. Burden of anemia and its underlying causes in 204 countries and territories, 1990-2019: results from the Global Burden of Disease Study 2019. J. Hematol. Oncol. 2021, 14, 185. [Google Scholar] [CrossRef] [PubMed]
- Saini, M.; Trehan, K.; Thakur, S.; Modi, A.; Jain, S.K. Advances in Iron Deficiency Anaemia Management: Exploring Novel Drug Delivery Systems and Future Perspectives. Curr. Drug Deliv. 2025, 22, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Kolarš, B.; Mijatović Jovin, V.; Živanović, N.; Minaković, I.; Gvozdenović, N.; Dickov Kokeza, I.; Lesjak, M. Iron Deficiency and Iron Deficiency Anemia: A Comprehensive Overview of Established and Emerging Concepts. Pharmaceuticals (Basel) 2025, 18, 1104. [Google Scholar] [CrossRef]
- Batchelor, E.K.; Kapitsinou, P.; Pergola, P.E.; Kovesdy, C.P.; Jalal, D.I. Iron Deficiency in Chronic Kidney Disease: Updates on Pathophysiology, Diagnosis, and Treatment. J. Am. Soc. Nephrol. 2020, 31, 456–468. [Google Scholar] [CrossRef]
- Moum, B.; Lindgren, S. Iron Deficiency and Iron Deficiency Anemia in Chronic Disease-Common, Important, and Treatable. J. Clin. Med. 2025, 14, 4519. [Google Scholar] [CrossRef]
- Pan, M.-L.; Chen, L.-R.; Tsao, H.-M.; Chen, K.-H. Iron Deficiency Anemia as a Risk Factor for Osteoporosis in Taiwan: A Nationwide Population-Based Study. Nutrients 2017, 9, 616. [Google Scholar] [CrossRef]
- Tari, E.; Vörhendi, N.; Kiss, S.; Teutsch, B.; Váradi, A.; Sisák, K.; Alizadeh, H.; Hegyi, P.; Erőss, B. Anaemia Is Associated with an Increased Risk of Fractures, a Systematic Review, and Meta-Analysis. Gerontology 2023, 69, 1–13. [Google Scholar] [CrossRef]
- Chuang, M.H.; Chuang, T.L.; Koo, M.; Wang, Y.F. Low Hemoglobin Is Associated With Low Bone Mineral Density and High Risk of Bone Fracture in Male Adults: A Retrospective Medical Record Review Study. Am. J. Mens Health 2019, 13, 1557988319850378. [Google Scholar] [CrossRef] [PubMed]
- Lichtler, R.; Cowley, M. Environmental Contaminants, Iron Deficiency, and Iron-Deficiency Anemia: A Review of the Literature. Scientifica (Cairo) 2025, 2025, 5007983. [Google Scholar] [CrossRef]
- Steinbicker, A.U.; Pantopoulos, K. Oral and Intravenous Iron Therapy. Adv. Exp. Med. Biol. 2025, 1480, 371–386. [Google Scholar]
- Pozzessere, S. Iron-Induced Hypophosphatemia: A Review of Pathophysiology, Drug Safety, and Pharmacogenomic Perspectives. J. Hematol. 2025, 14, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Strubbe, M.; David, K.; Peene, B.; Eeckhout, B.; Van der Schueren, B.; Decallonne, B.; Vangoitsenhoven, R.; Vanderschueren, D.; Antonio, L. No longer to be ignored: Hypophosphatemia following intravenous iron administration. Rev. Endocr. Metab. Disord. 2025, 26, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; David, V. Fibroblast growth factor 23 is pumping iron: C-terminal-fibroblast growth factor 23 cleaved peptide and its function in iron metabolism. Curr. Opin. Nephrol. Hypertens. 2024, 33, 368–374. [Google Scholar] [CrossRef]
- Cifuentes, A.; Laskar-Marchesseau, Z.; Courbon, G. FGF23: A player not only in bone diseases. Joint Bone Spine 2025, 93, 105988. [Google Scholar] [CrossRef]
- Cantoral, A.; Collado-López, S.; Betanzos-Robledo, L.; Lamadrid-Figueroa, H.; García-Martínez, B.A.; Ríos, C.; Díaz-Ruiz, A.; Mariscal-Moreno, R.M.; Téllez-Rojo, M.M. Dietary Risk Assessment of Cadmium Exposure Through Commonly Consumed Foodstuffs in Mexico. Foods 2024, 13, 3649. [Google Scholar] [CrossRef]
- Zhu, H.; Tang, X.; Gu, C.; Chen, R.; Liu, Y.; Chu, H.; Zhang, Z. Assessment of human exposure to cadmium and its nephrotoxicity in the Chinese population. Sci. Total Environ. 2024, 918, 170488. [Google Scholar] [CrossRef]
- Kolbaum, A.E.; Jung, C; Jaeger, A; Libuda, L; Lindtner, O. Assessment of long-term dietary cadmium exposure in children in Germany: Does consideration of data from total diet studies reduce uncertainties from food monitoring programmes? Food Chem. Toxicol. 2024, 184, 114404. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.T.; Jandev, V.; Petroni, M.; Atallah-Yunes, N.; Bendinskas, K.; Brann, L.S.; Heffernan, K.; Larsen, D.A.; MacKenzie, J.A.; Palmer, C.D.; Parsons, PJ; Gump, BB; Collins, MB; et al. Airborne levels of cadmium are correlated with urinary cadmium concentrations among young children living in the New York state city of Syracuse, USA. Environ. Res. 2023, 223, 115450. [Google Scholar] [CrossRef] [PubMed]
- Almerud, P.; Zamaratskaia, G.; Lindroos, A.K.; Bjermo, H.; Andersson, E.M.; Lundh, T.; Ankarberg, E.H.; Lignell, S. Cadmium, total mercury, and lead in blood and associations with diet, sociodemographic factors, and smoking in Swedish adolescents. Environ. Res. 2021, 197, 110991. [Google Scholar] [CrossRef]
- Fagerberg, B.; Barregard, L. Review of cadmium exposure and smoking-independent effects on atherosclerotic cardiovascular disease in the general population. J. Intern Med. 2021, 290, 1153–1179. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Song, H.; Lee, J.; Kim, Y.J.; Chung, H.S.; Yu, J.M.; Jang, G.; Park, R.; Chung, W.; et al. Smoking and passive smoking increases mortality through mediation effect of cadmium exposure in the United States. Sci. Rep. 2023, 13, 3878. [Google Scholar] [CrossRef]
- Satarug, S. Is Chronic Kidney Disease Due to Cadmium Exposure Inevitable and Can It Be Reversed? Biomedicines 2024, 12, 718. [Google Scholar] [CrossRef]
- Kunioka, C.T.; Manso, M.C.; Carvalho, M. Association between Environmental Cadmium Exposure and Osteoporosis Risk in Postmenopausal Women: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 20, 485. [Google Scholar] [CrossRef]
- Pouillot, R.; Santillana Farakos, S.; Van Doren, J.M. Modeling the risk of low bone mass and osteoporosis as a function of urinary cadmium in U.S adults aged 50-79 years. Environ. Res. 2022, 212 Pt B, 113315. [Google Scholar] [CrossRef]
- Nogawa, K.; Sakurai, M.; Ishizaki, M.; Kido, T.; Nakagawa, H.; Suwazono, Y. Threshold limit values of the cadmium concentration in rice in the development of itai-itai disease using benchmark dose analysis. J. Appl. Toxicol. 2017, 37, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Phelps, K. R. Cadmium exposure and toxicity. In Metal Toxicology Handbook; Bagchi, D, Bagchi, M, Eds.; CRC Press: New York, 2020; Volume Ch 14, pp. pp 219–272. [Google Scholar]
- Kasuya, M. Recent epidemiological studies on itai-itai disease as a chronic cadmium poisoning in Japan. Water Sci. Technol. 2000, 42, 147–154. [Google Scholar] [CrossRef]
- Baba, H.; Tsuneyama, K.; Kumada, T.; Aoshima, T.; Imura, J. Histopathological analysis for osteomalacia and tubulopathy in itai-itai disease. J. Toxicol. Sci. 2014, 39, 91–96. [Google Scholar] [CrossRef]
- Aoshima, K. Itai-itai disease: Renal tubular osteomalacia induced by environmental exposure to cadmium—historical review and perspectives. Soil Sci. Plant Nutr. 2016, 62, 319–326. [Google Scholar] [CrossRef]
- Sasaki, T.; Horiguchi, H.; Matsukawa, T.; Kobayashi, M.; Omori, Y.; Oguma, E.; Komatsuda, A. A suspected case of "itai-itai disease" in a cadmium-polluted area in Akita prefecture, Japan. Environ. Health Prev. Med. 2024, 29, 40. [Google Scholar]
- Kurata, Y.; Katsuta, O.; Doi, T.; Kawasuso, T.; Hiratsuka, H.; Tsuchitani, M.; Umemura, T. Chronic cadmium treatment induces tubular nephropathy and osteomalacic osteopenia in ovariectomized cynomolgus monkeys. Vet. Pathol. 2014, 51, 919–931. [Google Scholar] [CrossRef]
- Kido, S.; Fujihara, M.; Nomura, K.; Sasaki, S.; Mukai, R.; Ohnishi, R.; Kaneko, I.; Segawa, H.; Tatsumi, S.; Izumi, H.; et al. Molecular mechanisms of cadmium-induced fibroblast growth factor 23 upregulation in osteoblast-like cells. Toxicol. Sci. 2014, 139, 301–316. [Google Scholar] [CrossRef]
- Aranami, F.; Segawa, H.; Furutani, J.; Kuwahara, S.; Tominaga, R.; Hanabusa, E.; Tatsumi, S.; Kido, S.; Ito, M.; Miyamoto, K. Fibroblast growth factor 23 mediates the phosphaturic actions of cadmium. J. Med. Invest. 2010, 57, 95–108. [Google Scholar] [CrossRef]
- Nishito, Y.; Kambe, T. Absorption mechanisms of iron, copper, and zinc: An overview. J. Nutr. Sci. Vitaminol. (Tokyo) 2018, 64, 1–7. [Google Scholar] [CrossRef]
- Kondaiah, P.; Yaduvanshi, P.S.; Sharp, P.A.; Pullakhandam, R. Iron and zinc homeostasis and interactions: Does enteric zinc excretion cross-talk with intestinal iron absorption? Nutrients 2019, 11, 1885. [Google Scholar] [CrossRef]
- Okazaki, Y. Iron from the gut: The role of divalent metal transporter 1. J. Clin. Biochem. Nutr. 2024, 74, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Danko, T.; Bergeron, M.J.; Balazs, B.; Suzuki, Y.; Zsembery, A.; Hediger, M.A. Heavy metal cations permeate the TRPV6 epithelial cation channel. Cell Calcium 2011, 49, 43–55. [Google Scholar] [CrossRef]
- Kovacs, G.; Montalbetti, N.; Franz, M.C.; Graeter, S.; Simonin, A.; Hediger, M.A. Human TRPV5 and TRPV6: Key players in cadmium and zinc toxicity. Cell Calcium 2013, 54, 276–286. [Google Scholar] [CrossRef]
- Aydemir, T.B.; Cousins, R.J. The multiple faces of the metal transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef]
- Schneider, S.N.; Liu, Z.; Wang, B.; Miller, M.L.; Afton, S.E.; Soleimani, M.; Nebert, D.W. Oral cadmium in mice carrying 5 versus 2 copies of the Slc39a8 gene: Comparison of uptake, distribution, metal content, and toxicity. Int. J. Toxicol. 2014, 33, 14–20. [Google Scholar] [CrossRef]
- Nebert, D.W. Comparing gene expression during cadmium uptake and distribution: Untreated versus oral Cd-treated wild-type and ZIP14 knockout mice. Toxicol. Sci. 2015, 143, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Park, J.D.; Cherrington, N.J.; Klaassen, C.D. Intestinal absorption of cadmium is associated with divalent metal transporter 1 in rats. Toxicol. Sci. 2002, 68, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; el Belbasi, H.I.; Min, K.S.; Onosaka, S.; Okada, Y.; Matsumoto, Y.; Mutoh, N.; Tanaka, K. Fate of cadmium bound to phytochelatin in rats. Res. Commun. Chem. Pathol. Pharmacol. 1993, 82, 357–365. [Google Scholar]
- Langelueddecke, C.; Roussa, E.; Fenton, R.A.; Thévenod, F. Expression and function of the lipocalin-2 (24p3/NGAL) receptor in rodent and human intestinal epithelia. PLoS ONE 2013, 8, e71586. [Google Scholar] [CrossRef]
- Langelueddecke, C.; Lee, W.K.; Thévenod, F. Differential transcytosis and toxicity of the hNGAL receptor ligands cadmium-metallothionein and cadmium-phytochelatin in colon-like Caco-2 cells: Implications for in vivo cadmium toxicity. Toxicol. Lett. 2014, 226, 228–235. [Google Scholar] [CrossRef]
- Mitchell, C.J.; Shawki, A.; Ganz, T.; Nemeth, E.; Mackenzie, B. Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. Am. J. Physiol. Cell Physiol. 2014, 306, C450–C459. [Google Scholar] [CrossRef] [PubMed]
- Frazer, D.M.; Anderson, G.J.; Collins, J.F. Dietary Iron Absorption: Biochemical and Nutritional Aspects. Adv. Exp. Med. Biol. 2025, 1480, 75–87. [Google Scholar] [PubMed]
- Hoch, E.; Lin, W.; Chai, J.; Hershfinkel, M.; Fu, D.; Sekler, I. Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity. Proc. Natl. Acad. Sci. USA 2012, 109, 7202–7207. [Google Scholar] [CrossRef]
- Järup, L.; Rogenfelt, A.; Elinder, C.G.; Nogawa, K.; Kjellström, T. Biological half-time of cadmium in the blood of workers after cessation of exposure. Scand. J. Work Environ. Health 1983, 9, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Elinder, C.G.; Lind, B.; Kjellström, T.; Linnman, L.; Friberg, L. Cadmium in kidney cortex, liver, and pancreas from Swedish autopsies. Estimation of biological half time in kidney cortex, considering calorie intake and smoking habits. Arch. Environ. Health 1976, 31, 292–302. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kido, T.; Nakagawa, H.; Nishijo, M.; Honda, R.; Kobayashi, E.; Dochi, M.; Nogawa, K. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 2009, 14, 77–81. [Google Scholar] [CrossRef]
- Ishizaki, M.; Suwazono, Y.; Kido, T.; Nishijo, M.; Honda, R.; Kobayashi, E.; Nogawa, K.; Nakagawa, H. Estimation of biological half-life of urinary cadmium in inhabitants after cessation of environmental cadmium pollution using a mixed linear model. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 1273–1276. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Ruangyuttikarn, W.; Nishijo, M.; Gobe, G.C.; Phelps, K.R. The Source and Pathophysiologic Significance of Excreted Cadmium. Toxics 2019, 7, 55. [Google Scholar] [CrossRef]
- Sun, H.; Wang, D.; Zhou, Z.; Ding, Z.; Chen, X.; Xu, Y.; Huang, L.; Tang, D. Association of cadmium in urine and blood with age in a general population with low environmental exposure. Chemosphere 2016, 156, 392–397. [Google Scholar] [CrossRef]
- Orlowski, C.; Piotrowski, J.K.; Subdys, J.K.; Gross, A. Urinary cadmium as indicator of renal cadmium in humans: an autopsy study. Hum. Exp. Toxicol. 1998, 17, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Baker, J.R.; Reilly, P.E.; Moore, M.R.; Williams, D.J. Cadmium levels in the lung, liver, kidney cortex, and urine samples from Australians without occupational exposure to metals. Arch. Environ. Health 2002, 57, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Akerstrom, M.; Barregard, L.; Lundh, T.; Sallsten, G. The relationship between cadmium in kidney and cadmium in urine and blood in an environmentally exposed population. Toxicol. Appl. Pharmacol. 2013, 268, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Wallin, M.; Sallsten, G.; Lundh, T.; Barregard, L. Low-level cadmium exposure and effects on kidney function. Occup. Environ. Med. 2014, 71, 848–854. [Google Scholar] [CrossRef]
- Tokumoto, M.; Lee, J.-Y.; Fujiwara, Y.; Satoh, M. Long-Term Exposure to Cadmium Causes Hepatic Iron Deficiency through the Suppression of Iron-Transport-Related Gene Expression in the Proximal Duodenum. Toxics 2023, 11, 641. [Google Scholar] [CrossRef]
- Zhang, K.; Long, M.; Dong, W.; Li, J.; Wang, X.; Liu, W.; Huang, Q.; Ping, Y.; Zou, H.; Song, R.; et al. Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats. Int. J. Mol. Sci. 2024, 25, 763. [Google Scholar] [CrossRef]
- Giri, S.; Roy, A.; Kumar, A.; Ghosh, S.; Bhunia, A.; Patra, S. Cadmium toxicity-related metabolic bone disease: a clinical conundrum of five cases. Osteoporos. Int. 2025. [Google Scholar] [CrossRef]
- Samões, B.; Silva, B.; Martins, A.; Oliveira, D.; Rajão Martins, F.; Fonseca, D.; Costa, L.; Bernardes, M. Hypophosphatemic osteomalacia induced by intravenous iron therapy: a case report. Joint Bone Spine 2023, 90, 105586. [Google Scholar] [CrossRef]
- Vilaca, T.; Velmurugan, N.; Smith, C.; Abrahamsen, B.; Eastell, R. Osteomalacia as a Complication of Intravenous Iron Infusion: A Systematic Review of Case Reports. J. Bone Miner. Res. 2022, 37, 1188–1199. [Google Scholar] [CrossRef]
- Bartko, J.; Roschger, P.; Zandieh, S.; Brehm, A.; Zwerina, J.; Klaushofer, K. Hypophosphatemia, Severe Bone Pain, Gait Disturbance, and Fatigue Fractures After Iron Substitution in Inflammatory Bowel Disease: A Case Report. J. Bone Miner. Res. 2018, 33, 534–539. [Google Scholar] [CrossRef]
- Baban, Y.N.; Edicheria, C.M.; Joseph, J.; Kaur, P.; Mostafa, J.A. Osteoporosis Complications in Crohn's Disease Patients: Factors, Pathogenesis, and Treatment Outlines. Cureus 2021, 13, e20564. [Google Scholar] [CrossRef] [PubMed]
- Siffledeen, J.S.; Fedorak, R.N.; Siminoski, K.; Jen, H.; Vaudan, E.; Abraham, N.; Seinhart, H.; Greenberg, G. Bones and Crohn's: risk factors associated with low bone mineral density in patients with Crohn's disease. Inflamm. Bowel Dis. 2004, 10, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, D.M.; Stoecker, B.; Plattner, A.; Jennings, D.; Haub, M. Iron Deficiency Negatively Affects Vertebrae and Femurs of Rats Independently of Energy Intake and Body Weight. J. Nutr. 2004, 134, 3061–3067. [Google Scholar] [CrossRef]
- Díaz-Castro, J.; López-Frías, M.R.; Campos, M.S.; López-Frías, M.; Alférez, M.J.; Nestares, T.; Ojeda, M.L.; López-Aliaga, I. Severe nutritional iron-deficiency anaemia has a negative effect on some bone turnover biomarkers in rats. Eur. J. Nutr. 2012, 51, 241–247. [Google Scholar] [CrossRef]
- Katsumata, S.; Katsumata-Tsuboi, R.; Uehara, M.; Suzuki, K. Severe iron deficiency decreases both bone formation and bone resorption in rats. J. Nutr. 2009, 139, 238–243. [Google Scholar] [CrossRef]
- Katsumata, S.; Tsuboi, R.; Uehara, M.; Suzuki, K. Dietary iron deficiency decreases serum osteocalcin concentration and bone mineral density in rats. Biosci. Biotechnol. Biochem. 2006, 70, 2547–2550. [Google Scholar] [CrossRef]
- Bárány, E.; Bergdahl, I.A.; Bratteby, L.E.; Lundh, T.; Samuelson, G.; Skerfving, S.; Oskarsson, A. Iron status influences trace element levels in human blood and serum. Environ. Res. 2005, 98, 215–223. [Google Scholar] [CrossRef]
- Lee, B.K.; Kim, Y. Iron deficiency is associated with increased levels of blood cadmium in the Korean general population: analysis of 2008-2009 Korean National Health and Nutrition Examination Survey data. Environ. Res. 2012, 112, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.; Kim, S.H.; Kim, N.S.; Ham, J.O.; Kim, Y. Iron deficiency increases blood cadmium levels in adolescents surveyed in KNHANES 2010-2011. Biol. Trace Elem. Res 2014, 159, 52–58. [Google Scholar] [CrossRef]
- Cirovic, A.; Denic, A.; Clarke, B.L.; Vassallo, R.; Cirovic, A.; Landry, G.M. A hypoxia-driven occurrence of chronic kidney disease and osteoporosis in COPD individuals: New insights into environmental cadmium exposure. Toxicol. 2022, 482, 153355. [Google Scholar] [CrossRef]
- Roy, A.; Saha, T.; Sahoo, J.; Das, A. Hypophosphatemic osteomalacia due to cadmium toxicity in silverware industry: A curious case of aches and pains. J. Fam. Med. Prim. Care 2024, 13, 2516–2519. [Google Scholar] [CrossRef] [PubMed]
- Balusikova, K.; Dostalikova-Cimburova, M.; Tacheci, I.; Kovar, J. Expression profiles of iron transport molecules along the duodenum. J. Cell. Mol. Med. 2022, 26, 2995–3004. [Google Scholar] [CrossRef]
- Wang, C.Y.; Jenkitkasemwong; Duarte, S.; Sparkman, S.; Shawki, B.K.; Mackenzie, A.; Knutson, B.M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Ohba, K. Involvement of metal transporters in the intestinal uptake of cadmium. J. Toxicol. Sci. 2020, 45, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Asaad, S.; Econs, M.; Rubin, J.E. Severe FGF23-based hypophosphataemic osteomalacia due to ferric carboxymaltose administration. BMJ Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Saljooghi, A.S.; Fatemi, S.J. Cadmium transport in blood serum. Toxicol. Ind. Health 2010, 26, 195–201. [Google Scholar]
- Ledesma-Colunga, M.G.; Weidner, H.; Vujic Spasic, M.; Hofbauer, L.C.; Baschant, U.; Rauner, M. Shaping the bone through iron and iron-related proteins. Semin. Hematol. 2021, 58, 188–200. [Google Scholar] [CrossRef]
- Watts, D.; Gaete, D.; Rodriguez, D.; Hoogewijs, D.; Rauner, M.; Sormendi, S.; Wielockx, B. Hypoxia Pathway Proteins are Master Regulators of Erythropoiesis. Int. J. Mol. Sci. 2020, 21, 8131. [Google Scholar] [CrossRef]
- Luppen, C.A.; Chandler, R.L.; Noh, T.; Mortlock, D.P.; Frenkel, B. BMP-2 vs. BMP-4 expression and activity in glucocorticoid-arrested MC3T3-E1 osteoblasts: Smad signaling, not alkaline phosphatase activity, predicts rescue of mineralization. Growth Factors 2008, 26, 226–237. [Google Scholar] [CrossRef]
- Cirovic, A.; Cirovic, A. Letter to the editor for the "relationship between iron deficiency and expression of genes involved in iron metabolism in human myocardium and skeletal muscle. Int. J. Cardiol. 2023, 384, 75. [Google Scholar] [CrossRef]
- Cabrera, C.; Frisk, C.; Löfström, U.; Lyngå, P.; Linde, C.; Hage, C.; Persson, H.; Eriksson, M.J.; Wallén, H.; Persson, B.; Ekström, M. Relationship between iron deficiency and expression of genes involved in iron metabolism in human myocardium and skeletal muscle. Int. J. Cardiol. 2023, 379, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Toxqui, L.; Vaquero, M.P. Chronic iron deficiency as an emerging risk factor for osteoporosis: a hypothesis. Nutrients 2015, 7, 2324–2344. [Google Scholar] [CrossRef]
- Teng, Y.; Teng, Z.; Xu, S.; Zhang, X.; Liu, J.; Yue, Q.; Zhu, Y.; Zeng, Y. The Analysis for Anemia Increasing Fracture Risk. Med. Sci. Monit. 2020, 26, e925707. [Google Scholar] [CrossRef]
- Jiang, Y.; Lin, X.; Wang, Y.; Li, J.; Wang, G.; Meng, Y.; Li, M.; Li, Y.; Luo, Y.; Gao, Z. Preoperative Anemia and Risk of In-hospital Postoperative Complications in Patients with Hip Fracture. Clin. Interv. Aging 2023, 18, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.A.; Shin, D.W.; Yoo, J.H.; Ko, H.Y.; Jeong, S.M. Anemia and risk of fractures in older Korean adults: a nationwide population-based study. J. Bone Min. Res. 2019, 34, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Wallin, M.; Andersson, E.M.; Engström, G. Blood cadmium is associated with increased fracture risk in never-smokers - results from a case-control study using data from the Malmö Diet and Cancer cohort. Bone 2024, 179, 116989. [Google Scholar] [CrossRef]
- Kristjansdottir, H.L.; Mellström, D.; Johansson, P.; Karlsson, M.; Vandenput, L.; Lorentzon, M.; Herlitz, H.; Ohlsson, C.; Lerner, U.H.; Lewerin, C. Anemia is associated with increased risk of non-vertebral osteoporotic fractures in elderly men: the MrOS Sweden cohort. Arch. Osteoporos. 2022, 17, 85. [Google Scholar] [CrossRef]
- Valderrábano, R.J.; Lee, J.; Lui, L.Y.; Hoffman, A.R.; Cummings, S.R.; Orwoll, E.S.; Wu, J.Y. Osteoporotic Fractures in Men (MrOS) Study Research Group. Older Men With Anemia Have Increased Fracture Risk Independent of Bone Mineral Density. J. Clin. Endocrinol. Metab. 2017, 102, 2199–2206. [Google Scholar] [CrossRef]
- Jørgensen, L.; Skjelbakken, T; Løchen, ML; Ahmed, L; Bjørnerem, A; Joakimsen, R; Jacobsen, BK. Anemia and the risk of non-vertebral fractures: the Tromsø Study. Osteoporos. Int. 2010, 21, 1761–1768. [Google Scholar] [CrossRef]
- Chen, Z.; Thomson, C.A.; Aickin, M.; Nicholas, J.S.; Van Wyck, D.; Lewis, C.E.; Cauley, J.A.; Bassford, T. Short list of Women's Health Initiative Investigators. The relationship between incidence of fractures and anemia in older multiethnic women. J. Am. Geriatr. Soc. 2010, 58, 2337–2344. [Google Scholar] [CrossRef]
- Chwalba, A.; Orłowska, J.; Słota, M.; Jeziorska, M.; Filipecka, K.; Bellanti, F.; Dobrakowski, M.; Kasperczyk, A.; Zalejska-Fiolka, J.; Kasperczyk, S. Effect of Cadmium on Oxidative Stress Indices and Vitamin D Concentrations in Children. J. Clin. Med. 2023, 12, 1572. [Google Scholar] [CrossRef]
- Moon, J. The role of vitamin D in toxic metal absorption: a review. J. Am. Coll. Nutr. 1994, 13, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Schwalfenberg, G.K.; Genuis, S.J. Vitamin D, Essential Minerals, and Toxic Elements: Exploring Interactions between Nutrients and Toxicants in Clinical Medicine. Sci. World J. 2015, 2015, 318595. [Google Scholar] [CrossRef] [PubMed]
- Hanudel, M.R.; Chua, K.; Rappaport, M.; Gabayan, V.; Valore, E.; Goltzman, D.; Ganz, T.; Nemeth, E.; Salusky, I.B. Effects of dietary iron intake and chronic kidney disease on fibroblast growth factor 23 metabolism in wild-type and hepcidin knockout mice. Am. J. Physiol. Renal. Physiol. 2016, 311, F1369–F1377. [Google Scholar] [CrossRef] [PubMed]
- Clinkenbeard, E.L.; Farrow, E.G.; Summers, L.J.; Cass, T.A.; Roberts, J.L.; Bayt, C.A.; Lahm, T.; Albrecht, M.; Allen, M.R.; Peacock, M.; White, K.E. Neonatal iron deficiency causes abnormal phosphate metabolism by elevating FGF23 in normal and ADHR mice. J. Bone Miner. Res. 2014, 29, 361–369. [Google Scholar] [CrossRef]
- Li, X.; Lozovatsky, L.; Tommasini, S.M.; Fretz, J.; Finberg, K.E. Bone marrow sinusoidal endothelial cells are a site of Fgf23 upregulation in a mouse model of iron deficiency anemia. Blood Adv. 2023, 7, 5156–5171. [Google Scholar] [CrossRef]
- Bożentowicz-Wikarek, M.; Kocełak, P.; Owczarek, A.; Olszanecka-Glinianowicz, M.; Mossakowska, M.; Skalska, A.; Więcek, A.; Chudek, J. Plasma fibroblast growth factor 23 concentration and iron status. Does the relationship exist in the elderly population? Clin. Biochem. 2015, 48, 431–436. [Google Scholar] [CrossRef]
- Lewerin, C.; Ljunggren, Ö.; Nilsson-Ehle, H.; Karlsson, M.K.; Herlitz, H.; Lorentzon, M.; Ohlsson, C.; Mellström, D. Low serum iron is associated with high serum intact FGF23 in elderly men: The Swedish MrOS study. Bone 2017, 98, 1–8. [Google Scholar] [CrossRef]
- Al-Ghafari, A.; Elmorsy, E.; Fikry, E.; Alrowaili, M.; Carter, W.G. The heavy metals lead and cadmium are cytotoxic to human bone osteoblasts via induction of redox stress. PLoS One 2019, 14, e0225341. [Google Scholar] [CrossRef]
- Chen, X.; Zhu, G.; Gu, S.; Jin, T.; Shao, C. Effects of cadmium on osteoblasts and osteoclasts in vitro. Environ. Toxicol. Pharmacol. 2009, 28, 232–236. [Google Scholar] [CrossRef]
- Wan, Y.; Mo, L.J.; Wu, L.; Li, D.L.; Song, J.; Hu, Y.K.; Huang, H.B.; Wei, Q.Z.; Wang, D.P.; Qiu, J.M.; et al. Bone morphogenetic protein 4 is involved in cadmium-associated bone damage. Toxicol. Sci. 2023, 191, 201–211. [Google Scholar] [CrossRef]
- Lademann, F.; Hofbauer, L.C.; Rauner, M. The bone morphogenetic protein pathway: the osteoclastic perspective. Front. Cell Develop. Biol. 2020, 8, 586031. [Google Scholar] [CrossRef]
- Luppen, C.A.; Chandler, R.L.; Noh, T.; Mortlock, D.P.; Frenkel, B. BMP-2 vs. BMP-4 expression and activity in glucocorticoid-arrested MC3T3-E1 osteoblasts: Smad signaling, not alkaline phosphatase activity, predicts rescue of mineralization. Growth Factors 2008, 26, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dai, N.; Wang, Y.; Xu, C.; Zhao, H.; Xia, P.; Gu, J.; Liu, X.; Bian, J.; Yuan, Y.; et al. Role of autophagy in cadmium-induced apoptosis of primary rat osteoblasts. Sci. Rep. 2016, 6, 20404. [Google Scholar] [CrossRef] [PubMed]
- He, T.; Shen, H.; Zhu, J.; Zhu, Y.; He, Y.; Li, Z.; Lu, H. Geniposide attenuates cadmium-induced oxidative stress injury via Nrf2 signaling in osteoblasts. Mol. Med. Rep. 2019, 20, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Ran, D.; Zhou, D.; Liu, G.; Ma, Y.; Ali, W.; Yu, R.; Wang, Q.; Zhao, H.; Zhu, J.; Zou, H.; et al. Reactive oxygen species control osteoblast apoptosis through SIRT1/PGC-1α/P53Lys382 signaling, mediating the onset of Cd-induced osteoporosis. J. Agric. Food Chem. 2023, 71, 5991–6002. [Google Scholar] [CrossRef]
- Nan, W.; Zhou, W.M.; Zi, J.L.; Shi, Y.Q.; Dong, Y.B.; Song, W.; Ma, Y.C.; Zhang, H.H. Ferroptosis and bone metabolic diseases: the dual regulatory role of the Nrf2/HO-1 signaling axis. Front. Cell Dev. Biol. 2025, 13, 1615197. [Google Scholar] [CrossRef]
- Zhou, Y.S.; Huang, J.; Cao, W.X.; Yu, A.X.; Li, P.; Liang, J.L.; Leng, X.Y.; Jin, J.; Yu, P.; Liu, J. The therapeutic mechanism of Compound Lurong Jiangu Capsule for the treatment of cadmium-induced osteoporosis: network pharmacology and experimental verification. Front. Endocrinol. (Lausanne) 2024, 15, 1331488. [Google Scholar] [CrossRef]
- Noda, M.; Yasuda, M.; Kitagawa, M. Iron as a possible aggravating factor for osteopathy in itai-itai disease, a disease associated with chronic cadmium intoxication. J. Bone Miner. Res. 1991, 6, 245–255. [Google Scholar] [CrossRef]
- Hiratsuka, H.; Katsuta, O.; Toyota, N.; Tsuchitani, M.; Akiba, T.; Marumo, F.; Umemura, T. Iron deposition at mineralization fronts and osteoid formation following chronic cadmium exposure in ovariectomized rats. Toxicol. Appl. Pharmacol. 1997, 143, 348–356. [Google Scholar] [CrossRef]
- Gallio, A.E.; Marson, N.A.; Heesom, K.J.; Lewis, P.A.; Alibhai, D.; Dugdale, C.A.; Herman, A.; Basran, J.; Hudson, A.J.; Raven, EL. An extended network for regulation of heme homeostasis in cells. Proc. Natl. Acad. Sci. USA 2025, 122, e2508237122. [Google Scholar] [CrossRef] [PubMed]
- Simmons, S.O.; Fan, C.Y.; Yeoman, K.; Wakefield, J.; Ramabhadran, R. NRF2 Oxidative Stress Induced by Heavy Metals is Cell Type Dependent. Curr. Chem. Genomics 2011, 5, 1–12. [Google Scholar] [CrossRef]
- Takeda, K.; Ishizawa, S.; Sato, M.; Yoshida, T.; Shibahara, S. Identification of a cis-acting element that is responsible for cadmium-mediated induction of the human heme oxygenase gene. J. Biol. Chem. 1994, 269, 22858–22867. [Google Scholar] [CrossRef]
- Stewart, D.; Killeen, E.; Naquin, R.; Alam, S.; Alam, J. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J. Biol. Chem. 2003, 278, 2396–2402. [Google Scholar] [CrossRef]
- Suzuki, H.; Tashiro, S.; Sun, J.; Doi, H.; Satomi, S.; Igarashi, K. Cadmium induces nuclear export of Bach1, a transcriptional repressor of heme oxygenase-1 gene. J. Biol. Chem. 2003, 278, 49246–49253. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.A.; Mu, A.; Tai, T.T.; Kitajima, S.; Taketani, S. Continuous de novo biosynthesis of haem and its rapid turnover to bilirubin are necessary for cytoprotection against cell damage. Sci. Rep. 2015, 5, 10488. [Google Scholar] [CrossRef]
- Kumagai, A.; Ando, R.; Miyatake, H.; Greimel, P.; Kobayashi, T.; Hirabayashi, Y.; Shimogori, T.; Miyawaki, A. A bilirubin-inducible fluorescent protein from eel muscle. Cell 2013, 153, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Hadley, K.B.; Newman, S.M.; Hunt, J.R. Dietary zinc reduces osteoclast resorption activities and increases markers of osteoblast differentiation, matrix maturation, and mineralization in the long bones of growing rats. J. Nutr. Biochem. 2010, 21, 297–303. [Google Scholar] [CrossRef]
- Nagata, M.; Lönnerdal, B. Role of zinc in cellular zinc trafficking and mineralization in a murine osteoblast-like cell line. J. Nutr. Biochem. 2011, 22, 172–178. [Google Scholar] [CrossRef]
- Suzuki, T.; Katsumata, S.; Matsuzaki, H.; Suzuki, K. Dietary zinc deficiency induces oxidative stress and promotes tumor necrosis factor-α- and interleukin-1β-induced RANKL expression in rat bone. J. Clin. Biochem. Nutr. 2016, 58, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Tamaru, E.; Kokubu, D.; Ushida, Y.; Itoh, K. Nrf2 induction potency of plant-derived compounds determined using an antioxidant response element luciferase reporter and conventional NAD(P)H-quinone acceptor oxidoreductase 1 activity assay. BMC Res Notes 2024, 17, 373. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, T.Y.; Yu, A.X.; Liang, J.L.; Zhou, Y.S.; Sun, H.Z.; Dai, Y.L.; Liu, J.; Yu, P. The Role of Ferroptosis in Osteoporosis and Advances in Chinese Herbal Interventions. Biology (Basel) 2025, 14, 367. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Specialized transport proteins and receptors expressed by the enterocytes. The metal transporters involved in Cd absorption are those for calcium (TRPV6), zinc (ZIP14), iron (DMT1). Cd bound metallothionine (CdMT) and phytochelatin (CdPC) are absorbed by transcytosis and endocytosis mediated by NGAL/lipocalin 2 receptor. Abbreviations: CYBRD1, cytochrome b reductase 1; FPN1, ferroportin1; TRPV6, transient receptor potential vanilloid6; ZIP14, Zrt- and Irt-related protein 14; DMT1, divalent metal transporter1; MT, metallothionine; PC, phytochelatin; NGAL, neutrophil gelatinase-associated lipocalin.
Figure 1.
Specialized transport proteins and receptors expressed by the enterocytes. The metal transporters involved in Cd absorption are those for calcium (TRPV6), zinc (ZIP14), iron (DMT1). Cd bound metallothionine (CdMT) and phytochelatin (CdPC) are absorbed by transcytosis and endocytosis mediated by NGAL/lipocalin 2 receptor. Abbreviations: CYBRD1, cytochrome b reductase 1; FPN1, ferroportin1; TRPV6, transient receptor potential vanilloid6; ZIP14, Zrt- and Irt-related protein 14; DMT1, divalent metal transporter1; MT, metallothionine; PC, phytochelatin; NGAL, neutrophil gelatinase-associated lipocalin.
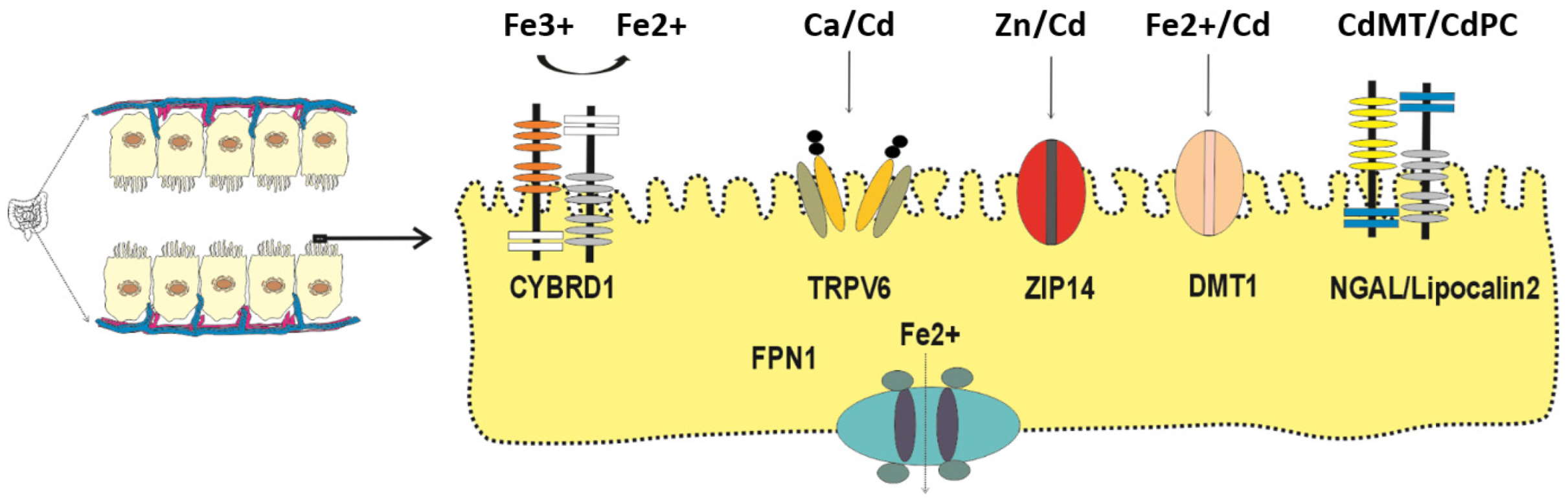
Figure 2.
Cd-induced iron deficiency and hypophosphatemia. Through the transferrin receptor 1 (TfR1), Cd readily enters the osteoblasts. In response, osteoblasts secrete fibroblast growth factor 23 (FGF23), which reduces phosphate reabsorption by kidney proximal tubular cells. Iron deficiency due to Cd may influence the cleavage of FGF23 to iFGF23 and Cter-FGF23.
Figure 2.
Cd-induced iron deficiency and hypophosphatemia. Through the transferrin receptor 1 (TfR1), Cd readily enters the osteoblasts. In response, osteoblasts secrete fibroblast growth factor 23 (FGF23), which reduces phosphate reabsorption by kidney proximal tubular cells. Iron deficiency due to Cd may influence the cleavage of FGF23 to iFGF23 and Cter-FGF23.
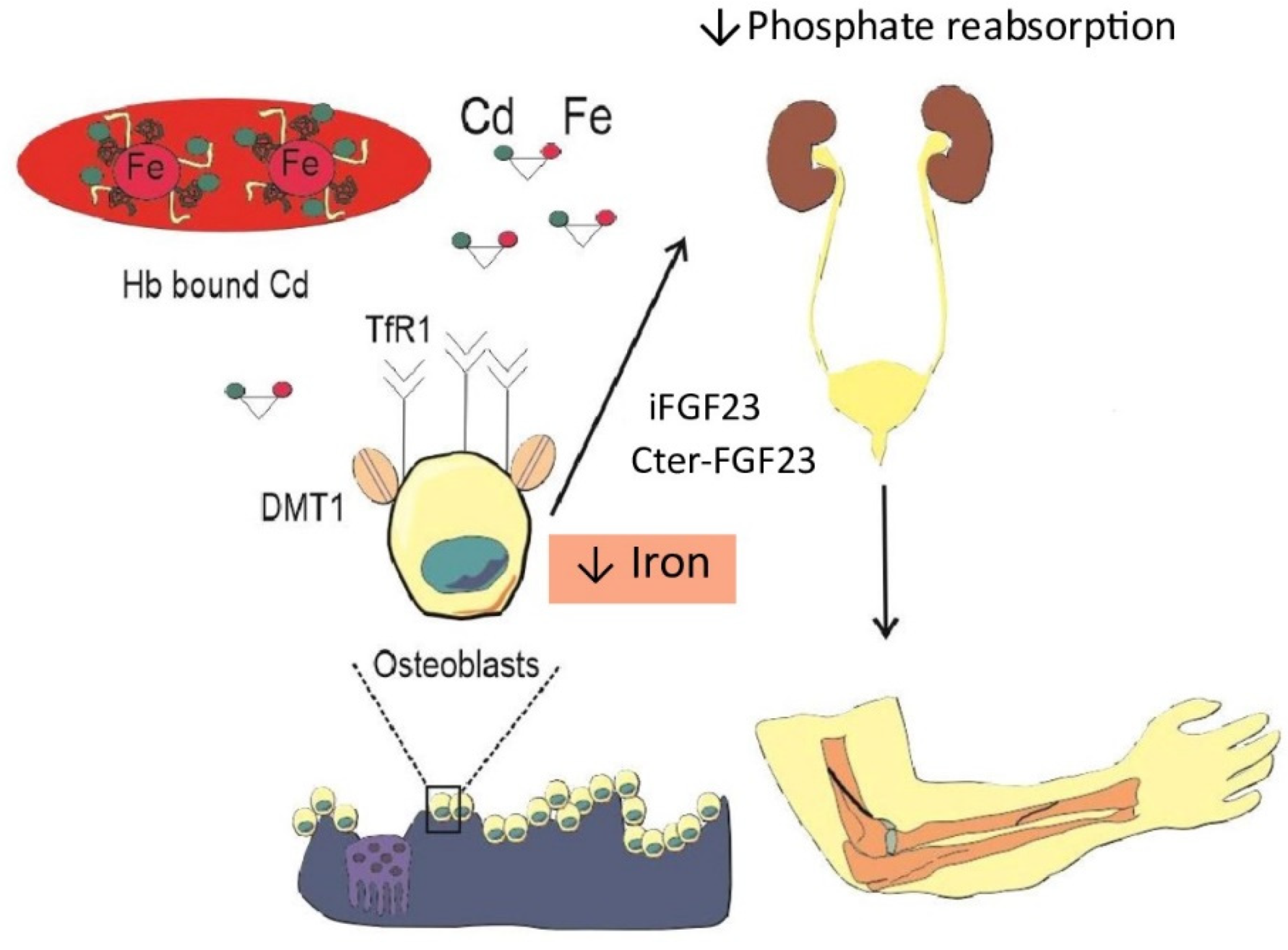
Figure 3.
The two-hit hypothesis of the cytotoxicity of Cd. Through the CdRE, Cd induces a massive increase in HO-1 activity with resultant release of Fe2+ without a concomitant increase in bilirubin synthesis, thereby depriving the ability to defend against lipid peroxidation due to ROS. Cd induces expression of ZnT1, a zinc specific efflux transporter which can lead to cellular zinc depletion.
Figure 3.
The two-hit hypothesis of the cytotoxicity of Cd. Through the CdRE, Cd induces a massive increase in HO-1 activity with resultant release of Fe2+ without a concomitant increase in bilirubin synthesis, thereby depriving the ability to defend against lipid peroxidation due to ROS. Cd induces expression of ZnT1, a zinc specific efflux transporter which can lead to cellular zinc depletion.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



