
Submitted:
30 December 2025
Posted:
31 December 2025
You are already at the latest version
Abstract
Quiescence is a reversible, non-proliferative cellular state that enables survival under nutrient limitation while preserving the capacity to resume growth. Rather than representing a passive default, quiescence is an actively regulated program conserved from unicellular eukaryotes to metazoans. This review focuses on the nuclear mechanisms underlying quiescence entry, maintenance, and exit, drawing on mechanistic insights from yeast models while highlighting conserved principles in multicellular systems. Across species, quiescence is characterized by global transcriptional repression, chromatin compaction, and extensive reorganization of nuclear architecture, coordinated by nutrient-sensing pathways centered on TOR/mTOR signaling. We discuss how transcriptional reprogramming is achieved through redistribution of RNA polymerases, dynamic transcription factor activities, and large-scale remodeling of histone modifications, alongside repressive chromatin formation. In parallel, post-transcriptional mechanisms—including intron retention, alternative polyadenylation, and accumulation of non-coding RNAs—fine-tune gene expression while limiting biosynthetic output. We further examine how changes in nuclear organization, such as nucleolar condensation, condensin-mediated chromosome rearrangements, and telomere hyperclusters, support long-term viability and genome stability. Collectively, this review highlights nuclear dynamics as an integrative regulatory layer that links metabolic state to cellular identity, adaptability, and long-term survival, with broad implications for development, stem cell function, and disease.
Keywords:
cellular quiescence
; nuclear organization
; chromatin remodeling
; transcriptional reprogramming
; post-transcriptional regulation
; epigenetic regulation
1. Introduction
Quiescence, the reversible arrest of cell proliferation, is the predominant state of life. Unlike terminally differentiated and senescent cells, quiescent cells maintain the full capacity to return from G0 and reenter the cell cycle. Unicellular organisms benefit from quiescence by surviving periods of growth limitation and resume active proliferation when favorable conditions return. Quiescence is also essential for various aspects of metazoan biology including reproduction, immunological memory, and tissue regeneration [1,2]. For example, unfertilized oocytes are generated during fetal development but can remain dormant for decades until release during ovulation. Even after fertilization, embryonic development can be temporally suspended: in some organisms, diapause, a form of developmental dormancy, is triggered to increase the chances of embryo survival under starvation or other stressors. In multicellular organisms, quiescent tissue-resident stem cells serve as reservoirs for tissue homeostasis and regeneration. At the same time, quiescent persister cells in tumors and fungal biofilms contribute to therapy resistance and disease recurrence, highlighting the clinical relevance of understanding quiescent cell biology.
Quiescence is believed to be an early evolutionary phenomenon that is universal across life. Periods of hardship and nutrient scarcity are inevitable companions of life – when conditions are favorable, life explodes, and intense competition for nutrients, and their depletion, are the consequence. It begs no question what an immense advantage the ability to withstand poor conditions must have posed from life’s inception. In the course of evolution, quiescence has been non-uniformly adapted to suit the specialized purpose of diverse biological contexts [1]. However, despite the diversity and complexity of eukaryotic cellular arrest models, cellular processes associated with transition, maintenance, and exit from the quiescent state are often shared. Characteristic features, including the reduction of cell size, chromosome condensation, decreased transcription, and reduced protein synthesis, are observed from yeast to metazoans. Across organisms, these changes are driven by starvation-responsive signaling pathways, prominently including inactivation of the highly conserved kinase complex Target of Rapamycin (TOR) complexes or its metazoan homolog Mechanistic Target of Rapamycin mTOR (for key terms and abbreviations, see Table 1). TOR and mTOR integrate information from multiple nutrient-sensing pathways and promote cell growth, proliferation, and survival when conditions are favorable; their inactivation initiates a conserved “hold and protect” program that gradually develops into quiescence [3,4] (Figure 1). The central nature of TOR signaling in quiescence establishment is underscored by the fact that pharmacological TOR inhibition, for example with rapamycin, recapitulates most hallmarks of quiescence even in complex systems, and has been used experimentally as a model of diapause [5].
Contrary to the initial view of quiescence as an inactive default state, its programmatic nature, marked by temporal dynamics, is now a widely accepted paradigm. Cells undergo a profound transformation, including global changes in gene expression and reorganization of cytoplasmic and nuclear structures, not only during entry and exit, but also throughout quiescence maintenance. This kind of rewiring is found in unicellular organisms as well as metazoan models, but can exhibit substantial heterogeneity across cell types and even among clonal cell populations [6,7,8,9,10,11,12,13]. This apparent contradiction between universal molecular pathways and diverse cellular responses raises critical questions: How can conserved molecular mechanisms give rise to such distinct phenotypic outcomes? And what cell type-specific factors shape quiescence dynamics?
Genetic screens have revealed the central importance of nuclear processes. Screens using yeast deletion collections or temperature-sensitive mutants have confirmed the central importance of TOR signaling, repression of protein biosynthesis, and an increase in autophagy in establishing quiescence [6], while also identifying numerous genes that are essential for survival during G0 or for re-entry into the cell cycle [14,15]. Notably, genes governing nuclear dynamics were frequently recovered in these screens, reflecting the extensive remodeling of nuclear architecture and chromatin reorganization that accompanies entry into quiescence across organisms [16]. This observation prompted Zahedi et al. to perform a targeted flow cytometry screen of known chromatin regulator deletion mutants in fission yeast [17], monitoring the proportion of cells in G0 as well as cell mortality over extended periods of quiescence to distinguish between genes essential for entering G0 and those required for long-term viability. Strikingly, among genes annotated with the Gene Ontology term “chromatin organization”, 68% of the mutants had defects in establishing or stably maintaining quiescence. Similarly high defect rates were observed for genes involved in “chromatin remodeling” (69%), “regulation of transcription” (71%), or “DNA repair” (76%), underscoring the importance of nuclear processes during cellular quiescence [17,18].
Scope and organization of this review. Despite the accumulating evidence for the critical role of nuclear remodeling during quiescence, our understanding of the underlying mechanisms remains incomplete. Here, we provide a comprehensive overview of current knowledge regarding nuclear dynamics during quiescence entry, maintenance, and exit, with an emphasis on features conserved across eukaryotes (Table 2). Because most mechanistic data derive from yeast models, we focus primarily on these systems, while highlighting similarities and differences in higher eukaryotes where data are available. Knowledge gaps and unresolved questions are highlighted throughout. For a comprehensive overview of genome stability and DNA damage repair, as well as non-nuclear aspects of quiescence, including signaling pathways, metabolic reprogramming, and the temporal dynamics of quiescence regulation, we refer the reader to recent reviews [2,8,19,20,21,22].
2. Established Models for Studying Nuclear Dynamics During Quiescence
2.1. Unicellular Eukaryotic Models
Yeast species are powerful models for studying both cellular proliferation and quiescence. Saccharomyces cerevisiae (budding yeast) and Schizosaccharomyces pombe (fission yeast) have been particularly instrumental in identifying essential conserved mechanisms involved in quiescence establishment. These species diverged 300-350 million years ago, yet they share fundamental cellular processes. Mechanisms conserved between these two evolutionarily distant yeasts are therefore likely to be relevant for metazoans as well. In both organisms, nutrient deprivation leads to a reversible cell cycle arrest and the transition into a quiescent state.
Budding yeast (Saccharomyces cerevisiae).S. cerevisiae has served as a leading eukaryotic model for quiescence research [19]. Early studies showed that cultures starved for carbon, nitrogen, or phosphate accumulate small G1-arrested cells [23], which were later referred to as G0 cells [24,25,26,27]. An extensive body of work has led to the development of established guidelines, toolkits and databases that facilitate experimental design and analysis. However, variation in strain background, chronological age, starvation protocols, and criteria to identify quiescent yeast cells has resulted in at least 48 possible experimental setups across published studies [28]. One commonly used approach induces quiescence through exhaustion of rich medium, resulting in gradual depletion of carbon the source. Exhaustion or abrupt starvation of other key nutrients (nitrogen, phosphate) also triggers a quiescent state, for which the specific cellular responses can differ. Importantly, the path used to trigger quiescence leads to very different functional outcomes, including gene expression profile, stress resistance, and lifespan [2]. For instance, exponentially growing cells abruptly starved from glucose will rapidly die, while cells from a stationary phase culture will survive in water for several weeks [29]. This is why quiescent cells resulting from the progressive exhaustion of rich medium are commonly used to study chronological aging [3]. It also emphasizes the importance of providing precise descriptions of experimental methods.
Work from the Werner-Washburne laboratory demonstrated that cells in stationary-phase cultures are heterogeneous and can be separated based on density. Most properties attributed to deep stationary-phase cells are, in fact, features of the dense fraction. This dense population is termed Q cells (or deep Q cells), whereas the less dense fraction is referred to as NQ cells (for non-Q). Q cells begin to differentiate as glucose levels decline [30,31], prior to the metabolic transition from fermentation to respiration known as the diauxic shift (DS). They are predominantly small G1-arrested daughter cells, whereas NQ cells are more heterogeneous in both size and cell-cycle phase. Q cells are also characterized by a thickened cell wall that enhances thermotolerance [29]. Notably, the relative abundance of Q and NQ cells varies across genetic backgrounds, contributing discrepancies in the literature. Strains with respiration defects, such as the commonly used BY background, produce lower yields of Q cells than W303 strains [32]. Auxotrophies—disruptions in key metabolic pathways introduced to facilitate genetic manipulation—also limit the ability of cells to enter the Q-cell state. It is therefore recommended to minimize auxotrophies and to culture cells under conditions that favor respiration [2].
Fission yeast (Schizosaccharomyces pombe).S. pombe is predominantly haploid and contains three chromosomes, with a genome size comparable to that of S. cerevisiae. Although these two yeast diverged early and differ substantially in genome organization, chromosome architecture, and regulatory pathways, they retain many conserved cellular processes; nonetheless, S. pombe offers complementary advantages as a quiescence model [16]. Quiescence in S. pombe is most commonly induced by nitrogen starvation. Starved cells undergo two more rounds of cell division without cellular growth and then arrest in G1, facing two possible outcomes: in the presence of opposite mating types, cells initiate a mating program that ultimately produces resilient spores. In contrast, heterothallic strains—which are fixed in one mating type and self-infertile—progressively enter the G0 state, in which they can remain viable for weeks or months in the presence of a carbon source [33]. Alternative conditions, such as glucose elimination or growth to saturation, cause G2 and G1 arrest, respectively, that resemble nitrogen starvation-induced quiescence but fail to support long-term cell survival [16]. Unlike S. cerevisiae, S. pombe does not have enzymes for the glyoxylate cycle required for ethanol metabolism and therefore does not undergo a diauxic shift (DS). An important experimental advantage of nitrogen starvation is that G0 entry and exit occur synchronously within the cell population [16,34].
2.2. Multicellular Models
Temporary entry into quiescent states in response to developmental or environmental cues is widespread in multicellular organisms, which are composed of complex tissues containing diverse cell types that must function in coordination. Quiescence programs can be executed by specific lineages, such as neuroblasts in Drosophila or the germline in C. elegans. Alternatively, quiescence can affect the entire organism, as exemplified by the C.elegans dauer diapause. These forms of quiescence have been extensively studied and are the subject of several recent reviews [35,36,37].
Adult stem cells. Many multicellular tissues harbor stem or progenitor cells that sustain tissue integrity under homeostatic conditions and, when required, mediate repair and regeneration [38]. In most cases, a subset of these cells remains in a dormant or quiescent G0 state and becomes activated only in response to defined physiological cues. These tissue-resident adult stem cells occupy specialized niches—microenvironments composed of neighboring cells, extracellular matrix components, and soluble factors—that protect them from cellular damage by restricting replication-associated and metabolic activity. Adult stem cells therefore constitute highly informative models for studying quiescence. They share characteristic features, including low RNA content, absence of proliferation markers, and frequent coexistence with more rapidly cycling progenitor pools within the same tissue compartment. Their behavior is tightly regulated by niche-derived signals, and disruption of these cues, for example during ageing, results in impaired stem cell activation and tissue decline. Across systems such as muscle stem cells (MuSC), hematopoietic stem cells (HSC), hair follicle cells (HFSC), and neural stem cells (NSC), quiescence is associated with a distinct epigenetic landscape. Among these, HSCs represent a paradigmatic example of adult stem cell quiescence. In adult mammals, HSCs reside in specialized bone-marrow niches, where a subset of long-term HSCs remains deeply quiescent for extended periods, dividing only rarely [39,40]. Lineage-tracing and label-retention studies indicate that these dormant HSCs contribute little to steady-state hematopoiesis, which is instead largely sustained by more actively cycling progenitors. However, in response to stresses such as inflammation or irradiation, quiescent HSCs reversibly re-enter the cell cycle and regenerate the hematopoietic system, underscoring quiescence as a protective yet readily deployable state [41,42,43]. This functional separation between dormant stem cells and active progenitors mirrors principles observed across adult stem cell systems, reinforcing their value as general models for reversible quiescence.
Dormant mouse embryo. Systemic dormancy in mammals is rare, occurring only in unfertilized oocytes and during early embryonic development. While adult stem cells maintain quiescence to support tissue homeostasis, repair, and regeneration, embryos employ dormancy as a reproductive strategy—either to optimize the timing of birth or to survive environmental challenges—a phenomenon known as embryonic diapause [44]. In the context of embryonic diapause, the period of dormancy can last from days to months, depending on the species [45]. In most cases, the embryo pauses at the blastocyst stage, delaying implantation, and further development. Documented to date in over 130 mammalian species, diapause remains a mystery due to the inaccessibility of embryos from most wildlife species. Mouse models have been instrumental in studying diapause, as it can be reliably induced through ovariectomy or hormone treatment [46,47]. Over the last decade, in vitro models of diapause have been developed and, together with advances in detection methods, have significantly propelled the field forward. For a detailed comparative analysis of dormancy in adult stem cells and embryos, readers are referred to [48].
Similar to unicellular organisms, the central cellular growth regulator mTOR has been shown to be a key factor controlling diapause in mammals [5,49]. Additionally, nutrient depletion, suppression of insulin signaling, Myc activity, and lipid metabolism have been documented to trigger a diapause-like response [5,50,51].
3. What Drives Transcriptional Reprogramming in Quiescent Cells?
3.1. Transcriptional Reprogramming During Metabolic Transitions in Yeast
As cells transition into quiescence, cellular processes shift from biomass production to maintenance. This is reflected in profound changes to the transcriptome and, to a lesser extent, the proteome. Global transcriptional repression results in a marked decrease in growth-related mRNAs, for example those encoding ribosomal proteins, while the relative expression of genes involved in nutrient uptake and scavenging increases. In S. cerevisiae, glucose deprivation results in an approximately 30-fold reduction in mean transcript abundance [52,53,54]. Similarly, measurements of absolute molecule numbers in fission yeast have revealed a global reduction in RNA levels upon nitrogen starvation, even when accounting for the smaller size of quiescent cells [55]. Recent studies have identified several mechanisms that likely act in concert to robustly limit mRNA production under these conditions.
In S. cerevisiae, quiescence is established over the course of several days, with quiescent (Q) cells exhibiting distinct transcriptional reprogramming. RNA polymerase II (RNAPII) distribution undergoes significant changes, characterized by reduced levels of both initiating and elongating forms of RNAPII [53]. Different studies have reported distinct RNAPII distributions, likely reflecting differences in the kinetics of quiescence entry arising from variations in growth conditions, such as aeration and medium pH [28,56]. Monitoring RNAPII over time revealed the evolution of RNAPII distribution during entry into quiescence [57]. Shortly after the DS (24 h of culture), RNAPII decreases in promoter proximal regions and accumulates near transcription end sites (TES), potentially reflecting regulatory changes in transcription termination [53]. RNAPII then accumulates slowly at upstream activating sequences of one third of coding genes in late quiescent cells [57,58,59]; intergenic enrichment correlated with fast resumption of transcription upon quiescence exit. As mentioned above, this decrease in RNAPII levels on chromatin accompanies a global transcription shut-down [52,53,54]. Nevertheless, the majority of transcripts remain detectable in mature Q cells, with 5,105 genes expressed compared to 6,205 in logarithmically growing (log) cells [53]. Notably, approximately 40% of these RNAs are found in an extraction-resistant compartment that is sensitive to protease treatment, indicative of sequestration in RNA-protein (RNP) complexes [53,60]. Indeed, two types of RNP granules are observed in Q-cells: processing bodies (P-bodies) and stress granules. While the former assemble after the DS upon PKA inactivation, the latter do not assemble until cells have reached the stationary phase [61]. Although most genes are shut down, approximately 3% of coding genes remain active, with seven detected in more than 80% of the cells in single-cell transcript analyses [57].
Similarly, S. pombe exhibits pronounced global repression of transcription during quiescence, with total mRNA levels reduced to approximately 15% of those in log cells [55]. This is accompanied by decreased levels of RNAPII engaged in transcription initiation and elongation [62]. Transcriptional reprogramming following nitrogen deprivation is dynamic, and distinct profiles are observed during the initial response (1–2 hr) and the establishment phase (10–24 hr) [63]. During establishment, several hundred genes, many of them involved in stress responses, detoxification, and recycling of amino acids and nucleotides, are transiently activated [55,63,64]. Once stable quiescence is achieved, these transiently induced genes are downregulated, and 97% of the genome becomes repressed. Only 16 ‘core quiescent genes’ remain highly expressed even after two weeks of nitrogen starvation [64].
3.2. Transcription Factor Dynamics Orchestrate Chromatin Remodeling
What then drives the extensive transcriptional reprogramming during quiescence entry? In S. cerevisiae, differential gene expression is coordinated by a network of transcriptional activators and repressors, whose activity is tightly regulated by the TOR and PKA nutrient-sensing pathways [2].
Global gene regulation through altered activities of transcriptional activators and repressors. In the presence of glucose, the C2H2-ZF transcription factors Mig1, Mig2, and Mig3 repress hundreds of genes. When glucose is depleted, Mig1 and M2 relocalize to the cytoplasm upon phosphorylation by the yeast AMPK Snf1, while Mig3 (and also Mig2) become transcriptionally repressed [2,65,66]. Upon the DS, the C2H2-family transcription factors Msn2 and Msn4 induce expression of a broad set of glucose-repressed genes through binding to specific stress-response elements (STREs) present in their promoter regions [2,67]. Msn2 also contributes to large-scale chromatin reorganization by promoting condensin recruitment to STRE-containing genes, thereby regulating higher-order chromatin structure (see 5.2). Msn2/4 activity is negatively regulated by the TORC1 and PKA pathways and positively regulated by the Rim15 protein kinase, which acts downstream of TORC1, through differential phosphorylation that controls nuclear localization of Msn2 and Msn4 [2,68].
Another layer of regulation is imposed by two transcriptional repressors, Xbp1 and Stb3. Xbp1 is transcriptionally induced by Msn2/4 during the DS and other forms of stress and becomes one of the most abundant transcripts in quiescent cells [2,31,69]. Its accumulation correlates with the frequency and duration of stress, suggesting that Xbp1 serves as a “stress memory” [2]. Xbp1 binds to specific promoter motifs present in over 500 genes, including the G1 cyclin CLN3 [2,31]. Stb3, in contrast, binds ribosomal RNA processing elements (RRPEs) and represses ribosomal biogenesis (RiBi) gene expression. Its recruitment to ribosomal protein gene promoters is inhibited by TOR-dependent phosphorylation, which prevents Stb3 nuclear localization [70,71]. Both repressors are intimately linked to dynamic changes in histone modifications.
Altered histone modifications and nucleosome density. Histone acetylation is typically associated with active transcription by modulating nucleosome structure and promoting the recruitment of effector proteins or ‘readers’. In addition, lysine methylation, occurring both at N-terminal tails and within core histone domains, is associated with transcriptional activation in budding yeast. Transcriptional repression in Q cells is accompanied by a global increase in histone density and a marked decrease in acetylation at multiple residues at histone H3 and H4 [2,52,54,72]. These chromatin changes are initiated early during the DS, particularly at ribosomal genes, but become fully established after six days in stationary culture. Although histone hypoacetylation occurs genome-wide, gene-specific patterns emerge: growth-related genes exhibit a pronounced decrease in acetylation, whereas stress-response genes upregulated in quiescence gain acetylation marks, indicating a tight correlation between histone acetylation and transcriptional activity [52].
Rpd3 is a key mediator of quiescence-specific chromatin remodeling. Stationary cells lacking the conserved class I histone deacetylase Rpd3 (HDAC1/2 in mammals) display histone acetylation levels and nucleosome density similar to proliferating cells or cells in early quiescence [52]. In log cells, Rpd3 primarily localizes to coding regions and is absent from promoters, whereas Rpd3 redistributes to the promoter regions of approximately 50% of genes during quiescence [52]. Promoter recruitment is mediated by Stb3 and Xbp1, each of them performing unique, non-redundant roles through targeting distinct sets of genes [52] (Figure 2). Xbp1 has been shown to physically interact with the Rpd3 complex [73], suggesting a direct targeting mechanism for histone deacetylation at specific promoters. Notably, while rpd3∆ cells do not show a major growth phenotype during logarithmic growth, deletion of RPD3 or XBP1 impairs the ability to enter quiescence and reduces chronological lifespan [52]. These findings support a critical role for Rdp3-mediated histone deacetylation in enabling transcriptional and chromatin remodeling as cells enter quiescence, although regulation of non-histone substrates cannot be excluded. Beyond its role in transcriptional repression, bulk histone deacetylation may also contribute to metabolic adaptation by generating free acetate for acetyl-CoA synthesis, thereby supporting energy production and anabolic processes under nutrient-limited conditions [74].
Restoration of histone acetylation during quiescence exit. Upon glucose addition, stationary S. cerevisiae cells reinitiate transcription in a few minutes and restore histone acetylation before DNA replication resumes, marking their exit from the quiescent state [54,72,75]. Histone H3 and H4 acetylation is mediated by the SAGA and NuA4 histone acetyltransferase complexes, respectively [75]. In particular, TOR signaling promotes NuA4 recruitment to ribosome protein gene promoters, ensuring that chromatin remodeling and transcriptional activation are tightly coupled to nutrient availability (for a detailed review, see [6]. The rapid re-initiation of transcription is largely dependent on the chromatin remodeling complex RSC that contributes to maintain NDRs upstream of specific genes and help the RNAPII to progress into gene bodies upon release [76]. The intergenic accumulation of RNAPII upstream of specific genes including RP genes was also proposed to contribute to their rapid and robust transcriptional activation within minutes of refeeding [57].
3.3. Establishment of Silent Chromatin Through Repressive Epigenetic Modifications
In quiescent S. cerevisiae cells, nearly half of the genome becomes targeted by Rpd3 [52], driving global histone deacetylation and transcriptional repression. While histone hypoacetylation characterizes quiescence across eukaryotes, the mechanisms guiding histone deacetylase recruitment differ substantially. Notably, neither Xbp1 nor Stb3 orthologs are found outside the Saccharomycetaceae clade, although functional analogs exist (e.g., the stress-responsive transcription factor Atf1 in S. pombe). Instead, fission yeast and many other fungi rely more heavily on repressive histone methylation, a mechanism broadly conserved across metazoans but absent in S. cerevisiae, to establish silent chromatin states.
Mechanisms of constitutive heterochromatin establishment in fission yeast. In S. pombe, silent chromatin is formed through locus-specific recruitment of the histone H3 lysine 9 (H3K9) methyltransferase Clr4, which is targeted through two complementary pathways [77]. Direct recruitment is mediated by sequence-specific DNA-binding factors, such as the heterodimeric transcription factor Atf1/Pcr1. RNA-guided recruitment involves the RNA interference (RNAi) machinery: Dcr1 (dicer) generates small interfering RNAs (siRNAs) that are loaded into Ago1, a core component of the RNA-induced transcriptional silencing (RITS) complex. Ago1-bound siRNAs guide RITS to nascent transcripts through base-paring, while the RNA-directed RNA polymerase Rdp1 amplifies siRNA production to reinforce the signal. Once recruited, Clr4 deposits H3K9me, which is recognized by Heterochromatin Protein 1 (HP1) family members (Swi6, Chp2) via their chromodomains. HP1 oligomerization, mediated by its chromoshadow domain, and additional Clr4 recruitment promote spreading of this histone mark across extended genomic regions. H3K9me is also recognized by RITS via its chromodomain protein Chp1, resulting in additional signal amplification and robust heterochromatin assembly. The resulting heterochromatin domains act as platforms for additional repressive factors, such as the class II HDAC Clr3. In log cells, this machinery establishes repressed chromatin at constitutive heterochromatin domains—pericentromeres, the silent mating type locus, rDNA, telomeres, and subtelomers—as well as at dispersed facultative heterochromatin islands [77,78].
Heterochromatin remodeling and function during quiescence. Emerging evidence indicates that heterochromatin factors play important roles in both quiescence entry and long-term viability during extended nutrient depletion. Multiple heterochromatin mutants display defects in G0 entry, and cells lacking Dcr1, Ago1, or Rpd3 show a progressive decline in viability following nitrogen starvation [17,63,79]. Reduced survival in G0 is also reported for mutants of the Clr6 HDAC complex—the S. pombe homolog of S. cerevisiae Rpd3L [17]— and for clr4∆ cells, although the severity varies across studies [63,79]. During quiescence entry, the heterochromatin landscape is extensively remodeled. Facultative heterochromatin is acquired at approximately 200 euchromatic loci across the genome, marked by de novo H3K9me deposition and accompanied by quiescence-specific siRNAs bound by Ago1 (Joh 2016; Figure 3). Many genes downregulated within 24 h of nitrogen starvation—including transcripts with functions in reproduction, carbohydrate metabolism, and transport—require Clr4 for silencing [63]. Consistent with the need to shut down protein biogenesis, the rDNA locus shows increased H3K9me levels and accounts for half of all siRNA production during quiescence, whereas other heterochromatin regions remain largely unchanged [63,79]. An exception are the subtelomeric regions that show a transient decrease in H3K9me levels, concomitant with the upregulation of several subtelomeric genes [63,80]. Consistently, among all surveyed histone methylation marks, both activating and repressive, H3K9me3 exhibits the most pronounced changes [62], underscoring the extent of heterochromatin remodeling.
Dcr1 functions beyond RNAi. While these findings support a key role for RNAi- and Clr4-dependent heterochromatin during early quiescence, genome-wide H3K9me remodeling at later stages, when nearly 97% of the genome is repressed [64], remain less well explored. Notably, Dcr1 performs an additional, RNAi-independent function at the rDNA locus that is linked to the release of stalled RNA polymerase I (Pol I) and is essential for long-term quiescence survival [79]. Cells lacking Dcr1 accumulate both Pol I and H3K9me at rDNA repeats at substantially higher levels than in WT during quiescence. Two non-mutually exclusive mechanisms may explain this H3K9me accumulation: First, stalled Pol I may directly recruit Clr4, in analogy to the recruitment of G9a by Pol I in mammalian cells [81]. Second, elevated H3K9me levels at rDNA repeats may arise through an indirect mechanisms in dcr1∆ cells. Specifically, this increase may result from the redistribution of silencing factors following their release from genomic regions that strictly depend on RNAi. This phenomenon has been documented in log cells, where loss of RNAi at one locus leads to enhanced heterochromatin formation at other genomic sites [82,83,84]. The viability defects of dcr1∆ can be rescued by mutations that impair heterochromatin formation (clr4∆, swi6∆) or by defects in Pol I recruitment or stability. Importantly, clr4∆ fails to suppress Pol I accumulation in dcr1∆, yet mutations that impair Pol I recruitment and stability nonetheless reduce H3K9me levels in this mutant [79]. This suggests that Pol I retention alone is insufficient to account for the viability defect. Rather, although heterochromatin normally accumulates at rDNA during quiescence, excessive H3K9me levels—such as those associated with heterochromatin redistribution or Pol I retention—are deleterious for survival and therefore require tight regulation.
Diverse repressive histone methylation patterns in metazoans. Similar regulatory principles, including transcriptional repression and histone hypoacetylation, operate in higher eukaryotes during quiescence, yet the accumulation and function of repressive histone marks vary substantially across cell types and physiological contexts. As in fission yeast, H3K9 methylation in metazoans is closely associated with constitutive heterochromatin at pericentromeric repeats and other repetitive elements. However, H3K9 methylation exhibits no uniform pattern during quiescence: several quiescent cell types, such as hair follicle stem cells and adult fibroblasts, show reduced H3K9me2/3, whereas others display moderate increases or little detectable change [72]. A similar heterogenous behavior is observed for H3K27me3. In some quiescent cell types, including fibroblasts, B lymphocytes, and hair follicle stem cells, H3K27me3 levels decrease together with reduced polycomb repressor complex 2 (PRC2) depositing this mark, whereas muscle stem cells maintain low promoter H3K27me3 in quiescence but gain this mark upon activation [72,85]. These divergent patterns likely reflect differential use of PRC2 paralogs, with EZH1 prevailing in quiescent cells and EZH2 associated with proliferative states [72,86].
In contrast, H40K20me3 displays a more consistent association with quiescence. H4K20me3 is a repressive mark found in metazoans at pericentromeric heterochromatin and other repetitive elements (including transposable elements and subtelomeric regions) but is absent in S. cerevisiae and S. pombe. During quiescence, H4K20me3 levels robustly increase across constitutive heterochromatin and can extend into facultative domains, contributing to the establishment of a deeply repressed state [72]. Primary human fibroblasts accumulate global levels of H4K20me2 and H4K20me3 during quiescence in association with enhanced chromatin compaction [87], and similar increases occur in serum-deprived mouse NIH 3T3 mouse fibroblasts, where H4K20me3 is enriched at rDNA and transposable elements [88]. Elevated H4K20me3 is likewise observed in quiescent muscle stem cells (MuSCs) compared with proliferating MuSCs [89]. Consistent with a broader role for H4K20me3 in deeply arrested cellular states, drug-tolerant persister cancer cells exhibit marked enrichment of H4K20me3 at promoters of stress-responsive genes, reinforcing transcriptional repression and supporting their low-activity, immune-evading phenotype [90].
Dynamic remodeling of epigenetic landscapes during mammalian embryonic diapause. Pluripotent cells in the early embryo, along with embryonic stem cells (ESCs), sustain highly adaptable transcriptional programs that maintain their undifferentiated state through a balance of global permissiveness and localized, Polycomb-mediated repression. Entry into dormancy during mouse embryonic diapause triggers a rapid, genome-wide decrease in transcriptional output [5,91] mirroring the suppression of other energy-intensive anabolic processes such as translation. The overall decrease in cellular activity during this state is accompanied by a loss of transcription-associated histone marks, such as H4K16ac and H3K9ac and increased accumulation of heterochromatin at the nuclear lamina in pluripotent epiblast cells[5,92,93,94]. DNA methylation, which is almost completely erased in the proliferative epiblast, likewise globally increases in the diapaused epiblast [95]. Despite this repressive epigenetic landscape that significantly diverges from the canonical state of proliferative epiblast cells, the diapaused epiblast retains the transcriptional signatures and functionality of naïve pluripotency [5,50]. Although many mechanisms that ensure fidelity of cell identity remain to be discovered, several signaling pathways have been shown to protect the epiblast from premature differentiation or cell death during diapause, including Wnt, LIF/Stat, Nodal/Smad, and Yap/Taz pathways [91,96,97]. In addition, TET DNA demethylases, in cooperation with the transcription factor TFE3, have been shown to specifically target cis-regulatory elements and maintain them in a lowly methylated state despite an overall increase in global DNA methylation [95]. Taken together, multiple regulatory layers from cellular signaling to targeted epigenetic maintenance ensure transcriptional fidelity in the face of increased genomic repression in dormant states.
3.4. Persistence and Redistribution of Active Histone Methylation Marks
In contrast to the drastic reduction in histone acetylation in S. cerevisiae, methylation marks associated with active transcription (H3K4me3, H36me3, and H3K79me3) largely persist when cells enter quiescence [2,53,54]. Genome-wide profiles of these marks correlate well between quiescent and log-phase cells, yet they differ at specific loci. In particular, all three methylation marks are strongly increased at stress-induced genes early in quiescence, coinciding with a sharp rise in transcription; in contrast, growth-related genes that are transcriptionally repressed exhibit a 50% decrease in H3K4me3 levels, without a comparable reduction of H3K36me3 or H3K79me3 [53]. As cells exit quiescence, histone methylation shifts from stress-related genes to growth genes, yet this redistribution exhibits slower kinetics than the rapid acetylation bursts and occurs only after DNA replication has resumed [54].
Notably, the histone methyltransferases responsible for H3K4me and H3K36me—Set1 and Set2, respectively—are both downregulated in Q cells, suggesting these histone marks are deposited during logarithmic growth or early in quiescence [53]. Histone demethylases remain expressed in Q cells but may be less active, and decreased replication-dependent and transcription-coupled nucleosome turnover may further contribute to the persistence of these marks [53]. Thus, rather than serving as direct indicators of transcriptional activity, these histone methylation marks may help maintain a chromatin environment that remains permissive to transcription [53,54].
Additional evidence from S. pombe further supports the view that H3K4me3 becomes selectively redistributed rather than globally erased during quiescence. Although H3K4me3 is broadly retained, its promoter-proximal peak is markedly diminished in quiescent cells, accompanied by extensive locus-specific remodeling: after 24 hours of nitrogen starvation, hundreds of genes show multi-fold increases or decreases in H3K4me3, even though most promoters lose the sharp TSS-centered peak characteristic of log cells [62]. A subset of quiescence-induced genes retains or gains H3K4me3 together with elevated levels of elongating RNAPII, particularly genes encoding metabolic enzymes, membrane transporters, and noncoding RNAs. Notably, H3K4me3 becomes enriched at ‘core quiescence’ genes, with several of them residing in subtelomeric regions. Their transcriptional induction partially depends on Set1, consistent with genetic evidence that Set1C/COMPASS components are required for efficient quiescence entry [17,62,64]. A comparable pattern is observed in quiescent adult stem cells of higher eukaryotes. However, unlike embryonic stem cells, adult stem cells display limited bivalent H3K4me3/H3K27me3 marks but retain widespread H3K4me3 at promoters, thereby maintaining a transcriptionally poised state that enables rapid activation upon exit from quiescence [38,72]. Together, these findings indicate that, despite global reduction of promoter-centered H3K4me3, the targeted retention and redeployment of this mark contribute to establishing a transcriptionally competent yet selectively reprogrammed chromatin landscape in quiescent cells.
H3K79me3: a timer for the duration in quiescence? While H3K79me3 remains detectable or even increases in quiescent cells, levels of H3K79me1 and H3K79me2 are strongly reduced, suggesting their conversion to the fully methylated state [53]. Intriguingly, this decrease is not observed in less dense phase of non-quiescent (NQ) cells, which are unable to reenter the cell cycle after prolonged quiescence when nutrients are restored, implying a Q cell-specific regulatory mechanism that remains unidentified [2,53].
3.5. Epigenetic Reprogramming of Metabolic Genes Through Altered Histone Dynamics and Nucleosome Remodeling
During quiescence establishment, numerous genes become transiently or persistently upregulated in S. pombe [17,55,63,64,80]. Many of these genes are located in subtelomeric regions and encode vacuolar transmembrane transporters essential for recycling amino acids and hexoses during the metabolic shift of quiescent cells. Multiple genetic studies converge on the conclusion that this transcriptional activation depends on regulated histone exchange and nucleosome remodeling, processes that remain active even when DNA replication—and thus replication-coupled histone turnover—is halted.
Ino80C mediates removal of histone variant H2A.Z. A major component of this regulatory network is the Ino80C chromatin-remodeling complex (Ino80C), which repositions nucleosomes and catalyzes the removal of the histone variant H2A.Z (Pht1). Loss of Ino80C subunits, such as Iec1 (ino eighty complex subunit 1), compromises survival during quiescence and induces premature, global transcriptional downregulation [17,64]. Accordingly, genes normally upregulated at quiescence onset in WT cells fail to be activated in iec1∆ and other Ino80C mutants. Concordantly, whereas H2A.Z is normally depleted from subtelomeric regions during quiescence entry, it remains aberrantly enriched in iec1∆, particularly at boundary elements that contain multiple long terminal repeat (LTR) sequences [64]. These findings imply that Ino80-mediated eviction of H2A.Z promotes the formation of a transcriptionally competent subtelomeric environment, facilitating activation of metabolic genes required for the establishment of the quiescent state.
This dependency is further modulated by inositol polyphosphate signaling: the inositol kinase Asp1, which influences Ino80C activity, is likewise required for survival under prolonged quiescence [64,98], underscoring the integration of metabolic cues with chromatin remodeling at this transition. Conversely, the H2A.Z-specific histone chaperone Swr1C is dispensable for viability during quiescence, indicating that H2A.Z removal, rather than its deposition, is critical at this stage [17]. Nonetheless, loss of H2A.Z itself also causes reduced viability [64], suggesting a distinct, Ino80C-independent for this variant.
Paf1C and histone turnover contribute to epigenetic reprogramming. The Paf1 complex (PafC) promotes histone exchange during vegetative growth and counteracts the accumulation of H3K9 methylation at constitutive and facultative heterochromatin [99]. Quiescent cells lacking Leo1 or other PafC components exhibit substantial viability defects [80], highlighting the importance of ongoing, DNA replication-independent histone replacement. In WT cells, heterochromatic H3K9me2/3 levels transiently decline during quiescence entry, particularly at subtelomeric regions; this decrease is attenuated in leo1Δ, resulting in elevated H3K9me and reduced expresino of transporter genes required for metabolic adaptation [80]. Conversely, other gene clusters, including those involved in DNA repair and chromatin organization, become aberrantly upregulated, reflecting broader disruptions to transcriptional control.
Genetic and pharmacological evidence further positions Leo1 downstream of the TORC2–Gad8Akt pathway: TORC2 inhibition by torin induces transcriptional changes that depend on Leo1, while Gad8Akt protein abundance transiently decreases during quiescence entry [100]. These relationships suggest that TORC2 signaling modulates PafC-dependent histone turnover to enable epigenetic reprogramming of metabolic pathways, particularly during the early phase of quiescence establishment [80]
Together, these findings illustrate that histone variant dynamics and replication-independent histone turnover are central determinants of gene regulation during quiescence. Ino80C-dependent H2A.Z eviction and Paf1-mediated histone replacement act in concert to remodel subtelomeric chromatin, permit activation of key metabolic genes, and maintain viability during long-term quiescence.
4. Post-Transcriptional Mechanisms Shape Quiescence-Specific Gene Expression
4.1. Intron Retention Regulates Protein Biogenesis Genes
Transcriptional adaptations during quiescence are accompanied by significant changes in co-transcriptional RNA processing, particularly splicing [101]. Although S. cerevisiae has largely lost splicing capacity during evolution, retaining introns in only 5% of its genes, introns are nevertheless strikingly preserved in ~73% of ribosomal protein genes (RPGs). During amino acid starvation, but not in response to other stress conditions, splicing of RPG mRNAs is selectively repressed, and strains lacking these introns exhibit reduced fitness during prolonged starvation [102,103,104]. This suggests that regulated intron retention within RPGs functions as a post-transcriptional mechanism to attenuate ribosome biogenesis, thereby limiting energy expenditure as cells enter quiescence. Notably, a subset of excised introns accumulates as a stable, linear form in saturated S. cerevisiae cultures or after TORC1 inhibition. These stable introns associate with spliceosomes and may themselves contribute to reduced splicing activity under starvation conditions [105]. A subset of stress-responsive genes escapes splicing inhibition and is even more efficiently spliced under starvation conditions, correlating with their induction. These transcripts selectively recruit auxiliary splicing factors, including the U1-associated proteins Nam8 and Mud1, to promote 5’ splice site recognition and intron removal [103].
In S. pombe, where splicing is more prevalent (~46% of genes contain introns), transcriptome profiling after 24 hours of nitrogen starvation reveals a global increase in intron retention, with RPGs again being particularly affected [55]; (Figure 4). This conservation across evolutionarily divergent yeast species indicates that splicing repression of RPGs represents a fundamental regulatory mechanism in quiescence establishment. Consistently, widespread intron retention is also a hallmark of mammalian quiescent adult stem cells (QSCs), with enrichment among transcripts encoding proteins involved in translation and splicing [106,107]; diapaused embryos likewise exhibit reduced splicing activity [49]. Functionally, intron retention appears to be essential for maintaining quiescence: activation of muscle QSCs requires quiescence exit, which can be blocked by removing the chromatin-associated factor Dek that mediates intron removal [106].
Beyond population-level changes, RPG splicing can also vary among individual cells within populations, creating phenotypic heterogeneity with distinct consequences for adaptation to stress. In S. cerevisiae, the RPG Rps22B exhibits a bimodal splicing pattern, with different subpopulations displaying different survival outcomes during starvation: cells with intron retention (low levels of Rps22B) show enhanced survival during prolonged starvation, while cells with efficient intron removal (high levels of Rps22B) resume growth more quickly after transient starvation [108]. Stochastic cell-to-cell variation may represent a bet-hedging strategy that exploits phenotypic diversity to maximize fitness in unpredictable environments. Remarkably, regulated splicing of an intron in the 5’ untranslated region (5’-UTR) is conserved in the homologous S. pombe gene, rps2202, where it activates a post-transcriptional decay mechanism that is specifically inactivated during nitrogen starvation in homothallic yeast [109,110].
4.2. 3’-UTR Lengthening Expands the Repertoire of Post-Transcriptional Regulation
Quiescence is characterized by widespread shifts in polyadenylation site (PAS) selection. While proliferating cells preferentially use proximal PAS and thereby generate transcripts with short 3’ untranslated regions (3’-UTRs), non-proliferating cells (including both differentiated and Q cells) more commonly use distal PAS, resulting in global 3’-UTR lengthening [111,112]. This shift toward distal PAS usage appears to be a conserved feature of quiescent states across eukaryotes, and has been documented in S. pombe and S. cerevisiae grown under nutrient-poor conditions [113], in S. pombe after 24 h and 7 d of nitrogen starvation [114], or upon rapamycin treatment [115].
Molecular mechanisms driving 3’-UTR extension. Kinetic coupling between transcription and 3’-end processing renders PAS selection highly sensitive to both the transcription elongation rate and the availability of the 3’-end processing machinery. Slow transcription and abundant 3’-end processing factors favor proximal PAS usage and 3’-UTR shortening, whereas faster elongation or reduced processing factor availability shifts selection towards distal sites [116,117]. Accordingly, chromatin changes that modulate elongation kinetics, together with the reduced expression of RNA cleavage and polyadenylation factors in Q cells, are expected to shift the equilibrium towards distal PAS usage and 3’-UTR lengthening [55,63,107]. Emerging evidence indicates that nutrient-sensing pathways help coordinate these shifts in PAS usage. In S. cerevisiae, rapamycin-induced alternative PAS selection requires the methyltransferases Set1 and Set2, linking TOR inhibition to transcription-dependent histone modifications (see above; [115]. In mammalian cells, mTOR activation also drives 3’-UTR shortening [118].
Regulatory consequences of 3’-UTR extension. Extended 3’-UTRs typically harbor additional binding sites for RBPs and microRNAs, thereby increasing the potential for post-transcriptional control. In differentiated cells, this expanded regulatory landscape generally attenuates gene expression by enhancing opportunities for transcript destabilization or translational repression. Whether this paradigm also applies to Q cells, however, remains unclear. RNA stability measurements in a dermal fibroblast model of quiescence revealed stabilization, rather than destabilization, of transcripts with extended 3’-UTRs [107], suggesting that Q cells may exploit 3’-UTR lengthening to achieve regulatory outcomes distinct from those observed in differentiated tissues. This observation aligns with recent analyses in S. cerevisiae showing that the impact of 3'-UTR isoforms on mRNA half-life is highly condition-dependent, with transcripts becoming progressively more stable when cells divide more slowly [119].
Suppression by microRNAs. In metazoans, microRNAs (miRNAs) provide an important layer of post-transcriptional control acting at the level of 3’-UTRs. These small RNAs function exclusively as repressors, either suppressing translation or inducing RNA cleavage in a sequence-specific manner after incorporation into RNA-induced silencing complexes (RISC) [120]. Interestingly, hypoxia-inducible factor 1α (HIF-1 α), a transcription factor involved in the generation and maintenance of quiescent cancer stem cells developing in hypoxic niches [121], appears to additionally dampen miRNA-dependent regulation. HIF-1α sequesters the miRNA processing component Dgcr8 and prevents its incorporation into microprocessors, thereby limiting miRNA production in the cancer stem cell niche [122]. However, evidence from multiple other mammalian quiescence models suggests that miRNAs actively regulate quiescent states. In muscle QSCs, for example, the quiescence-specific miRNA-489 maintains quiescence by repressing Dek along with other targets [123]. Similarly, entry into embryonic diapause involves extensive reprogramming of miRNA expression [49,124,125]. The upregulation of diapause-associated miRNAs is partly controlled by the nutrient-responsive transcription factors Tfe3/TfeB, which act downstream of mTOR signaling. Upon mTOR inhibition, these factors partially translocate to the nucleus and promote miRNA expression (Iyer et al. 2024). While miRNAs are dispensable for normal blastocyst development, they are critical for initiating diapause, evidenced by the inability of embryos and embryonic stem cells lacking Dgcr8 to enter dormancy [49,126]. One key miRNA, let-7—originally identified as a regulator of developmental timing in C. elegans—is upregulated during diapause and plays a role in modulating embryo implantation [124,127]. Let-7 inhibits Myc, mTOR signaling, and polyamine biosynthesis, thereby promoting a diapause-like state [127]. Given that embryonic diapause is predominantly regulated by maternal cues, it has been proposed that endometrial extracellular vesicles—key mediators of communication between the uterus and the embryo—deliver let-7 miRNA to the blastocyst to help establish the dormant state [127].
4.3. Regulatory Non-Coding RNAs Facilitate Rewiring of Gene Expression
A striking and conserved feature across diverse quiescence models is the widespread upregulation of non-coding RNAs (ncRNAs). Transcriptome-wide analyses in human somatic quiescence have identified numerous upregulated long non-coding RNAs (lncRNAs), and elevated ncRNA expression is similarly observed in both budding and fission yeast [55,56] (Figure 5A). This conservation across evolutionarily distant organisms suggests that ncRNA accumulation represents a fundamental feature of the quiescent state, rather than a species-specific adaptation.
What drives ncRNA accumulation in quiescence? Beyond transcriptional changes, reduced RNA decay rates are thought to contribute to the distinct lncRNA profiles observed in Q cells. In proliferating cells, nuclear RNA stability depends largely on susceptibility to degradation by the nuclear exosome, a conserved exonucleolytic complex central to RNA surveillance that is guided to its substrates by a modular system of targeting factors [128]. In Q cells, levels of many exosome substrates are substantially increased (Figure 5B). Three non-mutually exclusive mechanisms have been proposed to explain this phenomenon: (i) reduced exosome expression or activity (in fission yeast, exosome mRNA levels decrease to ~ 50%; Joh et al); (ii) altered exosome targeting, either through inactivation of specific targeting pathways or activation of quiescence-specific mechanisms (e.g., HIF1α-dependent retargeting of microprocessor protein Dgcr8 to the exosome [122,129]; or (iii) substrate competition, where retargeting of the exosome to a broader substrate pool, including incompletely processed mRNAs with retained introns or extended 3’ ends (see above), creates kinetic competition that stabilises ncRNAs [130,131].
Although the regulatory potential of quiescence-associated lncRNAs is clear, only few have been functionally characterized, sometimes in contexts unrelated to quiescence. LncRNAs, or the act of their transcription, can regulate cellular functions through various mechanisms, or a combination of them, including:
Recruitment of epigenetic modifiers. Some lncRNAs recruit chromatin-modifying enzymes to specific loci through direct interaction or via RNA-binding protein (RBP) intermediaries, establishing epigenetic signatures that regulate quiescence programs (Figure 6). This mechanism is conserved from yeast to mammals. In fission yeast, the lncRNA prt recruits H3K9 methyltransferase Clr4 to silence the adjacent pho1 locus under phosphate-replete conditions but is transcriptionally downregulated when cells are starved for phosphate [133]. This recruitment involves a sequence-specific RBP and a scaffold protein mediating the interaction [78]. In metazoans, similar lncRNA-dependent targeting mechanisms regulate chromatin compaction during quiescence. While the histone H4K20 methyltransferase Suv4-20h2 is typically recruited to constitutive heterochromatin in an HP1-dependent manner, quiescent mouse cells employ an alternative HP1-independent mechanism. Quiescence-induced lncRNAs produced in cis at ribosomal RNA repeats and other repetitive regions recruit Suv4-20h2, potentially through direct interaction with the enzyme, promoting chromatin compaction at repeat regions [88] (Figure 6).
ncRNA-mRNA interactions. Other lncRNAs modulate activity of individual target mRNAs through base-pairing interactions, leading to a range of potential outcomes. The lncRNA NR2F1-AS1, for example, is significantly upregulated in breast cancer stem-like cells, a type of cells known to exhibit metastatic dormancy in the lungs. NR2F2-AS1 functionally contributes to establishing or maintaining the quiescent state—its knock-down leads to increased proliferation [134]. Mechanistically, NR2F1-AS1 binds an inhibitory region within the 5’-UTR of NR2F1 mRNA, facilitating recruitment of the polypyrimidine tract-binding protein PTBP1 and thereby promoting translation of the transcription factor NR2F1. Similarly, the fission yeast lncRNA aal1 (ageing-associated lncRNA 1), has been suggested to suppress proliferation by interacting with mRNAs that encode ribosomal proteins, limiting their expression and decreasing the cellular ribosome content [135] (Figure 6).
Architectural lncRNAs and condensate nucleation. Many lncRNAs are large, structurally flexible molecules capable of interacting with multiple proteins through various binding sites. Such multivalent interactions can result in the formation of biomolecular condensates with high densities of RBPs that spatially or temporally regulate RNA-dependent processes [136] (Figure 6). The protein components of nuclear condensates often contain intrinsically disordered low-complexity regions that contribute to the network of multivalent interactions. A notable example of such condensates are paraspeckles—mammalian-specific nuclear structures that require a specific RNA scaffold, NEAT1 lncRNA, for their formation—which sequester specific RBPs and RNAs [137]. In S. pombe, certain quiescence-associated lncRNAs, such as omt3 [55] (Figure 5A), form condensates at the non-coding gene locus. During meiosis (when these lncRNAs are also induced), such condensates facilitate the pairing of homologous chromosomes through a tethering mechanism [138]. The proteins involved in these condensates are components of the RNA 3’-end processing machinery that, in log-phase cells, localize to cleavage bodies—structures thought to buffer nucleoplasmic concentrations of 3’-end formation factors and not known to be associated with lncRNAs [139]. Their behavior in Q cells remains uncharacterized.
Regulation through lncRNA transcription. The act of lncRNA transcription - rather than the lncRNA product itself - can strongly influence the expression of adjacent genes through distinct and context-dependent mechanisms. In transcriptional interference, passage of RNAPII through a promoter region can displace transcription factors necessary for initiation, effectively suppressing gene expression [140] (Figure 6). This repressive mechanism operates independently of gene orientation, and can occur when the lncRNA gene and the regulated gene are oriented in tandem or when they are in an antisense configuration. Silencing of the S. pombe pho1 locus by prt lncRNA also depends on this mechanism [133]. Transcriptional interference appears to be widely deployed during quiescence: in S. cerevisiae, even short periods of glucose or phosphate starvation induce distinct sets of antisense lncRNAs (asRNAs) [141,142]. In S. pombe, asRNAs are among the most strongly upregulated ncRNAs in Q cells and often accumulate to high levels [55,143]. This elevated expression typically coincides with downregulation of the corresponding sense gene, illustrating the regulatory impact of transcriptional interference (Figure 5A). Conversely, lncRNA transcription can facilitate gene activation through promoter remodeling. When RNAPII traverses compacted chromatin, transcription itself disrupts nucleosome organisation, rendering previously inaccessible promoters competent for transcription factor binding. At the S. pombe fbp1 locus, for example, glucose starvation induces cascading lncRNA transcription, which precedes and enables activation of fbp1 mRNA transcription [144]. Such pioneering lncRNA transcription, which primes chromatin for productive mRNA synthesis, has been observed at other genes responding to nutritional signals, and may represent a widespread mechanism for transcriptional reprogramming during quiescence entry and exit [145].
5. Reorganisation of Nuclear Structures and Genome Architecture
5.1. Biosynthetic Condensates
Nucleolar condensation as a conserved marker of quiescence. The biogenesis of core components of the gene expression machinery is spatially organized in specialized nuclear condensates whose morphology reflects cellular biosynthetic activity. Nucleoli serve as hubs for ribosome maturation, while spliceosome and small nucleolar ribonucleoprotein (snoRNP) production occurs in distinct compartments (see below). During quiescence, nucleoli undergo dramatic reorganization, condensing into compact structures as ribosomal RNA (rRNA) synthesis declines with growth arrest. This condensation is a highly conserved hallmark of quiescent cells, observed across phylogenetically distant organisms and triggered by different cues: in S. cerevisiae following DS [146]; in S. pombe cells during nitrogen starvation [33,147]; in C. elegans under insulin pathway inhibition [148]; and in mouse embryos during diapause [94]. This reorganization is reversible: upon exit from diapause, nucleoli decondense within 12 hours, coinciding with restored biosynthetic activity in the reactivating embryos [92,94]. To date, the molecular composition of condensed nucleoli remains largely unexplored. Ultrastructural analysis of quiescent S. pombe nuclei (nitrogen-starved for 24 h) revealed an accumulation of multi-mega Dalton molecular complexes in the condensed nucleolus [147], but their nature and functional role remain uncharacterised.
Cajal bodies coordinate small RNA biogenesis with growth state. Cajal bodies are nuclear structures essential for the maturation of small nuclear RNAs (snRNAs) and small nucleolar RNAs (snoRNAs) [137]. These RNAs are highly abundant components of the gene expression machinery: snRNAs form the catalytic core of the spliceosome, while snoRNAs guide the chemical modification of rRNA required for ribosome biogenesis. Because their production contributes significantly to the biosynthetic load, snRNA and snoRNA biogenesis is tightly coordinated with nutrient availability. In fission yeast, the Cajal body scaffold protein Mug174 (the orthologue of metazoan coilin) is required for viability during G0 and the subsequent return to proliferation, indicating that long-term survival depends on Cajal body-mediated coordination of small ribonucleoprotein biogenesis with the cellular growth state [149]. The molecular mechanisms underlying this nutrient-responsive regulation are better characterized in mammalian systems, where TOR signaling plays a central role: the survival of motor neurons (SMN) protein, responsible for assembling sn(o)RNPs from sn(o)RNAs and Sm proteins, is a direct mTOR target. Cajal body localization of SMN – which reflects its activity – is regulated by mTOR-dependent phosphorylation, providing a direct link between nutrient sensing and snoRNP biogenesis [150]. Intriguingly, not all organisms employ Cajal body-based regulation. S. cerevisiae lacks orthologues of coilin or SMN, and the site and mechanism of sn(o)RNP assembly in budding yeast remain unresolved. Assembly may occur diffusely within the nucleus, without discrete compartmentalization, raising the question of which alternative mechanisms coordinate small RNA biogenesis with nutrient availability in this organism.
5.2. Changes in Local and Higher-Order Chromatin Structures
Quiescence induces profound reorganization of genome architecture at both the local chromatin level and the genome-wide scale, resulting in a dramatic genome compaction and nuclear volume decrease across species [2]. In S. cerevisiae, these changes arise through two complementary mechanisms involving histone tail–dependent chromatin fibre compaction and condensin-mediated chromosome looping. Together, these processes generate a deeply compact, insulated, and transcriptionally restrained nuclear architecture tailored to the low-energy state of quiescent cells.
Local chromatin remodeling through chromatin fiber compaction. During G0 in budding yeast, neighboring nucleosomes engage in interactions dependent on the basic patch of the H4 histone tail, a process facilitated by global hypoacetylation of H4 [151]. This mode of fibre compaction is thought to broadly dampen transcription, largely independent of higher-order domain architecture. While mechanistically distinct, similar principles apply in human quiescent fibroblasts, where chromatin compaction is associated with increased H4K20me2/3 levels [87]. In S. cerevisiae, an additional contributor might be the histone H1 homolog Hho1. Hho1 chromatin occupancy is inversely correlated with gene expression, and it binds more strongly to chromatin in stationary-phase cells, with its loss leading to chromatin decompaction [152]. Notably, this decompaction does not result in widespread transcriptional derepression, indicating that chromatin compaction alone is not sufficient to enforce gene silencing [2,152].
Global chromosome reorganization by condensin. At a global scale, the quiescent genome is reorganized into a highly ordered structure defined by long-range interactions and condensin-mediated looping. Hi-C analyses of quiescent S. cerevisiae cells reveal a shift toward a more compact nuclear organization characterized by increased intrachromosomal contacts, reduced centromere clustering, and enhanced telomere–telomere interactions [146,153,154,155]. At the mesoscale, chromatin is organized into chromosomal interaction domains (CIDs), small genomic units encompassing 1–4 genes (0.5–8 kb) that engage in frequent local interactions and resemble metazoan topologically associating domains [156]. Superimposed on this architecture are larger chromatin interaction domains (L-CIDs) spanning 10–60 kb, which are present in both log and Q cell and whose boundaries remain largely unchanged. However, during quiescence, L-CIDs become more compact and topologically isolated, accompanied by enhanced loop formation [155]. Together, these global rearrangements indicate a transition to a condensed yet orderly chromatin state that is distinct from both log and stationary-phase cells.
A critical determinant in this process is the condensin complex, a ring-shaped protein complex that organizes higher-order chromosome structure and mediates sister chromatid cohesion [153,155]. During quiescence entry, condensin redistributes from its canonical locations at rDNA and centromeres to the 5′ ends of hundreds of stress-induced genes, aligning with L-CID boundaries. This relocalization is partially dependent on the transcription factor Msn2, which is involved in the activation of many stress-induced genes during the DS (see above). How Msn2 contributes to condensing binding is not well understood, but a role of transcription itself at Msn2-responsive genes has been discussed [59]. Loss of condensin in G₀ leads to decompaction of L-CIDs, a reduction in loop formation, increased inter-domain contacts, and transcriptional derepression of nearby genes [155], underscoring its role as an architectural organizer that enforces domain insulation during quiescence. How L-CID boundaries are established, which occurs cohesin-independently in log cells, remains unresolved.
5.3. Telomere Reorganization
Telomere hypercluster formation in S. cerevisiae. In budding yeast, telomere organization undergoes extensive remodeling during metabolic transitions. Following the diauxic shift, telomere foci become fewer and brighter while remaining associated with the nuclear periphery. Upon complete carbon source exhaustion and entry into stationary phase, telomeres undergo a more pronounced reorganization in long-lived quiescent cells (Q cells), coalescing into a large hypercluster positioned at the nuclear center [146]. This configuration is reversible: telomere hyperclusters are rapidly dismantled within 15 to 30 minutes upon return to growth, in a transcriptional-independent manner [57]. Importantly, telomere hypercluster formation correlates with survival upon quiescence exit and is specific to Q cells, as telomere organization remains unchanged in non-quiescent (NQ) cells [146].
The extent of telomere hyperclustering varies markedly between strain backgrounds. W303 strains exhibit robust hyperclustering, whereas BY strains display a strongly reduced capacity to form hyperclusters [154,157]. Although early work proposed that elevated Sir3 levels might account for this difference [154], this model has been excluded: moderate Sir3 overexpression (2–3.5-fold) in BY fails to rescue the telomere hyperclustering defect, and Sir3 protein levels remain stable across metabolic transitions in both backgrounds [146,157]. Instead, reduced hyperclustering in BY strains likely reflects respiration defects [158], consistent with respiration being a key requirement for telomere hypercluster formation [146]. Notably, Sir3 post-translational modifications, including sumoylation and phosphorylation, are dispensable for telomere hyperclustering in quiescent cells [157].
Recent physical modeling and genetic analyses identified telomere anchoring as a key negative regulator of telomere hyperclustering [157]. The inner nuclear membrane protein Esc1 acts as a major telomere anchor upon glucose exhaustion; in esc1∆ cells, telomere hyperclusters form prematurely, indicating that Esc1-mediated anchoring counteracts telomere clustering. Under glucose-replete conditions, however, hyperclusters do not form in esc1∆ cells, implying the existence of an Esc1-independent anchoring mechanism. This secondary anchoring pathway is regulated by glucose signaling, as glucose starvation or inactivation of the Ras/PKA pathway triggers hyperclustering in esc1∆ cells [157]. In wild-type Q cells, exhaustion of all carbon sources coincides with hypercluster formation, consistent with inactivation of Esc1 anchoring, whereas in NQ cells Esc1 remains active and prevents hypercluster formation [157].
Mechanistically, the Esc1-dependent anchoring pathway relies on phosphorylation of Esc1 at serine 1450, which enables interaction with the HBRCT domain of Sir4 [159,160]. Mutation of this residue phenocopies Esc1 loss, suggesting that reversible Esc1 phosphorylation controls telomere anchoring during metabolic transitions, although the upstream signaling cascade remains unknown [157].
Together, these findings indicate that budding yeast has evolved a fine-tuned mechanism to regulate telomere positioning in response to nutrient availability. While telomere hyperclustering promotes cell survival following exit from quiescence independently of transcriptional silencing, its precise protective function remains unknown [146]. Hyperclustering may enhance genome stability by protecting chromosome ends from degradation, end-to-end fusions, and aberrant recombination events. Alternatively, telomere cluster dynamics may influence the transcriptional regulation of subtelomeric genes, which are enriched in stress-response and metabolic genes, thereby coordinating cellular physiology with environmental conditions [161].
Telomere hypercluster formation in S. pombe. As in budding yeast, entry into quiescence in S. pombe is accompanied by a pronounced spatial reorganization of telomeres, most notably their collapse into a single hypercluster. In S. pombe log cells, telomeres form two to three nuclear envelope (NE)-associated clusters during interphase, an organization that persists through DNA replication [162,163,164]. The shelterin component Rap1 directly contributes to telomere tethering at the NE through interaction with the NE-associated Bqt3-Bqt4 complex [163,165]. NE localization is reinforced by subtelomeric anchoring mediated by the inner nuclear membrane LEM-domain proteins Lem2 and Man1, which associate with distinct subtelomeric regions [83,166,167]. Simultaneous loss of Bqt4 and the chromatin remodeler Fft3, which interacts with Man1, results in relocalization of telomeres toward the nuclear interior [166].
Upon quiescence entry, telomeres collapse into a single cluster. In contrast to S. cerevisiae, where telomere hyperclusters form in the center of the nucleus, fission yeast telomeres remain tightly anchored to the NE during nitrogen starvation, in a manner dependent on Rap1 and Bqt4. Nuclear polarity is maintained, with telomeres localizing opposite the spindle-pole body (SPB), where centromeres are anchored, thereby preserving the Rabl configuration [168]. Importantly, loss of Bqt4 does not alter the number of telomeric foci, indicating that NE tethering and telomere hyperclustering represent mechanistically distinct steps [168]. Disruption of NE attachment, either by loss of Bqt4 or telomere erosion, leads to increased telomeric transcription and predisposes cells to subtelomeric rearrangements via a quiescence-specific repair mechanism termed STEEx (STE1-expansion) [168,169]. Cells harboring rearranged telomeres fail to efficiently exit quiescence [169]. Telomere clustering has also been reported for G0 mouse and human lymphocytes [170], supporting the notion that telomere repositioning during quiescence may represent a conserved strategy to preserve genome stability in non-proliferating cells [171].
5.4. Dynamic Relocalization of Stress-Induced Gene Clusters
In S. pombe, stress-response genes are frequently organized into physical clusters along chromosomes, consistent with coordinated regulation shaped by chromatin architecture and nuclear organization [172]. Several clusters that are rapidly induced upon nitrogen starvation are located near subtelomeres and associate with the nuclear periphery in proliferating cells, but relocalize to the nuclear interior upon starvation, coincident with their activation [173]. Because the nuclear periphery is generally considered a repressive environment enriched in silencing factors [174,175], these observations raised the question of whether gene repositioning is a cause or a consequence of transcriptional activation. Notably, similar behavior is observed for the urg1–3 gene cluster, which is located in a euchromatic region rather than at subtelomeres. Despite this genomic context, the cluster associates with the nuclear periphery during growth and relocates to the nuclear interior upon nitrogen starvation, providing a useful model to study regulated gene repositioning independently of constitutive subtelomeric silencing.
Early experiments using the broad transcriptional inhibitor 1,10-phenanthroline suggested that cluster relocalization depends on transcription [173] However, given the pleiotropic effects of this compound, transcription-independent effects could not be excluded. More targeted perturbations support an alternative model, in which relocalization precedes transcription. The transcription factor Toe1 is required for the induction of genes within the urg cluster, yet deletion of toe1 abolishes transcriptional activation without preventing relocalization of the cluster to the interior in starved cells, arguing that repositioning occurs upstream, or independent, from transcription (S.B. and Vishnu N. Suma Screechekram, unpublished data).
Consistent with its euchromatic context, perinuclear tethering of the urg cluster does not require the inner nuclear membrane protein Lem2, a key factor in heterochromatin silencing and subtelomeric anchoring ([83]; S.B. and Vishnu N. Suma Screechekram, unpublished data). Instead, a putative NE-associated repressor complex, Ntu1-Ntu2, binds a promoter within this cluster, and loss of this complex phenocopies starvation-induced relocalization, implicating Ntu1-Ntur2 in anchoring the cluster at the nuclear periphery [176]. How nitrogen starvation signals trigger release of this tether, and whether additional NE proteins contribute to positioning of other stress-induced clusters, remain open questions.
6. Quiescence: Quo Vadis?
Work across yeast and metazoan models has firmly established quiescence as an actively regulated program rather than a passive cessation of proliferation. Nevertheless, major challenges remain in resolving its underlying mechanisms. A central open question concerns the physiological function of the diverse nuclear rearrangements observed in quiescent cells: Do these changes act as causal drivers of quiescence or instead arise as downstream consequences of altered transcription, metabolism, and chromatin state? This question has not yet been resolved for the repositioning of stress-responsive gene clusters, telomere hyperclustering, global chromosome compaction, remodeling of nuclear bodies, and extensive reprogramming of RNA metabolism. For many of these phenomena, their functional contribution to G0 entry, transcriptional adaption, and long-term survival during quiescence remain poorly defined.
A second major frontier is cellular heterogeneity, which complicates both mechanistic interpretation and conceptual definitions of quiescence. Quiescent populations typically comprise mixtures of quiescent (Q) and non-quiescent (NQ) cells, and even highly enriched Q populations likely span a continuum of states. This raises foundational questions including whether cells commit to quiescence long before proliferation ceases, whether previous experience of quiescence alters the speed of re-entry (is there a memory of quiescence?), when quiescence is firmly established, and whether there are differences in the “depth” of the quiescent state (see also this special issue [20]). Addressing these issues necessitates a shift from viewing quiescence as a single fixed endpoint toward a framework of layered or graded quiescence states, characterized by time-dependent changes in signaling output, chromatin dynamics, and gene expression, potentially relying on molecular timers that encode a physical memory of quiescence duration (see the review in this special issue by [7]). Progress in this area will rely on single-cell and longitudinal approaches capable of resolving transition trajectories during entry, maintenance, and exit.
A third emerging theme centers on RNA-mediated nuclear regulation, including non-coding RNAs and post-transcriptional control. Q cells typically globally reduce transcriptional output while selectively accumulating subsets of long non-coding RNAs, raising the possibility that components of the “dark transcriptome” exert regulatory functions that become apparent only in non-proliferative states. Beyond intron retention, future studies should systematically examine isoform usage, RNA stability control, nuclear-cytoplasmatic partitioning, and RNA-protein condensate dynamics as mechanisms that preserve readiness for renewed proliferation while suppressing energetically costly biogenesis.
Finally, quiescence-specific nuclear dynamics connect to development and disease. Quiescence underpins adult stem cell maintenance and immune homeostasis, while quiescence-like programs contribute to both therapy resistance of cancer persister cells and to drug tolerance of chronic or recurrent fungal infections. Quiescence research offers potential new therapies for these important medical challenges. A comparative perspective remains essential: how conserved are nuclear quiescence programs across species and triggers, and which features represent cell type-specific adaptations rather than universal molecular pathways. Can this heterogeneity be selectively targeted for therapeutic purposes? Addressing these questions will require systematic cross-system analyses to identify condition- and disease-specific nuclear implementations of quiescence.
Acknowledgments
We thank members of the ‘NuQuest’ consortium for fruitful discussions and valuable feedback. Schemes in figures were created with BioRender.com. This work was supported by the Agence Nationale pour la Recherche DeSynLE (ANR-22-CE120013-01) to A.T., the European Research Council (ERC; ERC-Starting Grant agreement no. 101117421, ‘DOR CODE’) and Max Planck Society to A.B.-K., the German Research Foundation (DFG) through the Heisenberg Program (project ID 464293512) to S.B., and the National Science Center, Poland, (OPUS grant no. 2024/53/B/NZ8/02259) to D.W.S.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created in this study. For the re-analysis of publicly available data, sources are indicated.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- O'Farrell, P.H. Quiescence: early evolutionary origins and universality do not imply uniformity. Philos. Trans. R. Soc. B: Biol. Sci. 2011, 366, 3498–3507. [Google Scholar] [CrossRef]
- Breeden, L.L.; Tsukiyama, T. Quiescence in Saccharomyces cerevisiae. Annu. Rev. Genet. 2022, 56, 253–278. [Google Scholar] [CrossRef]
- Gray, J.V.; Petsko, G.A.; Johnston, G.C.; Ringe, D.; Singer, R.A.; Werner-Washburne, M. “Sleeping Beauty”: Quiescence in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2004, 68, 187–206. [Google Scholar] [CrossRef]
- Laribee, R.N.; Weisman, R. Nuclear Functions of TOR: Impact on Transcription and the Epigenome. Genes 2020, 11, 641. [Google Scholar] [CrossRef]
- Bulut-Karslioglu, A.; Biechele, S.; Jin, H.; Macrae, T.A.; Hejna, M.; Gertsenstein, M.; Song, J.S.; Ramalho-Santos, M. Inhibition of mTOR induces a paused pluripotent state. Nature 2016, 540, 119–123. [Google Scholar] [CrossRef]
- De Virgilio, C. The essence of yeast quiescence. FEMS Microbiol. Rev. 2012, 36, 306–339. [Google Scholar] [CrossRef]
- Laporte, D.; Sagot, I. Quiescence Multiverse. Biomolecules 2025, 15, 960. [Google Scholar] [CrossRef] [PubMed]
- Sagot, I.; Laporte, D. The cell biology of quiescent yeast – a diversity of individual scenarios. J. Cell Sci. 2019, 132, jcs213025. [Google Scholar] [CrossRef]
- Jacquel, B.; Aspert, T.; Laporte, D.; Sagot, I.; Charvin, G. Monitoring single-cell dynamics of entry into quiescence during an unperturbed life cycle. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Medina-Menéndez, C.; Tirado-Melendro, P.; Li, L.; Rodríguez-Martín, P.; la Peña, E.M.-D.; Díaz-García, M.; Valdés-Bescós, M.; López-Sansegundo, R.; Morales, A.V. Sox5 controls the establishment of quiescence in neural stem cells during postnatal development. PLOS Biol. 2025, 23, e3002654. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wang, X.; Jiang, L.; Xu, J.; Zohar, Y.; Yao, G. Extracellular Fluid Flow Induces Shallow Quiescence Through Physical and Biochemical Cues. Front. Cell Dev. Biol. 2022, 10, 792719. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.S.; Everetts, N.J.; Wang, X.; Wang, W.; Della Croce, K.; Xing, J.; Yao, G. Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep. 2017, 20, 3223–3235. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Occean, J.R.; Melters, D.P.; Shi, C.; Wang, L.; Stransky, S.; Doyle, M.E.; Cui, C.-Y.; Delannoy, M.; Fan, J.; et al. A hyper-quiescent chromatin state formed during aging is reversed by regeneration. Mol. Cell 2023, 83, 1659–1676.e11. [Google Scholar] [CrossRef]
- Sajiki, K.; Hatanaka, M.; Nakamura, T.; Takeda, K.; Shimanuki, M.; Yoshida, T.; Hanyu, Y.; Hayashi, T.; Nakaseko, Y.; Yanagida, M. Genetic control of cellular quiescence in S. pombe. J. Cell Sci. 2009, 122, 1418–1429. [Google Scholar] [CrossRef]
- Sideri, T.; Rallis, C.; A Bitton, D.; Lages, B.M.; Suo, F.; Rodríguez-López, M.; Du, L.-L.; Bähler, J. Parallel Profiling of Fission Yeast Deletion Mutants for Proliferation and for Lifespan During Long-Term Quiescence. G3 Genes|Genomes|Genetics 2015, 5, 145–155. [Google Scholar] [CrossRef]
- Yanagida, M. Cellular quiescence: are controlling genes conserved? Trends Cell Biol. 2009, 19, 705–715. [Google Scholar] [CrossRef]
- Zahedi, Y.; Durand-Dubief, M.; Ekwall, K. High-Throughput Flow Cytometry Combined with Genetic Analysis Brings New Insights into the Understanding of Chromatin Regulation of Cellular Quiescence. Int. J. Mol. Sci. 2020, 21, 9022. [Google Scholar] [CrossRef]
- Rutherford, K.M.; Lera-Ramírez, M.; Wood, V. PomBase: a Global Core Biodata Resource—growth, collaboration, and sustainability. Genetics 2024, 227. [Google Scholar] [CrossRef]
- Sun, S.; Gresham, D. Cellular quiescence in budding yeast. Yeast 2020, 38, 12–29. [Google Scholar] [CrossRef]
- Marek, A.; Tomala, K.; Wloch-Salamon, D. Ecological History Shapes Transcriptome Variation in Quiescent Saccharomyces cerevisiae. Biomolecules 2025, 15, 1588. [Google Scholar] [CrossRef] [PubMed]
- de Morree, A.; Rando, T.A. Regulation of Adult Stem Cell Quiescence and Its Functions in the Maintenance of Tissue Integrity. Nat. Rev. Mol. Cell Biol. 2023, 24, 334–354. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, S.; Arcangioli, B. DNA repair and mutations during quiescence in yeast. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef]
- Johnston, G.C.; Pringle, J.R.; Hartwell, L.H. Coordination of Growth with Cell Division in the Yeast Saccharomyces Cerevisiae. Exp. Cell Res. 1977, 105, 79–98. [Google Scholar] [CrossRef]
- Lillie, S.H.; Pringle, J.R. Reserve carbohydrate metabolism in Saccharomyces cerevisiae: responses to nutrient limitation. J. Bacteriol. 1980, 143, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Werner-Washburne, M.; Braun, E.; Johnston, G.C.; A Singer, R. Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol. Rev. 1993, 57, 383–401. [Google Scholar] [CrossRef]
- Allen, C.; BütTner, S.; Aragon, A.D.; Thomas, J.A.; Meirelles, O.; Jaetao, J.E.; Benn, D.; Ruby, S.W.; Veenhuis, M.; Madeo, F.; et al. Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures. J. Cell Biol. 2006, 174, 89–100. [Google Scholar] [CrossRef]
- Yang, H.; Ren, Q.; Zhang, Z. Chromosome or chromatin condensation leads to meiosis or apoptosis in stationary yeast (Saccharomyces cerevisiae) cells. FEMS Yeast Res. 2006, 6, 1254–1263. [Google Scholar] [CrossRef]
- Opalek, M.; Tutaj, H.; Pirog, A.; Smug, B.J.; Rutkowska, J.; Wloch-Salamon, D. A Systematic Review on Quiescent State Research Approaches in S. cerevisiae. Cells 2023, 12, 1608. [Google Scholar] [CrossRef]
- Li, L.; Miles, S.; Melville, Z.; Prasad, A.; Bradley, G.; Breeden, L.L. Key events during the transition from rapid growth to quiescence in budding yeast require posttranscriptional regulators. Mol. Biol. Cell 2013, 24, 3697–3709. [Google Scholar] [CrossRef]
- Davidson, G.S.; Joe, R.M.; Roy, S.; Meirelles, O.; Allen, C.P.; Wilson, M.R.; Tapia, P.H.; Manzanilla, E.E.; Dodson, A.E.; Chakraborty, S.; et al. The proteomics of quiescent and nonquiescent cell differentiation in yeast stationary-phase cultures. Mol. Biol. Cell 2011, 22, 988–998. [Google Scholar] [CrossRef]
- Miles, S.; Li, L.; Davison, J.; Breeden, L.L. Xbp1 Directs Global Repression of Budding Yeast Transcription during the Transition to Quiescence and Is Important for the Longevity and Reversibility of the Quiescent State. PLOS Genet. 2013, 9, e1003854. [Google Scholar] [CrossRef]
- Miles, S.; Lee, C.; Breeden, L. BY4741 cannot enter quiescence from rich medium. MicroPubl. Biol. 2023, 2023. [Google Scholar] [CrossRef]
- Su, S.S.; Tanaka, Y.; Samejima, I.; Tanaka, K.; Yanagida, M. A nitrogen starvation-induced dormant G0 state in fission yeast: the establishment from uncommitted G1 state and its delay for return to proliferation. J. Cell Sci. 1996, 109, 109. [Google Scholar] [CrossRef]
- Roche, B.; Arcangioli, B.; Martienssen, R. Transcriptional reprogramming in cellular quiescence. RNA Biol. 2017, 14, 843–853. [Google Scholar] [CrossRef]
- Hubbard, E.J.A.; Schedl, T. Biology of the Caenorhabditis elegans Germline Stem Cell System. Genetics 2019, 213, 1145–1188. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Wong, C.; Roy, R. Developmental plasticity and the response to nutrient stress in Caenorhabditis elegans. Dev. Biol. 2021, 475, 265–276. [Google Scholar] [CrossRef]
- Gujar, M.R.; Wang, H. Signaling mechanisms in the reactivation of quiescent neural stem cells in Drosophila. Curr. Opin. Cell Biol. 2025, 96, 102566. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef]
- Trumpp, A.; Essers, M.; Wilson, A. Awakening dormant haematopoietic stem cells. Nat. Rev. Immunol. 2010, 10, 201–209. [Google Scholar] [CrossRef]
- Höfer, T.; Busch, K.; Klapproth, K.; Rodewald, H.-R. Fate Mapping and Quantitation of Hematopoiesis In Vivo. Annu. Rev. Immunol. 2016, 34, 449–478. [Google Scholar] [CrossRef]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic Stem Cells Reversibly Switch from Dormancy to Self-Renewal during Homeostasis and Repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.M.; Reth, M.; Höfer, T.; Rodewald, H.-R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Kucinski, I.; Campos, J.; Barile, M.; Severi, F.; Bohin, N.; Moreira, P.N.; Allen, L.; Lawson, H.; Haltalli, M.L.; Kinston, S.J.; et al. A time- and single-cell-resolved model of murine bone marrow hematopoiesis. Cell Stem Cell 2024, 31, 244–259.e10. [Google Scholar] [CrossRef]
- van der Weijden, V.A.; Bulut-Karslioglu, A. Molecular Regulation of Paused Pluripotency in Early Mammalian Embryos and Stem Cells. Front. Cell Dev. Biol. 2021, 9, 708318. [Google Scholar] [CrossRef] [PubMed]
- Renfree, M.B.; Fenelon, J.C. The enigma of embryonic diapause. Development 2017, 144, 3199–3210. [Google Scholar] [CrossRef]
- Hunter, S.M.; Evans, M. Non-surgical method for the induction of delayed implantation and recovery of viable blastocysts in rats and mice by the use of tamoxifen and Depo-Provera. Mol. Reprod. Dev. 1999, 52, 29–32. [Google Scholar] [CrossRef]
- Paria, B.C.; Huet-Hudson, Y.M.; Dey, S.K. Blastocyst's state of activity determines the "window" of implantation in the receptive mouse uterus. Proc. Natl. Acad. Sci. 1993, 90, 10159–10162. [Google Scholar] [CrossRef]
- zgüldez, H.Ö.; Bulut-Karslioğlu, A. Dormancy, Quiescence, and Diapause: Savings Accounts for Life. Annu. Rev. Cell Dev. Biol. 2024, 40, 25–49. [Google Scholar] [CrossRef]
- Iyer, D.P.; Khoei, H.H.; van der Weijden, V.A.; Kagawa, H.; Pradhan, S.J.; Novatchkova, M.; McCarthy, A.; Rayon, T.; Simon, C.S.; Dunkel, I.; et al. mTOR activity paces human blastocyst stage developmental progression. Cell 2024, 187, 6566–6583.e22. [Google Scholar] [CrossRef]
- Boroviak, T.; Loos, R.; Lombard, P.; Okahara, J.; Behr, R.; Sasaki, E.; Nichols, J.; Smith, A.; Bertone, P. Lineage-Specific Profiling Delineates the Emergence and Progression of Naive Pluripotency in Mammalian Embryogenesis. Dev. Cell 2015, 35, 366–382. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Cabezas-Wallscheid, N.; Thier, M.C.; Altamura, S.; Reyes, A.; Prendergast, Á.M.; Baumgärtner, D.; Carnevalli, L.S.; Atzberger, A.; Haas, S.; et al. Myc Depletion Induces a Pluripotent Dormant State Mimicking Diapause. Cell 2016, 164, 668–680. [Google Scholar] [CrossRef]
- McKnight, J.N.; Boerma, J.W.; Breeden, L.L.; Tsukiyama, T. Global Promoter Targeting of a Conserved Lysine Deacetylase for Transcriptional Shutoff during Quiescence Entry. Mol. Cell 2015, 59, 732–743. [Google Scholar] [CrossRef]
- Young, C.P.; Hillyer, C.; Hokamp, K.; Fitzpatrick, D.J.; Konstantinov, N.K.; Welty, J.S.; Ness, S.A.; Werner-Washburne, M.; Fleming, A.B.; Osley, M.A. Distinct histone methylation and transcription profiles are established during the development of cellular quiescence in yeast. BMC Genom. 2017, 18, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Zee, B.M.; Liu, S.; Donahue, G.; Garcia, B.A.; Berger, S.L. Histone Methylation Has Dynamics Distinct from Those of Histone Acetylation in Cell Cycle Reentry from Quiescence. Mol. Cell. Biol. 2014, 34, 3968–3980. [Google Scholar] [CrossRef]
- Marguerat, S.; Schmidt, A.; Codlin, S.; Chen, W.; Aebersold, R.; Bähler, J. Quantitative Analysis of Fission Yeast Transcriptomes and Proteomes in Proliferating and Quiescent Cells. Cell 2012, 151, 671–683. [Google Scholar] [CrossRef]
- Greenlaw, A.; Dell, R.; Tsukiyama, T. Initial acidic media promotes quiescence entry in Saccharomyces cerevisiae. 2024, 2024. [Google Scholar] [CrossRef]
- Pérez, M.B.; Laenen, G.; Loïodice, I.; Garnier, M.; Szachnowski, U.; Morillon, A.; Ruault, M.; Taddei, A. Intergenic accumulation of RNA polymerase II maintains the potential for swift transcriptional restart upon release from quiescence. Genome Res. 2025, 35, 2226–2239. [Google Scholar] [CrossRef]
- Radonjic, M.; Andrau, J.-C.; Lijnzaad, P.; Kemmeren, P.; Kockelkorn, T.T.; van Leenen, D.; van Berkum, N.L.; Holstege, F.C. Genome-Wide Analyses Reveal RNA Polymerase II Located Upstream of Genes Poised for Rapid Response upon S. cerevisiae Stationary Phase Exit. Mol. Cell 2005, 18, 171–183. [Google Scholar] [CrossRef]
- Swygert, S.G.; Tsukiyama, T. Unraveling quiescence-specific repressive chromatin domains. Curr. Genet. 2019, 65, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Aragon, A.D.; Rodriguez, A.L.; Meirelles, O.; Roy, S.; Davidson, G.S.; Tapia, P.H.; Allen, C.; Joe, R.; Benn, D.; Werner-Washburne, M. Characterization of Differentiated Quiescent and Nonquiescent Cells in Yeast Stationary-Phase Cultures. Mol. Biol. Cell 2008, 19, 1271–1280. [Google Scholar] [CrossRef]
- Shah, K.H.; Zhang, B.; Ramachandran, V.; Herman, P.K. Processing Body and Stress Granule Assembly Occur by Independent and Differentially Regulated Pathways inSaccharomyces cerevisiae. Genetics 2013, 193, 109–123. [Google Scholar] [CrossRef]
- Zeng, S.; Ekwall, K. Epigenome Mapping in Quiescent Cells Reveals a Key Role for H3K4me3 in Regulation of RNA Polymerase II Activity. Epigenomes 2024, 8, 39. [Google Scholar] [CrossRef]
- Joh, R.I.; Khanduja, J.S.; Calvo, I.A.; Mistry, M.; Palmieri, C.M.; Savol, A.J.; Sui, S.J.H.; Sadreyev, R.I.; Aryee, M.J.; Motamedi, M. Survival in Quiescence Requires the Euchromatic Deployment of Clr4/SUV39H by Argonaute-Associated Small RNAs. Mol. Cell 2016, 64, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Zahedi, Y.; Zeng, S.; Ekwall, K. An essential role for the Ino80 chromatin remodeling complex in regulation of gene expression during cellular quiescence. Chromosom. Res. 2023, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Westholm, J.O.; Nordberg, N.; Murén, E.; Ameur, A.; Komorowski, J.; Ronne, H. Combinatorial control of gene expression by the three yeast repressors Mig1, Mig2 and Mig3. BMC Genom. 2008, 9, 601–601. [Google Scholar] [CrossRef]
- Treitel, M.A.; Kuchin, S.; Carlson, M. Snf1 Protein Kinase Regulates Phosphorylation of the Mig1 Repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 1998, 18, 6273–6280. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Pastor, M.T.; Marchler, G.; Schüller, C.; Marchler-Bauer, A.; Ruis, H.; Estruch, F. The Saccharomyces cerevisiae zinc finger proteins Msn2p and Msn4p are required for transcriptional induction through the stress response element (STRE). EMBO J. 1996, 15, 2227–2235. [Google Scholar] [CrossRef]
- Santhanam, A.; Hartley, A.; DüvEl, K.; Broach, J.R.; Garrett, S. PP2A Phosphatase Activity Is Required for Stress and Tor Kinase Regulation of Yeast Stress Response Factor Msn2p. Eukaryot. Cell 2004, 3, 1261–1271. [Google Scholar] [CrossRef]
- Mai, B.; Breeden, L. Xbp1, a Stress-Induced Transcriptional Repressor of the Saccharomyces cerevisiae Swi4/Mbp1 Family. Mol. Cell. Biol. 1997, 17, 6491–6501. [Google Scholar] [CrossRef]
- Huber, A.; French, S.L.; Tekotte, H.; Yerlikaya, S.; Stahl, M.; Perepelkina, M.P.; Tyers, M.; Rougemont, J.; Beyer, A.L.; Loewith, R. Sch9 regulates ribosome biogenesis via Stb3, Dot6 and Tod6 and the histone deacetylase complex RPD3L. EMBO J. 2011, 30, 3052–3064. [Google Scholar] [CrossRef]
- Hirai, H.; Ohta, K. Comparative Research: Regulatory Mechanisms of Ribosomal Gene Transcription in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Biomolecules 2023, 13, 288. [Google Scholar] [CrossRef]
- Bonitto, K.; Sarathy, K.; Atai, K.; Mitra, M.; Coller, H.A. Is There a Histone Code for Cellular Quiescence? Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Tao, R.; Chen, H.; Gao, C.; Xue, P.; Yang, F.; Han, J.-D.J.; Zhou, B.; Chen, Y.-G. Xbp1-mediated histone H4 deacetylation contributes to DNA double-strand break repair in yeast. Cell Res. 2011, 21, 1619–1633. [Google Scholar] [CrossRef]
- Nirello, V.D.; de Paula, D.R.; Araújo, N.V.; Varga-Weisz, P.D. Does chromatin function as a metabolite reservoir? Trends Biochem. Sci. 2022, 47, 732–735. [Google Scholar] [CrossRef] [PubMed]
- Friis, R.M.N.; Wu, B.P.; Reinke, S.N.; Hockman, D.J.; Sykes, B.D.; Schultz, M.C. A glycolytic burst drives glucose induction of global histone acetylation by picNuA4 and SAGA. Nucleic Acids Res. 2009, 37, 3969–3980. [Google Scholar] [CrossRef] [PubMed]
- E Cucinotta, C.; Dell, R.H.; Braceros, K.C.; Tsukiyama, T. RSC primes the quiescent genome for hypertranscription upon cell-cycle re-entry. eLife 2021, 10. [Google Scholar] [CrossRef]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2017, 19, 229–244. [Google Scholar] [CrossRef]
- Zofall, M.; Yamanaka, S.; Reyes-Turcu, F.E.; Zhang, K.; Rubin, C.; Grewal, S.I.S. RNA Elimination Machinery Targeting Meiotic mRNAs Promotes Facultative Heterochromatin Formation. Science 2012, 335, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Roche, B.; Arcangioli, B.; Martienssen, R.A. RNA interference is essential for cellular quiescence. Science 2016, 354. [Google Scholar] [CrossRef]
- Oya, E.; Durand-Dubief, M.; Cohen, A.; Maksimov, V.; Schurra, C.; Nakayama, J.-I.; Weisman, R.; Arcangioli, B.; Ekwall, K. Leo1 is essential for the dynamic regulation of heterochromatin and gene expression during cellular quiescence. Epigenetics Chromatin 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Yuan, X.; Feng, W.; Imhof, A.; Grummt, I.; Zhou, Y. Activation of RNA Polymerase I Transcription by Cockayne Syndrome Group B Protein and Histone Methyltransferase G9a. Mol. Cell 2007, 27, 585–595. [Google Scholar] [CrossRef]
- Tadeo, X.; Wang, J.; Kallgren, S.P.; Liu, J.; Reddy, B.D.; Qiao, F.; Jia, S. Elimination of shelterin components bypasses RNAi for pericentric heterochromatin assembly. Genes Dev. 2013, 27, 2489–2499. [Google Scholar] [CrossRef]
- Barrales, R.R.; Forn, M.; Georgescu, P.R.; Sarkadi, Z.; Braun, S. Control of heterochromatin localization and silencing by the nuclear membrane protein Lem2. Genes Dev. 2016, 30, 133–148. [Google Scholar] [CrossRef]
- Muhammad, A.; Sarkadi, Z.; Mazumder, A.; Saada, A.A.; van Emden, T.; Capella, M.; Fekete, G.; Sreechakram, V.N.S.; Al-Sady, B.; A E Lambert, S.; et al. A systematic quantitative approach comprehensively defines domain-specific functional pathways linked to Schizosaccharomyces pombe heterochromatin regulation. Nucleic Acids Res. 2024, 52, 13665–13689. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cheung, T.H.; Charville, G.W.; Hurgo, B.M.C.; Leavitt, T.; Shih, J.; Brunet, A.; Rando, T.A. Chromatin Modifications as Determinants of Muscle Stem Cell Quiescence and Chronological Aging. Cell Rep. 2013, 4, 189–204. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 Maintain Repressive Chromatin through Different Mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Evertts, A.G.; Manning, A.L.; Wang, X.; Dyson, N.J.; Garcia, B.A.; Coller, H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 2013, 24, 3025–3037. [Google Scholar] [CrossRef]
- Bierhoff, H.; Dammert, M.A.; Brocks, D.; Dambacher, S.; Schotta, G.; Grummt, I. Quiescence-Induced LncRNAs Trigger H4K20 Trimethylation and Transcriptional Silencing. Mol. Cell 2014, 54, 675–682. [Google Scholar] [CrossRef]
- Boonsanay, V.; Zhang, T.; Georgieva, A.; Kostin, S.; Qi, H.; Yuan, X.; Zhou, Y.; Braun, T. Regulation of Skeletal Muscle Stem Cell Quiescence by Suv4-20h1-Dependent Facultative Heterochromatin Formation. Cell Stem Cell 2016, 18, 229–242. [Google Scholar] [CrossRef]
- Ramponi, V.; Richart, L.; Kovatcheva, M.; Attolini, C.S.-O.; Capellades, J.; Lord, A.E.; Yanes, O.; Ficz, G.; Serrano, M. H4K20me3-Mediated Repression of Inflammatory Genes Is a Characteristic and Targetable Vulnerability of Persister Cancer Cells. Cancer Res. 2024, 85, 32–51. [Google Scholar] [CrossRef]
- Chen, R.; Fan, R.; Chen, F.; Govindasamy, N.; Brinkmann, H.; Stehling, M.; Adams, R.H.; Jeong, H.-W.; Bedzhov, I. Analyzing embryo dormancy at single-cell resolution reveals dynamic transcriptional responses and activation of integrin-Yap/Taz prosurvival signaling. Cell Stem Cell 2024, 31, 1262–1279.e8. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Wang, B.; Wang, S.; Wu, W.; Wang, Q.; Chen, Y.; Kong, S.; Lu, J.; Tang, Z.; Ran, H.; et al. Integral Proteomic Analysis of Blastocysts Reveals Key Molecular Machinery Governing Embryonic Diapause and Reactivation for Implantation in Mice. Biol. Reprod. 2014, 90, 52. [Google Scholar] [CrossRef]
- Hussein, A.M.; Wang, Y.; Mathieu, J.; Margaretha, L.; Song, C.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic Control over mTOR-Dependent Diapause-like State. Dev. Cell 2020, 52, 236–250.e7. [Google Scholar] [CrossRef]
- van der Weijden, V.A.; Bulut-Karslioğlu, A. Embryos Burn Fat in Standby. Trends Cell Biol. 2024, 34, 700–702. [Google Scholar] [CrossRef]
- Stötzel, M.; Cheng, C.-Y.; Iiik, I.A.; Kumar, A.S.; Omgba, P.A.; van der Weijden, V.A.; Zhang, Y.; Vingron, M.; Meissner, A.; Aktaş, T.; et al. TET activity safeguards pluripotency throughout embryonic dormancy. Nat. Struct. Mol. Biol. 2024, 31, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Kim, Y.S.; Wu, J.; Chen, R.; Zeuschner, D.; Mildner, K.; Adachi, K.; Wu, G.; Galatidou, S.; Li, J.; et al. Wnt/Beta-catenin/Esrrb signalling controls the tissue-scale reorganization and maintenance of the pluripotent lineage during murine embryonic diapause. Nat. Commun. 2020, 11, 5499. [Google Scholar] [CrossRef] [PubMed]
- Furlan, G.; Martin, S.B.; Cho, B.; McClymont, S.A.; Robertson, E.J.; Collignon, E.; Ramalho-Santos, M. Nodal/Smad2 Signaling Sustains Developmental Pausing by Repressing Pparg-Mediated Lipid Metabolism. bioRxiv 2025. [Google Scholar] [CrossRef]
- Sajiki, K.; Tahara, Y.; Uehara, L.; Sasaki, T.; Pluskal, T.; Yanagida, M. Genetic regulation of mitotic competence in G 0 quiescent cells. Sci. Adv. 2018, 4, eaat5685. [Google Scholar] [CrossRef]
- Sadeghi, L.; Prasad, P.; Ekwall, K.; Cohen, A.; Svensson, J.P. The Paf1 Complex Factors Leo1 and Paf1 Promote Local Histone Turnover to Modulate Chromatin States in Fission Yeast. EMBO Rep. 2015, 16, 1673–1687. [Google Scholar]
- Oya, T.; Tanaka, M.; Hayashi, A.; Yoshimura, Y.; Nakamura, R.; Arita, K.; Murakami, Y.; Nakayama, J. Characterization of the Swi6/ HP1 binding motif in its partner protein reveals the basis for the functional divergence of the HP1 family proteins in fission yeast. FASEB J. 2025, 39, e70387. [Google Scholar] [CrossRef]
- Jacob, A.G.; Smith, C.W.J. Intron Retention as a Component of Regulated Gene Expression Programs. Hum. Genet. 2017, 136, 1043–1057. [Google Scholar] [CrossRef]
- Pleiss, J.A.; Whitworth, G.B.; Bergkessel, M.; Guthrie, C. Rapid, Transcript-Specific Changes in Splicing in Response to Environmental Stress. Mol. Cell 2007, 27, 928–937. [Google Scholar] [CrossRef]
- Parenteau, J.; Tsang, J.; Downs, S.R.; Zhou, D.; Scott, M.S.; A Pleiss, J.; Elela, S.A. Cells resist starvation through a nutrient stress splice switch. Nucleic Acids Res. 2025, 53. [Google Scholar] [CrossRef]
- Parenteau, J.; Maignon, L.; Berthoumieux, M.; Catala, M.; Gagnon, V.; Elela, S.A. Introns are mediators of cell response to starvation. Nature 2019, 565, 612–617. [Google Scholar] [CrossRef]
- Morgan, J.T.; Fink, G.R.; Bartel, D.P. Excised linear introns regulate growth in yeast. Nature 2019, 565, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Wan, R.; Luan, S.; Zeng, W.; Cheung, T.H. Dek Modulates Global Intron Retention during Muscle Stem Cells Quiescence Exit. Dev. Cell 2020, 53, 661–676.e6. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Johnson, E.L.; Swamy, V.S.; E Nersesian, L.; Corney, D.C.; Robinson, D.G.; Taylor, D.G.; Ambrus, A.M.; Jelinek, D.; Wang, W.; et al. Alternative polyadenylation factors link cell cycle to migration. Genome Biol. 2018, 19, 176. [Google Scholar] [CrossRef]
- Lukačišin, M.; Espinosa-Cantú, A.; Bollenbach, T. Intron-mediated induction of phenotypic heterogeneity. Nature 2022, 605, 113–118. [Google Scholar] [CrossRef]
- Kilchert, C.; Wittmann, S.; Passoni, M.; Shah, S.; Granneman, S.; Vasiljeva, L. Regulation of mRNA Levels by Decay-Promoting Introns that Recruit the Exosome Specificity Factor Mmi1. Cell Rep. 2015, 13, 2504–2515. [Google Scholar] [CrossRef]
- Harigaya, Y.; Tanaka, H.; Yamanaka, S.; Tanaka, K.; Watanabe, Y.; Tsutsumi, C.; Chikashige, Y.; Hiraoka, Y.; Yamashita, A.; Yamamoto, M. Selective elimination of messenger RNA prevents an incidence of untimely meiosis. Nature 2006, 442, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating Cells Express mRNAs with Shortened 3' Untranslated Regions and Fewer MicroRNA Target Sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3′ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. 2009, 106, 7028–7033. [Google Scholar] [CrossRef]
- Liu, X.; Hoque, M.; Larochelle, M.; Lemay, J.-F.; Yurko, N.; Manley, J.L.; Bachand, F.; Tian, B. Comparative Analysis of Alternative Polyadenylation in S. Cerevisiae and S. Pombe. Genome Res. 2017, 27, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Schlackow, M.; Marguerat, S.; Proudfoot, N.J.; Bähler, J.; Erban, R.; Gullerova, M. Genome-wide analysis of poly(A) site selection in Schizosaccharomyces pombe. RNA 2013, 19, 1617–1631. [Google Scholar] [CrossRef]
- Michaels, K.K.; Mostafa, S.M.; Capella, J.R.; Moore, C.L. Regulation of alternative polyadenylation in the yeast Saccharomyces cerevisiae by histone H3K4 and H3K36 methyltransferases. Nucleic Acids Res. 2020, 48, 5407–5425. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2016, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Geisberg, J.V.; Moqtaderi, Z.; Struhl, K. Chromatin regulates alternative polyadenylation via the RNA polymerase II elongation rate. Proc. Natl. Acad. Sci. USA 2024, 121, e2405827121. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.-W.; Zhang, W.; Yeh, H.-S.; De Jong, E.P.; Jun, S.; Kim, K.-H.; Bae, S.S.; Beckman, K.; Hwang, T.H.; Kim, K.-S.; et al. mRNA 3′-UTR shortening is a molecular signature of mTORC1 activation. Nat. Commun. 2015, 6, 7218. [Google Scholar] [CrossRef]
- Geisberg, J.V.; Moqtaderi, Z.; Struhl, K. Condition-specific 3′ mRNA isoform half-lives and stability elements in yeast. Proc. Natl. Acad. Sci. USA 2023, 120, e2301117120. [Google Scholar] [CrossRef]
- Iwakawa, H.-O.; Tomari, Y. Life of RISC: Formation, action, and degradation of RNA-induced silencing complex. Mol. Cell 2022, 82, 30–43. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. Role of hypoxia inducible factor-1 in cancer stem cells (Review). Mol. Med. Rep. 2020, 23, 1–1. [Google Scholar] [CrossRef]
- Macias, S.; Cordiner, R.A.; Gautier, P.; Plass, M.; Cáceres, J.F. DGCR8 Acts as an Adaptor for the Exosome Complex to Degrade Double-Stranded Structured RNAs. Mol. Cell 2015, 60, 873–885. [Google Scholar] [CrossRef]
- Cheung, T.H.; Quach, N.L.; Charville, G.W.; Liu, L.; Park, L.; Edalati, A.; Yoo, B.; Hoang, P.; Rando, T.A. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef]
- Liu, W.-M.; Pang, R.T.K.; Cheong, A.W.Y.; Ng, E.H.Y.; Lao, K.; Lee, K.-F.; Yeung, W.S.B. Involvement of microRNA Lethal-7a in the Regulation of Embryo Implantation in Mice. PLOS ONE 2012, 7, e37039. [Google Scholar] [CrossRef]
- Wang, Y.; Hussein, A.M.; Somasundaram, L.; Sankar, R.; Detraux, D.; Mathieu, J.; Ruohola-Baker, H. MicroRNAs Regulating Human and Mouse Naïve Pluripotency. Int. J. Mol. Sci. 2019, 20, 5864. [Google Scholar] [CrossRef]
- Wang, Y.; Medvid, R.; Melton, C.; Jaenisch, R.; Blelloch, R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007, 39, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Cheng, R.R.; Niu, Z.R.; Chen, A.C.; Ma, M.Y.; Li, T.; Chiu, P.C.; Pang, R.T.; Lee, Y.L.; Ou, J.P.; et al. Let-7 derived from endometrial extracellular vesicles is an important inducer of embryonic diapause in mice. Sci. Adv. 2020, 6, eaaz7070. [Google Scholar] [CrossRef] [PubMed]
- Kilchert, C. RNA Exosomes and Their Cofactors. Methods Mol. Biol. 2020, 2062, 215–235. [Google Scholar]
- Li, J.-N.; Wang, M.-Y.; Ruan, J.-W.; Lyu, Y.-J.; Weng, Y.-H.; Brindangnanam, P.; Coumar, M.S.; Chen, P.-S. A transcription-independent role for HIF-1α in modulating microprocessor assembly. Nucleic Acids Res. 2024, 52, 11806–11821. [Google Scholar] [CrossRef]
- Greenlaw, A.C.; Alavattam, K.G.; Tsukiyama, T. Post-transcriptional regulation shapes the transcriptome of quiescent budding yeast. Nucleic Acids Res. 2023, 52, 1043–1063. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, S.R.; Marguerat, S.; Bitton, D.A.; Rodríguez-López, M.; Rallis, C.; Lemay, J.-F.; Cotobal, C.; Malecki, M.; Smialowski, P.; Mata, J.; et al. Long noncoding RNA repertoire and targeting by nuclear exosome, cytoplasmic exonuclease, and RNAi in fission yeast. RNA 2018, 24, 1195–1213. [Google Scholar] [CrossRef]
- Birot, A.; Kus, K.; Priest, E.; Al Alwash, A.; Castello, A.; Mohammed, S.; Vasiljeva, L.; Kilchert, C. RNA-binding protein Mub1 and the nuclear RNA exosome act to fine-tune environmental stress response. Life Sci. Alliance 2021, 5, e202101111. [Google Scholar] [CrossRef]
- Shah, S.; Wittmann, S.; Kilchert, C.; Vasiljeva, L. lncRNA recruits RNAi and the exosome to dynamically regulate pho1 expression in response to phosphate levels in fission yeast. Genes Dev. 2014, 28, 231–244. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, P.; Wu, Q.; Fang, H.; Wang, Y.; Xiao, Y.; Cong, M.; Wang, T.; He, Y.; Ma, C.; et al. Long non-coding RNA NR2F1-AS1 induces breast cancer lung metastatic dormancy by regulating NR2F1 and ΔNp63. Nat. Commun. 2021, 12, 5233. [Google Scholar] [CrossRef]
- Anver, S.; Sumit, A.F.; Sun, X.-M.; Hatimy, A.; Thalassinos, K.; Marguerat, S.; Alic, N.; Bähler, J. Ageing-associated long non-coding RNA extends lifespan and reduces translation in non-dividing cells. Embo Rep. 2024, 25, 4921–4949. [Google Scholar] [CrossRef]
- Hirose, T.; Fujiwara, N.; Ninomiya, K.; Yamamoto, T.; Nakagawa, S.; Yamazaki, T. Architectural RNAs: blueprints for functional membraneless organelle assembly. Trends Genet. 2025, 41, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Ninomiya, K.; Nakagawa, S.; Yamazaki, T. A guide to membraneless organelles and their various roles in gene regulation. Nat. Rev. Mol. Cell Biol. 2022, 24, 288–304. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.-Q.; Okamasa, K.; Katou, Y.; Oya, E.; Nakayama, J.-I.; Chikashige, Y.; Shirahige, K.; Haraguchi, T.; Hiraoka, Y. Chromosome-associated RNA–protein complexes promote pairing of homologous chromosomes during meiosis in Schizosaccharomyces pombe. Nat. Commun. 2019, 10, 5598. [Google Scholar] [CrossRef]
- Aydin, E.; Schreiner, S.; Böhme, J.; Keil, B.; Weber, J.; Žunar, B.; Glatter, T.; Kilchert, C. DEAD-box ATPase Dbp2 is the key enzyme in an mRNP assembly checkpoint at the 3’-end of genes and involved in the recycling of cleavage factors. Nat. Commun. 2024, 15, 6829. [Google Scholar] [CrossRef]
- Shuman, S. Transcriptional interference at tandem lncRNA and protein-coding genes: an emerging theme in regulation of cellular nutrient homeostasis. Nucleic Acids Res. 2020, 48, 8243–8254. [Google Scholar] [CrossRef]
- Nguyen, L.A.C.; Mori, M.; Yasuda, Y.; Galipon, J. Functional Consequences of Shifting Transcript Boundaries in Glucose Starvation. Mol. Cell. Biol. 2023, 43, 611–628. [Google Scholar] [CrossRef]
- Nishizawa, M.; Komai, T.; Katou, Y.; Shirahige, K.; Ito, T.; Toh-E, A. Nutrient-Regulated Antisense and Intragenic RNAs Modulate a Signal Transduction Pathway in Yeast. PLOS Biol. 2008, 6, 2817–2830. [Google Scholar] [CrossRef]
- Miki, A.; Galipon, J.; Sawai, S.; Inada, T.; Ohta, K. RNA decay systems enhance reciprocal switching of sense and antisense transcripts in response to glucose starvation. Genes Cells 2016, 21, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Miyoshi, T.; Kugou, K.; Hoffman, C.S.; Shibata, T.; Ohta, K. Stepwise chromatin remodelling by a cascade of transcription initiation of non-coding RNAs. Nature 2008, 456, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Oda, A.; Takemata, N.; Hirata, Y.; Miyoshi, T.; Suzuki, Y.; Sugano, S.; Ohta, K. Dynamic transition of transcription and chromatin landscape during fission yeast adaptation to glucose starvation. Genes Cells 2015, 20, 392–407. [Google Scholar] [CrossRef]
- Guidi, M.; Ruault, M.; Marbouty, M.; Loïodice, I.; Cournac, A.; Billaudeau, C.; Hocher, A.; Mozziconacci, J.; Koszul, R.; Taddei, A. Spatial reorganization of telomeres in long-lived quiescent cells. Genome Biol. 2015, 16, 1–15. [Google Scholar] [CrossRef]
- Tan, Z.Y.; ShujunCai; Paithankar, S.A.; Liu, T.; Nie, X.; Shi, J. LuGan(甘露) Macromolecular and cytological changes in fission yeast G0 nuclei. J. Cell Sci. 2025, 138. [Google Scholar] [CrossRef]
- Tiku, V.; Jain, C.; Raz, Y.; Nakamura, S.; Heestand, B.; Liu, W.; Späth, M.; Suchiman, H.E.D.; Müller, R.-U.; Slagboom, P.E.; et al. Small nucleoli are a cellular hallmark of longevity. Nat. Commun. 2017, 8, 16083. [Google Scholar] [CrossRef]
- Deng, X.; Yao, Q.; Horvath, A.; Jiang, Z.; Zhao, J.; Fischer, T.; Sugiyama, T. The fission yeast ortholog of Coilin, Mug174, forms Cajal body-like nuclear condensates and is essential for cellular quiescence. Nucleic Acids Res. 2024, 52, 9174–9192. [Google Scholar] [CrossRef]
- Schilling, M.; Prusty, A.B.; Boysen, B.; Oppermann, F.S.; Riedel, Y.L.; Husedzinovic, A.; Rasouli, H.; König, A.; Ramanathan, P.; Reymann, J.; et al. TOR signaling regulates liquid phase separation of the SMN complex governing snRNP biogenesis. Cell Rep. 2021, 35, 109277. [Google Scholar] [CrossRef]
- Swygert, S.G.; Lin, D.; Portillo-Ledesma, S.; Lin, P.-Y.; Hunt, D.R.; Kao, C.-F.; Schlick, T.; Noble, W.S.; Tsukiyama, T. Local chromatin fiber folding represses transcription and loop extrusion in quiescent cells. eLife 2021, 10. [Google Scholar] [CrossRef]
- Schäfer, G.; McEvoy, C.R.E.; Patterton, H.-G. The Saccharomyces cerevisiae linker histone Hho1p is essential for chromatin compaction in stationary phase and is displaced by transcription. Proc. Natl. Acad. Sci. USA 2008, 105, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Rutledge, M.T.; Russo, M.; Belton, J.-M.; Dekker, J.; Broach, J.R. The yeast genome undergoes significant topological reorganization in quiescence. Nucleic Acids Res. 2015, 43, 8299–8313. [Google Scholar] [CrossRef]
- Laporte, D.; Courtout, F.; Tollis, S.; Sagot, I. QuiescentSaccharomyces cerevisiaeforms telomere hyperclusters at the nuclear membrane vicinity through a multifaceted mechanism involving Esc1, the Sir complex, and chromatin condensation. Mol. Biol. Cell 2016, 27, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Swygert, S.G.; Kim, S.; Wu, X.; Fu, T.; Hsieh, T.-H.; Rando, O.J.; Eisenman, R.N.; Shendure, J.; McKnight, J.N.; Tsukiyama, T. Condensin-Dependent Chromatin Compaction Represses Transcription Globally during Quiescence. Mol. Cell 2019, 73, 533–546.e4. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.-H.S.; Weiner, A.; Lajoie, B.; Dekker, J.; Friedman, N.; Rando, O.J. Mapping Nucleosome Resolution Chromosome Folding in Yeast by Micro-C. Cell 2015, 162, 108–119. [Google Scholar] [CrossRef]
- Ruault, M.; Loïodice, I.; Keister, B.D.; Even, A.; Garnier, M.; Baquero-Pérez, M.; Waterman, D.; Haber, J.E.; Blagoev, K.B.; Scolari, V.F.; et al. Esc1-Mediated Anchoring Regulates Telomere Clustering in Response to Metabolic Changes. bioRxiv 2025. [Google Scholar] [CrossRef]
- Ocampo, A.; Liu, J.; Schroeder, E.A.; Shadel, G.S.; Barrientos, A. Mitochondrial Respiratory Thresholds Regulate Yeast Chronological Life Span and its Extension by Caloric Restriction. Cell Metab. 2012, 16, 55–67. [Google Scholar] [CrossRef]
- Faure, G.; Jézéquel, K.; Roisné-Hamelin, F.; Bitard-Feildel, T.; Lamiable, A.; Marcand, S.; Callebaut, I. Discovery and Evolution of New Domains in Yeast Heterochromatin Factor Sir4 and Its Partner Esc1. Genome Biol. Evol. 2019, 11, 572–585. [Google Scholar] [CrossRef]
- Deshpande, I.; Keusch, J.J.; Challa, K.; Iesmantavicius, V.; Gasser, S.M.; Gut, H. The Sir4 H- BRCT domain interacts with phospho-proteins to sequester and repress yeast heterochromatin. EMBO J. 2020, 39, e103976. [Google Scholar] [CrossRef]
- Hocher, A.; Taddei, A. Subtelomeres as Specialized Chromatin Domains. BioEssays 2020, 42, e1900205. [Google Scholar] [CrossRef]
- Funabiki, H.; Hagan, I.; Uzawa, S.; Yanagida, M. Cell cycle-dependent specific positioning and clustering of centromeres and telomeres in fission yeast. J. Cell Biol. 1993, 121, 961–976. [Google Scholar] [CrossRef]
- Chikashige, Y.; Yamane, M.; Okamasa, K.; Tsutsumi, C.; Kojidani, T.; Sato, M.; Haraguchi, T.; Hiraoka, Y. Membrane proteins Bqt3 and -4 anchor telomeres to the nuclear envelope to ensure chromosomal bouquet formation. J. Cell Biol. 2009, 187, 413–427. [Google Scholar] [CrossRef]
- Ebrahimi, H.; Masuda, H.; Jain, D.; Cooper, J.P. Distinct ‘safe zones’ at the nuclear envelope ensure robust replication of heterochromatic chromosome regions. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Inoue, H.; Sun, W.; Takeshita, Y.; Huang, Y.; Xu, Y.; Kanoh, J.; Chen, Y. The Inner Nuclear Membrane Protein Bqt4 in Fission Yeast Contains a DNA-Binding Domain Essential for Telomere Association with the Nuclear Envelope. Structure 2019, 27, 335–343.e3. [Google Scholar] [CrossRef] [PubMed]
- Steglich, B.; Strålfors, A.; Khorosjutina, O.; Persson, J.; Smialowska, A.; Javerzat, J.-P.; Ekwall, K. The Fun30 Chromatin Remodeler Fft3 Controls Nuclear Organization and Chromatin Structure of Insulators and Subtelomeres in Fission Yeast. PLOS Genet. 2015, 11, e1005101. [Google Scholar] [CrossRef]
- Tange, Y.; Chikashige, Y.; Takahata, S.; Kawakami, K.; Higashi, M.; Mori, C.; Kojidani, T.; Hirano, Y.; Asakawa, H.; Murakami, Y.; et al. Inner nuclear membrane protein Lem2 augments heterochromatin formation in response to nutritional conditions. Genes Cells 2016, 21, 812–832. [Google Scholar] [CrossRef]
- Maestroni, L.; Reyes, C.; Vaurs, M.; Gachet, Y.; Tournier, S.; Géli, V.; Coulon, S. Nuclear envelope attachment of telomeres limits TERRA and telomeric rearrangements in quiescent fission yeast cells. Nucleic Acids Res. 2020, 48, 3029–3041. [Google Scholar] [CrossRef]
- Maestroni, L.; Audry, J.; Matmati, S.; Arcangioli, B.; Géli, V.; Coulon, S. Eroded telomeres are rearranged in quiescent fission yeast cells through duplications of subtelomeric sequences. Nat. Commun. 2017, 8, 1684–1684. [Google Scholar] [CrossRef] [PubMed]
- Weierich, C.; Brero, A.; Stein, S.; von Hase, J.; Cremer, C.; Cremer, T.; Solovei, I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosom. Res. 2003, 11, 485–502. [Google Scholar] [CrossRef]
- Pennarun, G.; Picotto, J.; Bertrand, P. Close Ties between the Nuclear Envelope and Mammalian Telomeres: Give Me Shelter. Genes 2023, 14, 775. [Google Scholar] [CrossRef]
- Mata, J.; Lyne, R.; Burns, G.; Bähler, J. The transcriptional program of meiosis and sporulation in fission yeast. Nat. Genet. 2002, 32, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson-Timmins, J.; Kristell, C.; Henningson, F.; Lyckman, S.; Bjerling, P. Reorganization of chromatin is an early response to nitrogen starvation in Schizosaccharomyces pombe. Chromosoma 2008, 118, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Taddei, A.; Gasser, S.M. Structure and Function in the Budding Yeast Nucleus. Genetics 2012, 192, 107–129. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Barrales, R.R. Beyond Tethering and the LEM domain: MSCellaneous functions of the inner nuclear membrane Lem2. Nucleus 2016, 7, 523–531. [Google Scholar] [CrossRef]
- Skribbe, M.; Soneson, C.; Stadler, M.B.; Schwaiger, M.; Sreechakram, V.N.S.; Iesmantavicius, V.; Hess, D.; Moreno, E.P.F.; Braun, S.; Seebacher, J.; et al. A comprehensive Schizosaccharomyces pombe atlas of physical transcription factor interactions with proteins and chromatin. Mol. Cell 2025, 85, 1426–1444.e8. [Google Scholar] [CrossRef]
Figure 1.
mTOR signaling integrates information on nutrient availability to regulate cell growth and proliferation. Signaling from different nutrient-sensing pathways converges on the mechanistic target of rapamycin (mTOR) kinase, which assembles into two different complexes, mTORC1 and mTORC2, to regulate central cellular processes related to growth and survival. In the presence of amino acids, guanine nucleotide exchange enables the Rag A/B-C/D complex to recruit mTORC1 to the lysosomal surface, where it can be activated by the small GTPase Rheb in its GTP-bound state. High Rheb-GTP levels are maintained in response to growth factor and insulin signaling via protein kinase cascades (including the kinases AKT and ERK) that inhibit the tuberous sclerosis complex (TSC1/2), a GTPase activating protein for Rheb. Activated mTORC1 phosphorylates cytoplasmic substrates and regulates nuclear transcriptional programs to promote growth. Key targets include Unc-51 Like Autophagy Activating Kinase 1 (ULK1; inhibitory phosphorylation suppresses autophagy); eIF4E-binding proteins (4E-BPs; phosphorylation prevents inhibition of cap-dependent translation); and S6 kinase (S6K), which phosphorylates diverse substrates including ribosomal protein S6 and transcription factors. In the nucleus, mTORC1 signaling maintains chromatin in an active state permissive for transcription. mTORC1 signaling is further modulated by cellular energy status via AMP-activated protein kinase (AMPK), which is activated when ATP levels drop. Growth factor signaling through phosphoinositide-3 kinase (PI3K) and AKT also activates mTORC2 at the plasma membrane, which regulates cell survival and cytoskeletal organization. mTORC2-dependent phosphorylation retains the key transcription factor FOXO in the cytoplasm, and its regulation of acetyl-CoA pools modulates histone acetylation levels. Inhibition of mTOR signaling—either by nutrient deprivation or chemical inhibitors such as rapamycin or Torin1—triggers entry into quiescence. Created in https://BioRender.com.
Figure 1.
mTOR signaling integrates information on nutrient availability to regulate cell growth and proliferation. Signaling from different nutrient-sensing pathways converges on the mechanistic target of rapamycin (mTOR) kinase, which assembles into two different complexes, mTORC1 and mTORC2, to regulate central cellular processes related to growth and survival. In the presence of amino acids, guanine nucleotide exchange enables the Rag A/B-C/D complex to recruit mTORC1 to the lysosomal surface, where it can be activated by the small GTPase Rheb in its GTP-bound state. High Rheb-GTP levels are maintained in response to growth factor and insulin signaling via protein kinase cascades (including the kinases AKT and ERK) that inhibit the tuberous sclerosis complex (TSC1/2), a GTPase activating protein for Rheb. Activated mTORC1 phosphorylates cytoplasmic substrates and regulates nuclear transcriptional programs to promote growth. Key targets include Unc-51 Like Autophagy Activating Kinase 1 (ULK1; inhibitory phosphorylation suppresses autophagy); eIF4E-binding proteins (4E-BPs; phosphorylation prevents inhibition of cap-dependent translation); and S6 kinase (S6K), which phosphorylates diverse substrates including ribosomal protein S6 and transcription factors. In the nucleus, mTORC1 signaling maintains chromatin in an active state permissive for transcription. mTORC1 signaling is further modulated by cellular energy status via AMP-activated protein kinase (AMPK), which is activated when ATP levels drop. Growth factor signaling through phosphoinositide-3 kinase (PI3K) and AKT also activates mTORC2 at the plasma membrane, which regulates cell survival and cytoskeletal organization. mTORC2-dependent phosphorylation retains the key transcription factor FOXO in the cytoplasm, and its regulation of acetyl-CoA pools modulates histone acetylation levels. Inhibition of mTOR signaling—either by nutrient deprivation or chemical inhibitors such as rapamycin or Torin1—triggers entry into quiescence. Created in https://BioRender.com.
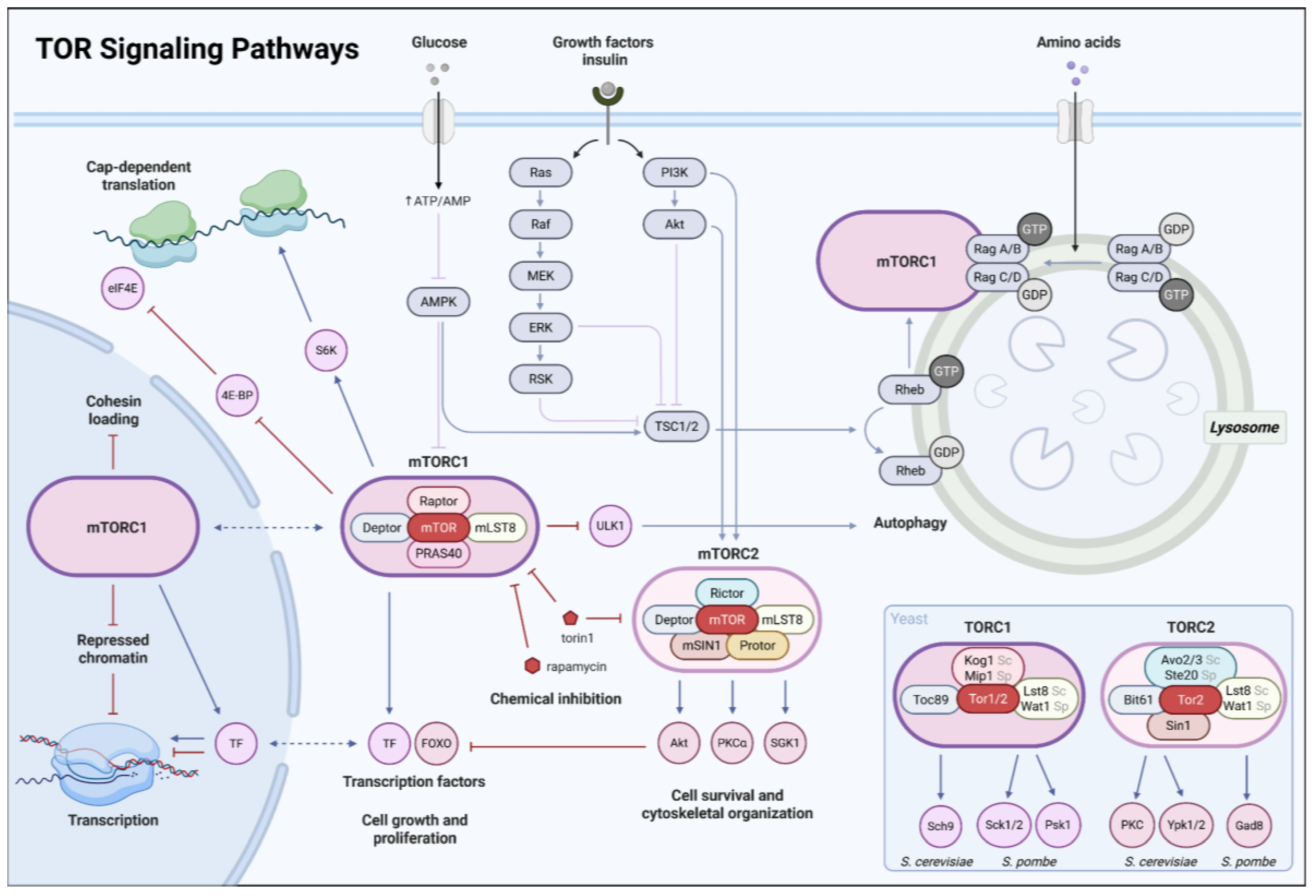
Figure 2.
Mechanisms of transcriptional reprogramming through concerted actions of repressor proteins. Histone deacetylation drives quiescence-associated transcriptional repression in S. cerevisiae. In log cells (top), high default levels of histone acetylation maintain open promoters and a chromatin structure permissive for transcription, as TORC1 and PKA signaling keep the transcriptional repressors Stb3 phosphorylated and inactive. In Q cells (bottom), TORC1 inactivation activates Stb3, which binds rRNA-processing elements (RRPEs) at ribosome biogenesis genes, and directly recruits the histone deacetylase complex Rpd3L. Concurrently, differential phosphorylation promotes nuclear localization of the stress-regulated transcription factors Msn2 and Msn4, which in return induce XBP1 expression. Xbp1 recruits Rpd3L to Xbp1-response elements. Created in https://BioRender.com.
Figure 2.
Mechanisms of transcriptional reprogramming through concerted actions of repressor proteins. Histone deacetylation drives quiescence-associated transcriptional repression in S. cerevisiae. In log cells (top), high default levels of histone acetylation maintain open promoters and a chromatin structure permissive for transcription, as TORC1 and PKA signaling keep the transcriptional repressors Stb3 phosphorylated and inactive. In Q cells (bottom), TORC1 inactivation activates Stb3, which binds rRNA-processing elements (RRPEs) at ribosome biogenesis genes, and directly recruits the histone deacetylase complex Rpd3L. Concurrently, differential phosphorylation promotes nuclear localization of the stress-regulated transcription factors Msn2 and Msn4, which in return induce XBP1 expression. Xbp1 recruits Rpd3L to Xbp1-response elements. Created in https://BioRender.com.
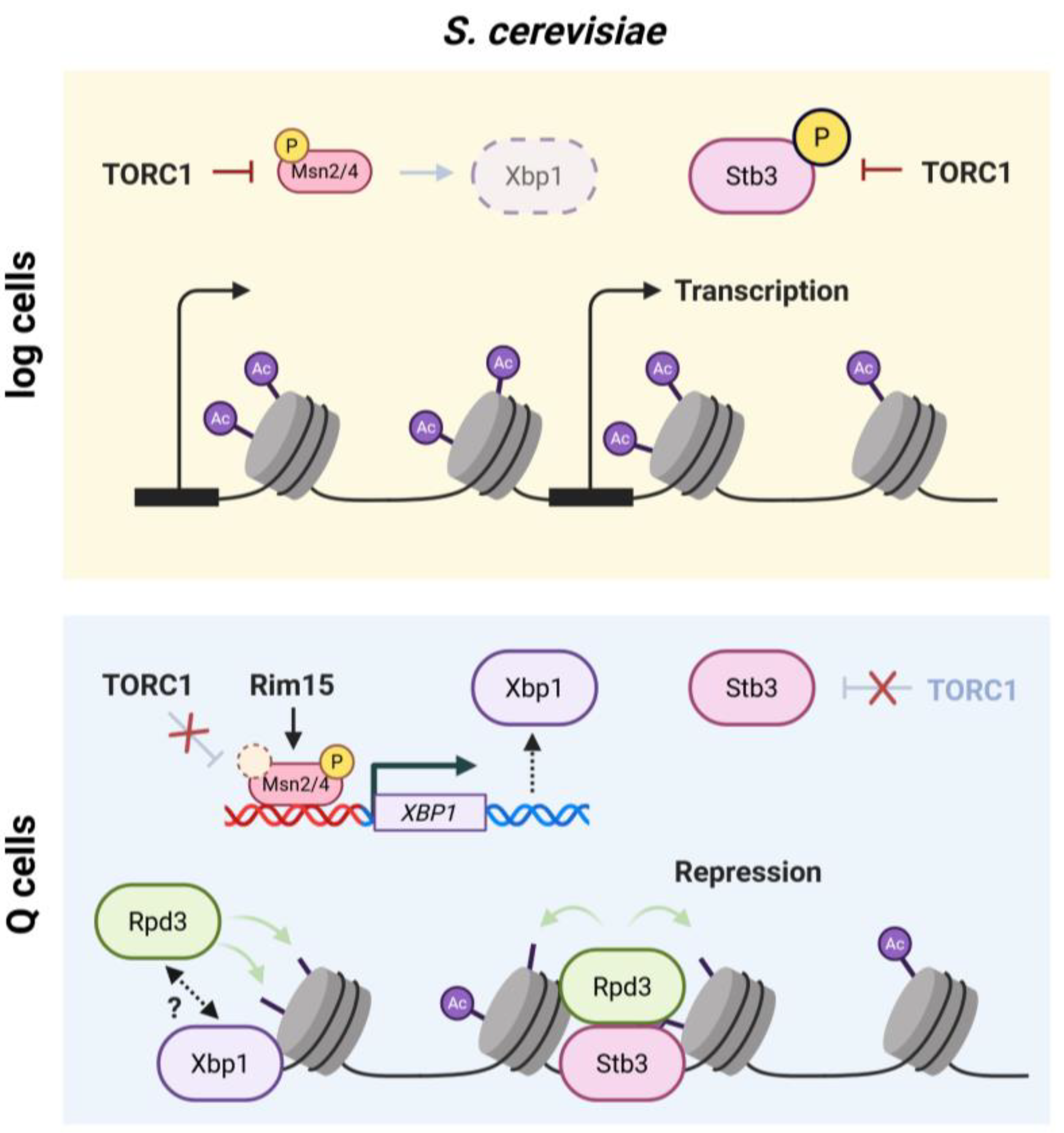
Figure 3.
Remodeling of repressive chromatin structures Repressive histone methylation contributes to transcriptional reprogramming in S. pombe. In log cells (top), silencing factors primarily act on constitutive heterochromatin (left). In Q cells, the RNase III enzyme Dicer (Dcr1) redistributes to euchromatic loci to generate small interfering RNAs (siRNAs), supported by RNA-dependent RNA polymerase (Rdp1). These siRNAs guide the RNA-induced transcriptional silencing complex (RITS) to nascent RNA, where it recruits the H3K9 methyltransferase Clr4, depositing repressive marks to form facultative heterochromatin at the silenced loci. RITS and Clr4 (not shown) are stabilized on heterochromatin through recognition of H3K9me via their chromodomains, thereby reinforcing heterochromatin assembly. For simplicity, assembly of HP1 proteins is not shown. The initial mechanism targeting Dcr1 and RITS to euchromatic loci remains unknown, and Clr4 may also be recruited by RNAi-independent mechanisms. Created in https://BioRender.com.
Figure 3.
Remodeling of repressive chromatin structures Repressive histone methylation contributes to transcriptional reprogramming in S. pombe. In log cells (top), silencing factors primarily act on constitutive heterochromatin (left). In Q cells, the RNase III enzyme Dicer (Dcr1) redistributes to euchromatic loci to generate small interfering RNAs (siRNAs), supported by RNA-dependent RNA polymerase (Rdp1). These siRNAs guide the RNA-induced transcriptional silencing complex (RITS) to nascent RNA, where it recruits the H3K9 methyltransferase Clr4, depositing repressive marks to form facultative heterochromatin at the silenced loci. RITS and Clr4 (not shown) are stabilized on heterochromatin through recognition of H3K9me via their chromodomains, thereby reinforcing heterochromatin assembly. For simplicity, assembly of HP1 proteins is not shown. The initial mechanism targeting Dcr1 and RITS to euchromatic loci remains unknown, and Clr4 may also be recruited by RNAi-independent mechanisms. Created in https://BioRender.com.
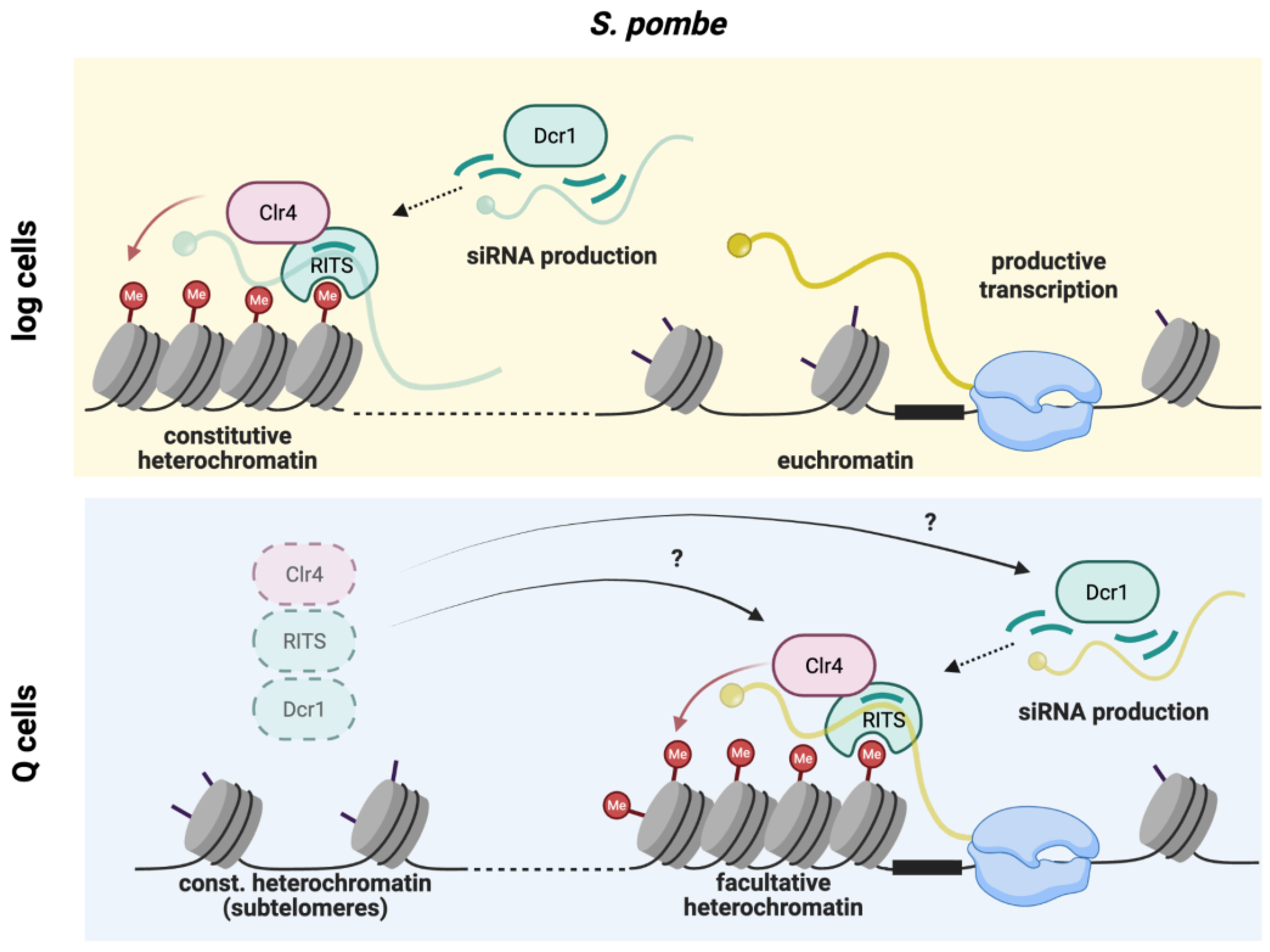
Figure 4.
Quiescence-induced intron retention in ribosomal protein mRNAs is conserved in S. pombe. Comparison of intron retention rates during log phase (+N) and after 24h of nitrogen starvation (-N) for mRNAs encoding ribosomal proteins and all other spliced mRNAs. Intron retention index based on published RNA-seq data (E-MTAB-1154; reference [55]) calculated as the ratio of intronic reads over exonic reads for each transcript.
Figure 4.
Quiescence-induced intron retention in ribosomal protein mRNAs is conserved in S. pombe. Comparison of intron retention rates during log phase (+N) and after 24h of nitrogen starvation (-N) for mRNAs encoding ribosomal proteins and all other spliced mRNAs. Intron retention index based on published RNA-seq data (E-MTAB-1154; reference [55]) calculated as the ratio of intronic reads over exonic reads for each transcript.
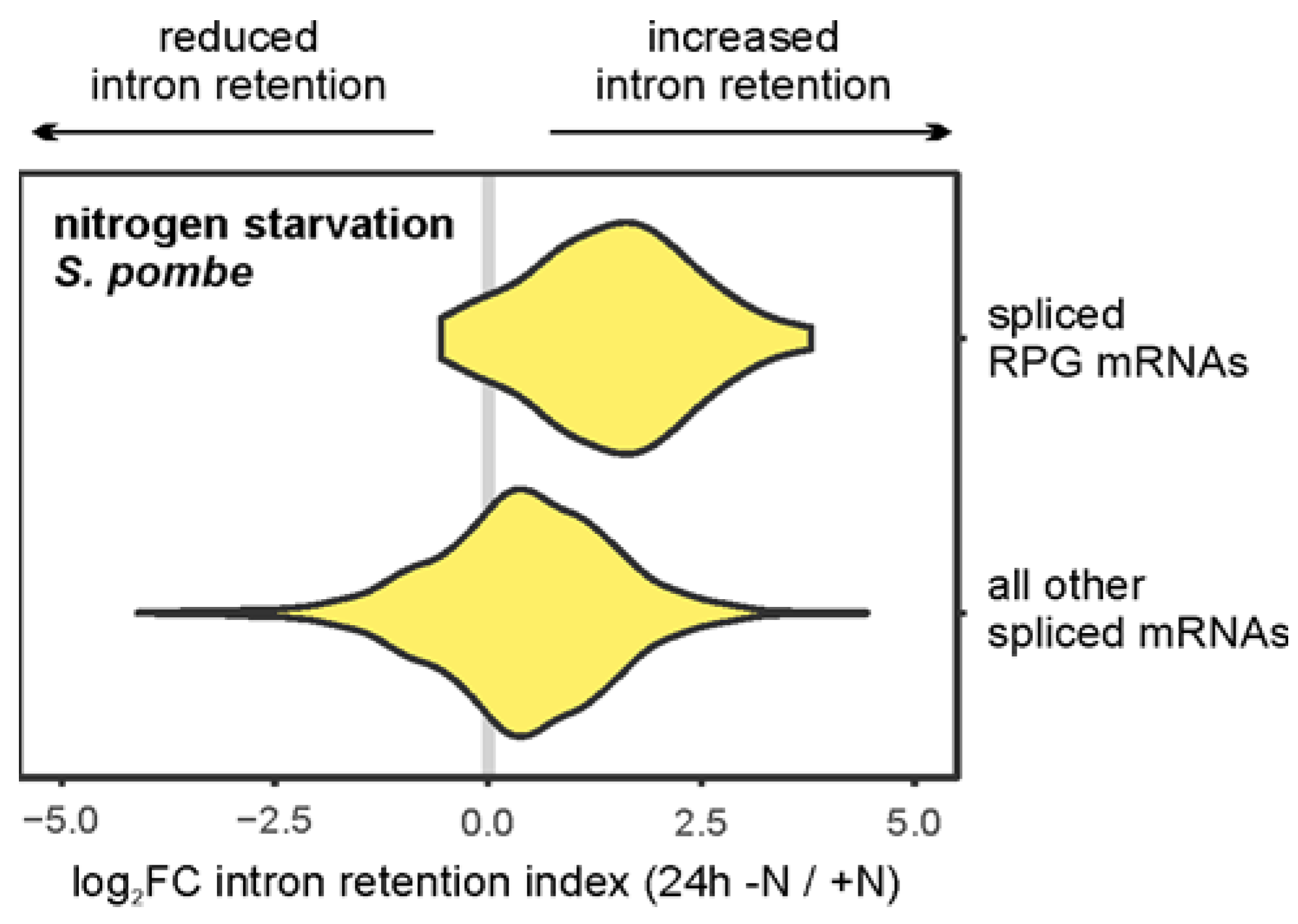
Figure 5.
Non-coding RNAs accumulate in quiescent fission yeast. A. Differential gene expression analysis comparing RNA levels during log phase (+N) and after 24h of nitrogen starvation (-N) for mRNAs (yellow) and ncRNAs (turquoise) (DESeq2). P-values adjusted for multiple testing with the Benjamini-Hochberg procedure to control the false discovery rate (FDR). Size of circles indicates absolute expression levels in nitrogen-starved cells (read counts per kilobase). RNA-seq data from E-MTAB-1154 (reference [55]; n = 2). B. Comparison of RNA expression level changes in nitrogen-starved cells and cells that lack the exonuclease Rrp6, a catalytic subunit of nuclear exosome for mRNAs (yellow) and ncRNAs (turquoise). Size of circles indicates statistical significance of the expression level change (FDR-corrected p-values). Contours of the 2d density estimates for both RNA biotypes are shown in yellow and turquoise, respectively. RNA-seq data from E-MTAB-1154 (reference [55]; n = 2) and GSE148799 (reference [132]; n = 3).
Figure 5.
Non-coding RNAs accumulate in quiescent fission yeast. A. Differential gene expression analysis comparing RNA levels during log phase (+N) and after 24h of nitrogen starvation (-N) for mRNAs (yellow) and ncRNAs (turquoise) (DESeq2). P-values adjusted for multiple testing with the Benjamini-Hochberg procedure to control the false discovery rate (FDR). Size of circles indicates absolute expression levels in nitrogen-starved cells (read counts per kilobase). RNA-seq data from E-MTAB-1154 (reference [55]; n = 2). B. Comparison of RNA expression level changes in nitrogen-starved cells and cells that lack the exonuclease Rrp6, a catalytic subunit of nuclear exosome for mRNAs (yellow) and ncRNAs (turquoise). Size of circles indicates statistical significance of the expression level change (FDR-corrected p-values). Contours of the 2d density estimates for both RNA biotypes are shown in yellow and turquoise, respectively. RNA-seq data from E-MTAB-1154 (reference [55]; n = 2) and GSE148799 (reference [132]; n = 3).
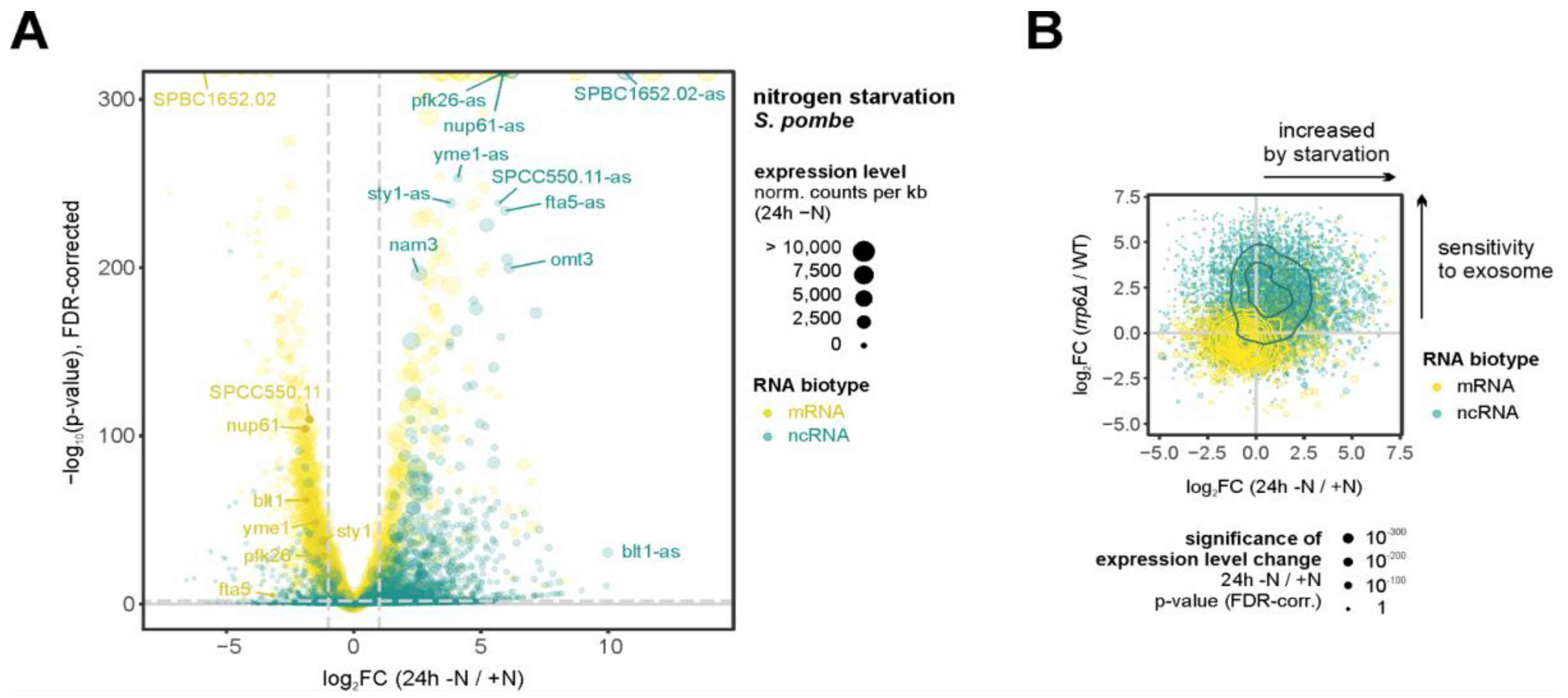
Figure 6.
Mechanisms of lncRNA-dependent regulation. lncRNA expression can regulate gene expression through mechanisms mediated by the RNA molecule itself (left). These include recruiting epigenetic modifiers to chromatin to alter local histone marks and transcription (top); base-pairing with a complementary RNA to alter protein binding and its expression (middle); or nucleating biomolecular condensates that control the stability or activity of sequestered mRNAs (bottom). Alternatively, the act of transcription can be regulatory independent of the lncRNA product (right). This can involve transcriptional interference, where lncRNA synthesis competes with and represses an adjacent gene (top), or promoter remodelling, where the passage of RNA polymerase opens an occluded promoter to activate a nearby gene (bottom). RNA biotype indicated by colour - mRNAs (yellow) and ncRNAs (turquoise). Created in https://BioRender.com.
Figure 6.
Mechanisms of lncRNA-dependent regulation. lncRNA expression can regulate gene expression through mechanisms mediated by the RNA molecule itself (left). These include recruiting epigenetic modifiers to chromatin to alter local histone marks and transcription (top); base-pairing with a complementary RNA to alter protein binding and its expression (middle); or nucleating biomolecular condensates that control the stability or activity of sequestered mRNAs (bottom). Alternatively, the act of transcription can be regulatory independent of the lncRNA product (right). This can involve transcriptional interference, where lncRNA synthesis competes with and represses an adjacent gene (top), or promoter remodelling, where the passage of RNA polymerase opens an occluded promoter to activate a nearby gene (bottom). RNA biotype indicated by colour - mRNAs (yellow) and ncRNAs (turquoise). Created in https://BioRender.com.
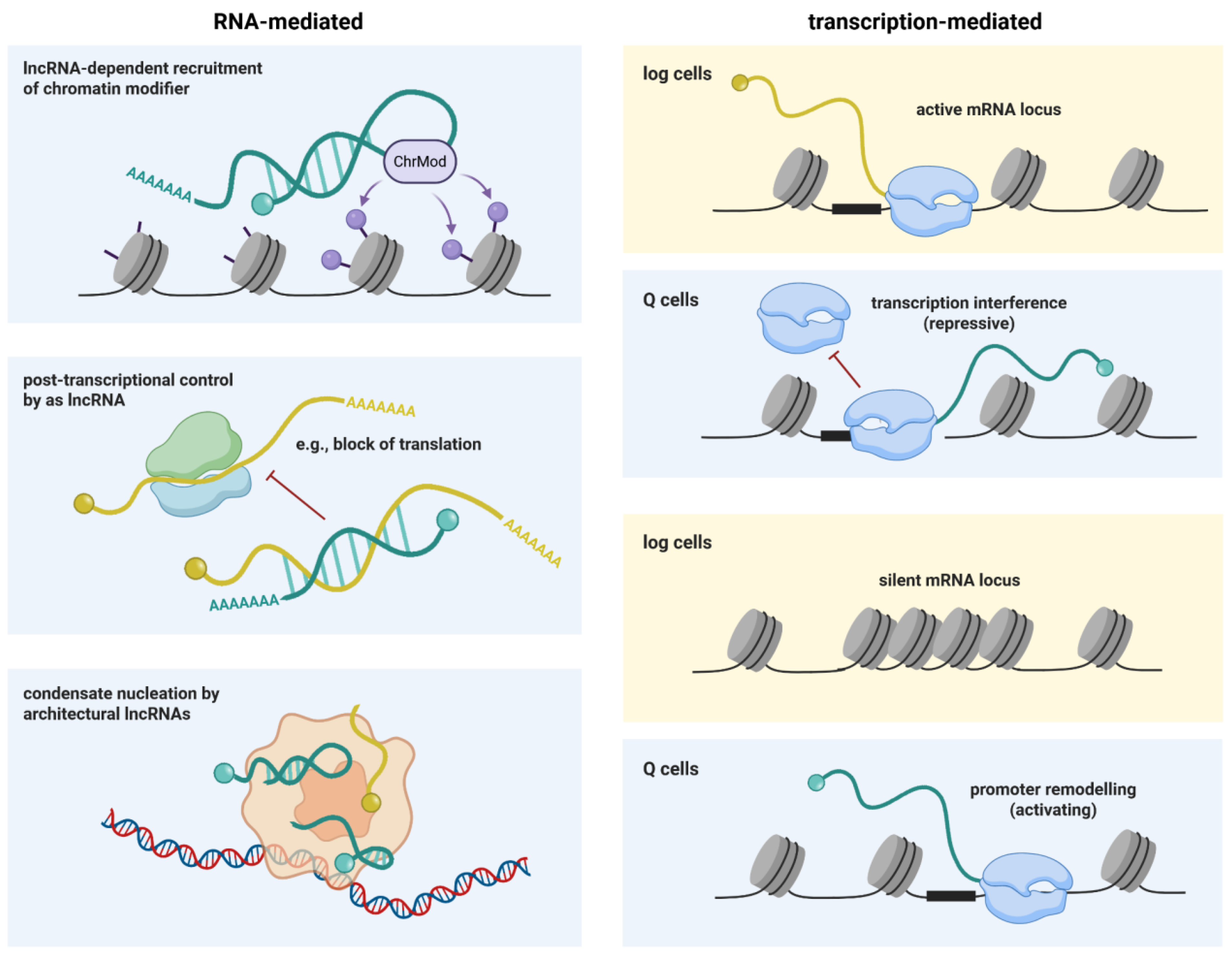

Table 1.
Glossary of key terms and abbreviations.
| Abbreviation | Definition | Explanation | Species context |
|---|---|---|---|
| G0 | G0 phase | Reversible cell-cycle arrest in which cells stop dividing but retain the ability to re-enter proliferation. | conserved |
| Q cells | Quiescent cells | Cells in a stable, non-proliferative state with reduced biosynthesis and extensive nuclear remodeling. | conserved |
| Diapause | Embryonic diapause | Reversible developmental arrest of the mammalian embryo, typically at the blastocyst stage. | metazoans (mammals) |
| TOR | Target of Rapamycin | Nutrient-sensing kinase pathway coordinating growth, metabolism, and quiescence. | yeast |
| mTOR | Mechanistic Target of Rapamycin | Metazoan TOR pathway integrating nutrient and growth-factor signaling to regulate dormancy and growth. | metazoans |
| TORC1 | TOR complex 1 | TOR-containing complex promoting ribosome biogenesis and translation; inhibited during quiescence. | conserved |
| TORC2 | TOR complex 2 | TOR-containing complex regulating stress responses and survival pathways. | conserved |
| PKA | Protein kinase A | cAMP-dependent kinase promoting growth-associated transcription and antagonizing quiescence. | conserved |
| DS | Diauxic shift | Metabolic transition in which yeast cells switch from fermentative growth on glucose to respiratory metabolism of alternative carbon sources after glucose depletion. | S. cerevisiae |
| RNAPII | RNA polymerase II | Enzyme transcribing mRNAs and many non-coding RNAs; redistributes during quiescence. | conserved |
| Pol I | RNA polymerase I | Enzyme transcribing ribosomal RNA genes at the rDNA locus; tightly linked to growth state. | conserved |
| RiBi | Ribosome biogenesis | Coordinate transcriptional program encoding ribosomal proteins, rDNA processing factors, and ribosome assembly component. | S. cerevisiae (conserved in eukaryotes) |
| RRPE | rRNA processing element | Cis-regulatory promoter motif enriched in ribosome biogenesis genes that mediates coordinated transcriptional repression via binding of Stb3. | S. cerevisiae |
| RP | Ribosomal proteins | Structural and functional components of ribosomes; their expression is tightly repressed during quiescence. | conserved |
| Stb3 | Sin3-binding protein 3 | Nutrient regulated transcriptional repressor of RiBi genes that recruit Rpd3 to promote chromatin compaction and biosynthetic shutdown | S. cerevisiae |
| Xbp1 | XhoI site- binding protein 1 | Stress-induced transcriptional repressor that binds promoter regions of growth-associated genes | S. cerevisiae |
| HDAC | Histone deacetylase | Enzymes removing acetyl groups from histones, promoting chromatin compaction and repression. | conserved |
| Rpd3 | Reduced potassium dependency 3 | Class I HDAC broadly retargeted to promoters during budding yeast quiescence. | S. cerevisiae |
| Set1C / COMPASS | Set1 complex | H3K4 methyltransferase complex depositing H3K4me3 at promoters. | conserved |
| SAGA | Spt-Ada-Gcn5 acetyltransferase complex | Transcriptional coactivator complex with histone acetyltransferase and deubiquitylation activities that promotes transcription initiation at stress- and growth-related genes. | conserved |
| H3K4me3 | H3 lysine 4 trimethylation | Active promoter mark selectively retained or redistributed during quiescence. | conserved |
| H3K36me3 | H3 lysine 36 trimethylation | Histone modification associated with transcription elongation and gene body marking, largely maintained during quiescence (in yeast). | conserved |
| Ino80C | Ino80 chromatin remodeling complex | ATP-dependent remodeler evicting H2A.Z and regulating transcription during quiescence. | conserved |
| H2A.Z | H2A.Z histone variant | Histone variant modulating promoter responsiveness and chromatin boundaries. | conserved |
| Hho1 | Histone H1 homolog | Linker histone contributing to chromatin compaction without enforcing full silencing. | S. cerevisiae |
| Pac1C | Polymerase-associated factor 1 complex | Transcription elongation complex that associates with RNAPII and regulates histone modifications linked to active transcription | conserved |
| Clr3 / Clr6 | Cryptic loci regulators 3/6 | HDACs contributing to heterochromatin formation and quiescence maintenance. | S. pombe |
| Clr4 | Cryptic loci regulators 4 | Histone H3K9 methyltransferase/Suvar 39h homolog, heterochromatin initiation and spreading. | S. pombe |
| H3K9me2/3 | H3 lysine 9 methylation (di-/trimethylation) |
Repressive histone mark defining constitutive and facultative heterochromatin. | conserved (absent in S. cerevisiae) |
| H3K27me3 | H3 lysine 27 trimethylation | Polycomb-associated repressive mark controlling facultative repression. | metazoans |
| H4K20me3 | H4 lysine 20 trimethylation | Deeply repressive mark associated with chromatin compaction and long-term arrest. | metazoans |
| PRC2 | Polycomb repressive complex 2 | Complex depositing H3K27me3 to mediate stable transcriptional repression. | metazoans |
| EZH1 | Enhancer of zeste homolog 1 | PRC2 catalytic subunit prevalent in non-proliferative and quiescent cells. | metazoans |
| EZH2 | Enhancer of zeste homolog 2 | PRC2 catalytic subunit associated with proliferation and activation. | metazoans |
| HP1 | Heterochromatin protein 1 | Chromodomain protein binding H3K9me and promoting heterochromatin spreading. | conserved (functional analogs in S. pombe) |
| RNAi | RNA interference | Small RNA–guided silencing pathway coupling transcripts to heterochromatin formation. | S. pombe, metazoans |
| Dcr1 | Dicer | RNase III enzyme producing siRNAs; also performs RNAi-independent roles in quiescence survival. | S. pombe |
| Ago1 | Argonaute 1 | Small-RNA effector protein targeting transcripts and chromatin. | S. pombe, metazoans |
| RITS | RNA-induced transcriptional silencing complex | RNAi effector complex linking siRNAs to chromatin-based repression. | S. pombe |
| lncRNA | Long non-coding RNA | >200-nt RNAs regulating chromatin, transcription, and nuclear organization. | conserved |
| asRNA | Antisense RNA | Non-coding RNA transcribed opposite a gene, often mediating transcriptional interference. | conserved |
| PAS | Polyadenylation site | RNA cleavage site determining 3′-UTR length and post-transcriptional regulation. | conserved |
| 3′-UTR | 3′ untranslated region | mRNA region controlling stability, localization, and translation efficiency. | conserved |
| CID | Chromosomal interaction domain | Local self-interacting chromatin unit detected by Hi-C. | yeast (conceptually conserved) |
| L-CID | Large chromosomal interaction domain | Larger chromatin domains that become more insulated during quiescence. | S. cerevisiae |
| Condensin | Condensin complex | SMC complex organizing higher-order chromosome structure via loop formation. | conserved |
| Cajal body | Cajal body | Nuclear condensate coordinating snRNA and snoRNA maturation with growth state. | metazoans, S. pombe |
| SMN | Survival of motor neurons protein | snRNP/sn(o)RNP assembly factor regulated by mTOR signaling. | metazoans |
| Myc | Myelocytomatosis oncogene family transcription factor | Global regulator of cell growth and proliferation, repressed during quiescence. | metazoans |
Table 2.
Nuclear hallmarks and molecular effectors of the quiescent state in unicellular and multicellular organisms described in this review. Superscript 1: yeast (S. cerevisiae or S. pombe); subscript 2: metazoans or mammalian cells.
Table 2.
Nuclear hallmarks and molecular effectors of the quiescent state in unicellular and multicellular organisms described in this review. Superscript 1: yeast (S. cerevisiae or S. pombe); subscript 2: metazoans or mammalian cells.
| Cellular process | Hallmarks/phenotype | Nuclear mechanism/factors |
|---|---|---|
| Cell cycle | Stable arrest with ability to re-enter proliferation | TOR1/mTOR2 inhibition, PKA1 signaling, Myc2 downregulation |
| Growth and metabolism | Reduced protein synthesis and metabolic activity | Repression of ribosomal- RNA and protein biogenesis1,2, translational control1,2, intron retention1,2 |
| Autophagy and survival | Increased autophagy and stress resistance | TOR1 inactivation, nutrient sensing pathways1,2 |
| Transcriptional regulation | Global transcriptional repression with selective gene activation | RNAPII redistribution)1,2 transcriptional repressors (Xbp11, Stb31), chromatin remodeling1,2 |
| Chromatin state | Increased chromatin compaction and heterochromatin formation | HDACs (Rpd31, Clr31, Clr61), histone methyltransferases (Clr41, PRC22) |
| Histone modifications | Global hypoacetylation, accumulation of repressive methylation marks | Histone deacetylation1,2, H3K91,2 / H3K272 / H4K202 methylation pathways |
| Genome organization | Nuclear and 3D genome reorganization | Condensin1,2, heterochromatin dynamics1,2, telomere clustering1, stress-induced gene cluster relocalization1 |
| RNA abundance | Strong reduction in total mRNA levesl | Reduced RNAP II initiation and elongation1, RNA decay modulation1,2 |
| RNA processing | Widespread intron retention | Splicing repression1,2, spliceosome retention1, TOR signaling1 |
| Post-transcriptional regulation | 3′-UTR lengthening, altered RNA stability | Polyadenylation site selection bias1, reduced cleavage1 |
| Transcript storage | Accumulation of RNAs in condensates | RNA-Protein phase separation1,2 |
| Non-coding RNA levels | Increased lncRNAs and other ncRNAs | Reduced nuclear exosome activity1,2, lncRNA-mediated recruitment of chromatin modifiers2 |
| Stress and metabolic genes |
Relative enrichment of stress-response and nutrient scavenging transcripts | Ino80C1, PafC1, histone turnover1, replication-independent chromatin remodeling1 |
| Re-proliferation capacity | Transcriptional reactivation upon nutrient readdition | Histone acetyltransferases (SAGA1, NuA41), TOR reactivation1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



