
Submitted:
30 December 2025
Posted:
01 January 2026
You are already at the latest version
Abstract
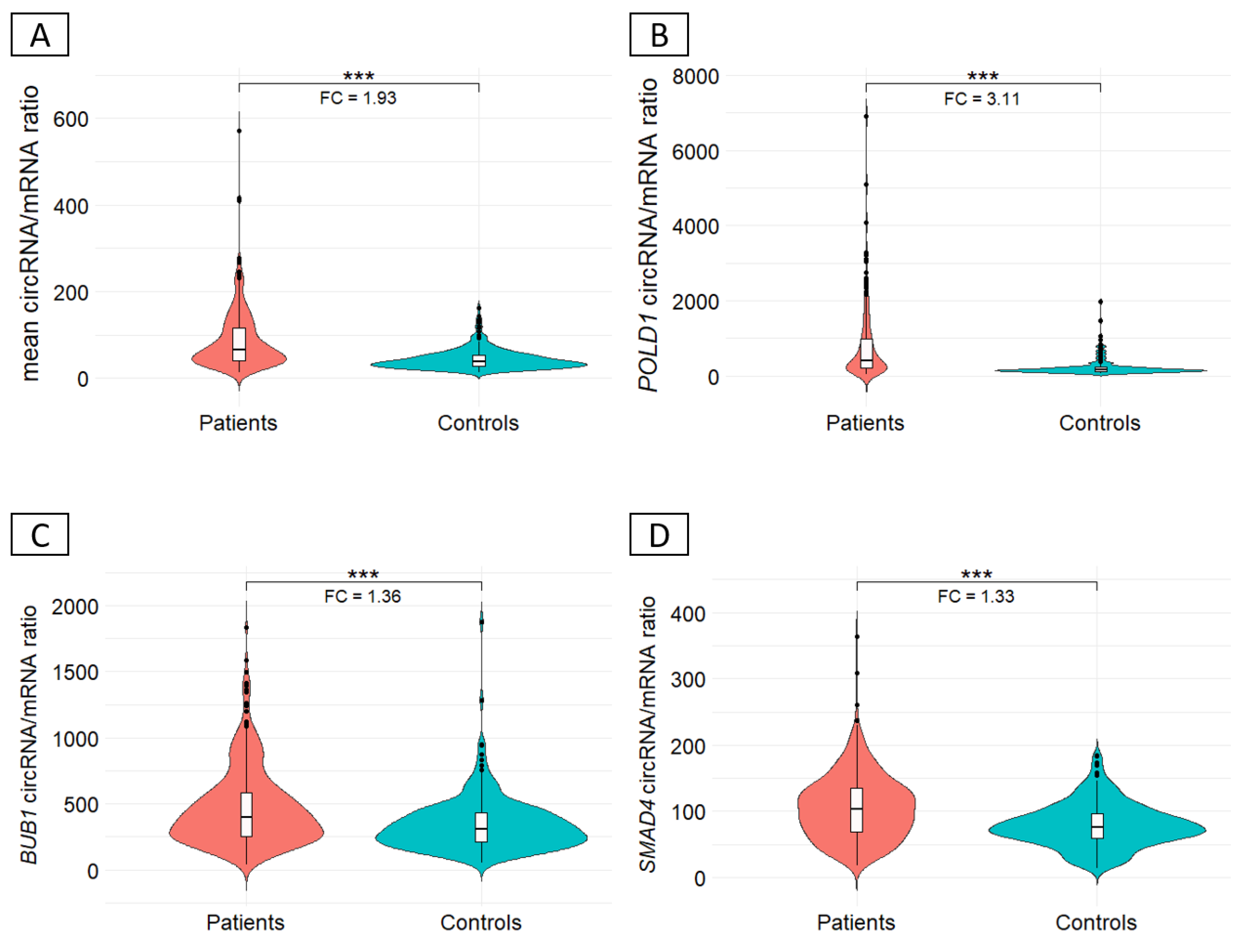
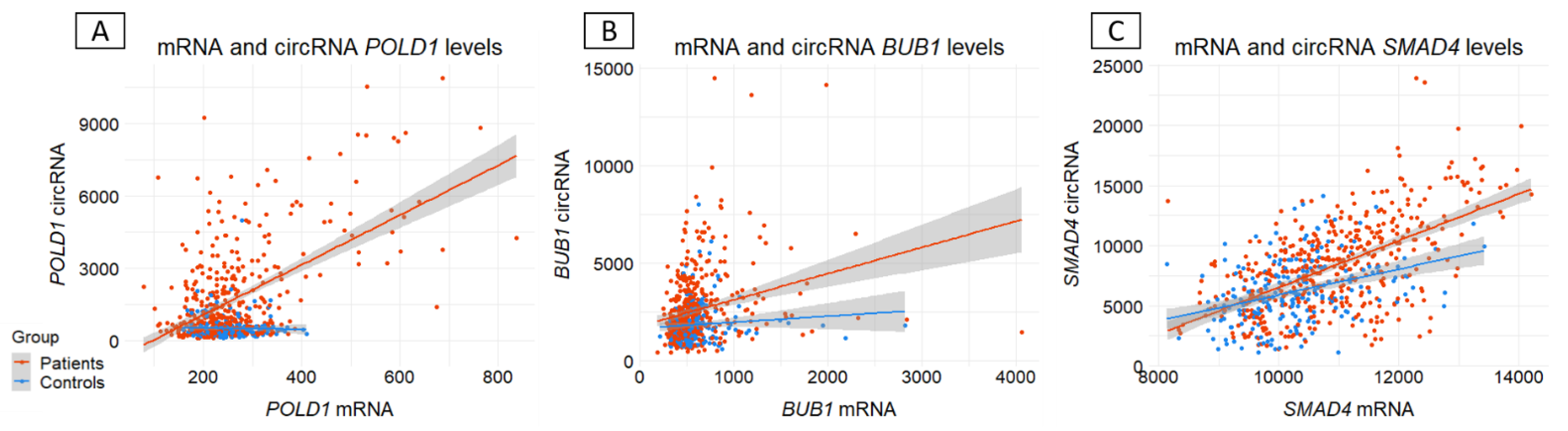
Background/Objectives: Circular RNAs (circRNAs) are emerging players in human diseases, with function as part of competing endogenous networks. Given the importance of messenger RNA (mRNA) regulation in human diseases and the potential of circRNAs in this regulation, we studied the circRNA-mRNA couple in blood within a cohort of 712 patients suspected of having hereditary colorectal cancer (CRC) and 249 matched controls. Methods: The circRNA-mRNA couple was studied by SEALigHTS (Splice and Expression Analyses by exon Ligation and High-Throughput Sequencing) using a panel of 23 genes involved in CRC predisposition, i.e., 788 probes designed at exon ends, enabling the exploration of all exon-exon junctions. Following reverse transcription and probe hybridization on cDNA, nearby probes are ligated, and the number of ligations quantified using unique molecular identifiers and sequencing. Results: We described 220 circular junctions, including 47 novel ones. The circRNA/mRNA ratio was 1.93-fold higher in patients compared to controls (p<2x10-16), irrespective of age of cancer onset. This increase was mainly driven by POLD1 (fold change 3.11) and a single circPOLD1 with oncogenic potential. Conclusions: This study supports the idea of a physiological balance between circRNA and mRNA that can be disrupted under pathological conditions. It rules out a competitive mechanism between circular and linear transcripts in CRC predisposition and raises questions about the role of specific circRNAs in the development of CRC, either as a cause or a consequence.
Keywords:
1. Introduction
2. Materials and Methods
- CRC in two first-degree relatives, one being diagnosed before 61 years of age
- CRC diagnosed before 51 years of age or advanced colorectal adenoma (diameter over 1 cm, and/or tubulovillous or villous and/or with high-grade dysplasia) before 41 years of age
- Multiple primary CRCs in the same individual, the first one being diagnosed before 61 years of age
- Lynch syndrome, as defined by the presence in the patient of a germline MMR gene pathogenic variant and/or a MSI tumour, in the context of a suggestive presentation (familial history, early age of onset)
- Adenomatous polyposis, as defined by more than 10 histologically proven adenomas
- Hamartomatous polyposis, as defined by the presence of histologically proven hamartomas
3. Results
3.1. General considerations

3.2. Backsplicing landscape
3.3. Ratio circRNA/mRNA between CRC patients and controls
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brenner, S.; Jacob, F.; Meselson, M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Gros, F.; Hiatt, H.; Gilbert, W.; Kurland, C.G.; Risebrough, R.W.; Watson, J.D. Unstable Ribonucleic Acid Revealed by Pulse Labelling of Escherichia Coli. Nature 1961, 190, 581–585. [Google Scholar] [CrossRef]
- Bentley, D.L. Coupling mRNA Processing with Transcription in Time and Space. Nat Rev Genet 2014, 15, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; den Dunnen, J.T.; et al. Full-Length mRNA Sequencing Uncovers a Widespread Coupling between Transcription Initiation and mRNA Processing. Genome Biol 2018, 19, 46. [Google Scholar] [CrossRef]
- Rodríguez-Molina, J.B.; West, S.; Passmore, L.A. Knowing When to Stop: Transcription Termination on Protein-Coding Genes by Eukaryotic RNAPII. Mol Cell 2023, 83, 404–415. [Google Scholar] [CrossRef]
- Mironov, A.; Petrova, M.; Margasyuk, S.; Vlasenok, M.; Mironov, A.A.; Skvortsov, D.; Pervouchine, D.D. Tissue-Specific Regulation of Gene Expression via Unproductive Splicing. Nucleic Acids Res 2023, 51, 3055–3066. [Google Scholar] [CrossRef]
- Passmore, L.A.; Coller, J. Roles of mRNA Poly(A) Tails in Regulation of Eukaryotic Gene Expression. Nat Rev Mol Cell Biol 2022, 23, 93–106. [Google Scholar] [CrossRef]
- Aregger, M.; Cowling, V.H. Regulation of mRNA Capping in the Cell Cycle. RNA Biol 2017, 14, 11–14. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. mRNAs, Proteins and the Emerging Principles of Gene Expression Control. Nat Rev Genet 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Das, A.; Rout, P.K.; Gorospe, M.; Panda, A.C. Rolling Circle cDNA Synthesis Uncovers Circular RNA Splice Variants. IJMS 2019, 20, 3988. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The Multilayered Complexity of ceRNA Crosstalk and Competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-Coding RNA Networks in Cancer. Nat Rev Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Wu, Y.-Y.; Kuo, H.-C. Functional Roles and Networks of Non-Coding RNAs in the Pathogenesis of Neurodegenerative Diseases. J Biomed Sci 2020, 27, 49. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, Y.; Zhang, X.; Dang, Y.; Cheng, Y.; Hua, W.; Teng, M.; Wang, S.; Lu, X. Novel lncRNA-miRNA-mRNA Competing Endogenous RNA Triple Networks Associated Programmed Cell Death in Heart Failure. Front Cardiovasc Med 2021, 8, 747449. [Google Scholar] [CrossRef]
- Huang, Z.; Kuang, N. Construction of a ceRNA Network Related to Rheumatoid Arthritis. Genes (Basel) 2022, 13, 647. [Google Scholar] [CrossRef]
- Qu, S.; Liu, Z.; Yang, X.; Zhou, J.; Yu, H.; Zhang, R.; Li, H. The Emerging Functions and Roles of Circular RNAs in Cancer. Cancer Letters 2018, 414, 301–309. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs Are Abundant, Conserved, and Associated with ALU Repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef]
- Wen, X.; Huang, C.; Xie, H.; Hu, D.; Luo, J.; Li, K. The Applications of CircRNA in the Diagnosis and Treatment of Alzheimer’s Disease. Mol Neurobiol 2024. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Liu, D.-L.; Xiong, X.-D. Circular RNAs as Competing Endogenous RNAs in Cardiovascular and Cerebrovascular Diseases: Molecular Mechanisms and Clinical Implications. Front Cardiovasc Med 2021, 8, 682357. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Jun, E.; Okugawa, Y.; Toiyama, Y.; Borazanci, E.; Bolton, J.; Taketomi, A.; Kim, S.C.; Shang, D.; Von Hoff, D.; et al. A Circulating Panel of circRNA Biomarkers for the Noninvasive and Early Detection of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2024, 166, 178–190.e16. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient microRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Huang, D.; Zhu, X.; Ye, S.; Zhang, J.; Liao, J.; Zhang, N.; Zeng, X.; Wang, J.; Yang, B.; Zhang, Y.; et al. Tumour Circular RNAs Elicit Anti-Tumour Immunity by Encoding Cryptic Peptides. Nature 2024, 625, 593–602. [Google Scholar] [CrossRef]
- Zeng, K.; Peng, J.; Xing, Y.; Zhang, L.; Zeng, P.; Li, W.; Zhang, W.; Pan, Z.; Zhou, C.; Lin, J. A Positive Feedback Circuit Driven by m6A-Modified Circular RNA Facilitates Colorectal Cancer Liver Metastasis. Mol Cancer 2023, 22, 202. [Google Scholar] [CrossRef]
- Li, Z.; Huang, C.; Bao, C.; Chen, L.; Lin, M.; Wang, X.; Zhong, G.; Yu, B.; Hu, W.; Dai, L.; et al. Exon-Intron Circular RNAs Regulate Transcription in the Nucleus. Nat Struct Mol Biol 2015, 22, 256–264. [Google Scholar] [CrossRef]
- Rossi, F.; Beltran, M.; Damizia, M.; Grelloni, C.; Colantoni, A.; Setti, A.; Di Timoteo, G.; Dattilo, D.; Centrón-Broco, A.; Nicoletti, C.; et al. Circular RNA ZNF609/CKAP5 mRNA Interaction Regulates Microtubule Dynamics and Tumorigenicity. Mol Cell 2022, 82, 75–89.e9. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Molecular Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Singh, R.N. How RNA Structure Dictates the Usage of a Critical Exon of Spinal Muscular Atrophy Gene. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2019, 1862, 194403. [Google Scholar] [CrossRef] [PubMed]
- Bachmayr-Heyda, A.; Reiner, A.T.; Auer, K.; Sukhbaatar, N.; Aust, S.; Bachleitner-Hofmann, T.; Mesteri, I.; Grunt, T.W.; Zeillinger, R.; Pils, D. Correlation of Circular RNA Abundance with Proliferation – Exemplified with Colorectal and Ovarian Cancer, Idiopathic Lung Fibrosis and Normal Human Tissues. Sci Rep 2015, 5, 8057. [Google Scholar] [CrossRef]
- Levacher, C.; Viennot, M.; Drouet, A.; Beaussire, L.; Coutant, S.; Théry, J.-C.; Baert-Desurmont, S.; Laé, M.; Ruminy, P.; Houdayer, C. Disequilibrium between BRCA1 and BRCA2 Circular and Messenger RNAs Plays a Role in Breast Cancer. Cancers (Basel) 2023, 15, 2176. [Google Scholar] [CrossRef]
- Trsova, I.; Hrustincova, A.; Krejcik, Z.; Kundrat, D.; Holoubek, A.; Staflova, K.; Janstova, L.; Vanikova, S.; Szikszai, K.; Klema, J.; et al. Expression of Circular RNAs in Myelodysplastic Neoplasms and Their Association with Mutations in the Splicing Factor Gene SF3B1. Mol Oncol 2023, 17, 2565–2583. [Google Scholar] [CrossRef]
- Wedge, E.; Ahmadov, U.; Hansen, T.B.; Gao, Z.; Tulstrup, M.; Côme, C.; Nonavinkere Srivatsan, S.; Ahmed, T.; Jespersen, J.S.; Schlotmann, B.C.; et al. Impact of U2AF1 Mutations on Circular RNA Expression in Myelodysplastic Neoplasms. Leukemia 2023, 37, 1113–1125. [Google Scholar] [CrossRef]
- Baert-Desurmont, S.; Charbonnier, F.; Houivet, E.; Ippolito, L.; Mauillon, J.; Bougeard, M.; Abadie, C.; Malka, D.; Duffour, J.; Desseigne, F.; et al. Clinical Relevance of 8q23, 15q13 and 18q21 SNP Genotyping to Evaluate Colorectal Cancer Risk. Eur J Hum Genet 2016, 24, 99–105. [Google Scholar] [CrossRef]
- Amiot, J.; Levacher, C.; Thibaut, L.M.; Lienard, G.; Vasseur, S.; Quenez, O.; Coutant, S.; Fourneaux, S.; Charbonnier, F.; Thorn, H.; et al. Bridging the Diagnostic Gap in Hereditary Cancers with Simple, Cost-Effective, High-Throughput RNA Splicing Analysis. J Mol Diagn 2025, 27, 954–968. [Google Scholar] [CrossRef] [PubMed]
- Lanic, M.-D.; Le Loarer, F.; Rainville, V.; Sater, V.; Viennot, M.; Beaussire, L.; Viailly, P.-J.; Angot, E.; Hostein, I.; Jardin, F.; et al. Detection of Sarcoma Fusions by a Next-Generation Sequencing Based–Ligation-Dependent Multiplex RT-PCR Assay. Modern Pathology 2022, 35, 649–663. [Google Scholar] [CrossRef]
- Bobée, V.; Drieux, F.; Marchand, V.; Sater, V.; Veresezan, L.; Picquenot, J.-M.; Viailly, P.-J.; Lanic, M.-D.; Viennot, M.; Bohers, E.; et al. Combining Gene Expression Profiling and Machine Learning to Diagnose B-Cell Non-Hodgkin Lymphoma. Blood Cancer J. 2020, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Schubert, S.A.; Morreau, H.; van Wezel, T. The Missing Heritability of Familial Colorectal Cancer. 2020, 35, 11. [Google Scholar]
- Li, Q.; Lai, H.; Li, Y.; Chen, B.; Chen, S.; Li, Y.; Huang, Z.; Meng, Z.; Wang, P.; Hu, Z.; et al. RJunBase: A Database of RNA Splice Junctions in Human Normal and Cancerous Tissues. Nucleic Acids Res 2021, 49, D201–D211. [Google Scholar] [CrossRef]
- Viscardi, M.J.; Arribere, J.A. Poly(a) Selection Introduces Bias and Undue Noise in Direct RNA-Sequencing. BMC Genomics 2022, 23, 530. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, J.; Zhao, F. Full-Length Circular RNA Profiling by Nanopore Sequencing with CIRI-Long. Nat Protoc 2023, 18, 1795–1813. [Google Scholar] [CrossRef]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of Intron Sequences Reveals Hallmarks of Circular RNA Biogenesis in Animals. Cell Reports 2015, 10, 170–177. [Google Scholar] [CrossRef]
- Liang, D.; Tatomer, D.C.; Luo, Z.; Wu, H.; Yang, L.; Chen, L.-L.; Cherry, S.; Wilusz, J.E. The Output of Protein-Coding Genes Shifts to Circular RNAs When the Pre-mRNA Processing Machinery Is Limiting. Molecular Cell 2017, 68, 940–954.e3. [Google Scholar] [CrossRef]
- Conn, V.M.; Gabryelska, M.; Toubia, J.; Kirk, K.; Gantley, L.; Powell, J.A.; Cildir, G.; Marri, S.; Liu, R.; Stringer, B.W.; et al. Circular RNAs Drive Oncogenic Chromosomal Translocations within the MLL Recombinome in Leukemia. Cancer Cell 2023, 41, 1309–1326.e10. [Google Scholar] [CrossRef]
- Zhang, Y.; Xue, W.; Li, X.; Zhang, J.; Chen, S.; Zhang, J.-L.; Yang, L.; Chen, L.-L. The Biogenesis of Nascent Circular RNAs. Cell Rep 2016, 15, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Okholm, T.L.H.; Venø, M.T.; Kjems, J. Circular RNAs Are Abundantly Expressed and Upregulated during Human Epidermal Stem Cell Differentiation. RNA Biol 2018, 15, 280–291. [Google Scholar] [CrossRef]
- Alonso-García, M.; Liaubet, L.; Faraut, T.; Dehais, P.; Kühn, C.; Demars, J.; Gutiérrez-Gil, B.; Robic, A. Decoupling Transcriptome Layers: The Distinct and Variable Nature of Circular RNAs. BMC Biol 2025, 23, 269. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.-B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline Mutations Affecting the Proofreading Domains of POLE and POLD1 Predispose to Colorectal Adenomas and Carcinomas. Nat Genet 2013, 45, 136–144. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, X.; Zhang, Y.; Cen, Y.; Zhu, T.; Wang, L.; Xia, L.; Li, Y.; Cheng, X.; Xie, X.; et al. The Biomarker Potential of circPOLD1 and Its Binding Protein YBX1 in Cervical Carcinogenesis. J Transl Med 2025, 23, 506. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, B.P.; Engels, S.; Aglialoro, F.; van den Akker, E.; von Lindern, M.; Wolkers, M.C. Circular RNA Expression in Human Hematopoietic Cells Is Widespread and Cell-Type Specific. Nucleic Acids Res 2018, 46, 8168–8180. [Google Scholar] [CrossRef]
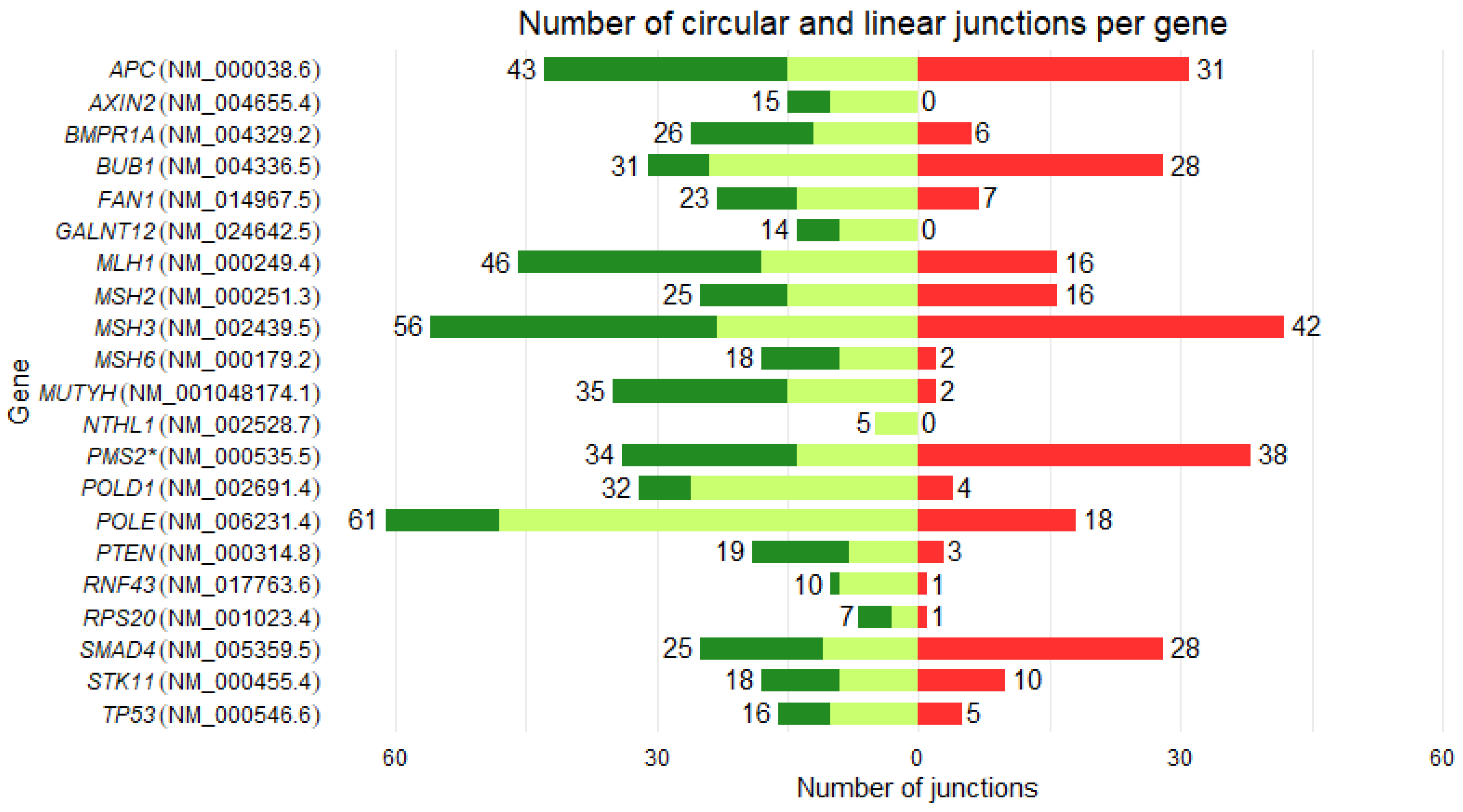

| Gene | Reference transcript |
Number of exons | Number of linear junctions | Number of circular junctions | Number of new circular RNAs | Circular / linear junctions (%) | Circular / linear UMIs (%) |
|---|---|---|---|---|---|---|---|
| POLD1 | NM_002691.4 | 27 | 32 | 4 | 3 | 12,50 | 547,28 |
| BUB1 | NM_004336.5 | 25 | 31 | 28 | 8 | 90,32 | 426,02 |
| PMS2 * | NM_000535.7 | 15 | 34 | 38 | 17 | 111,76 | 134,81 |
| SMAD4 | NM_005359.6 | 12 | 25 | 28 | 6 | 112,00 | 96,61 |
| POLE | NM_006231.4 | 49 | 61 | 18 | 6 | 29,51 | 40,91 |
| MSH3 | NM_002439.5 | 24 | 56 | 42 | 1 | 75,00 | 31,02 |
| RNF43 | NM_017763.6 | 10 | 10 | 1 | 0 | 10,00 | 18,37 |
| FAN1 | NM_014967.5 | 15 | 23 | 7 | 0 | 30,43 | 15,58 |
| MLH1 | NM_000249.4 | 19 | 46 | 16 | 6 | 34,78 | 12,58 |
| MSH2 | NM_000251.3 | 16 | 25 | 16 | 2 | 64,00 | 11,38 |
| APC | NM_000038.5 | 16 | 43 | 31 | 3 | 72,09 | 9,65 |
| MUTYH | NM_001048174.2 | 16 | 35 | 2 | 1 | 5,71 | 2,93 |
| STK11 | NM_000455.5 | 10 | 18 | 10 | 4 | 55,56 | 2,31 |
| BMPR1A | NM_004329.3 | 13 | 26 | 6 | 2 | 23,08 | 1,74 |
| MSH6 | NM_000179.3 | 10 | 18 | 2 | 2 | 11,11 | 1,68 |
| TP53 | NM_000546.6 | 11 | 16 | 5 | 2 | 31,25 | 1,54 |
| PTEN | NM_000314.8 | 9 | 19 | 3 | 0 | 15,79 | 0,17 |
| RPS20 | NM_001023.4 | 4 | 7 | 1 | 1 | 14,29 | 0,03 |
| AXIN2 | NM_004655.4 | 11 | 15 | 0 | 0 | 0,00 | 0,00 |
| GALNT12 | NM_024642.5 | 10 | 14 | 0 | 0 | 0,00 | 0,00 |
| NTHL1 | NM_002528.7 | 6 | 5 | 0 | 0 | 0,00 | 0,00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



