
Submitted:
29 December 2025
Posted:
30 December 2025
You are already at the latest version
Abstract
Engineered microbes are emerging as a new class of living immunotherapeutics capable of sensing, interpreting, and actively reshaping host immune systems. Unlike conventional biologics or cell therapies with fixed mechanisms of action, engineered microbial platforms operate as dynamic systems that integrate environmental, metabolic, and immunological cues, process these inputs through programmable biological circuits, and execute context-dependent immune modulation with spatial and temporal precision. This review presents an immune-first framework that conceptualizes engineered microbes as distributed immune-computational systems defined by coordinated sensing, signal processing, memory, and effector functions embedded within host immune networks. Organizing the field around immune logic rather than microbial taxonomy or disease category, we examine how engineered microbes detect tissue-specific and immune-state signals, translate these inputs through synthetic processing modules, and generate immune outputs that activate, suppress, educate, or reprogram immunity across cancer, autoimmunity, and infectious disease. We further define immune safety architecture as a core design principle governing inflammatory control, tolerance preservation, adaptive immunity, and therapeutic termination, and discuss the translational and regulatory implications of immune-state–resolved clinical evaluation. Together, this framework positions engineered microbes as programmable immune systems and establishes a unifying conceptual foundation for their development as next-generation living immunotherapies.
Keywords:
engineered microbial therapeutics
; living immunotherapies
; immune computation
; systems immunology
; cancer immunomodulation
; immune tolerance engineering
; immune safety architecture
; immune-state endpoints
1. Introduction: Microbes as a New Class of Immunotherapeutics
The field of immunotherapy has expanded from soluble biologics and small molecules to engineered immune cells and complex biomaterials [1,2]. A parallel and increasingly influential development is the emergence of engineered microbes as programmable agents capable of sensing, interpreting, and reshaping host immunity. These living systems are more than delivery vehicles or microbiome supplements [3,4]. They possess intrinsic immunological functions that can be redesigned, amplified, or redirected through synthetic biology to achieve precise modulation of local and systemic immune responses.
Several features make immunity the central axis through which engineered microbes should be understood. Microbes naturally interact with host immune pathways at nearly every tissue surface. They engage pattern recognition receptors, tune cytokine and chemokine networks, influence antigen presentation, and remodel metabolic landscapes that affect T cell and myeloid cell function [5,6]. When redesigned with synthetic circuits, these immune interactions can be converted into intentional therapeutic programs. Engineered bacteria can sense inflammatory cues, adjust their behavior in response to environmental signals, and deliver tailored immunomodulatory outputs that reshape specific pathways involved in cancer, autoimmunity, or infection [3,7]. This positions microbes not as passive colonizers but as programmable immune actuators.
The conceptual shift from microbial vectors to microbial immune engineering is essential. Early approaches relied on microbes as carriers for antigens or therapeutic proteins. The new generation of engineered strains incorporates logic gates, feedback circuits, and metabolite-responsive modules that allow real-time regulation of immune activity. These systems can control the intensity, timing, and spatial localization of immune responses with a degree of adaptability that non-living platforms cannot achieve [3,7,8]. For example, bacteria engineered to express pro-inflammatory cytokines only within hypoxic tumor niches can activate cytotoxic immunity while avoiding systemic toxicity. Conversely, strains engineered to promote tolerogenic signals in the gut can suppress inappropriate inflammation without weakening host defense [9,10,11].
A central advantage of microbial immunotherapeutics is their ability to unify multiple disease contexts under a single technological framework. Cancer requires localized activation of cytotoxic and innate inflammatory pathways [3,11]. Autoimmune and inflammatory diseases require restoration of tolerance, enhancement of regulatory networks, and repair of mucosal barriers [12]. Infectious diseases benefit from targeted augmentation of antimicrobial immunity without provoking immunopathology [13]. Engineered microbes can be adapted to each of these immune states by altering sensing modules, circuit topology, and effector outputs [3,14]. This cross-indication flexibility is distinctive among therapeutic platforms and reflects the modularity of immune pathway architecture itself.
Contemporary immunotherapies remain limited by several constraints that living microbial systems are positioned to overcome. Monoclonal antibodies and cytokine therapies lack spatial precision and often produce systemic adverse effects [15,16]. Cell therapies require complex manufacturing and can be difficult to deliver to mucosal sites or deep tissue microenvironments [17,18]. Small molecules have limited ability to encode conditional or adaptive behavior [19]. Engineered microbes inherently solve many of these challenges. They can autonomously navigate tissue landscapes, respond to microenvironmental cues, sustain therapeutic activity over time, and terminate activity through built-in circuit logic or immune-mediated clearance [20,21]. Their ability to operate continuously within the host, adjusting their activity as immune states evolve, provides a dynamic mode of intervention absent from conventional therapeutics.
This review adopts an immune design logic that organizes the field not by disease category or microbial chassis but by the mechanisms through which engineered microbes interface with immunity. This perspective provides a coherent framework to understand how microbial sensing, signal processing, and effector delivery can be tuned to achieve specific immune outcomes. The following sections examine the design principles of microbial immunotherapeutics, the intrinsic immune behaviors of different microbial platforms, and their application across cancer, autoimmunity, and infectious disease. Together, these concepts illustrate how engineered microbes are transforming immune modulation and establishing a new class of living immunotherapies.
2. The Immune Logic of Engineered Microbes as a New Framework for Living Immunotherapies
Engineered microbes function as living computational systems that sense defined features of the host environment, process these signals through synthetic biological circuits, and generate targeted immunological outputs. Their therapeutic value comes not only from the molecules they deliver but from the logic that governs their behavior. Unlike conventional immunotherapies with static modes of action, engineered microbes integrate real-time information about tissue physiology and immune activation, adjusting their responses with precision that is difficult to achieve through non-living platforms [4,22,23]. Understanding these organisms as programmable immune actuators requires a structured framework that links the signals they detect, the genetic circuits they deploy, and the immune pathways they modulate. This section establishes such a framework, defining engineered microbes through the interplay of immune inputs, synthetic processing modules, and immunological outputs (Figure 1).
Taken together, these properties position engineered microbes as distributed immune-computational systems rather than passive therapeutic carriers. They continuously sense immune-relevant signals, integrate these inputs through programmable biological logic, store contextual information through circuit-encoded memory, and execute immune-modulatory actions with spatial and temporal precision. In this view, engineered microbes function analogously to immune cells or synthetic immune circuits, implementing sensing, decision-making, memory, and actuation directly within host immune networks. This computational framing provides a unifying logic for understanding how microbial platforms can be rationally designed, evaluated, and deployed across cancer, autoimmunity, and infectious disease. The application of this immune-computational framework to distinct disease contexts, including cancer, autoimmunity, and infection (Figure 2).
2.1. Immune Inputs and the Signals Engineered Microbes Sense
Engineered microbes rely on their ability to detect the biochemical and immunological signatures that define specific tissue states. These sensory inputs determine when and where a microbe should activate a therapeutic program. Inflammation-associated molecules provide important contextual information. Reactive oxygen species, nitric oxide, and hypoxic conditions are characteristic of tumors, inflamed mucosa, and infected tissues [20,23,24,25]. Microbes naturally possess sensing systems for oxidative and nitrosative stress, and these can be redirected to control the activation of therapeutic circuits [24,26]. Hypoxia-responsive promoters can restrict immune-stimulatory outputs to tumor cores, while nitric oxide sensors can initiate anti-inflammatory functions at sites of mucosal damage [27,28]. Such environmental signals give engineered microbes spatial and contextual precision.
Host cytokines serve as another major category of immune input. These molecules encode dominant immune states ranging from acute inflammation to regulatory tolerance. Engineered microbes can detect cytokines such as interferons, TNF, IL-6, or IL-1 family members through synthetic receptors or transcriptional sensors [4,23,27]. This allows microbes to adjust their behavior according to the surrounding immune landscape. In cancer, inflammatory cytokines can be used as triggers for local immune activation [11,29], while in autoimmune disease the same molecules can initiate circuits that reduce inflammation or promote tolerance [30]. Cytokine sensing enables microbes to behave as adaptive elements within immune networks rather than simple vehicles of therapeutic molecules [31].
Metabolic cues provide a further layer of immune-relevant information. Metabolites such as lactate, short-chain fatty acids, tryptophan derivatives, and bile acids are tightly linked to specific immune phenotypes [5]. High lactate concentrations reflect immunosuppressive tumor niches and can be used to activate circuits that enhance antigen presentation or promote cytotoxic responses [32]. SCFAs and tryptophan metabolites influence Treg and Th17 differentiation, offering opportunities to engineer microbes that stabilize immune tolerance [33]. By sensing metabolites and in some cases chemically transforming them, engineered microbes can directly influence immunometabolic pathways that regulate T cell and myeloid function [9].
Inputs can also originate from pathogens. Microbes can detect pathogen-associated signals such as quorum-sensing molecules, virulence factors, or cell wall components [34,35]. These signals can trigger engineered bacteria to deliver immune stimulants that recruit phagocytes or enhance innate responses. Detection of pathogen-associated cues situates engineered microbes within the host’s broader defense architecture and enables targeted amplification of antimicrobial immunity only where needed.
Finally, engineered microbes can sense features of the microbiota itself. Patterns of microbial composition, nutrient availability, or interspecies metabolites indicate whether mucosal ecosystems are balanced or perturbed [31,36]. Dysbiosis can therefore serve as an activation signal for circuits that reinforce barrier function, secrete tolerogenic mediators, or restore colonization resistance [6,37]. In this way, engineered strains can participate in the preservation of immune homeostasis and intervene at early stages of disease development.
Together, these inputs allow engineered microbes to operate as context-aware immune modulators. They respond selectively to pathological conditions rather than acting indiscriminately. They sense inflammation, immune activation, metabolic stress, pathogen presence, or microbiota imbalance, and they convert these signals into defined immunological actions. This sensory layer forms the foundation of the immune logic that shapes the behavior of living microbial therapeutics.
2.2. Processing Modules and the Interpretation of Immune-Relevant Signals
Once immune inputs are detected, engineered microbes rely on internal processing modules that determine how those signals are interpreted and translated into action. These modules constitute the computational core of microbial immunotherapeutics. They allow microbes to make logical decisions, regulate amplitude and timing of responses, and coordinate multicellular behavior [3,8,22,23]. Their design draws heavily on synthetic biology but aligns with principles found in immune cell signaling, where signal integration, feedback regulation, and memory shape functional outcomes.
Logical gate systems form one of the foundational modules. AND gates allow microbial responses only when multiple conditions are simultaneously met, such as inflammation plus hypoxia or cytokine elevation plus a tumor-specific metabolic cue. This architecture improves specificity and prevents off-target immune activation [23,38,39]. OR gates, by contrast, allow outputs when any of several pathological signals are present. Using combinations of these modules, engineered microbes can execute decision-making programs that mirror the conditional activation seen in T cells or antigen-presenting cells [3,38].
Feedback and negative feedback controls modulate response stability and prevent runaway activation. Positive feedback can amplify weak signals to ensure robust therapeutic action within pathological niches. Negative feedback can limit excessive production of cytokines or immune ligands, protecting against toxic inflammatory overshoot [3,14,40]. These regulatory modules parallel the natural homeostatic loops that govern immune activation and tolerance, giving engineered microbes the capacity to shape immunological dynamics rather than deliver static stimuli.
More complex circuit designs incorporate band-pass filters that activate only within a defined range of signal intensity. This prevents responses at very low or very high levels of environmental cues, which is particularly important in tissues where gradients of cytokines, metabolites, or oxygen reflect distinct immune states [41,42,43]. Band-pass logic enables nuanced behaviors such as targeting premalignant inflammatory foci without activating in healthy tissue or responding only within the immunosuppressive threshold of a tumor microenvironment.
Memory modules, including toggle switches or recombinase-based circuits, allow microbes to record prior exposure to specific cues and maintain altered states over time. This property enables persistent immune modulation even after the initial trigger disappears. Microbial memory can support long-term tolerance induction, sustained anti-tumor activity, or prolonged metabolic reprogramming of immune cells [31,44,45,46]. By embedding memory into microbial behavior, engineered strains can achieve a temporal dimension of immunotherapy that resembles trained immunity in innate cells or long-lived responses in adaptive immunity.
Quorum-sensing networks, a natural communication system among bacteria, can be repurposed to coordinate collective behavior across microbial populations. These systems allow microbes to regulate output intensity based on population density, synchronize therapeutic activities, or create spatially patterned responses [47,48,49]. Quorum sensing also enables division of labor, where different subsets of microbes produce complementary immune factors only when an entire community is present within the lesion or mucosal environment.
Spatial logic further refines microbial therapeutic action. Engineered microbes can form biofilms or microcolonies in defined tissue regions, allowing localized immune modulation. Circuits responsive to mucosal adhesion molecules, epithelial glycan patterns, or gradients of oxygen and metabolites can position microbes at the exact sites where immune intervention is needed [22,50,51]. Spatial logic is particularly valuable in mucosal diseases, where microbe localization determines whether cytokines or tolerogenic factors engage epithelial cells, stromal cells, or resident immune populations.
Together, these processing modules form the computational architecture through which engineered microbes translate complex environmental information into precise therapeutic actions. They enable living therapeutics to behave less like passive vectors and more like programmable immune cells that adjust their function according to evolving tissue states.
2.3. Immune Outputs and the Modulation of Host Immunity
The final tier of microbial immune logic involves the therapeutic outputs that engineered microbes generate to influence host immune pathways. These outputs define the immunological phenotype that each microbe produces, spanning responses that promote cytotoxic activation as well as those that reinforce regulatory tolerance [3,11,31,35]. When these outputs are integrated with the sensory and processing modules described above, engineered microbes are able to exert highly targeted effects on immune networks relevant to cancer, autoimmunity, and infection.
A major class of outputs includes cytokines and chemokines. Microbes engineered to secrete molecules such as IL-12, IL-2, GM-CSF, or IFN family members can activate cytotoxic immunity within tumors or infected tissues. Localized microbial production of Th1 or NK-activating cytokines can convert immunologically cold tumors into inflamed lesions capable of supporting T cell infiltration and antigen spreading [28,52,53]. Conversely, secretion of IL-10 or TGF-β can suppress excessive inflammation, promote regulatory T cell differentiation, and restore mucosal tolerance in autoimmune disease [54,55]. These cytokine programs provide high potency with low systemic exposure because engineered microbes confine their activity to defined microenvironments.
Antigen display systems represent another powerful immunological output. Microbes can present tumor antigens, autoantigens, or pathogen-derived epitopes on their surface or within secreted vesicles. These displays can drive antigen-specific T cell priming, enhance cross-presentation by dendritic cells, or induce tolerance depending on the co-delivered signals [56,57,58]. Antigen display enables microbial therapeutics to function as living vaccines or as tools for restoring immune recognition in cancer and chronic infection.
Expression of engineered immune ligands extends microbial influence to co-stimulatory and co-inhibitory pathways that shape T cell activity. Microbes can be engineered to express PD-L1 decoys that sequester inhibitory signals, or ligands such as CD40L, OX40L, and 4-1BBL that enhance T cell activation [53,59,60]. These ligands can remodel T cell dynamics within tissue niches, reversing exhaustion or enhancing cytotoxic potential without requiring systemic administration of immune agonists. Microbial delivery of these molecules provides spatial precision and reduces toxicity often associated with systemic checkpoint or co-stimulatory therapies.
Tolerogenic factors constitute a distinct therapeutic class especially relevant to autoimmune diseases and inflammatory disorders. Engineered microbes can produce IL-10, TGF-β, retinoic acid, or specialized immunoregulatory metabolites to promote regulatory T cell expansion, stabilize tolerogenic dendritic cells, and repair mucosal barriers [61,62,63]. These outputs directly counteract pathogenic inflammation and restore homeostatic immune balance in the gut, skin, and other barrier tissues.
Metabolic rewiring is an emerging category of immune output. Many immune responses depend on metabolic pathways related to arginine, tryptophan, adenosine, or lactate. Engineered microbes can consume immunosuppressive metabolites such as lactate within tumors, degrade adenosine to relieve T cell inhibition, or regulate tryptophan pathways that control Treg and Th17 differentiation [64,65,66,67]. By reshaping metabolic microenvironments, microbes can influence immune cell function in ways that traditional cytokine therapies cannot achieve.
Together, these outputs demonstrate the versatility of engineered microbes as immunomodulatory agents. They can locally activate, suppress, educate, or redirect immunity depending on their design. When combined with the input detection and processing modules described earlier, these outputs complete a full systems logic through which engineered microbes can be rationally programmed as living immunotherapies tailored to distinct disease contexts (Table 1) (Figure 1).
This immune-computational framing also highlights a growing misalignment between living immunotherapeutics and existing regulatory paradigms. Current approval frameworks remain largely optimized for static agents defined by fixed molecular composition, dose–exposure relationships, and linear pharmacokinetics. Engineered microbes instead act by reshaping immune system state over time, with therapeutic efficacy and risk emerging from dynamic immune network reorganization rather than from single-target engagement. As a result, conventional endpoints such as peak exposure, systemic concentration, or isolated biomarker suppression often fail to capture the true mechanism of action of these systems. Immune-state–resolved endpoints, including cytokine topology, regulatory balance, spatial immune architecture, and memory formation, are therefore not ancillary measurements but central descriptors of both efficacy and safety. Explicitly articulating immune architecture and control logic is likely to become essential for regulatory evaluation, providing a common language through which dynamic, adaptive, and self-limiting living therapies can be meaningfully assessed.
3. Microbial Chassis Platforms and Their Intrinsic Immune Personalities
Engineered microbes are often classified according to taxonomic lineage or ease of genetic manipulation. However, for immunotherapeutic applications, a more informative organizing principle is the intrinsic immune personality of the microbial chassis. Each microbial species enters the host with a characteristic pattern of immune recognition, tissue localization, metabolic interaction, and signaling consequence [56,68,69]. These native immune behaviors fundamentally constrain and enable the types of therapeutic programs that can be safely and effectively implemented.
Classifying microbial chassis by immune behavior rather than taxonomy reveals why certain strains are naturally suited for tolerogenic applications while others are predisposed to inflammatory immune activation. Commensal organisms that have co-evolved with host mucosal immune systems tend to reinforce barrier integrity and immune tolerance [70,71]. In contrast, attenuated pathogens retain strong immunostimulatory capacities that can be harnessed for tumor targeting and antimicrobial immunity [22]. Beyond single strains, engineered microbial consortia introduce an additional layer of immune complexity by enabling distributed immune control across multiple microbial actors [72].
This immune-centric framework provides a rational basis for selecting, engineering, and deploying microbial platforms according to desired immunological outcomes rather than convenience of genetic engineering alone. These intrinsic immune personalities define the baseline inflammatory risk, regulatory capacity, and therapeutic suitability of different microbial chassis, independent of taxonomic classification (Figure 3).
3.1. Commensal Chassis and Their Tolerogenic Immune Profiles
Commensal bacteria such as Lactobacillus, Bacteroides, and members of the Clostridia class exhibit immune behaviors that are strongly biased toward tolerance, homeostasis, and barrier support. These organisms establish long-term residence at mucosal surfaces and have evolved to coexist with the immune system without provoking sustained inflammation [70,73,74]. Their surface structures, secreted metabolites, and metabolic byproducts engage host immune pathways in ways that favor regulatory immune tone rather than defensive activation.
A defining feature of these commensal chassis is their capacity to promote mucosal tolerance. Many Clostridia species induce regulatory T cell differentiation through the production of short-chain fatty acids and through epigenetic modulation of immune cells within the lamina propria [75,76]. Bacteroides species can stimulate tolerogenic dendritic cell phenotypes and support interleukin-10–dominated immune responses [70,77]. Lactobacillus strains modulate epithelial signaling and reinforce tight junction integrity, limiting inflammatory translocation of luminal antigens [78]. These properties make commensal microbes naturally aligned with therapeutic strategies aimed at dampening pathological inflammation rather than amplifying immune activation.
Commensal chassis also interface closely with antigen sampling and IgA-mediated immune regulation. They actively shape the education of mucosal B cells, influence the specificity and breadth of secretory IgA repertoires, and regulate antigen presentation through interactions with specialized epithelial and dendritic cell subsets [79,80,81]. By occupying niches at the interface between the microbiota and the immune system, commensals play a central role in calibrating immune recognition of luminal antigens. When engineered to display defined antigens or secrete specific immunomodulators, these organisms can leverage existing IgA and mucosal antigen-sampling pathways to induce either antigen-specific tolerance or controlled immune deviation [82].
These attributes confer unique advantages for applications in autoimmunity and inflammatory bowel disease. In such settings, therapeutic goals center on restoring immune balance, reinforcing epithelial barriers, suppressing aberrant effector responses, and expanding regulatory cell populations. Engineered commensal microbes can deliver tolerogenic cytokines, regulatory metabolites, or disease-relevant antigens within a biological context that already favors immune restraint. Their intrinsic low inflammatory profile reduces the risk of systemic immune activation, making them especially well-suited for chronic immune modulation in the gut and other mucosal tissues.
3.2. Attenuated Pathogens and Their Pro-Inflammatory Immune Architectures
Attenuated pathogenic bacteria such as Salmonella, Listeria, Shigella, and Escherichia coli Nissle retain strong immunostimulatory properties even after virulence attenuation. Unlike commensal chassis that favor immune tolerance, these organisms are intrinsically wired to activate innate immune surveillance pathways and to provoke robust inflammatory responses [83]. This immune personality makes them uniquely suited for applications in which strong immune activation is required, particularly in cancer immunotherapy and in the control of difficult infections [84].
A defining feature of attenuated pathogens is their high inflammatory capacity. Their cell wall components, flagellar proteins, secretion systems, and nucleic acids are efficiently detected by pattern recognition receptors expressed on epithelial cells, macrophages, dendritic cells, and innate lymphoid populations [85]. Engagement of Toll-like receptors such as TLR4 and TLR5, cytosolic DNA sensors linked to the STING pathway, and additional inflammasome-associated receptors results in rapid induction of pro-inflammatory cytokines, type I interferons, and chemokines that recruit and activate effector immune cells [86,87]. Even when virulence factors are weakened, these organisms retain the molecular signatures required to trigger strong host defense pathways.
This intrinsic propensity for immune activation provides important advantages for cancer applications. Attenuated Salmonella and Listeria preferentially accumulate within tumors due to aberrant vasculature, immune suppression, and necrotic tissue architecture. Once localized, their natural ability to trigger innate immune signaling can convert immunologically silent tumors into inflamed microenvironments that support antigen presentation, T cell infiltration, and cytotoxic effector function [28,88,89]. When further engineered to deliver cytokines, immune ligands, or tumor antigens, these microbes act as localized immune amplifiers capable of reshaping tumor immunity without inducing widespread systemic inflammation.
Attenuated pathogens are also well suited for infectious disease models in which immune augmentation is desirable [90]. Their strong engagement of innate immune sensors enhances antimicrobial effector pathways, promotes robust adaptive immune priming, and supports durable immune memory [91]. Engineered derivatives of these organisms can be programmed to secrete cytokines that boost macrophage bactericidal activity, enhance interferon signaling for antiviral defense, or recruit neutrophils to sites of persistent infection [92]. In this context, the inflammatory immune personality of the chassis is a therapeutic asset rather than a liability.
Nevertheless, the same properties that make attenuated pathogens powerful immune activators also impose constraints. Excessive inflammation, systemic cytokine release, and unintended tissue damage represent persistent risks. Effective clinical deployment therefore depends on precise attenuation strategies, stringent control of microbial replication, and integration of safety circuits that limit immune activation to defined spatial and temporal windows [22,93]. When these safeguards are properly implemented, attenuated pathogens offer a uniquely potent platform for immune amplification where strong activation is therapeutically required.
3.3. Consortium-Based Engineered Communities and Distributed Immune Control
Beyond single-strain platforms, engineered microbial consortia introduce a higher-order level of immune regulation by distributing therapeutic functions across multiple microbial species. In these systems, distinct strains are programmed with complementary sensing, processing, and effector roles, enabling collective behaviors that surpass the capabilities of any individual organism [72,94,95]. This distributed architecture mirrors key features of immune system organization, in which multiple cell types cooperate to generate coordinated responses.
A central advantage of consortia is the division of labor among strains. One strain may specialize in sensing inflammatory metabolites, another in antigen display, and a third in cytokine or tolerogenic factor production. By separating these tasks, each microbial component can be optimized for a specific function without overburdening cellular resources [96]. This modularity allows complex immune programs to be assembled from simpler biological parts and provides flexibility in reconfiguring therapeutic outputs for different disease contexts [48,97].
Consortium designs also enable multi-output immune orchestration. Rather than delivering a single immune signal, a coordinated community can release balanced combinations of cytokines, chemokines, metabolic modulators, and immune ligands. Such multi-dimensional immune programming is particularly valuable in diseases where pathological states arise from network-level dysregulation rather than single molecular defects [98,99,100]. In cancer, consortia can simultaneously promote antigen presentation, relieve immune suppression, and enhance effector recruitment. In autoimmunity, they can combine barrier repair, regulatory T cell induction, and suppression of pro-inflammatory cytokine axes within a unified therapeutic program [101].
Consortia further provide enhanced robustness and safety. Functional redundancy among strains can buffer against stochastic loss of individual members, while cross-regulatory quorum circuits can coordinate population-level activity and prevent uncontrolled expansion [94]. Importantly, safety features can also be distributed, with certain strains dedicated to containment, self-limitation, or immune-dependent clearance [102,103]. This architecture reduces the likelihood that any single genetic failure compromises the overall safety of the system.
Through division of labor, coordinated immune orchestration, and layered safety, consortium-based platforms extend the immune logic of engineered microbes from single-cell decision-making to community-level computation [104,105,106]. They represent a critical step toward therapeutic ecosystems capable of sustained, adaptive, and highly regulated immune modulation.
4. Engineered Microbes for Cancer Immunomodulation
Engineered microbes have emerged as a powerful class of cancer immunotherapeutics because of their unique ability to integrate tumor-specific environmental cues with programmable immune outputs [28,107]. Unlike conventional cancer therapies that rely on systemic drug exposure or ex vivo cell manipulation, microbial systems operate directly within the tumor microenvironment. This enables immune modulation to occur precisely where malignant cells evade immune surveillance [84]. The defining feature of this approach is not microbial presence within tumors alone, but the capacity to execute context-dependent immune activation that reshapes tumor–immune interactions with spatial and temporal control [108].
Tumor-homing behavior provides the foundational targeting layer for microbial immunotherapy. Solid tumors present abnormal vasculature, hypoxia, necrotic regions, and immunosuppressive metabolic conditions that preferentially support microbial accumulation [109,110]. Engineered bacteria exploit these features through both passive trapping and active chemotaxis toward tumor-derived metabolites. However, tumor homing alone is insufficient for therapeutic efficacy [3,84]. Its true value lies in enabling highly localized immune modulation that avoids the systemic toxicities associated with traditional cytokine therapies. When tumor localization is linked to conditional immune activation circuits, microbial therapies achieve spatial precision that few other platforms can match [3,84,110].
One of the most potent immune activation strategies enabled by engineered microbes is targeted engagement of the STING pathway [111,112]. The cGAS–STING axis plays a central role in innate immune detection of cytosolic DNA and in the induction of type I interferon responses critical for antitumor immunity. Engineered microbes can be programmed to synthesize and release cyclic dinucleotide STING agonists selectively within tumors. This local activation stimulates dendritic cell maturation, enhances cross-presentation of tumor antigens, and promotes recruitment of cytotoxic lymphocytes [113,114]. Unlike systemic STING agonist delivery, which is limited by severe inflammatory toxicity, microbial delivery confines activation to the tumor microenvironment and allows controlled signal amplitude.
Localized expression of cytokines represents another key dimension of microbial cancer immunotherapy. Cytokines such as IL-2, IL-12, and GM-CSF are among the most powerful immune activators known, yet their systemic use is severely constrained by life-threatening toxicities [115]. Engineered microbes circumvent this limitation by producing these cytokines only within tumors through hypoxia-responsive or inflammation-responsive promoters. Local IL-12 expression promotes Th1 polarization and interferon-dependent antitumor immunity [116]. IL-2 enhances intratumoral T cell expansion, while GM-CSF stimulates dendritic cell recruitment and antigen presentation [117]. The result is a focused immune amplification confined to the malignant niche.
Microbial immunotherapy also enables efficient antigen spreading, a critical requirement for durable cancer immunity. Tumor necrosis induced by microbial inflammation exposes a broader repertoire of tumor antigens to the immune system. Engineered microbes further enhance this process by delivering adjuvants, cytokines, and antigen-presentation scaffolds that support cross-priming [56,118]. This promotes diversification of T cell responses beyond a single dominant epitope and reduces the likelihood of immune escape through antigen loss [119]. Antigen spreading is a key mechanism through which localized microbial therapy generates systemic and long-lasting antitumor immune memory [120].
A particularly innovative application of engineered microbes is the direct expression of immune checkpoint modulators within tumors. Rather than administering systemic antibodies against PD-1, PD-L1, CTLA-4, or related pathways, microbes can be engineered to secrete checkpoint nanobodies or decoy receptors locally [56,118,121]. This approach uncouples therapeutic benefit from systemic immune dysregulation. Local sequestration of inhibitory ligands enhances effector T cell activity specifically within the tumor, while minimizing autoimmune complications in peripheral tissues. Microbial checkpoint modulation thus reframes immune checkpoint blockade as a spatially constrained intervention [11].
Cold tumors characterized by immune exclusion, low antigen presentation, and suppressive myeloid populations represent one of the most difficult challenges in oncology. Engineered microbes offer a direct solution by reconditioning these immunologically silent microenvironments [67]. Through combined delivery of STING agonists, pro-inflammatory cytokines, and metabolic modulators, microbes can convert cold tumors into inflamed lesions that support dendritic cell infiltration, lymphocyte trafficking, and effector function [87]. This immune reprogramming function is distinct from direct tumor cell killing and instead focuses on reconstructing the immune architecture of the tumor microenvironment [56].
Microbial modulation of tumor metabolism further amplifies immunotherapeutic efficacy. Tumors accumulate lactate, adenosine, and kynurenine that suppress cytotoxic immunity and promote regulatory cell populations [65]. Engineered microbes can be programmed to degrade these immunosuppressive metabolites directly within tumors. Lactate consumption relieves T cell exhaustion [122]. Adenosine degradation removes a potent inhibitory signal on lymphocytes. Interference with tryptophan metabolism alters regulatory T cell differentiation [66]. Through these mechanisms, microbes reshape the biochemical terrain of the tumor to favor immune dominance.
The precision of microbial cancer immunotherapy arises from the integration of sensing, processing, and output logic rather than from brute-force immune activation [3]. Tumor-associated hypoxia, inflammatory metabolites, and cytokines serve as input signals that gate microbial activity [123,124]. Genetic circuits implement conditional expression of immunostimulatory payloads. Feedback control prevents excessive cytokine release. This creates a quantitative immune control system embedded directly within the tumor [52]. Such programmable control distinguishes microbial platforms from all existing immunotherapeutic modalities.
Importantly, microbial cancer immunotherapy can operate synergistically with established treatment modalities. Local microbial immune activation sensitizes tumors to immune checkpoint inhibitors by increasing antigen presentation and effector infiltration. It enhances the immunogenicity of radiotherapy and chemotherapy by amplifying damage-associated molecular patterns. It also complements adoptive cell transfer by conditioning the tumor niche to support transferred T cells [118,125,126,127]. These interactions position engineered microbes not only as standalone immunotherapies but as immune amplifiers for combinatorial oncology.
Safety remains a defining constraint for all cancer-directed immune activation strategies [123]. The same inflammatory capacity that enables tumor destruction also carries the risk of systemic immune toxicity. Engineered microbes address this challenge through multi-layered attenuation strategies, kill switches, metabolic dependencies, and immune-mediated clearance circuits [103,128]. Importantly, the immune system itself becomes part of the safety architecture. As tumors are eliminated and inflammatory cues dissipate, microbial activation diminishes and the organisms are cleared, providing an intrinsic self-limiting feature absent from many drug-based platforms [129].
Together, these properties redefine how microbial therapies function in oncology. Their primary role is not tumor colonization or cytotoxic delivery, but precision immune activation driven by programmable biological logic. By transforming tumors into sites of controlled innate and adaptive immune engagement, engineered microbes establish a fundamentally new paradigm for cancer immunomodulation that is spatially restricted, dynamically regulated, and adaptable across tumor types.
5. Engineered Microbes for Autoimmunity and Mucosal Immune Tolerance
Autoimmune and chronic inflammatory diseases arise from persistent failures of immune tolerance rather than from simple immune deficiency [70,130]. In these conditions, the immune system retains its capacity for activation but loses the ability to restrain destructive effector responses against self or harmless environmental antigens. Unlike cancer, where immune amplification is the primary objective, autoimmunity requires precise suppression, redirection, and re-education of immune networks. Engineered microbes are uniquely suited for this task because they operate at mucosal interfaces where immune tolerance is physiologically established and maintained [131,132,133]. Their ability to deliver localized regulatory signals in a continuous and adaptive manner positions them as an entirely new class of tolerogenic immunotherapies.
One of the most direct strategies for microbial tolerance induction is the engineered secretion of immunoregulatory cytokines such as IL-10 and TGF-β. These cytokines play central roles in limiting effector T cell activation, promoting regulatory T cell differentiation, and stabilizing tolerogenic dendritic cell phenotypes [134,135,136]. When delivered systemically, both molecules are associated with broad immunosuppression and unacceptable side effects. Engineered microbes overcome this limitation by confining cytokine release to mucosal tissues, particularly the intestinal lamina propria. This localized delivery supports immune regulation at disease-relevant sites while preserving systemic immune competence.
In addition to soluble cytokines, engineered microbes can release extracellular vesicles enriched in tolerogenic signals [137,138]. These vesicles can carry regulatory cytokines, microRNAs, and metabolites that influence antigen-presenting cells and lymphocyte differentiation [139]. Vesicle-mediated delivery allows microbes to exert immunomodulatory effects without direct physical contact with immune cells and provides a mechanism for extending regulatory signals deeper into mucosal tissues [140,141]. This form of indirect communication further expands the spatial reach of microbial tolerance programs.
Antigen-specific tolerance represents one of the most important and underdeveloped goals in autoimmune therapy [142,143,144]. Instead of globally suppressing immunity, the aim is to silence only those immune responses directed against defined autoantigens. Engineered microbes provide a natural platform for achieving this specificity. By displaying autoantigens on their surface or secreting antigenic peptides in tolerogenic contexts, microbes can drive deletion, anergy, or regulatory differentiation of autoreactive T cell clones. This approach has direct relevance for diseases such as type 1 diabetes, multiple sclerosis, celiac disease, and rheumatoid arthritis, where dominant autoantigens have been well characterized.
The tolerogenic outcome of antigen delivery is critically dependent on the immune context in which antigens are encountered. Engineered microbes exploit this principle by coupling antigen presentation to the simultaneous release of IL-10, TGF-β, retinoic acid, or regulatory metabolites [142,143]. This combined signal environment biases antigen-presenting cells toward non-inflammatory phenotypes and drives the expansion of antigen-specific regulatory T cells rather than pathogenic effector populations [145,146]. In this way, engineered microbes recapitulate key features of oral tolerance that are often defective in autoimmune states.
Restoration of the regulatory T cell and Th17 cell balance is another central objective of microbial tolerance engineering [147]. Many autoimmune and inflammatory bowel diseases are characterized by excess Th17-driven inflammation and insufficient regulatory control. Engineered commensal microbes can be programmed to promote regulatory T cell differentiation through multiple complementary mechanisms [63,143]. These include secretion of IL-10, production of short-chain fatty acids, modulation of tryptophan metabolism, and delivery of tolerogenic antigens [148]. By simultaneously suppressing inflammatory differentiation and expanding regulatory networks, microbial platforms act at the level of immune network topology rather than single signaling pathways [63].
Barrier integrity represents a second critical axis of immune tolerance. Breakdown of epithelial barriers allows translocation of microbial products and dietary antigens into underlying tissues, perpetuating chronic inflammation [149]. Engineered microbes can actively participate in barrier repair by stimulating mucus production, enhancing tight junction protein expression, and promoting epithelial regeneration [150]. Some strains can be designed to secrete growth factors, antimicrobial peptides, or mucin-stabilizing enzymes that reinforce the physical separation between luminal contents and immune cells [151]. Restoration of barrier function reduces inflammatory antigen exposure and indirectly supports immune tolerance.
Metabolic regulation forms a further layer through which engineered microbes influence autoimmune immunity. Tryptophan metabolism through the aryl hydrocarbon receptor pathway is a powerful regulator of mucosal immune balance [61]. Microbial-derived tryptophan metabolites promote regulatory T cell differentiation, suppress pathogenic Th17 responses, and stabilize epithelial homeostasis [152]. Engineered microbes can be programmed to enhance beneficial tryptophan metabolites or reduce pathways that favor inflammation. Similarly, bile acid derivatives modulate innate lymphoid cell activity, T cell differentiation, and macrophage function [153]. By reshaping these metabolic circuits, microbes exert deep control over immune fate decisions.
A defining advantage of microbial tolerance platforms is their capacity for continuous and adaptive immune modulation. Autoimmune diseases typically involve fluctuating inflammatory states driven by environmental, microbial, and metabolic factors. Engineered microbes respond dynamically to these fluctuations by adjusting their regulatory output in proportion to local cues such as cytokines, metabolites, or tissue stress signals [31,63,154]. This adaptive behavior contrasts sharply with static drug dosing regimens and allows tolerance programs to scale with disease activity.
Engineered microbes also offer a route to regional immune specialization. Different autoimmune diseases affect distinct mucosal compartments, including the gut, skin, airway, and urogenital tract [101]. Microbial chassis can be selected or engineered for preferential colonization of these tissues, allowing disease-specific targeting. For example, gut-adapted commensals are particularly suited for inflammatory bowel disease and systemic autoimmunity driven by intestinal dysbiosis, whereas skin-associated strains may be applied to inflammatory dermatoses [59,155,156]. This tissue-specific targeting adds another level of therapeutic precision.
The immune safety profile of microbial tolerance strategies differs fundamentally from that of systemic immunosuppressive drugs [157]. Because regulatory signals are concentrated at mucosal sites and are tightly coupled to local immune inputs, the risk of global immune paralysis is reduced [63,134]. Moreover, microbes can be eliminated through standard antimicrobial strategies if adverse effects arise. This controllability is particularly important for chronic autoimmune conditions that require long-term therapy [104].
Despite these advantages, multiple translational challenges remain. Stability of antigen expression, maintenance of regulatory output under selective pressures, interactions with the native microbiota, and variability between individual host immune environments all influence therapeutic performance [158,159]. Addressing these challenges requires integration of synthetic biology with systems immunology and longitudinal immune profiling in patients [160]. These efforts are essential for moving microbial tolerance engineering from proof-of-concept studies to durable clinical interventions [155].
Taken together, engineered microbes provide a fundamentally new route for re-establishing immune tolerance in autoimmunity and chronic inflammatory disease. Their ability to combine antigen specificity, regulatory cytokine delivery, barrier repair, and metabolic immune control within a single living platform distinguishes them from all existing immunomodulatory technologies. By shifting the therapeutic objective from immune suppression to immune re-education, microbial tolerance engineering opens a new chapter in precision immunotherapy for autoimmune disease.
6. Engineered Microbes for Infection Control and Immune Enhancement
Infectious diseases are traditionally approached through antimicrobial strategies that aim to eliminate pathogens directly [22,134]. While these approaches remain essential, their effectiveness is increasingly compromised by antimicrobial resistance, pathogen immune evasion, and host immunopathology. An alternative paradigm is emerging in which engineered microbes are deployed not as antimicrobial agents but as immune enhancers that amplify, reprogram, or coordinate host defense mechanisms [20,161,162]. This immune-forward strategy reframes infection control as a problem of optimizing host immunity rather than solely suppressing microbial growth.
Engineered microbes can be designed to deliver cytokines that selectively amplify anti-pathogen immune responses at sites of infection [163,164]. Localized production of interferons, IL-12, IL-18, or GM-CSF can enhance macrophage microbicidal activity, promote dendritic cell maturation, and accelerate cytotoxic lymphocyte recruitment [165,166]. Because these cytokines are produced only within infected tissues in response to inflammatory or pathogen-derived inputs, immune activation is spatially constrained. This reduces the risk of systemic cytokine toxicity while maximizing immune pressure on the pathogen.
Targeted enhancement of mucosal immunity represents another major opportunity for microbial immune engineering. Many infections establish initial footholds at barrier surfaces such as the gut, respiratory tract, or urogenital mucosa [162,163,167]. Engineered commensal microbes can reinforce local IgA production, stimulate mucosal dendritic cells, and promote tissue-resident memory T cell formation [168]. By shaping immune readiness at these portals of entry, engineered microbes can raise the threshold for pathogen invasion and reduce the likelihood of systemic dissemination.
Trained immunity offers a further dimension for microbial immune enhancement. Innate immune cells such as monocytes, macrophages, and NK cells can acquire long-lasting functional reprogramming following exposure to defined microbial ligands [44,169]. Engineered microbes can be designed to deliver these ligands in a controlled manner, inducing beneficial epigenetic and metabolic changes that enhance future antimicrobial responses [91,170,171]. Unlike traditional vaccines that act primarily through adaptive immunity, trained immunity-based microbial platforms prime the innate immune system for heightened responsiveness across a broad range of pathogens.
Engineered microbial ligands that engage pattern recognition receptors provide a powerful mechanism for controlling trained immunity. Selective activation of receptors such as NOD2, dectin-1, or Toll-like receptors can bias myeloid cells toward enhanced cytokine production, increased phagocytic capacity, and improved antigen presentation [172,173,174]. When these signals are delivered repetitively at low levels by colonizing engineered microbes, they can maintain a state of heightened immune vigilance without inducing overt inflammation.
Synergy between engineered microbes and bacteriophages introduces an additional immune-forward strategy for infection control. Phages can selectively reduce pathogenic bacterial populations, while engineered microbes simultaneously stimulate immune clearance of infected cells and residual pathogens[175,176]. This combination allows direct pathogen reduction to be coupled with immune activation, reducing the probability of relapse and suppressing the emergence of resistant strains [177]. Importantly, phage-induced bacterial lysis also releases pathogen-associated molecular patterns that further amplify host immune responses.
Myeloid cell programming is a central target for microbial immune enhancement. Macrophages and dendritic cells orchestrate the balance between tolerance and immunity and determine the quality of downstream adaptive responses [22,110,178,179,180]. Engineered microbes can deliver cytokines, metabolites, or ligands that bias myeloid differentiation toward pro-inflammatory, antimicrobial phenotypes. This includes promotion of classically activated macrophages, enhancement of antigen presentation capacity, and increased production of reactive oxygen and nitrogen species for microbial killing.
Natural killer cell programming represents a parallel axis of immune enhancement [163]. NK cells play critical roles in early antiviral defense and in controlling intracellular pathogens. Engineered microbes can be designed to deliver IL-15, IL-12, or IL-18 locally, driving NK cell activation, expansion, and cytotoxic function within infected tissues [181,182,183]. This localized modulation avoids the systemic toxicity associated with cytokine-based NK cell therapies and strengthens early containment of infection before adaptive immunity is fully engaged.
Importantly, immune-forward microbial strategies also address the problem of immune exhaustion that arises during chronic infection [20,84]. Persistent antigen exposure and inhibitory signaling can progressively dampen effector function in T cells and innate populations [184,185]. Engineered microbes can counteract these suppressive circuits by locally delivering co-stimulatory ligands, inhibitory checkpoint decoys, or metabolic modulators that restore immune competence. This capacity to recondition immune responses distinguishes microbial immune enhancement from conventional vaccine or antimicrobial approaches.
The adaptability of engineered microbes further enables context-sensitive immune control across different stages of infection [22,186]. Early in infection, microbes can bias toward immune amplification and effector recruitment. As pathogen burden declines, the same platforms can shift toward resolution programs that promote tissue repair and limit immunopathology. This dynamic regulation reflects the natural progression of host defense and is difficult to reproduce with static pharmacological interventions [4,162,187].
Together, engineered microbes establish a new framework for infection control grounded in immune augmentation rather than direct antimicrobial action. By amplifying innate and adaptive defense mechanisms, inducing trained immunity, coordinating phage-mediated pathogen reduction, and reprogramming myeloid and NK cell function, these systems offer a versatile and resistance-resilient approach to infectious disease therapy. This immune-forward paradigm positions engineered microbes as active architects of host defense rather than passive antimicrobial tools.
7. Immune Safety Architecture for Living Immunotherapeutics
The therapeutic power of engineered microbes derives from their ability to engage and reshape immune circuits with high potency, spatial precision, and temporal adaptability [22]. These same properties, however, generate unique safety challenges that differ fundamentally from those associated with conventional biologics, small molecules, or cell therapies. Living immunotherapeutics are capable of sustained replication, autonomous decision-making, and dynamic immune engagement [188,189]. As a result, their safety cannot be reduced to questions of genetic stability or microbial containment alone. Instead, it must be understood through a comprehensive immune safety architecture that governs inflammatory magnitude, tolerance preservation, host–microbe immune memory, and controlled therapeutic termination (Figure 4).
Most existing discussions of microbial safety emphasize the need for genetic kill switches or auxotrophic containment [69,190]. While these measures are essential, they address only one layer of risk. Immune safety operates at a higher systems level and determines whether engineered microbes provoke pathological inflammation, breach tolerance checkpoints, induce deleterious immune memory, or interact unpredictably with host defense programs. The following subsections define immune safety as a multi-dimensional design problem that must be addressed with the same rigor as therapeutic efficacy.
7.1. Immunotoxicity and Unwanted Inflammation
The most immediate and potentially life-threatening risk of microbial immunotherapy is uncontrolled inflammation. Engineered microbes that deliver cytokines, immune ligands, or innate immune agonists are intentionally designed to activate powerful host defense pathways [87]. If this activation exceeds physiological limits or escapes spatial confinement, it can trigger systemic cytokine release, vascular leakage, tissue damage, and organ failure. Unlike conventional drugs, which dilute and decay with predictable pharmacokinetics, living microbes can amplify their own activity through replication and positive feedback within inflammatory microenvironments [116,123]. This creates the possibility of nonlinear immune escalation.
Overshoot inflammation arises when immune outputs exceed the regulatory capacity of local tissue networks. For example, local microbial production of IL-12 or type I interferons may successfully activate cytotoxic immunity within a tumor but can also propagate inflammatory cascades through dendritic cell migration and systemic T cell activation. Similarly, STING agonist delivery that is beneficial within a tumor can induce widespread interferonopathy if signal diffusion is not constrained [123,191,192]. These dynamics make quantitative control of immune output an essential design requirement rather than a secondary optimization step.
Avoidance of systemic cytokine spillover represents one of the core objectives of immune safety engineering. Microbial immunotherapies achieve therapeutic selectivity by exploiting spatial confinement, but spatial control is not absolute. Cytokines and chemokines produced locally can enter circulation, particularly in tissues with high vascular permeability such as tumors or inflamed mucosa [116]. Engineered control strategies must therefore integrate both spatial and quantitative regulation. These include promoter architectures that scale output with local signal intensity, feedback circuits that attenuate production once defined thresholds are reached, and signal-dependent degradation systems that terminate cytokine synthesis in the absence of sustained pathological cues [118,193].
Another critical dimension of immunotoxicity involves the interaction between engineered microbial outputs and endogenous inflammatory loops. Many immune pathways exhibit strong positive feedback, particularly those involving interferons, NF-κB signaling, and inflammasome activation [181,194]. When microbial outputs engage these loops, they can convert localized immune activation into self-propagating systemic inflammation. Effective immune safety architecture must therefore explicitly model and constrain feedback interactions between engineered signals and host immune amplification circuits [22].
Tissue-specific vulnerability further complicates the inflammatory safety landscape. The gut, lung, and central nervous system exhibit markedly different thresholds for inflammatory injury. Engineered microbes that are well tolerated in the intestinal lumen may be catastrophic if translocated into systemic circulation or ectopic tissues [195,196]. Similarly, mucosal cytokine exposure that supports tolerance can provoke pathology if redirected toward deeper immune compartments [28]. This necessitates tissue-aware immune safety design that accounts for regional immune thresholds, vascular permeability, and resident immune cell composition.
Importantly, immunotoxicity is not limited to acute cytokine release syndromes. Chronic low-grade inflammatory activation can also produce long-term pathology through fibrosis, epithelial barrier erosion, immune exhaustion, and dysregulated tissue remodeling. Engineered microbes designed for long-term colonization must therefore balance sustained therapeutic output against cumulative inflammatory burden [31,156,197]. This balance cannot be achieved through static genetic designs alone and requires dynamic immune-responsive regulation.
Collectively, these considerations establish immunotoxicity as a central and defining constraint in the development of microbial immunotherapeutics. Effective control of overshoot inflammation and prevention of systemic cytokine spillover require integration of spatial targeting, quantitative feedback regulation, tissue-specific immune thresholds, and host–microbe signaling dynamics. Immune safety at this level is not an auxiliary feature but a core architectural principle of living immunotherapy design.
7.2. Maintaining Immune Tolerance
A central challenge in the deployment of engineered microbial immunotherapeutics is preserving immune tolerance while deliberately manipulating immune activation. The immune system is organized around layered checkpoints that distinguish harmful from harmless stimuli, and disruption of these checkpoints can result in autoimmunity, chronic inflammation, or tissue-specific immunopathology [128,198,199]. Because engineered microbes operate at the interface of innate sensing, antigen presentation, and metabolic immune regulation, they occupy a position of exceptional influence over tolerance mechanisms. Immune safety therefore demands that tolerance preservation be treated as a primary design constraint rather than an emergent side effect.
Prevention of autoimmunity represents one of the most critical objectives of tolerance-aware microbial engineering. Microbial delivery of immune stimulatory molecules, whether cytokines, ligands, or metabolites, carries the intrinsic risk of lowering activation thresholds in autoreactive lymphocyte populations. This is particularly relevant in genetically predisposed hosts or in tissues with pre-existing low-grade inflammation [56,101,200]. If regulatory networks fail to counterbalance engineered immune activation, autoreactive clones that were previously restrained by peripheral tolerance can expand and initiate sustained tissue damage.
A second major tolerance hazard arises from excessive engagement of pattern recognition receptors (PRR). Toll-like receptors, NOD-like receptors, and cytosolic nucleic acid sensors evolved to detect microbial danger signals with high sensitivity [201,202]. Engineered microbes, even when attenuated, retain molecular structures that engage these receptors. Overactivation of PRR signaling can override regulatory circuits, amplify inflammatory cascades, and drive pathological immune activation independently of antigen specificity. This risk is magnified when microbial load increases or when engineered output reinforces PRR signaling through positive feedback.
Tolerance-preserving microbial design therefore requires quantitative control of PRR activation rather than simple presence or absence of stimulation. This can be achieved by modifying cell wall components, attenuating flagellar or lipopolysaccharide signaling strength, and engineering conditional expression of pro-inflammatory motifs [203,204]. By tuning the magnitude and duration of PRR engagement, engineered microbes can remain immunologically visible without overwhelming tolerance thresholds.
An additional layer of tolerance control involves deliberate co-delivery of regulatory signals alongside immune-stimulatory outputs. Simultaneous secretion of IL-10, TGF-β, or regulatory metabolites can buffer the effects of PRR activation and preserve regulatory T cell dominance [63,205]. This dual-output strategy reflects natural host responses to commensal microbes, which continuously stimulate innate sensors while maintaining tolerance through compensatory regulatory signaling [206,207]. Engineered systems that fail to incorporate such counter-regulation risk driving immune imbalance even when individual components appear safe in isolation.
Antigen context further modulates tolerance outcomes. When engineered microbes display antigens, the immune interpretation of those antigens depends entirely on the accompanying signal environment. The same antigen presented in an inflammatory context can provoke pathogenic immunity, whereas presentation in a regulatory context induces tolerance. Immune safety therefore requires that antigen display be explicitly coupled to tolerogenic circuit states and never be allowed to operate under uncontrolled inflammatory conditions [56,198,205].
Host heterogeneity adds another layer of complexity [208,209]. Baseline immune tone varies widely among individuals due to genetics, microbiota composition, metabolic status, prior infections, and age. A microbial design that preserves tolerance in one host may provoke autoimmunity in another. This underscores the need for immune-adaptive circuits that sense host inflammatory state and dynamically adjust output to remain within safe regulatory boundaries.
Taken together, maintaining immune tolerance in the context of microbial immunotherapy is not a passive property but an active engineering problem. It requires simultaneous control of PRR engagement, quantitative regulation of immune outputs, coordinated delivery of regulatory signals, and adaptive responsiveness to host immune state. Tolerance preservation emerges not from microbial attenuation alone but from dynamic systems-level immune design.
7.3. Adaptive Immunity Against the Therapeutic Microbe
While innate immune interactions dominate early host responses to engineered microbes, adaptive immunity ultimately determines long-term therapeutic efficacy and safety [210,211]. Host immune systems are highly efficient at recognizing, remembering, and eliminating microbial antigens. Once adaptive immune memory is established against a therapeutic microbe, repeated administrations may fail due to rapid clearance, neutralization of microbial products, or destructive immune responses against colonized tissues. This adaptive barrier represents one of the most underappreciated challenges in living immunotherapy.
Loss of efficacy is often the first observable consequence of anti-microbial adaptive immunity. Antibody-mediated neutralization can block microbial colonization, prevent adherence to mucosal surfaces, and eliminate circulating bacteria before they reach their therapeutic target. Cytotoxic T lymphocytes can recognize and destroy host cells harboring engineered microbes, particularly in intracellular platforms [212,213,214,215]. These processes can eliminate the therapeutic agent long before sufficient immune modulation is achieved.
Adaptive immunity also imposes evolutionary pressure on microbial platforms. Once a microbe is recognized as immunogenic, selection favors variants that escape immune recognition. This can destabilize engineered circuits, eliminate control modules, or alter immune outputs in unpredictable ways [104,216,217]. Immune-driven selection therefore introduces a unique form of functional drift that is not present in conventional pharmacological therapies.
Design strategies for evading anti-microbial immunity must operate within ethical and safety boundaries. Complete immune invisibility is neither achievable nor desirable, as controlled immune recognition is essential for safety and clearance [218]. Instead, microbial platforms can be engineered to reduce dominant antigenic epitopes, limit surface exposure of immunogenic structures, or cloak themselves using host-derived molecules [219,220]. Additional approaches include intermittent expression of therapeutic functions, phase-variable surface antigens, or shielding strategies that reduce antibody accessibility.
Predictable immunological memory further complicates repeated dosing regimens [129]. Once immune memory is established, subsequent microbial administrations may provoke faster and more intense inflammatory responses than the initial exposure [221]. In some contexts, this can enhance therapeutic efficacy. In others, it can produce excessive inflammation or rapid microbial elimination [222]. Immune safety architecture must therefore incorporate strategies for managing secondary exposure dynamics rather than focusing exclusively on primary responses.
Microbial platforms that rely on long-term colonization face particularly stringent adaptive immunity challenges. Even commensal-like strains can eventually be targeted when engineered outputs alter immune perception [198,223]. Maintenance of therapeutic populations over extended periods may require immune tolerance induction toward the microbe itself. This creates a complex design tradeoff in which the microbe must remain visible enough for immune clearance if needed, yet tolerated enough to persist therapeutically [224].
Host pre-existing immunity adds another layer of variability. Many chassis organisms belong to species that naturally colonize humans or cause prior infections. Pre-existing antibodies and memory T cells may eliminate engineered derivatives before they can exert any effect [215,223,225]. This variability further supports the need for personalized or stratified microbial immunotherapeutic design.
Ultimately, adaptive immunity is not merely an obstacle to microbial therapy but an integral component of immune safety. It provides long-term clearance, establishes population-level containment, and limits uncontrolled microbial persistence. Effective immune safety architecture must therefore balance immune evasion for short-term efficacy with immune recognition for long-term safety, treating adaptive immunity as a controlled feature rather than an external threat.
7.4. Kill Switches, Containment, and Immune-Clearance Logic
Genetic kill switches and containment systems are often presented as the primary safety solution for engineered microbes [103]. While essential, these mechanisms represent only the mechanical layer of a broader immune safety architecture. True safety emerges not simply from microbial self-destruction but from coordinated interaction between engineered termination logic and host immune clearance pathways [128,226]. Living immunotherapeutics must be designed to exit the host environment in a predictable, controllable, and immunologically benign manner [104].
Immune-dependent kill switches represent a powerful safety strategy that directly integrates microbial survival with host immune state. These systems activate microbial self-destruction in response to defined immune signals such as interferons, inflammatory cytokines, or antibody binding. In this configuration, the immune system itself becomes the trigger for therapeutic termination [26,93,227]. As inflammatory cues dissipate or immune memory develops, microbial populations are selectively eliminated, creating a dynamic self-limiting loop.
Environmental containment provides an orthogonal layer of safety [128]. Engineered microbes can be made dependent on synthetic nutrients, non-natural amino acids, or defined metabolic substrates that are absent outside controlled environments [228,229]. This prevents long-term survival outside the host and limits environmental dissemination. In gastrointestinal applications, containment can be achieved by restricting survival to specific pH ranges, bile acid profiles, or nutrient niches that exist only within targeted mucosal compartments [230].
Biosafety levels must be explicitly integrated into chassis selection and design logic. Commensal organisms with long histories of safe human exposure offer intrinsically lower biosafety risk than attenuated pathogens [136,188]. However, even commensal chassis can become hazardous when armed with immune stimulatory circuits. Biosafety classification therefore depends not only on the species but on the immune outputs encoded within the engineered genome [69]. Immune potency, not microbial taxonomy, becomes the dominant determinant of biosafety category.
Clearance logic also governs how microbes are removed when therapy is complete. Passive clearance through immune recognition, active clearance through kill switches, and pharmacological clearance through antimicrobial sensitivity must be coordinated to avoid incomplete elimination [104,128]. Incomplete clearance can promote chronic immune stimulation, biofilm persistence, or selection of escape variants [226,231]. Conversely, overly aggressive clearance can terminate therapy prematurely and provoke collateral inflammation.
An important but underexplored dimension of kill switch architecture is how microbial death itself influences immunity. Lysis releases pathogen-associated molecular patterns, DNA, and intracellular contents that can trigger secondary inflammatory responses [232]. Kill switches must therefore be designed to avoid synchronized mass lysis that could resemble septic shock at the molecular level [233]. Controlled, staggered, or apoptotic-like microbial death programs represent safer alternatives [53].
Horizontal gene transfer further complicates containment. Engineered immune circuits must be insulated from transfer to native microbiota or environmental microbes [234]. Genetic safeguards such as split circuits, non-canonical codon usage, and self-deleting payloads reduce the probability of functional transfer [104,235,236]. This containment layer is essential for preventing ecological propagation of immune-modulatory functions beyond the intended host.
In sum, kill switches, containment systems, and clearance logic form the mechanical backbone of microbial immune safety. However, their effectiveness depends on seamless integration with host immune recognition, adaptive memory, tissue-specific clearance pathways, and ecological containment. Safety in living immunotherapy is not achieved by a single genetic trigger but by a coordinated termination architecture embedded within host–microbe immune interactions.
8. Clinical Translation Landscape Focused on Immune Endpoints
Clinical translation in immunotherapy has undergone a fundamental shift from symptom-based and imaging-based outcome measures toward immune-state–resolved evaluation. Across oncology, autoimmunity, metabolic disease, and inflammatory disorders, therapeutic success is now increasingly defined by the capacity to reprogram immune network structure rather than by short-term suppression of isolated disease markers [128,165,169,237]. This transformation reflects a growing recognition that durable clinical benefit emerges only when immune system organization, regulatory balance, and inflammatory topology are stably reshaped. Representative immune-state–resolved endpoints, safety readouts, and translational implications across cancer, autoimmunity, infection, and cross-indication applications (Table 2).
In cancer trials, immune endpoints now occupy a central position in response assessment. Longitudinal profiling of cytotoxic T cell activation, interferon signaling, antigen presentation capacity, and myeloid suppressor cell dynamics consistently outperforms radiographic measurements in predicting durable remission [92,104]. Responders typically exhibit coordinated elevation of CD8-positive effector function, restoration of dendritic cell cross-presentation, and suppression of TGF-β–driven immune exclusion [238,239,240]. These immune signatures often precede measurable tumor regression and persist as indicators of long-term immune control.
Cytokine landscapes have emerged as unifying translational metrics across therapeutic platforms. Independent of whether the intervention involves checkpoint inhibition, cell therapy, microbial therapeutics, or gene modulation, successful clinical responses consistently align with specific shifts in cytokine topology [241,242,243]. Activation of interferon networks, controlled induction of IL-12 signaling, normalization of GM-CSF gradients, and suppression of chronic IL-6 and TNF inflammatory loops recur across effective trials. These cytokine reorganizations represent conserved immune attractor states associated with disease control [244,245,246].
In inflammatory bowel disease, the clinical meaning of immune tolerance reconstruction has become increasingly precise. Trials now routinely quantify regulatory T cell expansion, FOXP3 expression stability, and restoration of Th17 to regulatory T cell ratios as core efficacy endpoints [123,136,247]. Durable mucosal healing correlates more closely with reestablishment of immune regulatory dominance than with transient reductions in inflammatory cytokines alone [248,249]. This immune-centered interpretation has clarified why many anti-inflammatory drugs achieve short-term symptom relief without inducing lasting remission.
Stratification of patients according to microbiome engraftment status has emerged as a critical immune determinant of therapeutic response. In both autoimmune and oncologic studies, responders frequently demonstrate stable incorporation of therapeutic microbial strains, accompanied by downstream expansion of short-chain fatty acid–producing taxa and tolerogenic dendritic cell phenotypes [92,244,250,251,252]. Non-responders often display persistent immune rejection of administered microbes driven by unresolved type 1 inflammatory tone, preventing establishment of functional microbial–immune circuits.
Metabolic disease trials have similarly converged on immune inflammation as the dominant axis of clinical translation. Improvements in insulin sensitivity, lipid handling, and hepatic steatosis increasingly correlate with attenuation of macrophage-driven adipose inflammation, suppression of IL-1β signaling, and restoration of anti-inflammatory adipokine profiles [253,254,255]. These immune shifts frequently precede and predict metabolic normalization more accurately than changes in body mass or caloric intake alone. This has reframed metabolic disease as an immunometabolic disorder governed by inflammatory network stability.
Clinical safety assessment has likewise transitioned toward immune-centered surveillance. Systemic inflammatory markers such as C-reactive protein, ferritin, IL-6, interferon gamma, and neutrophil-to-lymphocyte ratios now serve as early indicators of therapeutic risk long before overt organ toxicity develops [256,257]. Expanded immune cell panel profiling further identifies shifts in activation thresholds, exhaustion states, and regulatory capacity that forecast adverse immune events. Immune monitoring has thus become the primary early warning system for clinical risk [258,259].
Immune correlates of response now function as the translational bridge between preclinical models and human disease. Animal systems that replicate human cytokine architecture, myeloid polarization states, and T cell differentiation dynamics exhibit markedly higher predictive value than those that mimic phenotypic disease features alone [260,261,262]. Translational fidelity is increasingly defined by immune-state equivalence rather than by anatomical or histological resemblance.
In the context of engineered microbial therapeutics, immune readouts define the core clinical logic. Therapeutic success is determined by coordinated shifts in innate immune training, antigen presentation efficiency, regulatory macrophage polarization, and systemic cytokine normalization. The engineered organism itself is no longer interpreted as the therapeutic endpoint [56,68,263]. The immune network that it reconstructs represents the true object of clinical intervention.
Neuroinflammatory and neuroimmune trials increasingly apply the same immune-centric translational logic. Microglial activation states, peripheral cytokine gradients, and blood–brain barrier immune signaling now serve as primary response metrics in neurodegenerative disease, depression, and chronic pain syndromes. Clinical improvement consistently aligns with normalization of microglial inflammatory tone and astrocytic metabolic coupling rather than with isolated neurotransmitter changes [264,265,266].
Across cancer, autoimmunity, metabolic disease, and neuroinflammation, a unifying principle has emerged in clinical translation [267,268,269]. Durable therapeutic benefit correlates with multi-axis immune network reorganization rather than with suppression of single effector pathways. Successful therapies reshape innate training programs, adaptive polarization states, metabolic immune coupling, and regulatory feedback architecture simultaneously. Short-lived responses arise when this coordinated immune restructuring fails to occur.
This immune-first framework also resolves many historical failures in clinical development. Numerous trials that achieved impressive short-term biomarker suppression but failed to induce durable remission now appear mechanistically incomplete. Without stabilization of regulatory balance, inflammatory containment, and immune memory architecture, long-term therapeutic success becomes statistically improbable. Immune architecture, not single target inhibition, governs clinical durability [270,271].
The central translational conclusion is therefore unavoidable. Modern therapeutic success is no longer explained by molecular targeting alone. It is explained by the capacity of an intervention to re-encode immune system state across tissues, time, and disease domains. Immune phenotyping has become the primary explanatory framework of clinical translation and the dominant lens through which the future of living immunotherapeutics must be interpreted.
9. Future Directions Toward Personalized and AI-Designed Immune Microbial Therapies
The future of engineered microbial immunotherapy lies not in incremental optimization of existing platforms but in the emergence of a fully integrated design ecosystem that unites systems immunology, artificial intelligence, and programmable biology. As immune phenotyping, spatial biology, and computational modeling converge, microbial therapeutics are poised to transition from generalized immune modulators to precisely individualized immune reprogramming agents. This transformation reflects a broader shift in medicine from population-averaged intervention toward dynamic, patient-specific immune state engineering.
Single-cell and spatial multi-omics technologies now permit direct measurement of immune cell states at unprecedented resolution across tissues and disease stages. These datasets reveal not only which immune cells are present, but how their transcriptional programs, metabolic states, ligand–receptor interactions, and spatial organization evolve during health and disease. In the future, microbial immunotherapies will be designed not to target abstract disease categories but to drive patient immune systems toward defined target immune attractor states identified through these high-dimensional maps.
Spatial immunology adds a critical architectural layer to this personalization paradigm. Immune function is not uniform across tissues but emerges from the geometric arrangement of immune cells, stromal compartments, vascular interfaces, and microbial niches. Engineered microbes will increasingly be designed to engage specific spatial immune microdomains such as perivascular niches, epithelial interfaces, lymphoid aggregates, or tumor margins. Spatially targeted immune reprogramming will replace uniform immune stimulation as the dominant therapeutic logic.
Artificial intelligence and machine learning will become essential design engines for microbial immunotherapy. The combinatorial complexity of microbial chassis selection, sensing module integration, circuit topology, and immune output tuning exceeds what can be explored through human intuition alone. Machine learning models trained on immune multi-omics and microbial genotype–phenotype relationships will be used to predict optimal chassis–circuit–output combinations for specific immune phenotypes. This will shift microbial design from artisanal genetic engineering toward algorithmically guided therapeutic construction.
Digital twins of the immune–microbe ecosystem represent a particularly powerful future development. These computational replicas of patient immune systems, continuously updated with clinical immune profiling data, will allow in silico testing of engineered microbial interventions before physical administration. Digital twins will simulate how microbial circuits interact with host immune networks, predict inflammatory risk, estimate durability of tolerance induction, and forecast interactions with concurrent therapies. This will radically compress development cycles and reduce clinical uncertainty.
Designer microbial consortia will increasingly replace single-strain therapeutics. Complex immune disorders rarely arise from defects in a single pathway and instead reflect network-level dysregulation across innate, adaptive, metabolic, and stromal axes. Microbial communities engineered with division of labor, coordinated quorum logic, and layered immune outputs will allow distributed immune control across multiple biological dimensions simultaneously. These systems will function as living immune control networks rather than as isolated molecular effectors.
Organoid and organ-on-chip immune model systems will serve as the experimental substrates for this next generation of microbial design. Human intestinal, hepatic, pulmonary, and tumor organoids integrated with immune cells and microbiota will allow direct measurement of microbial immune reprogramming in physiologically realistic contexts. These platforms will bridge the gap between animal models and human immune architecture, enabling iterative refinement of microbial circuits under clinically relevant conditions.
Integration of microbial immunotherapies with cell-based immunotherapies will define a major convergence trajectory. Engineered microbes will increasingly be used to precondition tissue immune landscapes prior to adoptive T cell transfer, CAR-T therapy, or natural killer cell infusion. By modulating antigen presentation, suppressive myeloid populations, and metabolic barriers, microbes can establish immune niches that enhance the persistence, functionality, and safety of cellular therapies.
A parallel convergence will occur between microbial immunotherapy and next-generation vaccine platforms. Microbes will be designed to act as living adjuvants that continuously shape mucosal and systemic immunity after antigen exposure. Rather than serving solely as antigen carriers, engineered microbes will define the immunological context within which vaccine-induced memory forms, influencing durability, breadth, and tissue distribution of protective immunity.
The regulatory science of living immunotherapeutics will likewise evolve toward immune-state–based approval frameworks. Instead of relying solely on classical toxicity and pharmacokinetic metrics, future regulatory evaluation will incorporate immune network remodeling, tolerance stability, trained immunity induction, and memory architecture as formal safety and efficacy endpoints. Regulatory definitions of therapeutic mechanism will increasingly reflect immune systems behavior rather than molecular target engagement.
Ethical and societal considerations will become inseparable from technical progress. As microbial immunotherapies acquire the capacity for long-term immune reprogramming and potential heritable ecological effects within the microbiome, questions of reversibility, intergenerational immune influence, and ecological containment will intensify. Immune safety architecture, digital surveillance, and transparent governance will be required to preserve public trust in these living technologies.
The dominant paradigm emerging from these converging trends is clear. Future microbial immunotherapies will no longer be standardized biological products administered uniformly across patient populations. They will be algorithmically designed, immune-state–resolved, spatially targeted, dynamically regulated, and continuously monitored living systems. They will function as programmable extensions of the immune system itself rather than as external pharmacological tools.
In this forthcoming therapeutic landscape, disease will be interpreted not as a static molecular abnormality but as a dynamic immune network configuration that can be measured, modeled, and re-encoded. Engineered microbes will serve as the living interface through which this immune recoding is executed. The era of generic immune modulation is therefore giving way to the era of personalized immune system programming through living microbial computation.
Author Contributions
O. A. and M.N. contributed equally to the conceptualization, design, and writing of the main manuscript.
Acknowledgments
N/A.
Conflict of Interest
The authors declare no conflict of interest.
References
- June, CH; O'Connor, RS; Kawalekar, OU; et al. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Yang, F; Shi, K; Jia, YP; et al. Advanced biomaterials for cancer immunotherapy. Acta Pharmacol Sin 2020, 41, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Gurbatri, CR; Arpaia, N; Danino, T. Engineering bacteria as interactive cancer therapies. Science 2022, 378, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, TH; Cho, BK; Lee, DH. Synthetic Biology-Driven Microbial Therapeutics for Disease Treatment. J Microbiol Biotechnol 2024, 34, 1947–1958. [Google Scholar] [CrossRef]
- Rooks, MG; Garrett, WS. Gut microbiota, metabolites and host immunity. Nat Rev Immunol 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Belkaid, Y; Hand, TW. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Chien, T; Doshi, A; Danino, T. Advances in bacterial cancer therapies using synthetic biology. Curr Opin Syst Biol 2017, 5, 1–8. [Google Scholar] [CrossRef]
- Lavrador, P; Gaspar, VM; Mano, JF. Engineering mammalian living materials towards clinically relevant therapeutics. EBioMedicine 2021, 74, 103717. [Google Scholar] [CrossRef]
- Isabella, VM; Ha, BN; Castillo, MJ; et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat Biotechnol 2018, 36, 857–864. [Google Scholar] [CrossRef]
- Zheng, JH; Nguyen, VH; Jiang, SN; et al. Two-step enhanced cancer immunotherapy with engineered Salmonella typhimurium secreting heterologous flagellin. Sci Transl Med 2017, 9. [Google Scholar] [CrossRef]
- Chowdhury, S; Castro, S; Coker, C; et al. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat Med 2019, 25, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Round, JL; Mazmanian, SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y; Harrison, OJ. Homeostatic Immunity and the Microbiota. Immunity 2017, 46, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Omer, R; Mohsin, MZ; Mohsin, A; et al. Engineered Bacteria-Based Living Materials for Biotherapeutic Applications. Front Bioeng Biotechnol 2022, 10, 870675. [Google Scholar] [CrossRef]
- Lee, DW; Gardner, R; Porter, DL; et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Scott, AM; Wolchok, JD; Old, LJ. Antibody therapy of cancer. Nat Rev Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Rafii, S; Mukherji, D; Komaranchath, AS; et al. Advancing CAR T-Cell Therapy in Solid Tumors: Current Landscape and Future Directions. In Cancers (Basel); 2025; Volume 17. [Google Scholar]
- Li, YR; Zhu, Y; Fang, Y; et al. Emerging trends in clinical allogeneic CAR cell therapy. Med 2025, 6, 100677. [Google Scholar] [CrossRef]
- Vincent, F; Gianni, D. The limitations of small molecule and genetic screening in phenotypic drug discovery. Cell Chem Biol 2025, 32, 1310–1320. [Google Scholar] [CrossRef]
- Xiang, X; Zhou, Z; Rao, X; et al. Recent advances of engineered bacteria for therapeutic applications. Mol Ther. 2025. [Google Scholar] [CrossRef]
- Zhou, T; Wu, J; Tang, H; et al. Enhancing tumor-specific recognition of programmable synthetic bacterial consortium for precision therapy of colorectal cancer. NPJ Biofilms Microbiomes 2024, 10, 6. [Google Scholar] [CrossRef]
- Zhao, L; Xin, J; Hu, M; et al. Programmable microbial therapeutics: advances in engineered bacteria for targeted in vivo delivery and precision medicine. J Adv Res. 2025. [Google Scholar]
- Armstrong, A; Isalan, M. Engineering bacterial theranostics: from logic gates to in vivo applications. Front Bioeng Biotechnol 2024, 12, 1437301. [Google Scholar] [PubMed]
- Shi, XR; Zhang, YF; Zhou, Y; et al. Engineered microbial sensors: providing a new paradigm for disease detection. J Mater Chem B 2025, 13, 14535–14555. [Google Scholar] [PubMed]
- Zhou, Y; Han, Y. Engineered bacteria as drug delivery vehicles: Principles and prospects. Eng Microbiol 2022, 2, 100034. [Google Scholar]
- Qin, Y; You, SH; Zhang, Y; et al. Genetic Programming by Nitric Oxide-Sensing Gene Switch System in Tumor-Targeting Bacteria. In Biosensors (Basel); 2023; Volume 13. [Google Scholar]
- Tanniche, I; Behkam, B. Engineered live bacteria as disease detection and diagnosis tools. J Biol Eng 2023, 17, 65. [Google Scholar] [CrossRef]
- Niu, L; Deng, Z; Jin, Y; et al. Engineering oncolytic bacteria as precision cancer therapeutics: design principles, therapeutic strategies, and translational perspectives. In Protein Cell; 2025. [Google Scholar]
- Spranger, S; Spaapen, RM; Zha, Y; et al. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013, 5, 200ra116. [Google Scholar]
- Smolen, JS; Aletaha, D; McInnes, IB. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Zou, ZP; Du, Y; Fang, TT; et al. Biomarker-responsive engineered probiotic diagnoses, records, and ameliorates inflammatory bowel disease in mice. Cell Host Microbe 2023, 31, 199–212 e195. [Google Scholar]
- Brand, A; Singer, K; Koehl, GE; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 2016, 24, 657–671. [Google Scholar]
- Arpaia, N; Campbell, C; Fan, X; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Hwang, IY; Tan, MH; Koh, E; et al. Reprogramming microbes to be pathogen-seeking killers. ACS Synth Biol 2014, 3, 228–237. [Google Scholar] [PubMed]
- Cubillos-Ruiz, A; Guo, T; Sokolovska, A; et al. Engineering living therapeutics with synthetic biology. Nat Rev Drug Discov 2021, 20, 941–960. [Google Scholar] [PubMed]
- Fischbach, MA; Segre, JA. Signaling in Host-Associated Microbial Communities. Cell 2016, 164, 1288–1300. [Google Scholar] [CrossRef] [PubMed]
- Round, JL; Palm, NW. Causal effects of the microbiota on immune-mediated diseases. Sci Immunol 2018, 3. [Google Scholar] [CrossRef]
- Roybal, KT; Rupp, LJ; Morsut, L; et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef]
- Nissim, L; Wu, MR; Pery, E; et al. Synthetic RNA-Based Immunomodulatory Gene Circuits for Cancer Immunotherapy. Cell 2017, 171, 1138–1150 e1115. [Google Scholar] [CrossRef]
- Del Vecchio, D; Dy, AJ; Qian, Y. Control theory meets synthetic biology. J R Soc Interface 2016, 13. [Google Scholar] [CrossRef]
- Sohka, T; Heins, RA; Phelan, RM; et al. An externally tunable bacterial band-pass filter. Proc Natl Acad Sci U S A 2009, 106, 10135–10140. [Google Scholar]
- Shui, S; Scheller, L; Correia, BE. Protein-based bandpass filters for controlling cellular signaling with chemical inputs. Nat Chem Biol 2024, 20, 586–593. [Google Scholar]
- Binnewies, M; Roberts, EW; Kersten, K; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Netea, MG; Joosten, LAB. Trained innate immunity: Concept, nomenclature, and future perspectives. J Allergy Clin Immunol 2024, 154, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Kalvapalle, PB; Sridhar, S; Silberg, JJ; et al. Long-duration environmental biosensing by recording analyte detection in DNA using recombinase memory. Appl Environ Microbiol 2024, 90, e0236323. [Google Scholar] [CrossRef] [PubMed]
- Huang, BD; Kim, D; Yu, Y; et al. Engineering intelligent chassis cells via recombinase-based MEMORY circuits. Nat Commun 2024, 15, 2418. [Google Scholar] [CrossRef] [PubMed]
- Scott, SR; Hasty, J. Quorum Sensing Communication Modules for Microbial Consortia. ACS Synth Biol 2016, 5, 969–977. [Google Scholar] [CrossRef]
- Guo, Y; Gao, M; Jiang, L; et al. Synthetic microbial consortia based on quorum-sensing for disease therapy. Bioresour Bioprocess 2025, 12, 125. [Google Scholar] [CrossRef]
- Fala, AK; Alvarez-Ordonez, A; Filloux, A; et al. Quorum sensing in human gut and food microbiomes: Significance and potential for therapeutic targeting. Front Microbiol 2022, 13, 1002185. [Google Scholar] [CrossRef]
- Tan, Q; Cao, X; Zou, F; et al. Spatial Heterogeneity of Intratumoral Microbiota: A New Frontier in Cancer Immunotherapy Resistance. Biomedicines 2025, 13. [Google Scholar] [CrossRef]
- Pignon, E; Schaerli, Y. Deciphering microbial spatial organization: insights from synthetic and engineered communities. ISME Commun 2025, 5, ycaf107. [Google Scholar] [CrossRef]
- Wang, H; Zheng, L; Yang, C; et al. Probiotic-mediated tumor microenvironment reprogramming with protease-sensitive interleukin-15 and photothermal therapy. Cell Rep Med 2025, 6, 102191. [Google Scholar] [CrossRef]
- Chen, J; Chen, J; Chen, Y; et al. Engineering Bacteria as Living Therapeutics in Cancer Therapy. Adv Sci (Weinh) 2025, 12, e07820. [Google Scholar] [CrossRef]
- Campos, GM; Americo, MF; Dos Santos Freitas, A; et al. Lactococcus lactis as an Interleukin Delivery System for Prophylaxis and Treatment of Inflammatory and Autoimmune Diseases. Probiotics Antimicrob Proteins 2024, 16, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Zou, ZP; Zhang, XP; Zhang, Q; et al. Genetically engineered bacteria as inflammatory bowel disease therapeutics. Eng Microbiol 2024, 4, 100167. [Google Scholar] [CrossRef] [PubMed]
- Redenti, A; Im, J; Redenti, B; et al. Probiotic neoantigen delivery vectors for precision cancer immunotherapy. Nature 2024, 635, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y; Xin, Q; Zhu, Y; et al. Probiotic-based oral vaccine mucosal delivery system enabling genetically encoded dual-antigen arrays. Nat Commun. 2025. [Google Scholar] [CrossRef]
- Johnston, CD; Wargo, JA. Engineering immunity: bacterial delivery of cancer neoantigen vaccines. Trends Immunol 2024, 45, 931–933. [Google Scholar] [CrossRef]
- Chen, YE; Bousbaine, D; Veinbachs, A; et al. Engineered skin bacteria induce antitumor T cell responses against melanoma. Science 2023, 380, 203–210. [Google Scholar] [CrossRef]
- Omolekan, TO; Folahan, JT; Tesfay, MZ; et al. Viral warfare: unleashing engineered oncolytic viruses to outsmart cancer's defenses. Front Immunol 2025, 16, 1618751. [Google Scholar] [CrossRef]
- Kouno, T; Zeng, S; Wang, Y; et al. Engineered bacteria producing aryl-hydrocarbon receptor agonists protect against ethanol-induced liver disease in mice. Alcohol Clin Exp Res (Hoboken) 2023, 47, 856–867. [Google Scholar] [CrossRef]
- Chen, W; Guo, Q; Li, H; et al. Engineered Probiotics Mitigate Gut Barrier Dysfunction Induced by Nanoplastics. Adv Sci (Weinh) 2025, 12, e2417283. [Google Scholar] [CrossRef]
- Wu, YQ; Zou, ZP; Zhou, Y; et al. Dual engineered bacteria improve inflammatory bowel disease in mice. Appl Microbiol Biotechnol 2024, 108, 333. [Google Scholar] [CrossRef]
- Wang, C; Feng, Q; Shi, S; et al. The Rational Engineered Bacteria Based Biohybrid Living System for Tumor Therapy. Adv Healthc Mater 2024, 13, e2401538. [Google Scholar] [CrossRef] [PubMed]
- Wang, H; Xu, F; Yao, C; et al. Engineering bacteria for cancer immunotherapy by inhibiting IDO activity and reprogramming CD8+ T cell response. Proc Natl Acad Sci U S A 2024, 121, e2412070121. [Google Scholar] [CrossRef] [PubMed]
- Wang, J; Wang, J; Yu, Z; et al. Targeting the Adenosine-Mediated Metabolic Immune Checkpoint with Engineered Probiotic for Enhanced Chemo-Immunotherapy. Adv Sci (Weinh) 2025, 12, e2411813. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y; Hu, Y; Liu, H; et al. High-Lactate-Metabolizing Photosynthetic Bacteria Reprogram Tumor Immune Microenvironment. Adv Mater 2024, 36, e2405930. [Google Scholar] [CrossRef]
- Frutos-Grilo, E; Ana, Y; Gonzalez-de Miguel, J; et al. Bacterial live therapeutics for human diseases. Mol Syst Biol 2024, 20, 1261–1281. [Google Scholar] [CrossRef]
- Tseng, CH; Wong, S; Yu, J; et al. Development of live biotherapeutic products: a position statement of Asia-Pacific Microbiota Consortium. Gut 2025, 74, 706–713. [Google Scholar] [CrossRef]
- Almansour, N; Al-Rashed, F; Choudhry, K; et al. Gut microbiota: a promising new target in immune tolerance. Front Immunol 2025, 16, 1607388. [Google Scholar]
- Wang, Q; Meng, Q; Chen, Y; et al. Interaction between gut microbiota and immunity in health and intestinal disease. Front Immunol 2025, 16, 1673852. [Google Scholar]
- Mkilima, T. Engineering artificial microbial consortia for personalized gut microbiome modulation and disease treatment. Ann N Y Acad Sci 2025, 1548, 29–55. [Google Scholar]
- Kim, S; Ndwandwe, C; Devotta, H; et al. Role of the microbiome in regulation of the immune system. Allergol Int 2025, 74, 187–196. [Google Scholar] [CrossRef]
- Zhang, X; Pan, Q; Yao, G; et al. Bacteroides fragilis and propionate synergize with low-dose methimazole to treat Graves' disease. Microbiol Spectr 2025, 13, e0318624. [Google Scholar] [CrossRef] [PubMed]
- Bui, TNY; Paul, A; Guleria, S; et al. Short-chain fatty acids-a key link between the gut microbiome and T-lymphocytes in neonates? Pediatr Res. 2025. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A; Sharma, G; Im, SH. Gut microbiota in regulatory T cell generation and function: mechanisms and health implications. Gut Microbes 2025, 17, 2516702. [Google Scholar] [CrossRef] [PubMed]
- Tian, K; Jing, D; Lan, J; et al. Commensal microbiome and gastrointestinal mucosal immunity: Harmony and conflict with our closest neighbor. Immun Inflamm Dis 2024, 12, e1316. [Google Scholar] [CrossRef]
- Kopczynska, J; Kowalczyk, M. The potential of short-chain fatty acid epigenetic regulation in chronic low-grade inflammation and obesity. Front Immunol 2024, 15, 1380476. [Google Scholar] [CrossRef]
- Tang, W; Wei, Y; Ni, Z; et al. IgA-mediated control of host-microbial interaction during weaning reaction influences gut inflammation. Gut Microbes 2024, 16, 2323220. [Google Scholar] [CrossRef]
- Mooser, C; Ronchi, F; Limenitakis, JP; et al. Diet-derived LPS determines intestinal IgA induction and repertoire characteristics independently of the microbiota. Immunity 2025, 58, 1778–1793 e1777. [Google Scholar] [CrossRef]
- Pietrzak, B; Tomela, K; Olejnik-Schmidt, A; et al. Secretory IgA in Intestinal Mucosal Secretions as an Adaptive Barrier against Microbial Cells. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Chowdhury, MR; Islam, A; Yurina, V; et al. Genetically Modified Lactic Acid Bacteria: a Promising Mucosal Delivery Vector for Vaccines. Probiotics Antimicrob Proteins 2025. [Google Scholar] [CrossRef]
- Zhou, M; Tang, Y; Xu, W; et al. Bacteria-based immunotherapy for cancer: a systematic review of preclinical studies. Front Immunol 2023, 14, 1140463. [Google Scholar] [CrossRef]
- Liu, S; Zhong, Z; Tu, Q; et al. Synthetic biology approaches to enhance cancer immune responses. Front Immunol 2025, 16, 1698158. [Google Scholar]
- Chen, R; Zou, J; Chen, J; et al. Pattern recognition receptors: function, regulation and therapeutic potential. Signal Transduct Target Ther 2025, 10, 216. [Google Scholar] [PubMed]
- Chen, YH; Wu, KH; Wu, HP. Unraveling the Complexities of Toll-like Receptors: From Molecular Mechanisms to Clinical Applications. Int J Mol Sci 2024, 25. [Google Scholar]
- Huang, Y; Piao, L; Liu, X. Enhancing Tumor-Specific immunity with SL(dacA): A attenuated Salmonella-mediated c-di-AMP delivery system targeting the STING pathway. Int J Pharm 2024, 666, 124759. [Google Scholar] [PubMed]
- Al-Saafeen, BH; Al-Sbiei, A; Bashir, G; et al. Attenuated Salmonella potentiate PD-L1 blockade immunotherapy in a preclinical model of colorectal cancer. Front Immunol 2022, 13, 1017780. [Google Scholar]
- Mowday, AM; van de Laak, JM; Fu, Z; et al. Tumor-targeting bacteria as immune stimulants - the future of cancer immunotherapy? Crit Rev Microbiol 2024, 50, 955–970. [Google Scholar]
- Li, W; Li, YA; Zhang, Y; et al. Engineered Salmonella Enteritidis vector targeting innate immune molecules provides protection against Salmonella Enteritidis and Salmonella Typhimurium. Poult Sci 2025, 104, 105724. [Google Scholar] [CrossRef]
- Damani-Yokota, P; Khanna, KM. Innate immune memory: The evolving role of macrophages in therapy. Elife 2025, 14. [Google Scholar]
- Zhang, S; Li, R; Xu, Y; et al. Engineered bacteria: Strategies and applications in cancer immunotherapy. Fundam Res 2025, 5, 1327–1345. [Google Scholar] [CrossRef]
- Kato, Y; Mori, H. Genetically stable kill-switch using "demon and angel" expression construct of essential genes. Front Bioeng Biotechnol 2024, 12, 1365870. [Google Scholar]
- Guo, Y; Gao, M; Wang, L; et al. An Engineered Probiotic Consortium Based on Quorum-Sensing for Colorectal Cancer Immunotherapy. Adv Sci (Weinh) 2025, 12, e12744. [Google Scholar] [CrossRef] [PubMed]
- Matuszynska, A; Ebenhoh, O; Zurbriggen, MD; et al. A new era of synthetic biology-microbial community design. Synth Biol (Oxf) 2024, 9, ysae011. [Google Scholar] [CrossRef] [PubMed]
- Wu, S; Zhou, Y; Dai, L; et al. Assembly of functional microbial ecosystems: from molecular circuits to communities. FEMS Microbiol Rev 2024, 48. [Google Scholar] [CrossRef] [PubMed]
- Chen, X; He, C; Zhang, Q; et al. Modularized Design and Construction of Tunable Microbial Consortia with Flexible Topologies. ACS Synth Biol 2024, 13, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Savage, TM; Vincent, RL; Rae, SS; et al. Chemokines expressed by engineered bacteria recruit and orchestrate antitumor immunity. Sci Adv 2023, 9, eadc9436. [Google Scholar] [CrossRef]
- Guo, P; Wang, W; Xiang, Q; et al. Engineered probiotic ameliorates ulcerative colitis by restoring gut microbiota and redox homeostasis. Cell Host Microbe 2024, 32, 1502–1518 e1509. [Google Scholar] [CrossRef]
- Furuichi, M; Kawaguchi, T; Pust, MM; et al. Commensal consortia decolonize Enterobacteriaceae via ecological control. Nature 2024, 633, 878–886. [Google Scholar] [CrossRef]
- Li, M; Liu, N; Zhu, J; et al. Engineered probiotics with sustained release of interleukin-2 for the treatment of inflammatory bowel disease after oral delivery. Biomaterials 2024, 309, 122584. [Google Scholar] [CrossRef]
- Li, X; Zhou, Z; Li, W; et al. Design of stable and self-regulated microbial consortia for chemical synthesis. Nat Commun 2022, 13, 1554. [Google Scholar] [CrossRef]
- Rottinghaus, AG; Ferreiro, A; Fishbein, SRS; et al. Genetically stable CRISPR-based kill switches for engineered microbes. Nat Commun 2022, 13, 672. [Google Scholar] [CrossRef]
- Nguyen, N; Wang, M; Li, L; et al. A genetic safeguard for eliminating target genes in synthetic probiotics in a gut environment. iScience 2025, 28, 113027. [Google Scholar] [CrossRef] [PubMed]
- Wang, S; Zhan, Y; Jiang, X; et al. Engineering Microbial Consortia as Living Materials: Advances and Prospectives. ACS Synth Biol 2024, 13, 2653–2666. [Google Scholar] [CrossRef] [PubMed]
- Quiroga-Centeno, AC; Atanasova, K; Ebert, MP; et al. Emerging microbiome-directed therapies in inflammatory bowel disease: beyond diet modification and FMT. Semin Immunopathol 2025, 47, 42. [Google Scholar] [CrossRef] [PubMed]
- Zalatan, JG; Petrini, L; Geiger, R. Engineering bacteria for cancer immunotherapy. Curr Opin Biotechnol 2024, 85, 103061. [Google Scholar]
- Akbariqomi, M; Kheirandish Zarandi, P; Abedi, A; et al. Bacteria-based immunosuppressive tumor microenvironment reprogramming: a promising dawn in cancer therapy. Microb Cell Fact 2025, 24, 207. [Google Scholar] [CrossRef]
- Zi, G; Zhou, W; Zhou, L; et al. Engineered Bacteria-Nano Hybrid System: The Intelligent Drug Factory for Next-Generation Cancer Immunotherapy. Pharmaceutics 2025, 17. [Google Scholar] [CrossRef]
- Fan, S; Zhu, S; Wang, W; et al. Current status and future perspectives of multi-modal bacteria-based cancer therapies. Clin Transl Med 2025, 15, e70485. [Google Scholar] [CrossRef]
- Lu, Y; Li, Z; Xu, H; et al. In situ enrichment and delivery of STING agonists by protamine-modified Salmonella for cancer immunotherapy. J Control Release 2025, 388, 114284. [Google Scholar] [CrossRef]
- Wang, B; Yu, W; Jiang, H; et al. Clinical applications of STING agonists in cancer immunotherapy: current progress and future prospects. Front Immunol 2024, 15, 1485546. [Google Scholar] [CrossRef]
- Nerdinger, YG; Binder, AK; Bremm, F; et al. STINGing Cancer: Development, Clinical Application, and Targeted Delivery of STING Agonists. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Leventhal, DS; Sokolovska, A; Li, N; et al. Immunotherapy with engineered bacteria by targeting the STING pathway for anti-tumor immunity. Nat Commun 2020, 11, 2739. [Google Scholar] [CrossRef] [PubMed]
- Liao, J; Pan, H; Huang, G; et al. T cell cascade regulation initiates systemic antitumor immunity through living drug factory of anti-PD-1/IL-12 engineered probiotics. Cell Rep 2024, 43, 114086. [Google Scholar] [CrossRef] [PubMed]
- Tumas, S; Meldgaard, TS; Vaaben, TH; et al. Engineered E. coli Nissle 1917 for delivery of bioactive IL-2 for cancer immunotherapy. Sci Rep 2023, 13, 12506. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z; Meng, F; Ma, Y; et al. In situ immunomodulation of tumors with biosynthetic bacteria promote anti-tumor immunity. Bioact Mater 2024, 32, 12–27. [Google Scholar] [CrossRef]
- Nguyen, DH; You, SH; Ngo, HT; et al. Reprogramming the tumor immune microenvironment using engineered dual-drug loaded Salmonella. Nat Commun 2024, 15, 6680. [Google Scholar] [CrossRef]
- Wang, S; Cheng, M; Chen, CC; et al. Salmonella immunotherapy engineered with highly efficient tumor antigen coating establishes antigen-specific CD8+ T cell immunity and increases in antitumor efficacy with type I interferon combination therapy. Oncoimmunology 2024, 13, 2298444. [Google Scholar] [CrossRef]
- Chen, X; Zhao, J; Yue, S; et al. An oncolytic virus delivering tumor-irrelevant bystander T cell epitopes induces anti-tumor immunity and potentiates cancer immunotherapy. Nat Cancer 2024, 5, 1063–1081. [Google Scholar] [CrossRef]
- Zhu, J; Sun, Y; Qian, X; et al. Intranodal injection of neoantigen-bearing engineered Lactococcus lactis triggers epitope spreading and systemic tumor regressions. Acta Pharm Sin B 2025, 15, 2217–2236. [Google Scholar] [CrossRef]
- Mao, L; Xarpidin, B; Shi, R; et al. Natural Enzyme-Loaded Polymeric Stealth Coating-Armed Engineered Probiotics by Disrupting Tumor Lactate Homeostasis to Synergistic Metabolism-Immuno-Enzyme Dynamic Therapy. Adv Sci (Weinh) 2025, 12, e2417172. [Google Scholar] [CrossRef]
- Luke, JJ; Piha-Paul, SA; Medina, T; et al. Phase I Study of SYNB1891, an Engineered E. coli Nissle Strain Expressing STING Agonist, with and without Atezolizumab in Advanced Malignancies. Clin Cancer Res 2023, 29, 2435–2444. [Google Scholar] [CrossRef]
- Xie, XT; Guan, M; Cheng, K; et al. Programmable engineered bacteria as sustained-releasing antibody factory in situ for enhancing tumor immune checkpoint therapy. Sci Adv 2025, 11, eadt7298. [Google Scholar] [CrossRef] [PubMed]
- Li, F; Yang, Z; Savage, TM; et al. Programmable bacteria synergize with PD-1 blockade to overcome cancer cell-intrinsic immune resistance mechanisms. Sci Immunol 2024, 9, eadn9879. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C; Liu, C; Wu, Q; et al. Remolding the tumor microenvironment by bacteria augments adoptive T cell therapy in advanced-stage solid tumors. Signal Transduct Target Ther 2024, 9, 307. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z; Lim, SH; Min, JJ; et al. Synergistic Antitumor Effect of Combined Radiotherapy and Engineered Salmonella typhimurium in an Intracranial Sarcoma Mouse Model. Vaccines (Basel) 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N; Lai, Y; Fuerte-Stone, J; et al. Cas9-assisted biological containment of a genetically engineered human commensal bacterium and genetic elements. Nat Commun 2024, 15, 2096. [Google Scholar] [CrossRef]
- Singer, ZS; Pabon, J; Huang, H; et al. Engineered bacteria launch and control an oncolytic virus. Nat Biomed Eng. 2025. [Google Scholar] [CrossRef]
- Yurtseven, B; Aydemir, E; Ayaz, F. The Role of Intestinal Microbiota and Immune System Interactions in Autoimmune Diseases. Immunotargets Ther 2025, 14, 1347–1372. [Google Scholar] [CrossRef]
- Liu, Y; Cao, X; Liu, H; et al. The crosstalk between probiotics and T cell immunity. Front Immunol 2025, 16, 1695840. [Google Scholar]
- Song, Y; Li, J; Wu, Y. Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders. Signal Transduct Target Ther 2024, 9, 263. [Google Scholar] [CrossRef]
- Omar, TM; Alfarttoosi, KH; Sanghvi, G; et al. Engineering the Microbiome: a Novel Approach to Managing Autoimmune Diseases. Neuromolecular Med 2025, 27, 63. [Google Scholar] [CrossRef]
- Duan, S; Wang, Y; Zhan, S; et al. Engineered probiotics: a new era in treating inflammatory bowel disease. J Transl Med 2025, 23, 1223. [Google Scholar] [CrossRef]
- Han, K; Xu, J; Xie, F; et al. Engineering Strategies to Modulate the Gut Microbiome and Immune System. J Immunol 2024, 212, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Sadhu, S; Paul, T; Yadav, N. Therapeutic engineering of the gut microbiome using synthetic biology and metabolic tools: a comprehensive review with E. coli Nissle 1917 as a model case study. Arch Microbiol 2025, 207, 213. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Paradas, E; Torrecillas-Lopez, M; Barrera-Chamorro, L; et al. Microbiota-derived extracellular vesicles: current knowledge, gaps, and challenges in precision nutrition. Front Immunol 2025, 16, 1514726. [Google Scholar] [CrossRef] [PubMed]
- Melo-Marques, I; Cardoso, SM; Empadinhas, N. Bacterial extracellular vesicles at the interface of gut microbiota and immunity. Gut Microbes 2024, 16, 2396494. [Google Scholar] [CrossRef]
- Nie, X; Li, Q; Ji, H; et al. Bifidobacterium longum NSP001-derived extracellular vesicles ameliorate ulcerative colitis by modulating T cell responses in gut microbiota-(in)dependent manners. NPJ Biofilms Microbiomes 2025, 11, 27. [Google Scholar] [CrossRef]
- Kozakiewicz, D; Razim, A; Schabussova, I; et al. Immunomodulatory properties of probiotic extracellular vesicles- do they have the potential to fill the gap in therapeutic strategies for allergies? Cell Commun Signal. 2025. [Google Scholar] [CrossRef]
- Liu, BD; Akbar, R; Oliverio, A; et al. Bacterial Extracellular Vesicles in the Regulation of Inflammatory Response and Host-Microbe Interactions. Shock 2024, 61, 175–188. [Google Scholar] [CrossRef]
- Mathieu, C; Wiedeman, A; Cerosaletti, K; et al. A first-in-human, open-label Phase 1b and a randomised, double-blind Phase 2a clinical trial in recent-onset type 1 diabetes with AG019 as monotherapy and in combination with teplizumab. Diabetologia 2024, 67, 27–41. [Google Scholar] [CrossRef]
- Xie, M; Ma, C; Wang, X; et al. Transcriptomic Analysis of Immune Tolerance Induction in NOD Mice Following Oral Vaccination with GAD65-Lactococcus lactis. In Vaccines (Basel); 2025; Volume 13. [Google Scholar]
- Dwyer, AJ; Shaheen, ZR; Fife, BT. Antigen-specific T cell responses in autoimmune diabetes. Front Immunol 2024, 15, 1440045. [Google Scholar] [CrossRef]
- Kedmi, R; Littman, DR. Antigen-presenting cells as specialized drivers of intestinal T cell functions. Immunity 2024, 57, 2269–2279. [Google Scholar] [CrossRef] [PubMed]
- Antonini Cencicchio, M; Montini, F; Palmieri, V; et al. Microbiota-produced immune regulatory bile acid metabolites control central nervous system autoimmunity. Cell Rep Med 2025, 6, 102028. [Google Scholar] [CrossRef] [PubMed]
- He, L; Zhong, Z; Wen, S; et al. Gut microbiota-derived butyrate restores impaired regulatory T cells in patients with AChR myasthenia gravis via mTOR-mediated autophagy. Cell Commun Signal 2024, 22, 215. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y; Liu, C; Li, R; et al. Lactobacillus-derived indole-3-lactic acid ameliorates colitis in cesarean-born offspring via activation of aryl hydrocarbon receptor. iScience 2023, 26, 108279. [Google Scholar] [CrossRef]
- Chen, Y; Bi, S; Zhang, X; et al. Engineered probiotics remodel the intestinal epithelial barrier and enhance bacteriotherapy for inflammatory bowel diseases. Acta Biomater 2025, 198, 467–481. [Google Scholar] [CrossRef]
- Laguna, JG; Freitas, ADS; Barroso, FAL; et al. Recombinant probiotic Lactococcus lactis delivering P62 mitigates moderate colitis in mice. Front Microbiol 2024, 15, 1309160. [Google Scholar] [CrossRef]
- Yu, M; Kim, J; Ahn, JH; et al. Nononcogenic restoration of the intestinal barrier by E. coli-delivered human EGF. JCI Insight 4 2019. [Google Scholar] [CrossRef]
- Rankin, LC; Kaiser, KA; de Los Santos-Alexis, K; et al. Dietary tryptophan deficiency promotes gut RORgammat(+) Treg cells at the expense of Gata3(+) Treg cells and alters commensal microbiota metabolism. Cell Rep 2023, 42, 112135. [Google Scholar] [CrossRef]
- Xu, Y; Xiong, Z; Zhang, P; et al. Microbiota metabolite taurodeoxycholic acid maintains intestinal tissue residency of innate lymphoid cells via engagement with P2Y10 receptor. Sci Adv 2025, 11, eadt9645. [Google Scholar] [CrossRef]
- Zhu, D; Galley, J; Pizzini, J; et al. Microbial Biosensor for Sensing and Treatment of Intestinal Inflammation. Adv Sci (Weinh) 2025, 12, e2504364. [Google Scholar] [CrossRef]
- Liu, J; Wang, L; Pang, B; et al. Engineered probiotics platform for oral delivery of antibody as a high-compliance alternative for immune-mediated inflammatory diseases. Cell Rep Med 2025, 102523. [Google Scholar] [CrossRef] [PubMed]
- Weibel, N; Curcio, M; Schreiber, A; et al. Engineering a Novel Probiotic Toolkit in Escherichia coli Nissle 1917 for Sensing and Mitigating Gut Inflammatory Diseases. ACS Synth Biol 2024, 13, 2376–2390. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Luna, AJ; Carlson, TJ; Garey, KW. Review Article: Safety of Live Biotherapeutic Products Used for the Prevention of Clostridioides difficile Infection Recurrence. Clin Infect Dis 2023, 77, S487–S496. [Google Scholar] [CrossRef]
- Jiang, S; Zhang, C; Han, Z; et al. Native microbiome dominates over host factors in shaping the probiotic genetic evolution in the gut. NPJ Biofilms Microbiomes 2023, 9, 80. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S; Hasty, J. Stability, robustness, and containment: preparing synthetic biology for real-world deployment. Curr Opin Biotechnol 2023, 79, 102880. [Google Scholar] [CrossRef]
- Bjork, JR; Bolte, LA; Maltez Thomas, A; et al. Longitudinal gut microbiome changes in immune checkpoint blockade-treated advanced melanoma. Nat Med 2024, 30, 785–796. [Google Scholar] [CrossRef]
- Zhong, F; Liu, X; Wang, X; et al. An AI-Designed Antibody-Engineered Probiotic Therapy Targeting Urease to Combat Helicobacter pylori Infection in Mice. Microorganisms 2025, 13. [Google Scholar] [CrossRef]
- Carolak, E; Czajkowska, J; Stypulkowska, A; et al. Being a better version of yourself: genetically engineered probiotic bacteria as host defense enhancers in the control of intestinal pathogens. Gut Microbes 2025, 17, 2519696. [Google Scholar] [CrossRef]
- Kamble, NS; Thomas, S; Madaan, T; et al. Engineered bacteria as an orally administered anti-viral treatment and immunization system. Gut Microbes 2025, 17, 2500056. [Google Scholar] [CrossRef]
- Jin, K; Huang, Y; Che, H; et al. Engineered Bacteria for Disease Diagnosis and Treatment Using Synthetic Biology. Microb Biotechnol 2025, 18, e70080. [Google Scholar]
- Huang, L; Tang, W; He, L; et al. Engineered probiotic Escherichia coli elicits immediate and long-term protection against influenza A virus in mice. Nat Commun 2024, 15, 6802. [Google Scholar] [CrossRef] [PubMed]
- Deckers, J; Anbergen, T; Hokke, AM; et al. Engineering cytokine therapeutics. Nat Rev Bioeng 2023, 1, 286–303. [Google Scholar] [CrossRef] [PubMed]
- Carreto-Binaghi, LE; Sztein, MB; Booth, JS. Role of cellular effectors in the induction and maintenance of IgA responses leading to protective immunity against enteric bacterial pathogens. Front Immunol 2024, 15, 1446072. [Google Scholar] [CrossRef] [PubMed]
- De Ponte Conti, B; Marino, R; Rezzonico-Jost, T; et al. Secretory IgA amplification during immune checkpoint blockade enhances the control of tumor growth by enterotropic T cells. Sci Adv 2025, 11, eaeb5308. [Google Scholar] [CrossRef]
- Schluter, T; van Elsas, Y; Priem, B; et al. Trained immunity: induction of an inflammatory memory in disease. Cell Res 2025, 35, 792–802. [Google Scholar] [CrossRef]
- Li, P; Liu, M; Liu, Y; et al. Recent Advances of Trained immunity in Macrophages. Int J Biol Sci 2025, 21, 5258–5283. [Google Scholar] [CrossRef]
- Hao, D; McBride, MA; Bohannon, JK; et al. Metabolic adaptations driving innate immune memory: mechanisms and therapeutic implications. J Leukoc Biol 2025, 117. [Google Scholar] [CrossRef]
- Kawai, T; Ikegawa, M; Ori, D; et al. Decoding Toll-like receptors: Recent insights and perspectives in innate immunity. Immunity 2024, 57, 649–673. [Google Scholar] [CrossRef]
- Sanchez-Ramon, S; Conejero, L; Netea, MG; et al. Trained Immunity-Based Vaccines: A New Paradigm for the Development of Broad-Spectrum Anti-infectious Formulations. Front Immunol 2018, 9, 2936. [Google Scholar] [CrossRef]
- Alexopoulou, L; Irla, M. Toll-like receptors (TLRs) in the trained immunity era. Elife 14 2025. [Google Scholar] [CrossRef]
- Champagne-Jorgensen, K; Luong, T; Darby, T; et al. Immunogenicity of bacteriophages. Trends Microbiol 2023, 31, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Roach, DR; Leung, CY; Henry, M; et al. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38–47 e34. [Google Scholar] [CrossRef] [PubMed]
- Gibb, B; Hyman, P; Schneider, CL. The Many Applications of Engineered Bacteriophages-An Overview. In Pharmaceuticals (Basel); 2021; Volume 14. [Google Scholar]
- Lu, Y; Yuan, H; Liang, S; et al. Microbial metabolite-driven immune reprogramming in tumor immunotherapy: mechanisms and therapeutic perspectives. Front Immunol 2025, 16, 1603658. [Google Scholar] [CrossRef] [PubMed]
- Vuscan, P; Kischkel, B; Joosten, LAB; et al. Microbial-induced trained immunity for cancer immunotherapy. Pharmacol Rev 2025, 77, 100074. [Google Scholar] [CrossRef]
- Perl, M; Fante, MA; Herfeld, K; et al. Microbiota-derived metabolites: Key modulators of cancer immunotherapies. Med 2025, 6, 100773. [Google Scholar] [CrossRef]
- Abel, AM; Yang, C; Thakar, MS; et al. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front Immunol 2018, 9, 1869. [Google Scholar] [CrossRef]
- He, B; Mai, Q; Pang, Y; et al. Cytokines induced memory-like NK cells engineered to express CD19 CAR exhibit enhanced responses against B cell malignancies. Front Immunol 2023, 14, 1130442. [Google Scholar] [CrossRef]
- Poznanski, SM; Lee, AJ; Nham, T; et al. Combined Stimulation with Interleukin-18 and Interleukin-12 Potently Induces Interleukin-8 Production by Natural Killer Cells. J Innate Immun 2017, 9, 511–525. [Google Scholar] [CrossRef]
- Dadas, O; Ertay, A; Cragg, MS. Delivering co-stimulatory tumor necrosis factor receptor agonism for cancer immunotherapy: past, current and future perspectives. Front Immunol 2023, 14, 1147467. [Google Scholar] [CrossRef]
- Susiriwatananont, T; Eiamprapaporn, P; Vazquez Roque, M; et al. The Gut Microbiome as a Biomarker and Therapeutic Target of Immune Checkpoint Inhibitors: A Review for Oncologists. Cells 2025, 14. [Google Scholar] [CrossRef]
- Cai, Y; Wang, Y; Hu, S. Synthetic Gene Circuits Enable Sensing in Engineered Living Materials. In Biosensors (Basel); 2025; Volume 15. [Google Scholar]
- Naydich, AD; Nangle, SN; Bues, JJ; et al. Synthetic Gene Circuits Enable Systems-Level Biosensor Trigger Discovery at the Host-Microbe Interface. In mSystems; 4, 2019. [Google Scholar]
- Dey, S; Sankaran, S. Engineered bacterial therapeutics with material solutions. Trends Biotechnol 2024, 42, 1663–1676. [Google Scholar] [CrossRef]
- Khazem, A; Schmachtenberg, R; Weiand, A; et al. Engineered microbial living matter for diagnostics, prevention, and therapy. Curr Opin Biotechnol 2025, 92, 103269. [Google Scholar] [CrossRef] [PubMed]
- George, DR; Danciu, M; Davenport, PW; et al. A bumpy road ahead for genetic biocontainment. Nat Commun 2024, 15, 650. [Google Scholar] [CrossRef] [PubMed]
- Santollani, L; Maiorino, L; Zhang, YJ; et al. Local delivery of cell surface-targeted immunocytokines programs systemic antitumor immunity. Nat Immunol 2024, 25, 1820–1829. [Google Scholar] [CrossRef] [PubMed]
- Crow, YJ; Stetson, DB. The type I interferonopathies: 10 years on. Nat Rev Immunol 2022, 22, 471–483. [Google Scholar] [CrossRef]
- Xia, JY; Hepler, C; Tran, P; et al. Engineered calprotectin-sensing probiotics for IBD surveillance in humans. Proc Natl Acad Sci U S A 2023, 120, e2221121120. [Google Scholar] [CrossRef]
- Guo, Q; Jin, Y; Chen, X; et al. NF-kappaB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther 2024, 9, 53. [Google Scholar] [CrossRef]
- Neurath, MF; Artis, D; Becker, C. The intestinal barrier: a pivotal role in health, inflammation, and cancer. Lancet Gastroenterol Hepatol 2025, 10, 573–592. [Google Scholar] [CrossRef]
- Di Vincenzo, F; Del Gaudio, A; Petito, V; et al. Gut microbiota, intestinal permeability, and systemic inflammation: a narrative review. Intern Emerg Med 2024, 19, 275–293. [Google Scholar] [CrossRef]
- Woo, SG; Kim, SK; Lee, SG; et al. Engineering probiotic Escherichia coli for inflammation-responsive indoleacetic acid production using RiboJ-enhanced genetic circuits. J Biol Eng 2025, 19, 10. [Google Scholar] [CrossRef]
- Fu, L; Upadhyay, R; Pokrovskii, M; et al. PRDM16-dependent antigen-presenting cells induce tolerance to gut antigens. Nature 2025, 642, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y; Lichterman, JN; Coughlin, LA; et al. Immune checkpoint blockade induces gut microbiota translocation that augments extraintestinal antitumor immunity. Sci Immunol 2023, 8, eabo2003. [Google Scholar] [CrossRef] [PubMed]
- Yin, R; Melton, S; Huseby, ES; et al. How persistent infection overcomes peripheral tolerance mechanisms to cause T cell-mediated autoimmune disease. Proc Natl Acad Sci U S A 2024, 121, e2318599121. [Google Scholar] [CrossRef] [PubMed]
- Li, Z; Shang, D. NOD1 and NOD2: Essential Monitoring Partners in the Innate Immune System. Curr Issues Mol Biol 2024, 46, 9463–9479. [Google Scholar] [CrossRef]
- Movert, E; Bolarin, JS; Valfridsson, C; et al. Interplay between human STING genotype and bacterial NADase activity regulates inter-individual disease variability. Nat Commun 2023, 14, 4008. [Google Scholar] [CrossRef]
- Clasen, SJ; Bell, MEW; Borbon, A; et al. Silent recognition of flagellins from human gut commensal bacteria by Toll-like receptor 5. Sci Immunol 2023, 8, eabq7001. [Google Scholar] [CrossRef]
- Chen, MY; Cheng, TW; Pan, YC; et al. Endotoxin-Free Outer Membrane Vesicles for Safe and Modular Anticancer Immunotherapy. ACS Synth Biol 2025, 14, 148–160. [Google Scholar] [CrossRef]
- Vasquez Ayala, A; Hsu, CY; Oles, RE; et al. Commensal bacteria promote type I interferon signaling to maintain immune tolerance in mice. J Exp Med 2024, 221. [Google Scholar] [CrossRef]
- Brockmann, L; Tran, A; Huang, Y; et al. Intestinal microbiota-specific Th17 cells possess regulatory properties and suppress effector T cells via c-MAF and IL-10. Immunity 2023, 56, 2719–2735 e2717. [Google Scholar] [CrossRef]
- Montero-Blay, A; Blanco, JD; Rodriguez-Arce, I; et al. Bacterial expression of a designed single-chain IL-10 prevents severe lung inflammation. Mol Syst Biol 2023, 19, e11037. [Google Scholar] [CrossRef]
- Zhernakova, DV; Wang, D; Liu, L; et al. Host genetic regulation of human gut microbial structural variation. Nature 2024, 625, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Moorlag, S; Folkman, L; Ter Horst, R; et al. Multi-omics analysis of innate and adaptive responses to BCG vaccination reveals epigenetic cell states that predict trained immunity. Immunity 2024, 57, 171–187 e114. [Google Scholar] [CrossRef] [PubMed]
- Berkson, JD; Wate, CE; Allen, GB; et al. Phage-specific immunity impairs efficacy of bacteriophage targeting Vancomycin Resistant Enterococcus in a murine model. Nat Commun 2024, 15, 2993. [Google Scholar] [CrossRef] [PubMed]
- Tan, X; Chen, K; Jiang, Z; et al. Evaluation of the impact of repeated intravenous phage doses on mammalian host-phage interactions. J Virol 2024, 98, e0135923. [Google Scholar] [CrossRef]
- Raman, V; Howell, LM; Bloom, SMK; et al. Intracellular Salmonella delivery of an exogenous immunization antigen refocuses CD8 T cells against cancer cells, eliminates pancreatic tumors and forms antitumor immunity. Front Immunol 2023, 14, 1228532. [Google Scholar] [CrossRef]
- Muhammad, S; Li, M; Jia, Q; et al. Advances in the engineering of living probiotics for cancer immunotherapy. Theranostics 2026, 16, 1164–1226. [Google Scholar] [CrossRef]
- Bousbaine, D; Bauman, KD; Chen, YE; et al. Discovery and engineering of the antibody response to a prominent skin commensal. Nature 2025, 638, 1054–1064. [Google Scholar] [CrossRef]
- Gribonika, I; Band, VI; Chi, L; et al. Skin autonomous antibody production regulates host-microbiota interactions. Nature 2025, 638, 1043–1053. [Google Scholar] [CrossRef]
- Viney, M; Cheynel, L. Gut immune responses and evolution of the gut microbiome-a hypothesis. Discov Immunol 2023, 2, kyad025. [Google Scholar] [CrossRef]
- Arboleda-Garcia, A; Alarcon-Ruiz, I; Boada-Acosta, L; et al. Advancements in synthetic biology-based bacterial cancer therapy: A modular design approach. Crit Rev Oncol Hematol 2023, 190, 104088. [Google Scholar] [CrossRef]
- Mazzolini, R; Rodriguez-Arce, I; Fernandez-Barat, L; et al. Engineered live bacteria suppress Pseudomonas aeruginosa infection in mouse lung and dissolve endotracheal-tube biofilms. Nat Biotechnol 2023, 41, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z; Zhong, W; Xiong, H; et al. Dual-Membrane-Camouflaged Engineered Bacteria for Targeted Melanoma Therapy. In ACS Nano; 2025. [Google Scholar]
- May, KL; Akiyama, T; Parker, BG; et al. LPS O-antigen polysaccharide length impacts outer membrane permeability of enteric gram-negative bacteria. mBio 2025, 16, e0251825. [Google Scholar] [CrossRef] [PubMed]
- Asada, N; Ginsberg, P; Paust, HJ; et al. The integrated stress response pathway controls cytokine production in tissue-resident memory CD4(+) T cells. Nat Immunol 2025, 26, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M; Lacerda Mariano, L; Canton, T; et al. Tissue-resident memory T cells mediate mucosal immunity to recurrent urinary tract infection. Sci Immunol 2023, 8, eabn4332. [Google Scholar] [CrossRef]
- Verma, S; Dufort, MJ; Olsen, TM; et al. Antigen-level resolution of commensal-specific B cell responses can be enabled by phage display screening coupled with B cell tetramers. Immunity 2024, 57, 1428–1441 e1428. [Google Scholar] [CrossRef]
- Rusconi, B; Bard, AK; McDonough, R; et al. Intergenerational protective anti-gut commensal immunoglobulin G originates in early life. Proc Natl Acad Sci U S A 2024, 121, e2309994121. [Google Scholar] [CrossRef]
- Gutzeit, C; Grasset, EK; Matthews, DB; et al. Gut IgA functionally interacts with systemic IgG to enhance antipneumococcal vaccine responses. Sci Adv 2025, 11, eado9455. [Google Scholar] [CrossRef]
- Foo, GW; Uruthirapathy, AS; Zhang, CQ; et al. Genetic Entanglement Enables Ultrastable Biocontainment in the Mammalian Gut. ACS Synth Biol 2025, 14, 3696–3708. [Google Scholar] [CrossRef]
- Buss, MT; Zhu, L; Kwon, JH; et al. Probiotic acoustic biosensors for noninvasive imaging of gut inflammation. Nat Commun 2025, 16, 7931. [Google Scholar]
- Grome, MW; Nguyen, MTA; Moonan, DW; et al. Engineering a genomically recoded organism with one stop codon. Nature 2025, 639, 512–521. [Google Scholar] [CrossRef]
- Gomez-Tatay, L; Hernandez-Andreu, JM. Xenobiology for the Biocontainment of Synthetic Organisms: Opportunities and Challenges. In Life (Basel); 2024; Volume 14. [Google Scholar]
- Stirling, F; Naydich, A; Bramante, J; et al. Synthetic Cassettes for pH-Mediated Sensing, Counting, and Containment. Cell Rep 2020, 30, 3139–3148 e3134. [Google Scholar] [CrossRef] [PubMed]
- Liguori, F; Pellicciotta, N; Milanetti, E; et al. Dynamic Gene Expression Mitigates Mutational Escape in Lysis-Driven Bacteria Cancer Therapy. Biodes Res 2024, 6, 0049. [Google Scholar] [CrossRef] [PubMed]
- Gross, JL; Basu, R; Bradfield, CJ; et al. Bactericidal antibiotic treatment induces damaging inflammation via TLR9 sensing of bacterial DNA. Nat Commun 2024, 15, 10359. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q; Wang, L; Zhang, Z; et al. Synergistic Antibacteria and Anti-Inflammation with Reversible Redox Selenium-Functionalized Polypeptides for Efficient Gram-Negative Bacterial Sepsis Treatment. In Small; 2025; p. e09805. [Google Scholar]
- Nyerges, A; Vinke, S; Flynn, R; et al. A swapped genetic code prevents viral infections and gene transfer. Nature 2023, 615, 720–727. [Google Scholar] [CrossRef]
- Choi, YN; Kim, D; Lee, S; et al. Quadruplet codon decoding-based versatile genetic biocontainment system. Nucleic Acids Res 2025, 53. [Google Scholar] [CrossRef]
- Tran, KM; Nong, NT; Ren, J; et al. Genetic "expiry-date" circuits control lifespan of synthetic scavenger bacteria for safe bioremediation. Nucleic Acids Res 2025, 53. [Google Scholar] [CrossRef]
- Parra, ER; Zhang, J; Duose, DY; et al. Multi-omics Analysis Reveals Immune Features Associated with Immunotherapy Benefit in Patients with Squamous Cell Lung Cancer from Phase III Lung-MAP S1400I Trial. Clin Cancer Res 2024, 30, 1655–1668. [Google Scholar] [CrossRef]
- Barras, D; Ghisoni, E; Chiffelle, J; et al. Response to tumor-infiltrating lymphocyte adoptive therapy is associated with preexisting CD8(+) T-myeloid cell networks in melanoma. Sci Immunol 2024, 9, eadg7995. [Google Scholar] [CrossRef]
- Shiao, SL; Gouin, KH, 3rd; Ing, N; et al. Single-cell and spatial profiling identify three response trajectories to pembrolizumab and radiation therapy in triple negative breast cancer. Cancer Cell 2024, 42, 70–84 e78. [Google Scholar] [CrossRef]
- Lee, SY; Lee, Y. Fibroblast TGF-beta signaling defines spatial tumor ecosystems linked to immune checkpoint blockade resistance. Commun Biol 2025, 8, 1730. [Google Scholar] [CrossRef]
- Synnott, D; O'Reilly, D; De Freitas, D; et al. Cytokine release syndrome in solid tumors. Cancer 2025, 131, e70069. [Google Scholar] [CrossRef] [PubMed]
- Brennan, AM. Development of synthetic biotics as treatment for human diseases. Synth Biol (Oxf) 2021, 7, ysac001. [Google Scholar] [CrossRef] [PubMed]
- Grau Bejar, JF; Yaniz Galende, E; Zeng, Q; et al. Immune predictors of response to immune checkpoint inhibitors in mismatch repair-deficient endometrial cancer. J Immunother Cancer 2024, 12. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, AM; Alayli, FA; Okrah, K; et al. Immunologic signatures of response and resistance to nivolumab with ipilimumab in advanced metastatic cancer. J Exp Med 2024, 221. [Google Scholar] [CrossRef]
- Walsh, MJ; Ali, LR; Lenehan, P; et al. Blockade of innate inflammatory cytokines TNFalpha, IL-1beta, or IL-6 overcomes virotherapy-induced cancer equilibrium to promote tumor regression. Immunother Adv 2023, 3, ltad011. [Google Scholar] [CrossRef]
- Soler, MF; Abaurrea, A; Azcoaga, P; et al. New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. J Immunother Cancer 2023, 11. [Google Scholar] [CrossRef]
- Mennillo, E; Kim, YJ; Lee, G; et al. Single-cell and spatial multi-omics highlight effects of anti-integrin therapy across cellular compartments in ulcerative colitis. Nat Commun 2024, 15, 1493. [Google Scholar] [CrossRef]
- Thomas, T; Friedrich, M; Rich-Griffin, C; et al. A longitudinal single-cell atlas of anti-tumour necrosis factor treatment in inflammatory bowel disease. Nat Immunol 2024, 25, 2152–2165. [Google Scholar] [CrossRef]
- Siebert, K; Faro, T; Kohler, N; et al. Endoscopic healing in pediatric IBD perpetuates a persistent signature defined by Th17 cells with molecular and microbial drivers of disease. Cell Rep Med 2025, 6, 102236. [Google Scholar] [CrossRef]
- Mellman, I; Chen, DS; Powles, T; et al. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity 2023, 56, 2188–2205. [Google Scholar] [CrossRef]
- Baruch, EN; Youngster, I; Ben-Betzalel, G; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Davar, D; Dzutsev, AK; McCulloch, JA; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Li, D; Zhong, J; Zhang, Q; et al. Effects of anti-inflammatory therapies on glycemic control in type 2 diabetes mellitus. Front Immunol 2023, 14, 1125116. [Google Scholar] [CrossRef]
- Samovski, D; Smith, GI; Palacios, H; et al. Effect of Marked Weight Loss on Adipose Tissue Biology in People With Obesity and Type 2 Diabetes. Diabetes Care 2025, 48, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Miranda, AMA; McAllan, L; Mazzei, G; et al. Selective remodelling of the adipose niche in obesity and weight loss. Nature 2025, 644, 769–779. [Google Scholar] [CrossRef]
- Jara, M; Norlin, J; Kjaer, MS; et al. Modulation of metabolic, inflammatory and fibrotic pathways by semaglutide in metabolic dysfunction-associated steatohepatitis. Nat Med 2025, 31, 3128–3140. [Google Scholar] [CrossRef]
- Buldak, L; Boldys, A; Skudrzyk, E; et al. Liraglutide Therapy in Obese Patients Alters Macrophage Phenotype and Decreases Their Tumor Necrosis Factor Alpha Release and Oxidative Stress Markers-A Pilot Study. Metabolites 14 2024. [Google Scholar]
- Su, M; Chen, L; Xie, L; et al. Identification of early predictive biomarkers for severe cytokine release syndrome in pediatric patients with chimeric antigen receptor T-cell therapy. Front Immunol 2024, 15, 1450173. [Google Scholar] [CrossRef]
- Kim, GD; Shin, SI; Sun, P; et al. Single-cell RNA sequencing of baseline PBMCs predicts ICI efficacy and irAE severity in patients with NSCLC. J Immunother Cancer 13 2025. [Google Scholar] [CrossRef]
- Daugherty-Lopes, A; Perez-Guijarro, E; Gopalan, V; et al. Immune and Molecular Correlates of Response to Immunotherapy Revealed by Brain-Metastatic Melanoma Models. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chupp, DP; Rivera, CE; Zhou, Y; et al. A humanized mouse that mounts mature class-switched, hypermutated and neutralizing antibody responses. Nat Immunol 2024, 25, 1489–1506. [Google Scholar] [CrossRef] [PubMed]
- Sefik, E; Philbrick, W; Zhang, F; et al. Humanization of CD47 enables development of functional human neutrophils via postirradiation remodeling of the bone marrow. Proc Natl Acad Sci U S A 2025, 122, e2426546122. [Google Scholar] [CrossRef] [PubMed]
- Knight, HR; Kim, M; Kannan, N; et al. Bioengineering approaches to trained immunity: Physiologic targets and therapeutic strategies. Elife 14 2025. [Google Scholar] [CrossRef] [PubMed]
- Greene, C; Connolly, R; Brennan, D; et al. Blood-brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat Neurosci 2024, 27, 421–432. [Google Scholar] [CrossRef]
- Singhal, T; Cicero, S; Rissanen, E; et al. Glial Activity Load on PET Reveals Persistent "Smoldering" Inflammation in MS Despite Disease-Modifying Treatment: 18 F-PBR06 Study. Clin Nucl Med 2024, 49, 491–499. [Google Scholar] [CrossRef]
- Hirata, K; Matsuoka, K; Tagai, K; et al. Altered Brain Energy Metabolism Related to Astrocytes in Alzheimer's Disease. Ann Neurol. 2023. [Google Scholar] [CrossRef]
- An, M; Mehta, A; Min, BH; et al. Early Immune Remodeling Steers Clinical Response to First-Line Chemoimmunotherapy in Advanced Gastric Cancer. Cancer Discov 2024, 14, 766–785. [Google Scholar] [CrossRef]
- Chen, Y; Wang, D; Li, Y; et al. Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer. Cancer Cell 2024, 42, 1268–1285 e1267. [Google Scholar] [CrossRef]
- Wong, CK; McLean, BA; Baggio, LL; et al. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab 2024, 36, 130–143 e135. [Google Scholar] [CrossRef]
- Ramirez-Valle, F; Maranville, JC; Roy, S; et al. Sequential immunotherapy: towards cures for autoimmunity. Nat Rev Drug Discov 2024, 23, 501–524. [Google Scholar] [CrossRef]
- Greco, R; Alexander, T; Del Papa, N; et al. Innovative cellular therapies for autoimmune diseases: expert-based position statement and clinical practice recommendations from the EBMT practice harmonization and guidelines committee. EClinicalMedicine 2024, 69, 102476. [Google Scholar] [CrossRef]
Figure 1.
Engineered microbes as distributed immune-computational systems. Engineered microbial
immunotherapies function as living systems that integrate immune-relevant environmental information,
process these signals through programmable biological circuits, and generate context-dependent immune
modulation. These platforms sense diverse immune inputs, including inflammatory cues, cytokines, metabolic
signals, pathogen-associated factors, and microbiota or tissue state, and transform this information through
logical integration, feedback regulation, memory formation, population-level coordination, and spatial control.
The resulting immune outputs can activate, suppress, educate, or reprogram host immunity through cytokine
delivery, antigen presentation, immunometabolic modulation, and barrier repair. Together, these processes form
a closed-loop system embedded within host innate and adaptive immune networks, enabling engineered
microbes to dynamically sense, compute, and reshape immune system state across disease contexts.
Figure 1.
Engineered microbes as distributed immune-computational systems. Engineered microbial
immunotherapies function as living systems that integrate immune-relevant environmental information,
process these signals through programmable biological circuits, and generate context-dependent immune
modulation. These platforms sense diverse immune inputs, including inflammatory cues, cytokines, metabolic
signals, pathogen-associated factors, and microbiota or tissue state, and transform this information through
logical integration, feedback regulation, memory formation, population-level coordination, and spatial control.
The resulting immune outputs can activate, suppress, educate, or reprogram host immunity through cytokine
delivery, antigen presentation, immunometabolic modulation, and barrier repair. Together, these processes form
a closed-loop system embedded within host innate and adaptive immune networks, enabling engineered
microbes to dynamically sense, compute, and reshape immune system state across disease contexts.

Figure 2.
Disease-specific immune logic implemented by engineered microbes. Engineered microbial
immunotherapies apply a shared immune-computational framework to achieve distinct immunological
objectives across disease contexts. In cancer, engineered microbes sense tumor-associated cues such as hypoxia,
inflammatory cytokines, and altered metabolism and process this information through conditional activation
and amplification logic to drive immune outputs including STING pathway activation, localized cytokine
delivery, and antigen spreading, resulting in conversion of immunologically cold tumors into inflamed, T cell–
infiltrated microenvironments. In autoimmunity and chronic inflammation, microbial platforms detect
inflammatory cytokines, dysbiosis, and epithelial stress and implement tolerance-biased computation that
restrains inflammation, promotes regulatory T cell differentiation, delivers IL-10 and TGF-β, and reinforces
epithelial barrier integrity, leading to immune re-education and restoration of tolerance. During infection,
engineered microbes sense pathogen-associated and inflammatory signals and engage immune amplification
and trained immunity programs to drive interferon responses, activate myeloid and natural killer cells, and
enhance host defense and pathogen control. Together, these examples illustrate how a unified immunecomputational
architecture can be selectively tuned to activate, suppress, or enhance immunity according to
disease-specific immune requirements.
Figure 2.
Disease-specific immune logic implemented by engineered microbes. Engineered microbial
immunotherapies apply a shared immune-computational framework to achieve distinct immunological
objectives across disease contexts. In cancer, engineered microbes sense tumor-associated cues such as hypoxia,
inflammatory cytokines, and altered metabolism and process this information through conditional activation
and amplification logic to drive immune outputs including STING pathway activation, localized cytokine
delivery, and antigen spreading, resulting in conversion of immunologically cold tumors into inflamed, T cell–
infiltrated microenvironments. In autoimmunity and chronic inflammation, microbial platforms detect
inflammatory cytokines, dysbiosis, and epithelial stress and implement tolerance-biased computation that
restrains inflammation, promotes regulatory T cell differentiation, delivers IL-10 and TGF-β, and reinforces
epithelial barrier integrity, leading to immune re-education and restoration of tolerance. During infection,
engineered microbes sense pathogen-associated and inflammatory signals and engage immune amplification
and trained immunity programs to drive interferon responses, activate myeloid and natural killer cells, and
enhance host defense and pathogen control. Together, these examples illustrate how a unified immunecomputational
architecture can be selectively tuned to activate, suppress, or enhance immunity according to
disease-specific immune requirements.
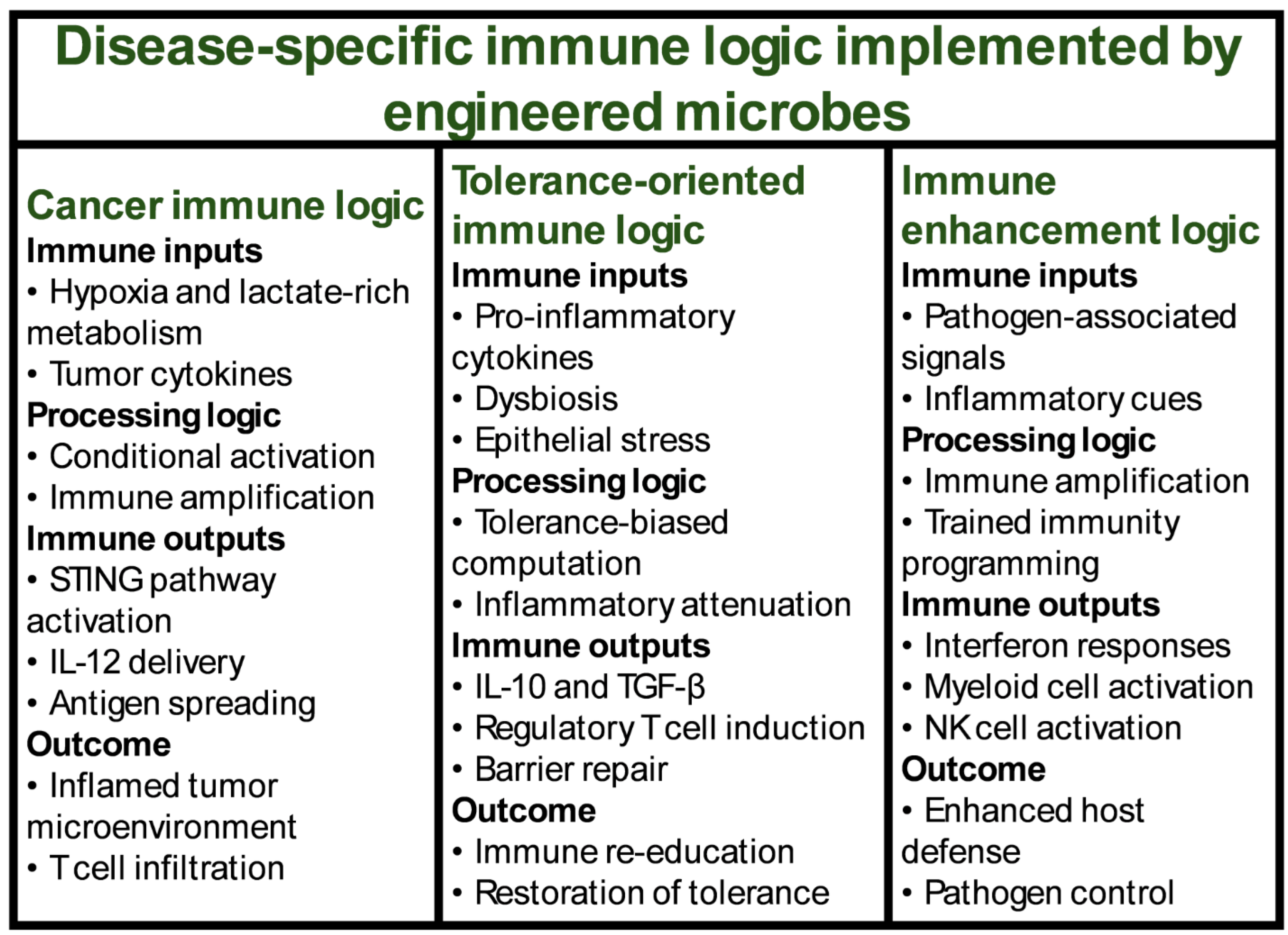
Figure 3.
Intrinsic immune personalities of microbial chassis. Microbial chassis differ fundamentally in their
baseline immune behaviors, which constrain and enable distinct immunotherapeutic applications independent
of genetic payload. Commensal chassis exhibit low pattern recognition receptor (PRR) activation and a
tolerogenic immune bias shaped by co-evolution with host mucosal immunity, making them well suited for
chronic immune modulation and tolerance induction. Attenuated pathogenic chassis retain strong innate
immune stimulatory capacity, characterized by high PRR engagement and pro-inflammatory signaling, which
can be exploited for immune amplification in cancer and infectious disease settings but requires stringent safety
control. Engineered microbial consortia distribute sensing, processing, and immune output functions across
multiple strains, enabling division of labor, coordinated population-level behavior, and layered safety
architectures that support network-level immune control. Together, these intrinsic immune personalities
demonstrate that therapeutic suitability is determined by immune behavior rather than microbial taxonomy,
underscoring the importance of chassis selection as a primary design decision in living immunotherapy.
Figure 3.
Intrinsic immune personalities of microbial chassis. Microbial chassis differ fundamentally in their
baseline immune behaviors, which constrain and enable distinct immunotherapeutic applications independent
of genetic payload. Commensal chassis exhibit low pattern recognition receptor (PRR) activation and a
tolerogenic immune bias shaped by co-evolution with host mucosal immunity, making them well suited for
chronic immune modulation and tolerance induction. Attenuated pathogenic chassis retain strong innate
immune stimulatory capacity, characterized by high PRR engagement and pro-inflammatory signaling, which
can be exploited for immune amplification in cancer and infectious disease settings but requires stringent safety
control. Engineered microbial consortia distribute sensing, processing, and immune output functions across
multiple strains, enabling division of labor, coordinated population-level behavior, and layered safety
architectures that support network-level immune control. Together, these intrinsic immune personalities
demonstrate that therapeutic suitability is determined by immune behavior rather than microbial taxonomy,
underscoring the importance of chassis selection as a primary design decision in living immunotherapy.
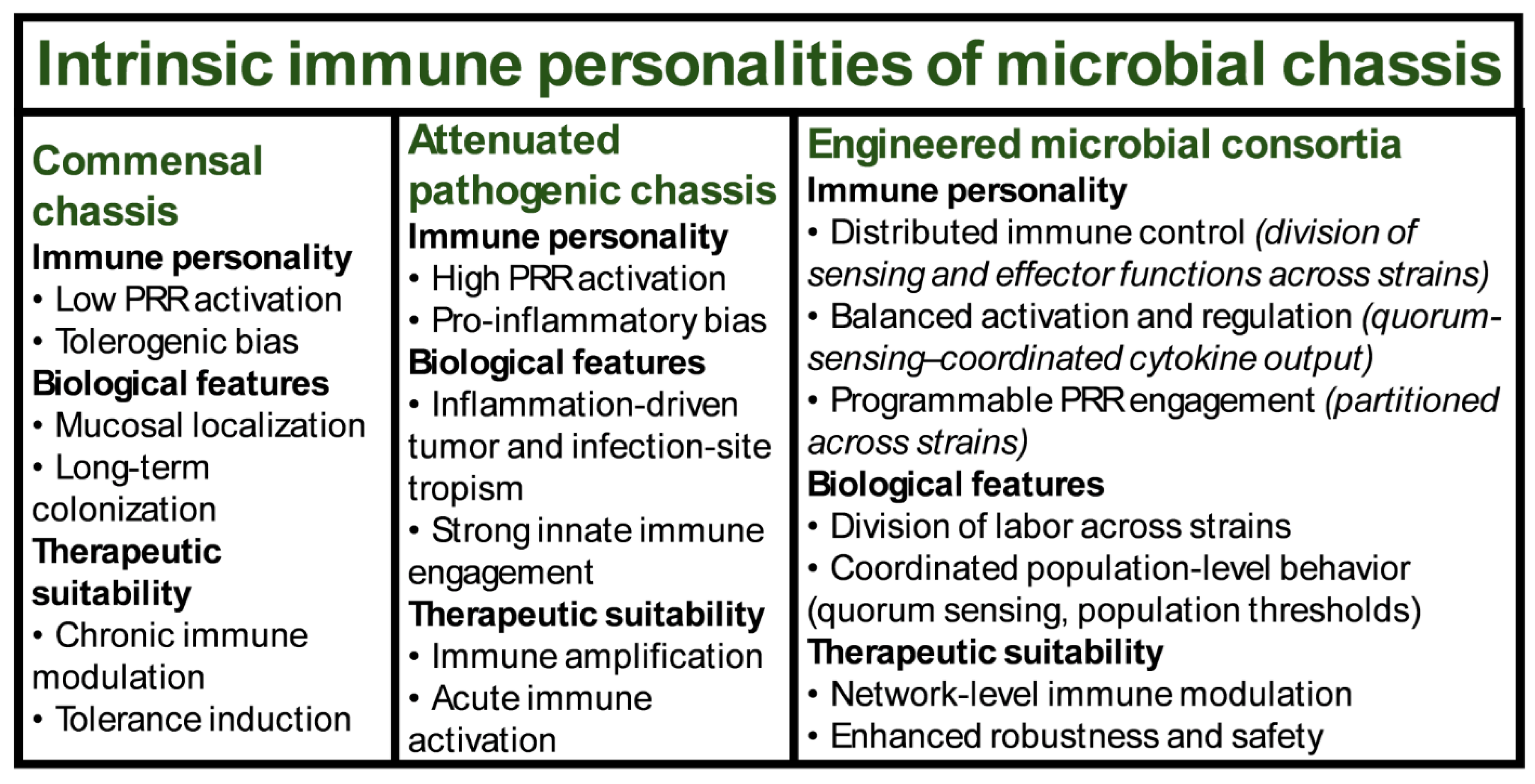
Figure 4.
Immune safety architecture for living immunotherapeutics. The safety of engineered microbial
immunotherapies arises from a multi-layered immune safety architecture that extends beyond genetic
containment alone. Layer 1 addresses immunotoxicity by regulating inflammatory magnitude through
quantitative control of cytokine output, spatial confinement of immune activity, and feedback attenuation to
prevent systemic immune escalation. Layer 2 focuses on tolerance preservation, incorporating pattern
recognition receptor (PRR) tuning, coordinated delivery of regulatory signals, and antigen-context control to
minimize disruption of host immune tolerance. Layer 3 manages adaptive immune responses to the therapeutic
microbe, including anti-microbial immunity, immune memory formation, clearance dynamics, and
considerations for repeat dosing. Layer 4 defines termination and containment strategies, integrating immunetriggered
kill switches, metabolic dependencies, and clearance logic to ensure predictable therapeutic exit.
Together, these layers establish immune safety as a systems-level design principle essential for the clinical
translation and regulatory evaluation of living immunotherapeutics.
Figure 4.
Immune safety architecture for living immunotherapeutics. The safety of engineered microbial
immunotherapies arises from a multi-layered immune safety architecture that extends beyond genetic
containment alone. Layer 1 addresses immunotoxicity by regulating inflammatory magnitude through
quantitative control of cytokine output, spatial confinement of immune activity, and feedback attenuation to
prevent systemic immune escalation. Layer 2 focuses on tolerance preservation, incorporating pattern
recognition receptor (PRR) tuning, coordinated delivery of regulatory signals, and antigen-context control to
minimize disruption of host immune tolerance. Layer 3 manages adaptive immune responses to the therapeutic
microbe, including anti-microbial immunity, immune memory formation, clearance dynamics, and
considerations for repeat dosing. Layer 4 defines termination and containment strategies, integrating immunetriggered
kill switches, metabolic dependencies, and clearance logic to ensure predictable therapeutic exit.
Together, these layers establish immune safety as a systems-level design principle essential for the clinical
translation and regulatory evaluation of living immunotherapeutics.


Table 1.
Immune inputs, processing logic, and immune outputs implemented by engineered microbes.
| Immune input type | Sensing mechanism | Processing logic | Immune output | Disease context |
|---|---|---|---|---|
| Hypoxia | Hypoxia-responsive promoters | AND gate with inflammatory cues | IL-12 delivery | Cancer |
| Tumor-associated cytokines | Cytokine-responsive transcriptional sensors | Signal amplification | STING pathway activation | Cancer |
| Lactate-rich metabolism | Metabolic sensing modules | Conditional activation | Antigen spreading | Cancer |
| Pro-inflammatory cytokines | Cytokine sensors (e.g., TNF, IL-6) | Tolerance-biased computation | IL-10 secretion | Autoimmunity |
| Dysbiosis | Microbiota-state sensing | Inflammatory restraint | Regulatory T cell induction | Autoimmunity |
| Epithelial stress | Stress-responsive promoters | Feedback attenuation | Barrier repair | Autoimmunity |
| Pathogen-associated molecular patterns | PRR-based sensing | Immune amplification | Interferon responses | Infection |
| Pathogen quorum signals | Quorum-sensing receptors | Population-level coordination | Myeloid cell activation | Infection |
| Inflammatory cues | Cytokine and metabolite sensing | Trained immunity programming | NK cell activation | Infection |
| SCFAs | Metabolic sensors | Tolerance logic | Regulatory T cell expansion | Autoimmunity |
| Nitric oxide | Redox-sensitive sensors | Band-pass filtering | Local immune modulation | Cancer |
| Microbiota nutrient state | Nutrient availability sensing | Adaptive regulation | Immune homeostasis | Chronic inflammation |
Table 2.
Clinical translation of engineered microbial immunotherapies through immune-state endpoints.
Table 2.
Clinical translation of engineered microbial immunotherapies through immune-state endpoints.
| Disease area | Immune objective | Immune endpoints measured | Safety readouts | Translational implication |
|---|---|---|---|---|
| Cancer | Immune activation | Cytokine topology; intratumoral T cell infiltration; antigen spreading | Cytokine release syndrome (CRS) markers; systemic cytokine levels | Defines efficacy through immune remodeling rather than tumor burden alone |
| Cancer | Reversal of immune suppression | STING activation; dendritic cell maturation; myeloid reprogramming | Local versus systemic inflammation; vascular leakage | Enables spatially confined immune activation with reduced systemic toxicity |
| Autoimmunity | Immune tolerance restoration | Treg frequency; Treg/Th17 balance; tolerogenic dendritic cell markers | Barrier integrity; absence of systemic immunosuppression | Supports antigen-specific and tissue-restricted tolerance induction |
| Autoimmunity | Suppression of chronic inflammation | Pro-inflammatory cytokine reduction; regulatory cytokine dominance | Infection susceptibility; immune competence markers | Demonstrates immune re-education rather than broad immune suppression |
| Chronic inflammatory disease | Barrier repair and homeostasis | Epithelial integrity; mucus production; IgA responses | Microbial translocation; systemic inflammation | Links mucosal immune architecture to clinical benefit |
| Infection | Immune enhancement | Interferon responses; myeloid activation; NK cell activity | Inflammatory overshoot; tissue damage markers | Enables immune-forward infection control without reliance on antimicrobials |
| Infection | Trained immunity induction | Epigenetic and metabolic markers of innate memory | Chronic inflammation markers; immune exhaustion | Supports durable, broad-spectrum host defense |
| Cross-indication | Durable immune remodeling | Immune state stability over time; memory formation | Long-term inflammatory sequelae | Shifts evaluation from transient exposure to sustained immune state change |
| Cross-indication | Therapeutic controllability | Immune-dependent clearance; reversibility of immune effects | Kill-switch activation; microbial clearance kinetics | Aligns immune dynamics with regulatory expectations for safety and control |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



