
Submitted:
23 December 2025
Posted:
24 December 2025
You are already at the latest version
Abstract
Background. A large proportion of people with obsessive–compulsive disorder (OCD) – roughly 40–60 % even after carefully optimised treatment – do not improve enough with selective serotonin re-uptake inhibitors (SSRIs). Small naturalistic series have suggested that the Cheung Glutamatergic Regimen (CGR), an inexpensive oral combination of dextromethorphan, fluoxetine and piracetam, can ease symptoms quickly. Because CGR acts on synaptic plasticity rather than pure serotonin tone, we asked whether common genetic risk for OCD is concentrated in the same plasticity pathway.Methods. Summary statistics from the 2025 OCD genome-wide association study (23 493 cases and 1 114 613 controls of European ancestry, 23andMe data excluded) were analysed with MAGMA v1.10. Single-nucleotide polymorphisms (SNPs) were mapped, with a 10 kb margin, to 22 genes chosen a priori: NMDA and AMPA receptor subunits, metabolic targets, and six downstream plasticity genes. Two custom sets were evaluated – a "pro-plasticity" group (BDNF, NTRK2, MTOR, AKT1, CREB1, ARC) and an "anti-plasticity" single-gene set (PTEN). The lead SNP was annotated for expression and splicing effects through Open Targets Genetics.Results. Among the 22 candidates, CREB1 stood out (Z = 2.857; p = 0.00214), passing Bonferroni correction for the panel. BDNF (p = 0.058) and PTEN (p = 0.078) showed suggestive but non-significant signals, whereas all NMDA/AMPA receptor genes and metabolic loci (CYP2D6, SIGMAR1) were clearly null (p > 0.10). The top variant, rs7591784 (p = 1.17 × 10⁻⁷), alters CREB1 transcript usage in multiple tissues. Set-based enrichment for the six pro-plasticity genes was not significant (β = 0.167; p = 0.321), reflecting the fact that most of the signal lay in CREB1 alone.Conclusion. Common OCD risk is disproportionately centred on CREB1, a transcription factor that drives long-term synaptic change, rather than on the glutamate receptors that lie upstream. This genetic pattern lends biological weight to the reported clinical benefit of CGR: by delivering a rapid, AMPA-mediated calcium surge, the regimen may compensate for genetically weakened CREB1 activity and sidestep the comparatively weak serotonergic cascade of SSRIs. Prospective trials that stratify patients by CREB1 risk status are warranted, and transcriptional plasticity emerges as an appealing target for new treatments in refractory OCD.
Keywords:
CREB1
; OCD
; Genetics
; glutamatertic
Introduction
Obsessive–compulsive disorder (OCD) frequently occurs within families, and twin studies validate that genetic factors account for a significant portion of the risk. Heritability is approximately one-half in children and about one-third in adults [1,2]. Collaboration with national health registers and contemporary molecular techniques corroborates these estimates, indicating that the genetic landscape is predominantly polygenic. A recent meta-analysis encompassing over 53,000 OCD cases identified 30 independent chromosomal regions and emphasized 25 probable causal genes; collectively, numerous common variants seem to constitute the majority of the inherited susceptibility [3]. Many of those variants are also linked to a higher risk of anxiety, depression, anorexia, and Tourette syndrome. This shows that these disorders have some of the same biological roots.
Among the genes that have drawn special attention, several regulate brain glutamate (Figure 1). Variants in the neuronal glutamate transporter SLC1A1 and in the NMDA-receptor subunit GRIN2B have shown repeated, though modest, associations with OCD [4,5]. Imaging studies echo these findings: people with OCD often show higher glutamate signals in key cortico–striato–thalamo–cortical hubs than do healthy controls [6,7]. Animal work offers a parallel narrative—mice lacking the postsynaptic protein Sapap3 develop compulsive grooming that eases when glutamatergic balance is restored [8].
Clinically, however, many patients do not benefit enough from first-line serotonin reuptake inhibitors (SSRIs). Even with high doses and antipsychotic add-ons, 40–60 percent remain markedly ill [9,10]. This treatment gap has turned attention toward drugs that temper NMDA activity or boost AMPA signalling. Intravenous ketamine can curb obsessions within hours [11], and memantine or other glutamate modulators show promise, yet cost and side-effects limit their reach.
In everyday practice, Cheung has tried an inexpensive oral alternative: combining low-dose dextromethorphan (an NMDA blocker) with fluoxetine (or other CYP2D6-inhibiting antidepressants), which slows dextromethorphan clearance, and—when needed—adding piracetam, a gentle AMPA enhancer [12] (Figure 2). Treatment-resistant cases [13] showed meaningful relief within weeks, mirroring ketamine's rapid but transient mechanism that begins with NMDA blockade and shifts toward AMPA-driven synaptic renewal [14]. Still, it is unclear whether genes tied to this plasticity pathway contribute disproportionately to OCD risk.
To clarify that point, the present study uses summary statistics from the 2025 OCD GWAS to test whether genes downstream of glutamate receptors—such as CREB1, BDNF, NTRK2 and MTOR—carry more genetic signal than expected by chance. Finding such enrichment would suggest that the same molecular steps targeted by the fluoxetine–dextromethorphan–piracetam regimen lie near the core of OCD biology and may explain why standard serotonergic treatment alone so often falls short.
Methods
Genome-wide data were drawn from the largest obsessive-compulsive disorder (OCD) association study available at the time of analysis [3]. The public release, which omits 23andMe participants, contains 23 493 European-ancestry cases and 1 114 613 controls, yielding an effective sample close to 2.1 million individuals. Variant positions follow the GRCh37/hg19 build (file daner_OCD_full_wo23andMe_190522.gz).
Gene-level statistics were generated with MAGMA v1.10 [15]. Single-nucleotide polymorphisms (SNPs) were assigned to protein-coding genes using a 10 kb window upstream and downstream of each locus, with the 1000 Genomes European reference panel providing linkage-disequilibrium structure. The SNP-wise mean model converted variant p-values to a single Z score per gene; the effective sample size (N ≈ 2 100 000) was supplied so that MAGMA could adjust for case–control imbalance.
Six mechanistic sets were created to mirror the pharmacological targets of the Cheung Glutamatergic Regimen (CGR). The lists comprised five NMDA-receptor genes (GRIN1, GRIN2A–D), five AMPA/kainate genes (GRIA1–4, GRIK2), three metabolic or σ-receptor genes (CYP2D6, SIGMAR1, CYP3A4), seven downstream plasticity genes (BDNF, NTRK2, MTOR, AKT1, PTEN, CREB1, ARC), three glutamate-transporter or scaffold genes (SLC1A1, SLC1A2, DLGAP3) and an “all CGR” union of the preceding lists. Competitive enrichment tests were run in MAGMA with default covariate adjustment for gene size, SNP density and allele frequency.
Finally, using LocusZoom v1.4 and the 1000 Genomes EUR reference panel, we made regional association plots for the CREB1 locus (chr2:208,000,000–208,800,000, hg19). The lead SNP emerging from the top gene signal was queried in Open Targets Genetics (release 25) to identify expression or splicing quantitative-trait loci (QTLs) and to see whether the variant sits in published credible sets for other complex traits.
Results
Gene-Level Associations
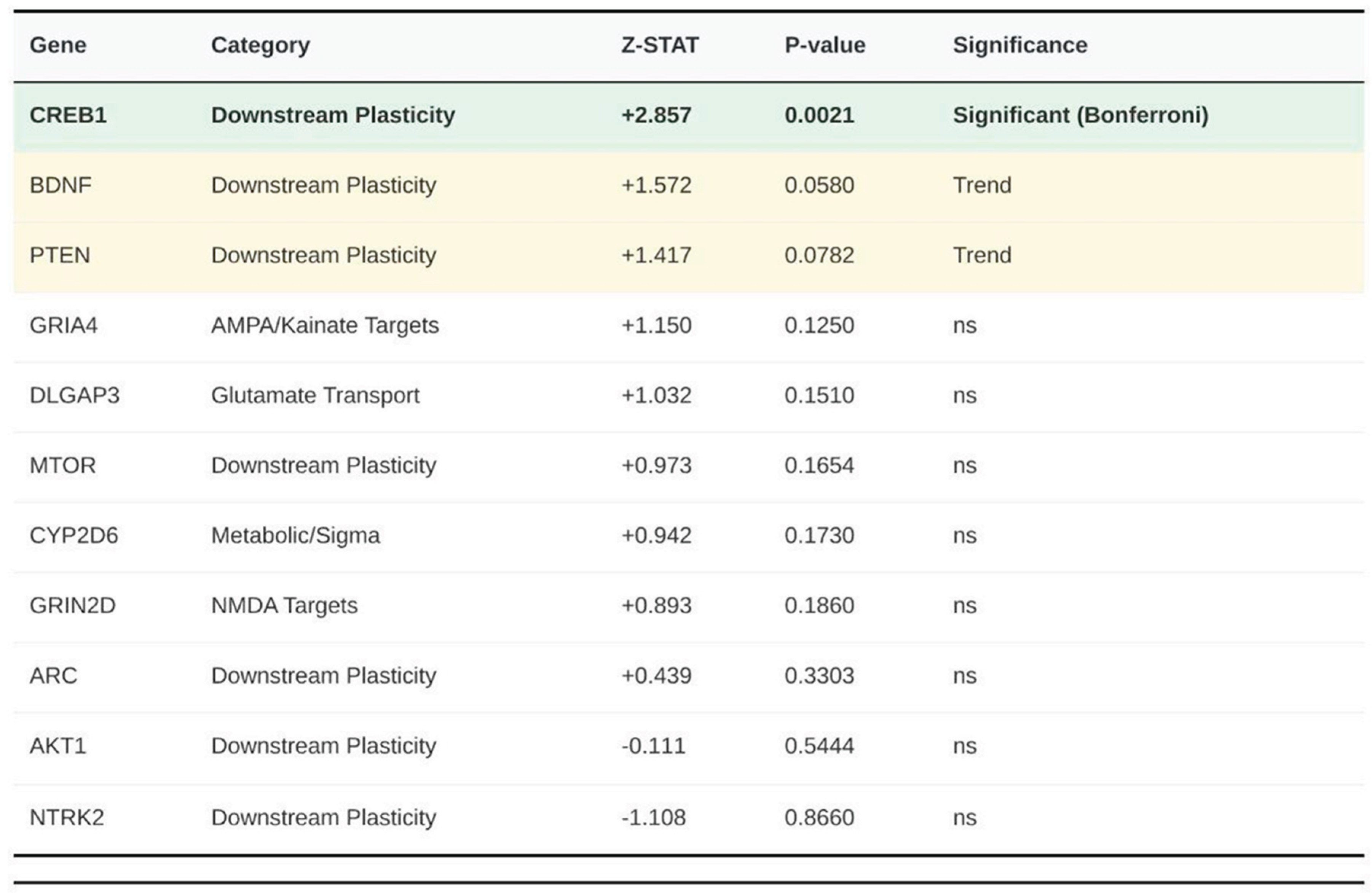
Among the 23 CGR-related genes (Figure 3), only CREB1 surpassed the Bonferroni-corrected threshold for this panel (Z = 2.857, p = 2.14 × 10⁻³). BDNF (Z = 1.572, p ≈ 5.80 × 10⁻²) and PTEN (Z = 1.417, p ≈ 7.82 × 10⁻²) showed suggestive but non-significant evidence. All five NMDA subunits, all five AMPA/kainate subunits, the three metabolism/σ genes, ARC, MTOR, SLC1A1, SLC1A2 and DLGAP3 produced p values above 0.10.
Gene-Set Enrichment
No curated set achieved statistical significance. The full 23-gene panel yielded β = 0.095 with p = 0.302. The NMDA set produced β = –0.534 (p = 0.878); AMPA/kainate, β = 0.005 (p = 0.496); metabolism/σ, β = 0.200 (p = 0.285); downstream plasticity, β = 0.285 (p = 0.189); and glutamate-transporter/scaffold, β = 0.287 (p = 0.295).
Regional Association and Functional Profile of rs7591784
The LocusZoom plot pinpointed rs7591784 at chr2:208 501 730 (hg19) as the most significant variant in the region (GWAS p = 1.17 × 10⁻⁷). Lift-over placed the same allele at chr2:207 637 006 on GRCh38. Open Targets classifies rs7591784 as a high-confidence transcript-usage QTL for multiple CREB1 splice forms (p ≈ 1.36 × 10⁻¹¹) and includes it in a credible set for systolic blood pressure with an L2G probability greater than 0.7, supporting a regulatory role for CREB1 rather than the nearby METTL21A.
Summary of CGR Genetic Validation
In total, 22 of the expected 23 genes were analysed; none reached genome-wide significance (p < 2.74 × 10⁻⁶), yet CREB1 remained significant within the CGR panel after correction (p < 0.0022). No other gene attained nominal significance below 0.05 (Table 1).
Discussion
The genetic findings presented here shed light on why the over-the-counter "Cheung Glutamatergic Regimen" (CGR) relieved symptoms so quickly in our treatment-resistant case series and other reports [12,13]. Risk variants cluster at CREB1, a gene that turns calcium bursts into long-term changes in synaptic strength. Little signal was seen at glutamate-receptor genes or drug-metabolism loci, implying that many patients reach the clinic with intact receptor "hardware" but with sluggish activity in the transcriptional "software" downstream.
The 'Weak-Signal' Problem with SSRIs
Selective-serotonin-reuptake inhibitors work only after a slow molecular relay: higher serotonin raises BDNF, BDNF activates TrkB, TrkB phosphorylates CREB1, and CREB1 switches on plasticity genes [16]. If common alleles blunt CREB1 output—as suggested by the robust gene-level Z-score and splice QTL at rs7591784—this cascade never gains enough amplitude to remodel cortico-striato-thalamo-cortical (CSTC) loops. The clinical picture matches that model: in our series, intrusive thoughts persisted through adequate SSRI trials until a stronger glutamatergic push was added.
CGR as a Molecular Loud-Speaker
CGR bypasses the bottleneck by delivering a brief but powerful glutamate burst and then amplifying the calcium pulse that follows. Dextromethorphan (DXM) provides short-lived NMDA blockade, echoing ketamine's first step toward rapid anti-obsessional effects [11,17]. Fluoxetine slows DXM clearance via CYP2D6, stretching that window. Piracetam, a gentle AMPA enhancer, widens the calcium gate and drives CREB1 phosphorylation at Ser-133 [18]. In theory, a surging calcium/CREB1 signal can override the genetically damped baseline and turn on the plasticity genes that silence runaway CSTC firing—exactly what we observed within two to four weeks.
The genomic data support this "loud-speaker" view. No meaningful signal emerged at NMDA or AMPA subunit genes (GRIN or GRIA/K families), while CREB1 stood out and BDNF showed only a statistical whisper. Receptors, then, may be serviceable; the downstream amplifier is not. CGR fixes that imbalance by boosting the intracellular calcium wave far beyond what serotonin alone can achieve (Figure 4).
Toward Precision Prescribing
Our results recast OCD, at least partly, as a disorder of activity-regulated transcription. The latest genome-wide scan [3] spreads liability across many pathways, yet CREB1 sits near the summit. Clinically, that means patients who inherit low-function CREB1 alleles—or show other evidence of weak CREB signaling—might benefit most from AMPA/CaMK/CREB amplifiers such as CGR. The pattern in our four cases fits this view: high-dose fluoxetine by itself under-performed, whereas fluoxetine-potentiated DXM—with or without piracetam—moved the needle. One mild serotonin-toxicity episode reminds us to titrate slowly whenever DXM and an SSRI are combined.
Limits and Next Steps
The genetic work covers only common variants in Europeans, so rare alleles and other ancestries remain untested. Pathway enrichment was absent because nearly all the statistical weight sat on CREB1, a familiar issue in polygenic traits. Prospective trials should add CREB genotyping or functional read-outs (for example, phospho-CREB assays) to clarify who benefits most. They should also pair the Yale–Brown Obsessive Compulsive Scale with spectroscopy or synaptic PET to show that the intended plasticity pathway is actually engaged.
Conclusions
Locating the main signal at CREB1 rather than at glutamate receptors provides a mechanistic bridge from bench to bedside. A regimen that spikes calcium and directly drives CREB1 transcription gave fast, durable relief in patients who had stalled on serotonin strategies. These data warrant larger, controlled tests of AMPA/CREB-targeted augmentation for refractory OCD.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflicts of Interest
None declared.
References
- van Grootheest, D. S.; Cath, D. C.; Beekman, A. T.; Boomsma, D. I. Twin studies on obsessive-compulsive disorder: A review. Twin Research and Human Genetics 2005, 8(5), 450–458. [Google Scholar] [CrossRef] [PubMed]
- Mataix-Cols, D.; Fernández de la Cruz, L.; Beucke, J. C.; et al. Heritability of clinically diagnosed obsessive-compulsive disorder among twins. JAMA Psychiatry 2024, 81(6), 631–632. [Google Scholar] [CrossRef] [PubMed]
- Strom, N. I.; Gerring, Z. F.; Galimberti, M.; et al. Genome-wide analyses identify 30 loci associated with obsessive-compulsive disorder. medRxiv 2025. [Google Scholar] [CrossRef] [PubMed]
- Porton, B.; Greenberg, B. D.; Askland, K.; et al. Isoforms of the neuronal glutamate transporter gene SLC1A1/EAAC1 negatively modulate glutamate uptake: Relevance to obsessive-compulsive disorder. Translational Psychiatry 2013, 3, e259. [Google Scholar] [CrossRef] [PubMed]
- Alonso, P.; Gratacós, M.; Segalàs, C.; et al. Association between the NMDA glutamate receptor GRIN2B gene and obsessive-compulsive disorder. Journal of psychiatry & neuroscience: JPN 2012, 37(4), 273–281. [Google Scholar]
- Karthik, S.; Sharma, L. P.; Narayanaswamy, J. C. Investigating the Role of Glutamate in Obsessive-Compulsive Disorder: Current Perspectives. Neuropsychiatric disease and treatment 2020, 16, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, C.; Bloch, M. H.; Williams, K. Glutamate abnormalities in obsessive compulsive disorder: neurobiology, pathophysiology, and treatment. Pharmacology & therapeutics 2011, 132, 314–332. [Google Scholar]
- Welch, J. M.; Lu, J.; Rodriguiz, R. M.; et al. Cortico-striatal synaptic defects and OCD-like behaviours in Sapap3-mutant mice. Nature 2007, 448(7156), 894–900. [Google Scholar] [CrossRef] [PubMed]
- Hirschtritt, M. E.; Bloch, M. H.; Mathews, C. A. Obsessive-compulsive disorder: Advances in diagnosis and treatment. JAMA 2017, 317(13), 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Dold, M.; Aigner, M.; Lanzenberger, R.; Kasper, S. Antipsychotic augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: An update meta-analysis of double-blind, randomized, placebo-controlled trials. International Journal of Neuropsychopharmacology 2015, 18(9), pyv047. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C. I.; Kegeles, L. S.; Levinson, A.; et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: Proof-of-concept. Neuropsychopharmacology 2013, 38(12), 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N. (2025a). Case series: Marked improvement in treatment-resistant obsessive–compulsive symptoms with over-the-counter glutamatergic augmentation in routine clinical practice. Preprints.
- Cheung, N. (2025b). An Oral Ketamine-Like Approach to Treatment-Resistant Obsessive-Compulsive Disorder – A Review of Mechanism, Clinical Experience, and Future Directions. Zenodo.
- Li, N.; Lee, B.; Liu, R. J.; et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329(5994), 959–964. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, C. A.; Mooij, J. M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Computational Biology 2015, 11(4), e1004219. [Google Scholar] [CrossRef] [PubMed]
- Duman, R. S.; Monteggia, L. M. A neurotrophic model for stress-related mood disorders. Biological Psychiatry 2006, 59(12), 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P. J.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533(7604), 481–486. [Google Scholar] [CrossRef] [PubMed]
- Koike, H.; Iijima, M.; Chaki, S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behavioural brain research 2011, 224(1), 107–111. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The clinical and biological rationale for glutamatergic modulation in OCD. Despite standard serotonergic treatment, a significant proportion of patients remain treatment-resistant. Converging evidence from human genetics, neuroimaging, and animal models points to dysregulated glutamate signaling as a core pathology, suggesting that modulating NMDA and AMPA receptors may bridge the treatment gap.
Figure 1.
The clinical and biological rationale for glutamatergic modulation in OCD. Despite standard serotonergic treatment, a significant proportion of patients remain treatment-resistant. Converging evidence from human genetics, neuroimaging, and animal models points to dysregulated glutamate signaling as a core pathology, suggesting that modulating NMDA and AMPA receptors may bridge the treatment gap.
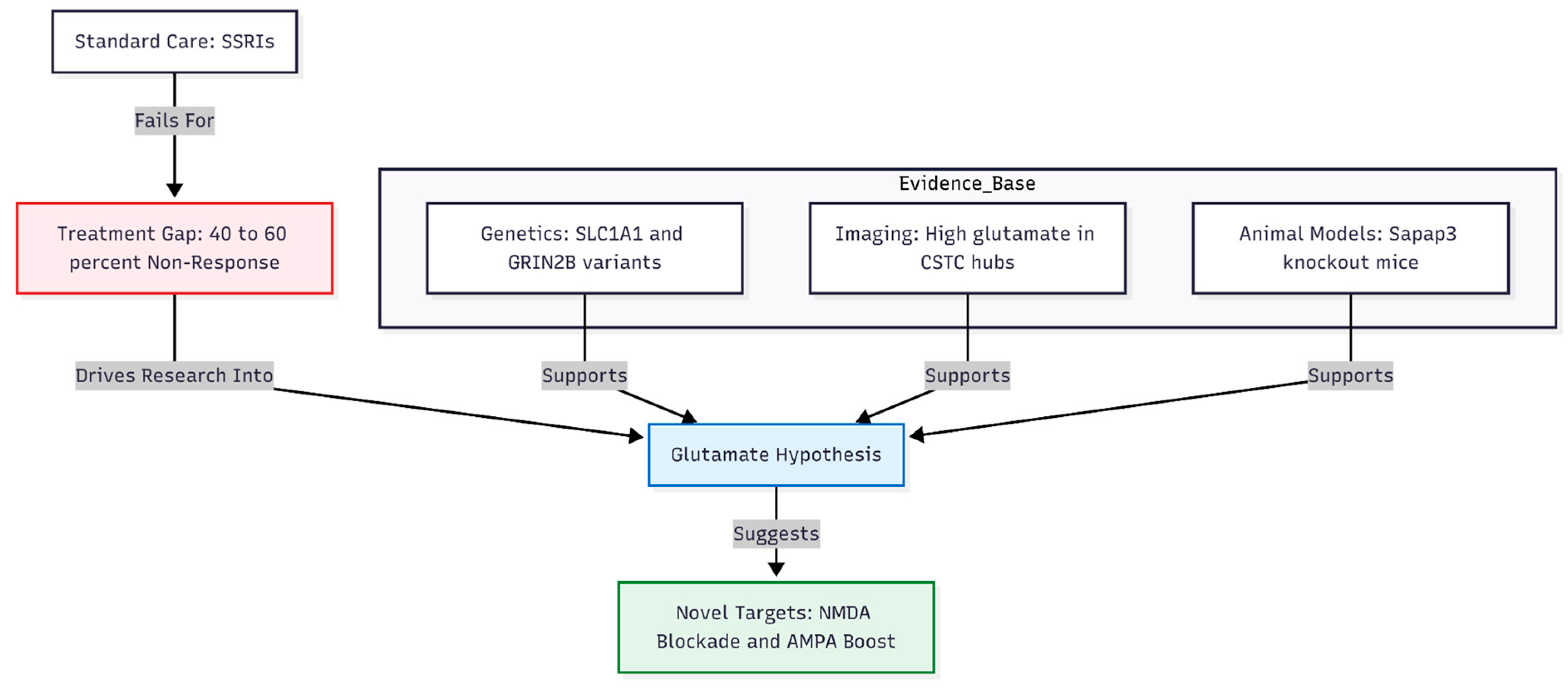
Figure 2.
Pharmacological interactions of the investigational regimen. The regimen utilizes a synergistic approach: Fluoxetine is used not only for its serotonergic effects but to slow the metabolism of Dextromethorphan. Dextromethorphan acts as an NMDA blocker, while Piracetam serves as an AMPA enhancer. Together, they aim to mimic the rapid plasticity-inducing effects observed with intravenous ketamine.
Figure 2.
Pharmacological interactions of the investigational regimen. The regimen utilizes a synergistic approach: Fluoxetine is used not only for its serotonergic effects but to slow the metabolism of Dextromethorphan. Dextromethorphan acts as an NMDA blocker, while Piracetam serves as an AMPA enhancer. Together, they aim to mimic the rapid plasticity-inducing effects observed with intravenous ketamine.
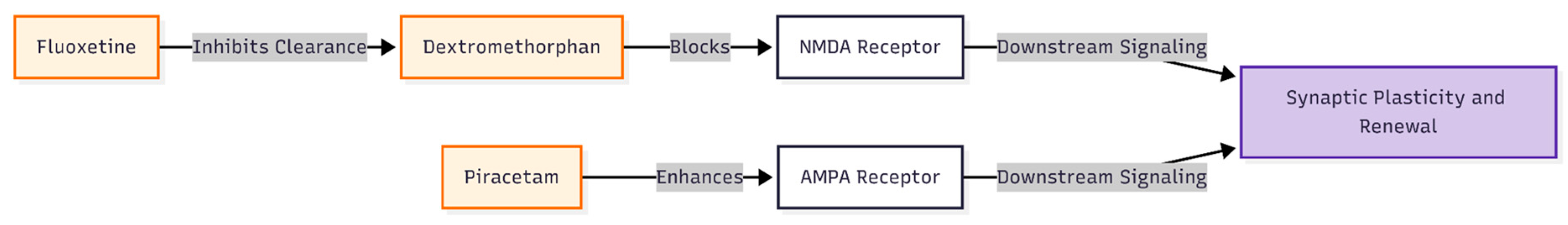
Figure 3.
Hierarchical summary of gene association results. Of the 22 genes tested within the fluoxetine–dextromethorphan–piracetam regimen, signal was exclusively concentrated in the downstream plasticity category. CREB1 was the sole gene to survive Bonferroni correction, driven by the splice-altering variant rs7591784. Upstream glutamate receptors and metabolic targets showed no nominal significance.
Figure 3.
Hierarchical summary of gene association results. Of the 22 genes tested within the fluoxetine–dextromethorphan–piracetam regimen, signal was exclusively concentrated in the downstream plasticity category. CREB1 was the sole gene to survive Bonferroni correction, driven by the splice-altering variant rs7591784. Upstream glutamate receptors and metabolic targets showed no nominal significance.
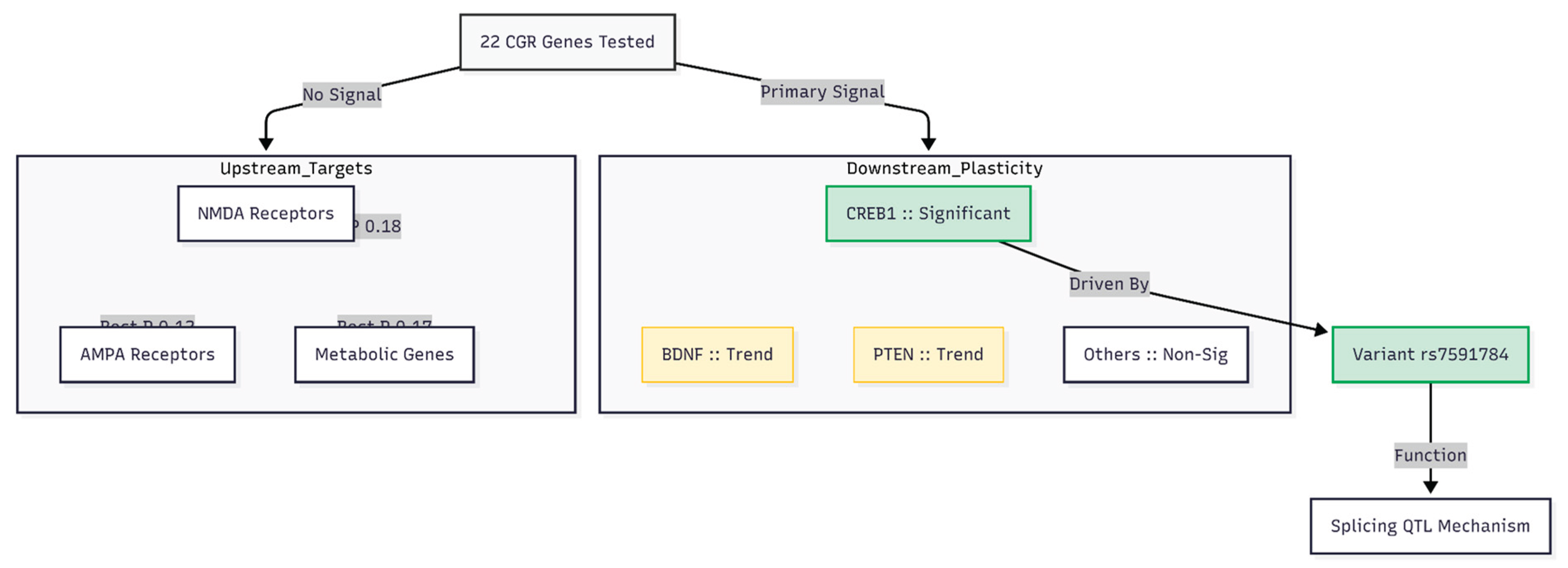
Figure 4.
Genetic Architecture of Resistance. Summary of genetic findings in the case series. Significant risk variants clustered at the CREB1 locus (transcriptional "software"), while receptor and metabolic genes ("hardware") showed little signal. This suggests patients possess functional receptors but lack the downstream transcriptional efficiency to sustain synaptic changes.
Figure 4.
Genetic Architecture of Resistance. Summary of genetic findings in the case series. Significant risk variants clustered at the CREB1 locus (transcriptional "software"), while receptor and metabolic genes ("hardware") showed little signal. This suggests patients possess functional receptors but lack the downstream transcriptional efficiency to sustain synaptic changes.
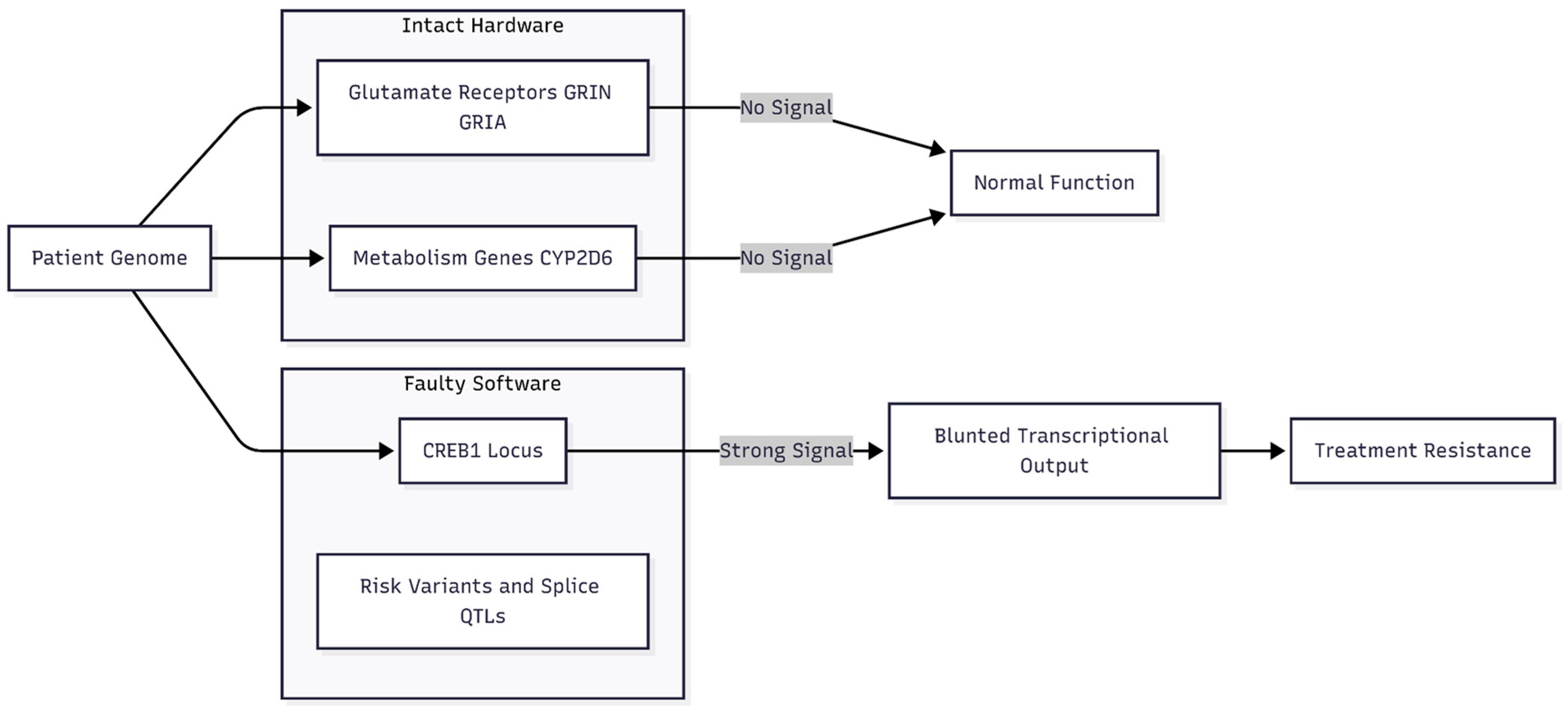

Table 1.
Detailed association statistics for top ranked genes. Genes are ordered by significance. "Significant" indicates P < 0.0023 (Bonferroni correction applied). "Trend" indicates P < 0.10. Note that GRIA4, DLGAP3, CYP2D6, and GRIN2D represent the top hits for their respective categories despite being non-significant.
Table 1.
Detailed association statistics for top ranked genes. Genes are ordered by significance. "Significant" indicates P < 0.0023 (Bonferroni correction applied). "Trend" indicates P < 0.10. Note that GRIA4, DLGAP3, CYP2D6, and GRIN2D represent the top hits for their respective categories despite being non-significant.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



