
Submitted:
19 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Analysis reproducibility has become one of the main challenges for scientific reporting as it is critical to ensure transparent and comparable results. Metagenomics studies are not exempted from these concerning aspects, and hence bioinformatics pipelines to handle this type of data have evolved rapidly as an attempt to equip the research community with reliable methodologies and procedures. Nonetheless, as these workflows grow in robustness and complexity, inexperienced users find it difficult to understand or customize the pipelines. To address this limitation, we developed an open, interactive and web-based tutorial (TaxoFlow) that guides scholars through the detailed development of a validated and reproducible Nextflow metagenomics profiling pipeline. This workflow integrates software to remove host sequences (Bowtie2), a taxonomic classification (Kraken2), a tool for species abundance re-estimation (Bracken) and different data visualization strategies. As important features, the tutorial emphasizes simplicity, modularity, and containerization, which empowers users with both conceptual understanding and practical implementation skills. Noteworthy, this tutorial provides all the required files, databases, dependencies, software and environment for the user to run without the need of local installation or computational adaptations elsewhere. Finally, by offering a fully reproducible pipeline with a step-by-step developing tutorial, this work aims to lower technical barriers in microbiome bioinformatics and promote best practices in metagenomics data analysis. TaxoFlow is freely available at https://taxoflow.work/.
Keywords:
metagenomics
; tutorial
; pipeline
; Nextflow
Introduction
The constant complexity growth of computational analyses in metagenomics research has brought the necessity of reproducible, scalable and transparent workflows [1]. In this sense, several authors have highlighted the central concern regarding reproducibility across various scientific disciplines as usually the same analysis with the same data yields different results, a situation that has escalated until a denominated reproducibility crisis [2]. The bioinformatics field is especially sensitive to a lack of reproducibility given the wide variety of inconsistent sources such as software versions, parameter settings, or execution environments [3]. Notwithstanding, to mitigate these issues, workflow management systems such as Nextflow [4] or Snakemake [5] have emerged as powerful tools for structuring, automating, and documenting analysis pipelines [6,7]. Specifically, Nextflow enables researchers to design modular, scalable, portable and controlled workflows that are fully reproducible across different computing environments using containers and/or dependency management systems. Being so, not only do these features facilitate scalability and reproducibility, but also enhance transparency, allowing analyses to be easily shared, inspected, and reused [7,8].
On the other hand, metagenomics provides unprecedented insight into microbial communities without the need for cultivation given the possibility to capture the entire genetic diversity present in a determined environment [9]. Moreover, the strategies to analyze metagenomics data are divided mainly into two categories: i) assembly-based approaches, which use the original reads to build longer sequences (contigs) and reconstruct afterwards Metagenome-Assembled Genomes (MAGs); and ii) read-based taxonomic profiling, which classifies sequencing reads directly by leveraging different reference or indexed databases [10]. In the case of assembly-based methodologies, they enable detailed genomic and functional characterization of individual microorganisms, albeit demanding substantial computational resources and high sequencing depth [11]. Meanwhile, taxonomic profiling allows a rapid and accurate community composition estimation especially for large-scale study implementation or complex environments given its efficiency in terms of CPU and memory usage [12].
Furthermore, there is a broad landscape of tools developed for short-read metagenomic taxonomic profiling that employ distinct algorithmic strategies. For instance, Kraken2 [13], a faster and more sensitive version of Kraken [14], classifies reads based on exact k-mer matches to a reference database. Frequently, Kraken2 is complemented with the execution of Bracken [15], a tool that refines the initial classifications by re-estimating species abundances through Bayesian reallocation of ambiguous reads. Other approaches to perform taxonomic profiling include mapping to clade-specific markers (MetaPhlAn4) [16], exact matches on the protein-level (Kaiju) [17], discriminative k-mers (CLARK) [18], space-optimized indexing schemes (Centrifuge) [19] and universal phylogenetic marker gene mapping (mOTUS2) [20]. Consistently, Kraken2 and Bracken rank among the top performers in terms of classification accuracy and runtime efficiency across simulated and real metagenomic datasets [10,21,22].
In order to promote reproducibility and scalability in to perform taxonomic profiling during metagenomics studies, some Nextflow-based pipelines that encompass Kraken2 and/or Bracken have been released, including nf-core/taxprofiler [23], kraken-nf [24], wf-metagenomics [25], nxf-kraken2 [26], 16S-Metatranscriptomic-Analysis [27] and specific modules within the Bactopia [28] and nf-core [29] suites. These workflows provide comprehensive implementations of metagenomic taxonomic classification software, integrating multiple profilers, database management steps, and reporting modules. Nonetheless, such pipelines feature a high number of parameters and an internal complex structure, making them challenging for inexperienced users to understand, modify or adapt to specific requirements. As a result, this “black box” nature of these useful and robust but sophisticated tools can hinder learning and flexibility. Furthermore, although there have been efforts to document training material to perform metagenomics data analysis [30,31] or to develop assembly-based Nextflow pipelines [32], there is a growing need for educational resources that provide the technical knowledge, materials, standardized computing environments and practical implementations to develop reproducible metagenomics-focused pipelines wrapped with workflow managers.
In this context, we developed an open, interactive and web-based tutorial (TaxoFlow) that guides users step-by-step through the creation of a simple yet complete Nextflow metagenomics pipeline. This tutorial is built on a reference protocol proposed by Lu et al. (2023) [33] that integrates the removal of host reads, taxonomic classification, species abundance re-estimation and the generation of an HTML report in a container-based environment. The tutorial emphasizes conceptual understanding, modular pipeline design, and reproducibility, providing a practical entry point for researchers seeking to build or customize their own workflows.
Content and Learning Objectives
TaxoFlow provides a practical and educational framework for developing a reproducible Nextflow workflow dedicated to metagenomics taxonomic profiling. Through this tutorial we are aiming at teaching both the conceptual foundations and technical implementation of reproducible data analysis, while producing biologically meaningful results using established tools such as Bowtie2 [34], Kraken2, Bracken and Krona plots [35]. The resource is hosted at https://taxoflow.work/ and offers an interactive, step-by-step learning experience complemented by commented code examples, schematic representations, and example datasets. It is worthy to mention that TaxoFlow assumes a basic familiarity with the command-line interface and provides links to complementary educational resources for users who wish to strengthen their background in Linux, Nextflow, or metagenomics data handling. In addition, it is designed for early-career researchers and students seeking to learn how workflow management systems can enhance reproducibility, scalability, and transparency in microbiome bioinformatics.
Educational Scope and Learning Objectives
TaxoFlow presents the process of building a linear pipeline that performs host read removal, taxonomic classification and species abundance re-estimation through a progressive learning path to introduce important workflow concepts. This initial exercise establishes a foundation for understanding process definition, parameterization, and file handling in Nextflow. Afterwards, the users then learn how to manage domain-specific outputs to ensure reproducible downstream analyses.
A central feature of TaxoFlow is the introduction of Nextflow’s dataflow paradigm, which enables dynamic parallelization of analyses across multiple samples. In this sense, learners first execute the pipeline for a single dataset and then generalize it to handle multiple input samples simultaneously, a process that depicts how channels and operators manage dependencies and data exchange between processes. Moreover, TaxoFlow shows the modularization of the workflows by separating individual processes into reusable components and assembling them into structured subworkflows; this modular design is aligned with nf-core guidelines, and it encompasses important considerations regarding workflow design and implementations [6,7,36]. Likewise, the tutorial is complemented with additional sections to demonstrate conditional execution and the use of logical operators to dynamically adapt the pipeline according to user-defined parameters or input availability. Also, TaxoFlow shows how to integrate custom scripts, maintaining portability through containerized environments. As a result, the tutorial follows and promotes the adoption of FAIR principles for research software [37], and it sticks to the general recommendations to organize computational biology projects [38].
Instructional Design
TaxoFlow is structured into three main parts with an additional orientation page to present the setup and materials provided to the students. As a result, it reflects a scaffolding approach that allows progressive learning:
Part 1: Pipeline
The workflow implemented during the tutorial development is presented in Figure 1. The example dataset used in the tutorials consists of paired-end reads recovered from an oligotrophic, phosphorus-deficient pond in Cuatro Ciénegas, Mexico [39]. The workflow takes as input raw FASTQ files from one or multiple metagenomic samples to remove host reads using Bowtie2 by aligning the reads against a reference genome. The filtered reads are then subjected to taxonomic classification with Kraken2, followed by species abundance re-estimation using Bracken, producing refined taxonomic profiles for each sample. TaxoFlow guides the downloading of an indexed genome of Arabidopsis thaliana, TAIR10, (only for the educational purpose) for Bowtie2, provides instructions to retrieve a custom database with 54 bacterial species for Kraken2 and Bracken (https://doi.org/10.5281/zenodo.17708950), and it suggests resources for the users to adapt their pipeline execution. Later, the resulting Bracken reports are visualized through Krona plots. The workflow is bifurcated if multiple samples are used as input to automatically concatenate Bracken outputs and convert them into a Biological Observation Matrix (BIOM) [40] file, which is subsequently imported into R as a Phyloseq [41] object for downstream analyses. The entire workflow concludes with the generation of an integrated HTML report that includes abundance visualization, α- and β-diversity estimation, and network construction. The pipeline execution relies on container images pulled and launched by Docker to achieve maximum reproducibility.
Part 2: Single Sample
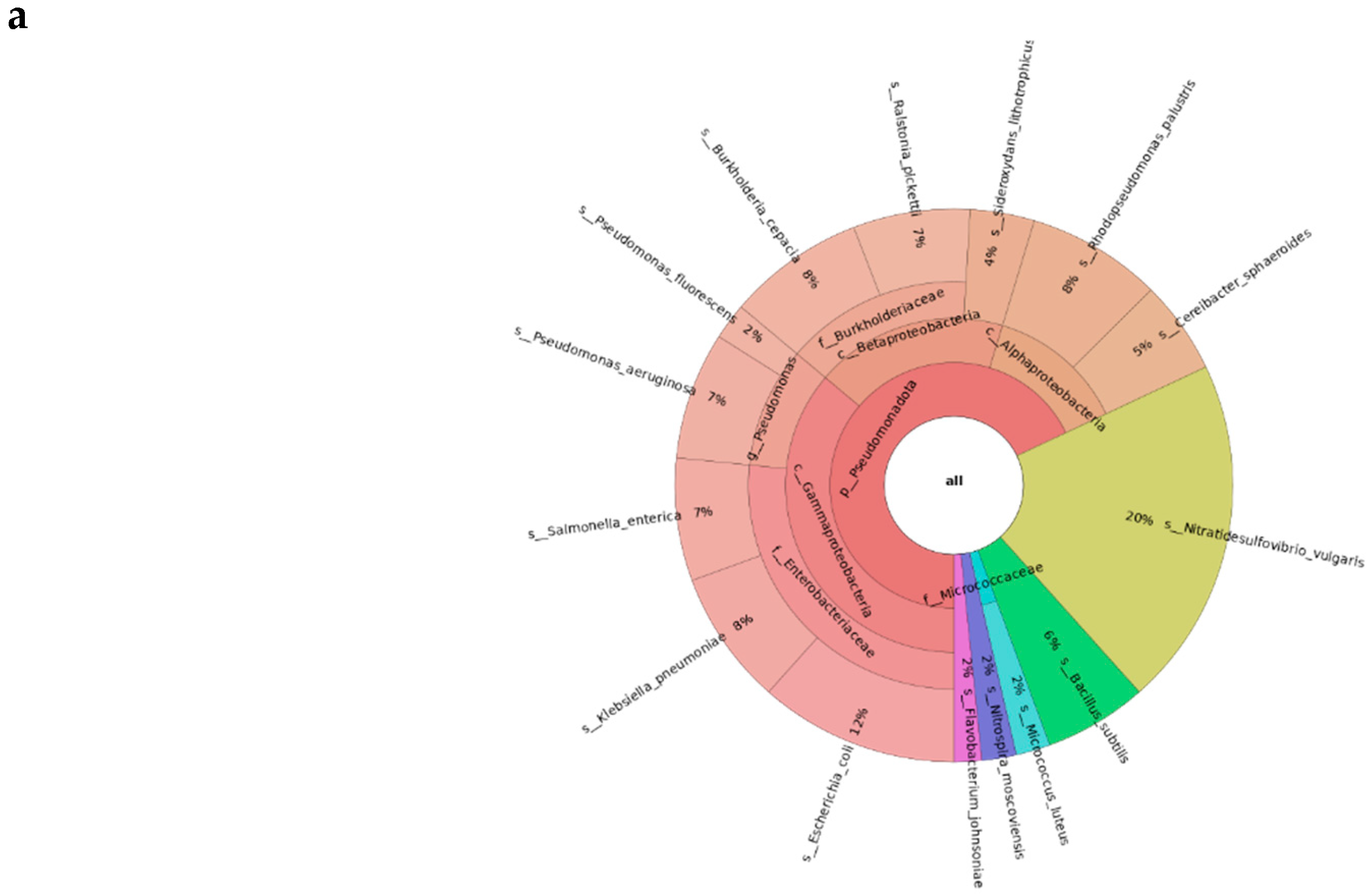
This section introduces the essentials of workflow design in Nextflow by walking learners through the construction of a metagenomics pipeline. Learners are presented with core concepts including processes, channels, configuration files, and the dataflow programming model, all framed within the minimal computational environment required to run the pipeline. The tutorial then transitions into hands-on development, where users build a functional workflow step by step. This includes defining modules for each process, centralizing the execution in a single workflow file that handles input channels for raw FASTQ files, databases and connection among processes. Each component is intentionally constructed incrementally to clarify how data moves between tasks, and how scripts run inside processes. Here, we also emphasize good workflow engineering practices by encouraging learners to modularize their code, name processes consistently, and adopt conventions inspired by nf-core. The section concludes with guidance on running the workflow for a single sample, ensuring that learners not only understand how the workflow functions but also how to adapt it. Figure 2a shows an example of the resulting output after single-sample execution of the developed pipeline.
Part 3: Multi-Sample
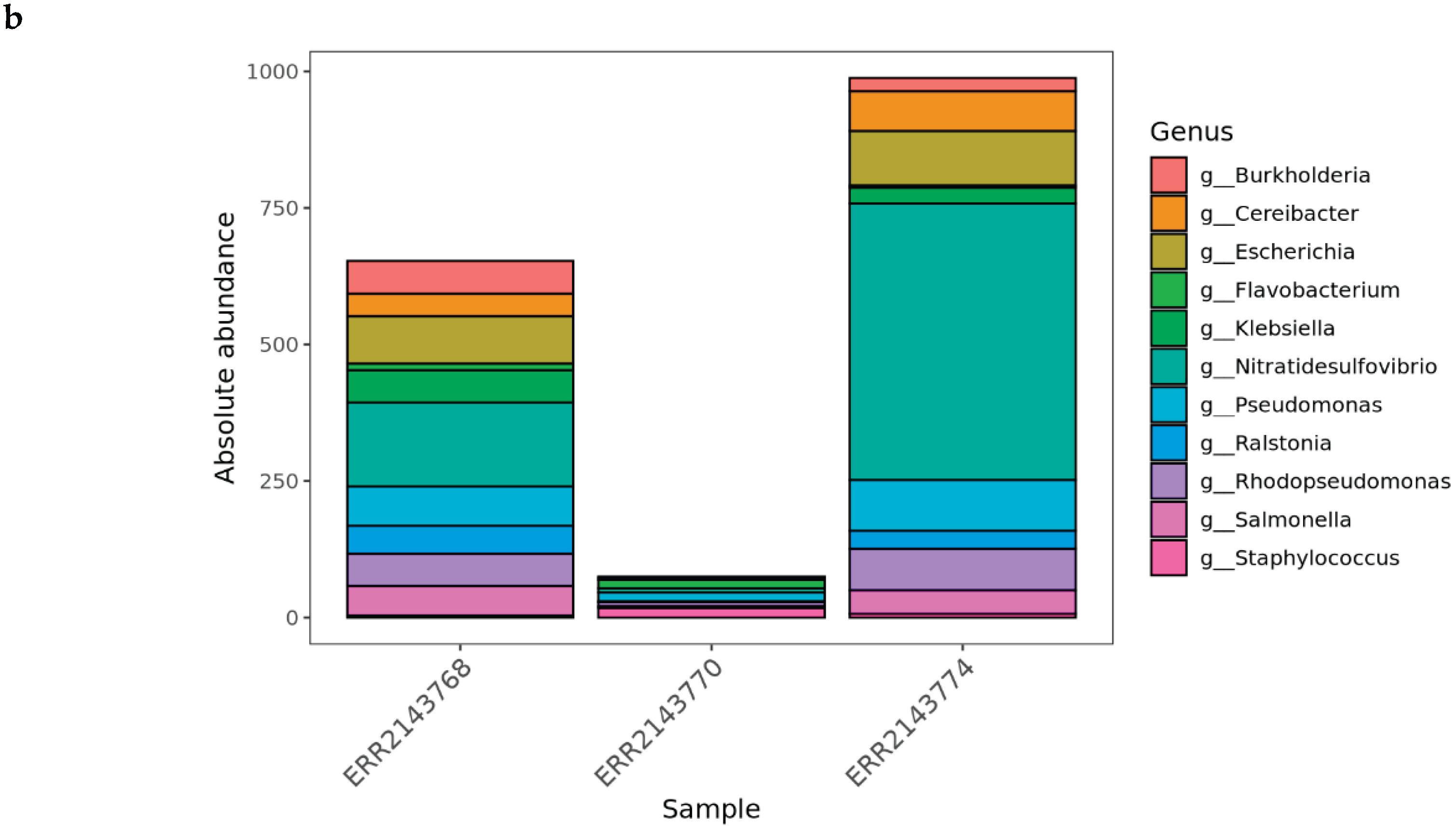
This part expands on the foundational skills developed in Part 2 by teaching learners how to scale the workflow to handle multiple samples and generate integrated metagenomics outputs. This section begins by explaining how the dataflow paradigm allows workflows to process many samples automatically and in parallel, without manually iterating through files. Learners modify their input definitions so that the workflow recognizes and processes an arbitrary number of FASTQ files. We also introduced more advanced workflow-control features, such as implementing conditional execution paths, using operators to coordinate outputs from different processes, and structuring pipeline logic to accommodate both optional and mandatory steps. Moreover, TaxoFlow shows how to perform pipeline enhancements by including the concatenation individual taxonomic reports into an abundance matrix, converting the matrix to BIOM format, generating a Phyloseq object, and producing a suite of ecological analyses such as alpha- and beta-diversity metrics. The workflow ultimately produces an automated HTML report that summarizes all results, illustrating how Nextflow can orchestrate complete end-to-end analyses. Alongside technical expansion, we aim at reinforcing best practices in workflow engineering by encouraging learners to organize processes into modules and incorporate custom scripts while maintaining portability and reproducibility. Figure 2b depicts the absolute abundance plot included in the HTML report generated after multi-sample analysis.
Conclusion
TaxoFlow serves both as an educational resource and a functional analytical tool, enabling scalable analysis from raw reads to ecological interpretation. The pipeline obtained through this tutorial facilitates reproducibility and provides an accessible entry point into the design principles of community standards such as nf-core guidelines. Likewise, the resulting workflow offers a lightweight, transparent, and customizable alternative for researchers who wish to understand or adapt taxonomic profiling pipelines from the ground up, while adhering to best practices in reproducible computational metagenomics. Finally, the tutorial demonstrates how accessible, well-documented pipelines can bridge the gap between learning and research, empowering users to develop and adapt reproducible bioinformatics tools for diverse metagenomics applications.
Availability of data and materials
The tutorial TaxoFlow is hosted under the domain https://taxoflow.work/. The source code for the tutorial is available at https://github.com/jeffe107/taxoflow_tutorial.
Acknowledgments
JYG specially thanks the Federal Commission for Scholarships for Foreign Students (FCS) for their support through the Swiss Government Excellence Scholarship. We also acknowledge Geraldine Van der Auwera from the Nextflow Training Team for her valuable contribution to conceive the idea of the tutorial and for her insightful feedback to implement it.
References
- Kim, N.; et al. Genome-resolved metagenomics: a game changer for microbiome medicine. Exp. Mol. Med. 2024, 56, 1501–1512. [Google Scholar] [CrossRef]
- Baker, M. 1,500 scientists lift the lid on reproducibility. Nature 2016, 533, 452–454. [Google Scholar] [CrossRef]
- Yang, C.; et al. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Computational and Structural Biotechnology Journal 2021, vol. 19, 6301–6314. [Google Scholar] [CrossRef]
- Tommaso, P. D.; et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Mölder, F.; et al. Sustainable data analysis with Snakemake. Research 2021, 10, 33. [Google Scholar]
- Roach, M. J.; et al. Ten simple rules and a template for creating workflows-as-applications. PLOS Comput. Biol. 2022, 18, e1010705. [Google Scholar] [CrossRef] [PubMed]
- Wratten, L.; Wilm, A.; Göke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat. Methods 2021, 18, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A. E.; et al. Design considerations for workflow management systems use in production genomics research and the clinic. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Navgire, G. S.; et al. Analysis and Interpretation of metagenomics data: an approach. Biol. Proced. Online 2022, 24, 1–22. [Google Scholar] [CrossRef]
- Edwin, N. R.; Fitzpatrick, A. H.; Brennan, F.; Abram, F.; O’Sullivan, O. An in-depth evaluation of metagenomic classifiers for soil microbiomes. Environ. Microbiome 2024, 19, 19. [Google Scholar] [CrossRef]
- Wajid, B.; et al. Music of metagenomics—a review of its applications, analysis pipeline, and associated tools. Funct. Integr. Genomics 2022, 22, 3–26. [Google Scholar] [CrossRef]
- Liu, Y.-X.; et al. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef]
- Wood, D. E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wood, D. E.; Salzberg, S. L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Breitwieser, F. P.; Thielen, P.; Salzberg, S. L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, e104. [Google Scholar] [CrossRef]
- Blanco-Míguez, A.; et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023, 1–12. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K. L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Ounit, R.; Wanamaker, S.; Close, T. J.; Lonardi, S. CLARK: fast and accurate classification of metagenomic and genomic sequences using discriminative k-mers. BMC Genomics 2015, 16, 236. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F. P.; Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- Milanese, A.; et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Timilsina, M.; Chundru, D.; Pradhan, A. K.; Blaustein, R. A.; Ghanem, M. Benchmarking Metagenomic Pipelines for the Detection of Foodborne Pathogens in Simulated Microbial Communities. J. Food Prot. 2025, 88, 100583. [Google Scholar] [CrossRef] [PubMed]
- Pusadkar, V.; Azad, R. K. Benchmarking Metagenomic Classifiers on Simulated Ancient and Modern Metagenomic Data. Microorganisms 2023, 11, 2478. [Google Scholar] [CrossRef] [PubMed]
- Stamouli, S.; et al. nf-core/taxprofiler: highly parallelised and flexible pipeline for metagenomic taxonomic classification and profiling. Preprint 2023. [Google Scholar] [CrossRef]
- Borry, M. Source code for: kraken-nf - Simple Kraken2 Nextflow pipeline. 2024. Available online: https://github.com/maxibor/kraken-nf.
- EPI2ME. Source code for: wf-metagenomics - Metagenomic classification of long-read sequencing data. 2025. Available online: https://github.com/epi2me-labs/wf-metagenomics.
- Angelov, A. Source code for: nxf-kraken2 - A simple nextflow pipeline for running Kraken2 and bracken in a docker container. 2025. Available online: https://github.com/angelovangel/nxf-kraken2.
- Terrón-Camero, L. C.; Gordillo-González, F.; Salas-Espejo, E.; Andrés-León, E. Comparison of Metagenomics and Metatranscriptomics Tools: A Guide to Making the Right Choice. Genes 2022, 13, 2280. [Google Scholar] [CrossRef] [PubMed]
- Petit, R. A.; Read, T. D. Bactopia: a Flexible Pipeline for Complete Analysis of Bacterial Genomes. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Langer, B. E.; et al. Empowering bioinformatics communities with Nextflow and nf-core. Genome Biol. 2025, 26, 228. [Google Scholar] [CrossRef]
- Zirión-Martínez, C.; et al. A Data Carpentry- Style Metagenomics Workshop. J. Open Source Educ. 2024, 7, 209. [Google Scholar] [CrossRef]
- Kruchten, A. E. A Curricular Bioinformatics Approach to Teaching Undergraduates to Analyze Metagenomic Datasets Using R. Front. Microbiol. 2020, 11. [Google Scholar]
- Telatin, A. Source code for: nextflow-example - A simple DSL2 workflow: tutorial. 2022. Available online: https://github.com/telatin/nextflow-example.
- Lu, J.; et al. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Ondov, B. D.; Bergman, N. H.; Phillippy, A. M. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 2011, 12, 1–10. [Google Scholar] [CrossRef]
- Kadri, S.; Sboner, A.; Sigaras, A.; Roy, S. Containers in Bioinformatics: Applications, Practical Considerations, and Best Practices in Molecular Pathology. J. Mol. Diagn 2022, 24, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Barker, M.; et al. Introducing the FAIR Principles for research software. Sci. Data 2022, 9, 622. [Google Scholar] [CrossRef]
- Noble, W. S. A Quick Guide to Organizing Computational Biology Projects. PLOS Comput. Biol. 2009, 5, e1000424. [Google Scholar] [CrossRef] [PubMed]
- Okie, J. G.; et al. Genomic adaptations in information processing underpin trophic strategy in a whole-ecosystem nutrient enrichment experiment. eLife 2020, 9, e49816. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; et al. The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. GigaScience 2012, 1, 2047-217X-1–7. [Google Scholar] [CrossRef]
- McMurdie, P. J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the educational pipeline built through TaxoFlow.
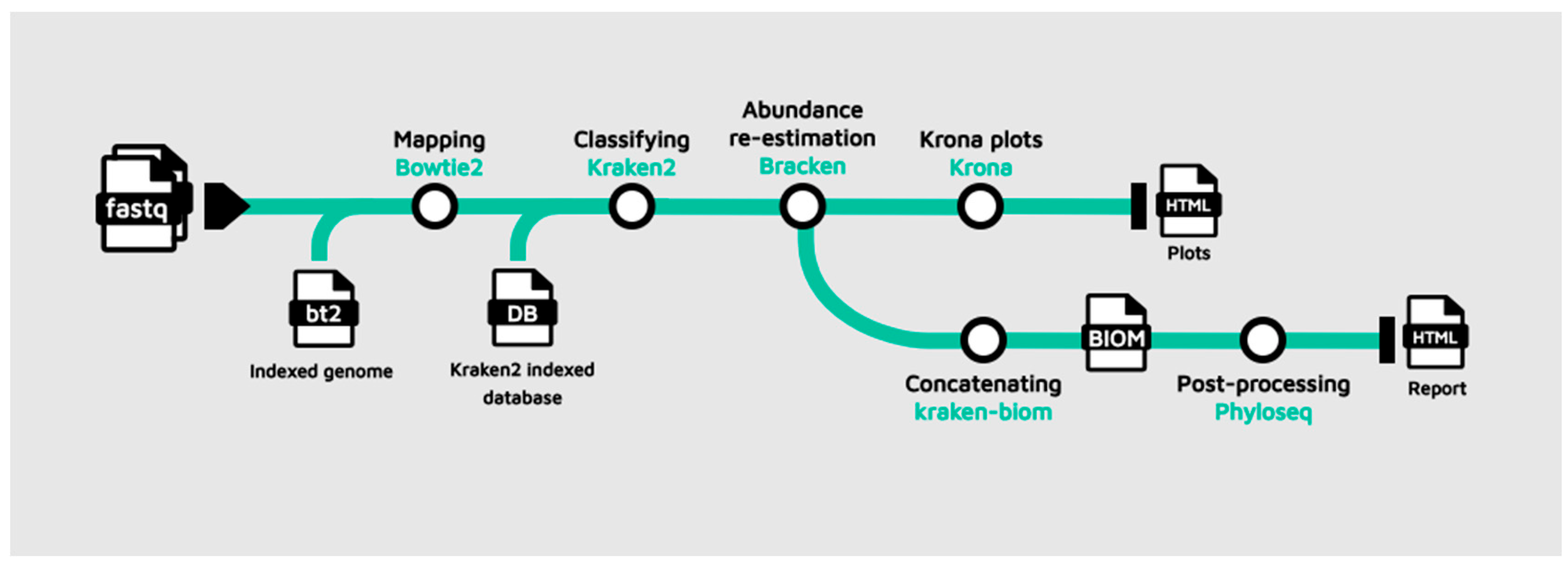
Figure 2.
a Snapshot of the Krona plot obtained through the single-sample execution of the pipeline during the Part 2 of the tutorial. b Absolute abundance plot encompassed by the final report depicting the genera present within the samples analyzed during the Part 3 execution of the tutorial.
Figure 2.
a Snapshot of the Krona plot obtained through the single-sample execution of the pipeline during the Part 2 of the tutorial. b Absolute abundance plot encompassed by the final report depicting the genera present within the samples analyzed during the Part 3 execution of the tutorial.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



