Submitted:
21 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Allogeneic (“off-the-shelf”) CAR-T cells promise faster treatment access, standardized manufacturing, and scalable supply compared with autologous products, but they face a core biological constraint: the recipient immune system recognizes allogeneic CAR-T as a graft and eliminates it, while residual donor T-cell receptor (TCR) activity can cause graft-versus-host disease (GvHD). Clinical progress over the last several years has validated feasibility, with encouraging response rates in B-cell malignancies and generally manageable safety profiles, yet persistence and repeat dosing remain limited by host-versus-graft (HvG) immunity. For example, a peer-reviewed report of allogeneic anti-CD19 CAR-T in LBCL described measurable expansion, persistence up to months, and objective responses in heavily pretreated patients. This mini-review focuses on an increasingly central barrier: humoral rejection (donor-specific antibodies, complement, and Fcγ receptor–dependent effector mechanisms), alongside cellular rejection (host T cells, NK cells, and macrophages) and the “missing-self” problem created by HLA class I ablation. We summarize the current engineering playbook—TCR disruption to prevent GvHD, HLA editing and inhibitory ligand strategies to evade cellular HvG, and emerging concepts to resist antibody-mediated killing—and propose a practical design logic for next-generation allogeneic CAR-T: multi-layer immune evasion without forfeiting antitumor function or manufacturability.
Keywords:
allogeneic CAR-T
; off-the-shelf
; host-versus-graft
; donor-specific antibodies
; complement
; ADCC
; Fcγ receptor
; B2M
; HLA-E
; missing self
; TCR knockout
; CD52 knockout
; hypoimmune engineering
1. Introduction
Allogeneic CAR-T aims to decouple CAR manufacturing from the individual patient by using healthy-donor (or banked) starting material to generate “ready-to-infuse” products. The appeal is compelling: shorter vein-to-vein time, reduced failure rates from poor patient T-cell quality, batch consistency, and the possibility of lower cost through scale. The challenge is equally fundamental: unlike autologous CAR-T, allogeneic CAR-T must survive bidirectional alloreactivity:
- GvHD risk: donor T cells recognize host alloantigens via the endogenous TCR unless the TCR is eliminated or functionally silenced.
- HvG rejection: recipient immunity (T cells, NK cells, macrophages, antibodies/complement) removes the infused allogeneic cells.
The field has moved from “can it be done?” to “how do we systematically manage rejection, persistence, and repeat dosing?” Recent reviews emphasize that humoral immunity—especially donor-specific antibodies (DSA) and Fc-/complement-mediated killing—may become a dominant limiter as products broaden beyond CD19 (where B-cell aplasia can reduce de novo antibody formation) and as repeat dosing is pursued.
2. Immune Rejection Mechanisms in Allogeneic CAR-T
2.1. Cellular HvG: Host T Cells, NK Cells, and Macrophages
Recipient T cells can recognize mismatched HLA on donor CAR-T and eliminate the graft. A common mitigation is HLA class I disruption (e.g., B2M knockout) to reduce CD8 T-cell recognition. However, this creates a predictable counter-problem: NK “missing-self” recognition of HLA-deficient cells.
Macrophage-mediated clearance is also increasingly recognized as relevant for off-the-shelf cell therapies, particularly when “don’t eat me” signals (e.g., CD47) are not present or when opsonizing antibodies appear.
2.2. Humoral HvG: DSA, Complement, and Fcγ Receptor Effector Killing
Figure 1 highlights a pathway that is sometimes underweighted in CAR-T discussions: recipient CD4 T-cell help to B cells, promoting DSA generation against donor antigens (HLA and non-HLA), which can then mediate rejection via:
- Complement-dependent cytotoxicity (CDC) initiated by C1q binding to antibody-decorated cells, culminating in membrane attack complex (MAC) formation.
- Fcγ receptor–dependent effector mechanisms, including antibody-dependent cellular cytotoxicity (ADCC) by NK cells and antibody-dependent cellular phagocytosis (ADCP) by macrophages.
These mechanisms are well-characterized in antibody-mediated rejection paradigms and have direct conceptual applicability to allogeneic cell therapeutics. Importantly, antibodies can be pre-existing (sensitized recipients) or treatment-induced, including antibodies against synthetic constructs (e.g., CAR binders) or process-related antigens.
2.3. The “B2M Paradox”: Evading T Cells Can Invite NK Killing
A major design tension is explicitly captured in Figure 1: MHC-I ablation can reduce host T-cell recognition but increase NK cytotoxicity through missing-self responses. This tradeoff is a central reason why current “stealth” strategies rarely rely on single edits and increasingly adopt combinatorial immune evasion.
3. Engineering Strategies: The Contemporary Design Playbook
3.1. Preventing GvHD: Eliminating Donor TCR Activity
Most leading allogeneic CAR-T platforms incorporate TCR disruption (e.g., TRAC knockout) to prevent GvHD. Clinical studies with genome-edited “universal” CAR19 T cells have demonstrated feasibility and provided longer-term outcome insights, often in treatment paradigms where allogeneic CAR-T serves as a bridge to transplantation in selected settings.
3.2. Extending Persistence: Managing Cellular HvG Without Losing Fitness
Common approaches include:
- HLA class I reduction (e.g., B2M knockout) to reduce host CD8 recognition, balanced against NK risk.
- NK-evasion add-ons, such as expression of minimally polymorphic inhibitory ligands (e.g., HLA-E) that can engage inhibitory NK receptors and suppress missing-self killing.
- Macrophage-evasion elements (e.g., CD47), often discussed as part of “hypoimmune” designs that aim to address both innate and adaptive rejection.
A key practical point: each additional edit introduces manufacturing complexity and potential fitness costs. Recent work also suggests that engineering choices that reduce immune recognition (e.g., adhesion or HLA modulation) can have non-obvious impacts on trafficking, synapse formation, and antitumor potency, underscoring the need for head-to-head functional benchmarking.
3.3. Countering Humoral Rejection: An Emerging Frontier
Humoral rejection is moving from theoretical risk to design driver, especially for products targeting antigens that do not delete host B cells. A notable engineering concept is to protect graft cells from antibody-mediated killing by shielding antibody Fc domains from complement and Fcγ receptor engagement. One published strategy is CD64 (Fc receptor) overexpression on therapeutic cells to capture monomeric IgG Fc and prevent CDC/ADCC/ADCP, with demonstration across multiple cell types including CAR-T in experimental systems.
While such approaches remain early, they directly map onto the DSA → CDC/ADCC pathways depicted in Figure 1 and represent a plausible route to improved persistence and repeat dosing in sensitized hosts.
3.4. Lymphodepletion as an Enabling Layer
Clinical allogeneic CAR-T regimens frequently rely on lymphodepletion to transiently suppress HvG responses and enable initial expansion. Intensified lymphodepletion has been explicitly discussed as a means to mitigate rejection and improve expansion/persistence in some allogeneic programs. However, lymphodepletion alone is unlikely to solve durable persistence once adaptive immunity (including antibodies) develops—hence the increasing emphasis on multi-layer immune evasion.
4. Clinical Landscape: Where the Field Stands
Peer-reviewed studies and late-stage updates collectively support three conclusions:
- Feasibility and safety: Multiple genome-edited allogeneic CAR-T products have demonstrated manageable safety profiles and antitumor activity in heavily pretreated patients.
- Persistence is variable and often limited: Detectable expansion is common, but long-term persistence frequently falls short of autologous benchmarks, consistent with HvG rejection biology.
- Durability can still be clinically meaningful: Even time-limited persistence may be sufficient to induce deep remissions in some contexts (e.g., as a bridge to transplant), as reflected in longer-term outcome analyses of universal CAR19 approaches.
A concrete example: a 2025 peer-reviewed report of anti-CD19 allogeneic CAR-T products in LBCL described objective responses (including complete responses) and persistence up to ~4 months in some patients, with hematologic toxicities prominent among adverse events. This pattern—activity with constrained persistence—fits the mechanistic picture in Figure 1.
5. A practical Framework for “Next” Allogeneic CAR-T Design
A useful way to operationalize this checklist is to treat allogeneic CAR-T design as a layered threat-modeling exercise in which each immune effector arm (host T cells, NK cells, macrophages, and antibodies/complement) is addressed with a dedicated mitigation that is then stress-tested for “cross-coupling” liabilities (Table 1). For example, reducing HLA class I to evade host CD8+ T-cell recognition predictably increases susceptibility to NK missing-self responses, which in turn necessitates an inhibitory ligand strategy (e.g., HLA-E) or alternative “hypoimmune” configurations; similarly, measures that extend residence time in vivo may increase the probability of de novo DSA formation, shifting the dominant rejection mode toward complement and FcγR-mediated mechanisms that are not solved by TCR/B2M edits alone. In this framing, the most informative preclinical package is not a single potency readout but a matrix of assays that explicitly probes each rejection axis—CDC/ADCC/ADCP in the presence of human serum and FcγR+ effectors, NK cytotoxicity under HLA perturbations, and T-cell alloreactivity under representative HLA mismatch scenarios—followed by a manufacturability assessment (edit burden, product fitness, stability after cryopreservation) to ensure that added immune-evasion layers do not erode antitumor function or scalability.
6. Manufacturing and Translational Considerations
Allogeneic CAR-T adds scale advantages but increases the importance of:
- Donor selection and banking (consistent phenotype, viral status, HLA considerations).
- Genome editing QC (on-/off-target assessment, karyotype/genomic stability where relevant, residual nuclease, product heterogeneity).
- Batch release comparability across large runs and extended storage (cryopreservation stability).
- Immunogenicity risk management, especially for non-CD19 targets where B cells remain intact and DSA/anti-CAR antibodies can emerge.
Given growing evidence that antibodies can compromise persistence in allogeneic cell therapeutics, antibody monitoring (pre-existing sensitization, treatment-induced antibodies) is likely to become routine in trial designs that aim for repeat dosing or prolonged exposure.
7. Conclusions and Outlook
Allogeneic CAR-T has crossed the feasibility threshold: multiple platforms can generate potent products with acceptable safety and clinically meaningful responses. The major remaining constraint is immune rejection, increasingly understood as a layered problem that includes cellular HvG and—critically—humoral mechanisms (DSA → complement/FcγR effector killing). The most credible path to durable, broadly applicable off-the-shelf CAR-T is not a single “magic edit,” but combinatorial immune evasion with explicit management of the B2M paradox (avoid T-cell recognition without triggering NK killing), coupled to strategies that address antibody-mediated killing for targets that do not remove host B cells. Early proof-of-concept for antibody-resistant engineering (e.g., Fc shielding via CD64-based approaches) suggests this frontier is tractable and likely to shape the next wave of allogeneic CAR-T development.
Funding
This work was funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (project number FZSM-2025-0001).
Conflicts of Interest
The author declares no conflicts of interest.
References
- Delpire, B.; van Loon, E.; Naesens, M. The Role of Fc Gamma Receptors in Antibody-Mediated Rejection of Kidney Transplants. Transplant International 2022, 35. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Fang, Y.; Niu, S.; Chen, Y.; Lyu, Z.; Yang, L. Managing Allorejection in Off-the-Shelf CAR-Engineered Cell Therapies. Molecular Therapy 2025, 33, 2368–2390. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, R.; Jain, N.; Maus, M. v; Boissel, N.; Graham, C.; Jozwik, A.; Yallop, D.; Konopleva, M.; Frigault, M.J.; Teshima, T.; et al. UCART19, a First-in-Class Allogeneic Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy for Adults with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukaemia (CALM): A Phase 1, Dose-Escalation Trial. Lancet Haematol 2022, 9, e833–e843. [Google Scholar] [CrossRef] [PubMed]
- Guardo, D.; Mishra, A.K.; Rashed, H.; Gilmour, K.; Adams, S.; Pinner, D.; Sauer, M.; Vora, A.; Veys, P.; Pavasovic, V.; et al. Long-Term Outcomes of Genome-Edited “Universal” CAR19 T Cells for Relapsed/Refractory B-ALL at a Single Pediatric Center. Blood Adv 2025, 9, 4750–4754. [Google Scholar] [CrossRef] [PubMed]
- Locke, F.L.; Munoz, J.L.; Tees, M.T.; Lekakis, L.J.; de Vos, S.; Nath, R.; Stevens, D.A.; Malik, S.A.; Shouse, G.P.; Hamadani, M.; et al. Allogeneic Chimeric Antigen Receptor T-Cell Products Cemacabtagene Ansegedleucel/ALLO-501 in Relapsed/Refractory Large B-Cell Lymphoma: Phase I Experience From the ALPHA2/ALPHA Clinical Studies. Journal of Clinical Oncology 2025, 43, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.-A.; Clegg, D.O.; et al. HLA-E-Expressing Pluripotent Stem Cells Escape Allogeneic Responses and Lysis by NK Cells. Nat Biotechnol 2017, 35, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Gravina, A.; Tediashvili, G.; Rajalingam, R.; Quandt, Z.; Deisenroth, C.; Schrepfer, S.; Deuse, T. Protection of Cell Therapeutics from Antibody-Mediated Killing by CD64 Overexpression. Nat Biotechnol 2023, 41, 717–727. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Humoral and innate mechanisms of allogeneic CAR-T rejection. Recipient CD4+ T-cell help promotes B-cell processing of donor-derived antigens and generation of donor-specific antibodies (DSA). DSAs mediate allogeneic CAR-T rejection through (i) complement-dependent cytotoxicity initiated by C1q binding and culminating in membrane attack complex (MAC) formation, and (ii) Fcγ receptor–dependent effector mechanisms (e.g., macrophage and NK-cell mediated ADCC/ADCP). This also highlights the “missing-self” vulnerability: MHC-I ablation on allogeneic CAR-T reduces T-cell recognition but can increase NK-cell cytotoxicity, creating a design tradeoff that motivates combinatorial immune-evasion engineering.
Figure 1.
Humoral and innate mechanisms of allogeneic CAR-T rejection. Recipient CD4+ T-cell help promotes B-cell processing of donor-derived antigens and generation of donor-specific antibodies (DSA). DSAs mediate allogeneic CAR-T rejection through (i) complement-dependent cytotoxicity initiated by C1q binding and culminating in membrane attack complex (MAC) formation, and (ii) Fcγ receptor–dependent effector mechanisms (e.g., macrophage and NK-cell mediated ADCC/ADCP). This also highlights the “missing-self” vulnerability: MHC-I ablation on allogeneic CAR-T reduces T-cell recognition but can increase NK-cell cytotoxicity, creating a design tradeoff that motivates combinatorial immune-evasion engineering.
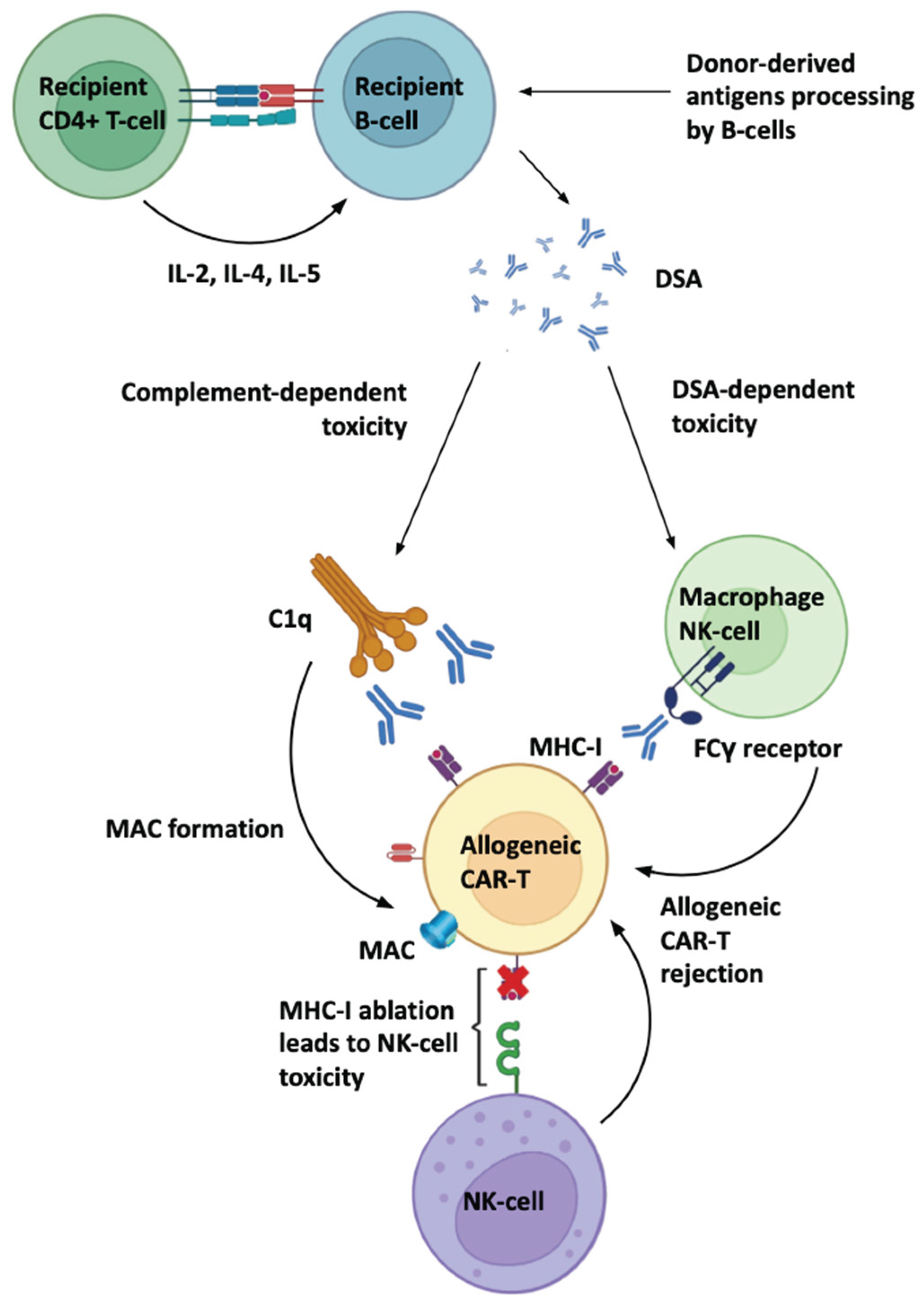

Table 1.
Dominant rejection mechanisms and representative mitigation strategies.
| Barrier (Figure 1 mapping) | Mechanistic driver | Typical mitigation concept |
|---|---|---|
| GvHD | Donor TCR recognizes host alloantigens | TRAC/TCR disruption; TCRαβ depletion (platform-dependent) |
| Host T-cell rejection | Recipient T cells recognize donor HLA | HLA class I reduction (e.g., B2M KO) ± adjunct edits |
| NK rejection after HLA loss (“missing self”) | NK activation against HLA-deficient graft | Add inhibitory ligands (e.g., HLA-E); “hypoimmune” combinations |
| Macrophage clearance | Innate phagocytosis, especially with opsonization | CD47-based approaches; innate-evasion layers |
| DSA-mediated CDC | Antibody → C1q → MAC | Reduce immunogenic targets; Fc/complement shielding concepts |
| DSA-mediated ADCC/ADCP | FcγR effector killing/phagocytosis | Fc shielding or decoy strategies; control of anti-CAR/anti-HLA responses |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



