
Submitted:
19 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Neurons have exceptionally high energy demands, sustained by thousands to millions of mitochondria per cell. Each mitochondrion depends on the integrity of its mitochondrial DNA (mtDNA), which encodes essential electron transport chain (ETC) subunits required for oxidative phosphorylation (OXPHOS). The continuous, high-level production of ATP through OXPHOS generates reactive oxygen species (ROS), posing a significant threat to the highly exposed mtDNA. To counter these insults, neurons rely on base excision repair (BER), the principal mechanism for removing oxidative and other small, non-bulky base lesions in nuclear and mtDNA. BER involves a coordinated enzymatic pathway that excises damaged bases and restores DNA integrity, helping maintain mitochondrial genome stability, which is vital for neuronal bioenergetics and survival. When mitochondrial BER is impaired, mtDNA becomes unstable, leading to ETC dysfunction and a self-perpetuating cycle of bioenergetic failure, elevated ROS levels, and continued mtDNA damage. Damaged mtDNA fragments can escape into the cytosol or extracellular space, where they act as damage-associated molecular patterns (DAMPs) that activate innate immune pathways and inflammasome complexes. Chronic activation of these pathways drives sustained neuroinflammation, exacerbating mitochondrial dysfunction and neuronal loss, and functionally links genome instability to innate immune signaling in neurodegenerative diseases. This review summarizes recent advancements in understanding how BER preserves mitochondrial genome stability, affects neuronal health when dysfunctional, and contributes to damage-driven neuroinflammation and neurodegenerative disease progression. We also explore emerging therapeutic strategies to enhance mtDNA repair, optimize its mitochondrial environment, and modulate neuroimmune pathways to counteract neurodegeneration.
Keywords:
mitochondrial DNA
; damage associated molecular patterns
; neuroinflammation
; neurodegeneration
; base excision repair
1. Introduction
Cells of the central nervous system are among the most energy-intensive in the human body, relying on a vast network of mitochondria to sustain the high ATP output required for synaptic transmission and cellular maintenance [1,2]. The essential role of mitochondrial DNA (mtDNA) in encoding crucial subunits of the electron transport chain (ETC) directly connects genome integrity to neuronal energy homeostasis [3]. Unlike nuclear DNA, mtDNA lacks protective histones and is located near sites of oxidative phosphorylation (OXPHOS), which continually exposes it to reactive oxygen species (ROS) produced during respiration [3,4,5,6]. Although many oxidative lesions are efficiently repaired or tolerated with minimal impact, persistent or unrepaired mutagenic damage can compromise ETC function and reduce ATP production when a detrimental threshold is reached, and this decline in bioenergetics activates cellular stress responses that can create a self-perpetuating cycle of mitochondrial dysfunction, elevated ROS generation, and progressive mtDNA instability [7,8,9,10,11,12,13]. The long life and post-mitotic nature of neurons prevent the dilution or replacement of damaged mitochondria through cell division. As a result, mtDNA lesions accumulate over time and progressively compromise oxidative phosphorylation, promoting neurodegeneration in disorders such as Alzheimer's disease and Parkinson's disease [14,15,16,17,18].
While mitochondria possess additional quality control mechanisms, such as rapid mtDNA turnover, dynamic fission and fusion remodeling, and targeted mitophagy, there is ongoing debate over the necessity of mtDNA repair [4,19,20]. Some groups propose that these quality control systems can compensate for mtDNA damage by eliminating or diluting dysfunctional genomes, reducing the need for robust DNA repair pathways, such as base excision repair (BER) [5,21]. This viewpoint suggests that rapid mitochondrial turnover may limit the long-term consequences of mtDNA lesions.
However, others argue that exclusive dependence on degradation and turnover is energetically expensive and ultimately unsustainable, particularly for long-lived, post-mitotic neurons. Constant mitophagy and biogenesis would present a significant metabolic burden on cells and risk depleting healthy mitochondrial pools [22]. Therefore, a finely tuned balance is required, in which active repair provides a first-line defense against oxidative damage. Among the known cellular DNA repair systems, BER is the only fully characterized and universally accepted functional DNA repair pathway within mitochondria, directly correcting oxidative and other non-bulky lesions, thereby preserving mtDNA integrity and supporting cellular longevity [23,24,25,26,27,28,29,30,31,32,33]. Damage that escapes repair can give rise to not only genomic instability but also to innate immune activation. Notably, unstable or fragmented mtDNA can be released into the cytosol or extracellular milieu, acting as damage-associated molecular patterns (DAMPs) that activate innate immune signaling pathways, such as the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) pathway and inflammasomes [34]. Chronic neuroinflammation driven by these signals exacerbates neuronal dysfunction and accelerates neurodegenerative processes.
Our goal in this review is to provide a comprehensive overview of recent advances in understanding the role of BER in maintaining mitochondrial genome integrity in neurons. We examine current perspectives on how BER dysfunction contributes to genetic instability, bioenergetic deficits, and neuroinflammation, and we highlight emerging therapeutic strategies targeting DNA repair and neuroimmune pathways. Finally, we identify outstanding questions and delineate promising directions for future research to interrupt or slow the progression of neurodegeneration.
2. Mitochondrial DNA: Structure, Function, and Instability in Neurons
As a relic of their bacterial ancestry, mitochondria retain a separate genome from the nuclear genome [35,36,37]. Human mtDNA is a double-stranded, circular molecule of approximately 16.5 kb that encodes 13 core subunits of respiratory complexes I, III, IV, and V (ATP synthase), along with two rRNAs and 22 tRNAs, supporting a dedicated mitochondrial translation system that uses a genetic code distinct from the nuclear genome [38,39,40,41] (Figure 1). Despite its essential role, mtDNA contributes only a small fraction of the mitochondrial proteome; more than 1,500 additional mitochondrial proteins, including other OXPHOS components and genome maintenance factors, are encoded by nuclear DNA, synthesized in the cytosol, and imported into mitochondria via several distinct pathways, including the presequence pathway for proteins bearing N-terminal mitochondrial targeting sequences, the carrier pathway for multi-pass inner membrane proteins, and specialized routes for β-barrel and intermembrane space proteins [42,43].
Our typical view of DNA organization is shaped mainly by what is observed in the nucleus, where DNA is tightly wrapped around histone octamers to form nucleosomes. These repeating units occur hundreds of thousands of times along each chromosome, creating dense protein shielding and highly ordered chromatin architecture [44,45]. However, mtDNA is packaged in a fundamentally different manner. Instead of nucleosomes, mtDNA is organized into protein-DNA complexes, called nucleoids, which lack histones [3,46,47,48,49]. Mammalian cells contain hundreds to approximately 100,000 copies of mtDNA, with neurons typically enriched in mitochondria [2,50,51]. Even so, total mtDNA copy number per neuron typically ranges from only a few thousand, which is lower than the estimated number of mitochondria per neuron [52]. This likely reflects the heterogeneous distribution of nucleoids, as individual nucleoids typically contain only one or a few mtDNA molecules, and, as a recent preprint reported [53], many axonal or distal neuronal mitochondria appear to lack detectable mtDNA, resulting in regions of the mitochondrial network that are effectively genome-poor [53,54,55,56]. This apparent mismatch highlights the decentralized, multi-copy organization of mtDNA, which contrasts sharply with chromosome-wide compaction in the nucleus and provides a more dynamic framework for genome distribution across the mitochondrial network [56].
Each mitochondrial nucleoid forms a compact, highly regulated, and dynamic complex that contains approximately 37 distinct proteins [57]. The central architectural component is mitochondrial transcription factor A (TFAM), which coats and bends mtDNA to promote tight packaging while simultaneously regulating transcription initiation [58,59,60]. Other key mitochondrial replisome proteins are enriched within nucleoids, including Twinkle, the main mtDNA helicase; DNA polymerase gamma (POLG), which synthesizes new mtDNA strands, and the mitochondrial single-stranded DNA-binding protein (mtSSB), which stabilizes single-stranded DNA and promotes processive replication [46]. Collectively, these proteins form the nucleoid's functional hub, mediating essential genome maintenance and expression.
Nucleoids are typically tethered to the inner mitochondrial membrane, positioning replication and transcription machinery adjacent to the ETC [61] (Figure 1). This localization, while enhancing the translation and membrane integration of OXPHOS proteins, also increases the exposure of mtDNA to an oxidative environment. The inner membrane is the leading site of OXPHOS and the primary source of ROS, making mtDNA much more prone to oxidative damage than nuclear DNA [6,62]. The absence of histones and an open DNA configuration further increases exposure to ROS, lipid peroxidation products, and environmental toxins [6]. Compared to the nucleus, the protective DNA repair activities in mitochondria are limited: while BER is well established, the overall repertoire of glycosylases and other downstream repair enzymes is restricted, and entire pathways such as nucleotide excision repair and mismatch repair are absent or incomplete [25,63,64,65].
The consequences of compromised mtDNA maintenance manifest clinically. Pathogenic mutations in mtDNA underlie a range of mitochondrial diseases, such as Leber hereditary optic neuropathy (LHON), mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), myoclonic epilepsy associated with ragged red fibers (MERRF), and neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP), which often present with severe neurological symptoms owing to the nervous system's heavy reliance on OXPHOS [66]. These pathogenic mutations frequently arise spontaneously during mtDNA replication or are maternally inherited [67,68,69,70]. Many other mitochondrial syndromes arise from mutations in nuclear-encoded mitochondrial genes, such as POLG [71]. As POLG's activities span replication, repair, and disease pathogenicity, our review will continually highlight its diverse roles across mitochondrial genome maintenance pathways. Disease-causing mutations in nuclear-encoded genes, such as POLG, are typically transmitted in an autosomal dominant or recessive pattern, resulting in defective mtDNA replication and repair following import of the abnormal polymerase into mitochondria [71,72].
In contrast to these inherited disorders, mtDNA damage most relevant to aging and neurodegeneration, especially those affecting post-mitotic neurons, is accumulated gradually over a lifetime [4,73,74,75]. Chronic exposure to ROS can cause mutagenic lesions and lead to deletions in or loss of mitochondrial genes [64,76]. These cumulative changes can lead to progressive mtDNA instability, which, instead of manifesting as rare inherited syndromes, results in persistent metabolic failure leading to neurodegeneration. Neuronal tissue is therefore acutely sensitive to mitochondrial dysfunction stemming from both inherited and acquired mtDNA instability. Thus, maintaining mtDNA integrity is essential for long-term neuronal survival. Among all genome maintenance mechanisms, BER is the key process safeguarding the mitochondrial genome against the damaging effects of oxidative stress. The degree to which BER preserves mtDNA stability and neuronal viability underlies much of the neurodegenerative risk discussed in subsequent sections.
3. Base Excision Repair in Mitochondria: Mechanisms and Enzymes
The persistent oxidative environment, associated with proximity to the ETC, gives rise to a broad spectrum of DNA lesions, including oxidative base modifications, deamination, and alkylation [77]. If left unrepaired, this damage disrupts essential mitochondrial processes and compromises cellular homeostasis [78,79]. To protect against these threats, mitochondria utilize BER, an evolutionarily conserved pathway that identifies chemically altered bases and restores DNA integrity by removing and replacing them [25].
3.1. Lesion Detection and Removal
The first and defining step of BER is damage recognition [25,78,80,81]. Because oxidative stress generates a wide variety of chemically modified bases, mitochondria rely on a set of lesion-specific DNA glycosylases to detect these abnormalities with high sensitivity [25,82]. Glycosylases continually scan the genome, probing each base for subtle structural distortions or the loss of chemical functionality [81,82,83]. Once a damaged base is identified, a DNA glycosylase recognizes and excises the lesion, cleaving the N-glycosidic bond to remove the altered base and generate an abasic site; the precise recognition and excision mechanism varies among individual BER glycosylases, which can use distinct lesion-flipping, active-site, and catalytic strategies [81,82,84]. These actions commit the lesion to repair and hand the substrate off to downstream enzymes that complete the restoration of the DNA backbone. Of the eleven mammalian DNA glycosylases, at least seven have confirmed or strongly supported mitochondrial localization and known function within the organelle [25]. These seven mtDNA glycosylases are distributed between all four mammalian glycosylase superfamilies: the uracil-DNA glycosylases (UDG), helix-hairpin-helix (HhH) superfamily, formamidopyrimidine (Fpg)/endonuclease eight (Nei) superfamily, and the alkyladenine DNA glycosylase (AAG) superfamily [25].
The UDG family includes three glycosylases that remove uracil arising from cytosine deamination or misincorporation [85]. The key mitochondrial member of this family is uracil DNA glycosylase isoform 1 (UNG1), which excises uracil and 5-fluorouracil generated by cytosine deamination or deoxyuridine triphosphate (dUTP) misincorporation, with high efficiency on single-stranded DNA and replication intermediates, making it the primary mitochondrial uracil-removal enzyme [85,86].
AAG, also known as methyl purine glycosylase (MPG), is the only member of this mammalian DNA glycosylase superfamily [87]. AAG has been reported to localize to mitochondria via an N-terminal targeting sequence; however, the extent of its mitochondrial contribution may vary among cell types [25,88]. AAG recognizes and removes a broad spectrum of alkylated and deaminated bases, including 3-methyladenine, 7-methylguanine, hypoxanthine, and etheno-adducts, in both single- and double-stranded DNA, providing defense against alkylation and lipid peroxidation-derived lesions within mtDNA [88,89].
The helix-hairpin-helix (HhH) superfamily comprises enzymes that eliminate a wide range of oxidative lesions [90,91]. Among the four members of this superfamily, three localize within the mitochondria: endonuclease three (Nth) like DNA glycosylase 1 (NTHL1), 8-oxoguanine glycosylase (OGG1), and the MutY DNA glycosylase (MUTYH) [25]. NTHL1 and OGG1 remove oxidized pyrimidines and 8-oxoguanine (8-oxoG), respectively [92,93]. NTHL1 removes oxidized pyrimidines, such as thymine glycol, 5-hydroxycytosine, and 5-hydroxyuracil, through its bifunctional glycosylase-lyase activity, thereby counteracting oxidative pyrimidine damage in mtDNA [92]. OGG1 primarily excises 8-oxoG and the related purine lesion FapyG, preventing mutagenic G-to-T transversions that would otherwise accumulate under mitochondrial oxidative stress [93]. MUTYH removes adenine mispaired with 8-oxoG and other oxidized purines, forming an essential component of the OGG1-MUTYH cooperative system that prevents G-to-T mutations in the mitochondrial genome [94].
Lastly, although humans lack a direct ortholog of bacterial Fpg, within the Fpg/Nei superfamily they express three Nei-like (NEIL) enzymes, NEIL1, NEIL2, and NEIL3, which possess distinct subcellular localizations and substrate profiles [95,96,97]. Notably, NEIL1 and NEIL2 function in both mitochondria and the nucleus, whereas studies thus far show that NEIL3 localization and functions are restricted to the nucleus [25]. NEIL1 excises a wide array of replication-blocking oxidative lesions, including thymine glycol (Tg), FapyG, FapyA, and hydantoin derivatives such as spiroiminodihydantoin (Sp) and guanidinohydantoin (Gh), with strong activity on single-stranded and forked DNA structures characteristic of mitochondrial replication intermediates [95,98,99,100,101]. NEIL2 preferentially targets oxidized pyrimidines, such as 5-hydroxyuracil and cytosine-derived lesions, and functions efficiently on bubble and transcription-associated DNA structures relevant to mitochondrial gene expression [96,101,102].
The monofunctional glycosylases that translocate to mitochondria (AAG, MUTYH, and UNG1) possess glycosylase activity only and generate abasic sites [24]. In contrast, the bifunctional glycosylases (NTHL1, OGG1, NEIL1, NEIL2) have intrinsic lyase activity, enabling them to cleave the DNA backbone at abasic sites and generate 3′-blocking ends [24]. After glycosylases remove a damaged base and create an abasic (AP) site or nick the backbone, subsequent end processing is necessary to allow accurate DNA synthesis and ligation.
3.2. End Processing
Once a damaged base has been removed, the second phase of BER is carried out by apurinic/apyrimidinic endonuclease 1 (APE1) or polynucleotide kinase/phosphatase (PNKP), which can process the DNA backbone immediately adjacent to the lesion to generate a single-stranded break with ligatable termini [103,104,105]. In monofunctional BER, APE1 incises the abasic site and removes blocking 3′-sugar residues to create a 3′-hydroxyl end suitable for gap filling [90]. In contrast, many bifunctional DNA glycosylases generate strand breaks with 3′-phosphate or 3′-phospho-α,β-unsaturated aldehyde groups that are poor substrates for polymerases and therefore require PNKP to restore a 3′-hydroxyl before ligation [90]. These end processing enzymes ensure that the ends of the DNA are correctly configured for repair synthesis by removing blocking groups, trimming residual sugar fragments, and producing a clean 3’-hydroxyl group required for nucleotide insertion by a DNA polymerase. During long-patch BER, the 5′-flap endonuclease 1 (FEN1) further processes the displaced flap to create a nick suitable for ligation [28,32,106]. By converting the AP site into a repair-ready substrate, this step maintains the pathway’s continuity and prevents accumulation of unstable intermediates that would otherwise threaten genome stability.
3.3. Gap Filling
With properly processed DNA ends, the next stage of BER restores the missing nucleotides. In mitochondria, gap filling is primarily carried out by polymerase gamma (POLG), which supports BER by inserting nucleotide(s) into the repair gap before ligation [107,108]. Under conditions of oxidative stress, mitochondrial polymerase beta (POLB) provides additional gap-filling and dRP-lyase capacity and is particularly important for processing BER intermediates that contain 5′-dRP groups or when POLG becomes limiting [109,110,111]. In mitochondrial BER, this synthesis step can proceed via short-patch (SP) or long-patch (LP) modes: SP-BER incorporates a single nucleotide and is generally sufficient for simple, non-bulky lesions, whereas LP-BER inserts 2-7 nucleotides and, together with FEN-1, removes an oxidized or otherwise unligatable 5′ terminus, enabling repair of more complex lesions such as oxidized AP sites [25,26,106,112].
3.4. Ligation
The final phase of BER restores DNA strand continuity by ligating the broken strand. Once the correct nucleotide(s) have been inserted by SP or LP processes, DNA ligase IIIα, which is predominantly localized to mitochondria, seals the remaining nick in the phosphodiester backbone, completing repair of the lesion [113]. By closing the repair intermediate and fully reestablishing strand integrity, ligation prevents single-strand breaks from persisting, thereby avoiding replication stalling, double-strand break formation, and mitochondrial genome instability, and allowing the mtDNA molecule to resume normal replication and transcription.
4. BER Dysfunction: A Delicate Balance in Mitochondrial Genome Maintenance
Understanding the crucial role of mitochondrial BER in maintaining neuronal genome stability and energy balance helps set the stage for evaluating the serious consequences that occur when this pathway fails. Impaired mitochondrial BER arises through multiple, often convergent mechanisms: age-related declines in BER enzyme expression, chronic oxidative stress that exceeds repair capacity, and mutations or epigenetic modifications in nuclear genes encoding BER proteins or proteins required for their mitochondrial import. Paradoxically, both insufficient and excessive BER activity can be detrimental to cellular health (Figure 2).
Loss or reduced activity of mitochondrial BER enzymes directly leads to mtDNA instability, as demonstrated across a broad range of animal models and in vitro studies. In mice, knockout of NEIL1 results in accumulation of mtDNA mutations and deletions, leading to a metabolic syndrome phenotype and increased oxidative sensitivity [114]. OGG1-deficient mice display elevated levels of 8-oxoG throughout mitochondrial genomes in multiple tissues compared to wild-type controls [115]. Silencing or mutating mitochondrial BER proteins, including OGG1, NEIL1, MUTYH, and POLG, in diverse cell models, including but not limited to neuronal systems, results in substantial accumulation of oxidized base lesions, increased mtDNA mutations and deletions, increased genome instability, and diminished mitochondrial respiration in response to oxidative challenges [116,117,118,119].
In neuronal systems specifically, neural stem cells (NSCs) isolated from OGG1-deficient mice show increased mtDNA 8-oxoG accumulation, impaired neuronal differentiation, and reduced neurite outgrowth after oxidative challenge compared to NSCs from wild-type mice [120]. These and other BER-compromised neural models exhibit a characteristic constellation of mitochondrial defects, including diminished basal and maximal respiration, decreased ATP production, mitochondrial membrane depolarization, and heightened susceptibility to apoptosis following ROS exposure [119,120]. In primary neurons and brain tissue from BER-deficient mice, these mitochondrial deficits are accompanied by synaptic loss, altered firing properties, and region-specific neurodegeneration, indicating that even moderate, chronic BER impairment can deplete the bioenergetic reserves of vulnerable neuronal populations and trigger progressive cell loss [119,120]. Together, these findings demonstrate that failure of mitochondrial BER drives persistent mtDNA instability and bioenergetic collapse in neuronal contexts, creating a feed-forward loop in which defective repair promotes mitochondrial dysfunction, mtDNA release, and neuroinflammatory signaling that converge on neurodegenerative disease. The consequences of impaired BER are especially evident when defects extend to widespread loss of some BER proteins. Biallelic loss of NTHL1 or MUTYH results in NTHL1-associated polyposis (NAP) and MUTYH-associated polyposis (MAP), respectively [121,122,123]. These are hereditary cancer disorders characterized by high colorectal adenoma burden and increased cancer risk, particularly colorectal cancer [121,122,123]. The severity of these syndromes underscores the essential role of BER in safeguarding genome integrity and preventing malignant transformation.
However, excessive or unregulated BER can also be deleterious. The natural progression of the BER pathway generates strand-break intermediates when lesion recognition and base excision occur faster than gap filling and ligation, producing toxic repair intermediates that accumulate and become a source of genome instability rather than protection [124]. Within mitochondria, where repair enzyme stoichiometry and nucleotide availability are tightly constrained, this imbalance can stall repair, collapse replication forks, and fragment the mitochondrial genome. In this context, mitochondrial poly(ADP-ribose) polymerase 1 (PARP1) has been proposed to be recruited to BER intermediates and, when overactivated or present in excess, can clamp onto mtDNA breaks, consume NAD⁺, and inhibit POLG-dependent gap filling, thereby exacerbating mtDNA damage and impairing mitochondrial function [125]. However, the extent of PARP1 localization and function within mitochondria remains a subject of debate. Overexpression of several BER enzymes, including the DNA glycosylases AAG, OGG1, and NEIL1, has been shown to increase DNA damage intermediates [126,127,128]. At the same time, targeted overexpression of mitochondrial OGG1 can enhance mtDNA repair capacity, limit oxidant-induced mtDNA damage and apoptosis, and improve mitochondrial function and metabolic homeostasis in specific settings, illustrating that increased glycosylase activity can be beneficial when matched to the relevant lesion burden [129]. These repair intermediates can trigger innate immune sensors and further amplify inflammatory signaling [128,130,131,132,133,134].
Taken together, proper coordination and proportional activity within the BER pathway are critical for mtDNA homeostasis. Insufficient BER allows oxidative lesions to persist, promoting mtDNA instability and chronic inflammation, whereas excessive BER generates an excess of repair intermediates that are equally destabilizing and immunostimulatory. Neurons are particularly vulnerable to mtDNA instability because of their high metabolic rate. As previously discussed, mature neurons are post-mitotic and cannot re-enter the cell cycle or be easily replenished in response to injury or during an energy crisis. Their survival and function depend on the careful regulation of mitochondrial homeostasis, including spatial distribution at synaptic sites and the continuous production of ATP required for neurotransmission, maintenance of ion gradients, and intracellular transport [1,2]. Due to these metabolic demands, neurons are especially sensitive to disruptions in mitochondrial OXPHOS. Loss or mutation of mtDNA-encoded subunits can impair ETC assembly and function, leading to decreased ATP production, increased oxidative stress, and the activation of degenerative pathways [77]. Because neurons cannot readily compensate for mitochondrial dysfunction, chronic bioenergetic deficits lead to synaptic dysfunction, reduced plasticity, and eventual cell death, making mtDNA instability a critical driver of neurodegenerative disease in the central nervous system.
5. mtDNA Instability as a Trigger for Neuroinflammation
In addition to impairing bioenergetic capacity, growing evidence shows that mitochondrial genome instability can lead to the release of mtDNA fragments, which act as potent DAMPs that trigger and amplify neuroinflammatory signaling [135,136,137,138,139,140]. Several stress-response pathways enable mtDNA to escape the mitochondrial matrix and reach the cytosol or the extracellular space, linking impaired mitochondrial quality control directly to inflammatory responses within the nervous system (Figure 3).
During severe mitochondrial stress or injury, the mitochondrial outer membrane becomes permeable (MOMP), often mediated by the pro-apoptotic BCL-2 family proteins BAX and BAK, which oligomerize into macropores during apoptosis and enable the extrusion of entire mitochondrial nucleoids or DNA fragments into the cytosol [141,142]. This breach has been observed during caspase-dependent and caspase-independent apoptosis and in certain non-apoptotic stress responses, indicating that mtDNA release via MOMP is not restricted to terminal cell death [142]. Excessive calcium or oxidative stress can induce opening of the mitochondrial permeability transition pore (mPTP), causing matrix swelling and inner-membrane permeabilization [143]. This permeabilization provides a route for smaller mtDNA fragments to cross the inner membrane into the intermembrane space and, ultimately, the cytosol [135]. Depending on the duration and extent of opening, mPTP activation can function as a reversible stress response or progress to catastrophic mitochondrial dysfunction and cell death [143]. Under more moderate stress, oligomerization of the voltage-dependent anion channel (VDAC) in the outer membrane has been proposed to support mtDNA fragment release without complete mitochondrial rupture or overt apoptosis [144]. In addition to membrane permeabilization, defects in mitochondrial turnover, including impaired mitophagy or lysosomal degradation, can allow damaged mitochondria to persist and eventually rupture, leading to mtDNA leakage even in the absence of apoptosis [145,146,147,148,149]. Thus, mitochondrial injury, disrupted clearance, or incomplete organelle degradation each create opportunities for mtDNA to escape regular compartmentalization. The specific location of mtDNA release (cytosol or extracellular matrix) determines which immune pathways are activated and how neuroinflammatory signaling is propagated throughout the nervous system.
Once in the cytosol, mtDNA fragments activate robust innate immune signaling within the originating neuronal cell. The cGAS-STING pathway is a key mechanism for detecting cytosolic mtDNA. cGAS binds cytosolic DNA to produce cGAMP, which activates STING and triggers transcriptional responses to stimulate interferon and proinflammatory cytokine production [34,140,150]. Emerging evidence indicates that oxidized forms of mtDNA are particularly immunostimulatory and more efficiently activate STING signaling [151]. Additionally, oxidized or fragmented mtDNA can directly engage inflammasome complexes, particularly NLR (NOD-like receptor) Family Pyrin Domain Containing 3 (NLRP3), leading to caspase-1 activation and the production of Interleukin-1 beta (IL-1β) and other cytokines, further amplifying inflammation and contributing to local tissue damage [152].
mtDNA DAMP signaling extends beyond the cytosol of the originating neuron or glial cell. Damaged neurons can actively expel mtDNA into the extracellular space via vesicle secretion, mitochondrial extrusion, or direct membrane rupture [138,153]. Cell-free mtDNA species can then be absorbed by neighboring microglia, astrocytes, or even peripheral immune cells recruited to the neuroinflammatory environment. As the brain's resident immune cells, microglia are especially sensitive to extracellular mtDNA. Microglia rapidly internalize mtDNA and activate their own cGAS-STING pathway responses, leading to increased tumor necrosis factor-alpha (TNFα), interleukin-6 (IL-6), and interferon signaling [154,155]. Astrocytes also respond to extracellular mtDNA but may exert region- and disease-specific influences on inflammatory progression [156]. Ultimately, persistent or recurrent mtDNA DAMP signaling establishes a self-perpetuating cycle: mitochondrial dysfunction promotes neuroinflammation, while inflammatory mediators further damage mitochondria and destabilize mtDNA. This reciprocal cycle establishes a direct molecular connection between impaired mtDNA repair, chronic innate immune activation, disrupted neuronal health, and accelerated neurodegenerative decline [154,155,156] (Figure 4).
Together, these mechanisms highlight a direct molecular link between defective mtDNA maintenance, from oxidative damage, impaired repair, or failed organelle quality control, and pathological inflammation. This connection is increasingly recognized as a key driver in the progression of disorders such as Alzheimer's disease, Parkinson's disease, traumatic brain injury, and other neuroinflammatory and neurodegenerative conditions.
6. Emerging Therapeutic Strategies
Therapeutic strategies aimed at limiting mtDNA instability can intervene at multiple levels: by directly supporting mitochondrial BER, by improving the mitochondrial environment in which BER operates, and by attenuating the downstream inflammatory and metabolic consequences of BER failure. In rodent models, delivery of BER enzymes, such as OGG1 or APE1, to neurons using AAV-mediated gene transfer and transgenic or mitochondrially targeted overexpression has been shown to reduce mtDNA damage, improve mitochondrial function, and restore neural viability [157,158]. Strategies developed for primary mitochondrial diseases can inform treatment approaches for neurodegenerative and neuroinflammatory conditions, in which mtDNA instability frequently arises secondary to aging-related oxidative stress and inflammation [159]. For example, nucleoside supplementation strategies developed for primary mtDNA maintenance disorders are discussed as a template for treating more common conditions, including neurodegenerative states [159,160,161]. Supplementation with nucleotides or their precursors may counteract nucleotide pool imbalances and support efficient repair and replication, particularly in conditions where POLG function is compromised [160,161].
The development of genome-editing tools further enables correcting pathogenic mtDNA mutations or removing mutant mtDNA genomes altogether, thereby reducing mitochondrial heteroplasmy and restoring the pool of healthy mtDNA [162,163,164,165,166,167,168,169]. Experimental mitochondrial transplantation and intercellular mitochondria transfer approaches likewise seek to repopulate stressed or dysfunctional neurons with respiration-competent organelles, improving bioenergetics and attenuating tissue injury in preclinical models [170,171,172,173,174,175,176,177,178]. Mitochondria-tagged antioxidants (MTA) reduce the oxidative burden on mtDNA and mitigate the formation of lesions that overwhelm BER capacity [179].
Therapeutic innovation is also targeting the downstream consequences of BER failure. Restoring global mitochondrial quality control is a significant focus, with compounds that stimulate mitophagy or promote mitochondrial biogenesis showing promise in reducing the accumulation of mutant mtDNA and limiting the release of proinflammatory signals [180,181,182,183,184,185,186,187]. However, while stimulated mitophagy generally protects against the accumulation of damaged mitochondria and proinflammatory signaling, excessive mitophagy has also been linked to programmed neuronal death and increased neuroinflammation in preclinical models, highlighting the need for careful modulation and therapeutic balance [188]. Inhibitors of the cGAS-STING pathway and the NLRP3 inflammasome are now being assessed in preclinical models for their ability to blunt DAMP-induced neuroinflammation [189,190,191,192,193,194,195,196,197]. PARP1 inhibitors provide an additional means to mitigate metabolic collapse during unbalanced BER by limiting PARP1-dependent consumption of NAD⁺ and ATP, which can secondarily compromise mitochondrial bioenergetics, and by potentially modulating PARP1 activity reported at mtDNA lesions [125,198,199,200,201,202,203].
Although many of these interventions remain at preclinical or early translational stages, they collectively highlight the multifaceted therapeutic opportunities that arise from understanding how mtDNA instability drives neurodegeneration. Ultimately, a combinational approach that directly augments mitochondrial BER, protects from oxidative challenge, and modulates maladaptive neuroimmune responses is likely to be most effective for restoring mitochondrial homeostasis and preventing neuronal loss in neurodegenerative conditions.
7. Conclusions and Future Directions
Mitochondrial BER stands as a fundamental guardian of neuronal genome stability and energy homeostasis, particularly in the context of the central nervous system's extraordinary metabolic demands and long-lived post-mitotic cells [24,25,26,27,74,78]. Decades of research have clarified that both insufficient and excessive mitochondrial BER disrupt the delicate balance required for effective DNA repair, leading to persistent mtDNA instability, bioenergetic collapse, and aberrant activation of innate immune pathways [114,115,116,117,118,119,120]. These consequences, amplified by the neural tissue's unique vulnerabilities, form a mechanistic bridge connecting inherited and acquired mtDNA defects to the pathogenesis of neurodegenerative and neuroinflammatory diseases.
Recent advances in molecular genetics, neuroimmunology, and mitochondrial medicine have expanded the therapeutic landscape, offering new ways to intervene at multiple points along the mtDNA damage-response spectrum. Gene and enzyme delivery approaches, mitochondrial genome editing, targeted antioxidants, mitochondrial transplantation, and precision immunomodulators provide promising routes to restore mitochondrial stability or limit downstream pathology [157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203]. However, meaningful clinical translation will require surmounting several challenges. Achieving neuronal and region-specific delivery, tuning repair activity without triggering harmful hyperactivation, and establishing biomarkers that reliably monitor mtDNA repair capacity and innate immune engagement in vivo will be essential steps. Likewise, integrating heterogeneity in neuronal subtypes, aging trajectories, and metabolic states into therapeutic design will be crucial for identifying which interventions are most beneficial and then deploying them.
Several outstanding research questions define the path ahead. How can BER activity be precisely calibrated to maintain fidelity without tipping into detrimental overactivity? Are there biomarkers for early mtDNA instability, and can they predict therapeutic windows before irreversible neurodegeneration begins? A recently developed blood-based mtDNA damage detection assay (Sanders and colleagues) stratifies Parkinson’s disease patients, and illustrates that such mtDNA-focused biomarkers are feasible and could be adapted to other neurodegenerative settings [204]. Further, which immune signaling pathways downstream of mtDNA DAMPs are most amenable to therapeutic modulation without unintended immunosuppression? And how do individual genetics, metabolic states, and mitochondrial dynamics shape the susceptibility of neurons to BER imbalance, mitochondrial quality control, and neuroimmune activation? Addressing these gaps will depend on multidisciplinary collaboration, the development of innovative tools, and seamless translation from experimental models to human systems. Ultimately, restoring the balance of mitochondrial BER and limiting pathological mtDNA instability holds the potential to significantly slow, or perhaps prevent, the progression of multiple neurodegenerative and neuroinflammatory disorders. By uniting insights from DNA repair, mitochondrial biology, and neuroimmunology, the field is poised to develop interventions that address the root cause of neuronal decline rather than its downstream consequences.
Funding
Research in the Prakash lab on DNA repair is funded by grants from the National Institutes of Health (NIH) [ES030084], the National Science Foundation (NSF) [NSF- 2450845], and the American Cancer Society (ACS) [RSG-25-1412630-01-DMC]. Support was also provided by start-up funds from the Mitchell Cancer Institute (to AP). MP, MBN, MKT, contributed to the writing and editing of this review article. MP and AP conceptualized the topic and AP also performed editing.
Conflicts of Interest
AP is a member of Canal House Biosciences, LLC. Canal House Biosciences was not involved in this study. The authors state that there is no conflict of interest.
References
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef]
- Bogenhagen, D.F. Mitochondrial DNA Nucleoid Structure. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 2012, 1819, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Druzhyna, N.M.; Wilson, G.L.; LeDoux, S.P. Mitochondrial DNA Repair in Aging and Disease. Mech. Ageing Dev. 2008, 129, 383–390. [Google Scholar] [CrossRef]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative Stress Induces Degradation of Mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA Damage Is More Extensive and Persists Longer than Nuclear DNA Damage in Human Cells Following Oxidative Stress. Proc. Natl. Acad. Sci. 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative Damage in Nucleic Acids and Parkinson’s Disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Llanos-González, E.; Henares-Chavarino, Á.A.; Pedrero-Prieto, C.M.; García-Carpintero, S.; Frontiñán-Rubio, J.; Sancho-Bielsa, F.J.; Alcain, F.J.; Peinado, J.R.; Rabanal-Ruíz, Y.; Durán-Prado, M. Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease. Front. Neurosci. 2020, 13, 1444. [Google Scholar] [CrossRef]
- Wallace, D.C. A Mitochondrial Etiology of Neuropsychiatric Disorders. JAMA Psychiatry 2017, 74, 863–864. [Google Scholar] [CrossRef]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.-P.; Letellier, T. Mitochondrial Threshold Effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial Genetic Medicine. Nat. Genet. 2018, 50, 1642–1649. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, K.J.; Reeve, A.K.; Turnbull, D.M. Do Mitochondrial DNA Mutations Have a Role in Neurodegenerative Disease? Biochem. Soc. Trans. 2007, 35, 1232–1235. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Lott, M.T.; Shoffner, J.M.; Ballinger, S. Mitochondrial DNA Mutations in Epilepsy and Neurological Disease. Epilepsia 1994, 35 Suppl 1, S43–50. [Google Scholar] [CrossRef] [PubMed]
- Elfawy, H.A.; Das, B. Crosstalk between Mitochondrial Dysfunction, Oxidative Stress, and Age Related Neurodegenerative Disease: Etiologies and Therapeutic Strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Cruz, A.C.P.; Ferrasa, A.; Muotri, A.R.; Herai, R.H. Frequency and Association of Mitochondrial Genetic Variants with Neurological Disorders. Mitochondrion 2019, 46, 345–360. [Google Scholar] [CrossRef]
- Gonzalez-Hunt, C.P.; Sanders, L.H. DNA Damage and Repair in Parkinson’s Disease: Recent Advances and New Opportunities. J. Neurosci. Res. 2021, 99, 180–189. [Google Scholar] [CrossRef]
- Kai, Y.; Takamatsu, C.; Tokuda, K.; Okamoto, M.; Irita, K.; Takahashi, S. Rapid and Random Turnover of Mitochondrial DNA in Rat Hepatocytes of Primary Culture. Mitochondrion 2006, 6, 299–304. [Google Scholar] [CrossRef]
- Zhao, L. Mitochondrial DNA Degradation: A Quality Control Measure for Mitochondrial Genome Maintenance and Stress Response. The Enzymes 2019, 45, 311–341. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Persistent Damage Induces Mitochondrial DNA Degradation. DNA Repair 2013, 12, 488–499. [Google Scholar] [CrossRef]
- Wilhelm, L.P.; Ganley, I.G. Eating Your Mitochondria-When Too Much of a Good Thing Turns Bad. EMBO J. 2023, 42, e114542. [Google Scholar] [CrossRef]
- Rong, Z.; Tu, P.; Xu, P.; Sun, Y.; Yu, F.; Tu, N.; Guo, L.; Yang, Y. The Mitochondrial Response to DNA Damage. Front. Cell Dev. Biol. 2021, 9, 669379. [Google Scholar] [CrossRef] [PubMed]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the Damage: Repair Pathways Keep Mitochondrial DNA Intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublié, S. Base Excision Repair in the Mitochondria. J. Cell. Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R. DNA Damage and Base Excision Repair in Mitochondria and Their Role in Aging. J. Aging Res. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Pinz, K.G.; Perez-Jannotti, R.M. Enzymology of Mitochondrial Base Excision Repair. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 257–271. [Google Scholar] [CrossRef]
- Akbari, M.; Visnes, T.; Krokan, H.E.; Otterlei, M. Mitochondrial Base Excision Repair of Uracil and AP Sites Takes Place by Single-Nucleotide Insertion and Long-Patch DNA Synthesis. DNA Repair 2008, 7, 605–616. [Google Scholar] [CrossRef]
- Szczesny, B.; Tann, A.W.; Longley, M.J.; Copeland, W.C.; Mitra, S. Long Patch Base Excision Repair in Mammalian Mitochondrial Genomes. J. Biol. Chem. 2008, 283, 26349–26356. [Google Scholar] [CrossRef]
- Karahalil, B.; Hogue, B.A.; de Souza-Pinto, N.C.; Bohr, V.A. Base Excision Repair Capacity in Mitochondria and Nuclei: Tissue-Specific Variations. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1895–1902. [Google Scholar] [CrossRef]
- Stuart, J.A.; Hashiguchi, K.; Wilson, D.M.; Copeland, W.C.; Souza-Pinto, N.C.; Bohr, V.A. DNA Base Excision Repair Activities and Pathway Function in Mitochondrial and Cellular Lysates from Cells Lacking Mitochondrial DNA. Nucleic Acids Res. 2004, 32, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early Steps in the DNA Base Excision/Single-Strand Interruption Repair Pathway in Mammalian Cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Jun, Y.W.; Albarran, E.; Wilson, D.L.; Ding, J.; Kool, E.T. Fluorescence Imaging of Mitochondrial DNA Base Excision Repair Reveals Dynamics of Oxidative Stress Responses. Angew. Chem. Int. Ed Engl. 2022, 61, e202111829. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, H.-S.; Chung, J.H. Molecular Mechanisms of Mitochondrial DNA Release and Activation of the cGAS-STING Pathway. Exp. Mol. Med. 2023, 55, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Nass, M.M.; Nass, S. INTRAMITOCHONDRIAL FIBERS WITH DNA CHARACTERISTICS. I. FIXATION AND ELECTRON STAINING REACTIONS. J. Cell Biol. 1963, 19, 593–611. [Google Scholar] [CrossRef]
- Luck, D.J.; Reich, E. DNA IN MITOCHONDRIA OF NEUROSPORA CRASSA. Proc. Natl. Acad. Sci. U. S. A. 1964, 52, 931–938. [Google Scholar] [CrossRef]
- Schatz, G.; Haslbrunner, E.; Tuppy, H. DEOXYRIBONUCLEIC ACID ASSOCIATED WITH YEAST MITOCHONDRIA. Biochem. Biophys. Res. Commun. 1964, 15, 127–132. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and Organization of the Human Mitochondrial Genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Chomyn, A.; Mariottini, P.; Cleeter, M.W.; Ragan, C.I.; Matsuno-Yagi, A.; Hatefi, Y.; Doolittle, R.F.; Attardi, G. Six Unidentified Reading Frames of Human Mitochondrial DNA Encode Components of the Respiratory-Chain NADH Dehydrogenase. Nature 1985, 314, 592–597. [Google Scholar] [CrossRef]
- Chomyn, A.; Cleeter, M.W.; Ragan, C.I.; Riley, M.; Doolittle, R.F.; Attardi, G. URF6, Last Unidentified Reading Frame of Human mtDNA, Codes for an NADH Dehydrogenase Subunit. Science 1986, 234, 614–618. [Google Scholar] [CrossRef]
- Suzuki, T.; Nagao, A.; Suzuki, T. Human Mitochondrial tRNAs: Biogenesis, Function, Structural Aspects, and Diseases. Annu. Rev. Genet. 2011, 45, 299–329. [Google Scholar] [CrossRef] [PubMed]
- Neupert, W.; Herrmann, J.M. Translocation of Proteins into Mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Wiedemann, N. Molecular Machineries and Pathways of Mitochondrial Protein Transport. Nat. Rev. Mol. Cell Biol. 2025, 26, 848–867. [Google Scholar] [CrossRef] [PubMed]
- Cutter, A.R.; Hayes, J.J. A Brief Review of Nucleosome Structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Bogenhagen, D.F.; Rousseau, D.; Burke, S. The Layered Structure of Human Mitochondrial DNA Nucleoids. J. Biol. Chem. 2008, 283, 3665–3675. [Google Scholar] [CrossRef]
- Chen, X.J.; Butow, R.A. The Organization and Inheritance of the Mitochondrial Genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef]
- Malka, F.; Lombès, A.; Rojo, M. Organization, Dynamics and Transmission of Mitochondrial DNA: Focus on Vertebrate Nucleoids. Biochim. Biophys. Acta 2006, 1763, 463–472. [Google Scholar] [CrossRef]
- Kucej, M.; Butow, R.A. Evolutionary Tinkering with Mitochondrial Nucleoids. Trends Cell Biol. 2007, 17, 586–592. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking Outside the Nucleus: Mitochondrial DNA Copy Number in Health and Disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Wai, T.; Ao, A.; Zhang, X.; Cyr, D.; Dufort, D.; Shoubridge, E.A. The Role of Mitochondrial DNA Copy Number in Mammalian Fertility. Biol. Reprod. 2010, 83, 52–62. [Google Scholar] [CrossRef]
- Rath, S.P.; Gupta, R.; Todres, E.; Wang, H.; Jourdain, A.A.; Ardlie, K.G.; Calvo, S.E.; Mootha, V.K. Mitochondrial Genome Copy Number Variation across Tissues in Mice and Humans. Proc. Natl. Acad. Sci. U. S. A. 2024, 121, e2402291121. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Lewis, T.L.; Du, Y.; Virga, D.M.; Decker, A.M.; Coceano, G.; Alvelid, J.; Paul, M.A.; Hamilton, S.; Kneis, P.; et al. Most Axonal Mitochondria in Cortical Pyramidal Neurons Lack Mitochondrial DNA and Consume ATP. BioRxiv Prepr. Serv. Biol. 2024, 2024.02.12.579972. [Google Scholar] [CrossRef]
- Fuke, S.; Kubota-Sakashita, M.; Kasahara, T.; Shigeyoshi, Y.; Kato, T. Regional Variation in Mitochondrial DNA Copy Number in Mouse Brain. Biochim. Biophys. Acta 2011, 1807, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Keeney, P.M.; Algarzae, N.K.; Ladd, A.C.; Thomas, R.R.; Bennett, J.P. Mitochondrial DNA Copy Numbers in Pyramidal Neurons Are Decreased and Mitochondrial Biogenesis Transcriptome Signaling Is Disrupted in Alzheimer’s Disease Hippocampi. J. Alzheimers Dis. JAD 2014, 40, 319–330. [Google Scholar] [CrossRef]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-Resolution Microscopy Reveals That Mammalian Mitochondrial Nucleoids Have a Uniform Size and Frequently Contain a Single Copy of mtDNA. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Udeshi, N.D.; Deerinck, T.J.; Svinkina, T.; Ellisman, M.H.; Carr, S.A.; Ting, A.Y. Proximity Biotinylation as a Method for Mapping Proteins Associated with mtDNA in Living Cells. Cell Chem. Biol. 2017, 24, 404–414. [Google Scholar] [CrossRef]
- Kaufman, B.A.; Durisic, N.; Mativetsky, J.M.; Costantino, S.; Hancock, M.A.; Grutter, P.; Shoubridge, E.A. The Mitochondrial Transcription Factor TFAM Coordinates the Assembly of Multiple DNA Molecules into Nucleoid-like Structures. Mol. Biol. Cell 2007, 18, 3225–3236. [Google Scholar] [CrossRef]
- Song, Y.; Wang, W.; Wang, B.; Shi, Q. The Protective Mechanism of TFAM on Mitochondrial DNA and Its Role in Neurodegenerative Diseases. Mol. Neurobiol. 2024, 61, 4381–4390. [Google Scholar] [CrossRef] [PubMed]
- Alam, T.I.; Kanki, T.; Muta, T.; Ukaji, K.; Abe, Y.; Nakayama, H.; Takio, K.; Hamasaki, N.; Kang, D. Human Mitochondrial DNA Is Packaged with TFAM. Nucleic Acids Res. 2003, 31, 1640–1645. [Google Scholar] [CrossRef]
- Macuada, J.; Molina-Riquelme, I.; Eisner, V. How Are Mitochondrial Nucleoids Trafficked? Trends Cell Biol. 2025, 35, 194–204. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal Oxidative Damage to Mitochondrial and Nuclear DNA Is Extensive. Proc. Natl. Acad. Sci. U. S. A. 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- King, D.E.; Copeland, W.C. DNA Repair Pathways in the Mitochondria. DNA Repair 2025, 146, 103814. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of Replication and Repair in Mitochondrial DNA Deletion Formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Takao, M.; Aburatani, H.; Kobayashi, K.; Yasui, A. Mitochondrial Targeting of Human DNA Glycosylases for Repair of Oxidative DNA Damage. Nucleic Acids Res. 1998, 26, 2917–2922. [Google Scholar] [CrossRef]
- Wen, H.; Deng, H.; Li, B.; Chen, J.; Zhu, J.; Zhang, X.; Yoshida, S.; Zhou, Y. Mitochondrial Diseases: From Molecular Mechanisms to Therapeutic Advances. Signal Transduct. Target. Ther. 2025, 10, 9. [Google Scholar] [CrossRef]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal Inheritance of Human Mitochondrial DNA. Proc. Natl. Acad. Sci. U. S. A. 1980, 77, 6715–6719. [Google Scholar] [CrossRef]
- Cree, L.M.; Samuels, D.C.; Chinnery, P.F. The Inheritance of Pathogenic Mitochondrial DNA Mutations. Biochim. Biophys. Acta 2009, 1792, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Vadakedath, S.; Kandi, V.; Ca, J.; Vijayan, S.; Achyut, K.C.; Uppuluri, S.; Reddy, P.K.K.; Ramesh, M.; Kumar, P.P. Mitochondrial Deoxyribonucleic Acid (mtDNA), Maternal Inheritance, and Their Role in the Development of Cancers: A Scoping Review. Cureus 2023, 15, e39812. [Google Scholar] [CrossRef]
- Copeland, W.C. Defects of Mitochondrial DNA Replication. J. Child Neurol. 2014, 29, 1216–1224. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. MtDNA-Maintenance Defects: Syndromes and Genes. J. Inherit. Metab. Dis. 2017, 40, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Inherited Mitochondrial Diseases of DNA Replication. Annu. Rev. Med. 2008, 59, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA Oxidative Damage and Repair in Aging and Alzheimer’s Disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Cha, M.-Y.; Kim, D.K.; Mook-Jung, I. The Role of Mitochondrial DNA Mutation on Neurodegenerative Diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef]
- Mecocci, P.; MacGarvey, U.; Kaufman, A.E.; Koontz, D.; Shoffner, J.M.; Wallace, D.C.; Beal, M.F. Oxidative Damage to Mitochondrial DNA Shows Marked Age-Dependent Increases in Human Brain. Ann. Neurol. 1993, 34, 609–616. [Google Scholar] [CrossRef]
- Damas, J.; Samuels, D.C.; Carneiro, J.; Amorim, A.; Pereira, F. Mitochondrial DNA Rearrangements in Health and Disease--a Comprehensive Study. Hum. Mutat. 2014, 35, 1–14. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA Damage and Reactive Oxygen Species in Neurodegenerative Disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjoras, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583–a012583. [Google Scholar] [CrossRef]
- Kang, D.; Hamasaki, N. Mitochondrial Oxidative Stress and Mitochondrial DNA. Clin. Chem. Lab. Med. 2003, 41, 1281–1288. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and Decay of the Primary Structure of DNA. Nature 1993, 362, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Mol, C.D.; Parikh, S.S.; Putnam, C.D.; Lo, T.P.; Tainer, J.A. DNA Repair Mechanisms for the Recognition and Removal of Damaged DNA Bases. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 101–128. [Google Scholar] [CrossRef]
- Stivers, J.T.; Jiang, Y.L. A Mechanistic Perspective on the Chemistry of DNA Repair Glycosylases. Chem. Rev. 2003, 103, 2729–2759. [Google Scholar] [CrossRef]
- Seeberg, E.; Eide, L.; Bjørås, M. The Base Excision Repair Pathway. Trends Biochem. Sci. 1995, 20, 391–397. [Google Scholar] [CrossRef]
- Jacobs, A.L.; Schär, P. DNA Glycosylases: In DNA Repair and Beyond. Chromosoma 2012, 121, 1–20. [Google Scholar] [CrossRef]
- Schormann, N.; Ricciardi, R.; Chattopadhyay, D. Uracil-DNA Glycosylases-Structural and Functional Perspectives on an Essential Family of DNA Repair Enzymes. Protein Sci. Publ. Protein Soc. 2014, 23, 1667–1685. [Google Scholar] [CrossRef] [PubMed]
- Bharati, S.; Krokan, H.E.; Kristiansen, L.; Otterlei, M.; Slupphaug, G. Human Mitochondrial Uracil-DNA Glycosylase Preform (UNG1) Is Processed to Two Forms One of Which Is Resistant to Inhibition by AP Sites. Nucleic Acids Res. 1998, 26, 4953–4959. [Google Scholar] [CrossRef] [PubMed]
- Dalhus, B.; Helle, I.H.; Backe, P.H.; Alseth, I.; Rognes, T.; Bjørås, M.; Laerdahl, J.K. Structural Insight into Repair of Alkylated DNA by a New Superfamily of DNA Glycosylases Comprising HEAT-like Repeats. Nucleic Acids Res. 2007, 35, 2451–2459. [Google Scholar] [CrossRef]
- van Loon, B.; Samson, L.D. Alkyladenine DNA Glycosylase (AAG) Localizes to Mitochondria and Interacts with Mitochondrial Single-Stranded Binding Protein (mtSSB). DNA Repair 2013, 12, 177–187. [Google Scholar] [CrossRef]
- O’Connor, T.R. Purification and Characterization of Human 3-Methyladenine-DNA Glycosylase. Nucleic Acids Res. 1993, 21, 5561–5569. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base Excision Repair of Oxidative DNA Damage: From Mechanism to Disease. Front. Biosci. Landmark Ed. 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [PubMed]
- Trasviña-Arenas, C.H.; Demir, M.; Lin, W.-J.; David, S.S. Structure, Function and Evolution of the Helix-Hairpin-Helix DNA Glycosylase Superfamily: Piecing Together the Evolutionary Puzzle of DNA Base Damage Repair Mechanisms. DNA Repair 2021, 108, 103231. [Google Scholar] [CrossRef]
- Ikeda, S.; Biswas, T.; Roy, R.; Izumi, T.; Boldogh, I.; Kurosky, A.; Sarker, A.H.; Seki, S.; Mitra, S. Purification and Characterization of Human NTH1, a Homolog of Escherichia Coli Endonuclease III. Direct Identification of Lys-212 as the Active Nucleophilic Residue. J. Biol. Chem. 1998, 273, 21585–21593. [Google Scholar] [CrossRef] [PubMed]
- Aburatani, H.; Hippo, Y.; Ishida, T.; Takashima, R.; Matsuba, C.; Kodama, T.; Takao, M.; Yasui, A.; Yamamoto, K.; Asano, M. Cloning and Characterization of Mammalian 8-Hydroxyguanine-Specific DNA Glycosylase/Apurinic, Apyrimidinic Lyase, a Functional mutM Homologue. Cancer Res. 1997, 57, 2151–2156. [Google Scholar] [PubMed]
- Fromme, J.C.; Banerjee, A.; Huang, S.J.; Verdine, G.L. Structural Basis for Removal of Adenine Mispaired with 8-Oxoguanine by MutY Adenine DNA Glycosylase. Nature 2004, 427, 652–656. [Google Scholar] [CrossRef]
- Bandaru, V. A Novel Human DNA Glycosylase That Removes Oxidative DNA Damage and Is Homologous to Escherichia Coli Endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and Characterization of a Novel Human DNA Glycosylase for Repair of Cytosine-Derived Lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar] [CrossRef]
- Liu, M.; Doublié, S.; Wallace, S.S. Neil3, the Final Frontier for the DNA Glycosylases That Recognize Oxidative Damage. Mutat. Res. 2013, 743–744, 4–11. [Google Scholar] [CrossRef]
- Zhao, X.; Krishnamurthy, N.; Burrows, C.J.; David, S.S. Mutation versus Repair: NEIL1 Removal of Hydantoin Lesions in Single-Stranded, Bulge, Bubble, and Duplex DNA Contexts. Biochemistry 2010, 49, 1658–1666. [Google Scholar] [CrossRef]
- Hailer, M.K.; Slade, P.G.; Martin, B.D.; Rosenquist, T.A.; Sugden, K.D. Recognition of the Oxidized Lesions Spiroiminodihydantoin and Guanidinohydantoin in DNA by the Mammalian Base Excision Repair Glycosylases NEIL1 and NEIL2. DNA Repair 2005, 4, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Minko, I.G.; Christov, P.P.; Li, L.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. Processing of N5-Substituted Formamidopyrimidine DNA Adducts by DNA Glycosylases NEIL1 and NEIL3. DNA Repair 2019, 73, 49–54. [Google Scholar] [CrossRef]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential Repair of Oxidized Base Damage in the Transcribed Genes of Mammalian Cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Schurman, S.H.; Harboe, C.; de Souza-Pinto, N.C.; Bohr, V.A. Base Excision Repair of Oxidative DNA Damage and Association with Cancer and Aging. Carcinogenesis 2009, 30, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Tahbaz, N.; Subedi, S.; Weinfeld, M. Role of Polynucleotide Kinase/Phosphatase in Mitochondrial DNA Repair. Nucleic Acids Res. 2012, 40, 3484–3495. [Google Scholar] [CrossRef]
- Li, M.; Zhong, Z.; Zhu, J.; Xiang, D.; Dai, N.; Cao, X.; Qing, Y.; Yang, Z.; Xie, J.; Li, Z.; et al. Identification and Characterization of Mitochondrial Targeting Sequence of Human Apurinic/Apyrimidinic Endonuclease 1. J. Biol. Chem. 2010, 285, 14871–14881. [Google Scholar] [CrossRef]
- Liu, P.; Qian, L.; Sung, J.-S.; de Souza-Pinto, N.C.; Zheng, L.; Bogenhagen, D.F.; Bohr, V.A.; Wilson, D.M.; Shen, B.; Demple, B. Removal of Oxidative DNA Damage via FEN1-Dependent Long-Patch Base Excision Repair in Human Cell Mitochondria. Mol. Cell. Biol. 2008, 28, 4975–4987. [Google Scholar] [CrossRef]
- Copeland, W.C. The Mitochondrial DNA Polymerase in Health and Disease. Subcell. Biochem. 2010, 50, 211–222. [Google Scholar] [CrossRef]
- Krasich, R.; Copeland, W.C. DNA Polymerases in the Mitochondria: A Critical Review of the Evidence. Front. Biosci. Landmark Ed. 2017, 22, 692–709. [Google Scholar] [CrossRef]
- Sykora, P.; Kanno, S.; Akbari, M.; Kulikowicz, T.; Baptiste, B.A.; Leandro, G.S.; Lu, H.; Tian, J.; May, A.; Becker, K.A.; et al. DNA Polymerase Beta Participates in Mitochondrial DNA Repair. Mol. Cell. Biol. 2017, 37, e00237-17. [Google Scholar] [CrossRef]
- Prasad, R.; Çağlayan, M.; Dai, D.-P.; Nadalutti, C.A.; Zhao, M.-L.; Gassman, N.R.; Janoshazi, A.K.; Stefanick, D.F.; Horton, J.K.; Krasich, R.; et al. DNA Polymerase β: A Missing Link of the Base Excision Repair Machinery in Mammalian Mitochondria. DNA Repair 2017, 60, 77–88. [Google Scholar] [CrossRef]
- Baptiste, B.A.; Baringer, S.L.; Kulikowicz, T.; Sommers, J.A.; Croteau, D.L.; Brosh, R.M.; Bohr, V.A. DNA Polymerase β Outperforms DNA Polymerase γ in Key Mitochondrial Base Excision Repair Activities. DNA Repair 2021, 99, 103050. [Google Scholar] [CrossRef]
- Liu, P.; Demple, B. DNA Repair in Mammalian Mitochondria: Much More than We Thought? Environ. Mol. Mutagen. 2010, 51, 417–426. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Campbell, C. A Novel Interaction between DNA Ligase III and DNA Polymerase Gamma Plays an Essential Role in Mitochondrial DNA Stability. Biochem. J. 2007, 402, 175–186. [Google Scholar] [CrossRef]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The Metabolic Syndrome Resulting from a Knockout of the NEIL1 DNA Glycosylase. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1864–1869. [Google Scholar] [CrossRef]
- de Souza-Pinto, N.C.; Eide, L.; Hogue, B.A.; Thybo, T.; Stevnsner, T.; Seeberg, E.; Klungland, A.; Bohr, V.A. Repair of 8-Oxodeoxyguanosine Lesions in Mitochondrial Dna Depends on the Oxoguanine Dna Glycosylase (OGG1) Gene and 8-Oxoguanine Accumulates in the Mitochondrial Dna of OGG1-Defective Mice. Cancer Res. 2001, 61, 5378–5381. [Google Scholar]
- Ruchko, M.V.; Gorodnya, O.M.; Zuleta, A.; Pastukh, V.M.; Gillespie, M.N. The DNA Glycosylase Ogg1 Defends against Oxidant-Induced mtDNA Damage and Apoptosis in Pulmonary Artery Endothelial Cells. Free Radic. Biol. Med. 2011, 50, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, P.; Zhou, K.; Zhang, W.; Liu, S.; Ouyang, J.; Bai, M.; Ding, G.; Huang, S.; Jia, Z.; et al. HUWE1-Mediated Degradation of MUTYH Facilitates DNA Damage and Mitochondrial Dysfunction to Promote Acute Kidney Injury. Adv. Sci. Weinh. Baden-Wurtt. Ger. 2025, 12, e2412250. [Google Scholar] [CrossRef]
- Lloyd, R.S. Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA 2022, 2, 279–301. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Schomacher, L.; Schüle, K.M.; Mallick, M.; Musheev, M.U.; Karaulanov, E.; Krebs, L.; von Seggern, A.; Niehrs, C. NEIL1 and NEIL2 DNA Glycosylases Protect Neural Crest Development against Mitochondrial Oxidative Stress. eLife 2019, 8, e49044. [Google Scholar] [CrossRef]
- Wang, W.; Esbensen, Y.; Kunke, D.; Suganthan, R.; Rachek, L.; Bjørås, M.; Eide, L. Mitochondrial DNA Damage Level Determines Neural Stem Cell Differentiation Fate. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 9746–9751. [Google Scholar] [CrossRef]
- Weren, R.D.A.; Ligtenberg, M.J.L.; Kets, C.M.; de Voer, R.M.; Verwiel, E.T.P.; Spruijt, L.; van Zelst-Stams, W.A.G.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A Germline Homozygous Mutation in the Base-Excision Repair Gene NTHL1 Causes Adenomatous Polyposis and Colorectal Cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited Variants of MYH Associated with Somatic G. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Sieber, O.M.; Lipton, L.; Crabtree, M.; Heinimann, K.; Fidalgo, P.; Phillips, R.K.S.; Bisgaard, M.-L.; Orntoft, T.F.; Aaltonen, L.A.; Hodgson, S.V.; et al. Multiple Colorectal Adenomas, Classic Adenomatous Polyposis, and Germ-Line Mutations in MYH. N. Engl. J. Med. 2003, 348, 791–799. [Google Scholar] [CrossRef]
- Simonelli, V.; Narciso, L.; Dogliotti, E.; Fortini, P. Base Excision Repair Intermediates Are Mutagenic in Mammalian Cells. Nucleic Acids Res. 2005, 33, 4404–4411. [Google Scholar] [CrossRef]
- Herrmann, G.K.; Russell, W.K.; Garg, N.J.; Yin, Y.W. Poly(ADP-Ribose) Polymerase 1 Regulates Mitochondrial DNA Repair in an NAD-Dependent Manner. J. Biol. Chem. 2021, 296, 100309. [Google Scholar] [CrossRef]
- Teoh, P.J.; An, O.; Chung, T.-H.; Chooi, J.Y.; Toh, S.H.M.; Fan, S.; Wang, W.; Koh, B.T.H.; Fullwood, M.J.; Ooi, M.G.; et al. Aberrant Hyperediting of the Myeloma Transcriptome by ADAR1 Confers Oncogenicity and Is a Marker of Poor Prognosis. Blood 2018, 132, 1304–1317. [Google Scholar] [CrossRef]
- Fishel, M.L.; Seo, Y.R.; Smith, M.L.; Kelley, M.R. Imbalancing the DNA Base Excision Repair Pathway in the Mitochondria; Targeting and Overexpressing N-Methylpurine DNA Glycosylase in Mitochondria Leads to Enhanced Cell Killing. Cancer Res. 2003, 63, 608–615. [Google Scholar] [PubMed]
- Wang, R.; Li, C.; Qiao, P.; Xue, Y.; Zheng, X.; Chen, H.; Zeng, X.; Liu, W.; Boldogh, I.; Ba, X. OGG1-Initiated Base Excision Repair Exacerbates Oxidative Stress-Induced Parthanatos. Cell Death Dis. 2018, 9, 628. [Google Scholar] [CrossRef]
- Dobson, A.W.; Grishko, V.; LeDoux, S.P.; Kelley, M.R.; Wilson, G.L.; Gillespie, M.N. Enhanced mtDNA Repair Capacity Protects Pulmonary Artery Endothelial Cells from Oxidant-Mediated Death. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L205–210. [Google Scholar] [CrossRef]
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-Ribose) Signals to Mitochondrial AIF: A Key Event in Parthanatos. Exp. Neurol. 2009, 218, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M. Deadly Conversations: Nuclear-Mitochondrial Cross-Talk. J. Bioenerg. Biomembr. 2004, 36, 287–294. [Google Scholar] [CrossRef]
- Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. [Google Scholar] [CrossRef]
- Zhao, S.; Goewey Ruiz, J.A.; Sebastian, M.; Kidane, D. Defective DNA Polymerase Beta Invoke a Cytosolic DNA Mediated Inflammatory Response. Front. Immunol. 2022, 13, 1039009. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Chen, T.; Fang, H.; Shen, X.; Tang, Z.; Zhao, J. Mitochondrial DNA Release via the Mitochondrial Permeability Transition Pore Activates the cGAS-STING Pathway, Exacerbating Inflammation in Acute Kawasaki Disease. Cell Commun. Signal. CCS 2024, 22, 328. [Google Scholar] [CrossRef] [PubMed]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial Permeability Transition Triggers the Release of mtDNA Fragments. Cell. Mol. Life Sci. CMLS 2004, 61, 3100–3103. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Lin, M.; Liu, N.; Qin, Z.; Wang, Y. Mitochondrial-Derived Damage-Associated Molecular Patterns Amplify Neuroinflammation in Neurodegenerative Diseases. Acta Pharmacol. Sin. 2022, 43, 2439–2447. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-Derived Damage-Associated Molecular Patterns in Neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Quan, S.; Fu, X.; Cai, H.; Ren, Z.; Xu, Y.; Jia, L. The Neuroimmune Nexus: Unraveling the Role of the mtDNA-cGAS-STING Signal Pathway in Alzheimer’s Disease. Mol. Neurodegener. 2025, 20, 25. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX Macropores Facilitate Mitochondrial Herniation and mtDNA Efflux during Apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef]
- Crompton, M. The Mitochondrial Permeability Transition Pore and Its Role in Cell Death. Biochem. J. 1999, 341 Pt 2, 233–249. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC Oligomers Form Mitochondrial Pores to Release mtDNA Fragments and Promote Lupus-like Disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Takamatsu, H. Mitochondrial DNA: Leakage, Recognition and Associated Human Diseases. J. Biochem. (Tokyo) 2025, 178, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhu, Y.; Liu, W.; Xiang, W.; He, S.; Hayashi, T.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Ikejima, T. Impaired Mitophagy Causes Mitochondrial DNA Leakage and STING Activation in Ultraviolet B-Irradiated Human Keratinocytes HaCaT. Arch. Biochem. Biophys. 2023, 737, 109553. [Google Scholar] [CrossRef]
- Zhong, W.; Rao, Z.; Xu, J.; Sun, Y.; Hu, H.; Wang, P.; Xia, Y.; Pan, X.; Tang, W.; Chen, Z.; et al. Defective Mitophagy in Aged Macrophages Promotes Mitochondrial DNA Cytosolic Leakage to Activate STING Signaling during Liver Sterile Inflammation. Aging Cell 2022, 21, e13622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Wei, H.; Zhao, A.; Yan, X.; Zhang, X.; Gan, J.; Guo, M.; Wang, J.; Zhang, F.; Jiang, Y.; et al. Mitochondrial DNA Leakage: Underlying Mechanisms and Therapeutic Implications in Neurological Disorders. J. Neuroinflammation 2025, 22, 34. [Google Scholar] [CrossRef]
- Newman, L.E.; Weiser Novak, S.; Rojas, G.R.; Tadepalle, N.; Schiavon, C.R.; Grotjahn, D.A.; Towers, C.G.; Tremblay, M.-È.; Donnelly, M.P.; Ghosh, S.; et al. Mitochondrial DNA Replication Stress Triggers a Pro-Inflammatory Endosomal Pathway of Nucleoid Disposal. Nat. Cell Biol. 2024, 26, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, Z.; Hu, X. cGAS-STING-Mediated Inflammation and Neurodegeneration as a Strategy for the Treatment of Neurodegenerative Diseases. Acta Biochim. Biophys. Sin. 2024, 56, 148–150. [Google Scholar] [CrossRef]
- Li, Q.; Yang, L.; Wang, K.; Chen, Z.; Liu, H.; Yang, X.; Xu, Y.; Chen, Y.; Gong, Z.; Jia, Y. Oxidized Mitochondrial DNA Activates the cGAS-STING Pathway in the Neuronal Intrinsic Immune System after Brain Ischemia-Reperfusion Injury. Neurother. J. Am. Soc. Exp. Neurother. 2024, 21, e00368. [Google Scholar] [CrossRef] [PubMed]
- Xian, H.; Watari, K.; Sanchez-Lopez, E.; Offenberger, J.; Onyuru, J.; Sampath, H.; Ying, W.; Hoffman, H.M.; Shadel, G.S.; Karin, M. Oxidized DNA Fragments Exit Mitochondria via mPTP- and VDAC-Dependent Channels to Activate NLRP3 Inflammasome and Interferon Signaling. Immunity 2022, 55, 1370–1385.e8. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, B.; Xu, L.; Yu, S.; Fu, J.; Wang, J.; Yan, X.; Su, J. ROS-Induced mtDNA Release: The Emerging Messenger for Communication between Neurons and Innate Immune Cells during Neurodegenerative Disorder Progression. Antioxid. Basel Switz. 2021, 10, 1917. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, B.; Duan, R.; Liu, Y. Mitochondrial DNA Leakage and cGas/STING Pathway in Microglia: Crosstalk Between Neuroinflammation and Neurodegeneration. Neuroscience 2024, 548, 1–8. [Google Scholar] [CrossRef]
- Huang, P.; Li, L.; Chen, Y.; Li, Y.; Zhu, D.; Cui, J. Mitochondrial DNA Drives Neuroinflammation through the cGAS-IFN Signaling Pathway in the Spinal Cord of Neuropathic Pain Mice. Open Life Sci. 2024, 19, 20220872. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of Neuroinflammation in Neurodegeneration Development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Dai, K.; Wang, Z.; Gao, B.; Li, L.; Gu, F.; Tao, X.; You, W.; Wang, Z. APE1 Regulates Mitochondrial DNA Damage Repair after Experimental Subarachnoid Haemorrhage in Vivo and in Vitro. Stroke Vasc. Neurol. 2024, 9, 230–242. [Google Scholar] [CrossRef]
- Hussain, M.; Chu, X.; Duan Sahbaz, B.; Gray, S.; Pekhale, K.; Park, J.-H.; Croteau, D.L.; Bohr, V.A. Mitochondrial OGG1 Expression Reduces Age-Associated Neuroinflammation by Regulating Cytosolic Mitochondrial DNA. Free Radic. Biol. Med. 2023, 203, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Yan, S.S. Mitochondrial Medicine for Neurodegenerative Diseases. Int. J. Biochem. Cell Biol. 2010, 42, 560–572. [Google Scholar] [CrossRef]
- Garone, C.; Viscomi, C. Towards a Therapy for Mitochondrial Disease: An Update. Biochem. Soc. Trans. 2018, 46, 1247–1261. [Google Scholar] [CrossRef]
- Blázquez-Bermejo, C.; Carreño-Gago, L.; Molina-Granada, D.; Aguirre, J.; Ramón, J.; Torres-Torronteras, J.; Cabrera-Pérez, R.; Martín, M.Á.; Domínguez-González, C.; de la Cruz, X.; et al. Increased dNTP Pools Rescue mtDNA Depletion in Human POLG-Deficient Fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 7168–7179. [Google Scholar] [CrossRef]
- Yi, Z.; Zhang, X.; Tang, W.; Yu, Y.; Wei, X.; Zhang, X.; Wei, W. Strand-Selective Base Editing of Human Mitochondrial DNA Using mitoBEs. Nat. Biotechnol. 2024, 42, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Joore, I.P.; Shehata, S.; Muffels, I.; Castro-Alpízar, J.; Jiménez-Curiel, E.; Nagyova, E.; Levy, N.; Tang, Z.; Smit, K.; Vermeij, W.P.; et al. Correction of Pathogenic Mitochondrial DNA in Patient-Derived Disease Models Using Mitochondrial Base Editors. PLoS Biol. 2025, 23, e3003207. [Google Scholar] [CrossRef]
- Falkenberg, M.; Hirano, M. Editing the Mitochondrial Genome. N. Engl. J. Med. 2020, 383, 1489–1491. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Baek, G.; Kim, J.-S. Precision Mitochondrial DNA Editing with High-Fidelity DddA-Derived Base Editors. Nat. Biotechnol. 2023, 41, 378–386. [Google Scholar] [CrossRef]
- Lim, K. Mitochondrial Genome Editing: Strategies, Challenges, and Applications. BMB Rep. 2024, 57, 19–29. [Google Scholar] [CrossRef]
- Moraes, C.T. Tools for Editing the Mammalian Mitochondrial Genome. Hum. Mol. Genet. 2024, 33, R92–R99. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.H.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R.; et al. Genome Editing in Mitochondria Corrects a Pathogenic mtDNA Mutation in Vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Gammage, P.A.; Gaude, E.; Van Haute, L.; Rebelo-Guiomar, P.; Jackson, C.B.; Rorbach, J.; Pekalski, M.L.; Robinson, A.J.; Charpentier, M.; Concordet, J.-P.; et al. Near-Complete Elimination of Mutant mtDNA by Iterative or Dynamic Dose-Controlled Treatment with mtZFNs. Nucleic Acids Res. 2016, 44, 7804–7816. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Jiang, P.; Wang, Z.; Kong, W.; Feng, L. Mitochondrial Transplantation: A Novel Therapeutic Approach for Treating Diseases. MedComm 2025, 6, e70253. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Del Nido, P.J.; Emani, S.M. Mitochondrial Transplantation: The Advance to Therapeutic Application and Molecular Modulation. Front. Cardiovasc. Med. 2023, 10, 1268814. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Yáñez-Sepúlveda, R.; Tornero-Aguilera, J.F. Mitochondrial Transfer as a Novel Therapeutic Approach in Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 8848. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Lee, S.; Kim, W.-K.; Han, B.-S. Mitochondrial Transplantation: An Overview of a Promising Therapeutic Approach. BMB Rep. 2023, 56, 488–495. [Google Scholar] [CrossRef]
- Zhang, T.-G.; Miao, C.-Y. Mitochondrial Transplantation as a Promising Therapy for Mitochondrial Diseases. Acta Pharm. Sin. B 2023, 13, 1028–1035. [Google Scholar] [CrossRef]
- Khan, M.M.; Paez, H.G.; Pitzer, C.R.; Alway, S.E. The Therapeutic Potential of Mitochondria Transplantation Therapy in Neurodegenerative and Neurovascular Disorders. Curr. Neuropharmacol. 2023, 21, 1100–1116. [Google Scholar] [CrossRef]
- Eo, H.; Yu, S.-H.; Choi, Y.; Kim, Y.; Kang, Y.C.; Lee, H.; Kim, J.H.; Han, K.; Lee, H.K.; Chang, M.-Y.; et al. Mitochondrial Transplantation Exhibits Neuroprotective Effects and Improves Behavioral Deficits in an Animal Model of Parkinson’s Disease. Neurother. J. Am. Soc. Exp. Neurother. 2024, 21, e00355. [Google Scholar] [CrossRef]
- Riou, A.; Broeglin, A.; Grimm, A. Mitochondrial Transplantation in Brain Disorders: Achievements, Methods, and Challenges. Neurosci. Biobehav. Rev. 2025, 169, 105971. [Google Scholar] [CrossRef]
- Li, S.-J.; Zheng, Q.-W.; Zheng, J.; Zhang, J.-B.; Liu, H.; Tie, J.-J.; Zhang, K.-L.; Wu, F.-F.; Li, X.-D.; Zhang, S.; et al. Mitochondria Transplantation Transiently Rescues Cerebellar Neurodegeneration Improving Mitochondrial Function and Reducing Mitophagy in Mice. Nat. Commun. 2025, 16, 2839. [Google Scholar] [CrossRef]
- Fields, M.; Marcuzzi, A.; Gonelli, A.; Celeghini, C.; Maximova, N.; Rimondi, E. Mitochondria-Targeted Antioxidants, an Innovative Class of Antioxidant Compounds for Neurodegenerative Diseases: Perspectives and Limitations. Int. J. Mol. Sci. 2023, 24, 3739. [Google Scholar] [CrossRef]
- Charmpilas, N.; Fang, E.F.; Palikaras, K. Mitophagy and Neuroinflammation: A Compelling Interplay. Curr. Neuropharmacol. 2023, 21, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, J.; Yu, L.; Qiao, G.; Qin, D.; Yuen-Kwan Law, B.; Ren, F.; Wu, J.; Wu, A. Mitophagy and cGAS-STING Crosstalk in Neuroinflammation. Acta Pharm. Sin. B 2024, 14, 3327–3361. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy Inhibits Amyloid-β and Tau Pathology and Reverses Cognitive Deficits in Models of Alzheimer’s Disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Lagunas-Rangel, F.A. Sirtuins in Mitophagy: Key Gatekeepers of Mitochondrial Quality. Mol. Cell. Biochem. 2025, 480, 5877–5896. [Google Scholar] [CrossRef]
- Tang, M.-B.; Liu, Y.-X.; Hu, Z.-W.; Luo, H.-Y.; Zhang, S.; Shi, C.-H.; Xu, Y.-M. Study Insights in the Role of PGC-1α in Neurological Diseases: Mechanisms and Therapeutic Potential. Front. Aging Neurosci. 2024, 16, 1454735. [Google Scholar] [CrossRef]
- Xu, H.; Liu, Y.-Y.; Li, L.-S.; Liu, Y.-S. Sirtuins at the Crossroads between Mitochondrial Quality Control and Neurodegenerative Diseases: Structure, Regulation, Modifications, and Modulators. Aging Dis. 2023, 14, 794–824. [Google Scholar] [CrossRef] [PubMed]
- Golpich, M.; Amini, E.; Mohamed, Z.; Azman Ali, R.; Mohamed Ibrahim, N.; Ahmadiani, A. Mitochondrial Dysfunction and Biogenesis in Neurodegenerative Diseases: Pathogenesis and Treatment. CNS Neurosci. Ther. 2017, 23, 5–22. [Google Scholar] [CrossRef]
- Rakshe, P.S.; Dutta, B.J.; Chib, S.; Maurya, N.; Singh, S. Unveiling the Interplay of AMPK/SIRT1/PGC-1α Axis in Brain Health: Promising Targets against Aging and NDDs. Ageing Res. Rev. 2024, 96, 102255. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhang, J.; Deng, Q.; Chen, X. Mitophagy-Associated Programmed Neuronal Death and Neuroinflammation. Front. Immunol. 2024, 15, 1460286. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, B.; Sinha, S.C.; Amin, S.; Gan, L. Mechanism and Therapeutic Potential of Targeting cGAS-STING Signaling in Neurological Disorders. Mol. Neurodegener. 2023, 18, 79. [Google Scholar] [CrossRef]
- Hinkle, J.T.; Patel, J.; Panicker, N.; Karuppagounder, S.S.; Biswas, D.; Belingon, B.; Chen, R.; Brahmachari, S.; Pletnikova, O.; Troncoso, J.C.; et al. STING Mediates Neurodegeneration and Neuroinflammation in Nigrostriatal α-Synucleinopathy. Proc. Natl. Acad. Sci. U. S. A. 2022, 119, e2118819119. [Google Scholar] [CrossRef]
- Chung, S.; Jeong, J.-H.; Park, J.-C.; Han, J.W.; Lee, Y.; Kim, J.-I.; Mook-Jung, I. Blockade of STING Activation Alleviates Microglial Dysfunction and a Broad Spectrum of Alzheimer’s Disease Pathologies. Exp. Mol. Med. 2024, 56, 1936–1951. [Google Scholar] [CrossRef]
- Fryer, A.L.; Abdullah, A.; Mobilio, F.; Jobling, A.; Moore, Z.; de Veer, M.; Zheng, G.; Wong, B.X.; Taylor, J.M.; Crack, P.J. Pharmacological Inhibition of STING Reduces Neuroinflammation-Mediated Damage Post-Traumatic Brain Injury. Br. J. Pharmacol. 2024, 181, 3118–3135. [Google Scholar] [CrossRef]
- Zhang, H.; He, Z.; Yin, C.; Yang, S.; Li, J.; Lin, H.; Hu, G.; Wu, A.; Qin, D.; Hu, G.; et al. STING-Mediated Neuroinflammation: A Therapeutic Target in Neurodegenerative Diseases. Front. Aging Neurosci. 2025, 17, 1659216. [Google Scholar] [CrossRef]
- Han, Q.-Q.; Le, W. NLRP3 Inflammasome-Mediated Neuroinflammation and Related Mitochondrial Impairment in Parkinson’s Disease. Neurosci. Bull. 2023, 39, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Auger, A.; Faidi, R.; Rickman, A.D.; Martinez, C.P.; Fajfer, A.; Carling, J.; Hilyard, A.; Ali, M.; Ono, R.; Cleveland, C.; et al. Post-Symptomatic NLRP3 Inhibition Rescues Cognitive Impairment and Mitigates Amyloid and Tau Driven Neurodegeneration. NPJ Dement. 2025, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, L.; Liu, Y.-Z.; Shen, X.-L.; Wu, T.-Y.; Zhang, T.; Wang, W.; Wang, Y.-X.; Jiang, C.-L. NLRP3 Inflammasome Mediates Chronic Mild Stress-Induced Depression in Mice via Neuroinflammation. Int. J. Neuropsychopharmacol. 2015, 18, pyv006. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, V. NLRP3 Inhibitors: Unleashing Their Therapeutic Potential against Inflammatory Diseases. Biochem. Pharmacol. 2023, 218, 115915. [Google Scholar] [CrossRef]
- Szczesny, B.; Brunyanszki, A.; Olah, G.; Mitra, S.; Szabo, C. Opposing Roles of Mitochondrial and Nuclear PARP1 in the Regulation of Mitochondrial and Nuclear DNA Integrity: Implications for the Regulation of Mitochondrial Function. Nucleic Acids Res. 2014, 42, 13161–13173. [Google Scholar] [CrossRef]
- Gallyas, F.; Sumegi, B. Mitochondrial Protection by PARP Inhibition. Int. J. Mol. Sci. 2020, 21, 2767. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, G. The Role of PARP1 in Neurodegenerative Diseases and Aging. FEBS J. 2022, 289, 2013–2024. [Google Scholar] [CrossRef]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ Consumption by PARP1 in Response to DNA Damage Triggers Metabolic Shift Critical for Damaged Cell Survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef] [PubMed]
- Felici, R.; Cavone, L.; Lapucci, A.; Guasti, D.; Bani, D.; Chiarugi, A. PARP Inhibition Delays Progression of Mitochondrial Encephalopathy in Mice. Neurother. J. Am. Soc. Exp. Neurother. 2014, 11, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Schriewer, J.M.; Peek, C.B.; Bass, J.; Schumacker, P.T. ROS-Mediated PARP Activity Undermines Mitochondrial Function after Permeability Transition Pore Opening during Myocardial Ischemia-Reperfusion. J. Am. Heart Assoc. 2013, 2, e000159. [Google Scholar] [CrossRef] [PubMed]
- Qi, R.; Sammler, E.; Gonzalez-Hunt, C.P.; Barraza, I.; Pena, N.; Rouanet, J.P.; Naaldijk, Y.; Goodson, S.; Fuzzati, M.; Blandini, F.; et al. A Blood-Based Marker of Mitochondrial DNA Damage in Parkinson’s Disease. Sci. Transl. Med. 2023, 15, eabo1557. [Google Scholar] [CrossRef]
Figure 1.
Mitochondrial nucleoid arrangement and genome content. Left: Schematic representation of a mitochondrion, highlighting nucleoids tethered to the inner mitochondrial membrane (IMM). A transparent nucleoid visually reveals the mitochondrial DNA (mtDNA). Adjacent to the nucleoid, electron transport chain (ETC) subunits are shown embedded in the IMM. Right: The human mitochondrial genome map, showing the distribution of protein-coding genes for ETC complexes I, III, IV, and ATP synthase, plus mitochondrial tRNAs and rRNAs needed for local translation within the organelle..
Figure 1.
Mitochondrial nucleoid arrangement and genome content. Left: Schematic representation of a mitochondrion, highlighting nucleoids tethered to the inner mitochondrial membrane (IMM). A transparent nucleoid visually reveals the mitochondrial DNA (mtDNA). Adjacent to the nucleoid, electron transport chain (ETC) subunits are shown embedded in the IMM. Right: The human mitochondrial genome map, showing the distribution of protein-coding genes for ETC complexes I, III, IV, and ATP synthase, plus mitochondrial tRNAs and rRNAs needed for local translation within the organelle..
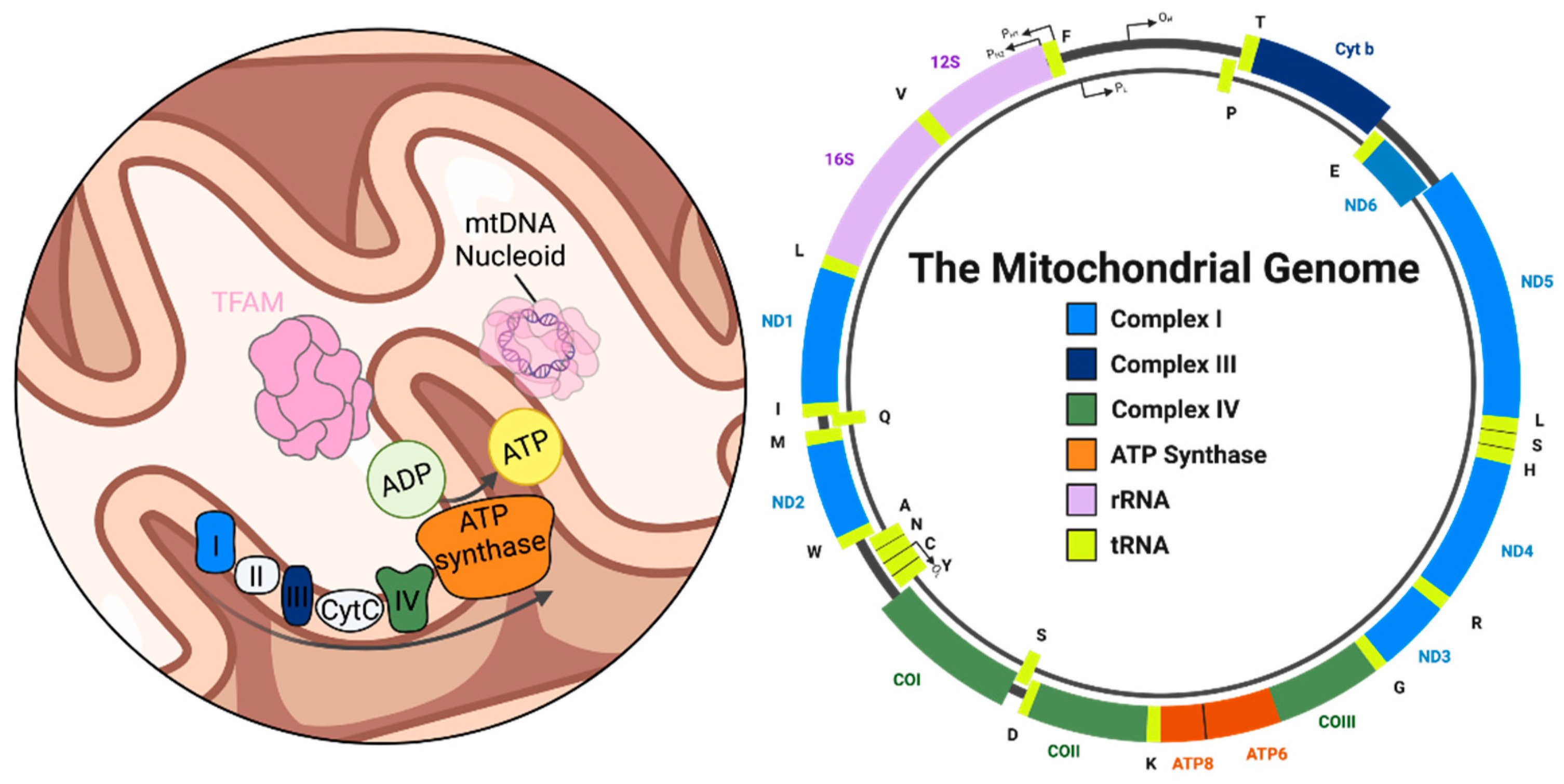
Figure 2.
Schematic of mitochondrial base excision repair (BER) activity levels and their respective cellular outcomes. Balanced BER maintains mitochodnrial DNA (mtDNA) integrity. Impaired BER leads to accumulation of mtDNA lesions, genome instability, and energy failure. Excessive BER generates toxic repair intermediates, triggers poly(ADP-ribose) polymerase 1 (PARP1) overactivation, genome instability, and energy failure.
Figure 2.
Schematic of mitochondrial base excision repair (BER) activity levels and their respective cellular outcomes. Balanced BER maintains mitochodnrial DNA (mtDNA) integrity. Impaired BER leads to accumulation of mtDNA lesions, genome instability, and energy failure. Excessive BER generates toxic repair intermediates, triggers poly(ADP-ribose) polymerase 1 (PARP1) overactivation, genome instability, and energy failure.
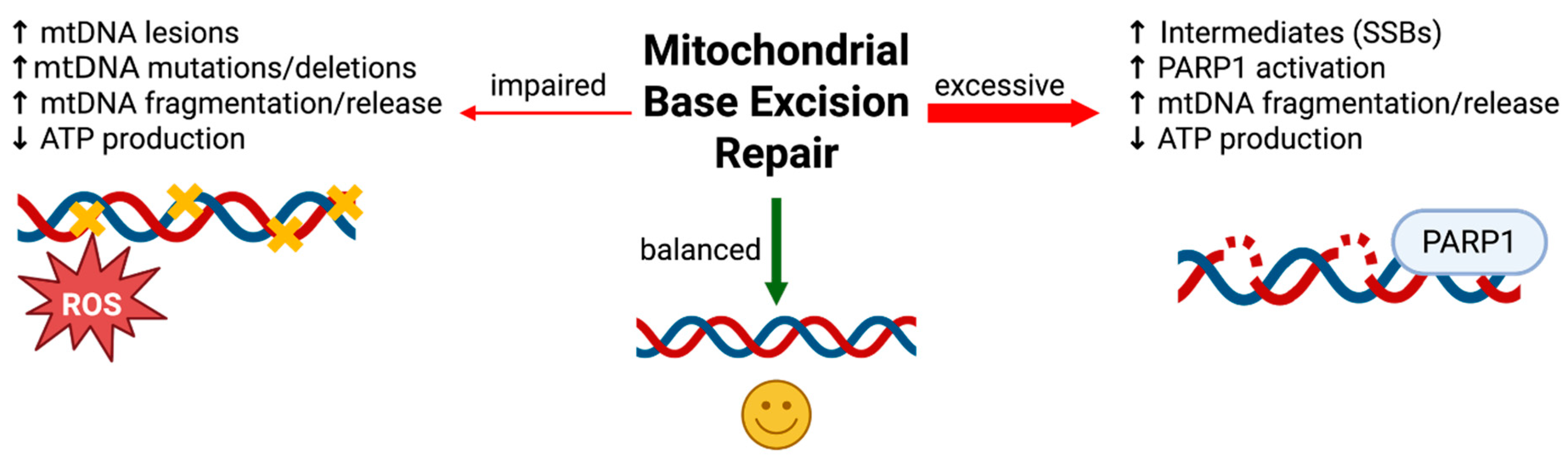
Figure 3.
Mechanisms of mitochondrial DNA (mtDNA) release under cellular stress conditions. Apoptotic mtDNA release: Mitochondrial outer membrane permeabilization (MOMP) is induced by the pro-apoptotic BCL-2 family proteins BAX/BAK macropore formation during apoptosis, enabling the escape of mtDNA and entire nucleoids into the cytosol; mPTP-dependent mtDNA release: The mitochondrial permeability transition pore (mPTP) opens in response to excessive calcium or oxidative stress, allowing small mtDNA fragments to exit the matrix. Persistent mPTP opening leads to mitochondrial swelling and outer membrane rupture, facilitating release of matrix contents into the cytosol; VDAC oligomer-driven mtDNA release: Under moderate cellular stress, voltage-dependent anion channel (VDAC) oligomerization promotes the release of small mtDNA fragments into the cytosol. This process requires prior permeabilization of the inner membrane, often via mPTP; mtDNA release from failed mitophagy or lysosomal rupture: Ineffective removal of damaged mitochondria by mitophagy or lysosomal rupture results in the leakage of mtDNA from permeabilized or ruptured organelles into the cytosol.
Figure 3.
Mechanisms of mitochondrial DNA (mtDNA) release under cellular stress conditions. Apoptotic mtDNA release: Mitochondrial outer membrane permeabilization (MOMP) is induced by the pro-apoptotic BCL-2 family proteins BAX/BAK macropore formation during apoptosis, enabling the escape of mtDNA and entire nucleoids into the cytosol; mPTP-dependent mtDNA release: The mitochondrial permeability transition pore (mPTP) opens in response to excessive calcium or oxidative stress, allowing small mtDNA fragments to exit the matrix. Persistent mPTP opening leads to mitochondrial swelling and outer membrane rupture, facilitating release of matrix contents into the cytosol; VDAC oligomer-driven mtDNA release: Under moderate cellular stress, voltage-dependent anion channel (VDAC) oligomerization promotes the release of small mtDNA fragments into the cytosol. This process requires prior permeabilization of the inner membrane, often via mPTP; mtDNA release from failed mitophagy or lysosomal rupture: Ineffective removal of damaged mitochondria by mitophagy or lysosomal rupture results in the leakage of mtDNA from permeabilized or ruptured organelles into the cytosol.
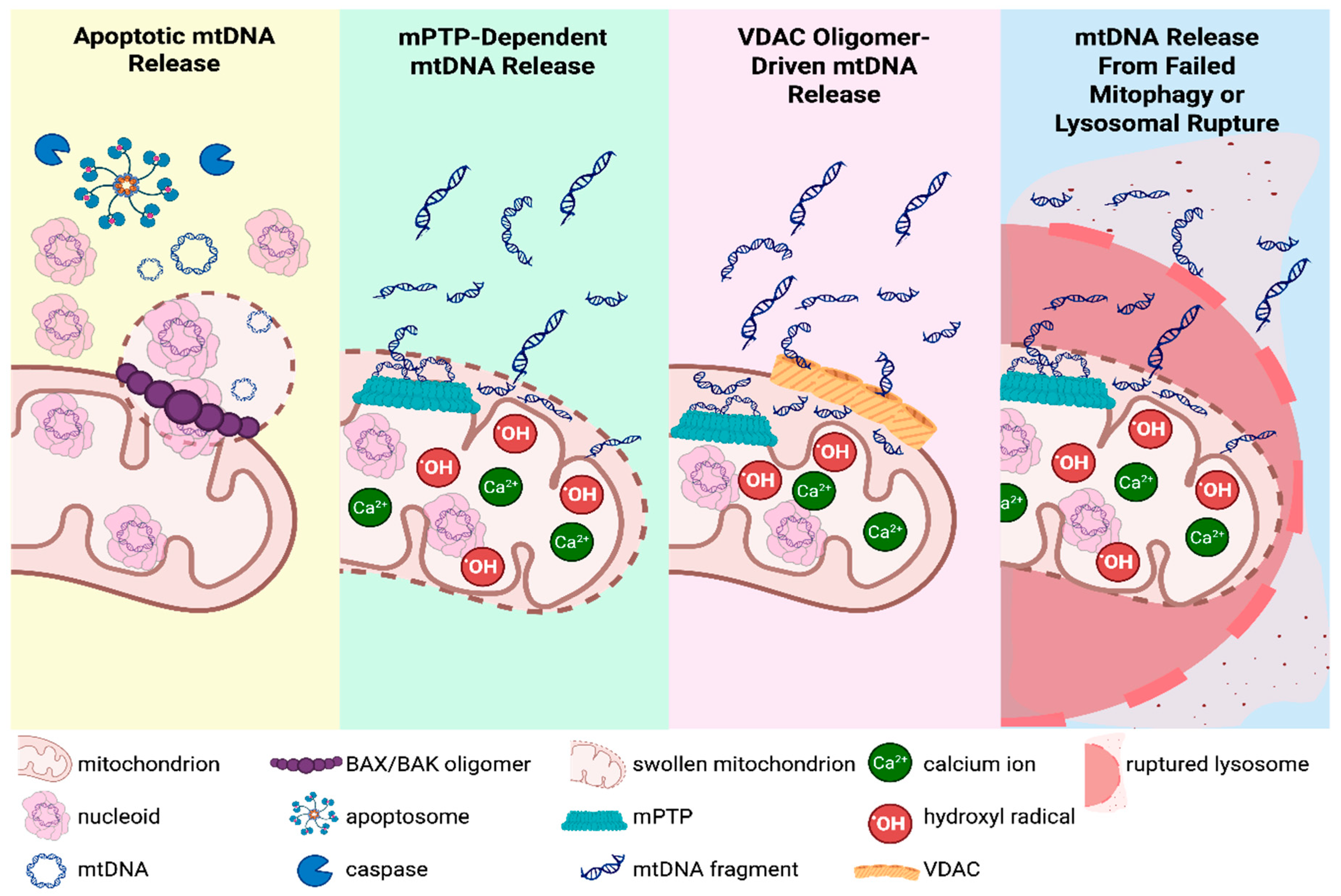
Figure 4.
Cascade of Neurodegeneration Initiated by mitochondrial DNA (mtDNA) Damage. Unrepaired mtDNA damage in neurons impairs electron transport chain function, reduces ATP production, and increases reactive oxygen species (ROS). Damaged mitochondria accumulate when mitophagy is impaired or overwhelmed, further elevating ROS levels and releasing mtDNA fragments. These fragments act as damage-associated molecular patterms (DAMPs) that activate microglial innate immune pathways, driving neuroinflammation, neuronal dysfunction, and progressive neurodegeneration.
Figure 4.
Cascade of Neurodegeneration Initiated by mitochondrial DNA (mtDNA) Damage. Unrepaired mtDNA damage in neurons impairs electron transport chain function, reduces ATP production, and increases reactive oxygen species (ROS). Damaged mitochondria accumulate when mitophagy is impaired or overwhelmed, further elevating ROS levels and releasing mtDNA fragments. These fragments act as damage-associated molecular patterms (DAMPs) that activate microglial innate immune pathways, driving neuroinflammation, neuronal dysfunction, and progressive neurodegeneration.
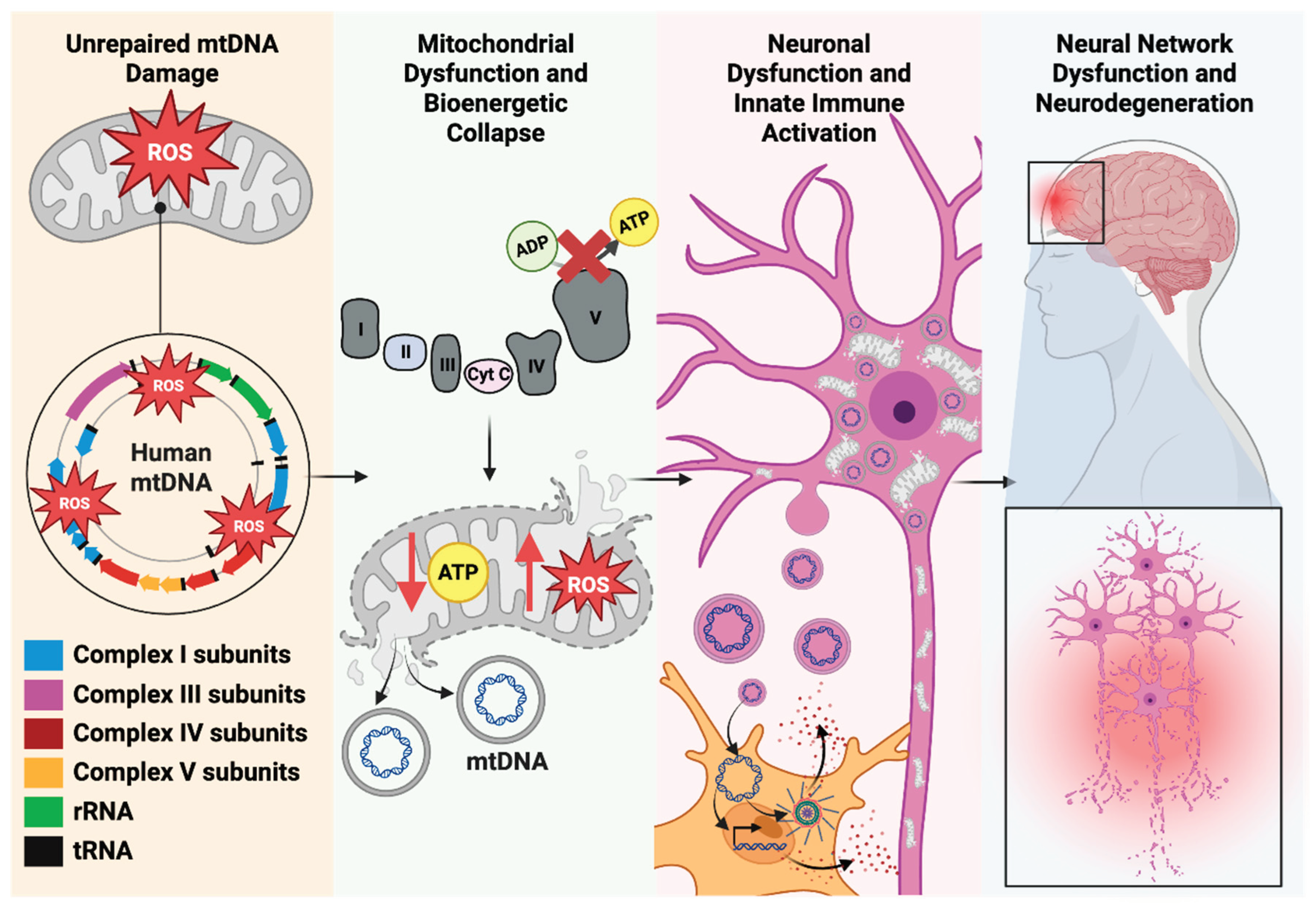
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



