Introduction
Sleep is a physiological state essential for maintaining the health of our brain and our body. It is characterized by the cyclic change in state between Non-Rapid Eye Movement (NREM) Sleep and Rapid Eye Movement (REM) Sleep every 80-110 minutes over the duration of sleep [
4]. NREM stages are associated with restoration, synaptic pruning, and clearance of neurotoxic materials such as β-amyloid [
32,
48], whereas REM Sleep contributes to emotional regulation, memory consolidation, and neuroplasticity [
4].
Sleep deprivation (SD) is a major public health concern with growing prevalence in all age groups. Chronic sleep loss is linked to adverse effects such as an increased risk of Alzheimer’s disease [
8,
34], anxiety disorders [
29,
48], cognitive decline [
39,
46,
49], cardiovascular dysfunction, and other systemic inflammatory conditions [
7,
9,
44].
The emerging evidence shows that sleep is not only a neurobehavioral phenomenon but a critical modulator of immune, endocrine, and autonomic function. Even partial sleep restriction has shown to disrupt circadian homeostasis, elevates sympathetic drive, and induces a persistent low-grade inflammatory state. These changes are associated with the dysregulation of pro-inflammatory cytokines, particularly Tumor Necrosis Factor-α (TNF-α), Interleukin-1β (IL-1β), and Interleukin-6 (IL-6), Hypothalamic-pituitary-adrenal axis (HPA axis) dysregulation and altered activity of microglia.
Given these findings, it is critical to understand the molecular mechanisms through which sleep deprivation drives inflammatory pathology. This Systematic Review aims to find the molecular mechanisms by which sleep deprivation induces neuroinflammation in humans and animal models, with a focus on pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) and microglial activation.
Methods
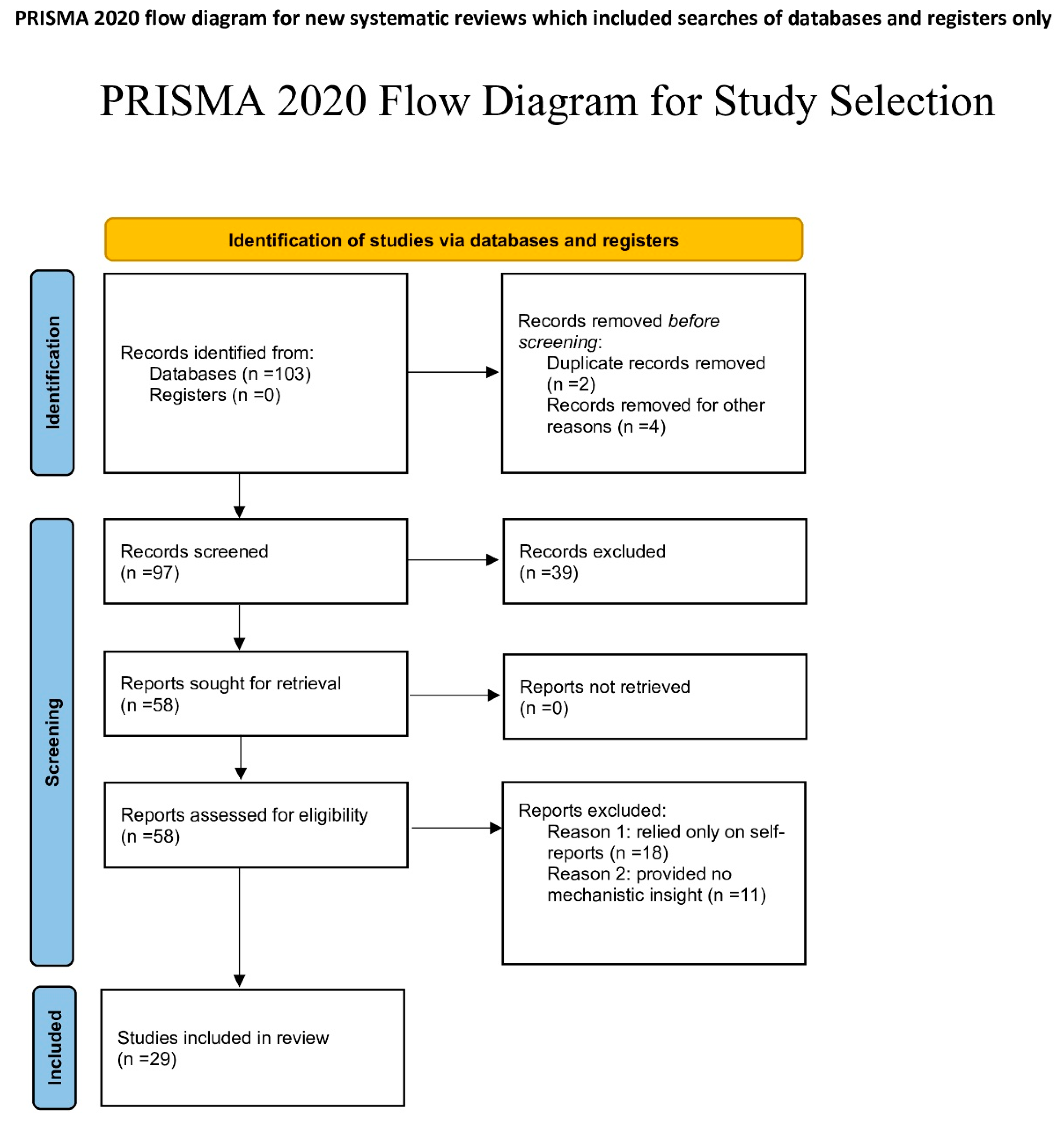
A systematic review was conducted to investigate how sleep deprivation drives inflammation via neuroimmune mechanisms, with a particular focus on microglial activation and TNF-α signaling in mice and humans. All screening and extraction were performed by a single reviewer due to scope constraints.
Search Strategy:
A manual and systematic search was performed to identify relevant scientific articles on PubMed, Google Scholar, and ResearchRabbit from 1995 to July 30th 2025, using the following keywords in titles and abstracts: ("sleep deprivation" OR "sleep fragmentation") AND ("microglia" OR "TNF-α") AND ("inflammation"). Due to platform limitations, exact reproducible queries could not be exported from Google Scholar or ResearchRabbit. This limitation is acknowledged, and results should be interpreted accordingly. Searches were limited to English-language articles only.
Eligibility Criteria:
Experimental studies (in vivo or in vitro) assessing neuroimmune or inflammatory outcomes following sleep deprivation, and mechanistic reviews providing molecular or cellular insights into TNF-α and microglial pathways were included. Human experimental or observational studies were also eligible when they measured systemic or neural inflammatory markers following sleep deprivation, even if they did not include microglial endpoints. Clinical trials, animal studies, and in vitro studies were all treated equally throughout the process. Mechanistic reviews were included only when they contributed essential molecular context not accessible from primary data alone. Studies had to mention at least two of the following core terms: sleep deprivation or sleep fragmentation, microglia, TNF-α, inflammation, to be included. Studies relying solely on self-reported sleep measures or not meeting the minimum keyword threshold were excluded from this review.
Study Selection:
A total of 97 records were identified through database searching across Google Scholar and ResearchRabbit (literature discovery platform). After title and abstract screening, 47 records were excluded for not meeting the inclusion criteria. To ensure consistency, a random sample of 20% of the excluded records at the title/abstract stage (n = 9 out of 47) was re-screened one week later by the same author to confirm the initial decision, demonstrating high intra-rater reliability. The full texts of 50 articles were assessed for eligibility, and 29 were included in the final synthesis. Study selection was conducted by a single reviewer.
Data Extraction and Assessment:
Key study characteristics (species, SD method and duration, brain region, TNF-α and microglial outcomes, assay methods) were extracted using a standardized chart. Although no formal risk-of-bias tool was applied, as the included studies were primarily mechanistic and heterogeneous in design, the studies were informally evaluated for clarity of experimental design, consistency of SD protocols, and transparency of outcome reporting. Informal evaluation considered study clarity, reproducibility of SD protocols, appropriateness of outcome assays, and general methodological transparency.
Synthesis:
Although a structured thematic systematic review or meta-analysis was considered, due to the heterogeneity in study designs, species, and outcome measures, a narrative synthesis was performed. Findings were grouped by species (mouse vs human) and type/duration of sleep deprivation (acute vs chronic), with emphasis on TNF-α modulation, microglial activation, and associated signaling pathways. Human studies primarily contributed cytokine and systemic inflammatory data, while microglial mechanisms were derived mainly from rodent and in vitro models. Species-specific differences were retained, but conserved pathways (TNF-α signaling, ATP-mediated microglial activation, HPA-axis effects) were synthesized together when appropriate. Some studies contributed data to multiple mechanistic categories (cytokines, microglia, HPA axis, oxidative stress). These were analyzed under each relevant domain.
Results
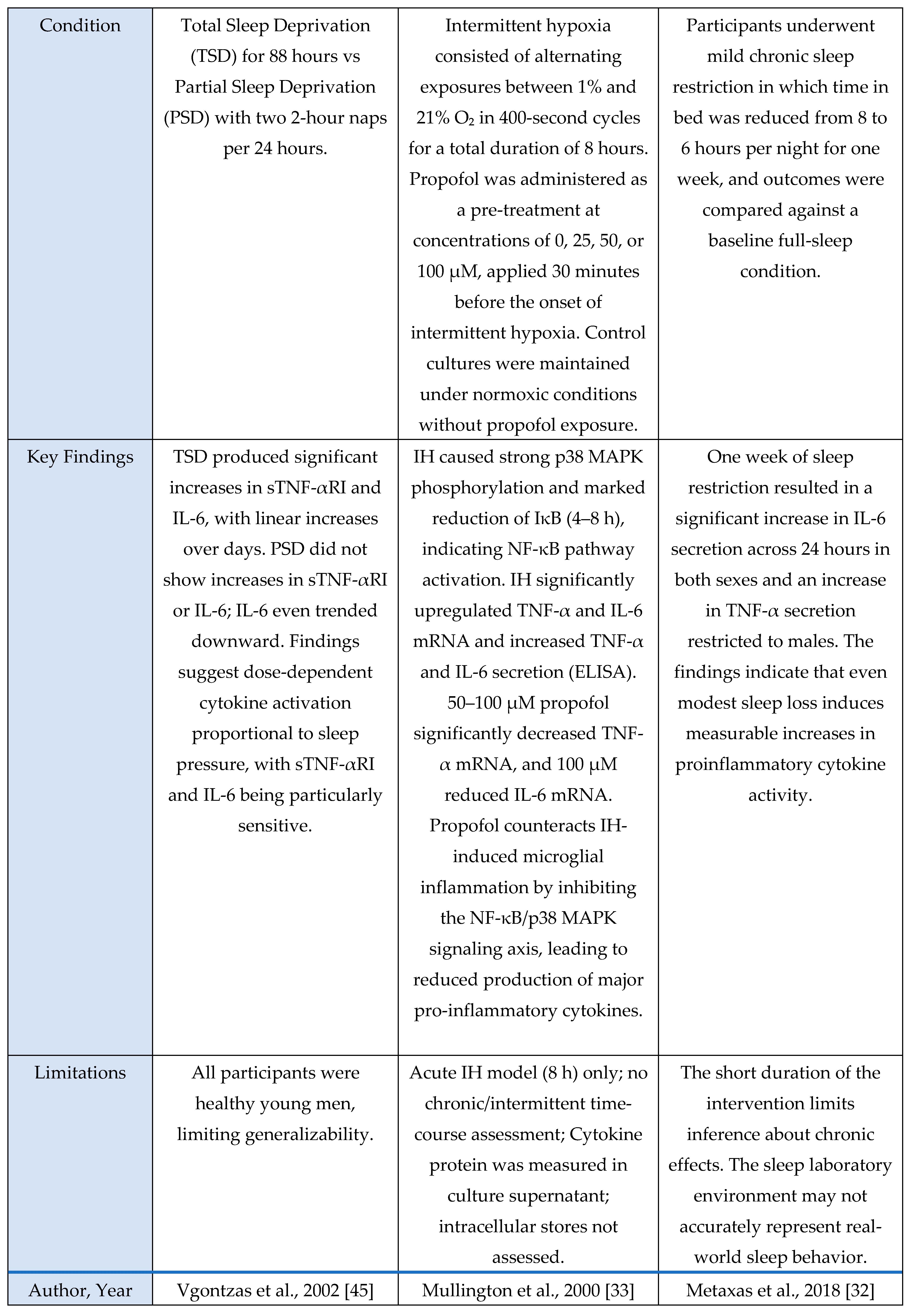
Cytokine aspect
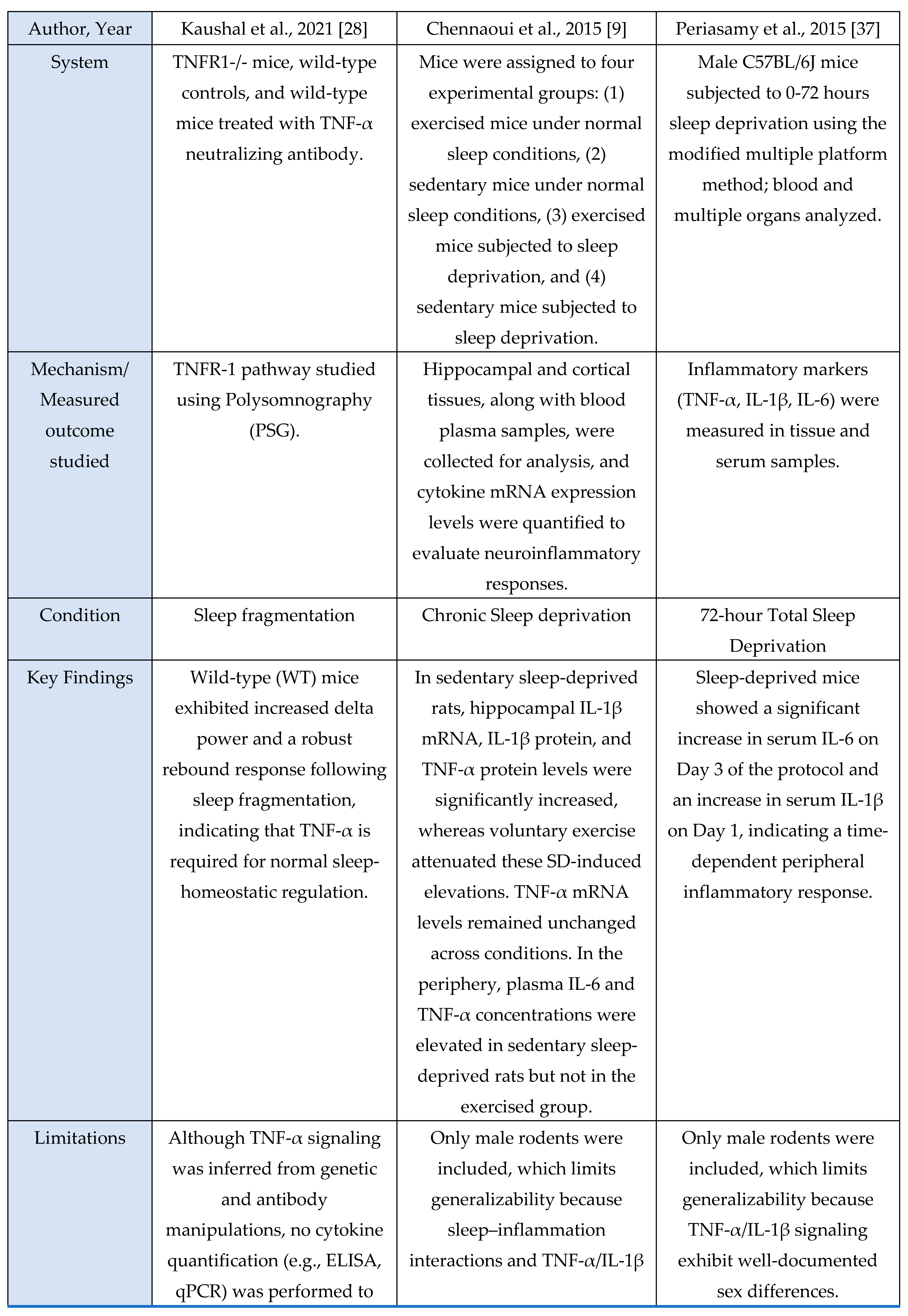
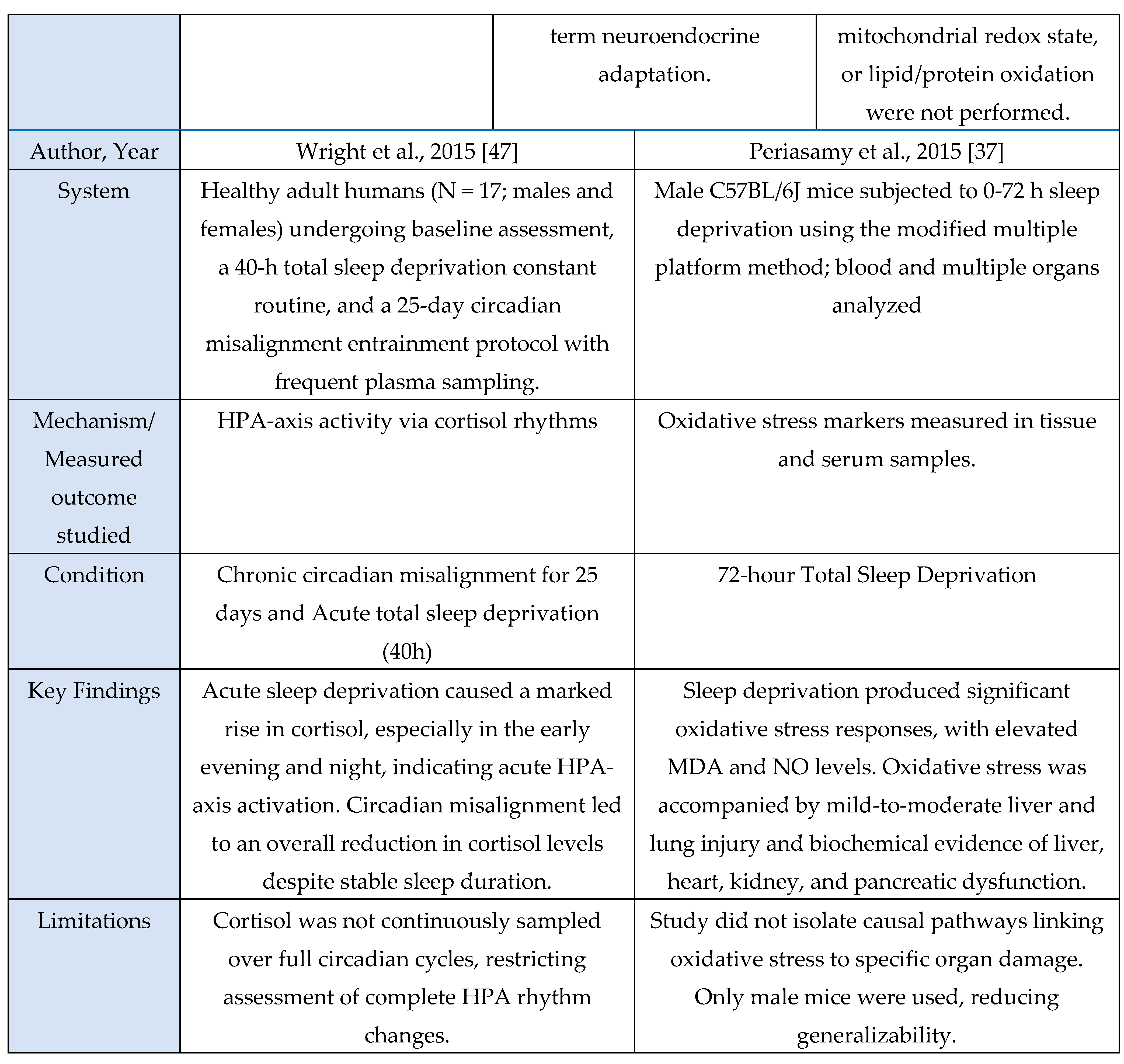
A total of 23 studies met inclusion criteria for the Cytokine Axis, comprising 13 human studies, 9 murine models, and 1 in vitro investigation. Across methodologies, a consistent pattern was observed in which sleep loss, circadian misalignment, or stress exposure induced elevations in pro-inflammatory cytokines including IL-6, TNF-α and IL-1β. These cytokine shifts were observed in both central and peripheral compartments, indicating that sleep stress perturbations engage conserved inflammatory pathways across species. This axis is mechanistically important because these cytokines serve as upstream initiators and amplifiers of neuroimmune signaling that subsequently alter microglial activation states, synaptic homeostasis, and sleep-regulatory circuits.
Evidence Synthesis
This section summarizes the characteristics and principal findings of the included studies shown in
Figure 1. Across the 23 included studies, 13 were human investigations, 9 were murine experimental models, and 1 used an in-vitro microglial system. Human studies comprised experimental sleep-manipulation paradigms, cross-sectional observational analyses, and behavioral or lifestyle interventions. Experimental paradigms most often employed total or partial sleep deprivation to induce changes in inflammatory signaling [
1,
7,
13,
18,
23,
25,
33,
36,
42,
44,
47]. Cross-sectional work focused on sleep-dependent redistribution of circulating immune cells [
7], diurnal cytokine rhythms [
18,
23,
45], and inflammatory profiles during circadian misalignment or chronic sleep disruption [
36,
47]. Several studies incorporated polysomnography [
1,
25,
44,
45] or dense sampling protocols [
33,
42] to relate sleep architecture with circulating IL-6, TNF-α, IL-10, cortisol, and soluble TNF receptors. Two human trials tested interventions: weight-loss programs [
1] and behavioral sleep therapies (CBT-I, Tai Chi Chih, sleep-seminar control) [
22], both reporting measurable immunomodulatory effects.
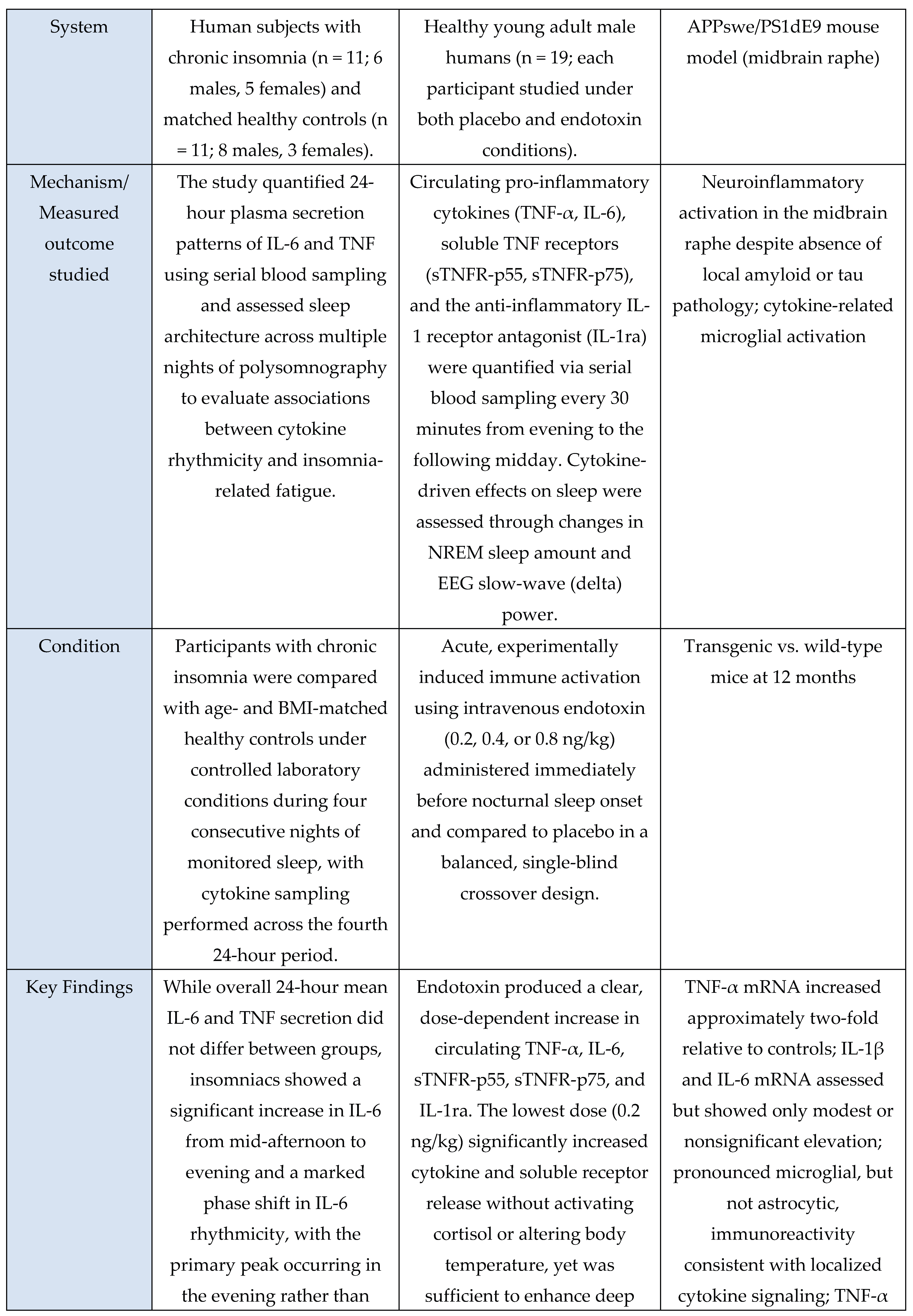
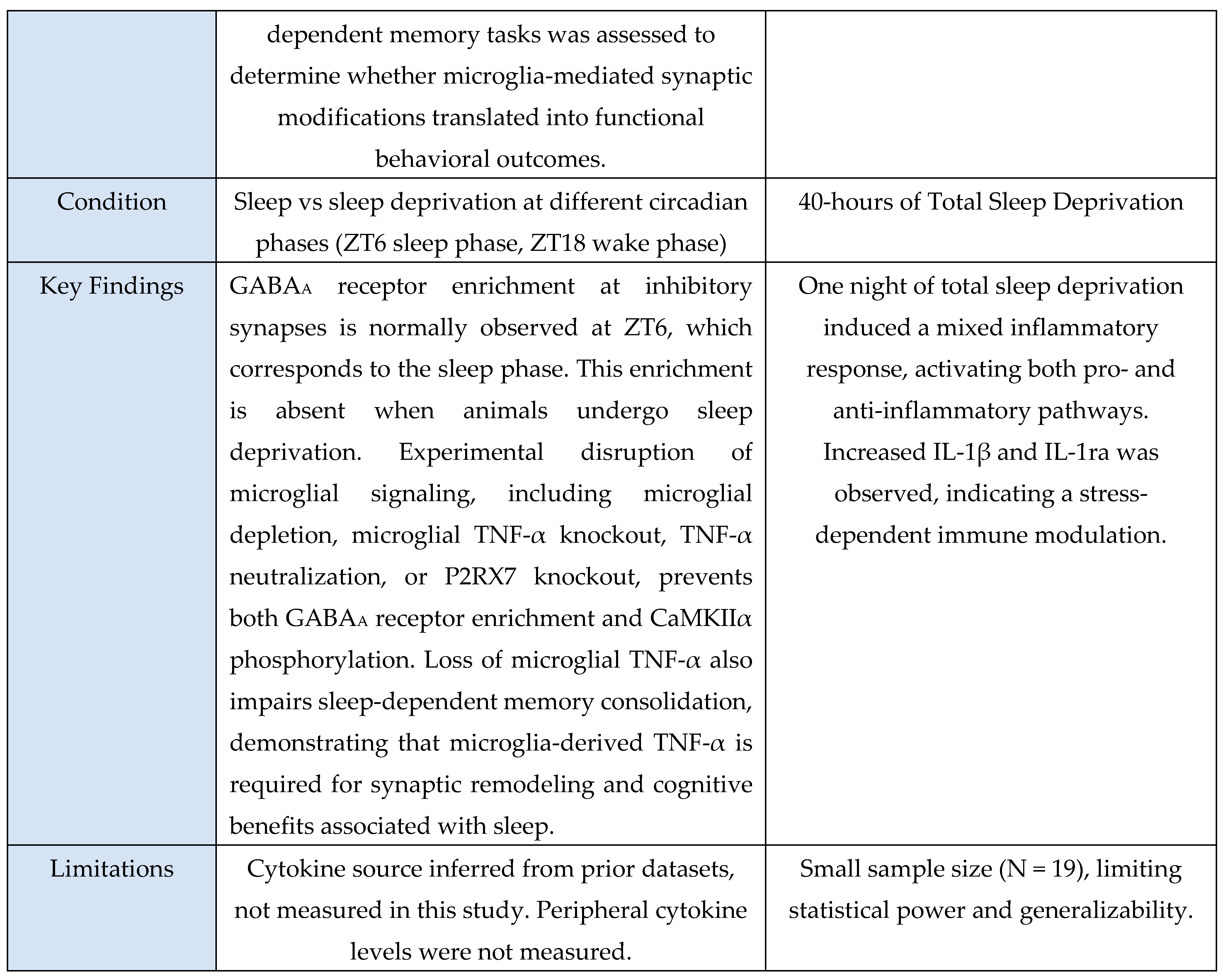
Murine studies uniformly used controlled experimental manipulations to examine mechanistic links between sleep, microglial activation, and cytokine signaling. These included chronic sleep fragmentation [
28], acute or total sleep deprivation [
9,
37,
39], genetic perturbations of microglial receptors [
46], TNF-α or P2RX7 knockouts [
39], and LPS-induced neuroinflammation to probe cytokine transport across the Blood-Brain-Barrier (BBB) [
35]. Additional studies used transcriptomic analyses to identify neuroinflammatory pathway enrichment [
29] or characterized sleep-deprivation-induced microglial changes in specific brain regions such as the midbrain raphe [
32]. Rodent models consistently quantified TNF-α, IL-1β, IL-6, Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB)/Mitogen-activated protein kinase (MAPK) activity, oxidative stress markers, and synaptic proteins using qPCR, ELISA, Western blotting, and immunohistochemistry.
The sole in-vitro study [
30] examined NF-κB and MAPK activation under inflammatory stimulation, measuring IκB degradation, p38 MAPK phosphorylation, and microglia-derived cytokine release.
Across human, murine, and in-vitro evidence, a consistent pattern was observed: sleep deprivation, circadian disruption, or immune challenge reliably increased pro-inflammatory signaling. Human studies [
1,
7,
13,
18,
22,
23,
25,
33,
36,
42,
44,
45,
47] repeatedly observed elevations in IL-6 and TNF-α following sleep loss, in some cases persisting beyond recovery sleep [
25] or manifesting as disrupted diurnal rhythmicity in insomnia [
45]. Murine studies [
9,
28,
29,
32,
35,
37,
39,
46] showed parallel activation of central and peripheral cytokine pathways, with receptor-specific requirements demonstrated in models using TNF-α or P2RX7 deletion [
39] and BBB-transport mechanisms identified through LPS models [
35]. Lifestyle factors such as voluntary exercise modulated neuroinflammatory responses [
9]. The in-vitro findings [
30] supported pathway-level mechanisms, confirming microglial NF-κB and p38 MAPK activation and pharmacological suppression of cytokine release.
Overall, human studies detailed systemic inflammatory responses, whereas murine and in-vitro models provided mechanistic resolution, identifying microglial and receptor-level pathways that drive cytokine changes. Across evidence domains, the findings consistently support a strong association between sleep disruption and activation of microglia-dependent cytokine signaling.
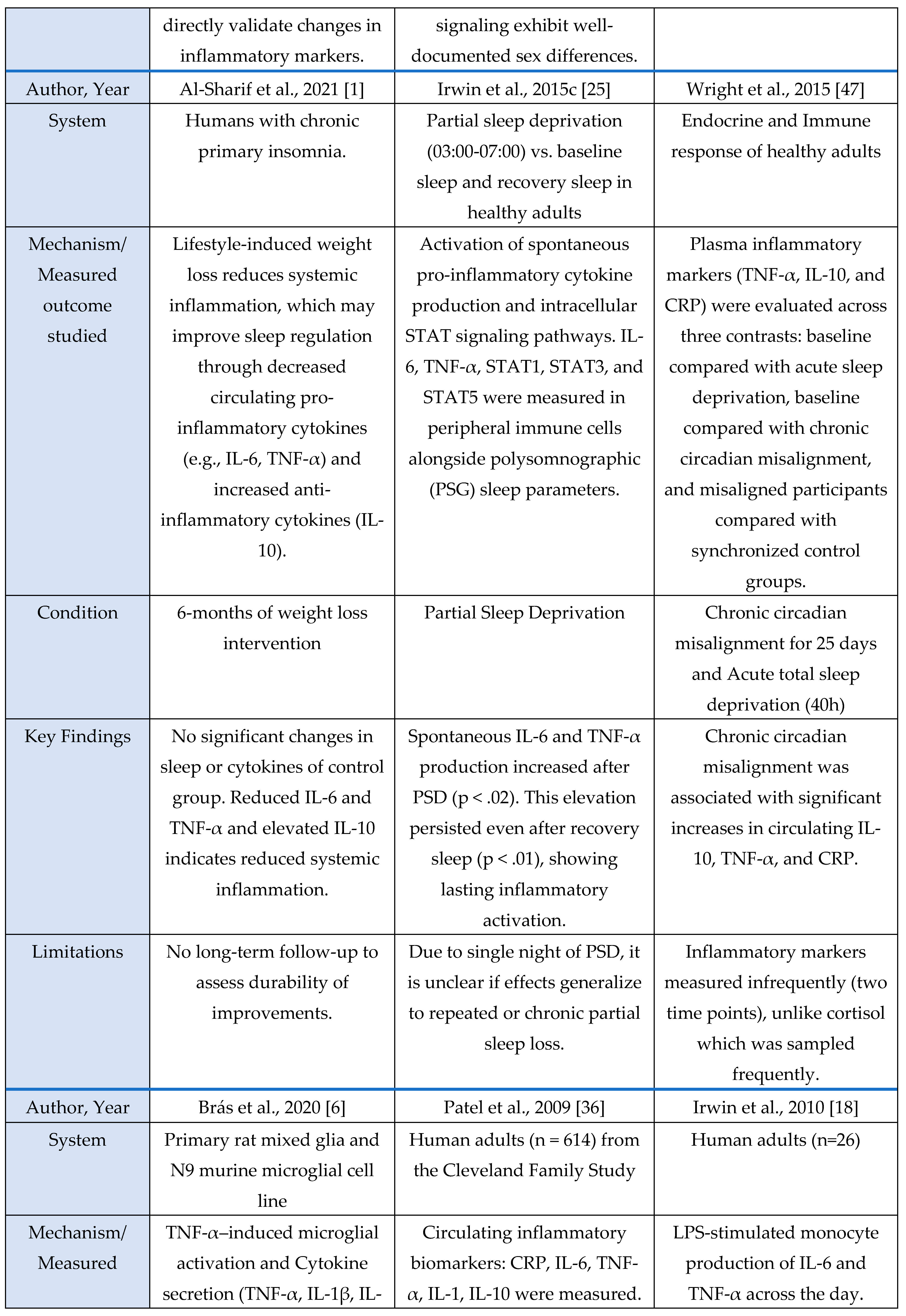
Figure 2.
Overview of mechanistic cytokine studies showing how sleep deprivation alters inflammatory signaling pathways, including IL-6, TNF-α, and related immune mediators, across animal, human, and in vitro models.
Figure 2.
Overview of mechanistic cytokine studies showing how sleep deprivation alters inflammatory signaling pathways, including IL-6, TNF-α, and related immune mediators, across animal, human, and in vitro models.
Mechanistic Narrative-
During sleep deprivation, wake-associated neural activity increases extracellular ATP accumulation in cortical and subcortical circuits [
31,
39]. Microglia are the primary sensors of this danger-associated ATP signal [
39]. Two major purinergic receptors: P2X7 and P2Y12
, mediate distinct but complementary inflammatory and sleep-regulatory responses.
Elevated extracellular ATP binds to P2X7, a low-affinity ligand-gated ion channel expressed on microglia. P2X7 activation produces a rapid K⁺ efflux that triggers assembly of the NLRP3 inflammasome, leading to cleavage and release of IL-1β, a cytokine strongly linked to sleep pressure and homeostatic slow-wave activity [
3,
39,
50].
Simultaneously, P2X7 stimulation promotes microglial TNF-α release. TNF-α then binds on TNFR-1, activating intracellular cascades such as CaMKII phosphorylation. CaMKII modifies synaptic availability of GABA
AA receptors and glutamatergic components, facilitating a shift toward increased slow-wave sleep (SWS), a compensatory response that aids synaptic enrichment and metabolic clearance following sleep loss [
39,
50].
In the context of sleep loss, P2Y12 signaling induces controlled Ca²⁺ oscillations within microglia, facilitates ATP-to-adenosine conversion via microglial enzymatic pathways. Adenosine accumulates extracellularly and acts on neuronal A1 receptors to suppress wake-promoting circuits and enhance restorative sleep [
31].
- 2.
NF-κB as a Primary Amplifier in Microglial Pro-inflammatory Signaling
Sleep deprivation increased microglial release of TNF-α and IL-1β, which in turn activated their respective receptors (TNFR1 and IL-1R1) on microglia. TNF-α–TNFR1 signaling rapidly promoted NF-κB nuclear translocation, while IL-1β amplified this effect through NLRP3-dependent maturation and MyD88-mediated signaling [
14,
28,
30,
33,
35,
49]. This stimulation rapidly engaged the canonical NF-κB pathway, leading to IκBα degradation and nuclear translocation of p65/p50. The resulting NF-κB activation boosted transcription of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), creating a self-reinforcing loop that sustained microglial activation [
6,
30,
33,
35]. Because both TNF-α and IL-1β converged on the same NF-κB–dependent program, their combined action amplified inflammatory signaling and likely contributed to the observed effects on synaptic regulation and increased sleep pressure.
- 3.
STAT proteins and MAPK as Secondary Amplifiers in Microglial Pro-inflammatory Signaling
Pro-inflammatory signaling is further amplified through STAT and MAPK cascades downstream of cytokine and TNF-family receptors. IL-6 and IFN-γ activate their receptors (IL-6R/GP130), leading to JAK-mediated phosphorylation of STAT1/STAT3 [
25,
38], which then dimerize and enter the nucleus to induce secondary pro-inflammatory genes (e.g., IL-6, CCL2, iNOS) and reinforce NF-κB–dependent programs [
2,
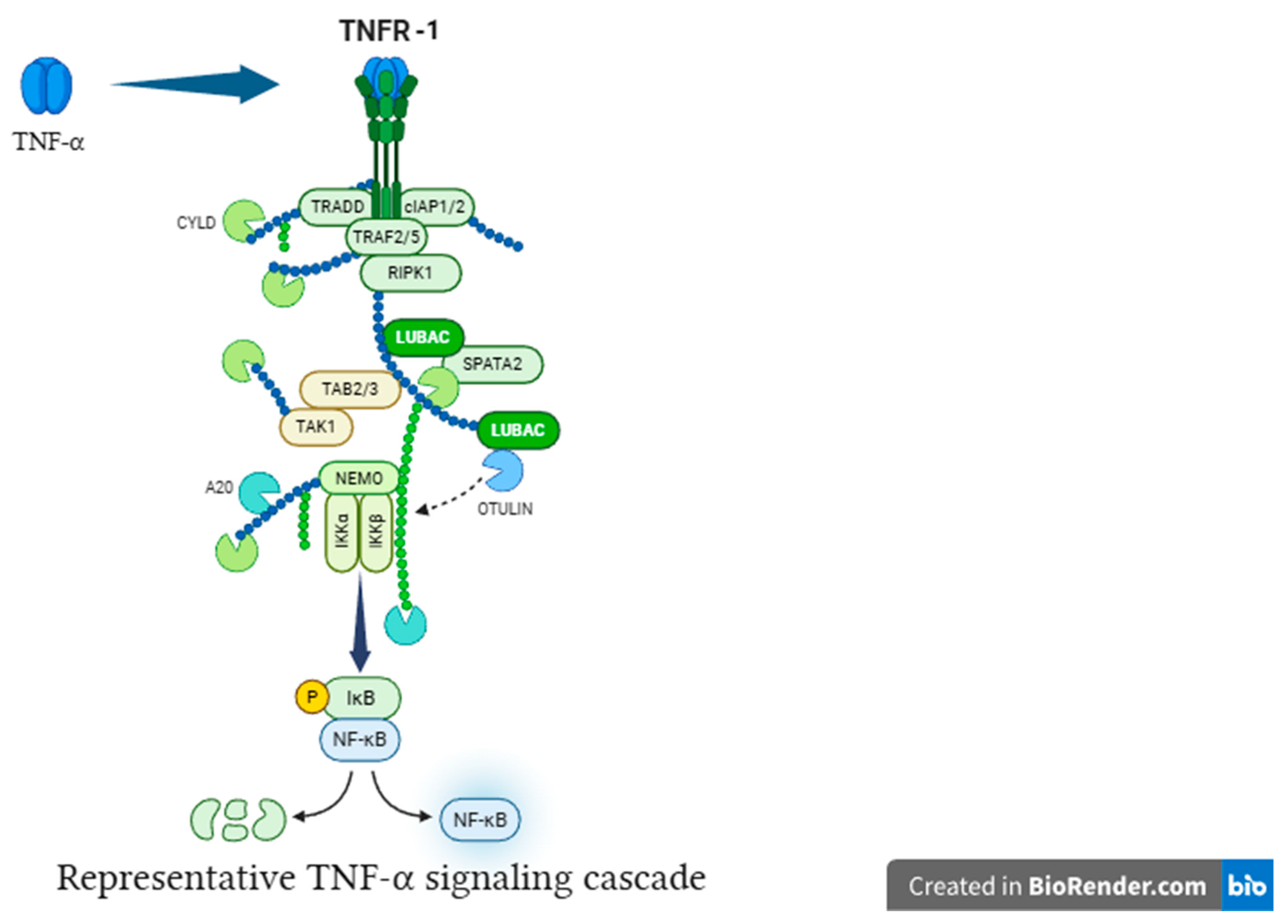
25]. TNF-α binding to TNFR1 initiates TRADD-dependent assembly of Complex I (TRAF2, RIPK-1, LUBAC), which engages MAPK pathways [
26]. These MAPK signals further increase transcription of TNF-α, IL-1β, and IL-6, thereby acting as parallel amplifiers that sustain and escalate microglial activation [
30].
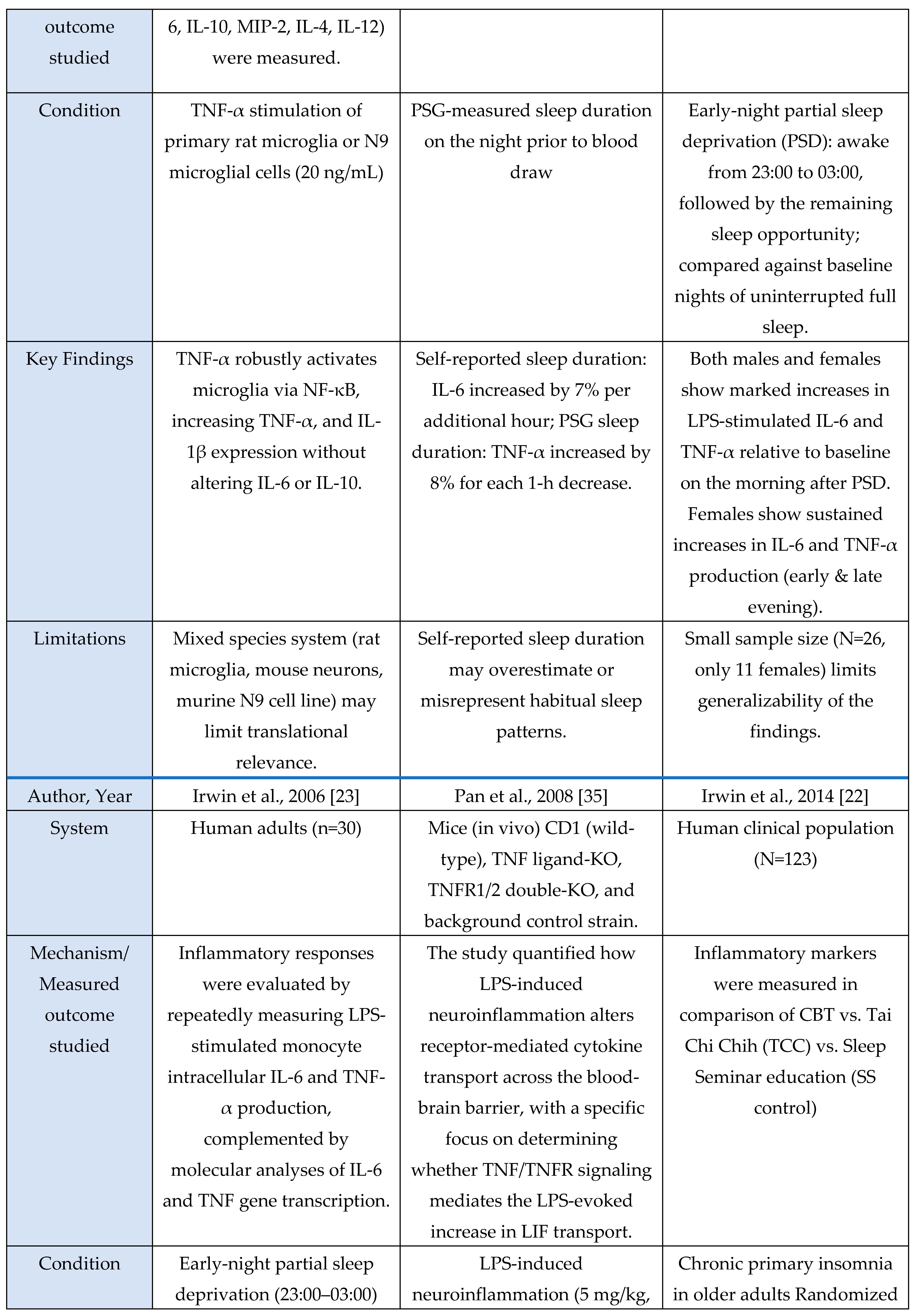
Figure 3.
TNF-α activates NF-κB via TNFR signaling. TNF-α binding to TNFR triggers downstream signaling, resulting in IκB degradation, NF-κB nuclear translocation, and transcriptional activation of target genes.
Figure 3.
TNF-α activates NF-κB via TNFR signaling. TNF-α binding to TNFR triggers downstream signaling, resulting in IκB degradation, NF-κB nuclear translocation, and transcriptional activation of target genes.
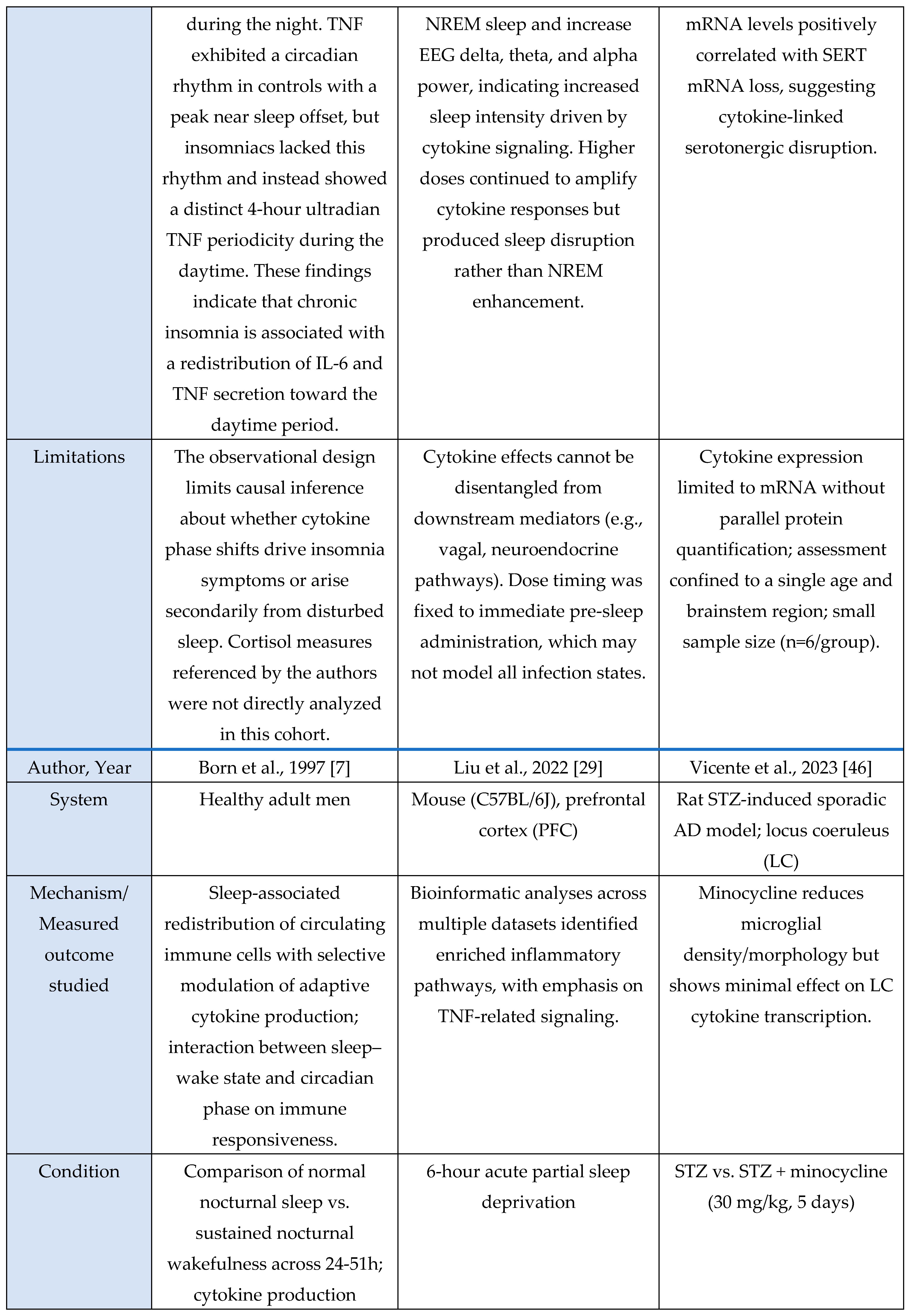
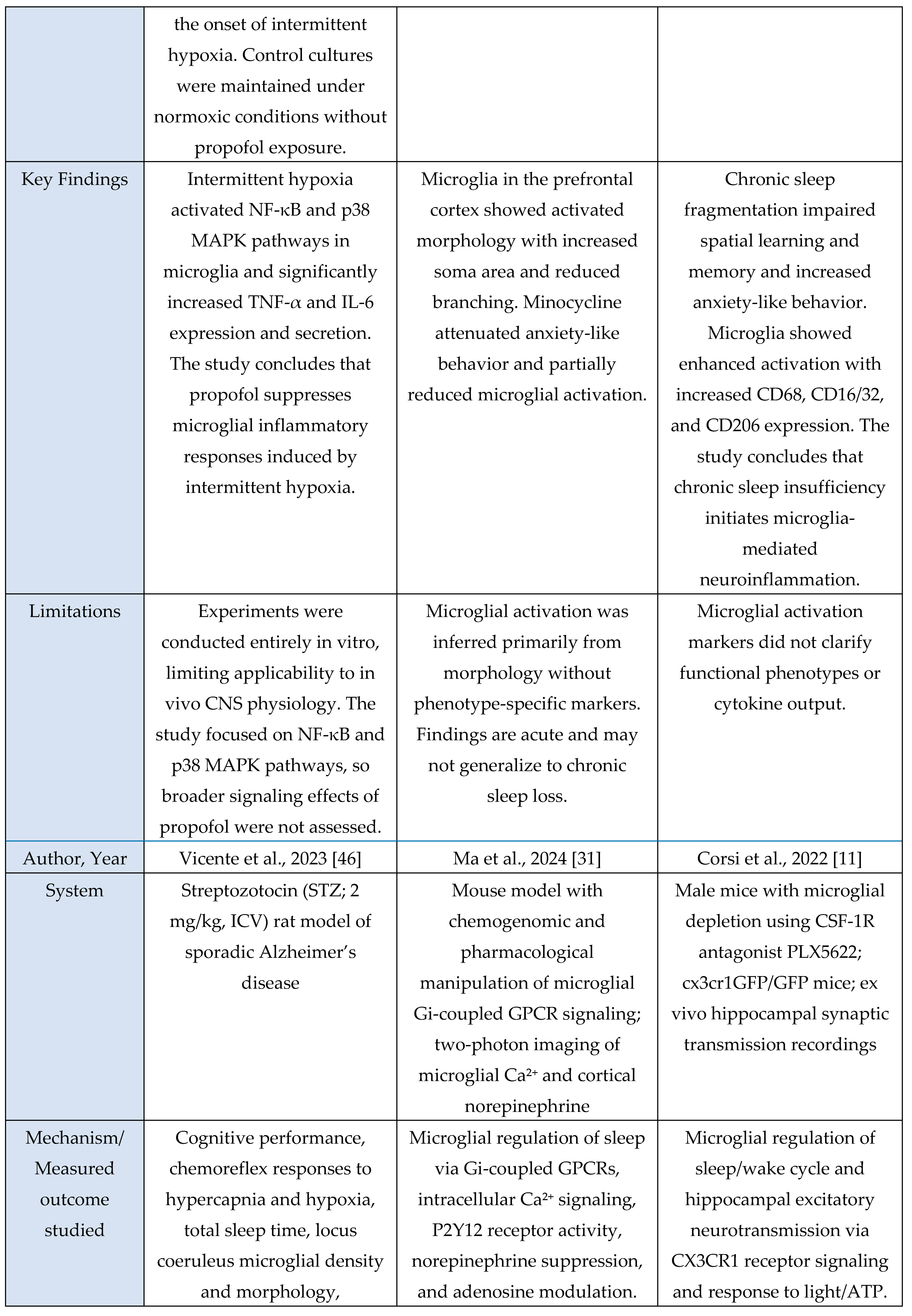
Oxidative stress-HPA axis
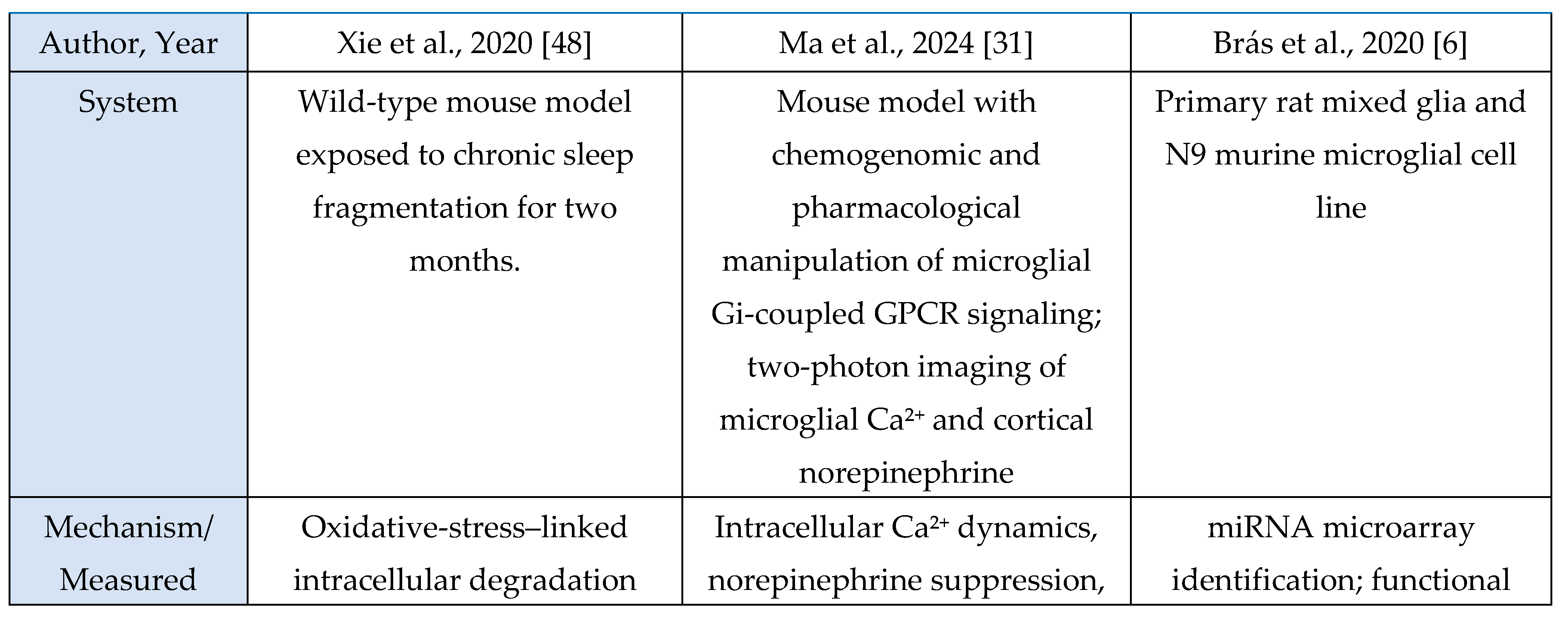
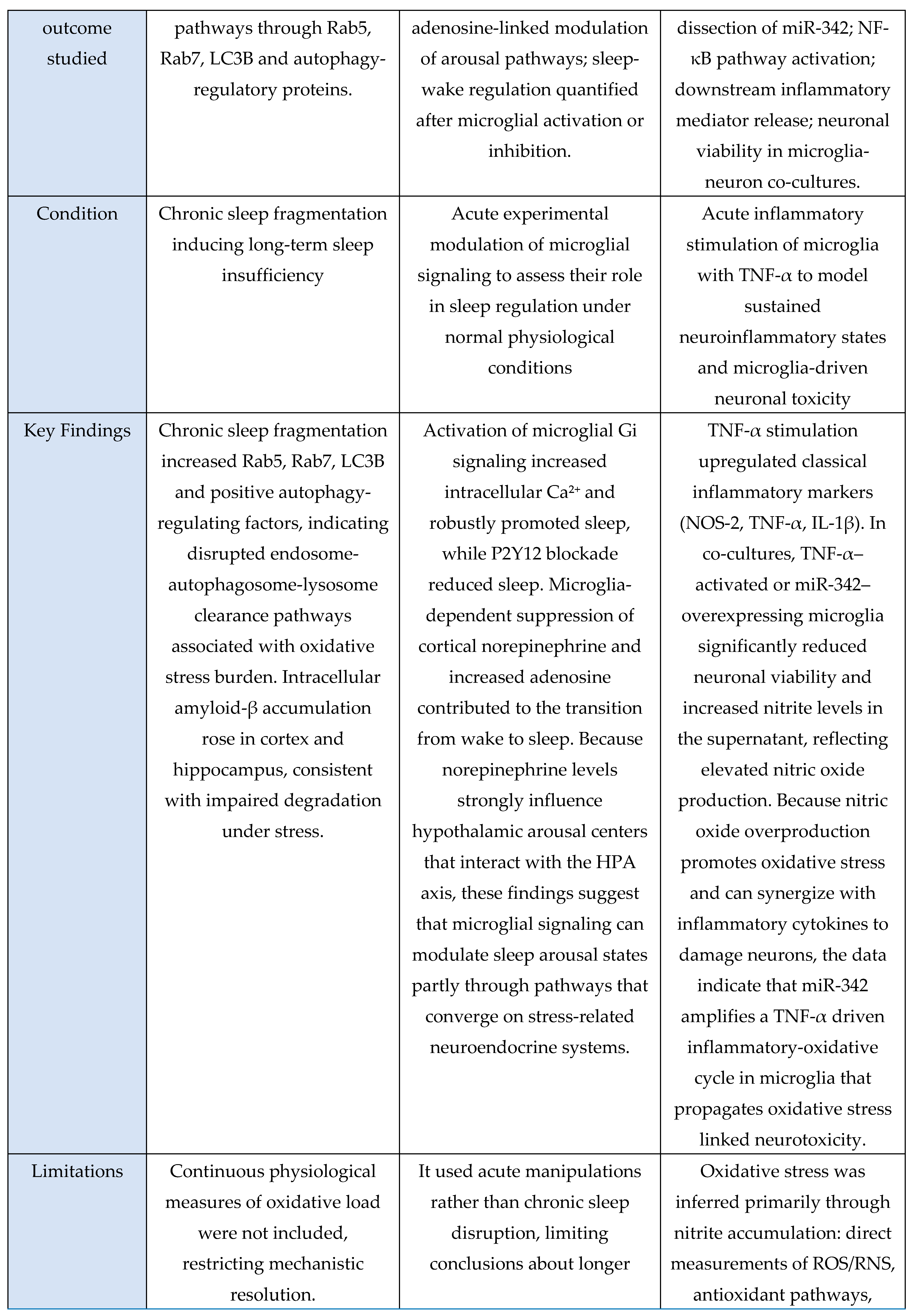
Across the oxidative-stress and HPA-axis evidence base, four studies were murine experimental models and one was a human circadian-disruption paradigm. Murine investigations examined chronic sleep fragmentation and its effects on oxidative-stress–linked intracellular degradation pathways involving Rab5, Rab7, LC3B, and autophagy regulators [
48]; acute microglial activation or inhibition to assess Ca²⁺ dynamics and neuromodulatory changes in norepinephrine and adenosine signaling during sleep–wake regulation [
31]; miRNA-mediated inflammatory amplification via miR-342 and NF-κB activation with downstream effects on neuronal viability in microglia–neuron co-cultures [
6]; and extended total sleep deprivation (72 h) to quantify oxidative-stress markers in brain and serum [
37]. The sole human study used 25 days of circadian misalignment combined with 40 h of total sleep deprivation, demonstrating altered cortisol rhythmicity and increased systemic oxidative stress [
47]. Outcomes across studies included molecular assays (Rab5, Rab7, LC3B, autophagy proteins, NOS-2, MDA, NO), neuronal-viability assays, hormonal profiling (cortisol), and behavioral sleep metrics. Together, these studies indicate that sleep and circadian disruption converge on microglia-linked oxidative-stress pathways and HPA-axis dysregulation, with murine models providing mechanistic insights and the human model confirming physiological relevance.
Across murine and cellular models, all studies consistently reported oxidative-stress-linked disruptions to intracellular homeostasis. Chronic sleep fragmentation impaired endosome-autophagosome-lysosome trafficking via Rab5, Rab7, and LC3B dysregulation and increased amyloid-β accumulation [
48]. Microglial activation amplified oxidative and metabolic load through Ca²⁺ dynamics and neuromodulatory shifts that promote sleep pressure and arousal suppression [
31]. TNF-α-activated microglia increased NOS-2 expression, nitrite release, and miR-342-dependent neurotoxicity [
6]. Acute sleep deprivation produced systemic oxidative injury with elevated MDA and NO and multi-organ biochemical dysfunction [
37]. The human study showed that extreme sleep disruption acutely activated the HPA axis via elevated evening-night cortisol during total sleep deprivation, whereas circadian misalignment suppressed overall cortisol output [
47]. Together, these findings demonstrate that sleep disruption and inflammatory activation reliably induce oxidative-stress responses and modulate stress-axis output.
Murine models provided mechanistic resolution, revealing disrupted autophagy-lysosomal trafficking [
48], microglia-driven nitric-oxide overproduction [
6,
37], neuronal vulnerability, and Ca²⁺/adenosine-mediated modulation of sleep–arousal circuits [
31]. The human model highlighted physiologically measurable HPA-axis consequences: acute cortisol elevation during sleep loss and reduced output during circadian misalignment, without cellular-level mechanistic access [
47].
Overall, the evidence is strengthened by convergence between mechanistic rodent data and physiologically relevant human endocrine alterations. Despite methodological heterogeneity, the evidence supports a consistent association between sleep disruption, microglia-linked oxidative stress, and HPA-axis dysregulation.
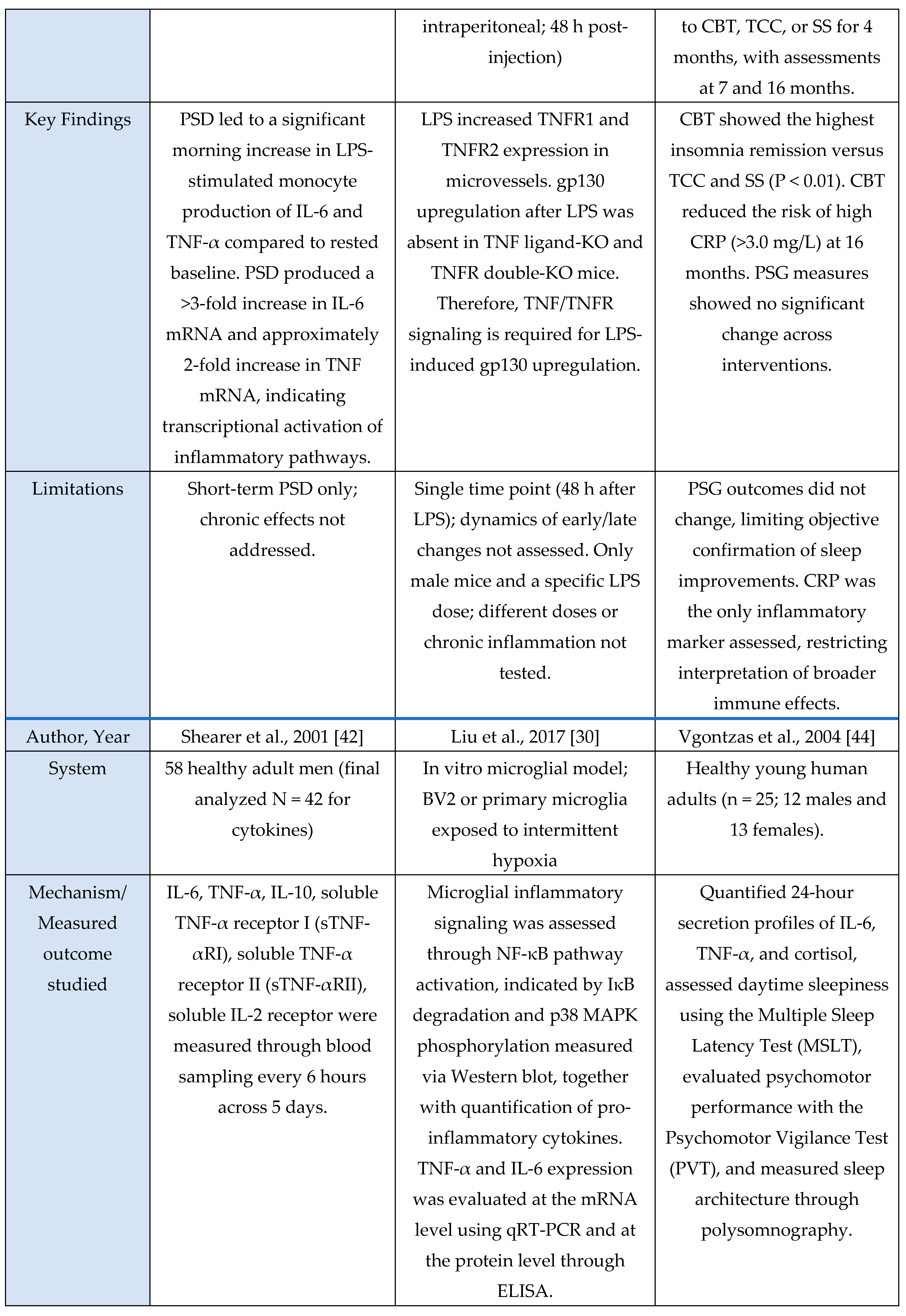
Figure 4.
Summary of mechanistic studies evaluating oxidative stress and HPA-axis responses to sleep deprivation across animal, human, and in vitro models.
Figure 4.
Summary of mechanistic studies evaluating oxidative stress and HPA-axis responses to sleep deprivation across animal, human, and in vitro models.
Mechanistic narrative
Across included studies, sleep deprivation consistently triggered early oxidative stress responses, reflecting one of the most rapid physiological consequences of sustained wakefulness. SD increased Nitric Oxide generation in neural and peripheral tissues [
6,
37]. This burden of oxidative stress disrupted endosomal-autophagy pathways, as reflected by elevated Rab5, Rab7, LC3B, and other autophagy-related markers, indicating impaired clearance of damaged proteins and organelles under prolonged sleep disruption [
48]. This redox imbalance is mechanistically important because oxidative stress acts as an upstream amplifier of inflammatory signaling, mitochondrial dysfunction, and synaptic vulnerability.
In parallel, sleep deprivation activated the HPA axis, leading to elevated cortisol levels [
47]. Glucocorticoid elevations further exacerbated oxidative load by promoting mitochondrial dysfunction and weakening antioxidant systems, thereby amplifying OS-driven cellular stress. Together, these findings indicate that sleep loss initiates a dual oxidative pathway, jointly contributing to cumulative oxidative and metabolic strain in sleep-disrupted conditions.
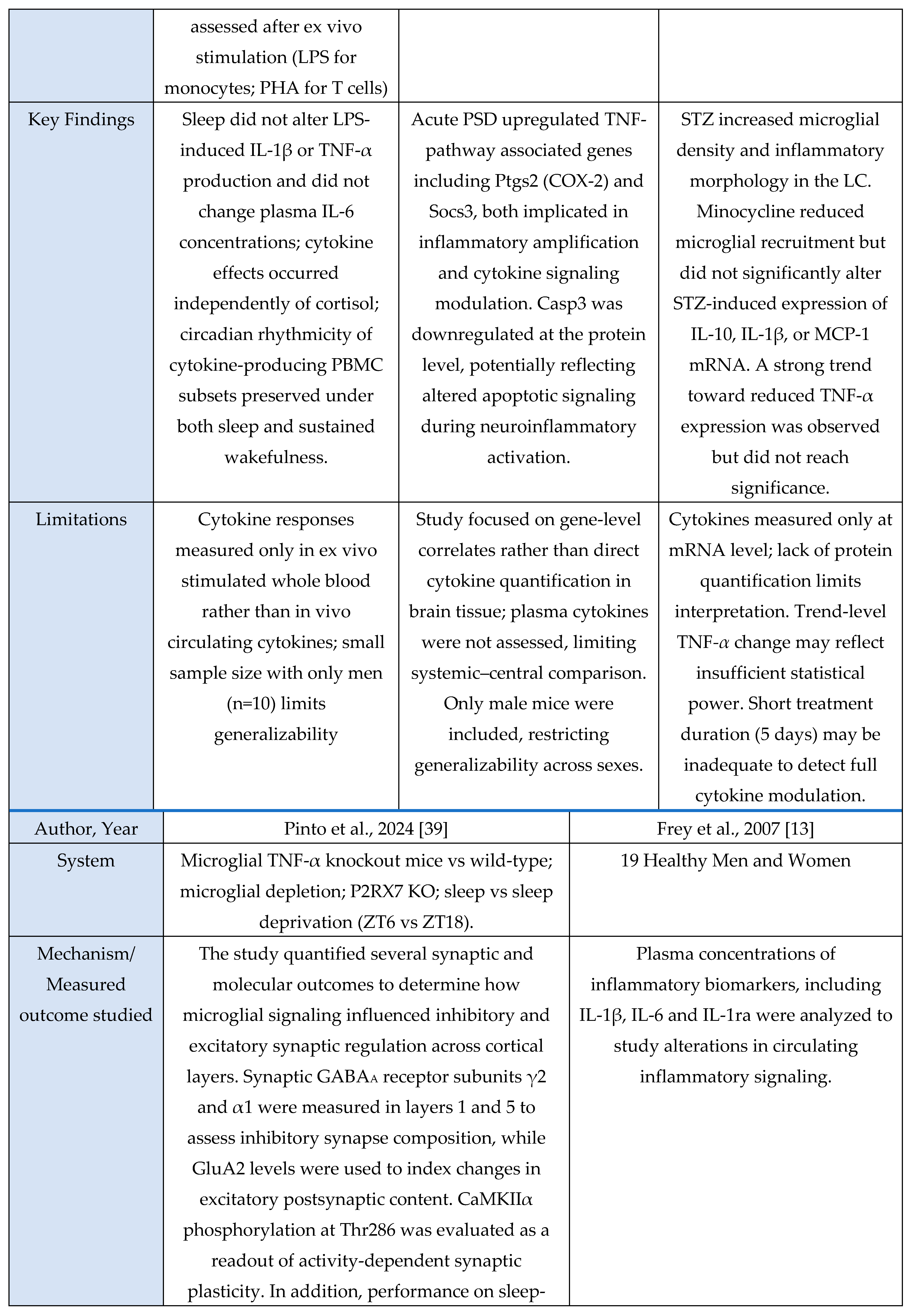
Microglial Dynamics
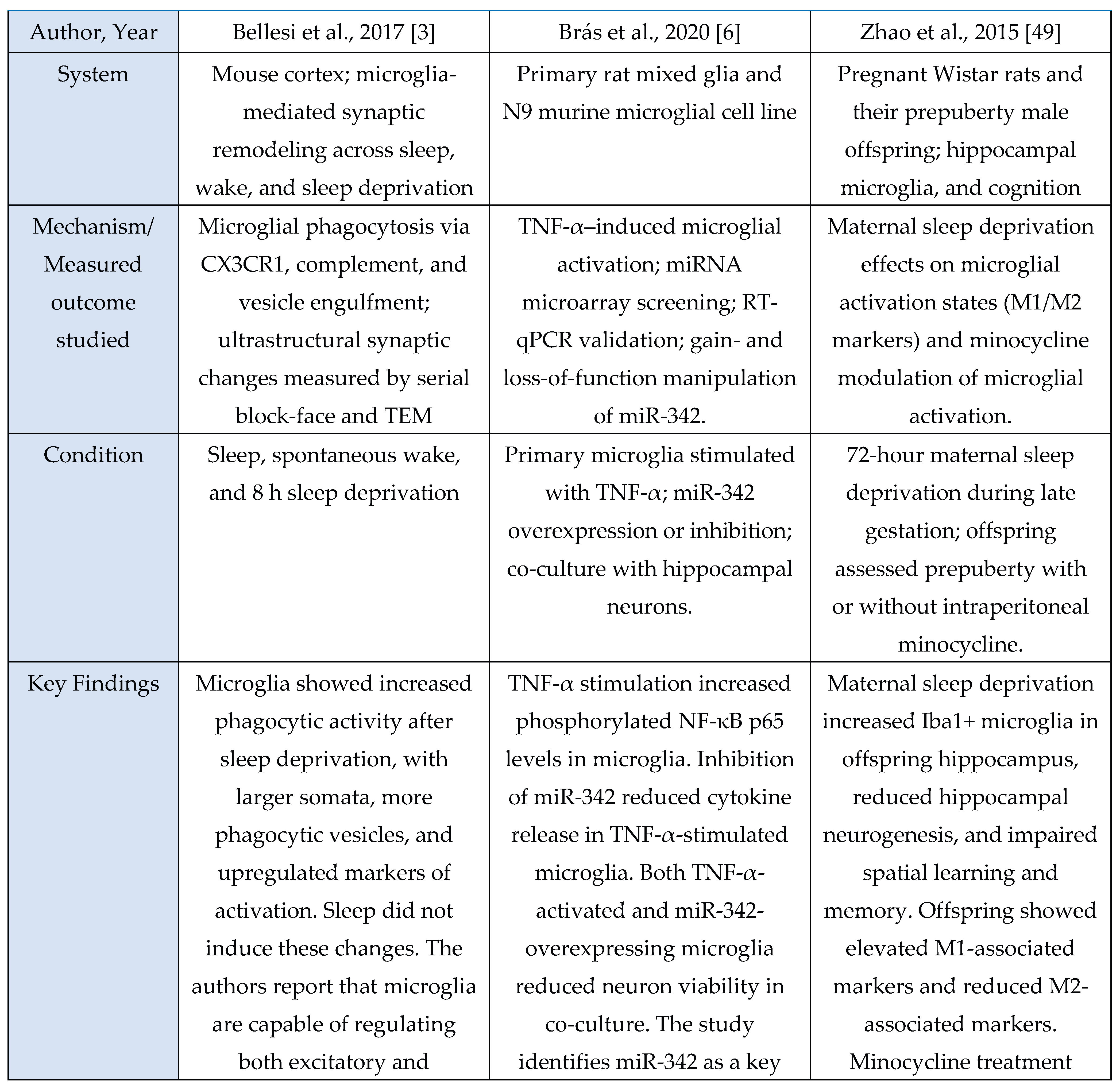
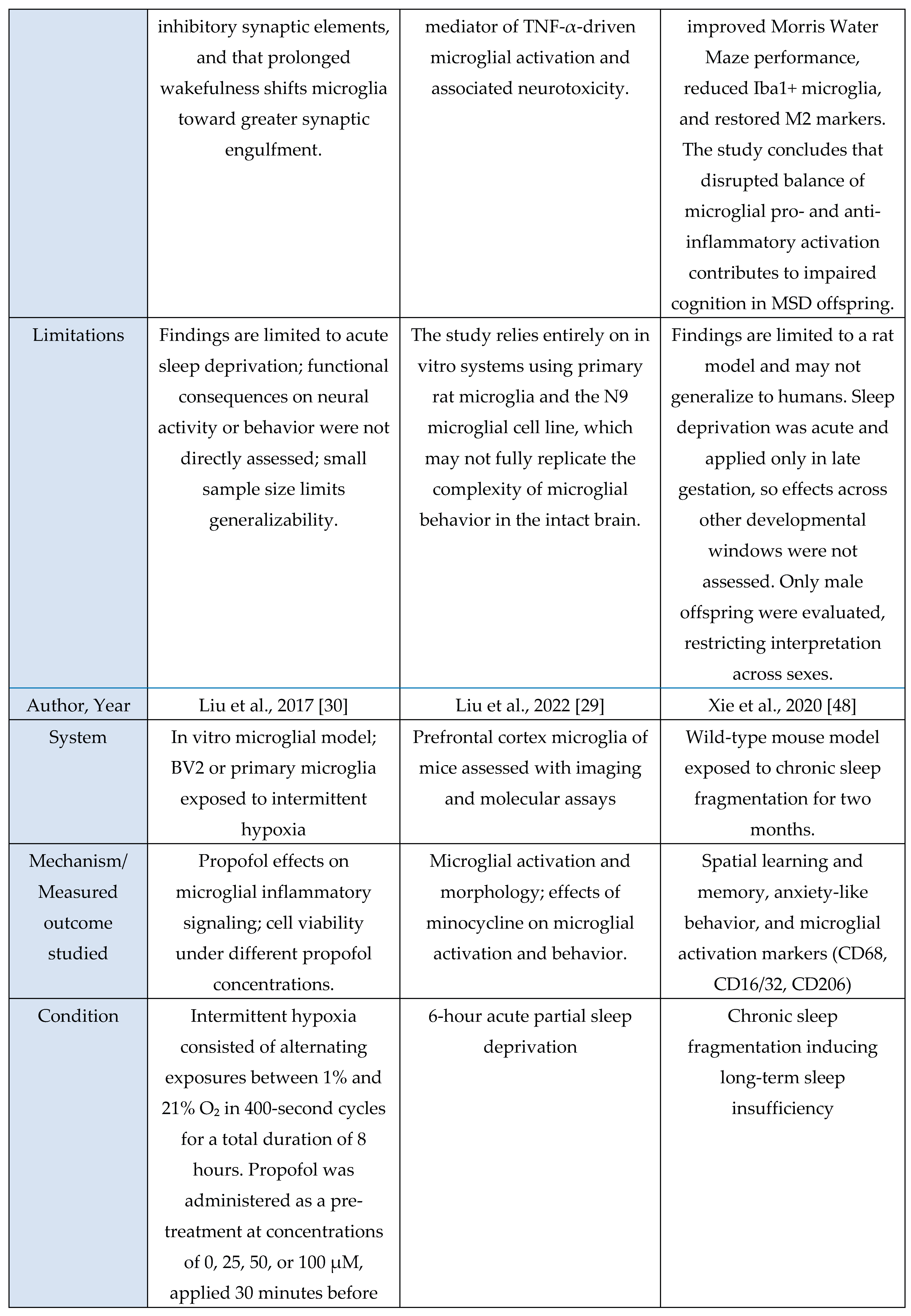
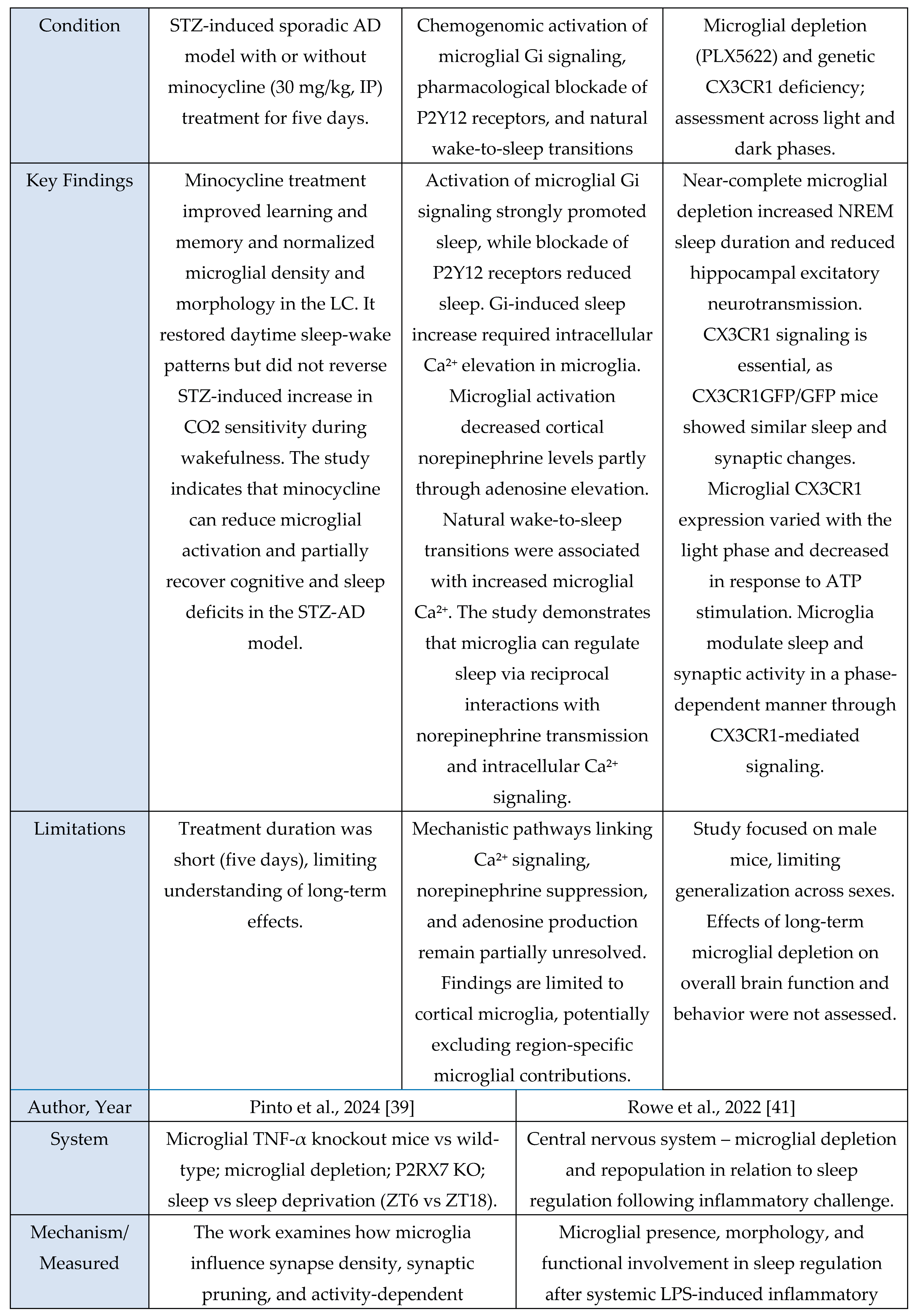
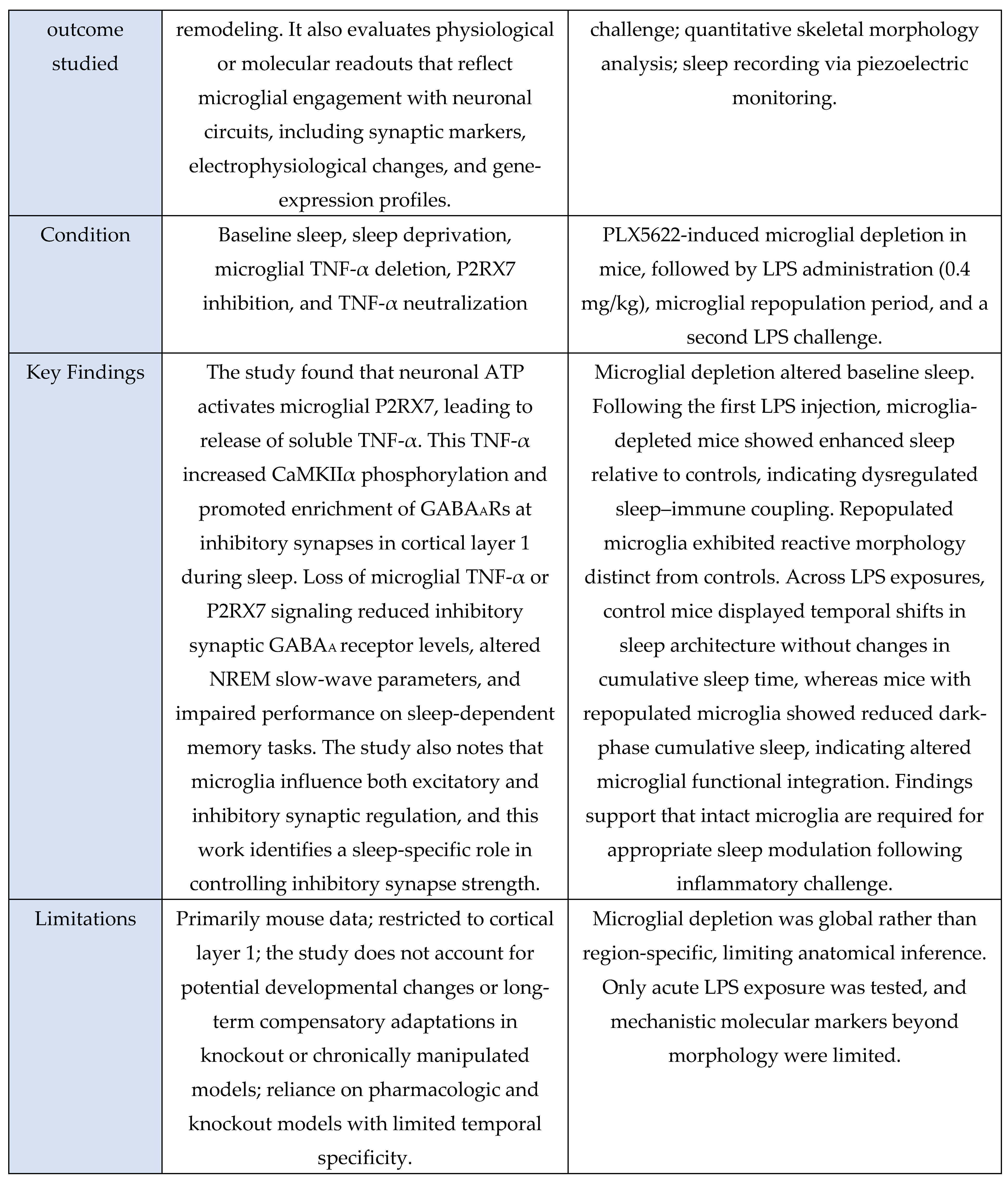
A total of 11 studies met inclusion criteria for the Microglial Axis, comprising 9 murine models and 2 in vitro investigations. Across paradigms involving stress exposure, a convergent pattern emerged in which microglia transitioned toward a more reactive or phagocytic state. These studies consistently reported increased expression of activation-associated markers (e.g., CD68, CD16/32) and enhanced synaptic contact, surveillance, or engulfment behaviors. Collectively, these findings indicate that sleep stress perturbations reliably recruit microglial pathways that modulate synaptic architecture and contribute to downstream changes in sleep homeostasis.
Evidence summary
Among the included microglia studies (n = 11), none used human cohorts, nine employed rodent models, and two used in vitro systems. Study designs were predominantly experimental and interventional, spanning acute sleep deprivation [
3,
29], chronic sleep fragmentation [
48], maternal sleep deprivation [
49], intermittent hypoxia [
30], inflammatory challenge with LPS [
41], and disease-relevant stressors such as STZ-induced sporadic Alzheimer’s disease [
46]. Interventions also included pharmacological manipulation (minocycline [
29,
46,
49], propofol [
30], PLX5622-mediated microglial depletion [
11,
41]) and genetic or chemogenomic perturbations (microglial TNF-α knockout, P2RX7 KO, CX3CR1 deficiency, Gi-coupled GPCR activation [
11,
31,
39]). Microglial outcomes were quantified through immunohistochemistry and morphology (Iba1, CD68, CD16/32, CD206) [
29,
48,
49], ultrastructural imaging (serial block-face EM, TEM) [
3], two-photon Ca²⁺ imaging [
31], electrophysiology [
11,
39], transcriptomic or microRNA analysis [
6], and biochemical assays of microglial activation or secretory activity [
6,
30]. These methods consistently revealed sleep- or challenge-induced microglial alterations such as enlarged somata, increased phagocytic vesicles [
3], miR-342 dependent inflammatory signaling [
6], shifts toward M1-like (pro-inflammatory) activation after maternal sleep deprivation [
49], and Gi-mediated modulation of sleep via microglial Ca²⁺ dynamics [
31].
The dominant pattern across these studies is that manipulations disrupting sleep, inducing systemic or local inflammatory challenge, or directly perturbing microglial signaling produce measurable changes in microglial morphology, functional state, or engagement with synapses. 10/11 (91%) studies reported measurable microglial changes in response to sleep perturbation, inflammatory challenge, hypoxia, or direct manipulation of microglial signaling [
3,
6,
29,
30,
31,
39,
41,
46,
48,
49]. A consistent pattern of increased microglial activation or altered morphology emerged across paradigms, including greater phagocytic activity after sleep deprivation [
3], NF-κB and p38 MAPK activation under intermittent hypoxia [
30], increased Iba1 and M1 markers in offspring following maternal sleep deprivation [
49], and heightened CD68/CD16/32 expression after chronic sleep fragmentation [
48]. Studies targeting specific microglial pathways (P2RX7, TNF-α, CX3CR1, Gi-coupled GPCRs) converged on the finding that sleep or its disruption reliably engages microglial mechanisms that influence synaptic structure or function [
11,
31,
39]. Both in vitro studies (2/2, 100%) replicated core features of microglial inflammatory activation, including NF-κB and MAPK pathway induction, increased TNF-α and IL-6 production, and microglia-induced neuronal toxicity [
6,
30]. A subset of 6/11 (55%) studies explicitly reported increases in canonical activation markers or secreted inflammatory mediators following the experimental manipulation (for example TNF-α/IL-6 increases with intermittent hypoxia, upregulation of CD68/CD16/32 with chronic fragmentation, and elevated Iba1 and M1 markers after maternal sleep deprivation) [
6,
29,
30,
46,
48,
49]. Although methodological heterogeneity existed, the dominant pattern indicated a consistent directional shift toward increased microglial activation, heightened cytokine-linked signaling, and enhanced microglial engagement with synaptic substrates following sleep disruption or related stressors.
Rodent models provided the clearest systems-level evidence linking microglial state to sleep and circuit function, showing alterations in NREM sleep duration [
11], slow-wave parameters [
39], cortical norepinephrine levels [
31], hippocampal synaptic transmission [
11], and sleep-dependent memory performance [
39,
46,
49]. In vitro systems (primary microglia and microglial cell lines) reproduced cell-autonomous mechanisms such as TNF-α–induced NF-κB activation, miR-342–regulated cytokine secretion [
6], and hypoxia-triggered inflammatory signaling [
30], supporting mechanistic plausibility but lacking behavioral endpoints. Studies involving microglial depletion and repopulation revealed altered baseline sleep and exaggerated or dysregulated responses to repeated inflammatory challenge, indicating that microglial history and state strongly shape sleep–immune coupling [
11,
41]. Genetic and chemogenomic approaches further demonstrated pathway specificity: P2RX7–TNF-α signaling was required for sleep-associated GABAAR enrichment at inhibitory synapses [
39], CX3CR1 regulated phase-dependent synaptic transmission and NREM sleep [
11], and Gi-coupled GPCR activation promoted sleep via Ca²⁺-dependent suppression of cortical norepinephrine [
31]. The body of evidence provides moderate support for a causal contribution of microglia to sleep synapse interactions. This conclusion is grounded in convergent interventional data showing that targeted disruption of microglial pathways (for example microglial TNF-α deletion [
39], P2RX7 blockade [
39], CX3CR1 deficiency [
11], microglial depletion [
11,
41], Gi-pathway activation [
31]) produces predictable changes in sleep architecture, synaptic receptor composition, electrophysiological responses, or behavior. Overall, findings across the included studies consistently suggest that microglia respond to and influence sleep-related processes, with moderate-strength evidence supporting a functional and mechanistic role.
Figure 5.
Summary of microglia-focused mechanistic studies evaluating neuroimmune responses to sleep deprivation across animal and cellular models.
Figure 5.
Summary of microglia-focused mechanistic studies evaluating neuroimmune responses to sleep deprivation across animal and cellular models.
Mechanistic narrative-
According to the literature, extracellular ATP accumulation during sleep deprivation due to heightened neuronal activity was found to act as a key trigger for microglial state transitions [
50]. ATP engagement of P2X7 reliably increased microglial phagocytic activity [
3,
10], reflected by elevated CD68, and CD16/32 consistent with a phagocytic profile, and coincided with enhanced synaptic pruning responses [
10,
48,
49]. This P2X7-driven shift occurred alongside inflammasome-dependent cytokine release [
39], indicating that ATP functions as both a danger signal and a pruning cue in sleep-loss relevant conditions.
Simultaneously, ATP sensing through P2Y12, together with parallel CX3CR1 signaling, supported microglial process motility and targeted engulfment of synaptic elements. ATP-driven P2Y12 activation, occurring alongside CX3CR1-mediated signaling promoted process extension toward active synapses and maintained similar phagocytic profile, indicating coordinated receptor-specific contributions to microglial engulfment behavior [
3,
11,
31]. Together, these results demonstrate that ATP engages two separable microglial pathways: a purinergic mechanism and a CX3CR1 pathway, each contributing differentially to microglial modulation of synaptic architecture.
Discussion
This systematic synthesis identifies convergent neuroimmune and cellular pathways activated by sleep deprivation, spanning cytokine signaling, microglial state transitions, oxidative stress responses, and HPA-axis activity. Together, these results delineate a coordinated multi-system response linking sleep loss to neuroinflammatory and synaptic alterations. Across 29 included studies, we found consistent activation of pro-inflammatory cytokines, convergent microglial morphological and functional changes, and reproducible alterations in sleep pressure following targeted perturbations. Although methodological heterogeneity was present, a common directional pattern emerged in which sleep disruption caused an inflammatory state in the brain and peripheral tissue. Findings across the cytokine, microglial, and Oxidative Stress-HPA axes converged on a shared set of upstream processes involving extracellular ATP [
3,
31,
39,
50], inflammasome activation [
48,
50], and glucocorticoid or catecholamine dependent modulation of inflammatory tone [
15,
16,
18,
19,
31,
47]. Elevated ATP consistently activated P2RX7/P2Y12-mediated microglial pathways, resulting in altered phagocytic profiles and increased engagement with synaptic substrates. In parallel, TNF-α and IL-1β signaling activated canonical (e.g., NF-κB / MAPK) pathways, shaping downstream transcriptional responses that aligned with observed morphological changes [
2,
6,
25,
26,
30,
33,
35]. Together, these mechanistic threads point toward a coordinated multi-axis response rather than isolated pathway activation. The patterns identified in this review are broadly consistent with prior work demonstrating immune involvement in sleep regulation, microglial contributions to synaptic remodeling, and the sensitivity of cytokine networks to physiological stressors. The observed modulation of TNF-α during sleep disruption aligns with longstanding evidence linking these cytokines to sleep intensity and homeostatic regulation. The convergent role of P2RX7 and CX3CR1 signaling in microglial process dynamics resembles findings from developmental synaptic pruning and disease models, indicating that sleep loss induced mechanisms leverage conserved microglial pathways [
3,
11,
39]. However, relative to earlier descriptive work, the present synthesis more clearly separates purinergic, cytokine, and neuroendocrine contributions and highlights their intersection in shaping sleep synapse coupling. This distinction may refine theoretical models describing how immune signals reorganize neural activity during sleep loss. A key strength of the included literature is the use of multimodal approaches, combining ultrastructural imaging; electrophysiology; genetic perturbation; and molecular profiling, to dissect microglial and cytokine mechanisms with high resolution. Interventional designs such as microglial depletion, pathway-specific knockouts, and targeted receptor manipulation provide causal leverage that strengthens mechanistic inference. The reproducible directionality of cytokine and microglial responses across diverse paradigms suggests robustness of the core biological signal. Future work should include standardized sleep-disruption paradigms to improve cross-study comparability, alongside longitudinal designs that identify how microglial and cytokine dynamics evolve over time. Expanded investigation of the Oxidative Stress and HPA-axis, including Glucocorticoid Resistance and β-adrenergic contributions, would strengthen mechanistic models of neuroimmune integration. Ideally, translational work utilizing human biosamples or polysomnography-linked inflammatory [
8] profiling will be needed to establish clinical relevance and refine therapeutic targets.
Limitations
This review has several methodological and conceptual limitations that should be considered when interpreting the synthesized evidence. First, the literature included displays substantial heterogeneity in experimental paradigms, with studies differing in outcome measures, tissue sources, and analytical platforms. Such variability constrains direct comparison and complicates efforts to draw unified mechanistic conclusions. A second limitation is the predominant reliance of animal and in vitro studies on interpreting mechanistic insights and translational value of conclusions, particularly with respect to microglial activation states, oxidative stress pathways, and systems-level neuroimmune interactions. Relatedly, there remains limited human data on microglial dynamics and oxidative stress axes in the context of sleep deprivation, which restricts the ability to evaluate whether the mechanisms observed in preclinical models accurately reflect human neurobiology. Finally, this review employed single-reviewer screening and did not conduct a formal risk-of-bias assessment, which may introduce selection bias and limits the methodological rigor compared to systematic reviews that adhere to multi-reviewer and validated appraisal protocols. As a result, the conclusions presented should be interpreted as integrative trends rather than definitive causal statements.
Conclusion
This systematic review demonstrates that sleep disruption consistently engages cytokine, microglial, and neuroendocrine mechanisms that converge on synaptic and circuit-level processes. Across models, elevated ATP signaling, cytokine activation, and microglial remodeling represent robust biological pathways through which sleep loss shapes neural function. Although further work is needed to establish translational relevance, the integrated evidence base supports a multi-axis framework for understanding how immune processes regulate sleep and its associated cognitive functions.
Funding
The author has no funding to report.
Data Availability Statement
The data supporting the findings of this study are available within the article. The author can be contacted for further inquiries at akshadareddy@gmail.com.
Acknowledgments
I thank the authors of the studies included in this systematic review, as their work made this synthesis possible. Figures in this manuscript were created with BioRender.com. No specific funding was received for this study.
References
- Al-Sharif, FM; El-Kader, SMA. Inflammatory cytokines and sleep parameters response to lifestyle intervention in subjects with obese chronic insomnia syndrome. Afr Health Sci. 2021, 21(3), 1223–9. [Google Scholar] [CrossRef]
- Awasthi, N; Liongue, C; Ward, AC. STAT proteins: A kaleidoscope of canonical and non-canonical functions in immunity and cancer. J Hematol Oncol. 2021, 14(1), 198. [Google Scholar] [CrossRef]
- Bellesi, M; de Vivo, L; Chini, M; Gilli, F; Tononi, G; Cirelli, C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J Neurosci. 2017, 37(21), 5263–73. [Google Scholar] [CrossRef] [PubMed]
- Besedovsky, L; Lange, T; Haack, M. The sleep-immune crosstalk in health and disease. Physiol Rev. 2019, 99(3), 1325–80. [Google Scholar] [CrossRef]
- Bilbo, SD; Schwarz, JM. Early-life programming of later-life brain and behavior: A critical role for the immune system. Front Behav Neurosci. 2009, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Brás, JP; Bravo, J; Freitas, J; Barbosa, MA; Santos, SG; Summavielle, T; et al. TNF-alpha-induced microglia activation requires miR-342: Impact on NF-κB signaling and neurotoxicity. Cell Death Dis. 2020, 11(6), 415. [Google Scholar] [CrossRef]
- Born, J; Lange, T; Hansen, K; Mölle, M; Fehm, HL. Effects of sleep and circadian rhythm on human circulating immune cells. Journal of immunology (Baltimore, Md.: 1950) 1997, 158(9), 4454–64. [Google Scholar] [CrossRef]
- Carpi, M; Fernandes, M; Mercuri, NB; Liguori, C. Sleep biomarkers for predicting cognitive decline and Alzheimer's disease: A systematic review of longitudinal studies. J Alzheimers Dis. 2024, 97(1), 121–43. [Google Scholar] [CrossRef]
- Chennaoui, M; Gomez-Merino, D; Drogou, C; Geoffroy, H; Dispersyn, G; Langrume, C; et al. Effects of exercise on brain and peripheral inflammatory biomarkers induced by total sleep deprivation in rats. J Inflamm. 2015, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, ME; Miyanishi, K; Takeda, H; Tanaka, J. Microglia and the aging brain: Are geriatric microglia linked to poor sleep quality? Int J Mol Sci. 2021, 22(15), 7824. [Google Scholar] [CrossRef]
- Corsi, G; Picard, K; di Castro, MA; Garofalo, S; Tucci, F; Chece, G; et al. Microglia modulate hippocampal synaptic transmission and sleep duration along the light/dark cycle. Glia 2022, 70(1), 89–105. [Google Scholar] [CrossRef] [PubMed]
- Falvo, JV; Tsytsykova, AV; Goldfeld, AE. Transcriptional control of the TNF gene. Curr Dir Autoimmun. 2010, 11, 27–60. [Google Scholar] [CrossRef]
- Frey, DJ; Fleshner, M; Wright, KP. The effects of 40 hours of total sleep deprivation on inflammatory markers in healthy young adults. Brain Behav Immun. 2007, 21(8), 1050–7. [Google Scholar] [CrossRef]
- Hayden, MS; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin Immunol. 2014, 26(3), 253–66. [Google Scholar] [CrossRef]
- Irwin, MR. Why sleep is important for health: A psychoneuroimmunology perspective. Annu Rev Psychol. 2015a, 66, 143–72. [Google Scholar] [CrossRef]
- Irwin, MR. Sleep and inflammation: Partners in sickness and in health. Nat Rev Immunol 2019, 19, 702–15. [Google Scholar] [CrossRef] [PubMed]
- Irwin, MR; Cole, SW. Reciprocal regulation of the neural and innate immune systems. Nat Rev Immunol. 2011, 11(9), 625–32. [Google Scholar] [CrossRef]
- Irwin, MR; Carrillo, C; Olmstead, R. Sleep loss activates cellular markers of inflammation: Sex differences. Brain Behav Immun. 2010, 24(1), 54–7. [Google Scholar] [CrossRef]
- Irwin, MR; Opp, MR. Sleep health: Reciprocal regulation of sleep and innate immunity. Neuropsychopharmacology 2017, 42(1), 129–55. [Google Scholar] [CrossRef]
- Irwin, MR; Olmstead, R; Breen, EC; Witarama, T; Carrillo, C; Sadeghi, N; et al. Cognitive behavioral therapy and tai chi reverse cellular and genomic markers of inflammation in late-life insomnia: A randomized controlled trial. Biol Psychiatry 2015b, 78(10), 721–9. [Google Scholar] [CrossRef] [PubMed]
- Irwin, MR; Olmstead, R; Carroll, JE. Sleep disturbance, sleep duration, and inflammation: A systematic review and meta-analysis of cohort studies and experimental sleep deprivation. Biol Psychiatry 2016, 80(1), 40–52. [Google Scholar] [CrossRef]
- Irwin, MR; Olmstead, R; Carrillo, C; Sadeghi, N; Breen, EC; Witarama, T; et al. Cognitive behavioral therapy vs. Tai Chi for late-life insomnia and inflammatory risk: A randomized controlled comparative efficacy trial. Sleep 2014, 37(9), 1543–52. [Google Scholar] [CrossRef]
- Irwin, MR; Wang, M; Campomayor, CO; Collado-Hidalgo, A; Cole, S. Sleep deprivation and activation of morning levels of cellular and genomic markers of inflammation. Arch Intern Med. 2006, 166(16), 1756–62. [Google Scholar] [CrossRef] [PubMed]
- Irwin, MR; Wang, M; Ribeiro, D; Cho, HJ; Olmstead, R; Breen, EC; et al. Sleep loss activates cellular inflammatory signaling. Biol Psychiatry 2008, 64(6), 538–40. [Google Scholar] [CrossRef]
- Irwin, MR; Witarama, T; Caudill, M; Olmstead, R; Breen, EC. Sleep loss activates cellular inflammation and signal transducer and activator of transcription (STAT) family proteins in humans. Brain Behav Immun 2015c, 47, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Jang, DI; Lee, AH; Shin, HY; Song, HR; Park, JH; Kang, TB; et al. The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int J Mol Sci. 2021, 22(5), 2719. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B; Helweg, LP; Greiner, JFW; Kaltschmidt, C. NF-κB in neurodegenerative diseases: Recent evidence from human genetics. Front Mol Neurosci. 2022, 15, 954541. [Google Scholar] [CrossRef]
- Kaushal, N; Ramesh, V; Gozal, D. TNF-α and temporal changes in sleep architecture in mice exposed to sleep fragmentation. PLoS One 2012, 7(9), e45610. [Google Scholar] [CrossRef]
- Liu, H; Huang, X; Li, Y; Xi, K; Han, Y; Mao, H; et al. TNF signaling pathway-mediated microglial activation in the PFC underlies acute paradoxical sleep deprivation-induced anxiety-like behaviors in mice. Brain Behav Immun. 2022, 100, 254–66. [Google Scholar] [CrossRef]
- Liu, S; Sun, JY; Ren, LP; Chen, K; Xu, B. Propofol attenuates intermittent hypoxia-induced up-regulation of proinflammatory cytokines in microglia through inhibiting the activation of NF-κB/p38 MAPK signaling. Folia Neuropathol 2017, 55(2), 124–31. [Google Scholar] [CrossRef]
- Ma, C; Li, B; Silverman, D; Ding, X; Li, A; Xiao, C; et al. Microglia regulate sleep through calcium-dependent modulation of norepinephrine transmission. Nat Neurosci. 2024, 27, 249–58. [Google Scholar] [CrossRef]
- Metaxas, A; Vaitheeswaran, R; Jensen, KT; Thygesen, C; Ilkjaer, L; Darvesh, S; et al. Reduced serotonin transporter levels and inflammation in the midbrain raphe of 12-month-old APPswe/PSEN1dE9 mice. Curr Alzheimer Res. 2018, 15(5), 420–8. [Google Scholar] [CrossRef]
- Mullington, J; Korth, C; Hermann, DM; Orth, A; Galanos, C; Holsboer, F; et al. Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul Integr Comp Physiol. 2000, 278(4), R947–55. [Google Scholar] [CrossRef]
- Nolan, E; Sun, Y; Shi, H; Archer, D; Perry, A; Pechman, K; et al. The association between poor sleep health and Alzheimer's disease structural neuroimaging biomarkers. Alzheimers Dement. 2025, 21(6), e70364. [Google Scholar] [CrossRef]
- Pan, W; Yu, C; Hsuchou, H; Zhang, Y; Kastin, AJ. Neuroinflammation facilitates LIF entry into brain: Role of TNF. Am J Physiol Cell Physiol. 2008, 294(6), C1436–42. [Google Scholar] [CrossRef]
- Patel, SR; Zhu, X; Storfer-Isser, A; Mehra, R; Jenny, NS; Tracy, R; et al. Sleep duration and biomarkers of inflammation. Sleep 2009, 32(2), 200–4. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, S; Hsu, DZ; Fu, YH; Liu, MY. Sleep deprivation-induced multi-organ injury: Role of oxidative stress and inflammation. EXCLI J 2015, 14, 672–83. [Google Scholar] [CrossRef] [PubMed]
- Piber, D; Olmstead, R; Cho, JH; Irwin, MR. Disturbance of sleep maintenance, but not sleep duration, activates nuclear factor-κB and signal transducer and activator of transcription family proteins in older adults: Sex differences. Sleep 2023, 46(10), zsad130. [Google Scholar] [CrossRef] [PubMed]
- Pinto, MJ; Bizien, L; Fabre, JMJ; Ðukanović, N; Lepetz, V; Henderson, F; et al. Microglial TNFα controls daily changes in synaptic GABAARs and sleep slow waves. J Cell Biol. 2024, 223(7), e202401041. [Google Scholar] [CrossRef]
- Page, MJ; McKenzie, JE; Bossuyt, PM; Boutron, I; Hoffmann, TC; Mulrow, CD; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Rowe, RK; Green, TRF; Giordano, KR; Ortiz, JB; Murphy, SM; Opp, MR. Microglia are necessary to regulate sleep after an immune challenge. Biology 2022, 11(8), 1241. [Google Scholar] [CrossRef]
- Shearer, WT; Reuben, JM; Mullington, JM; Price, NJ; Lee, BN; Smith, EO; et al. Soluble TNF-alpha receptor 1 and IL-6 plasma levels in humans subjected to the sleep deprivation model of spaceflight. J Allergy Clin Immunol. 2001, 107(1), 165–70. [Google Scholar] [CrossRef]
- Theoharides, TC; Tsilioni, I; Bawazeer, M. Mast cells, neuroinflammation and pain in fibromyalgia syndrome. Front Cell Neurosci. 2019, 13, 353. [Google Scholar] [CrossRef]
- Vgontzas, AN; Zoumakis, E; Bixler, EO; Lin, HM; Follett, H; Kales, A; et al. Adverse effects of modest sleep restriction on sleepiness, performance, and inflammatory cytokines. J Clin Endocrinol Metab. 2004, 89(5), 2119–26. [Google Scholar] [CrossRef]
- Vgontzas, AN; Zoumakis, M; Papanicolaou, DA; Bixler, EO; Prolo, P; Lin, HM; et al. Chronic insomnia is associated with a shift of interleukin-6 and tumor necrosis factor secretion from nighttime to daytime. Metabolism 2002, 51(7), 887–92. [Google Scholar] [CrossRef] [PubMed]
- Vicente, MC; Paneghini, JL; Stabile, AM; Amorim, M; Anibal Silva, CE; Patrone, LGA; et al. Inhibition of pro-inflammatory microglia with minocycline improves cognitive and sleep-wake dysfunction under respiratory stress in a sporadic model for Alzheimer's disease. J Alzheimers Dis. 2023, 95(1), 317–37. [Google Scholar] [CrossRef]
- Wright, KP, Jr.; Drake, AL; Frey, DJ; Fleshner, M; Desouza, CA; Gronfier, C; et al. Influence of sleep deprivation and circadian misalignment on cortisol, inflammatory markers, and cytokine balance. Brain Behav Immun. 2015, 47, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y; Ba, L; Wang, M; Deng, SY; Chen, SM; Huang, LF; et al. Chronic sleep fragmentation shares similar pathogenesis with neurodegenerative diseases: Endosome-autophagosome-lysosome pathway dysfunction and microglia-mediated neuroinflammation. CNS Neurosci Ther. 2020, 26(2), 215–27. [Google Scholar] [CrossRef]
- Zhao, Q; Xie, X; Fan, Y; Zhang, J; Jiang, W; Wu, X; et al. Phenotypic dysregulation of microglial activation in young offspring rats with maternal sleep deprivation-induced cognitive impairment. Sci Rep. 2015, 5, 9513. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, MR; Gibbons, AJ. Neuroinflammation, sleep, and circadian rhythms. Front Cell Infect Microbiol 2022, 12, 853096. [Google Scholar] [CrossRef]
- Zielinski, MR; Systrom, DM; Rose, NR. Fatigue, sleep, and autoimmune and related disorders. Front Immunol 2019, 10, 1827. [Google Scholar] [CrossRef] [PubMed]
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).