Submitted:
17 December 2025
Posted:
18 December 2025
You are already at the latest version
Abstract
Human non-polio enteroviruses (NPEV) cause a plethora of infections in humans, ranging from mild to severe neurological diseases including aseptic meningitis. NPEV are the leading cause of aseptic meningitis in both children and adults worldwide. In Russia, reports of NPEV infections have surged, especially in the post-COVID era starting in 2022, with elevated infection rates into 2023. A comprehensive examination of the complete genome is crucial for understanding the evolution of NPEV genes and for predicting potential outbreaks. The study focused on to identify the circulating NPEV strains in the Ural Federal District and Western Siberia, using Sanger sequencing and next-generation sequencing (NGS) methodologies. Biological samples were collected from (n = 225) patients diagnosed with aseptic. Bioinformatics analysis targeted the nucleotide sequences of the VP1 gene fragment, and the assembly of complete NPEV genomes. 159 NPEV were characterized, representing 70.7% of the collected samples. The main capsid variants forming the predominant genotypic profile included E30 (n = 39, 24.3%), E6 (n = 31, 19.3%), and CVA9 (n = 25, 15.6%). Using NGS sequencing, we successfully assembled 13 complete genomes for E6, E30, EV-B80, CVA9, CVB5, E11 and EV-A71. This molecular-genetic analysis provides contemporary insights into the genotypic composition, circulation patterns, and evolutionary dynamics of the dominant NPEV associated with aseptic meningitis in the Ural Federal District and Western Siberia. The laboratory-based monitoring and epidemiological surveillance for genetic changes and evolutionary studies are important for improving prevention and healthcare.
Keywords:
aseptic meningitis
; NPEV
; genotypic composition
; next-generation sequencing (NGS)
; E30
; E6
; CVA9
; phylogenetic analysis
1. Introduction
Non-polio enteroviruses (NPEV) constitute the largest genus within the Picornaviridae family. These are small naked single-stranded positive RNA viruses of approximately 7,500 nucleotides grouped into four species: Enterovirus alphacoxsackie (EV-A), Enterovirus betacoxsackie (EV-B), Enterovirus coxsackiepol (EV-C), and Enterovirus deconjuncti (EV-D) and more than 100 distinct genotypes [1,2]. Most viruses in this genus cause infections in humans, ranging from mild respiratory illnesses to severe neurological diseases [3]. Clinical manifestations may develop after an incubation period of 3 to 21 days. NPEV are distributed worldwide and have a seasonal incidence pattern in temperate regions during summer and fall, while they occur year-round in tropical regions [4].
NPEV are associated with a range of central nervous system infections and are the leading cause of aseptic meningitis in adults and children worldwide [5,6]. Aseptic meningitis is a clinical entity characterized by an elevated leukocyte count in the cerebrospinal fluid (CSF), typically manifesting as pleocytosis. This condition is distinguished by the absence of positive findings on Gram stain and bacterial culture. Furthermore, aseptic meningitis is defined by the lack of a discernible parameningeal focus or systemic illness, underscoring its etiological heterogeneity and the challenges in identifying its causative agents. This leads to favorable clinical outcomes [7] and manifests differently depending on the patient’s age and immune health [8]. Nearly 50 NPEV types have been identified as causes of aseptic meningitis globally, distributed across the four species [9]. EV-B types including CV-B3-5, CV-A9, E6, 9 and 30 are the most identified in patients with aseptic meningitis [5] with E30 the most common cause of viral meningitis and reported in outbreaks worldwide [8,10].
The NPEV genome includes a long single open reading frame (ORF) flanked by 5’UTR and 3’UTR. The ORF encodes a single polyprotein that can be cleaved into mature viral capsid proteins P1 (VP4, VP2, VP3 and VP1) and non-structural proteins P2 (2A–C) and P3 (3A–D). VP1, VP2 and VP3 are on the surface of the viral capsid and are under immune pressure, whereas VP4 is inside the capsid [11]. VP1 is the most external and immunodominant capsid protein that contain the neutralization epitopes, and can be used for NPEV genotyping [12].
Genetic recombination is a major process in NPEV evolution, which makes it essential to determine the complete genomes to detect the occurrence of these recombination events for surveillance and public health purposes [13]. Currently, Sanger sequencing of the VP1 capsid protein gene is the gold standard for NPEV genotyping. But whole-genome sequencing became increasingly affordable, accessible and cost-effective, which will likely allow improved typing in the near future [13].
There is limited information on circulating NPEV genotypes in aseptic meningitis patients from the Ural Federal District (UFD) and Western Siberia (WS). Such knowledge is essential for laboratory diagnostics, patient management and future outbreak responses. To gain a deeper understanding of the epidemiology of aseptic meningitis for the period spanning 2022 to 2024, this study aimed to identify the NPEV circulating in the Ural Federal District and Western Siberia population using VP1 gene typing and whole-genome sequencing (WGS). Additionally, this research aimed to investigate the correlation between NPEV types and the genomic evolution of the disease.
2. Materials and Methods
2.1. Ethical Consideration
The research was performed as part of the Russian state program for NPEV surveillance. Since 2017, the national enterovirus surveillance has been collecting laboratory data and biological samples from enteroviral infections in the UFD and WS. The participating centers were asked to report NPEV meningitis/encephalitis detections monthly to the Ural-Siberian regional scientific and methodological center for the study of enteroviral infections. According to national regulations, the use of anonymous samples and data from state epidemiological surveillance does not require informed consent. The research was performed with a waiver of consent under IRB № 4 of the Local Ethics Committee of the State Scientific Center of Virology and Biotechnology “Vector” (date of approval 24 June 2022).
2.2. Clinical Samples and Data Collection
A total of 281 positive samples from 225 patients with NPEV meningitis were received in the Laboratory of Enteric Infections from January 2022 to the end of December 2024. These samples included 119 residual CSF samples positive for NPEV from patients with confirmed aseptic meningitis, that were collected by participating centers and transported to our laboratory at approximately 4°C (Table 1). Additionally, pharyngeal swabs (n=58) and stool specimens (n=48) were included in our study and genotyped when CSF samples were not available or untypable. All samples were stored at -20°C until processing. For laboratory studies, stool suspensions were prepared by adding approximately 0.1-0.2 g of each stool sample to polypropylene test tubes, containing 1 - 1.5 ml of PBS.
2.3. Enterovirus Diagnosis and Molecular Genotyping
First, the samples were centrifuged for 2 minutes at 10,000 xg at room temperature in order to remove the solid particles. Viral RNA genome was isolated from CSF, pharyngeal swabs and stool suspensions employing the RIBO-prep kit (InterLabService Ltd., Moscow, Russia) according to manufacturer’s protocol. cDNA synthesis was carried out using the REVERTA-L-100 kit (InterLabService Ltd., Moscow, Russia). Next, two rounds of PCR targeting the VP1 gene were performed according to Nix et al. with minor modifications as previously described [14]. The reaction products were separated and visualized on 1.5% agarose gels and extracted by using the Cleanup Standard reagent kit (Evrogen JSC, Moscow, Russia). Purified products were sequenced in both directions (forward and reverse) using the deoxy sequencing Sanger method with the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Austin, USA) on an automatic genetic analyzer 3130 Genetic Analyzer (Thermo Fisher Scientific, Waltham, USA), following manufacturers’ protocol.
2.4. Virus Isolation, Preparation of Libraries, Whole Genome Sequencing
All the samples that were attempted for WGS sequencing, were inoculated on RD (Rhabdomyosarcoma) for the detection of non-polio enteroviruses as per the WHO non-polio enteroviruses Guidelines (Figure 1). Briefly, A volume of 0.2 ml of treated fecal sample, CSF or pharyngeal swab suspension was used for the RD inoculation. The inoculated tubes were then incubated at 37 °C for a period of 1–10 days, until a complete cytopathic effect was observed under ordinary light microscope. Uninfected cells were used as negative control. NPEV strains isolated on RD cell lines were directed to WGS. Cells were frozen and thawed three times and then subjected to RNA extraction procedure. A volume of 200 µl of the cell culture supernatant were used for total RNA extraction with ExtractRNA reagent (Evrogen JSC, Moscow, Russia) for further molecular biological analysis. The RNA concentration was determined on a Qubit 4.0 fluorimeter using the Qubit RNA Assay kit (Qubit™ RNA BR Assay, Thermo Fisher Scientific, Waltham, USA). To confirm the isolation of NPEV RNA, real-time PCR amplification was performed using a set of reagents “AmpliSens Enterovirus-FL” (InterLabService Ltd., Moscow, Russia ) in a StepOnePlus amplifier (Applied Biosystems, Austin, USA).
cDNA synthesis was performed using the Mint cDNA synthesis kit (Evrogen JSC, Moscow, Russia). DNA libraries were obtained with the NEB Next Ultra II RNA Library Prep Kit (New England Biolabs, IUSA) and DNA concentration was measured after library preparation on a Qubit 4.0 fluorimeter using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, USA) for the quantitative determination of double-stranded DNA. The quality of each library was assessed by analyzing a volume of 2 µl using a Qsep One Plus fragment analyzer (BioOptic, Taiwan) with a high-resolution cartridge and by visualizing on a 2% agarose gel (TBE buffer, 180 V for 30 minutes). Prepared libraries were pooled into a single tube pool of 4 nM by dilution in sterile water. The concentration of the final library pool was measured using the QubitTM dsDNA HS Assay Kit. Genomic libraries were sequenced on the Illumina MiSeq platform (Illumina, San Diego, USA), utilizing the MiSeq v2 cartridge, which generates paired readings of 2 × 250 cycles. All kits were used following manufacturer’s instructions.
2.5. Bioinformatic Analysis for Sanger and WGS
All VP1 sequences were visualized using Chromas 2.6.6 (Technelysium, Pty, Ltd. version 2.6.6) and NPEV genotyping was carried out by comparing the results of direct genome sequencing of NPEV with the reference sequences presented in the international GenBank database of genetic data and the BLAST service (https://blast.ncbi.nih.gov/). In addition, genotyping was confirmed using the online RIVM program (https://www.rivm.nl/mpf/typingtool/enterovirus/, accessed on 2 July 2025).
For WGS, low-quality reads were filtered, and adaptors were trimmed with Trimmomatic. The quality of the read was visualized using FastQC before and after trimming. De novo assembly was performed using SPAdes genome assembler V3.9 programs with default parameters.
2.6. Phylogenetic and Statistical Analysis
Using MEGA software, nucleotide and amino acid sequences were aligned and subjected to preliminary analysis through ClustalW procedures. Next, random sets of additional whole-genome sequences, corresponding to each obtained genotype were added to alignments from the GenBank database. Then, alignments were analyzed in BEAST software v1.10.4 with parsing of collection years for each strain and performing calibrated strict molecular clock for EVA71 because of authors concern about its origin (strict clock was used due to prevent over-parameterization). The evolutionary models for all trees was GTR+G+I based on results of ModelTest-NG software v0.1.7. Obtained phylogenetic trees were inspected using Tracer software v1.7.2, finalized in TreeAnnotator software v1.10.4 and visualized in FigTree software v1.4.4 with minimal graphical adaptations.
3. Results
3.1. Incidence of Enterovirus Meningitis in the Ural Federal District and Western Siberia
From January 2019 to December 2024, 2,465 cases of NPEV-associated aseptic meningitis were recorded in the UFD and WS. (Figure 2). The average monthly incidence rate was 0,19 per 100,000 inhabitants with most cases occurring in the Sverdlovsk oblast (40%; 987/2465), Khanty-Mansi autonomous okrug (13.9%; 342/2465), and Novosibirsk oblast (12%, 295/2465). A notable increase in the number of aseptic meningitis NPEV-positive cases was observed in September-October 2023, declining in November 2023 and peaking again in the summer of 2024. The monthly incidence has shown a summer-autumn pattern with 94.85% of the cases (2338/2465) occurring from June until November, with a peak in August-September. The long-term dynamics of the incidence of NPEV aseptic meningitis were characterized by a large decrease in 2020, an increase in 2022 with a return to pre-pandemic values in 2023.
The most common clinical symptoms were fever, headache, and vomiting. A total of 281 positive samples from 225 patients with NPEV meningitis were received in the Laboratory of Enteric Infections from January 2022 to the end of December 2024. We analyzed and genotyped 225 samples (119 CSF, 58 pharyngeal, and 48 fecal samples). The age of the patients, ranged from 1 year to 57 years, with a median of 8 years and the male to female ratio was 1:0.63 (138 males / 87 females). Schoolchildren, from 7 to 17 years, constituted 57.3% of the patients (129/225). Preschool children, from 3 to 6 years, constituted the second largest age group 27.1% (61/225).
3.2. Partial VP1 Genotyping
From the 225 aseptic meningitis patients, NPEV genotyping was successfully performed in 159 (70.7%), and more specifically 84/119 CSF samples (70.6%), 42/48 stool samples (87.5%), and 33/58 pharyngeal swabs (56.9%), indicating that stool and CSF samples are the most suitable biological samples for meningitis diagnosis. CSF samples were prioritized for genotyping. The NPEV genotypes detected in the different available samples were identical in all cases. VP1 genotyping revealed twenty-one different NPEV genotypes circulating among patients with aseptic meningitis in the UFD and WS (Figure 2). Most genotypes belonged to the Enterovirus betacoxsackie (EV-B) genus (90%), and less to the Enterovirus alphacosackie (EV-A) genus (9.4%), and only one rhinovirus detected (0.6%; and which belongs to a different genus). Some Federal districts reported a low number of registered cases and sent few samples. Most samples were received and typed from the Sverdlovsk oblast, Khanty-Mansi autonomous okrug, and the Novosibirsk oblast (Table 2). Only two samples were successfully typed in the Omsk oblast (from two samples sent to our center) and one case was typed in the Tyumen oblast (from three samples sent). Three Federal district did not sent samples from aseptic meningitis patients (Yamalo-Nenets autonomous okrug, Kemerovo oblast and Altai krai). The most prevalent genotypes were E30 (24.3%, 39/160), E6 (19.3%, 31/160), CVA9 (15.6%, 25/160), CVB5 (6.25%,10/160), E25 (5.6%, 9/160) (Table 2 and Figure 3). EV-A species were mainly detected in children less than 6 years (12 out of 15 detected) and from the Sverdlovsk oblast (14 out of 15 detected). E30 was mainly detected in the schoolchildren group from 7 to 17 years (32 out of the 39 detected E30). While E6 was almost exclusively detected in patients from 3 to 18 years (28 out of the 31 detected E6). The sole EV-A71 strain was isolated in a patient with aseptic meningitis from a nasopharyngeal sample.
3.3. Whole Genome Sequencing (WGS) and Sequence Assembly
Forty NPEV strains (35 EV-B and 5 EV-A) from respective clinical cases were isolated initially by cell culture on RD cells. We chose the most predominant NPEV types found by VP1 genotyping: E30, E6, CVA9, CVB5, CVA2 and the potentially neurovirulent EV-A71. Due to the reduced sensitivity that is frequently encountered with conventional cell culture systems, repeated passages were required in most cases until the observation of a cytopathic effect. All these NPEV isolates were attempted by WGS sequencing on Illumina MiSeq and only 16 were successfully sequenced by the NGS method. A list of the sequenced genome is provided in Table 3.
3.4. Phylogenetic Analysis of Full-Length Sequences
Concerning the reconstruction of phylogenetic events for nucleotide sequences from two E30 full-length genomes, the topology of the cladogram demonstrates the formation of two distinct nodes (Figure 4A): i) the 832 strain clustered with E30 circulating in Spain in 2018 [13] forming a cluster (sub-genogroup II), ii) another cluster consisted of strain 1173 that formed a monophyletic cluster with strains from Germany and the Netherlands in 2018 (sub-genogroup V).
Phylogenetic analysis of E6 genovariants (6 isolates) showed that most of the identified enteroviruses circulated in the studied regions for some years. As shown on Figure 4B, strains of E6 clustered closely with strains circulating in Baltimore, USA from 2016 to 2017 [5] and strains that circulated in Spain during 2015 [13]. While they were distant from Chinese E9 strains reported between 2013 and 2019.
Phylogenetic reconstruction, involving nucleotide sequences from full-length genomes CVA9 circulating in the Sverdlovsk oblast, show the formation of an internal clade with a hypothetical common ancestor among CVA9 (Figure 4C). These CVA9 strains forming a common cluster with sequences from Kazakhstan, without polyphyletic relationships with sequences from China.
Finally, EVA71 strain were clustered with strains from Asian-Pacific region (Figure 4D), but estimated age of the most recent common ancestor (tMRCA) were 20 years (95% HPD 18-21).
4. Discussion
Aseptic meningitis, the most frequent infection of the central nervous system, is caused by viruses, bacteria, fungi, and parasites, with viruses being the predominant agents [15]. NPEV are the primary viral agents responsible for this condition. Hence, rapid NPEV identification and genotyping is critical for patients with aseptic meningitis. NPEV meningitis outbreaks have been frequently reported in the Russia Federation [16].
There is little current data on NPEV circulating in the UFD and WS population with aseptic meningitis. This study describes the NPEV types detected in samples from aseptic meningitis patients (CSF, Feces, NP), as part of the National Surveillance Program from January 2022 to December 2024. A total of 281 NPEV positive samples were received, and 225 samples were attempted by partial VP1 gene sequencing, and 40 samples were attempted by WGS.
The monthly incidence shows a summer-autumn pattern, which is typically observed in temperate regions. The COVID-19 pandemic halted the spread of NPEV, likely due to stringent infection control measures [5]. As these measures relaxed, the circulation and incidence of NPEV meningitis surged in our region. In our cohort, most patients with positive NPEV were schoolchildren (7 and 17 years). Over the three years study period, partial VP1 genotyping showed that EV-B species E30, E6 and CVA9 were the major identified types in aseptic meningitis patients. Minor genotypes recorded were CVB5, E25, and E9. EV-B species have been globally the most frequent in aseptic meningitis in Europe [17,18,19,20], in Africa [6,21,22,23], in Asia [2,8,12,24,25], in the Middle East [26,27], and in the US [5].
E30 was mainly detected in schoolchildren, which is consistent with a British study by Holmes et al. [17] but defers from previously published reports where E30 was mainly identified in adults [5,21]. E6 was the predominant type in 2024, which is in alignment with previous studies where E6 was the predominant types before the COVID-19 pandemic in 2018 and 2019 [8,24]. Also, CVA9 have been reported as a major type in aseptic meningitis cases worldwide including the USA [5]. In addition, the EV-A71 strain subgenogroup C4, mainly circulating in Asia, was identified in this study. This strain is considered highly neurovirulent and account for 80% of severe and 93% of fatal HFMD cases in China[28]. The EV-A71 subgroup C4 were reported in several European countries including France [29] and the city of Rostov-on-Don in Russia [16]. Nevertheless, identified EV-A71 strain has large evolutionary distance from tMRCA. The authors would interpret this result with caution because of possible selection bias during the building of alignment (only whole-genome sequences were included with its linkage to meningitis cases).
Because CSF contains low enteroviral copies, it is recommended to test and type additionally respiratory and stool samples [30]. In this study, VP1 genotyping was higher in stool samples (87.5%) compared to CSF (70.6%) and pharyngeal samples (56.9%). In all cases, the NPEV genotype was identical in all tested samples, when applied. This result is in accordance with a Greek study [20]. This finding is important for clinicians as CSF quantity is usually limited for molecular analysis especially in neonates and infants.
Schoolchildren accounted for the highest number of NPEV positive cases, making up about 60% of all infections. This finding contrasts with a study from Palestine [26], which reported the highest rates among infants under one year old.
The circulation of multiple NPEV genovariants may be affected by various factors such as viral evolution, and recombination events, population bottlenecks, virulence, host immunity and the introduction of novel types [31].
The success rate of direct NPEV typing was high (70.6%) which is significantly higher than in previous studies 21.2% [8], 50% [21]. The high detection rate of NPEV infections might be attributed to effective reporting practices, careful sample transportation, and the refined PCR conditions in the Nix nested-PCR protocol [32].
There are some limitations to this study. First, the number of samples is relatively small. Second, the epidemic circulation patterns of different types of NPEV, with years of high incidence (e.g., E30) followed by others with low or very low detection. Third, the NGS protocol required the growth of NPEV on cell culture which is a limiting factor as many aseptic meningitis viruses are characterized by poor growth (EV-A71) or no growth CVA1-A6 [22].
Limited data are available regarding circulating genotypes, which are associated with aseptic meningitis in Russia and in Europe during this period[33]. E30 was the predominant NPEV genotype in our area in 2023. E30 meningitis outbreaks have been reported to occur worldwide every 3 to 5 years [12,32]. Recent E30 meningitis outbreak occurred in Germany[19] and in Northern Europe [34].
Other similar studies reported totally different results with a predilection for the infant and adult age group [17,35]. This may be due to variation in the health-seeking behavior between different population. Outdoor activity is probably the main reason why the male population was more infected by NPEV than the female population, with more aseptic meningitis cases in males. These results are consistent with previous reports [12].
In this study, a preliminary phylogenetic analysis of E30, CVA9 and E6 strains was performed and indicted a possible correlation with other strains circulating in Spain (E6, E30), Germany and the Netherlands (E30), USA (E6) and Kazakhstan (CVA9) which could be attributed to migration and population movement.
A large proportion of the NPEV positive samples originated from preschool/schoolchildren from 3 to 17 years (190 out of the 225 included patients). An interesting observation of this data is that the number of cases in the infant and adult group remained low at 6.7% and 8.9% respectively. Within the predominant genotype, E30 E6 and CVA9 there was a surprising genetic diversity with full-length sequences like those obtained from Europe and Asia. This provides a reservoir from which novel strains may potentially emerge and perhaps as an outbreak event. Although E30, E6, and CVA9 strains detected in this study belonged to a single genogroup within each type, they clustered within the same genogroup.
The high mutation rate and the RNA recombination are responsible for genetic diversity of NPEV. The recombination increases viral pathogenicity, removes lethal mutations and increases viral fitness. Of the four NPEV species, Enterovirus exhibited the highest rates of recombination particularly between members of the same species. The continuous diversity of the VP1 coding region, which is involved in virus-cell interactions and antibody neutralization, might explain the high endemicity and infection of NPEV [26].
5. Conclusions
In conclusion, this study documents for the first time the WGS sequences of E6, CVA9, E30 and EVA71 circulating in parts of the Russian Federation. The public health impact of enteroviruses has been highlighted by outbreaks of severe neurological disease that mimicked polio but was linked to non-polio enteroviruses. Considering this, we hypothesize that NPEVs may predominate in their ability to cause paralysis after polio have been completely eradicated. Thus, investigating non-polio enteroviruses linked to neurological effects becomes essential.
Author Contributions
Conceptualization, A.V.S. and T.M.I.; methodology, molecular investigation, T.M.I., V.I.C., A.K.P., E.S.K.; epidemiological data: V.I.C.; data analysis, T.M.I. and V.I.C.; validation, A.V.S. and T.M.I.; resources, T.M.I. and V.I.C.; data curation, V.I.C.; writing—original draft preparation, T.M.I., V.I.C.; writing—review and editing, T.M.I., V.I.C.; A.V.S.; visualization, V.I.C.; supervision, A.V.S.All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
The study protocol was approved by the Local Ethics Committee of the State Scientific Center of Virology and Biotechnology “Vector” (№ 4, 24 June 2022).
Informed Consent Statement
The research was performed as part of the Russian state program for NPEV sur-veillance. According to national regulations, the use of anonymous samples and data from state epidemiological surveillance does not require informed consent.
Data Availability Statement
Not applicable.
Acknowledgments
We thank the Federal Service for Surveillance on Consumer Rights Protection and Human Wellbeing (Rospotrebnadzor) Centers for Hygiene and Epidemiology in Sverdlovsk Oblast, Chelyabinsk Oblast, Tyumen Oblast, Khanty-Mansi Autonomous Okrug, Kurgansk Oblast, Yamalo-Nenets Autonomous Okrug, Tomsk Oblast, Omsk Oblast, Novosibirsk Oblast, Kemerovo Oblast and Altai Krai for their assistance in the collection and delivery of the clinical specimens, and for providing epidemiological and clinical data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and Enterovirus Diversity with Associated Human Diseases. Infection, Genetics and Evolution 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Farshadpour, F.; Taherkhani, R. Molecular Epidemiology of Enteroviruses and Predominance of Echovirus 30 in an Iranian Population with Aseptic Meningitis. Journal of NeuroVirology 2021, 27, 444–451. [Google Scholar] [CrossRef]
- Zhou, F.; Kong, F.; Wang, B.; McPhie, K.; Gilbert, G.L.; Dwyer, D.E. Molecular Characterization of Enterovirus 71 and Coxsackievirus A16 Using the 5′ Untranslated Region and VP1 Region. Journal of Medical Microbiology 2011, 60, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Pons-Salort, M.; Parker, E.P.K.; Grassly, N.C. The Epidemiology of Non-Polio Enteroviruses: Recent Advances and Outstanding Questions. Current Opinion in Infectious Diseases 2015, 28, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Fall, A.; Forman, M.; Morris, C.P.; Gniazdowski, V.; Luo, C.H.; Hanlon, A.; Miller, H.; Bergman, Y.; Mostafa, H.H. Enterovirus Characterized from Cerebrospinal Fluid in a Cohort from the Eastern United States. Journal of Clinical Virology 2023, 161, 105401. [Google Scholar] [CrossRef]
- Nkosi, N.; Preiser, W.; Van Zyl, G.; Claassen, M.; Cronje, N.; Maritz, J.; Newman, H.; McCarthy, K.; Ntshoe, G.; Essel, V.; et al. Molecular Characterisation and Epidemiology of Enterovirus-Associated Aseptic Meningitis in the Western and Eastern Cape Provinces, South Africa 2018–2019. Journal of Clinical Virology 2021, 139, 104845. [Google Scholar] [CrossRef]
- Shukla, B.; Aguilera, E.A.; Salazar, L.; Wootton, S.H.; Kaewpoowat, Q.; Hasbun, R. Aseptic Meningitis in Adults and Children: Diagnostic and Management Challenges. Journal of Clinical Virology 2017, 94, 110–114. [Google Scholar] [CrossRef]
- Wang, J.; Meng, M.; Xu, H.; Wang, T.; Liu, Y.; Yan, H.; Liu, P.; Qin, D.; Yang, Q. Analysis of Enterovirus Genotypes in the Cerebrospinal Fluid of Children Associated with Aseptic Meningitis in Liaocheng, China, from 2018 to 2019. BMC Infect Dis 2021, 21, 405. [Google Scholar] [CrossRef]
- Hua, Y.; Lv, Z.; Zhou, Y.; Xiang, H.; Sun, M.; Kang, Y.-J. Metatranscriptomics Revealed the Molecular Characterization of Circulating Enterovirus Strains Causing Aseptic Meningitis in Children in Wuxi, China. Heliyon 2024, 10, e26847. [Google Scholar] [CrossRef]
- Yuan, F.; Wei, X.; Ma, X.; Ma, J.; Ma, X.; Sun, X.; Cao, M.; Zhou, J.; Zhang, W.; Chen, H.; et al. Genetic Characterizations and Molecular Epidemiology of Human Echovirus 30 Isolated from Ningxia, China. Biosafety and Health 2023, 5, 346–354. [Google Scholar] [CrossRef]
- Baggen, J.; Thibaut, H.J.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M. The Life Cycle of Non-Polio Enteroviruses and How to Target It. Nat Rev Microbiol 2018, 16, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhou, X.; Liu, J.; Xia, L.; Pan, Y.; Chen, J.; Luo, N.; Yin, J.; Ma, S. Molecular Identification of Human Enteroviruses Associated with Aseptic Meningitis in Yunnan Province, Southwest China. SpringerPlus 2016, 5, 1515. [Google Scholar] [CrossRef] [PubMed]
- Gámbaro, F.; Pérez, A.B.; Agüera, E.; Prot, M.; Martínez-Martínez, L.; Cabrerizo, M.; Simon-Loriere, E.; Fernandez-Garcia, M.D. Genomic Surveillance of Enterovirus Associated with Aseptic Meningitis Cases in Southern Spain, 2015–2018. Sci Rep 2021, 11, 21523. [Google Scholar] [CrossRef] [PubMed]
- Itani, T.M.; Chalapa, V.I.; Slautin, V.N.; Bykov, R.O.; Imangaliev, B.S.; Starikova, P.K.; Sergeev, A.G.; Semenov, A.V. Non-Polio Enterovirus Surveillance in the Ural Federal District and Western Siberia, 2022: Is There a Need for a Vaccine? Vaccines 2023, 11, 1588. [Google Scholar] [CrossRef]
- Kumar, R. Aseptic Meningitis: Diagnosis and Management. Indian J Pediatr 2005, 72, 57–63. [Google Scholar] [CrossRef]
- Akhmadishina, L.V.; Govorukhina, M.V.; Kovalev, E.V.; Nenadskaya, S.A.; Ivanova, O.E.; Lukashev, A.N. Enterovirus A71 Meningoencephalitis Outbreak, Rostov-on-Don, Russia, 2013. Emerg. Infect. Dis. 2015, 21, 1440–1443. [Google Scholar] [CrossRef]
- Holmes, C.W.; Koo, S.S.F.; Osman, H.; Wilson, S.; Xerry, J.; Gallimore, C.I.; Allen, D.J.; Tang, J.W. Predominance of Enterovirus B and Echovirus 30 as Cause of Viral Meningitis in a UK Population. Journal of Clinical Virology 2016, 81, 90–93. [Google Scholar] [CrossRef]
- Dilcher, M.; Howard, J.C.; Dalton, S.C.; Anderson, T.; Clinghan, R.T.; Werno, A.M. Clinical, Laboratory, and Molecular Epidemiology of an Outbreak of Aseptic Meningitis Due to a Triple-Recombinant Echovirus in Ashburton, New Zealand. Viruses 2022, 14, 658. [Google Scholar] [CrossRef]
- Graf, J.; Hartmann, C.J.; Lehmann, H.C.; Otto, C.; Adams, O.; Karenfort, M.; Schneider, C.; Ruprecht, K.; Bosse, H.M.; Diedrich, S.; et al. Meningitis Gone Viral: Description of the Echovirus Wave 2013 in Germany. BMC Infect Dis 2019, 19, 1010. [Google Scholar] [CrossRef]
- Posnakoglou, L.; Tatsi, E.-B.; Chatzichristou, P.; Siahanidou, T.; Kanaka-Gantenbein, C.; Syriopoulou, V.; Michos, A. Molecular Epidemiology of Enterovirus in Children with Central Nervous System Infections. Viruses 2021, 13, 100. [Google Scholar] [CrossRef]
- Rmadi, Y.; Elargoubi, A.; González-Sanz, R.; Mastouri, M.; Cabrerizo, M.; Aouni, M. Molecular Characterization of Enterovirus Detected in Cerebrospinal Fluid and Wastewater Samples in Monastir, Tunisia, 2014–2017. Virol J 2022, 19, 45. [Google Scholar] [CrossRef]
- Rai, A.; Ammi, Z.; Anes-Boulahbal, D.L.; Assadi, A.A.; Amrane, A.; Baaloudj, O.; Mouni, L. Molecular Amplification and Cell Culturing Efficiency for Enteroviruses’ Detection in Cerebrospinal Fluids of Algerian Patients Suffering from Meningitis. Viruses 2024, 16, 170. [Google Scholar] [CrossRef]
- Janse Van Rensburg, M.; Mans, J.; Mafuyeka, R.T.; Strydom, K.; Myburgh, M.; Van Zyl, W.B. Diversity of Enteroviruses in Cerebrospinal Fluid Specimens Collected from Hospitalised Patients in the Private and Public Sector in South Africa. Journal of Medical Virology 2024, 96, e29514. [Google Scholar] [CrossRef]
- Chen, X.; Guo, J.; Li, J.; Li, Q.; Ai, J.; Sun, S.; Xie, Z. Serotypes of Human Enteroviruses Causing Pediatric Viral Encephalitis and Meningitis in Hebei Province, China, from 2013 to 2015. Pediatric Investigation 2018, 2, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Anahita, I; Ali Akbar, R; Yousef, M; Sayed Yousef, M. Prevalence of Enterovirus Meningitis in Children: Report from a Tertiary Center. Maedica (Bucur) 2018, 13(3), 213–216. [Google Scholar] [CrossRef] [PubMed]
- Dumaidi, K.; Al-Jawabreh, A.; Samarah, F.; Zraiqi, A.; Yaseen, D. Genetic Diversity of the Enteroviruses Detected from Cerebrospinal Fluid (CSF) Samples of Patients with Suspected Aseptic Meningitis in Northern West Bank, Palestine in 2017. PLoS ONE 2018, 13, e0202243. [Google Scholar] [CrossRef] [PubMed]
- Fratty, I.S.; Kriger, O.; Weiss, L.; Vasserman, R.; Erster, O.; Mendelson, E.; Sofer, D.; Weil, M. Increased Detection of Echovirus 6-Associated Meningitis in Patients Hospitalized during the COVID-19 Pandemic, Israel 2021–2022. Journal of Clinical Virology 2023, 162, 105425. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, X.; Cui, A.; Mao, N.; Xu, S.; Zhu, Z.; Zhou, J.; Shi, J.; Zhao, Y.; Wang, X.; et al. Complete Genome Analysis of the C4 Subgenotype Strains of Enterovirus 71: Predominant Recombination C4 Viruses Persistently Circulating in China for 14 Years. PLoS ONE 2013, 8, e56341. [Google Scholar] [CrossRef]
- Schuffenecker, I.; Henquell, C.; Mirand, A.; Coste-Burel, M.; Marque-Juillet, S.; Desbois, D.; Lagathu, G.; Bornebusch, L.; Bailly, J.-L.; Lina, B. New Introductions of Enterovirus 71 Subgenogroup C4 Strains, France, 2012. Emerg. Infect. Dis. 2014, 20, 1343–1346. [Google Scholar] [CrossRef]
- Harvala, H.; Broberg, E.; Benschop, K.; Berginc, N.; Ladhani, S.; Susi, P.; Christiansen, C.; McKenna, J.; Allen, D.; Makiello, P.; et al. Recommendations for Enterovirus Diagnostics and Characterisation within and beyond Europe. Journal of Clinical Virology 2018, 101, 11–17. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Vakulenko, Y.A.; Turbabina, N.A.; Deviatkin, A.A.; Drexler, J.F. Molecular Epidemiology and Phylogenetics of Human Enteroviruses: Is There a Forest behind the Trees? Reviews in Medical Virology 2018, 28, e2002. [Google Scholar] [CrossRef]
- Itani, T.M.; Chalapa, V.I.; Slautin, V.N.; Imangaliev, B.S.; Kungurtseva, M.S.; Patrusheva, A.K.; Sergeev, A.G.; Semenov, A.V. Circulation of Non-Polio Enteroviruses in the Ural Federal District and Western Siberia in 2023: The Return of an Old Foe? Arch Virol 2025, 170, 110. [Google Scholar] [CrossRef]
- Yarmolskaya, M.S.; Shumilina, E.Yu.; Ivanova, O.E.; Drexler, J.F.; Lukashev, A.N. Molecular Epidemiology of Echoviruses 11 and 30 in Russia: Different Properties of Genotypes within an Enterovirus Serotype. Infection, Genetics and Evolution 2015, 30, 244–248. [Google Scholar] [CrossRef]
- Smura, T.; Blomqvist, S.; Kolehmainen, P.; Schuffenecker, I.; Lina, B.; Böttcher, S.; Diedrich, S.; Löve, A.; Brytting, M.; Hauzenberger, E.; et al. Aseptic Meningitis Outbreak Associated with Echovirus 4 in Northern Europe in 2013–2014. Journal of Clinical Virology 2020, 129, 104535. [Google Scholar] [CrossRef]
- Papadakis, G.; Chibo, D.; Druce, J.; Catton, M.; Birch, C. Detection and Genotyping of Enteroviruses in Cerebrospinal Fluid in Patients in Victoria, Australia, 2007-2013: Detection and Genotyping of Enteroviruses in CSF. J. Med. Virol. 2014, 86, 1609–1613. [Google Scholar] [CrossRef]
Figure 1.
Flow diagram of the study cohort and sequenced samples in aseptic meningitis patients from in the Ural Federal District and Western Siberia.
Figure 1.
Flow diagram of the study cohort and sequenced samples in aseptic meningitis patients from in the Ural Federal District and Western Siberia.
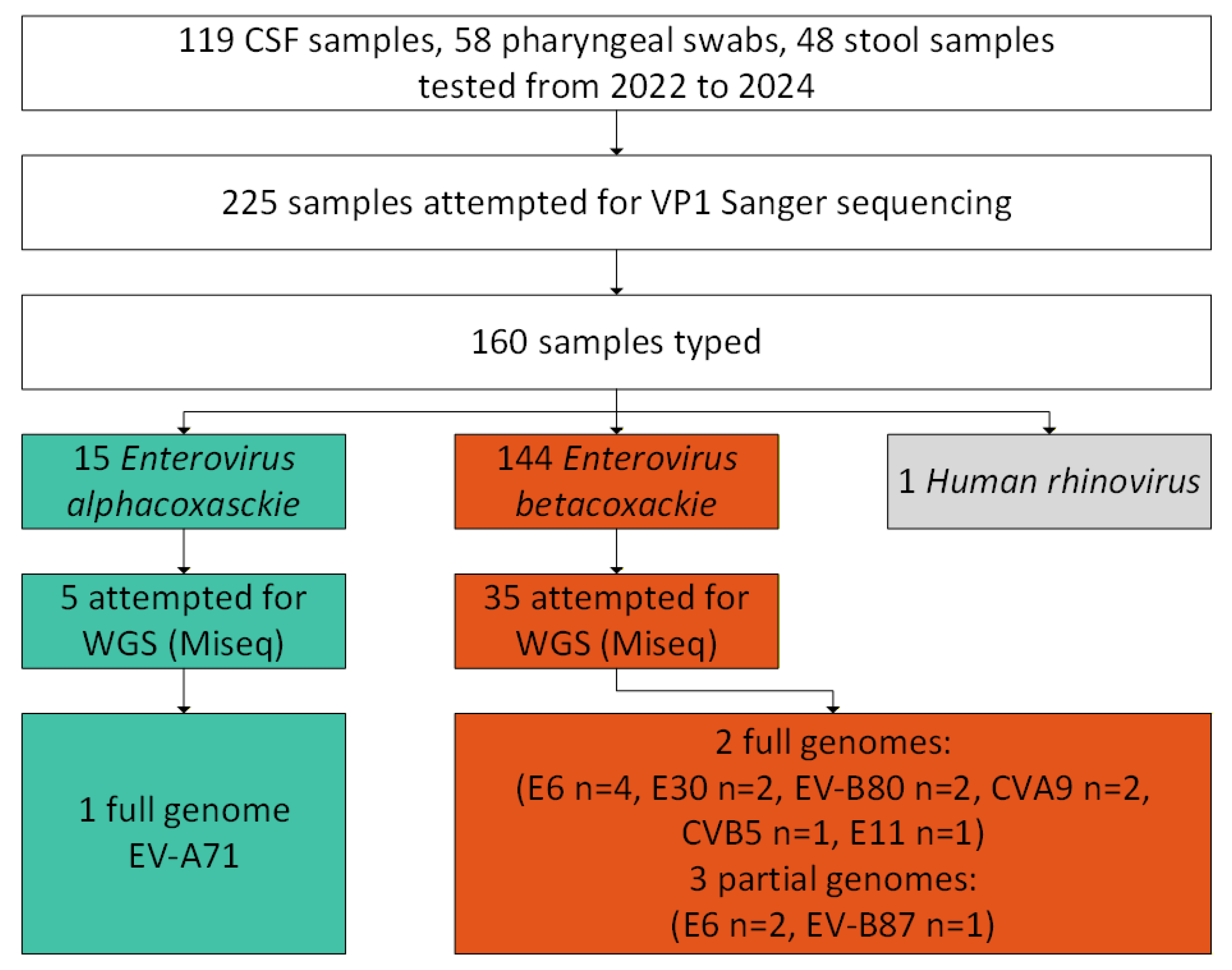
Figure 2.
Long-term dynamics of the incidence rates of NPEV aseptic meningitis in UFD and WS, 2019-2024: monthly incidence rates per 100,000 inhabitants and number of collected samples from 2022 to 2024.
Figure 2.
Long-term dynamics of the incidence rates of NPEV aseptic meningitis in UFD and WS, 2019-2024: monthly incidence rates per 100,000 inhabitants and number of collected samples from 2022 to 2024.
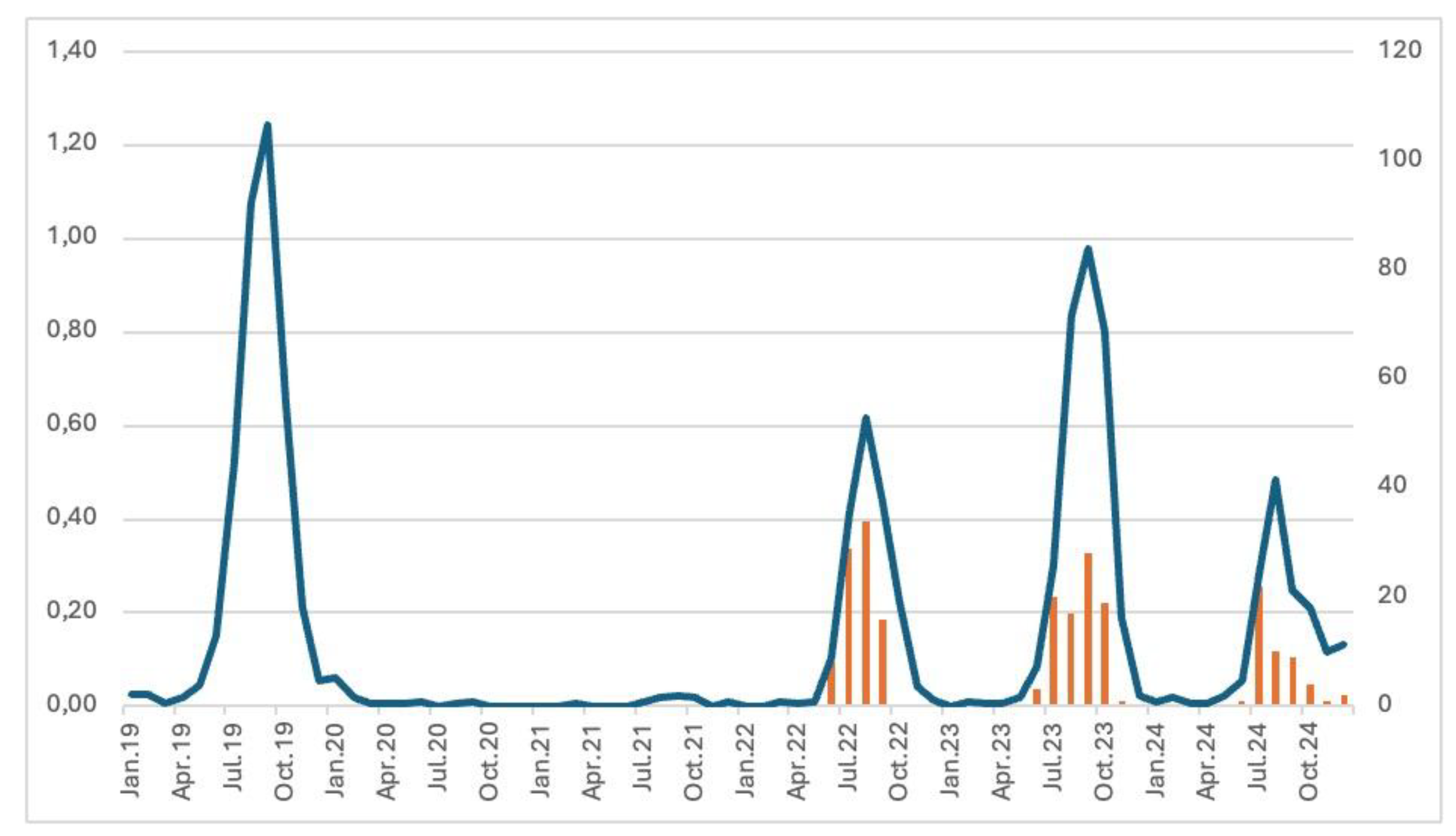
Figure 3.
Species and types of the NPEV isolated in aseptic meningitis in the Ural Federal District and Western Siberia (n= number of cases).
Figure 3.
Species and types of the NPEV isolated in aseptic meningitis in the Ural Federal District and Western Siberia (n= number of cases).
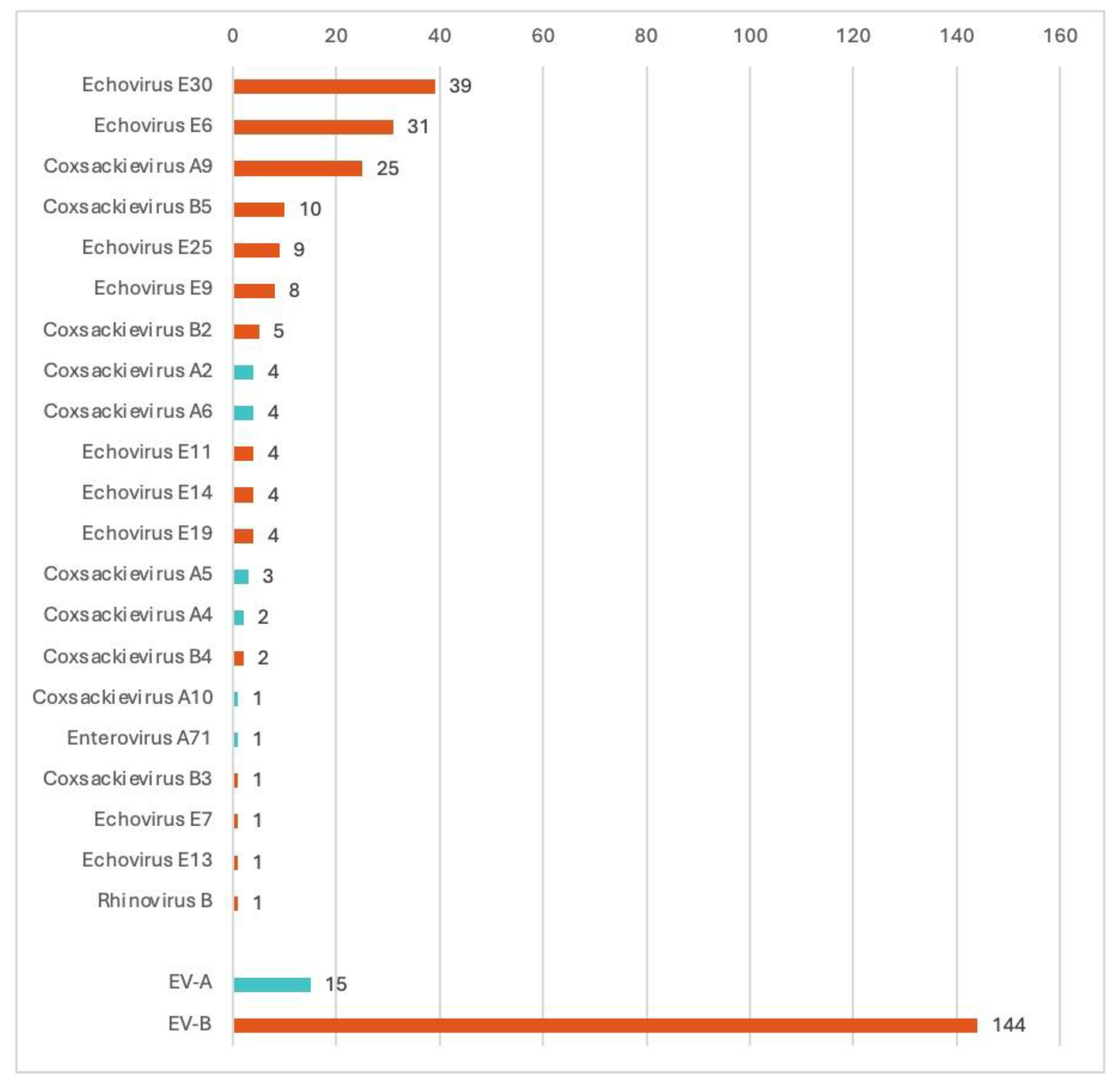
Figure 4.
Cladogram of nucleotide sequences from full-length genomes: A. six E6 strains. B. three E30 strains. C. one EV-A71 strain (blue bars corresponds to time after tMRCA (in years) depicted above the branch). D. Two CVA9 strains. Black arrows indicate full-length genome sequences NPEV strains identified in the Ural Federal District and Western Siberia. Numbers at each branch correspond to its posterior probability values.
Figure 4.
Cladogram of nucleotide sequences from full-length genomes: A. six E6 strains. B. three E30 strains. C. one EV-A71 strain (blue bars corresponds to time after tMRCA (in years) depicted above the branch). D. Two CVA9 strains. Black arrows indicate full-length genome sequences NPEV strains identified in the Ural Federal District and Western Siberia. Numbers at each branch correspond to its posterior probability values.
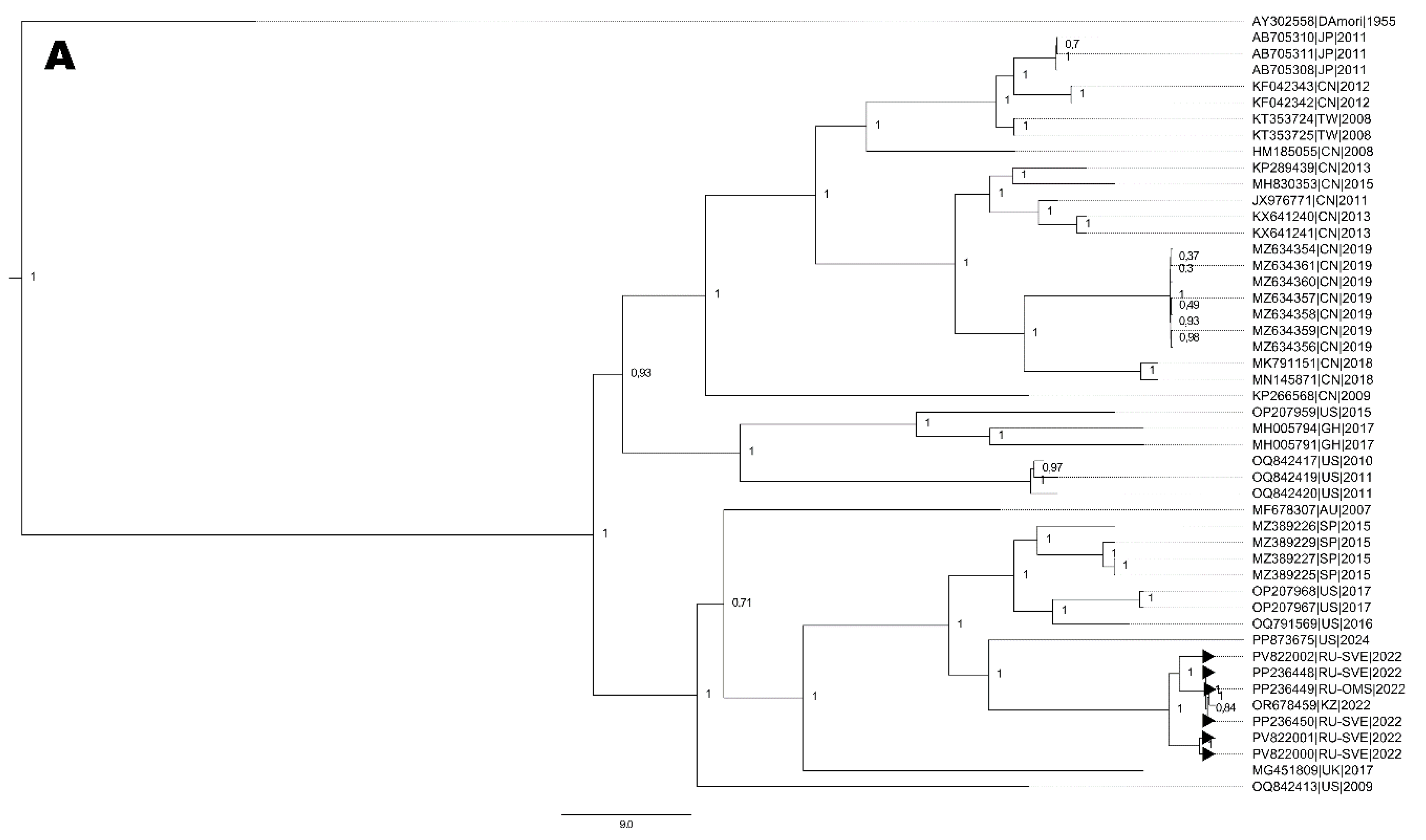
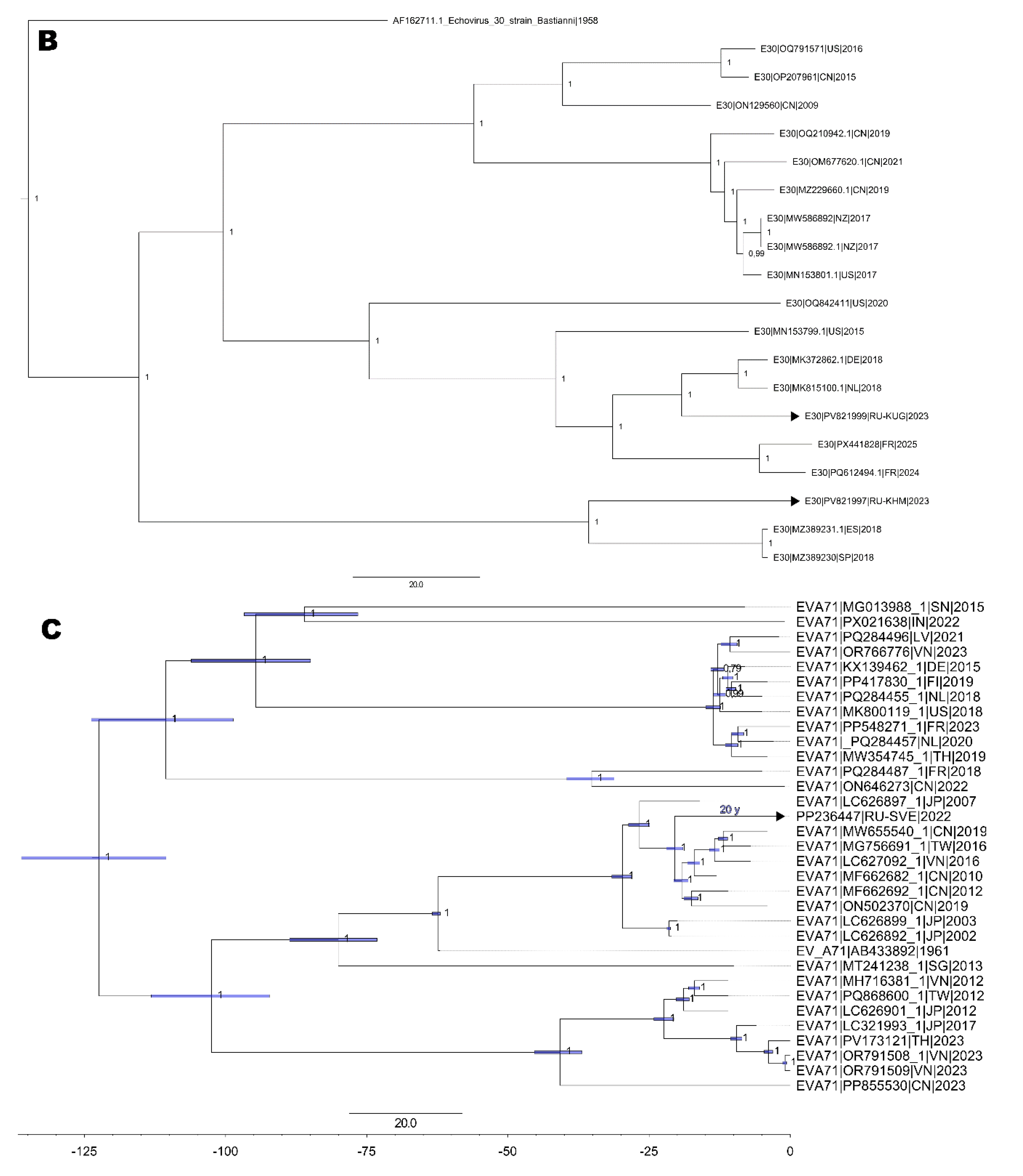
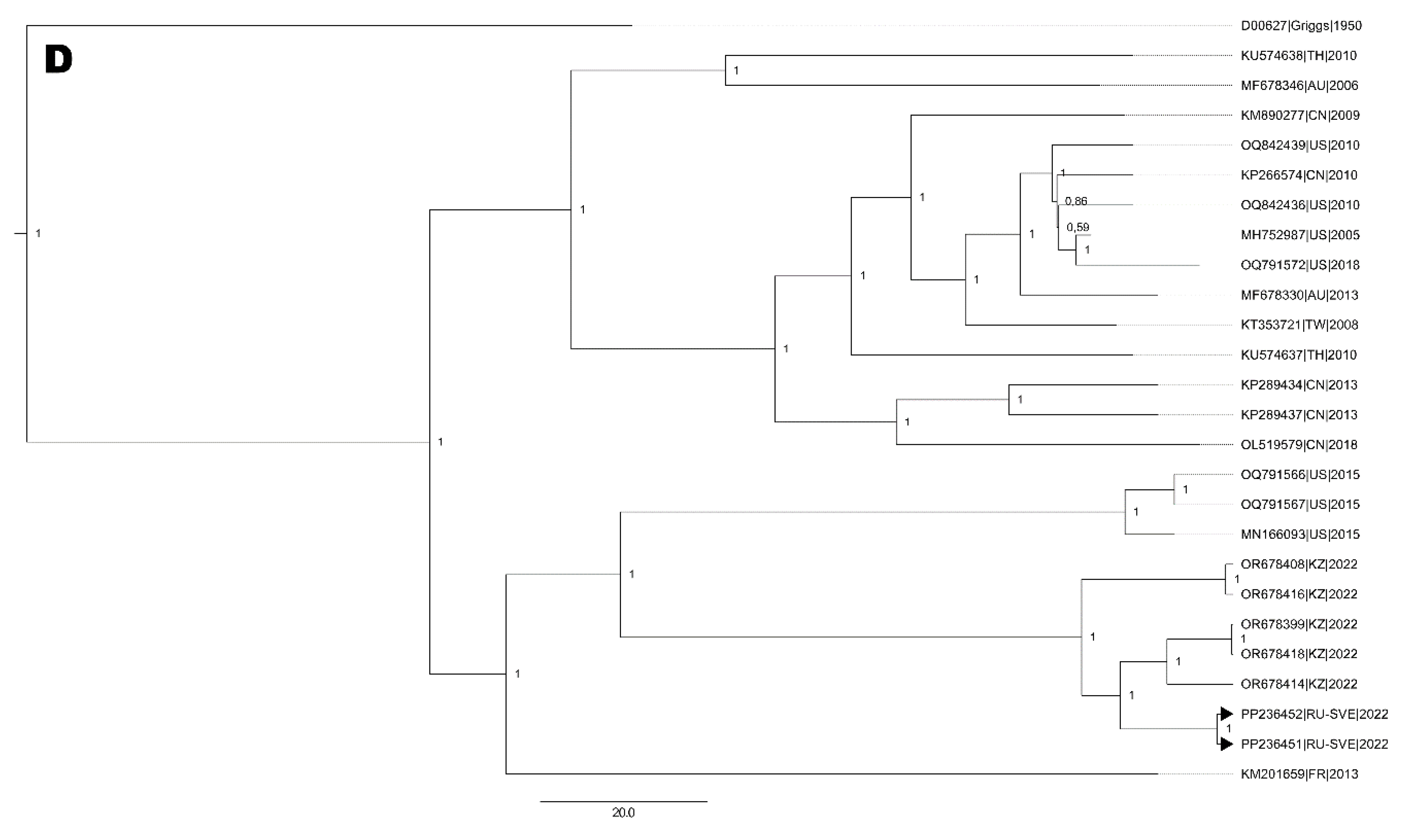

Table 1.
Patients and sample types.
| Infants group 0 to 2 y.o. |
Preschool children group 3 to 6 y.o. |
Schoolchildren group 7 to 17 y.o. |
Adults 18 + y.o. | Total | |
|---|---|---|---|---|---|
| Stool specimens | 3 | 15 | 26 | 4 | 48 |
| Pharyngeal swabs | 6 | 18 | 32 | 2 | 58 |
| CSF | 6 | 28 | 71 | 14 | 119 |
| Total | 15 (6,7%) |
61 (27,1%) |
129 (57,3%) |
20 (8,9%) |
225 |
CSF: Cerebrospinal fluid, y.o.: years old.
Table 2.
Species and types of the NPEV isolated in aseptic meningitis cases in different subjects of the Ural Federal District and Western Siberia, n (%).
Table 2.
Species and types of the NPEV isolated in aseptic meningitis cases in different subjects of the Ural Federal District and Western Siberia, n (%).
| Species/types | Sverdlovsk oblast | Chelyabinsk oblast | Tyumen oblast | Kurgansk oblast | Khanty-Mansi autonomous okrug | Omsk oblast | Tomsk oblast | Novosibirsk oblast | Total |
|---|---|---|---|---|---|---|---|---|---|
| Coxsackievirus A2 | 4 (2.5) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (2.5) |
| Coxsackievirus A4 | 2 (1.2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (1.2) |
| Coxsackievirus A5 | 3 (1.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (1.9) |
| Coxsackievirus A6 | 3 (1.9) | 0 | 0 | 0 | 1 (0.6) | 0 | 0 | 0 | 4 (2.5) |
| Coxsackievirus A10 | 1 (0.62) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.62) |
| Enterovirus A71 | 1 (0.62) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.62) |
| Enterovirus alphacoxsackie | 14 (8.8) | 0 | 0 | 0 | 1 (0.6) | 0 | 0 | 0 | 15 (9.4) |
| Coxsackievirus A9 | 17 (10.7) | 0 | 0 | 0 | 4 (2.5) | 2 (1.2) | 2 (1.2) | 0 | 25 (15.6) |
| Coxsackievirus B2 | 1 (0.62) | 3 (1.9) | 0 | 0 | 0 | 1 (0.62) | 0 | 0 | 5 (3.1) |
| Coxsackievirus B3 | 0 | 1 (0.62) | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.62) |
| Coxsackievirus B4 | 0 | 0 | 0 | 0 | 1 (0.62) | 0 | 0 | 1 (0.62) | 2 (1.2) |
| Coxsackievirus B5 | 1 (0.62) | 1 (0.62) | 0 | 2 (1.2) | 5 (3.1) | 0 | 0 | 1 (0.62) | 10 (6.2) |
| Echovirus E6 | 8 (5) | 0 | 0 | 0 | 10 (6.2) | 1 (0.62) | 0 | 12 (7.5) | 31 (19,4) |
| Echovirus E7 | 1 (0.62) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.6) |
| Echovirus E9 | 3 (1.9) | 0 | 0 | 0 | 4 (2.5) | 1 (0.62) | 0 | 0 | 8 (5) |
| Echovirus E11 | 1 (0.62) | 0 | 1 (0.62) | 0 | 0 | 1 (0.62) | 0 | 1 (0.62) | 4 (2.5) |
| Echovirus E13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (0.62) | 1 (0.62) |
| Echovirus E14 | 0 | 0 | 0 | 0 | 4 (2.5) | 0 | 0 | 0 | 4 (2.5) |
| Echovirus E19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 (2.5) | 4 (2.5) |
| Echovirus E25 | 0 | 0 | 0 | 3 (1.9) | 2 (1.2) | 0 | 0 | 4 (2.5) | 9 (5.6) |
| Echovirus E30 | 11 (6.9) | 0 | 0 | 3 (1.9) | 25 (15.6) | 0 | 0 | 0 | 39 (24.4) |
| Enterovirus betacoxsackie | 43 (26.9) | 5 (3.1) | 1 (0.62) | 8 (5) | 55 (34.4) | 6 (3.8) | 2 (1.2) | 24 (15) | 144 (90) |
| Rhinovirus B | 0 | 0 | 1 (0,62) | 0 | 0 | 0 | 0 | 0 | 1 (0.62) |
| Total | 57 (36) | 5 (3) | 2 (1) | 8 (5) | 56 (35) | 6 (4) | 2 (1) | 24 (15) | 160 |
Table 3.
Demographic data and clinical characteristic of NGS sequences NPEV strains.
| Sample No. | Accession No. | NPEV genotype | Sample type | Year of isolation | Age (years) | City, Federal subject | Genome size (bp) |
|---|---|---|---|---|---|---|---|
| 1086_22 | PP236447.1 | EV-A71 | Nasopharyngeal | 2022 | 5 | Ekaterinburg, SO | 7283 |
| 2831_22 | PV822004.1 | CVB5 | Feces | 2022 | 7 | Ekaterinburg, SO | 7236 |
| 2827_22 | PV822003.1 | E11 | Feces | 2022 | 13 | Ekaterinburg, SO | 7430 |
| 2826_22 | PV822002.1 | E6 | Feces | 2022 | 8 | Ekaterinburg, SO | 7381 |
| 2819_22 | PV822001.1 | E6 | CSF | 2022 | 4 | Ekaterinburg, SO | 7378 |
| 2808_22 | PV822000.1 | E6 | Feces | 2022 | 7 | Ekaterinburg, SO | 7410 |
| 2806_22 | PP236450.1 | E6 | Feces | 2022 | 3 | Ekaterinburg, SO | 6616 |
| 787_22 | PP236449.1 | E6 | CSF | 2022 | 5 | Omsk, OO | 5820 |
| 2813_22 | PP236448.1 | E6 | Feces | 2022 | 57 | Ekaterinburg, SO | 7482 |
| 1173_23 | PV821999.1 | E30 | CSF | 2023 | 12 | Kurgan, KO | 7266 |
| 832_23 | PV821997 | E30 | Nasopharyngeal | 2023 | 15 | Surgut, KMAO | 7414 |
| 2816_22 | PP236452 | CVA9 | Feces | 2022 | 7 | Ekaterinburg, SO | 7650 |
| 2805_22 | PP236451.1 | CVA9 | Nasopharyngeal | 2022 | 24 | Ekaterinburg, SO | 7410 |
| 1188_23 | PQ572232.1 | EV-B87 | CSF | 2023 | 7 | Surgut, KMAO | 6455 |
| 1326_23 | PQ572231.1 | EV-B80 | CSF | 2023 | 6 | Ekaterinburg, SO | 6943 |
| 1117_23 | PQ572230.1 | EV-B80 | CSF | 2023 | 12 | Kurgan, KO | 6858 |
Abbreviations: CSF= Cerebrospinal fluid, SO= Sverdlovsk oblast, KO= Kurgansk Oblast, KMAO= Khanty-Mansi autonomous okrug, pb= base pair.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



