
Submitted:
15 December 2025
Posted:
17 December 2025
You are already at the latest version
Abstract
Pregnant women undergo a myriad of physiological changes during this stage, including important hormonal variations. Pregnancy gingivitis is a condition that affects up to 30% to 100% of women, is related to hormonal modifications, and could play an important role in gestational gut colonization and immunological training in the newborn. Nonetheless, oral health is not always considered part of routine prenatal care. In this study, we collected saliva samples of pregnant women with and without pregnancy gingivitis and analyzed the oral microbiota through 16S sequencing. In addition, meconium from the infants of participating women was also analyzed. The oral microbiota of pregnant women with and without pregnancy gingivitis did not show significant diversity differences. However, significant differences in microbiome composition were observed. In addition, it appears that microbiome composition of the offspring of mothers with gingivitis may also differ from that of mothers without gingivitis, although the number of available samples did not allow definite conclusions. As such, a larger cohort and deeper sequencing methods are needed to demonstrate the differences in the oral microbiota of pregnant women with and without gingivitis and to explore the possibility of bacterial translocation from the maternal gingiva to the fetal gut.
Keywords:
oral microbiota
; microbiota
; metagenomics
; pregnancy gingivitis
; newborn
1. Introduction
During pregnancy, women experience multiple modifications at the hormonal, immunological, and metabolic levels. The maternal oral microbiota experiences notable changes during pregnancy, particularly in the context of pregnancy gingivitis (PG). Balan et al. (2018) highlighted that hormonal, metabolic, and immunological factors influence the oral microbiome, leading to dysbiosis associated with PG [1]. Their findings reveal a pathogenic shift in the oral microbiome during pregnancy, which reverts to a healthy state postpartum. This change underscores the potential role of oral dysbiosis in adverse pregnancy outcomes. La et al. (2022) further elucidated this transition by characterizing the oral microbiota from preconception to late pregnancy [2]. Their study indicates a significant reduction in microbial diversity during the third trimester, with an increase in pathogenic taxa such as Prevotella and Atopobium parvulum. Notably, women with better oral hygiene practices exhibited lower richness and diversity of pathogens, suggesting that oral hygiene may mitigate the adverse effects of dysbiosis. Collectively, these studies suggest that the maternal oral microbiota is susceptible to changes during pregnancy, particularly in the presence of gingivitis, emphasizing the importance of maintaining oral health to support both maternal and fetal well-being.
Alterations in maternal oral microbiota during pregnancy, especially those related to PG, could influence the neonatal gut colonization. Whether microbial colonization occurs during the fetal stage of not is still a debate. At present, there is no consensus about the origin of the infant’s intestinal colonization, and in this ongoing debate, bacteria from the intestinal, oral, vaginal, and skin from the maternal microbiota have been proposed as potential sources [1,2].
Recent studies have detected bacterial DNA in the placenta, umbilical cord blood, meconium, and amniotic fluid, indicating the presence of microbes before birth [3,4,5]. Some of these microbes, including species of Streptococcus, Fusobacterium, and Porphyromonas, are commonly found in the oral cavity, supporting the hypothesis of potential vertical transmission of the maternal oral microbiota to the fetus, especially in women with PG [6]. Maternal oral bacteria that colonize the fetus may prime the immune system, influencing susceptibility to infections, allergies, and metabolic conditions later in life. However, dysbiosis—an imbalance in microbial communities—may have negative consequences[7].
One possible mechanism of the origins of colonization was proposed by Zaura et al, who reported that moderate gingivitis during pregnancy is a physiological and non-pathological process that aims to translocate the oral bacteria via the bloodstream into the placenta, and finally to the fetus [8]. PG is the inflammation and bleeding of the gums during the gestational stage [9]. Of note, dysbiosis of the oral microbiota has been reported in the presence of gingivitis; if such changes occur during pregnancy, this might have a significant impact in the immune system training of the newborn [10]. However, the specific contribution of maternal oral microbiota on neonatal intestinal colonization remains poorly understood.
In this study, we describe the oral microbiota of pregnant women with and without PG. In addition, we analyzed the relationship between maternal oral microbiota and meconium microbiota from their respective newborns, to assess the potential role of the oral cavity as a source for the first bacterial colonization in infants.
2. Materials and Methods
We recruited 30 pregnant women (15 with PG and 15 without it (NPG group)) at the Obstetrics and Gynecology Service at Hospital Central “Dr. Ignacio Morones Prieto”. The study was approved by the Ethics and Research Committee from the hospital with approbation number: 35-19. The inclusion criteria were Mexican healthy pregnant women aged 18 or older, with a gestational age from 30 to 42 weeks. The non-inclusion criteria were women with systemic or chronic disease, gestational diabetes, preeclampsia or eclampsia, ongoing antibiotic treatment, orthodontic treatment, and periodontal disease.
2.1. Sample Collection
Unstimulated saliva was collected during prenatal care visits, in a Zymo Research DNA/RNA shield Saliva Collection Kit (Zymo Research Corp, United States) from the recruited women; also, shortly after birth a meconium sample was collected from the diaper in a Fecal Collection tube DNA/RNA Shield TM (Zymo Research Corp, United States) of their newborn infants.
2.2. DNA Extraction, 16s Amplicon PCR, and Sequencing
DNA was extracted from saliva and meconium samples using the ZymoBIOMICS Miniprep Kit (Zymo Research Corp, United States), following the manufacturer´s instructions.
A 16S rRNA PCR was performed using the Illumina primers 16S Amplicon PCR (Forward TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and Reverse GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC).
Cycle conditions were 95 °C (3 min), then 35 cycles of 95 °C (30 s), 55 °C (30 s), 72 °C (30 s), then a final extension of 72 °C (5 min). Libraries were purified using Select-a-Size DNA Clean & Concentrator MagBead Kit (Zymo Research Corp; United States) according to the Illumina 16S metagenomic sequencing library protocol. Dual indices and Illumina sequencing adapters from the Illumina Nextera XT index kits v2 B and C (Illumina, San Diego, USA) were added to the target amplicons in a second PCR step using GoTaq Colorless Master Mix (Promega, United States). The DNA sequences of the amplicon library were determined using the Illumina MiSeq platform at Laboratorio de Genómica Funcional y Comparativa at the Instituto Potosino de Ciencia y Tecnología (IPICYT).
2.3. Bioinformatic Analysis
To perform the analysis, we used the FASTQ files generated by the Illumina MiSeq platform. Then, a quality check was performed using the FASTQC tool [11], and then the R package DADA2 v1.16.0 [12] was used to analyze the 16S sequencing data obtained by first, trimming 10nt at the left and on the right sides of the fragments. The forward primer AGRGTTYGATYMTGGCTCAG and the reverse primer RGYTACCTTGTTACGACTT were removed by using the removePrimers () function, with default parameters.
By using the dada2 function the sample composition was inferred using default parameters. Chimeras were removed using the removeBimeraDenovo () function and then the assignTaxonomy () function for the taxonomy assignment. To obtain the amplicon sequence variant (ASV), we used the silva_nr99_v138 database.
2.4. Taxa Abundance Analysis
Taxa abundances were processed using custom R v4.0.2 [13] scripts.
2.5. Diversity
R package vegan v2.5.6 [14] was used to calculate the alpha diversity index with the diversity() function with default parameters.
2.6. Pathway’s Inference
We have used Tax4Fun2 [15] to perform the pathway prediction for the 16S sequencing data. First, we used the runRefBlast () and then the makeFunctionalPrediction() functions, with default parameters. The input to these functions was the ASV quantification profiles. We used the reference RF99NR in both steps. The resulting pathway prediction table was used to perform a sparse Partial Least Squares Discriminant Analysis (sPLS-DA). To perform this, we used the function spls-da() from the R package MixOmics with default parameters [16]. We used in-house R scripts to generate the plots to represent the results of the sPLS-DA [17].
2.7. Statistical Analysis
The frequency of detection of individual phyla and genera between mothers with and without PG were compared using Fisher’s exact test of the chi-square test, as appropriate. Analyses were carried out using OpenEpi, Version 3.01 (Dean AG, Sullivan KM, Soe MM. OpenEpi: Open Source Epidemiologic Statistics for Public Health, Version 3.01. Available at: www.OpenEpi.com; Accessed 2025/11/08)
3. Results
We collected 30 maternal salivary samples (15 with PG and 15 NPG). We also collected meconium samples from the corresponding babies, but only 6 samples from the NPG group and 3 from the PG group were suitable for sequencing of the 16S rRNA gene. We sequenced the 30 maternal samples, as well as the 9 neonatal samples to describe the characteristics of microbiota, at phylum and genera level, and the differences in diversity and abundance between the study groups of mothers and newborns. Maternal characteristics are shown in Table 1.
3.1. The Diversity Between the Two Groups
3.2. Relative Abundance of Bacteria at Phylum and Genus Level in Saliva Samples from Mothers
Phylum composition on saliva samples from women in the NPG group were: Bacillota (85.5%), Actinomycetota (9.1%), Pseudomonadota (3.15 %), and Bacteroidota (2.11%) (Figure 2). Women with PG showed a similar composition, although there were differences in the contribution of each phylum: Bacillota (74.3%), Bacteroidota (13.7%), Pseudomonadota (10.5%), and Actinomycetota (1.2%) (Figure 2). Bacillota was the predominant phylum in all, but two mothers; these two mothers belonged to the PG group. Pseudomonadota contributed significantly (>5% of phyla) in 7 of 15 (46.7%) PG mothers compared with 1 of 15 (6.7%) NPG mothers (P=0.035). Bacteroidota were present (>5% of phyla) in 9 of 15 (46.7%) PG mothers compared with 5 of 15 (26.7%) NPG mothers (P=0.45). Overall, microbiota was composed almost entirely by Bacillus and Actynomycetota (>90% of phyla) in 10 of 15 (66.7%) NPG mothers, while such composition was identified in 6 of 15 (40%) PG mothers (P=0.27)
The top 5 genera present in saliva samples from women from the NPG group were Streptococcus (85.5%), Rothia (8.1%), Haemophilus (3.1%) and Prevotella (2.1%), while in those having PG the top 5 were Streptococcus (73.5%), Prevotella (13.7%), Haeomophilus (10.5%), and Schaalia (1.2%) (Figure 3). Streptococcus was the predominant genus in most mothers (13 of 15 in both groups). Of note, Haemophilus contributed significantly to the microbiota (>5% of genera detected) of 7 of 15 (46.7%) mothers in the PG group compared with 1 of 15 (6.7%) mothers in the NPG group (P=0.035).
3.3. Relative Abundance of Bacteria at Phylum and Genus Level in Newborn Samples
In the meconium samples of newborns from NPG women, at phylum level the composition was: Bacillota (70.3%), Pseudomonadota (11.1%), Bacteroidota (10.2%), Actinomycetota (4.5%) and Fusobacteriota (0.3%), (Figure S1a), and in the meconium from the PG group: Pseudomonadota (64.5%), Bacillota (23.8%), Actinomycetota (6%), Bacteroidota (2.8%)and Cyanobacteriota (1%), (Figure S1b).
The five predominant genera in meconium samples of newborns from NPG mothers were Streptococcus (65.4%), Lactobacillus (15.1%), Prevotella (9.2%), Haemophilus (7.2%), and Rothia (2.1%) (Figure S2a). On the other hand, the predominant genera in the meconium samples of newborns from PG mothers were Streptococcus (52.1%), Enterobacter (33.3%), Haemophilus (14.2%), Enhydrobacter (11.3%), and Brevudimonas (7.6%) (Figure S3b).
3.4. Differential Genera Were Found Between Groups
By using permutation and sPLS-DA analysis we obtained the differential genera between study groups.
Comparing the results of saliva samples from women between the two groups (NPG vs PG), differences in microbiota composition were identified with the following genera present predominantly in the NPG: Actinomyces, Shwartzia, Rothia, Jeotbalibaca, Stomatobaculum, Campylobacter, Alloprevotela, Megasphaera, and Alloscardovia. In contrast, Catonella, Haemophilus, Mycoplasma, Peptococcus, Olsenella, Filifactor, Prevotela, Cutibacterium, Faucicola, Kingella, Odoribacter, Aggregatibacter, Neisseria, Phocaeicola, Bifidobacterium, Dialister, Tannerella, Lactobacillus, and Defluvitaleaceae UCG-10. were the genera present predominantly in the PG group (Figure 4).
Analysis of the meconium samples of newborns from both groups (meconium NPG vs meconium PG) showed the presence of Granulicatella, Streptococcus, Gemella, Filifactor, Prevotellla 7, Bifidobacterium, Parabacteroides, TM7x, Candidatus Scharimonas, Megasphaera, Acholeplasma, Rothia, Abiotrophia, Veillonella, Campylobacter, Oceanivirga, Stomatobaculum, Oribacterium, Neisseria, and Prevotella in the NPG group, while Streptomyces, Sneathia, Ralstonia, Lentimicrobium, Klebsiella, Hydrogenobacter, Enterobacter, Brevudimonas, Enhydrobacter, Streptobacillus, Solobacterium, Lachnoanaerobaculum, Catonella, and Bergeyella were identified in the PG group (Figure S4).
3.5. Phylum and Genera Coincidence Analysis by Study Group
At the phylum level, both at the meconium and saliva samples from the NPG and PG groups were characterized by presence of Bacillota, Bacteroidota, Actinomycetota and Pseudomonadota.
The coincidental genera present in both samples from at least one mother and infant pair were Streptococcus, Prevotella, Haemophilus, Rothia, and Schaalia in the NPG group. In the PG group, only Streptococcus, and Prevotella were found in both mother and infant in at least one pair.
3.6. Metabolic Pathway Prediction
Prediction of the metabolic pathways related to the main genera present in each study group was carried out using the Tax4Fun2 R package. The identified metabolic pathways were plotted using Sparse Partial Least Squares Discriminant Analysis (sPLS-DA).
Comparison between maternal NPG vs PG saliva samples showed that lysine biosynthesis, glucagon signaling pathway, D-alanine metabolism, phenylalanine metabolism, retinol metabolism, arginine biosynthesis, fructose and manose metabolism, peptidoglycan biosynthesis, two-component system, glycerolipid metabolism, biosynthesis of aminoacids, degradation of aromatic compounds, atrazine degradation, naphthalene degradation, and MAPK signaling pathway were more predominant in the NPG group, while glycerophospholipid metabolism, glutathione metabolism, pyruvate metabolism, phosphatidylinositol signaling system, cationic antimicrobial peptide (CAMP) resistance, bacterial secretion system, carbon fixation in photosynthetic organisms, monobactam biosynthesis, lipopolysaccharide biosynthesis, mismatch repair, biotin metabolism, oxidative phosphorylation, NOD-like receptor signaling pathway, one carbon pool by folate, sulfur relay system, fatty acid metabolism, fluid shear stress and atherosclerosis, riboflavin metabolism, and metabolic pathways were more frequent in the PG group (Figure 5).
Analyses based on results from newborn meconium samples showed that biofilm formation-vibrio cholerae related, biotin metabolism, plant-pathogen interaction, biofilm formation- pseudomonas aeruginosa, two-component system, prodigiosin biosynthesis, nitrogen metabolism, and bacterial secretion system pathways were more relevant in the meconium NPG group, while glucagon signaling, RNA polymerase, central carbon metabolism in cancer, lysine biosynthesis, nucleotide excision repair, base excision repair, longevity regulating pathway- multiple species, homologus recombination, staphylococcus aureus infection, D-alanine metabolism, aminoacyl-tRNA biosynthesis, purine metabolism, photosynthesis, DNA replication, ribosome, glycerophospholipid metabolism, pyrimidine metabolism, and D-glutamine and D-glutamate metabolism pathways were relevant in the PG group (Figure S5).
4. Discussion
In recent years, the composition of the microbiome has been associated with diverse health outcomes, and it is possible that these effects could begin from early infancy. Therefore, understanding the mechanisms leading to intestinal colonization in infants is of great interest. Diverse factors (such as mode of delivery) and potential sources (including vaginal secretions, breastmilk, skin, and saliva) may contribute to shaping each individual microbiome [18,19]. Several studies have suggested that enteral colonization by bacteria may start during the prenatal period, and the maternal oral microbiome may contribute significantly to this process [20,21,22,23,24]. While this hypothesis is still controversial, this would imply that conditions that alter the microbiome during pregnancy could have an important effect on the composition of the microbiome in neonates, with the consequent short and long-term health effects. In the present study we sought to determine if PG, a frequent condition during pregnancy, is associated with significant changes in the oral microbiome, and explore if this could have an impact on the offspring’s microbiome.
The oral microbiota is constantly changing due to environment exposures, through eating and drinking, and oral hygiene habits. During pregnancy, gingivitis has been associated with the presence of higher levels of the genera Prevotella, Streptococcus, Veilonella, and Neisseria [25]. Balan et al. previously described that pregnancy leads to significant changes in the oral microbiota, with pregnant women (with or without gingivitis) exhibiting higher diversity than healthy non pregnant women [9]. In addition, gingivitis (in general) is associated with microbiome changes characterized by dominance of species such as Streptococcus vestibularis, Treponema sp., Veillonella dispar, Fusobacterium naviformis, and Selenomonas sp.[26] However, microbiome composition during PG shows distinct characteristics, as it is most often a result of hormonal changes, in contrast with gingivitis outside of pregnancy which is usually the result of poor oral hygiene.
In the present study, we identified differences in the microbiome of women with and without PG. Streptococcus was the most abundant genus in both groups, but its abundance was lower in the PG group compared with the NPG group; in addition, there was also a lower abundance of Rothia, while there was a higher abundance of Haemophilus in the PG group. Among the dominant genera, Rothia and Schaalia (from the Actinomycetota phylum) are typically associated with a healthy microbiota [27]. In contrast, Haemophilus (from the Pseudomonadota phylum) is a gram negative aerobic genus linked to plaque formation [28]. Prevotella and Streptococcus are generally associated with gingivitis [29]. Overall, the NPG group showed mostly commensal bacteria, while the PG group has presence of opportunistic pathogens.
We also predicted and compared the metabolic pathways between saliva from women with and without PG. Interestingly, despite the similarity in the top phyla and genera, the pathway analysis revealed significant functional differences. This indicates that microbiome changes associated with PG have an impact on microbial activity, networks, and functions [1,7,30,31]. The microbiota in the NPG group, showed central metabolism, stress metabolism, and repair mechanisms, while the PG group had activated energy metabolism and bacterial growth, lipid metabolism, and virulence pathways. The PG group also showed activation of the NOD-like pathway, which would indicate a response of the host immune system to bacterial components like peptidoglycans [32]. This suggest a pro-inflammatory metabolic state in the PG group, consistent with previous reports on maternal oral microbiota associated with gingivitis [33].
Unfortunately, we obtained sufficient DNA for sequencing in only 6 meconium samples from infants in the NPG group and 3 from infants in the PG group; this was likely due to the composition of meconium, which is a complex matrix that includes mucus, amniotic fluid, and other substances, that act as PCR inhibitors [37]. Nevertheless, comparison of the microbiome of meconium of infants from mothers with and without PG showed interesting findings. While Streptococcus was present in infants of both groups, the frequency was lower in the PG group. In addition, Enterobacter was the 2nd most abundant genus in infants of the PG group, and Enhydrobacter and Brevudimonas wer identified only in this group. In contrast, Lactobacillus was found exclusively in the NPG group.
Overall, genera identified in each infant group showed important differential patterns: Streptococcus, Prevotella, Granulicatella, Gemella, Veilonella, Campylobacter and Sneatia distinguished newborn samples from the NPG group, while Ralstonia, Estreptomyces, Brevusimonas, Enterobacter and Lachnoanaerobaculum, were identified as differential in newborns from the maternal PG group. Streptococcus and Rothia, which are characteristic of the oral microbiota, have been previously described to be present in the meconium microbiota [34,35]. In women with oral dysbiosis, hematogenous transport across the placenta or by extracellular vesicles, could be a route of fetal exposure of these bacteria [35,36]. Prevotella, Granulicatella, Gemella, Veilonella, Campylobacter, and Sneatia are also commonly found in oral microbiota; however, they can be found in gut microbiota mostly as commensal bacteria that can transition to opportunistic pathogens [36,37,38,39]. On the other hand, Ralstonia has been found abundant in meconium microbiota in some preterm infants [40], Streptomyces is known for its antibiotic production and immunoregulation [41], Enterobacter is a well-known member of the meconium microbiota as an opportunistic member [42], Brevudimonas is considered a potential opportunistic microbe [43], and Lachnoanaerobaculum is a contributor to saccharolytic and butyrate-producing functions of the early gut [44]. Thus, meconium from the NPG group contained mostly commensal bacteria, while the PG group featured bacteria that could generate a pro-inflammatory environment.
Predicted metabolic pathways from the meconium samples further supported this idea. The PG group showed activated processes related with bacterial infection and pathogenicity, like the two-component system, biofilm formation, and biotin metabolism. The two component system regulates the expression of the virulence genes [45], while biotin metabolism is essential for activation of carboxylases, with synthesis vital for survival and pathogenicity in bacteria [46]. In contrast, the NPG group displayed activation of pathways like glucagon signaling pathway, RNA polymerase, central carbon metabolism, and lysine biosynthesis. The glucagon signaling pathway is specific to eukaryotic physiology [47], but in here, it could be related to the ability to respond to nutritional stress [48], the RNA polymerase pathway could be due to high transcriptional and metabolic activity[49], the central carbon metabolism [50] and lysine biosynthesis [51]could be related to metabolic versatility from the bacteria [52]. This aligns with the genera present, depicting a pro-inflammatory, potentially pathogenic ecosystem in the PG newborns, in contrast to a focus on growth and homeostasis in the NPG group.
Recent studies provide context to our findings, Park et al. (2023) published a study where they collected 160 meconium samples in a Korean hospital and described the microbiota, as well as the association with microbiota from several anatomic sites of the mother. The most frequent genera identified in meconium samples were Lactobacillus, Staphylococcus, and Ureaplasma [53]. A study performed in Taiwan, sequenced meconium samples to describe their composition. They reported that the top phylum levels were Proteobacteria, Firmicutes, Bacteroides, Actinobacteria, and Fusobacteria [54]. The observed differences with our study suggest that regional or population characteristics may also have a significant impact on bacterial gut colonization in the newborn period.
In conclusion, we observed a healthier oral microbiota in NPG mothers, and a potentially pro-inflammatory one in mothers with PG. This profile was consistent with the microbiota found in meconium of their newborns. This could be the result of vertical transmission of oral bacteria during pregnancy via bloodstream, as supported by previous studies [25,55,56]. While the number of meconium samples with available results was small, the potential impact of PG on microbial colonization of the fetus supports the need of additional studies to confirm our findings.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Authors Rocha-Viggiano AK, Aranda-Romo S, Noyola D, Ovando-Vázquez C, Brieño-Enriquez MA, Salgado-Bustamante M. contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Rocha-Viggiano AK, Gómez-Hernández N, Casas-Flores S, Ovando-Vázquez C and Salgado-Bustamante M. The first draft of the manuscript was written by Rocha-Viggiano AK and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
This work was founded by the FORDECYTPRONACES Ciencia de Frontera 2019/101732 to COV. AKRV acknowledges Secretaría de Ciencia, Humanidades, Tecnología e Innovación (SECIHTI) for the scholarship 2018-000068-02NACF-25236. COV acknowledges the Investigadores por México, Secretaría de Ciencia, Humanidades, Tecnología e Innovación (SECIHTI) program.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of Hospital Central “Dr. Ignacio Morones Prieto”, with the approval number: 36-19, accepted on April 26th 2019.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
The authors acknowledge the Instituto Potosino de Investigación Científica y Tecnológica (IPICYT) and National Supercomputing Center (CNS) computing resources, provided to develop this project under the project grant TKII-R2018- COV1.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Stinson LF, Payne MS, Keelan JA. Planting the seed: Origins, composition, and postnatal health significance of the fetal gastrointestinal microbiota. Crit Rev Microbiol 2017;43:352–69. [CrossRef]
- Zakis DR, Paulissen E, Kornete L, Kaan AM (Marije), Nicu EA, Zaura E. The evidence for placental microbiome and its composition in healthy pregnancies: A systematic review. J Reprod Immunol 2022;149. [CrossRef]
- Rocha-Viggiano AK, Aranda-Romo S, Salgado-Bustamante M, Ovando-Vázquez C. Meconium Microbiota Composition and Association with Birth Delivery Mode. Adv Gut Microbiome Res 2022;2022:1–18. [CrossRef]
- Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The Placenta Harbors a Unique Microbiome. Sci Transl Med 2014;6:237ra65-237ra65. [CrossRef]
- Tapiainen T, Paalanne N, Tejesvi MV, Koivusaari P, Korpela K, Pokka T, et al. Maternal influence on the fetal microbiome in a population-based study of the first-pass meconium. Pediatr Res 2018;84:371–9. [CrossRef]
- Zakis DR, Paulissen E, Kornete L, Kaan AM (Marije), Nicu EA, Zaura E. The evidence for placental microbiome and its composition in healthy pregnancies: A systematic review. J Reprod Immunol 2022;149. [CrossRef]
- Mishra A, Lai GC, Yao LJ, Aung TT, Shental N, Rotter-Maskowitz A, et al. Microbial exposure during early human development primes fetal immune cells. Cell 2021;184:3394-3409.e20. [CrossRef]
- Zaura E, Nicu EA, Krom BP, Keijser BJF. Acquiring and maintaining a normal oral microbiome: current perspective. Front Cell Infect Microbiol 2014. [CrossRef]
- Balan P, Brandt BW, Chong YS, Crielaard W, Wong ML, Lopez V, et al. Subgingival Microbiota during Healthy Pregnancy and Pregnancy Gingivitis. JDR Clin Transl Res 2020;XX:1–9. [CrossRef]
- Wu M, Chen S-W, Jiang S-Y. Relationship between gingival inflammation and pregnancy. Mediators Inflamm 2015;2015:623427. [CrossRef]
- LaMar D. FastQC 2015. https://qubeshub.org/resources/fastqc.
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 2016;13:581–3. [CrossRef]
- R Core Team (2020). — European Environment Agency n.d.
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, Mcglinn D, et al. Package “vegan” Title Community Ecology Package Version 2.5-7 2020.
- Wemheuer F, Taylor JA, Daniel R, Johnston E, Meinicke P, Thomas T, et al. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ Microbiomes 2020;15:1–12.
- Rohart F, Gautier B, Singh A, Cao KAL. mixOmics: An R package for ‘omics feature selection and multiple data integration 2017;13:e1005752.
- Worley B, Powers R. PCA as a predictor of OPLS-DA model reliability. Curr Metabolomics 2016;4:97–103.
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A 2010;107:11971–5. [CrossRef]
- Mueller NT, Differding MK, Østbye T, Hoyo C, Benjamin-Neelon SE. Association of birth mode of delivery with infant faecal microbiota, potential pathobionts, and short chain fatty acids: a longitudinal study over the first year of life. BJOG Int J Obstet Gynaecol 2021;128:1293–303. [CrossRef]
- Park JY, Yun H, Lee S been, Kim HJ, Jung YH, Choi CW, et al. Comprehensive characterization of maternal, fetal, and neonatal microbiomes supports prenatal colonization of the gastrointestinal tract. Sci Rep 2023;13:1–14. [CrossRef]
- Collado MC, Schwab C, Zurich E, Pilar SM, Nagpal R, Tsuji H, et al. Sensitive Quantitative Analysis of the Meconium Bacterial Microbiota in Healthy Term Infants Born Vaginally or by Cesarean Section. Front Microbiol 2016;7. [CrossRef]
- Stinson LF, Boyce MC, Payne MS, Keelan JA. The not-so-sterile womb: Evidence that the human fetus is exposed to bacteria prior to birth. Front Microbiol 2019;10:1–15. [CrossRef]
- Suárez-Martínez C, Santaella-Pascual M, Yagüe-Guirao G, Martínez-Graciá C. Infant gut microbiota colonization: influence of prenatal and postnatal factors, focusing on diet. Front Microbiol 2023;14. [CrossRef]
- Bearfield C, Davenport ES, Sivapathasundaram V, Allaker RP. Possible association between amniotic fluid micro-organism infection and microflora in the mouth. BJOG Int J Obstet Gynaecol 2002;109:527–33. [CrossRef]
- Balan P, Chong YS, Umashankar S, Swarup S, Loke WM, Lopez V, et al. Keystone Species in Pregnancy Gingivitis: A Snapshot of Oral Microbiome During Pregnancy and Postpartum Period. Front Microbiol 2018;9. [CrossRef]
- Han Y, Ding P-H. Advancing periodontitis microbiome research: integrating design, analysis, and technology. Front Cell Infect Microbiol 2025;15. [CrossRef]
- Abusleme L, Hoare A, Hong BY, Diaz PI. Microbial signatures of health, gingivitis, and periodontitis. Periodontol 2000 2021;86:57–78. [CrossRef]
- Sanz M, Beighton D, Curtis MA, Cury JA, Dige I, Dommisch H, et al. Role of microbial biofilms in the maintenance of oral health and in the development of dental caries and periodontal diseases. Consensus report of group 1 of the Joint EFP/ORCA workshop on the boundaries between caries and periodontal disease. J Clin Periodontol 2017;44 Suppl 18:S5–11. [CrossRef]
- Abusleme L, Hoare A, Hong BY, Diaz PI. Microbial signatures of health, gingivitis, and periodontitis. Periodontol 2000 2021;86:57–78. [CrossRef]
- Ihekweazu FD, Versalovic J. Development of the Pediatric Gut Microbiome: Impact on Health and Disease. Am J Med Sci 2018;356:413–23. [CrossRef]
- Korpela K, Renko M, Vänni P, Paalanne N, Salo J, Tejesvi MV, et al. Microbiome of the first stool and overweight at age 3 years: A prospective cohort study. Pediatr Obes 2020;15:e12680. [CrossRef]
- Almeida-da-Silva CLC, Savio LEB, Coutinho-Silva R, Ojcius DM. The role of NOD-like receptors in innate immunity. Front Immunol 2023;14:1122586. [CrossRef]
- Ye C, Kapila Y. Oral microbiome shifts during pregnancy and adverse pregnancy outcomes: Hormonal and Immunologic changes at play. Periodontol 2000 2021;87:276–81. [CrossRef]
- He Q, Kwok L-Y, Xi X, Zhong Z, Ma T, Xu H, et al. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes n.d.;12:1794266. [CrossRef]
- Moles L, Gómez M, Heilig H, Bustos G, Fuentes S, Vos W de, et al. Bacterial Diversity in Meconium of Preterm Neonates and Evolution of Their Fecal Microbiota during the First Month of Life. PLOS ONE 2013;8:e66986. [CrossRef]
- Taylor KD, Wood AC, Rotter JI, Guo X, Herrington DM, Johnson WC, et al. Metagenomic Study of the MESA: Detection of Gemella Morbillorum and Association With Coronary Heart Disease. J Am Heart Assoc 2024;13:e035693. [CrossRef]
- Iqbal NT, Chen RY, Griffin NW, Hibberd MC, Khalid A, Sadiq K, et al. A shared group of bacterial taxa in the duodenal microbiota of undernourished Pakistani children with environmental enteric dysfunction. mSphere 2024;9. [CrossRef]
- Zhang S-M, Huang S-L. The Commensal Anaerobe Veillonella dispar Reprograms Its Lactate Metabolism and Short-Chain Fatty Acid Production during the Stationary Phase. Microbiol Spectr 2023;11:e03558-22. [CrossRef]
- Theis KR, Florova V, Romero R, Borisov AB, Winters AD, Galaz J, et al. Sneathia: an emerging pathogen in female reproductive disease and adverse perinatal outcomes. Crit Rev Microbiol 2021;47:517–42. [CrossRef]
- Kim SY, Youn Y-A. Gut Dysbiosis in the First-Passed Meconium Microbiomes of Korean Preterm Infants Compared to Full-Term Neonates. Microorganisms 2024;12:1271. [CrossRef]
- Bolourian A, Mojtahedi Z. Streptomyces, shared microbiome member of soil and gut, as ‘old friends’ against colon cancer. FEMS Microbiol Ecol 2018;94:fiy120. [CrossRef]
- Bacterial Diversity in Meconium of Preterm Neonates and Evolution of Their Fecal Microbiota during the First Month of Life | PLOS One n.d. https://journals.plos.org/plosone/article?id=10.1371%2Fjournal.pone.0066986 (accessed November 2, 2025).
- Huang Z, Yu K, Xiao Y, Wang Y, Xiao D, Wang D. Comparative Genomic Analysis Reveals Potential Pathogenicity and Slow-Growth Characteristics of Genus Brevundimonas and Description of Brevundimonas pishanensis sp. nov. Microbiol Spectr 2022;10:e0246821. [CrossRef]
- Hedberg ME, Moore ERB, Svensson-Stadler L, Hörstedt P, Baranov V, Hernell O, et al. Lachnoanaerobaculum gen. nov., a new genus in the Lachnospiraceae: characterization of Lachnoanaerobaculum umeaense gen. nov., sp. nov., isolated from the human small intestine, and Lachnoanaerobaculum orale sp. nov., isolated from saliva, and reclassification of Eubacterium saburreum (Prévot 1966) Holdeman and Moore 1970 as Lachnoanaerobaculum saburreum comb. nov. Int J Syst Evol Microbiol 2012;62:2685–90. [CrossRef]
- Hirakawa H, Kurushima J, Hashimoto Y, Tomita H. Progress Overview of Bacterial Two-Component Regulatory Systems as Potential Targets for Antimicrobial Chemotherapy. Antibiotics 2020;9:635. [CrossRef]
- Sirithanakorn C, Cronan JE. Biotin, a universal and essential cofactor: synthesis, ligation and regulation. FEMS Microbiol Rev 2021;45:fuab003. [CrossRef]
- Janah L, Kjeldsen S, Galsgaard KD, Winther-Sørensen M, Stojanovska E, Pedersen J, et al. Glucagon Receptor Signaling and Glucagon Resistance. Int J Mol Sci 2019;20:3314. [CrossRef]
- Zeng Y, Wu Y, Zhang Q, Xiao X. Crosstalk between glucagon-like peptide 1 and gut microbiota in metabolic diseases. mBio 2023;15:e02032-23. [CrossRef]
- Murakami KS. Structural Biology of Bacterial RNA Polymerase. Biomolecules 2015;5:848–64. [CrossRef]
- Sudarsan S, Dethlefsen S, Blank LM, Siemann-Herzberg M, Schmid A. The Functional Structure of Central Carbon Metabolism in Pseudomonas putida KT2440. Appl Environ Microbiol 2014;80:5292–303. [CrossRef]
- Gillner DM, Becker DP, Holz RC. Lysine biosynthesis in bacteria: a metallodesuccinylase as a potential antimicrobial target. J Biol Inorg Chem JBIC Publ Soc Biol Inorg Chem 2013;18:10.1007/s00775-012-0965–1. [CrossRef]
- Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, et al. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol Mol Biol Rev 2017;81:10.1128/mmbr.00036-17. [CrossRef]
- Park JY, Yun H, Lee S been, Kim HJ, Jung YH, Choi CW, et al. Comprehensive characterization of maternal, fetal, and neonatal microbiomes supports prenatal colonization of the gastrointestinal tract. Sci Rep 2023;13:1–14. [CrossRef]
- Chang YS, Li CW, Chen L, Wang XA, Lee MS, Chao YH. Early Gut Microbiota Profile in Healthy Neonates: Microbiome Analysis of the First-Pass Meconium Using Next-Generation Sequencing Technology. Child Basel Switz 2023;10. [CrossRef]
- Yu K, Rodriguez M, Paul Z, Gordon E, Gu T, Rice K, et al. Transfer of oral bacteria to the fetus during late gestation. Sci Rep 2021;11:708. [CrossRef]
- Dodson B, Suner T, Haar ELV, Han YW. Oral Microbiome and Adverse Pregnancy Outcomes. Am J Reprod Immunol N Y N 1989 2025;93:e70107. [CrossRef]
Figure 1.
Alpha diversity. Diversity index. (a) Chao1 index (p value= 0.9). (b) ACE index (p value= 0.8). (c) Shannon index (0.7. (d) Simpson index (p value= 0.7).
Figure 1.
Alpha diversity. Diversity index. (a) Chao1 index (p value= 0.9). (b) ACE index (p value= 0.8). (c) Shannon index (0.7. (d) Simpson index (p value= 0.7).
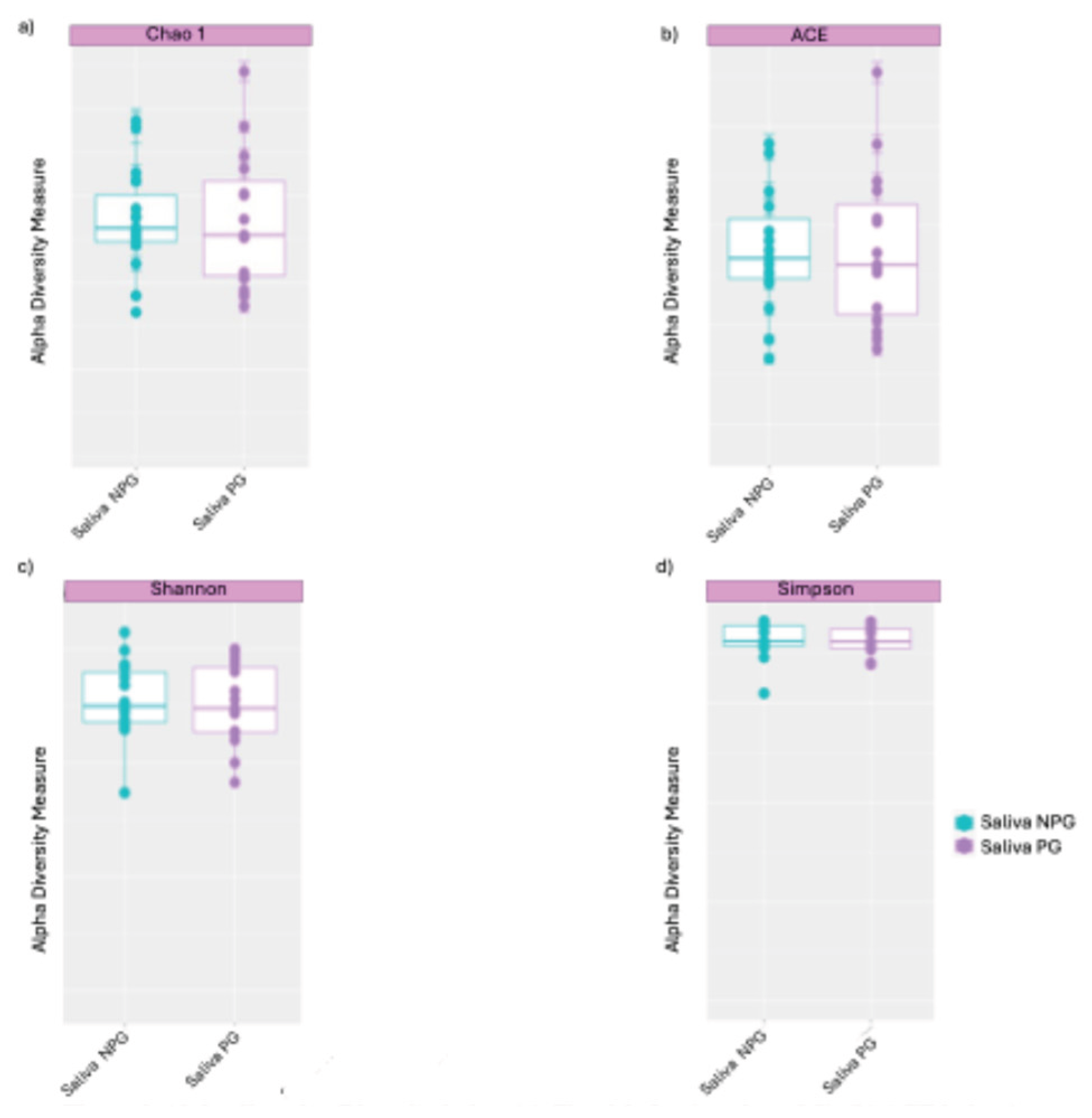
Figure 2.
Barchart (a) and Heatmap (b) showing the relative abundance at phylum level in maternal saliva samples.
Figure 2.
Barchart (a) and Heatmap (b) showing the relative abundance at phylum level in maternal saliva samples.
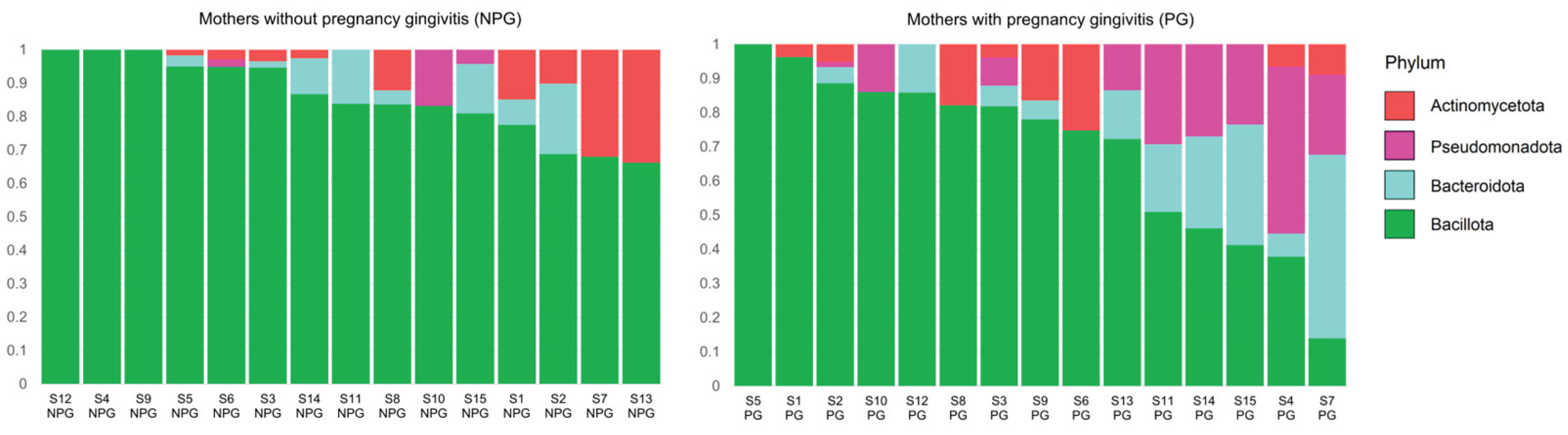
Figure 3.
Figure 3. Relative abundance at the genus level. Cumulative relative abundance from saliva from NPG and PG groups, respectively.
Figure 3.
Figure 3. Relative abundance at the genus level. Cumulative relative abundance from saliva from NPG and PG groups, respectively.
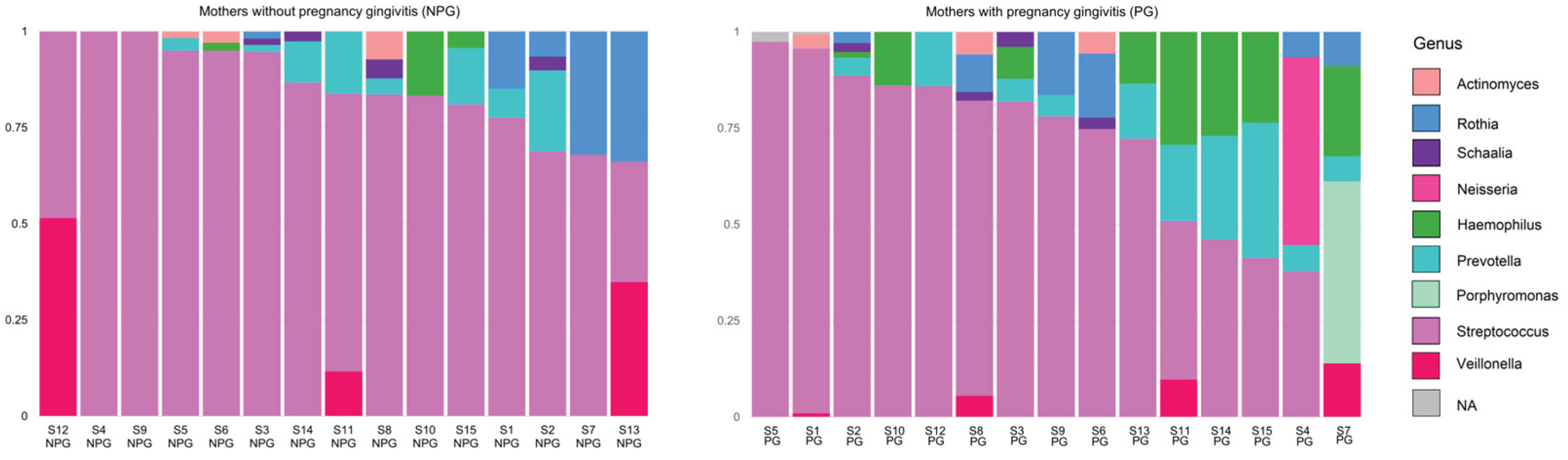
Figure 4.
sPLS-DA at genera level. Saliva PG (red) vs Saliva NPG (blue).
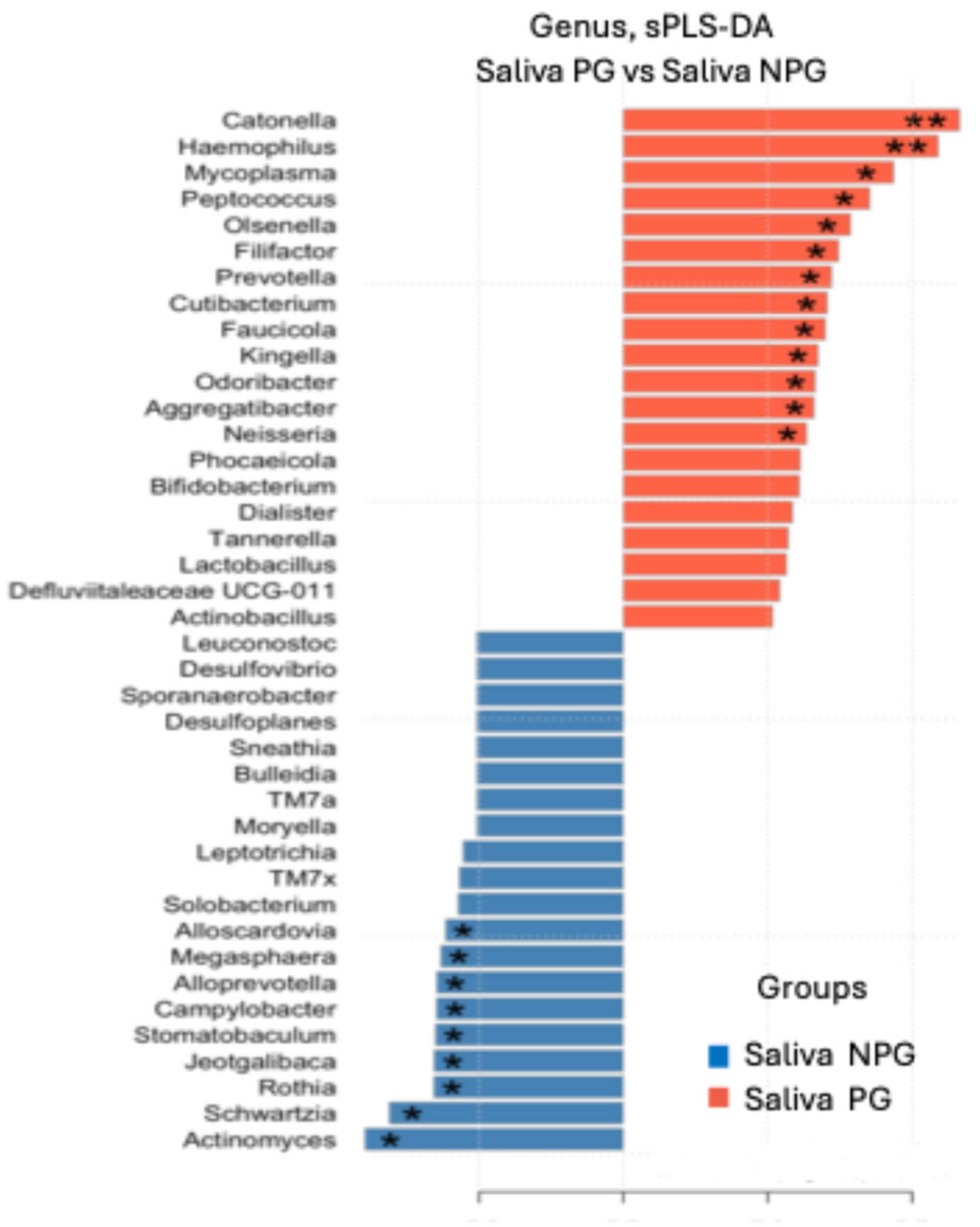
Figure 5.
Metabolic pathways. Saliva PG (red) vs Saliva NPG (blue). .
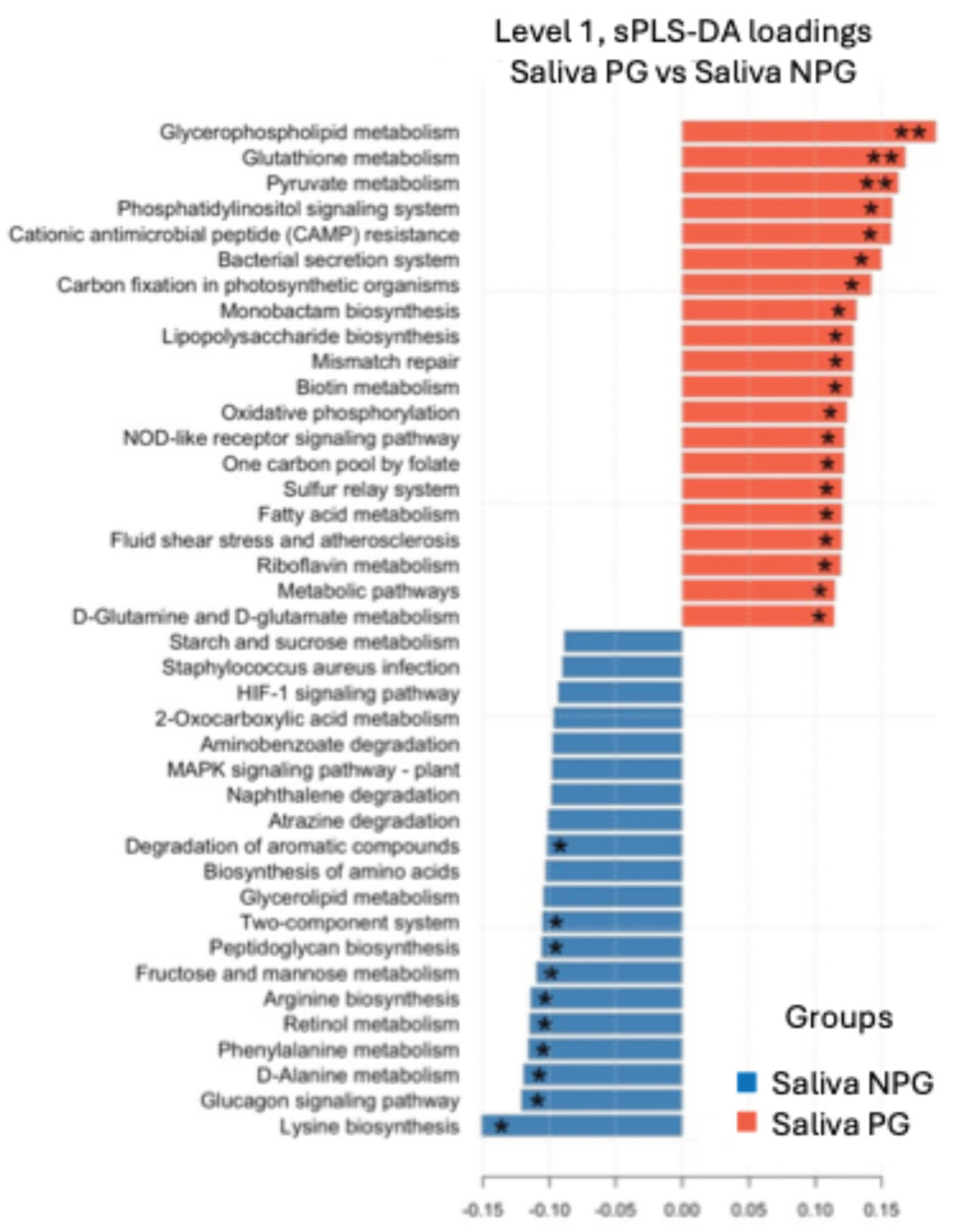

Table 1.
Baseline characteristics of study participants
| Mothers without gingivitis (NPG) (n=15) | Mothers with gingivitis (PG) (n=15) | p | |
|---|---|---|---|
| Age (years) | 26.21 (±6.2) | 28.13 (±5.9) | 0.35 |
| Gestation age at recruitment (weeks) | 33.5 (±1.9) | 35.4 (±1.9) | 0.77 |
| Pregnancy number | 2.5 (±1.2) | 2.3 (±1.2) | 0.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



