
Submitted:
15 December 2025
Posted:
16 December 2025
You are already at the latest version
Abstract
Despite decades of investigation, Aldose Reductase (AR; AKR1B1) -an enzyme that plays a key role in the metabolism of glucose and other carbonyl compounds and whose hy-peractivity contributes to oxidative stress and vascular dysfunction- inhibitors have failed to translate into clinical application for Diabetic Retinopathy (DR). We argue that these failures might arise from non-selective inhibition, which does not consider AR’s dual roles in pathology but also in retinal health, as AR is also an important detoxifying enzyme for aldehydes produced during oxidative stress, and discuss the missing structural infor-mation, despite the over one hundred crystal structures of AR in complex with inhibitors. Our review bridges this gap by proposing how recent advances in structural biology, namely, fragment-based drug discovery and MicroED, provide novel ways of selectively modulating AR functions, offering advantages in the detection of weak, allosteric, or conformation-dependent binding events. Despite past challenges, we suggest that therapeutic targeting or finding new-generation inhibitors for AR will become more effective once we have a clearer understanding of AR’s requirements for selective inhi-bition of its pathological and physiological functions. By integrating fragment screening and structural biology, we outline a strategy to reinvigorate AR modulation as a viable retina-specific approach for managing DR first, although potentially relevant across multiple diabetic microvascular complications later.
Keywords:
Aldose Reductase (AR)
; Diabetes Retinopathy (DR)
; Fragment-Based Drug Discovery (FBDD)
; Microcrystal Electron Diffraction (MicroED)
1. Introduction
Diabetes mellitus (DM) is one of the prominent health issues of the 21st century, contributing significantly to mortality, healthcare costs, and reduced life expectancy. The prevalence of diabetes is continuously increasing. According to The Diabetes Atlas, 589 million adults (20-79 years) are living with diabetes worldwide, and the number is predicted to rise to 853 million by 2050. Beyond its direct impact on health, diabetes has a strong socioeconomic burden, responsible for 3.4 million deaths in 2024, 1 every 9 seconds, and caused at least 1 trillion US dollars in health expenditure, a 338% increase over the last 17 years.
Diabetes is a metabolic disorder characterized by high blood glucose levels (e.g., hyperglycemia) over a prolonged period. Persistent hyperglycemia can lead to severe secondary complications [1,2]. Secondary complications of diabetes can be grouped into micro-vascular and macro-vascular complications, which are caused by damage to capillaries and arteries/veins, respectively [3]. Various cardiovascular, peripheral vascular, and cerebrovascular diseases come under macrovascular complications [3], and the main microvascular complications include neuropathies that afflict the nervous system (Diabetic neuropathy), the renal system (Diabetic nephropathy), and retinopathies that afflict the eye-Diabetic retinopathy (DR) with the most vision-threatening outcome [4].
Moreover, DR is particularly critical, being the most common diagnosis for 80% people who have DM for more than 20 years [5], and with DR figures for visual impairment, including blindness, steadily increasing and being reported in the millions [6]. However, timely diabetic therapy prevents the incidence and the progression of DR [6,7,8], but because diabetes often remains undiagnosed for a long time, that window of opportunity is not always used. The pathophysiology of DR is characterized by a number/cascade of events, including vascular endothelial dysfunction, breakdown of the blood-retinal barrier, microvascular occlusion, and ultimately retinal ischemia and neovascularization [6,7,9,10,11,12,13].
The molecular mechanisms contributing to these diabetic complications are multifaceted. Chronic hyperglycemia is a known initiator of microvascular problems in DR, and four molecular mechanisms have been suggested to converge in developing those complications of diabetes [14]: (1) the mechanisms involved are the increased flux through the polyol pathway, (2) oxidative stress, (3) the formation of advanced glycation end-products (AGEs) and (4) the activation of protein kinase C (PKC) [12,15,16]. Those mechanisms have been shown to affect each other, as will be demonstrated later, due to the glucose metabolites being substrates or cofactors for different metabolic pathways [15].
Among these, the polyol pathway (an alternative two-step glucose metabolic pathway that converts glucose into fructose via sorbitol), is initiated by the enzyme Aldo-keto reductase family 1, member B1 (AKR1B1), better known as Aldose Reductase (AR), the first enzyme involved in the polyol pathway, and plays an important role in the pathogenesis of DR and other diabetes related complications [17]. AR is a broad range oxidoreductase that catalyses the reduction of aldehydes, including glucose, to their corresponding alcohols.
In a normal physiological state, AR activity is relatively low, but in conditions like diabetes, where blood glucose levels are chronically elevated, the activity of AR increases, resulting in the conversion of glucose to sorbitol within the retinal cells, eye lens, kidney, and peripheral nerves, and others, leading to various complications [18,19,20] (Figure 1). The accumulation of sorbitol in the retinal cell generates osmotic stress, formation of advanced glycation end (AGE) products, and depletion of nicotinamide adenine dinucleotide phosphate (NADPH), resulting in oxidative damage and cellular dysfunction. A similar mechanism occurs in the eye lens, where polyols (sorbitol) start accumulating within lens fiber, triggering water influx and inducing both osmotic imbalance and oxidative stress, which ultimately contribute to the development of sugar cataracts [21,22]. Additionally, AR-derived fructose metabolism leads to the generation of diacylglycerol (DAG) and activation of protein kinase C (PKC), a signalling cascade which further promotes the inflammation, vascular permeability, and angiogenesis marker/indicator of diabetic retinopathy [19,23,24,25].
Accumulation of sorbitol leads to complications in multiple organs (retinopathy, nephropathy, neuropathy, etc.) [26]. However, the retina is particularly vulnerable because of its high glucose flux and sensitivity to osmotic and oxidative stress, making DR a key target for AR-focused therapies. As mentioned above, a chain of changes including microvascular damage, vascular leakage, retinal hemorrhages, upregulation of growth factors and inflammatory cytokines, abnormal angiogenesis and capillary closure drives disease progression in DR. DR is characterized by vascular injury and neo-vascularization leading to blindness [27], and retinal neovascularization is often associated with retinal detachment and macular edema, leading to partial or complete blindness [28]. Given its central role in various diabetic conditions/complications, AR has been considered as a promising pharmacological target, in addition to the currently available DR therapies, nicely reviewed elsewhere [6,7,10,12,19,28]. Therefore, in the last four decades, AR inhibitors (ARIs) have been developed and considered as a critical treatment option for the management of diabetic complications. As of the current date, there are many ARIs available, but none of them are effectively working against AR, clinically, as therapeutics. Extensive descriptions of the many ARIs and detailed analysis have been compiled elsewhere [29,30,31,32,33,34,35,36]. Despite the many trials, Epalrestat is the only ARI approved for clinical use and successfully marketed in a few countries, such as Japan, India, and China, for the treatment of diabetic neuropathy [37]. Importantly, its potential benefits extend beyond neuropathy. A re-analysis of the ARI-Diabetes Complications Trial by Hotta and colleagues (2012) demonstrated that Epalrestat could significantly delay the progression of diabetic retinopathy and nephropathy, particularly in patients with milder neuropathy at baseline. Interestingly, patients with less severe neuropathy at study completion showed greater protection against disease progression, suggesting that the benefits of Epalrestat may be influenced by the stage of neuropathy [38]. Complementing these clinical observations, an experimental study by Senthil Kumari and co-workers (2017) reported that glucose-induced toxicity in RPE cells was effectively inhibited by Epalrestat at nanomolar concentrations after 72 hours. These findings suggest that AR inhibition is not only a promising strategy for managing diabetic complications but may also help to suppress retinal neovascularization in proliferative diabetic retinopathy [39]. Epalrestat is generally safe in clinical trials with a moderate dosage of 50 mg three times a day. But, in clinical trials, some adverse effects have been also observed, such as elevations in liver enzyme levels, mainly aspartate aminotransferase, alanine aminotransferase, and gastrointestinal-related events such as nausea and vomiting but no life-threatening effects have been observed during the trials [40].
Prior attempts to inhibit AR have been hindered by nonselective, nonspecific inhibition, which resulted in limited efficacy and significant off-target safety effects. Off-target inhibition is attributed to high sequence identity between human AKR1 family (>40%), with AKR1A1 and AKR1B10 sharing 65 and 70% sequence identity with AR, respectively [41]. Preclinical evidence reinforces the relevance of AR as a retina-specific target [42]. For instance, genetic deletion of AR prevents early diabetes-induced damage in neural, glial, and vascular cells of the retina [43], while ARIs can mitigate a spectrum of retinal abnormalities more effectively than other drug classes [44]. However, clinical translation has been limited. For example, in the negative Sorbinil Retinopathy Trial [45], the dose used in humans was ~20-fold lower than that shown to prevent retinal polyol pathway activation and the development of retinopathy in diabetic rats [46,47]. An aspect of the toleration problem with drugs such as highlighting challenges related to tissue penetration, subtherapeutic dosing, and toxicity. A contributing factor to the tolerability issues with drugs like Sorbinil may be their limited selectivity for AR over aldehyde reductase (AKR1A1), a closely related enzyme involved in detoxifying reactive aldehydes [48]. Despite the setback, recent advances in structural biology, pharmacology, drug discovery, and targeted drug delivery domains offer a new prospective to inhibit the AR with a differential inhibition approach [49,50]. Given the limitations of classical ARIs, this review aims to address this challenge with a new approach coined Fragment-Based Drug Discovery (FBDD) to achieve differential inhibition of AR. FBDD is yet to be fully explored in the case of AR, but this approach is very popular and has already demonstrated success in tackling one of the most challenging drug classes-Protein kinase, where selectivity is a major hurdle. By exploiting subtle structural differences among kinase subpockets, FBDD has enabled the development of highly selective inhibitors, with several approved drugs such as Erdafitinib, Pexidartinib, and Vemurafenib originating from this approach [51]. These successes underscore FBDD’s power to achieve selectivity in difficult targets, providing a strong rationale for applying similar strategies to AR, where distinguishing between physiological and pathological functions is equally critical. Similarly, the major goal of this approach is to keep the potential physiological function of this protein intact while selectively targeting its pathological activity in diabetic complications, with a particular emphasis on DR, where retina-specific AR modulation may offer the most immediate therapeutic benefit.
2. Aldose Reductase: Function, and Pathological and Physiological Roles
Aldose Reductase (EC 1.1.1.21) is a cytosolic, monomeric enzyme and member of Aldo-keto reductase (AKR) superfamily that includes about 190 enzymes capable of reducing a wide range of carbonyl-containing substrates, including sugar and products deriving from lipid peroxidation [52]. Most of the members of this group are cytosolic and monomeric proteins with an average length of 320 amino acid residues, with a molecular weight ranging between 34 and 37 kDa.
AR was first observed in the eye lens, and also expressed in various tissues like the sciatic nerve, testicle, heart, and cornea [53,54]. The expression level of AR varies among different tissues and organs, such as the liver, stomach, spleen, lung, and small intestine, which express a low level of this protein [55]. Under normal physiological conditions, AR exhibits low catalytic activity toward glucose due to its high Km, reflecting its low affinity for glucose as a substrate. However, in diabetic conditions, when glucose concentration is high in blood, the glucose molecules move towards the polyol pathway [56]. Therefore, this enzyme and polyol pathway becomes the major component in the hyperglycaemic condition to catalyse the glucose molecules [57]. The physiological roles of AR are vast and involved in detoxification of aldehyde compounds such as 4-hydroxynonenal [58], steroid metabolism in the adrenal gland and reproductive organs [59], hormone regulation in the ovary [60], catecholamine metabolism and osmoregulation [61], and fructose production via the polyol pathway [62]. The enzyme is also thought to function in osmoregulation and contributes to several metabolic pathways [49], including the production of fructose [63], the biosynthesis of tetrahydrobiopterin [64,65], and the metabolism of corticosteroids [59,66].
2.1. The Polyol Pathway
The polyol pathway, which was initially identified in the seminal vesicles, is an alternative two-step glucose metabolic pathway that converts glucose into fructose [26] (Figure 1). Nonphosphorylated glucose is first reduced to sorbitol by the enzyme AR, using NADPH as a cofactor, and the resulting sorbitol is then changed to fructose by sorbitol dehydrogenase (SDH), using NAD+ and producing NADH as a by-product. Under normal glycemic conditions, glucose is primarily phosphorylated into glucose 6-phosphate by hexokinase and enters the glycolytic pathway, while only a small fraction of non-phosphorylated glucose (3%) is shunted into the polyol pathway [52]. This pathway plays an important role in metabolizing glucose, osmoregulation, and maintaining the redox balance by utilizing substrates such as NADPH and NAD+ to drive the reactions. However, under hyperglycemic conditions, elevated intracellular glucose results in one-third of the glucose entering the polyol pathway [26]. As a result, AR and Glutathione Reductase (GR), an important antioxidant enzyme, compete for the same cytoplasmic pool of NADPH [29]. Thus, the increased flux through the polyol pathway depletes NADPH, diminishing reduced glutathione (GSH) levels and elevating the generation of reactive oxygen species (ROS) and oxidative stress [67,68], one of the major causes of diabetic complications [17,19,35,36,69].
An additional point of concern regarding the polyol pathway is that not all tissues have the enzyme SDH [26]. Those include kidneys, the retina, and Schwann cells, and complications arise because of sorbitol accumulation and the fact that it is practically membrane impermeable [25,62,70].
Secondly, the increased flux through the polyol pathway leads to the imbalance of NAD+/NADH, which has been termed “pseudohypoxia” and linked to a multitude of metabolic and signalling changes known to alter cell function [18,71]. On one hand, NADH is overproduced by the glycolytic and Krab cycle, and on the other hand, the activation of the polyol pathway via SDH. Furthermore, NAD+ can be depleted by the overactivation of polyADP ribose polymerase (PARP), which uses NAD+ as substrate in response to oxidative DNA damage [72]. The imbalance of NAD+/NADH leads to oxidative stress and damage of macromolecules like DNA, protein, and lipids. This imbalance further leads to impairment in the biochemical reaction involved in lipid metabolism, growth formation, and PKC activity [26]. Collectively, these metabolic disturbances play a pivotal role in the pathogenesis of diabetic complications, including diabetic retinopathy, neuropathy, and nephropathy (Figure 1).
2.1.1. Polyol Pathway Activation and Sorbitol Accumulation
Sorbitol is an alcohol, polyhydroxylated and strongly hydrophilic, and therefore does not diffuse readily through cell membranes and accumulates intracellularly with possible osmotic consequences [25,73]. More recent studies have demonstrated that increased AR is localised in several retinal cells, such as pericytes, retinal endothelial cells, ganglion cells, Muller cells, retinal pigment epithelial cells, and neurons [43,46,74,75,76,77]. A study done on the mice proved that cataracts develop in diabetic and galactose-fed animals at a rate directly dependent on the amount of AR present in the lens [78]. In mice where lens AR levels are extremely low, cataracts do not develop in either of these metabolic states. In diabetic rat lenses, AR activity has been found to increase, whereas SDH activity decreases [79]. Using immunohistochemical techniques with antibodies against purified AR, there is increasing staining of the lens with the onset of diabetes [78]. In patients with nephropathy, AR mRNA expression was markedly upregulated as D-glucose levels increased from normal to hyperglycemic ranges (P < 0.0001). Conversely, no significant changes in AR mRNA levels were observed in patients without nephropathy or in healthy controls. Notably, among the nephropathy group, individuals carrying the Z–2/X susceptibility genotype exhibited the highest elevation in AR mRNA compared with those harbouring lower-risk genotypes (P = 0.007) [80]. In tissues with no or limited sorbitol dehydrogenase (SDH) activity, such as the retina, lens, and Schwann cells, the accumulation of sorbitol increases the osmotic stress and imbalances. Sorbitol is a membrane-impermeable substance that leads to the influx of water, resulting in cell swelling and ultimately leading to structural and functional damage. This mechanism underlies the pathogenesis of diabetic retinopathy, cataracts, and peripheral neuropathy [15,81].
2.1.2. Oxidative Stress Generation
The glucose flux through the polyol pathway consumes NADPH as a cofactor (Figure 1). It has been established that there is about a 15% decrease in NADPH in the diabetic lens [78,82]. This depletes intracellular NADPH reserves essential for regenerating reduced glutathione (GSH) via Glutathione reductase (GR) activity, a very important and critical antioxidant enzyme. The depletion of NADPH could further impair the GSH/GSSG redox balance, as GSSG (the oxidized glutathione) reduction by glutathione reductase requires NADPH as a cofactor [67,82]. NADPH is also involved in the biosynthesis of several biomolecules like fatty acids and nitric oxide, so the depletion of NADPH will impact many other anabolic pathways [83,84]. The one major consequence of this redox imbalance is an oversupply of electron donors to the mitochondrial electron transport chain [85]. One of the peculiar features of complex I electron transport is that it produces more superoxide, which is the precursor of all the Reactive oxygen species (ROS) [86]. Moreover, the increased NADH pool also stimulates oxidative stress via NADH oxidases [31,70,87], including by PKC regulation [16,88].
2.1.3. Formation of Advanced Glycation End Products (AGEs)
AGEs are nonenzymatically formed by reducing glucose, lipids, or amino acids. An alternate mechanism of AGE formation comprises the “carbonyl stress” pathway, where oxidation of sugar or lipids creates dicarbonyl intermediates that use highly reactive carbonyl groups to bind amino acids and form AGEs [89,90]. People with diabetes have higher levels of AGEs because of hyperglycemia and oxidative stress, both of which contribute to the accumulation of AGEs in the cell. Apart from the carbonyl stress pathway, the AR-mediated polyol pathway also plays an essential role in the formation of AGEs. Glucose entering the polyol pathway may directly form AGEs via 3-deoxyglucosone AGE intermediates, but this reaction also causes depletion of NADPH and glutathione, and the resultant oxidative stress indirectly increases the formation of AGEs [91]. The other factor to increase the formation of AGEs is fructose, as the polyol pathway consumes approx. 30% of blood glucose in diabetes, fructose is overproduced in the body [82]. It is known that fructose can be further metabolized to produce 3-deoxyglucose and fructose-3-phosphate, and both the products are nonenzymatic glycation agents [92]. Of note, AGE formation alters the properties of several extracellular matrix proteins, interfering with matrix-cell interactions. AGE also cross-links with type I and type IV collagen, thus altering the function of vessels and basement proteins, respectively [15,16]. Relevant here is also the fact that in diabetic animals, increased levels of MMP2, MMP9, and MMP14 have been detected, and cells treated with MMP2 and MMP9 showed gap junction alterations, thus possibly contributing to blood-retinal barrier breakdown [16], and it is long known abnormalities of extracellular matrix, both qualitative and quantitative, contribute to an irreversible vascular permeability [14]. Clinical studies in patients with diabetic retinopathy, as well as findings from animal models, have consistently reported elevated levels of MMP2 and MMP9 in both the retina and vitreous [93,94,95]. Similarly, diabetes has been associated with increased retinal mRNA expression of MMP2, MMP9, and MT1-MMP [96,97], while higher levels of the inactive pro-forms of MMP-2 and MMP-9 have been detected in neovascular retinal membranes [98]. These enzymes are thought to contribute to disease progression through multiple mechanisms. Notably, MMPs play a key role in preserving blood-retinal barrier (BRB) integrity, and BRB dysfunction is recognized as an early hallmark of diabetic retinopathy [99]. Elevated retinal MMP activity in diabetes can promote vascular leakage by degrading the tight junction protein occludin and destabilizing the junctional complex as a whole [95].
2.1.4. Activation of Protein Kinase C (PKC)
Increased NADH/NAD+ ratio leads to the formation of the lipid secondary messenger DAG and activates the PKC [32,100]. The DAG-PKC pathway is one of the most studied pathways in cellular signalling induced by diabetes. PKC is a group of enzymes that are members of the serine/threonine protein kinase family and have the ability to phosphorylate various target proteins [101,102]. The PKC family is involved in regulating several biological pathways like cell growth, differentiation, apoptosis, as well as transformation [102]. The activation of PKC is one of the major pathways involved in the pathogenesis of DR. Hyperglycemia and oxidative stress caused by the polyol pathway and AR increase the synthesis of DAG and cause an activation of classic isoforms of PKC-α, -β and -δ [101,103,104]. Also, some products of GR are reduced by AR, such as 3-glutahionyl 1,4-dihydroxynonanal (GS-HNE), which is reduced into 3-gluthathionyl-1,4-dihydroxynonane (GS-DHN) [105,106,107], which in turn activates the nuclear factor κB (NF-κB) signalling cascade, influencing the PKC pathway and thus proinflammation [105,108,109,110,111]. Additionally, PKC activation causes retinal vascular dysfunction by altering enzyme activities in Endothelial Cells (NO, ET-1, VEGF) and pericytes (platelet-derived growth factor, PDGF, reactive oxygen species) [112]. Alteration in nitric oxide (NO) production and endothelial nitric oxide synthase (eNOS) expression has a direct influence on the vascular hemodynamics, such as contraction and relaxation, which may affect retinal blood flow [101,113].
2.2. Clinical Manifestations
AR has a well-established role in retinopathy and cataractogenesis. Among diabetic eye complications, include corneal epitheliopathy, cataract, and retinopathy; retinopathy represents the most vision-threatening outcome, often leading to irreversible blindness despite interventions such as photocoagulation or vitreous surgery [114]. AR has been strongly implicated in the initiation of diabetic retinopathy, particularly through its role in pericyte degeneration [115,116], a hallmark of early disease, which also occurs in diabetic and galactose-fed dogs that develop retinopathy. Experimental studies in diabetic and galactose-fed dogs have shown that AR inhibition can dose-dependently prevent pericyte loss, reinforcing its causal involvement [117,118,119]. In humans, genetic studies link AR polymorphisms with variable retinopathy risk, where the Z−2 allele associates with higher erythrocyte AR levels and increased prevalence of DR, while the Z+2 allele appears protective [114,120]; however, to date, some studies have reported no association of these alleles [121,122,123]. These findings suggest that AR activity not only drives pericyte loss but may also serve as a biomarker of early-stage DR, highlighting the importance of selective AR inhibition alongside glycemic control as a preventive strategy [114]. Sugar cataracts form in transgenic diabetic mice expressing human AR in the lens but not in wild-type streptozotocin-induced diabetic mice, which may have low expression of AR [79,124]. Studies on slow cataract formation showed that metabolic imbalance caused by increased activity of AR plays a major role in slow cataract development in mature diabetic animals, which is more relevant to diabetic patients [42,77]. Two studies have shown that high glucose and diabetes induced impairment of lenticular signalling via AR-dependent oxidative stress mechanisms [125,126]. Therefore, AR activity is likely to contribute to diabetic cataract formation through an oxidative signalling mechanism. Beyond retinopathy, AR is also implicated in other diabetic complications, including neuropathy and nephropathy.
AR is also a potential target for diabetic neuropathy, and increased activity of AR is implicated in the pathogenesis of diabetic neuropathy. Increased AR activity leads to increased levels of oxidative stress, decreased levels of reduced glutathione (GSH), and results in axonal damage and nerve dysfunction [127,128]. Several studies demonstrated that hyperglycemia-induced oxidative stress led to the activation of mitogen-activated protein kinase (MAPK), which may further contribute to neuronal pathogenesis [129,130]. Importantly, both AR inhibition and genetic ablation reduce oxidative stress and prevent functional deficits in the peripheral nervous system of diabetic mice [131]. All the findings indicate that AR contributes to the pathogenesis of diabetic neuropathy via oxidative stress.
AR is differentially expressed in the human kidney, and expression is usually low in the kidney, but when hyperglycemia or a diabetic condition occurs, the level of AR is significantly raised [55,132]. Increased activity of AR in retinal cells is linked with aberrant activation of PKC, generation of AGE, increased expression of TGF- β, and generation of ROS [133,134]. A recent study using AR-deficient mice (except in the renal medulla) demonstrated a marked reduction in diabetic nephropathy, reinforcing the key role of AR in disease progression [135]. Thus, the data indicate that AR, by hyperglycemia in the renal glomeruli, contributes to the onset and progression of diabetic nephropathy.
2.3. Physiological Function of AR
As compared to a significant number of studies on AR focusing on the pathological role in glucose toxicity, less attention has been paid to the physiological function of AR in normal and glycemic conditions. AR serves important roles in cellular detoxification, osmoregulation, and metabolic regulation.
Through the polyol pathway, AR plays a key role in osmoregulation in the kidney and in fructose production in the male genital tract [136]. In the seminal vesicles, SDH converts fructose from sorbitol (Figure 1), and it is an energy source for sperm cells [137]. In the absence of hyperglycemia, hyperosmotic stress can induce AR activation and lead to sorbitol accumulation in renal papillary interstitial cells, the latter considered as the organic osmolytes that balance the osmotic pressure of extracellular sodium chloride (NaCl), and AR appears to play a key role in osmoregulation [138].
Another important role of AR is in aldehyde reduction/detoxification of reactive aldehydes, namely by reducing alkenals and alkanals, produced during oxidative stress conditions, thus contributing to the reduction of oxidative stress [29]. AR and other aldo-ketoreductases catalyse the reduction of reactive biogenic aldehydes such as methyglyoxal (MG), 3-deoxyglucosone (3-DG), acrolein, and 4HNE, which are the products of lipid peroxidation (with HNE been its preeminent product) and glycolysis [105,136]. AR may represent an important metabolic route for the detoxification of lipid aldehydes with a Km in the micro molar range [108] and in comparison, the Km of the enzyme for glucose is in the millimolar range (50-100 mM) [17].
AR also participates in steroid metabolism by catalyze the reduction of biogenic aldehydes derived from the catabolism of the steroid hormones, catecholamines, and serotonin. In steroid metabolism, isocorticosteroids and isocaproaldehyde are the preferred substrates for AR. Isocaproaldehyde, produced in large amounts in the adrenal cortex during steroidogenesis, displays cytotoxic actions in vitro, and AR can be a detoxifying enzyme in this tissue [136,139].
Understanding the role of AR in the diverse physiological and pathological conditions not only highlights its critical role in cellular homeostasis but also emphasizes the importance of its structural features, which lead to both substrate specificity and inhibitor selection [140]. While agreed and extensively persuaded, pharmacologically inhibition of AR contribution to the polyol pathway is therapeutic strategy to decrease oxidative stress and inflammation, such molecules need to be selective not only for the pathological function of AR, but also leaving the detox function operational (i.e., strong on inhibition of the reduction of sugars and GS-HNE, but weak, or ideally, unaffecting the reduction of HNE and other alkanals and alkenals [105]). Specially, when taking into account that no correlation between an altered level of AR gene expression in target tissue of diabetes complications and the onset or progression of pathogenesis was ever objectively demonstrated [141], stressing the fact that physiological AR functions must be preserved when selecting AR inhibitors, thus protecting cells from oxidative stress and damage, aiding osmoregulation and supporting proper fluid and electrolytic balance within cells [32].
The inhibitors also need to be specific for AR, leaving close homologues (such as for AKR1B10 and others) undisturbed. As they have a crucial function in detoxifying aldehydes and ketones, thereby influencing intracellular signaling that activates pro-inflammatory transcription factors such as NF-κB and activator protein 1 (AP-1), and in regulating lipid metabolism by promoting fatty acid synthesis, which can further amplify inflammatory responses [142]. The continuous oxidation of carbohydrates and lipids produces a variety of reactive carbonyl species (RCSs), including methylglyoxal, 3-deoxyglucosone, hexanal, acrolein, 4-hydroxynonenal, 4-oxo-2-nonenal, and phospholipid-derived aldehydes [143]. Within the AKR family, AKR1B10 plays a prominent role by catalyzing the reduction of these RCSs into their corresponding, less toxic alcohols [144]. Importantly, AKR1B10 exhibits particularly high efficiency in detoxifying the highly cytotoxic aldehydes 4-hydroxynonenal and 4-oxo-2-nonenal compared with other AKR isoforms.
Therefore, a key limitation of conventional AR inhibitors is their broad spectrum of activity. While they effectively suppress the polyol pathway, they also inhibit AR’s detoxification function and impact related aldo-keto reductases such as AKR1B10, which play important physiological roles in clearing reactive aldehydes. This lack of specificity can be counterproductive, as the reduction of detox activity may exacerbate oxidative stress, offsetting the benefits of polyol pathway inhibition. This paradox highlights the need for selective or differential inhibitors that can block AR’s pathological glucose flux without impairing its protective detoxifying activity [6,19].
3. Structural Characteristics of Aldose Reductase
To date, 176 crystal structures have been described with the help of the X-ray crystallography technique to understand their structure and mechanism of action in more detail. The first structures were published in 1992 and were heterologous in nature, being of human protein recombinantly expressed and of pig lens AR [145,146,147]. Over the years since, more detailed structures have been described in apo as well as in complex with various ligands, especially inhibitors and the co-enzyme (or analogues) [141]. These studies gave us the opportunity to look more into the catalytic mechanism as well as the binding and interaction of the ligands to it [141].
The human enzyme is 316 amino acids long and has been observed to have a molecular weight of 34 kDa, which can be due to protein primary sequence plus post-translational modifications, such as glycosylation and others. The enzyme is monomeric in nature and is composed of an eight-sheeted β/α barrel with the N-terminus capping the bottom of the barrel, composed of two short antiparallel β-strands. The barrel structural motif is characterized by parallel β-strands, with each alternating with α-helical segments that run anti-parallel to the β-sheet [141], and provides the rigid catalytic scaffold, while some loops between the α-helices and β-strands are the mobile and sequence diverse elements that shape the active site. The substrate binding site is in the C-terminal region of the barrel and gives a large and deep cleft in appearance. The pyridine NADP+ acts as a cofactor and is localized at the C-terminal end of the parallel strands of the barrel [145,146,147], in an extended form and with the nicotinamide ring positioned at the centre of the barrel, contributing to the active-site machinery, and the pyrophosphate straddling the lip of the barrel [141,147,148]. The active site relevant loops are named A, B and C and corresponds to residues 109 to 137 in between β4 and α4, residues 209 to 230 in between β7 and α7, and the C-terminal residues respectively protruding from the top of the barrel [145,146,147,149]. Two additional α-helices, namely, H1 and H2, are instrumental in anchoring the loops. H1 helix anchors the cofactor binding loop while the H2 helix helps in anchoring the C-terminus loop [150].
The region that helps to shelter the cofactor is known as the ‘safety-belt’ loop and is made of residues Gly 213 to Ser 226, which can be traced to the loop B [147,148]. While the cofactor is bound to the loop, it becomes partially covered by it and thus referred to as “closed” conformation and the absence of the cofactor is termed as the “open” conformation [148]. This safety belt is responsible for maintaining the conformational changes of the enzyme and for the release and binding of the cofactor. The “closed” and “open” conformations are thus central to the working of the enzyme [148]. Within the safety belt, Gly213, Ser214 and Ser226 are the hinge points and take part in the conversion of these closed and open states, while Trp219 acts as the “latch” bridge between Cys298 and Arg293 (via sulfur-aromatic or aromatic stacking interactions, respectively) to stabilize the whole conformation as well as interactions with ligands [150].
The active site of AR has been described in slightly different ways across the literature (Figure 2), but most structural studies agree on the presence of two principal regions: a rigid anion-binding pocket and a more flexible specificity that enables ligand diversity [29,34,151]. According to Maccari & Ottanà (2015), the anion-binding pocket is a polar, hydrogen-bonding subsite formed by residues such as Asp43, Tyr48, Lys77, His110, Ser159, Asn160, Gln183, and Tyr209 [34]. This anion-binding pocket anchors the nicotinamide ring of NADPH and ensures catalytic precision [152,153]. Among these residues, Tyr48 serves as a proton donor to the carbonyl oxygen atom of the substrate during the second step of the reduction, whereas His110 maintains the proper orientation of the substrate in the active site. This subsite can serve as a polar recognition region able to anchor anionic and/or hydrogen bond acceptor groups of various substrates and inhibitors [34]. Urzhumtsev and colleagues (1997) similarly highlighted these residues as central to a hydrophilic network dominated by hydrogen bonds, van der Waals forces, and water-mediated interactions [151]. Additional residues, including Trp20, Val47, and Trp111, also contribute to substrate stabilization at the catalytic site. That study highlighted that the anion-binding pocket, lined by residues such as Tyr48, His110, Trp20, Val47, and Trp111, anchors the catalytic machinery by stabilizing substrate interactions via hydrogen bonds and water-mediated contacts. Urzhumtsev and colleagues also indicated the hydrophobic substrate-binding cleft consisting of Trp20, Trp79, Trp111, Phe122, Pro218, Trp219, Cys298, Leu300, and Val47. Of those, Trp20, Phe122 and Trp219 fully face the active site, and are thus expected to make the major contacts with a potential substrate. The study further mentioned the existence of a specificity pocket, the residues of which are also in the second group of cleft-lining residues, and include Thr113, Phe115, Val130, Ser302 and Cys303. Residues Trp111, Phe122 and Leu300 are shared by both the active site and the specificity pockets [151]. This cavity can show a variety of flexible states and adopt conformations differently to accommodate different ligands accordingly [152,153].
The second region of the AR active site is a hydrophobic cleft situated above the coenzyme. This “specificity pocket” (or “induced cavity”) is lined by residues such as Trp20, Trp79, Trp111, Phe122, Pro218, Trp219, Cys298, and Leu300 [34]. It is highly flexible and capable of adopting different conformations depending on the size and nature of the ligand. While Maccari & Ottanà (2015) described this pocket as comprising Trp111, Thr113, Phe122, Ala299, Leu300, Ser302, and Cys303 [34]. In contrast, Balestri and colleagues (2022) stated that the specificity pocket (also referred to as the induced cavity) consists of Thr113, Phe115, Phe122, Cys303, and Tyr309, and contributes to ligand adaptability by undergoing conformational rearrangements [29]. Several residues, including Trp111, Phe122, and Leu300, are shared by both the anion-binding and specificity pockets, underscoring their dual role in substrate recognition [151].
With respect to the enzymatic reactions, it is known that AR follows an ordered reaction mechanism with first binding of NADPH, which induces conformational changes before the binding of substrate, followed by substrate binding, its conversion to the alcohol, and its release. After, and while still bound to NADP+, there is a second conformation change in the protein. The reaction finishes with the release of NADP+ [29,141,148]. This difference in classification reflects the resolution of structural analysis, but in both models, the rigid anion-binding region and the flexible specificity pocket remain the central determinants for substrate recognition and inhibitor selectivity. Importantly, many fragment-based and classical inhibitors exploit induced conformations of the specificity pocket, making its flexibility a key target for rational drug design.
3.1. Conformational Flexibility
When AR inhibitors such as minalrestat, zopolrestat, and tolrestat bind to the enzyme, it results in a change in the conformation of the loops containing the residues Phe122 and Leu300. This rearrangement opens the normally closed specificity pocket (apo state), allowing bulky hydrophobic groups (such as 4-bromo-2-fluorophenyl, benzothiazole, or naphthalene moieties) to fit into the cavity effectively [151,154,155]. Leu300 acts as a gate-keeper residue and controls the opening and accessibility of this pocket and also influence the orientation of nearby residues, including Phe122, Trp219, and Cys303 [152,156]. By contrast, sorbinil lacks a suitable hydrophobic group and therefore does not access the specificity pocket. In its bound state, the pocket remains closed [151]. These examples show that induced cavity adaptation can affect the binding efficacy of the ligands to the enzyme, but it is not the only condition. Notably, residues within the specificity pocket are less conserved across related enzymes compared with catalytic residues such as Tyr48 and His110, which may underlie the structural basis for selective inhibition of AR. The flexibility of the specificity pocket of the AR can also thus be a challenging target for co-crystallization due to the increase in the number of possible binding conformations as well as, can be the reason for an increased affinity towards a large group of small molecule ligands and substrates containing either hydrophilic (for example, glyceraldehyde and aldoses) or hydrophobic (for example, HNE) moieties [151,152,157].
3.1.1. Structural Homology and Challenges
The challenge in designing an effective ARI lies in the fact that AR shares more than 85% sequence homology with aldehyde reductase (AKR1A1), one of the most important aldo-keto reductases. Additionally, AKR1B10 (aldo-keto reductase family 1, member B10) also shares high sequence and structural homology with AR. The homology between AKR1B10 and AR is 71% [149,158,159]. The architecture of the active sites also shares similarities between these two enzymes. As common features of AKRs, AKR1B1 and AKR1B10 show a (α/β)8-barrel core motif and a highly conserved catalytic tetrad in the active site, which is composed of residues Asp43, Tyr48, Lys77, and His110 and the active site, the neighbouring Trp111 is Trp112 in AKR1B10 (AKR1B1 numbering) [149,160]. This provides a challenge in selectively identifying these two enzymes by the inhibitors. Aldehyde reductase is involved in the reduction of highly reactive toxic 2-oxalodehydes, for example, 3-deoxyglucosone and methylglyoxal. Under hyperglycemic conditions, these aldehydes are produced in high quantity and results in tissue and vascular damage if gone unchecked [161,162]. The similarity in the active sites, thus, present a high challenge to the inhibitors, since the inhibition of aldehyde reductase in place of AR will hamper the reduction of these harmful byproducts, which can ultimately result in hepatotoxicity, one of the most observed side-effects of the ARIs which have gone for trial.
Understanding the three-dimensional structure and catalytic landscape of AKRs is crucial for the rational design of selective inhibitors, especially in the case of AR (as promptly realized by the initial structures from 1992 and the immediately mention of rational design of specific inhibitors by Wilson and colleagues [147]), and its split between invariant (anion-binding pocket) and its more flexible part (“specificity” pocket) [152,153] (Figure 2). The failure of classical ARIs has largely been attributed to a lack of specificity and off-target interactions due to the conserved nature of the AKR active site, but taking into account the flexibility of the protein and the different bind modes for distinct ligands, selective inhibitors are -still- plausible and merit further research [29,105]. The following section reviews the limitations of current ARIs and explores how emerging strategies like fragment-based design may overcome these barriers.
3.2. Current AR Inhibitors
Due to the involvement of AR in various diseases, a great effort has been put in the last fifty years to design and the synthesis of the molecules that can inhibit the pathological activity of AR. The proposed inhibitors have multiple structures, but some features are constant. ARIs usually present a polar moiety that interacts with the “anion binding pocket” of the enzyme and a hydrophobic moiety that interacts with the non-polar region of AR active sites [33,151,155,163,164,165]. Most of the proposed inhibitors are divided into three broad spectrums: compounds that contain cyclic imides, carboxylic-acid derivatives, and polyphenol compounds, and these have been nicely reviewed in recent publications [29,30,31,33,166], and these categories include well-studied representatives such as Sorbinil and Fiderestat (cyclic imides), Epalrestat and Tolrestat (carboxylic acid derivatives), and Curcumin and Quercetin (polyphenols) [29]. Several synthetic ARIs have been developed since the 1970s, starting with alrestatin and sorbinil, but most were discontinued due to toxicity or limited efficacy [167,168]. The only ARI that remains in clinical use is epalrestat, approved in Japan since 1992 [169,170,171]. More recently, second- and third-generation ARIs such as fidarestat, ranirestat, and govorestat have been evaluated, though none have yet gained global approval [172,173,174,175].
3.2.1. Major Limitations of ARIs
Currently, the best therapeutics available for DR focus on antagonizers of the vascular endothelial growth factor (VEGF) signalling pathway and on laser photocoagulation that closes microaneurysms. However, these therapies address later stages of disease; thus, DR paradigm-shifting therapies will need to tackle early changes before outright microvascular lesions, and thus AR is still a worthy target [7]. Despite the continuous and considerable efforts in the direction of finding a suitable and effective inhibitor against AR, virtually all synthesized compounds failed as drugs for the treatment of diabetic complications. As we mentioned above to the date, the only drug marketed and approved for clinical use is Epalrestat, that is to sale only in eastern countries and approved for clinical use. There is some potential reason behind the failure of the proposed drugs, such as poor bioavailability, undesirable pharmacokinetics, and the occurrence of adverse side effects, such as hepatotoxicity. On the other side, there is a possible explanation behind the failure of the inhibitors is that the proposed inhibitors are inhibiting both pathological and physiological AR functions [29]. Furthermore, AR share overlapping substrate specificities, and the challenge of achieving selectivity is compounded by the high sequence homology with other aldo-keto reductases [176]. Structural studies highlight this complexity. In AR holoenzyme, Trp111 is located between the anion binding pocket and the specificity pocket, with its side chain always oriented toward the anion binding site (AR-position). In this conformation, Trp111 is stabilized through hydrophobic interactions with Leu300. By contrast, in AKR1B10 holoenzyme, the corresponding same Trp (Trp112) side chain points to the specificity pocket, and it is stabilized by a network of hydrogen bond centred on the Gln114 [177]. This complicates drug design, as inhibitors often cross-react. Importantly, off-target inhibition of AKR1B10 is especially problematic: this enzyme is crucial for detoxifying reactive carbonyls and for lipid metabolism, including retinoid and isoprenoid homeostasis [140]. Inhibiting AKR1B10 can disrupt lipid balance and promote carcinogenic processes. Similarly, AKR1A1 plays a protective role by reducing reactive dicarbonyls such as methylglyoxal; its inhibition may enhance glycation stress and tissue injury [178]. This overlap makes selective inhibition particularly challenging, as broad inhibition of AKR1B10 impairs detoxification and lipid homeostasis, while inhibition of AKR1A1 affects dicarbonyl metabolism. These limitations underscore the need for novel inhibitor design strategies that can overcome the twin challenges of selectivity and functional preservation of AR physiological roles, without disturbing the conserved catalytic core shared across the AKR family, and that will look into more than just inhibitory potency for the production of sorbitol [140].
3.2.2. New Strategies for Aldose Reductase Inhibitor Design
Several approaches have been used for ARIs or drug design, such as High-Throughput Screening (HTS), structure-based or ligand-based drug design (SBDD, LBDD), machine learning (ML) models and screening of natural compounds. Since the discovery of the 3D structure of AR, molecular modelling studies such as SBDD have been reported. Molecular modelling studies are useful to give insightful information into the structure of the enzyme-bound inhibitor and their inhibition mechanisms. For instance, Wang and colleagues (2013) adopted a structure-based virtual screening strategy using several crystal structures of AR to represent its flexible active site [179]. By docking compound libraries against multiple receptor conformations, they identified several novel inhibitors with IC₅₀ values in the low micromolar range and demonstrated favourable selectivity over the related enzyme AKR1A1. While this multi-conformational approach accounted for the enzyme’s dynamic pocket architecture, the identified compounds still fell short of progressing to clinical application, often limited by bioavailability, metabolic instability, or lack of efficacy in vivo [179]. Similarly, the study done by Michael Eisenmann and colleagues in 2009 used a structure-based drug design approach to optimize the novel ARIs identified through virtual screening [180]. They integrated both computational and experimental methods to enhance the potency and selectivity of ARIs to develop novel candidates for the treatment of diabetic complications. They employed ultrahigh-resolution crystal structures of AR complexed with inhibitors such as IDD-594 to guide structure-based optimization. Using these structural insights, they conducted virtual screening to identify new scaffolds with low micromolar to nanomolar IC₅₀ values. The initial hits were refined by simplifying chemical structures and synthesizing derivatives, with X-ray crystallography confirming their improved binding modes. Despite this rational and targeted approach, none of these candidates progressed successfully through preclinical development [180], maybe partially because the many conformations determined or generated, still do not capture the full bread of AR flexibility and adaptation to different ligands, significantly complicating structure-based design of inhibitors.
Similarly, other studies using combinatorial libraries, natural product screening, and rational design approaches have yielded promising leads in vitro but failed in later stages of development. This lack of clinical success, often due to off-target and toxicity effects, underscores the need for alternative strategies (such as FBDD and differential inhibition approaches) that aim to preserve the physiological function of AR while selectively inhibiting its pathological activity [49]. Unlike HTS, which relies on large, complex molecules, FBDD begins with small fragments that interact in an “atom-efficient” manner, enabling the detection of subtle but exploitable structural differences. Such differences, including those in sub-pocket plasticity and residue orientations (e.g., Trp111/Trp112 positioning), can be used during fragment elaboration to achieve differential inhibition of AR, while sparing homologous enzymes like AKR1A1 and AKR1B10. This strategy not only reduces chemical complexity and cost but also enhances the likelihood of achieving selective and physiologically compatible inhibition. FBDD has been very successful in discovering several drugs where traditional high-throughput screening methods have limitations [181,182]. In this review article, we are proposing a new strategy to screen the inhibitors with the FBDD approach, along with further structural validation of the proposed inhibitor with Microcrystal Electron Diffraction (MicroED). MicroED can resolve high-resolution conformational states of AR-fragment complexes that are difficult to capture through conventional crystallography, thereby guiding rational fragment growing, merging, or linking to achieve inhibitors with both selectivity and clinical translatability.
4. Emerging Tools in Rational Drug Design: Fragment-Based Drug Discovery and MicroED
To address the limitations of traditional strategies for AR inhibitor development below, we explore two emerging techniques that may overcome the previous challenges.
4.1. Fragment-Based Drug Discovery (FBDD)
FBDD usually starts with the screening of a relatively small compound library comprised of compounds with low molecular weights, up to 300 Da, called fragments. Usually, the small fragments from the library have fewer than 20 heavy atoms and lower molecular complexity. In the FBDD approach, the throughput compared to HTS is rather low; that’s why selection and construction of the library is very important. Fragment libraries are often constructed using commercially available compounds, and their diversity can be enhanced by including pharmacophore-based selection criteria [183]. The advantage of a fragment-based library is that one can include a set of natural products or natural-product-inspired fragments. Typically, fragments obey the rule of three (RO3), having the following peculiar properties: the molecular weight is ≤300 Da, the number of hydrogen-bond donors ≤3, the number of hydrogen-bond acceptors ≤3, and the logP is ≤3 [184] (Figure 3).
After constructing the diverse fragment library, the main challenge of the FBDD approach is to detect and select those fragments that are tightly bound to the target of interest/protein. Given the typically low binding affinities of fragments (in the micromolar to millimolar range), this process poses significant challenges. To have the best fragment molecule, researchers usually apply several orthogonal methods to confirm that the fragment is binding to the target and to characterize the fragment’s binding mode [185]. The orthogonal screening methods include biophysical and biochemical techniques which explore the ligand-target binding [186]. This enables the identification of the most promising fragment, which can be further used for the generation of the lead compound. Once the fragments are identified and validated, compound elaboration can proceed in three main ways: fragment growing, fragment merging, and fragment linking [187,188]. In fragment growing, the initial fragment is systematically expanded into adjacent sub-pockets within the binding site while retaining its original interactions, thereby enhancing inhibitory activity [189]. Fragment merging involves synthetically combining two fragments that overlap in the active site to preserve key interactions from both [190]. In fragment linking, fragments binding to distinct, non-overlapping subregions are tethered together, allowing their interactions to be simultaneously maintained [187]. Notably, fragment growing and linking have been shown to rapidly improve binding affinity with relatively few synthetic steps, often achieving comparable potencies. There are several strategies for finding the binding-affinity assay with their own advantages and disadvantages.
Virtual screening (VS), a computer-based method for predicting the binding compounds from a compound library to a target protein, can be used. The advancement in the field of computational approaches and AI virtual screening methods has become more common in the FBDD approach [191]. In this approach, VS allows one to reduce the number of compounds from up to ten million conceivable scaffolds down to a moderate number around one thousand. Usually, VS methods often used approaches involve purely ligand-based pharmacophore modelling and structure-based screening [192]. Ligand-based modelling identifies key molecular features required for target binding by analysing a set of active compounds without needing the target’s 3D structure. Structure-based virtual screening relies on the 3D structural information of the target protein and then computationally docks, and predicts how molecules bind. However, both approaches have its own advantages both these experiments are hypothetical and need further experimental validation and testing [193].
4.1.1. Fragment Screening Biophysical Detection
There are several Biophysical detection methods for fragment screening: Surface Plasmon Resonance (SPR), Thermal Shift Assay (TSA), Microscale Thermophoresis (MST), and X-ray methods (Figure 3). There are advantages and disadvantages of each and every method.
SPR is a powerful technique for the determination of binding events [194]. Importantly, this technique is well suited for the detection of very weak probe-target interactions mediated by fragments because of its high sensitivity. The real-time monitoring provides the possibility to conduct off-rate screening (ORS), which provides basic information about how long the molecules are interacting with each other, and this aspect is very important in FBDD to identify the new fragment hits [195]. Due to the requirement of a very low concentration of the protein, this technique is very cost-effective but direct information about the binding site or interacting groups of a fragment cannot be derived [196,197].
TSA is a reliable technique to measure the denaturation temperature of a protein. The stability of protein correlates/or depends on many parameters, and binding/interacting with ligands and fragments is one of them. Fluorescence-based TSA is the most common ones and is usually called thermofluor assays or differential scanning fluorimetry (DSF) [198]. In principle, the fluorescence of a protein is measured with respect to an increase in temperature, the fluorescence dye (e.g., SYPRO orange) should be added and this dye shows a different fluorescence signal in polar and non-polar environment [199]. TSA may not be suitable for all the target proteins due to the indirect readout of the melting temperature. Due to the high-throughput option and high sensitivity, this technique is very beneficial for the FBDD but researchers must be aware with the high number of false positives.
MST is a well-established biophysical technique used for any kind of biomolecular interactions [200,201]. In this technique, if a ligand binds to the target, the chemical environment is changed and leading to a change in the specific thermophoretic profile of the molecule [202]. In several studies, researchers have demonstrated that MST can detect weak binders such as fragments. The major advantage of MST is that one can record the measurement in solution; there is no need of immobilization of the protein [203]. Additionally, another advantage of MST is that the shape of the MST traces provides information about the protein aggregation or denaturation and give an indication of the specific or unspecific ligand-protein binding during the measurement, and this could be very helpful in FBDD [204].
Macromolecular X-ray analysis is a key method for FBDD, and it generates very detailed information about the protein-ligand interaction. By this method, opportunities are given to conduct structure-based design studies to improve the ligand affinity efficiently [205,206,207]. The crystal can be achieved by two different methods: co-crystallization or soaking. Both methods are challenging but achievable, since not all proteins are easily crystalized and some ligands can disrupt the crystal lattice [208]. By generating a co-crystal with each with a different fragment, the different modes of binding can enable rational fragment linking or merging.
4.2. Microcrystal Electron Diffraction (MicroED)
While X-ray crystallography remains the gold standard for high-resolution protein-ligand structural analysis, it often requires large, well-ordered crystals, therefor representing important bottlenecks in fragment screening campaigns. Many proteins or fragment-protein complexes fail to crystallize under traditional conditions, or only yield micro- or nanocrystals unsuitable for standard diffraction. To address these challenges, Microcrystal Electron Diffraction (MicroED) has emerged as a powerful complementary technique, capable of resolving atomic details from extremely small crystals that are otherwise unusable for conventional X-ray methods. Small macromolecular crystals may have fewer defects and lower mosaicity than larger crystals, and any external changes, such as ligand soaking or rapid flash freezing, can be easily and uniformly applicable [209,210]. Small crystal molecules have their own disadvantages like overall diffraction is much weaker, and radiation damage can limit the possibility of getting high resolution. But in recent years, MicroED has really emerged as a potential tool for structural biology, determining the structures of several macromolecules [211,212,213,214,215]. There is an additional advantage of MicroED that can complement existing methods in structural biology, that it really helps to overcome the size limitation and biomolecules that have low molecular weight, which are challenging for the single-particle cryoEM, but can be studied by MicroED [216,217]. Similarly, high-throughput screening of a large number of fragments for possible interactions with the target protein can reveal binding interactions that inform the design of novel inhibitors. The potential of MicroED in drug discovery is exponentially increasing; for instance, it has been explored in the study of binding of the inhibitor bevirimat (BVM) to the C-terminal domain of the HIV gag protein [218], resolving key structural interactions that were previously inaccessible. In 2020, Clabbers and colleagues, with the help of MicroED investigated the drug-binding interactions to the active site of human carbonic anhydrase isoform II (HCA II) [219], a key milestone demonstrating MicoED’s capacity to contribute directly to structure-guided inhibitor design. In 2013, the first protein structure was successfully determined from 3D microcrystals using MicroED [220,221], and since then, many protein structures have been determined from (sub-)micron-sized crystals [211,214,215,219,222]. In this context, combining FBDD with MicroED creates a powerful platform for fragment screening, structural validation, and lead optimization- especially for AR, where selective inhibition and precise characterization are essential for therapeutic aspects (Figure 3).
4.3. Proposed Strategy: Integrating FBDD and MicroED for Aldose Reductase Inhibitor Design
Drug discovery remains a critical aspect of structural biology and pharmaceutical development, where a detailed understanding of a protein’s active site and its interactions with small molecules is fundamental for rational inhibitor design. In the context of AR, we are proposing a fragment-based drug discovery (FBDD) approach aimed at identifying small molecules capable of differential binding, that is, selectively targeting pathological functions of the enzyme without impairing its essential physiological roles.
As described above, AR exhibits both pathological and physiological activities: under hyperglycemic conditions, it contributes to diabetic complications by catalyzing the reduction of glucose to sorbitol; conversely, it plays a protective role by detoxifying cytotoxic aldehydes such as 4-hydroxy-2-nonenal (HNE) and other alkenals and alkanals generated via lipid peroxidation [49,105]. Owing to its broad substrate specificity, AR can reduce both hydrophilic and hydrophobic carbonyl compounds and is capable of discriminating among substrates of the same chemical class [140,223]. Molecular modelling has already revealed distinct binding interactions for glucose, HNE, and glutathione-conjugated HNE (GS-HNE), suggesting multiple substrate-specific interaction modes within the active site [105]. These findings support the hypothesis that differential inhibition of AR is feasible, where a compound could selectively disrupt the enzyme’s glucose-reducing function while sparing its detoxifying activity. An ideal differential inhibitor of AR would, therefore, preferentially block its activity toward glucose or GS-HNE while maintaining or minimally affecting its ability to metabolize reactive aldehydes [49]. This dual outcome could attenuate the pro-inflammatory and pro-oxidant consequences of AR activation in diabetes, while preserving its physiological role in cellular protection against oxidative stress.
To identify the hit in FBDD, fragments undergo systematic optimization to enhance their binding affinity and drug-like properties. Fragments serve as highly efficient starting points due to their structural simplicity and ability to form high-quality interactions within a protein’s binding site. This allows for iterative optimization, frequently resulting in improved pharmacokinetic profiles compared to modifying larger, drug-like molecules, which may lack optimal binding features or contribute to unfavourable interactions (Figure 4).
4. 4. Fragment Optimization Strategies for Selective Aldose Reductase Inhibition
Following the identification of promising fragment hits that differentially bind to AR, the next critical step is fragment optimization. This step is crucial for enhancing potency, improving pharmacokinetic properties, and retaining selectivity between pathological and physiological targets. Fragment optimization is typically guided by high-resolution structural data and follows one of three strategies: growing, linking, or merging [224] (Figure 3 and Figure 4). As this is a new approach towards the generation of differential inhibitors, likely all three strategies must be tried.
Fragment growing is the commonly used strategy employed during fragment-based Drug/Lead discovery (FBDD/FBLD). It involves adding functional groups to an initial fragment hit to improve its interaction with the target protein [189]. In the context of AR, this strategy holds a particular significance for differential inhibition, as the active site possesses both hydrophilic and hydrophobic sites. Fragment growing can exploit specific sub-pocket interaction, such as π-π stacking interaction with aromatic amino acid Trp111 or hydrogen bonding between His110 and Tyr48, to exclusively binding to glucose related conformations while avoiding aldehyde-detoxification sites. A recent paper published by Strecker et al. (2019), targeting Human blood group B galactosyltransferase (GTB), is an example that demonstrated how the growing strategy can be used to improve binding affinity [225]. That study showed that a modification of a fragment phenyl moiety to a naphthyl allowed two new simultaneous π-π interactions, a parallel-displaced with Trp300 and an edge-to-face with His233. This minor modification led to a compound with a 3-fold improved binding affinity (Ki = 271 μM). The study also highlights the use of optimal growth vectors to introduce a rigid group, which led to an increased binding affinity [225]. During this strategy for lead compound generation for AR, attention need to be paid towards the number of rotatable bond because an increased number of rotatable bonds could impact negatively, due to entropic penalty, in the affinity. Importantly, such fragment growing should be guided by structural data at each step to avoid entropic penalties or misdirected growth. The presence of multiple binding conformations in AR, as shown in various molecular dynamics and crystallographic studies, makes structure-guided growth essential to preserve functional selectivity and optimize pharmacological properties.
Fragment linking describes the process of joining two non-competitive fragments (fragments which bind in two different sub-pockets of the binding site) with a chemical linker or spacer, and it could be another approach for optimizing and preparing the lead molecule/compound [187]. For AR, this approach could be really significant because this enzyme possesses different active sites. In AR, however, fragment linking is particularly promising due to the enzyme’s compartmentalized active site, which includes the anion-binding pocket, the hydrophobic substrate binding cleft and the specificity pocket, each of which interacts differently with glucose, HNE, and other endogenous ligands. By identifying fragment pairs that bind independently within these sub-pockets, one could design a linker that preserves the original binding modes while enhancing overall potency. However, care must be taken in such a design, for instance, linking fragments across the glucose-binding tunnel and another fragment occupying the hydrophobic pocket involved in aldehyde detoxification could risk blocking both physiological and pathological functions of AR. Thus, fragment-based strategies should ideally focus on exploiting sub-pockets associated with pathological activity.
Therefore, successful implementation of fragment linking in AR requires careful design of the linker’s length, rigidity, and orientation to avoid conformational strain, disrupt original binding geometries, or induce entropic penalties that reduce binding efficiency. Prior studies, such as Chung et al. (2009), on the human uracil DNA glycosylase, have shown how modifying linker chemistry (from oximes to monoamines or diamines) can influence molecular rigidity and binding efficacy [226]. Moreover, the impact of linker design on pharmacokinetics (PK) should not be underestimated. Linkers often introduce additional rotatable bonds, which may adversely affect oral bioavailability, permeability, and metabolic stability [227]. In the context of AR inhibitor development, balancing binding potency with favourable absorption, distribution, metabolism, excretion and toxicity (ADMET) profiles will be essential for lead optimization and translational success [228].
For generating the lead compound for AR, the merging approach can also be tried. Fragment merging involves combining two fragments that occupy overlapping or adjacent binding regions into a single, more potent ligand. Although merging is a simpler strategy than linking, as there is no need of join and design the spacer that joins two fragments together. However, it requires precise structural information to ensure that merged fragments retain the favourable interactions of both parent compounds [190,229]. In the case of AR, merging may be feasible when fragments hit partially share contact points within the catalytic site, such as overlapping regions of glucose and HNE binding. This strategy could generate molecules that mimic the favourable contacts of both substrates, selectively inhibiting glucose reduction while leaving detoxification largely intact [190]. In a recent example, which proves merging as a significant method, Nikiforov et al. (2016) achieved two orders of magnitude in potency for an inhibitor of flavin-dependent monooxygenase (EthA) transcriptional repressor (EthR), where the existence of overlapping groups within fragments bound to EthR allowed the use of merging as an optimization strategy [224,230]. Fragment merging strategy has been validated in other disease models, also like the utility of fragment merging has also been validated in oncology: an FBDD campaign against MCL-1 identified two independent fragments that bound to different protein sites. Through merging and subsequent optimization, highly selective, picomolar MCL-1 inhibitors were obtained, showing strong cellular potency, drug-like pharmacokinetics, and antitumor efficacy in hematologic malignancies and lung xenograft models [231,232].
The integration of fragment growing, linking, and merging offers a versatile toolkit for designing differential AR inhibitors. Each approach brings unique strengths and challenges, and its success hinges on high-quality structural data, careful biophysical validation, and attention to pharmacokinetic consequences. By strategically applying these optimization methods, it may be possible to overcome the limitations of classical AR inhibitors and develop next-generation therapeutics that preserve the enzyme’s protective roles while mitigating its contribution to diabetic complications.
4.5. Structural Validation of Optimized Leads Using MicroED
Once fragment hits are optimized through growing, linking, or merging strategies, confirming their precise binding mode is critical to ensure both efficacy and selectivity. This is particularly relevant for a target like AR, which has overlapping physiological and pathological roles. While traditional X-ray crystallography has long served this role in structure-based drug design, its reliance on large, well-ordered crystals remains a major limitation, especially when working with small fragments or structurally fragile protein-ligand complexes.
Traditionally, X-ray crystallography has been the gold standard for high-resolution structure determination of protein-ligand complexes. However, the method has several inherent limitations that restrict its utility in FBDD [233]. The requirement for large, well-ordered protein crystals poses a major bottleneck, especially when working with small fragments or with proteins that are difficult to crystallize under standard conditions [234]. Moreover, soaking optimized ligands into preformed crystals can disturb the lattice or fail to capture subtle conformational shifts necessary for differential inhibition [235]. Recent advances in microcrystallography [236], such as microfocus X-ray beams, serial femtosecond crystallography (SFX), and especially Microcrystal Electron Diffraction (MicroED), have dramatically improved the ability to study small crystals [236,237]. MicroED, in particular, enables high-resolution structure determination from crystals as small as a few hundred nanometers, overcoming key barriers in traditional methods [220,238]. It also allows for rapid and uniform soaking of ligands or fragments, essential for validating differential binding modes in dynamic enzymes like AR. This advantage can be explores also in dedicated microfocus X-ray beamlines such as VMXm in Diamond Light Source [236,239], or taking full advantage of their VMXi beamline and Xchem platform, that couples it with in situ diffraction and fragment screening [240], but electrons scattering is dictated by the electrostatic potential of electrons and nuclei while X-rays photons scattering is dictated be electron clouds around individual atoms, thus for drug development microED brings the added advantage of potential resolving electrostatic potential maps and hydrogen positioning, thus revealing binding modes [236,241,242]. Additionally, MicroED’s compatibility with routine cryo-EM instruments, that with the spread of cryoEM instrumentation increases its reach compared to microfocus-equipped synchrotrons [243], and its lower sample volume requirement (as electrons are charged particles and interact with matter much stronger than X-ray photons producing less damage per scattering event, allowing crystal volumes a billion times smaller [220,243]) make it highly suitable for fragment screening programs where obtaining large crystals is not feasible. Despite advances in precision crystal control growth and size targeting [236,244], techniques like FIB-SEM milling, cryogenic sample, and optimized grid preparation have further expanded MicroED’s applicability across a wide range of targets, when proteins of interest crystallize “just” with larger crystals [241,245,246]. Integrating MicroED into the FBDD workflow for AR offers a high-throughput structural validation of fragment binding in physiologically and pathologically relevant pockets. This insight is invaluable for designing selective inhibitors that modulate AR’s harmful hyperglycemic activity while preserving its detoxification function.
4.6. Application Workflow: Using MicroED to Guide Differential Inhibitor Design
In our proposed workflow, optimized AR binding fragments will be co-crystallized or soaked into pre-grown microcrystals of AR. These nanocrystals (often only a few hundred nanometres thick) can then be applied to cryo-electron microscopy (cryo-EM) grids, vitrified via plunge-freezing in cryogenic ethane, and a diffraction pattern can be collected/recorded in a cryoEM microscope under diffraction mode (Figure 4). Using continuous rotation data collection, MicroED can reveal atomic-level details of fragment binding even from tiny crystals. By visualizing how lead compounds engage specific residues, such as His110, Tyr48, or Trp111, and distinguishing between interactions with glucose or aldehyde-binding pockets, MicroED will help validate whether the designed inhibitors successfully achieve differential inhibition. These insights will guide further chemical modifications to enhance selectivity, minimize off-target effects, and inform the structure-activity relationship (SAR) of the leads [247]. Ultimately, integrating MicroED into the FBDD pipeline for AR allows rapid, high-resolution structural feedback in cases where conventional crystallographic approaches are limited, thus increasing the efficiency and accuracy of lead development.
5. Conclusions and Future Perspectives
Despite decades of effort, the development of clinically effective ARIs remains an unsolved challenge, primarily due to issues of selectivity, bioavailability, and adverse off-target effects. Classical approaches such as high-throughput screening (HTS) and rational drug design have identified promising leads, but these have rarely translated into viable therapies beyond epalrestat, which itself has limited availability. In this review, we listed the current short comes have highlighted how emerging tools such as FBDD and MicroED can be harnessed to overcome these limitations. FBDD offers a structurally efficient and chemically tractable way to identify fragment hits that may exhibit selective binding to distinct sub-pockets within the AR active site. When integrated with MicroED, a technique capable of resolving atomic-level detail from nanocrystals, this pipeline enables high-resolution structural validation of fragment binding even in cases where classical X-ray crystallography is not feasible.
Looking forward, this combined FBDD-MicroED approach opens new avenues for developing differential inhibitors of AR that preserve its physiological detoxification role while selectively suppressing its pathological activity in diabetic complications. Future work should focus on expanding structurally diverse fragment libraries tailored to the unique architecture of AR, validating binding through multi-technique pipelines, and iterating lead optimization using real-time structural feedback.
Author Contributions
Conceptualization, V.K. and H.F.; data extraction, V.K., S.K. and H.F.; Literature search, V.K. and H.F.; graphics, V.K.; writing—original draft preparation, V.K., S.K and H.F.; writing—review and editing, V.K. and H.F.; All authors have read and agreed to the published version of the manuscript.
Funding
The review relates to projects funded by the (i) National Science Centre, Poland, under project no. 2025/09/X/NZ1/00769 under the MINIATURA 9 call (to V.K.); (ii) European Union and the Foundation for Polish Science, under project no. FENG.02.01-IP.05-T005/23 under the IRAP FENG call; and (iii) European Union, under project no. 101136570 under the Teaming for Excellence call and Foundation for Polish Science, under project no. FENG.02.01-IP.05-T005/23 under IRAP FENG call.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- American Diabetes, A. Diagnosis and classification of diabetes mellitus. Diabetes Care 2010, 33 Suppl 1(Suppl 1), S62–9. [Google Scholar] [CrossRef]
- American Diabetes Association Professional Practice, C., 2. Diagnosis and Classification of Diabetes: Standards of Care in Diabetes-2025. Diabetes Care 2025, 48 (1 Suppl 1), S27–S49. [CrossRef]
- Cade, W.T. Diabetes-related microvascular and macrovascular diseases in the physical therapy setting. Phys Ther 2008, 88(11), 1322–35. [Google Scholar] [CrossRef]
- Vithian, K.; Hurel, S. Microvascular complications: pathophysiology and management. Clin Med (Lond) 2010, 10(5), 505–9. [Google Scholar] [CrossRef]
- Klein, R.; et al. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch Ophthalmol 1984, 102(4), 527–32. [Google Scholar] [CrossRef] [PubMed]
- Ansari, P.; et al. Diabetic Retinopathy: An Overview on Mechanisms, Pathophysiology and Pharmacotherapy. Diabetology 2022, 3(1), 159–175. [Google Scholar] [CrossRef]
- Honasoge, A.; et al. Emerging Insights and Interventions for Diabetic Retinopathy. Curr Diab Rep 2019, 19(10), p. 100. [Google Scholar] [CrossRef]
- Santiago, J.V. Lessons from the Diabetes Control and Complications Trial. Diabetes 1993, 42(11), 1549–54. [Google Scholar] [CrossRef]
- Hendrick, A.M.; Gibson, M.V.; Kulshreshtha, A. Diabetic Retinopathy. Prim Care 2015, 42(3), 451–64. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Wong, T.Y. Current concepts in diabetic retinopathy. Diabetes Metab J 2014, 38(6), 416–25. [Google Scholar] [CrossRef]
- Bortea, C.I.; et al. Risk Factors Associated with Retinopathy of Prematurity in Very and Extremely Preterm Infants. Medicina (Kaunas) 2021, 57(5). [Google Scholar] [CrossRef]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int J Mol Sci 2018, 19(6). [Google Scholar] [CrossRef]
- Antonetti, D.A.; Silva, P.S.; Stitt, A.W. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol 2021, 17(4), 195–206. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414(6865), 813–20. [Google Scholar] [CrossRef]
- Sheetz, M.J.; King, G.L. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 2002, 288(20), 2579–88. [Google Scholar] [CrossRef]
- Stewart, M.W. Pathophysiology of Diabetic Retinopathy, in Diabetic Retinopathy; 2010; pp. 1–30. [Google Scholar]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11(10). [Google Scholar] [CrossRef]
- Van den Enden, M.K.; et al. Elevated glucose levels increase retinal glycolysis and sorbitol pathway metabolism. Implications for diabetic retinopathy. Invest Ophthalmol Vis Sci 1995, 36(8), 1675–85. [Google Scholar] [PubMed]
- Danila, A.I.; et al. Aldose Reductase as a Key Target in the Prevention and Treatment of Diabetic Retinopathy: A Comprehensive Review. Biomedicines 2024, 12(4). [Google Scholar] [CrossRef] [PubMed]
- Vinores, S.A.; et al. Aldose reductase expression in human diabetic retina and retinal pigment epithelium. Diabetes 1988, 37(12), 1658–64. [Google Scholar] [CrossRef]
- Kinoshita, J.H.; et al. Aldose reductase in diabetic complications of the eye. Metabolism 1979, 28((4) Suppl 1, 462–9. [Google Scholar] [CrossRef]
- Mestry, S.N.; Juvekar, A.R. Aldose reductase inhibitory potential and anti-cataract activity of Punica granatum Linn. leaves against glucose-induced cataractogenesis in goat eye lens. Oriental Pharmacy and Experimental Medicine 2017, 17(3), 277–284. [Google Scholar] [CrossRef]
- Gabbay, K.H. Hyperglycemia, polyol metabolism, and complications of diabetes mellitus. Annu Rev Med 1975, 26, 521–36. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, J.H. A thirty year journey in the polyol pathway. Exp Eye Res 1990, 50(6), 567–73. [Google Scholar] [CrossRef]
- Steinmetz, P.R. C. Balko, and K.H. Gabbay, The Sorbitol Pathway and the Complications of Diabetes. New England Journal of Medicine 1973, 288(16), 831–836. [Google Scholar] [CrossRef]
- Srikanth, K.K.; Orrick, J.A. Biochemistry, Polyol Or Sorbitol Pathways, in StatPearls; Treasure Island (FL), 2025. [Google Scholar]
- Ali, T.K.; El-Remessy, A.B. Diabetic retinopathy: current management and experimental therapeutic targets. Pharmacotherapy 2009, 29(2), 182–92. [Google Scholar] [CrossRef]
- Julius, A.; Hopper, W. A non-invasive, multi-target approach to treat diabetic retinopathy. Biomed Pharmacother 2019, 109, 708–715. [Google Scholar] [CrossRef]
- Balestri, F.; et al. In Search of Differential Inhibitors of Aldose Reductase. Biomolecules 2022, 12(4). [Google Scholar] [CrossRef]
- Gulec, O.; et al. Novel spiroindoline derivatives targeting aldose reductase against diabetic complications: Bioactivity, cytotoxicity, and molecular modeling studies. Bioorg Chem 2024, 145, 107221. [Google Scholar] [CrossRef]
- Ahmad, S.; et al. Exploring aldose reductase inhibitors as promising therapeutic targets for diabetes-linked disabilities. Int J Biol Macromol 2024, 280 (Pt 2), 135761. [Google Scholar] [CrossRef]
- Kumari, P.; et al. Selectivity challenges for aldose reductase inhibitors: A review on comparative SAR and interaction studies. Journal of Molecular Structure 2024, 1318. [Google Scholar] [CrossRef]
- Sarges, R.; Oates, P.J. Aldose reductase inhibitors: recent developments. Prog Drug Res 1993, 40, 99–161. [Google Scholar] [PubMed]
- Maccari, R.; Ottana, R. Targeting aldose reductase for the treatment of diabetes complications and inflammatory diseases: new insights and future directions. J Med Chem 2015, 58(5), 2047–67. [Google Scholar] [CrossRef]
- Grewal, A.S.; et al. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-diabetic Diseases. Mini Rev Med Chem 2016, 16(2), 120–62. [Google Scholar] [CrossRef]
- Grewal, A.S.; et al. Natural Compounds as Source of Aldose Reductase (AR) Inhibitors for the Treatment of Diabetic Complications: A Mini Review. Curr Drug Metab 2020, 21(14), 1091–1116. [Google Scholar] [CrossRef]
- Gabbay, K.H. Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Curr Diab Rep 2004, 4(6), 405–8. [Google Scholar] [CrossRef]
- Hotta, N.; et al. Long-term clinical effects of epalrestat, an aldose reductase inhibitor, on progression of diabetic neuropathy and other microvascular complications: multivariate epidemiological analysis based on patient background factors and severity of diabetic neuropathy. Diabet Med 2012, 29(12), 1529–33. [Google Scholar]
- Senthilkumari, S.; et al. Epalrestat, an Aldose Reductase Inhibitor Prevents Glucose-Induced Toxicity in Human Retinal Pigment Epithelial Cells In Vitro. J Ocul Pharmacol Ther 2017, 33(1), 34–41. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.A.; Borja, N.L. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy 2008, 28(5), 646–55. [Google Scholar] [CrossRef]
- Petrash, J.M. All in the family: aldose reductase and closely related aldo-keto reductases. Cell Mol Life Sci 2004, 61(7-8), 737–49. [Google Scholar] [CrossRef]
- Sun, W.; et al. A selective aldose reductase inhibitor of a new structural class prevents or reverses early retinal abnormalities in experimental diabetic retinopathy. Diabetes 2006, 55(10), 2757–62. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.K.; et al. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 2005, 54(11), 3119–25. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; et al. Aspirin at low-intermediate concentrations protects retinal vessels in experimental diabetic retinopathy through non-platelet-mediated effects. Diabetes 2005, 54(12), 3418–26. [Google Scholar] [CrossRef]
- A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Sorbinil Retinopathy Trial Research Group. Arch Ophthalmol 1990, 108(9), 1234–44. [CrossRef]
- Dagher, Z.; et al. Studies of rat and human retinas predict a role for the polyol pathway in human diabetic retinopathy. Diabetes 2004, 53(9), 2404–11. [Google Scholar] [CrossRef]
- Asnaghi, V.; et al. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes 2003, 52(2), 506–11. [Google Scholar] [CrossRef] [PubMed]
- Barski, O.A.; et al. Mechanism of human aldehyde reductase: characterization of the active site pocket. Biochemistry 1995, 34(35), 11264–75. [Google Scholar] [CrossRef]
- Del-Corso, A.; et al. A new approach to control the enigmatic activity of aldose reductase. PLoS One 2013, 8(9), e74076. [Google Scholar] [CrossRef]
- Elimam, D.M.; et al. Olive and ginkgo extracts as potential cataract therapy with differential inhibitory activity on aldose reductase. Drug Discov Ther 2017, 11(1), 41–46. [Google Scholar] [CrossRef]
- Wang, Z.Z.; et al. Fragment-based drug design facilitates selective kinase inhibitor discovery. Trends Pharmacol Sci 2021, 42(7), 551–565. [Google Scholar] [CrossRef]
- Ramana, K.V. ALDOSE REDUCTASE: New Insights for an Old Enzyme. Biomol Concepts 2011, 2(1-2), 103–114. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Goldberg, I.J. Aldose reductase and cardiovascular diseases, creating human-like diabetic complications in an experimental model. Circ Res 2010, 106(9), 1449–58. [Google Scholar] [CrossRef]
- Rao, M.; Chang, K.C. Aldose reductase is a potential therapeutic target for neurodegeneration. Chem Biol Interact 2024, 389, 110856. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.; et al. Clinical analysis of aldose reductase for differential diagnosis of the pathogenesis of diabetic complication. Analytica Chimica Acta 1998, 365(1-3), 285–292. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005, 54(6), 1615–25. [Google Scholar] [CrossRef] [PubMed]
- Alwadani, F.; Saif, M. The role and prevalence of polyol pathway and oxidative stress markers as risk factors for diabetic cataract in adult type-I diabetic and diabetic cataract Saudi patients. J Clin Exp Ophthalmol 2016, 7(558), p. 2. [Google Scholar] [CrossRef]
- Doorn, J.A.; Srivastava, S.K.; Petersen, D.R. Aldose reductase catalyzes reduction of the lipid peroxidation product 4-oxonon-2-enal. Chem Res Toxicol 2003, 16(11), 1418–23. [Google Scholar] [CrossRef]
- Petrash, J.M.; Harter, T.M.; Murdock, G.L. A potential role for aldose reductase in steroid metabolism. Adv Exp Med Biol 1997, 414, 465–73. [Google Scholar]
- Iwata, N. Hormonal regulation of aldose reductase in rat ovary during the estrous cycle. Eur J Biochem 1996, 235(1-2), 444–8. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev 2004, 56(3), 331–49. [Google Scholar] [CrossRef]
- Niimi, N.; et al. Aldose Reductase and the Polyol Pathway in Schwann Cells: Old and New Problems. Int J Mol Sci 2021, 22(3). [Google Scholar] [CrossRef]
- Hers, H.G. The mechanism of the transformation of glucose in fructose in the seminal vesicles. Biochim Biophys Acta 1956, 22(1), 202–3. [Google Scholar] [CrossRef]
- Hirakawa, H.; et al. Expression analysis of the aldo-keto reductases involved in the novel biosynthetic pathway of tetrahydrobiopterin in human and mouse tissues. J Biochem 2009, 146(1), 51–60. [Google Scholar] [CrossRef]
- Park, Y.S.; et al. Human carbonyl and aldose reductases: new catalytic functions in tetrahydrobiopterin biosynthesis. Biochem Biophys Res Commun 1991, 175(3), 738–44. [Google Scholar] [CrossRef]
- Endo, S.; et al. Kinetic studies of AKR1B10, human aldose reductase-like protein: endogenous substrates and inhibition by steroids. Arch Biochem Biophys 2009, 487(1), 1–9. [Google Scholar] [CrossRef]
- Bravi, M.C.; et al. Polyol pathway activation and glutathione redox status in non-insulin-dependent diabetic patients. Metabolism 1997, 46(10), 1194–8. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.; et al. Hyperglycemia and Oxidative Stress: An Integral, Updated and Critical Overview of Their Metabolic Interconnections. Int J Mol Sci 2023, 24(11). [Google Scholar] [CrossRef]
- Shi, X.; et al. Hesperidin prevents retinal and plasma abnormalities in streptozotocin-induced diabetic rats. Molecules 2012, 17(11), 12868–81. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G. Increased sorbitol pathway activity generates oxidative stress in tissue sites for diabetic complications. Antioxid Redox Signal 2005, 7(11-12), 1543–52. [Google Scholar] [CrossRef] [PubMed]
- Luis-Rodriguez, D.; et al. Pathophysiological role and therapeutic implications of inflammation in diabetic nephropathy. World J Diabetes 2012, 3(1), 7–18. [Google Scholar] [CrossRef]
- Wu, J.; et al. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes 2016, 9, 145–53. [Google Scholar]
- Lorenzi, M. The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res 2007, 2007, 61038. [Google Scholar] [CrossRef]
- Hohman, T.C.; Nishimura, C.; Robison, W.G., Jr. Aldose reductase and polyol in cultured pericytes of human retinal capillaries. Exp Eye Res 1989, 48(1), 55–60. [Google Scholar] [CrossRef]
- Li, W.; et al. Non-competitive inhibition of myo-inositol transport in cultured bovine retinal capillary pericytes by glucose and reversal by Sorbinil. Biochim Biophys Acta 1986, 857(2), 198–208. [Google Scholar] [CrossRef]
- Chakrabarti, S.; et al. Aldose reductase in the BB rat: isolation, immunological identification and localization in the retina and peripheral nerve. Diabetologia 1987, 30(4), 244–51. [Google Scholar] [CrossRef]
- Drel, V.R.; et al. Aldose reductase inhibitor fidarestat counteracts diabetes-associated cataract formation, retinal oxidative-nitrosative stress, glial activation, and apoptosis. Int J Mol Med 2008, 21(6), 667–76. [Google Scholar] [CrossRef]
- Kador, P.F.; Kinoshita, J.H. Role of aldose reductase in the development of diabetes-associated complications. Am J Med 1985, 79(5A), 8–12. [Google Scholar] [CrossRef]
- Varma, S.D.; Kinoshita, J.H. The absence of cataracts in mice with congenital hyperglycemia. Exp Eye Res 1974, 19(6), 577–82. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, A.D.; et al. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int 2001, 60(1), 211–8. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.J. Diabetic Retinopathy. 2010. [Google Scholar] [PubMed]
- Yan, L.J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Animal Model Exp Med 2018, 1(1), 7–13. [Google Scholar] [CrossRef]
- Yan, L.J.; et al. Mouse heat shock transcription factor 1 deficiency alters cardiac redox homeostasis and increases mitochondrial oxidative damage. EMBO J 2002, 21(19), 5164–72. [Google Scholar] [CrossRef] [PubMed]
- Mapanga, R.F.; Essop, M.F. Damaging effects of hyperglycemia on cardiovascular function: spotlight on glucose metabolic pathways. Am J Physiol Heart Circ Physiol 2016, 310(2), H153–73. [Google Scholar] [CrossRef]
- Treberg, J.R.; Quinlan, C.L.; Brand, M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 2011, 286(31), 27103–10. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem J 2009, 417(1), 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Martin, K.A.; Hwa, J. Aldose reductase, oxidative stress, and diabetic mellitus. Front Pharmacol 2012, 3, 87. [Google Scholar] [CrossRef]
- Inoguchi, T.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49(11), 1939–45. [Google Scholar] [CrossRef]
- Miyata, T.; et al. Accumulation of carbonyls accelerates the formation of pentosidine, an advanced glycation end product: carbonyl stress in uremia. J Am Soc Nephrol 1998, 9(12), 2349–56. [Google Scholar] [CrossRef]
- Miyata, T.; et al. Alterations in nonenzymatic biochemistry in uremia: origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int 1999, 55(2), 389–99. [Google Scholar] [CrossRef]
- Kaneko, M.; et al. Aldose reductase and AGE-RAGE pathways: key players in myocardial ischemic injury. Ann N Y Acad Sci 2005, 1043, 702–9. [Google Scholar] [CrossRef]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv Nutr 2017, 8(1), 54–62. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Zhong, Q.; Santos, J.M. Matrix metalloproteinases in diabetic retinopathy: potential role of MMP-9. Expert Opin Investig Drugs 2012, 21(6), 797–805. [Google Scholar] [CrossRef]
- Jin, M.; et al. Matrix metalloproteinases in human diabetic and nondiabetic vitreous. Retina 2001, 21(1), 28–33. [Google Scholar] [CrossRef]
- Giebel, S.J.; et al. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab Invest 2005, 85(5), 597–607. [Google Scholar] [CrossRef]
- Mohammad, G.; Kowluru, R.A. Matrix metalloproteinase-2 in the development of diabetic retinopathy and mitochondrial dysfunction. Lab Invest 2010, 90(9), 1365–72. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Kanwar, M. Oxidative stress and the development of diabetic retinopathy: contributory role of matrix metalloproteinase-2. Free Radic Biol Med 2009, 46(12), 1677–85. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, S.; et al. MMP-2 and MMP-9 secretion by rpe is stimulated by angiogenic molecules found in choroidal neovascular membranes. Retina 2006, 26(4), 454–61. [Google Scholar] [CrossRef]
- Navaratna, D.; et al. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes 2007, 56(9), 2380–7. [Google Scholar] [CrossRef]
- Ramana, K.V.; et al. Requirement of aldose reductase for the hyperglycemic activation of protein kinase C and formation of diacylglycerol in vascular smooth muscle cells. Diabetes 2005, 54(3), 818–29. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res 2010, 106(8), 1319–31. [Google Scholar] [CrossRef] [PubMed]
- Das Evcimen, N.; King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol Res 2007, 55(6), 498–510. [Google Scholar] [CrossRef]
- Derubertis, F.R.; Craven, P.A. Activation of protein kinase C in glomerular cells in diabetes. Mechanisms and potential links to the pathogenesis of diabetic glomerulopathy. Diabetes 1994, 43(1), 1–8. [Google Scholar] [CrossRef]
- Xia, P.; et al. Characterization of the mechanism for the chronic activation of diacylglycerol-protein kinase C pathway in diabetes and hypergalactosemia. Diabetes 1994, 43(9), 1122–9. [Google Scholar] [CrossRef]
- Balestri, F.; et al. Aldose Reductase Differential Inhibitors in Green Tea. Biomolecules 2020, 10(7). [Google Scholar] [CrossRef]
- Srivastava, S.; et al. Lipid peroxidation product, 4-hydroxynonenal and its conjugate with GSH are excellent substrates of bovine lens aldose reductase. Biochem Biophys Res Commun 1995, 217(3), 741–6. [Google Scholar] [CrossRef] [PubMed]
- Vander Jagt, D.L.; et al. Substrate specificity of human aldose reductase: identification of 4-hydroxynonenal as an endogenous substrate. Biochim Biophys Acta 1995, 1249(2), 117–26. [Google Scholar] [CrossRef]
- Ramana, K.V.; Srivastava, S.K. Aldose reductase: a novel therapeutic target for inflammatory pathologies. Int J Biochem Cell Biol 2010, 42(1), 17–20. [Google Scholar] [CrossRef]
- Frohnert, B.I.; Bernlohr, D.A. Glutathionylated products of lipid peroxidation: A novel mechanism of adipocyte to macrophage signaling. Adipocyte 2014, 3(3), 224–9. [Google Scholar] [CrossRef] [PubMed]
- Frohnert, B.I.; et al. Glutathionylated lipid aldehydes are products of adipocyte oxidative stress and activators of macrophage inflammation. Diabetes 2014, 63(1), 89–100. [Google Scholar] [CrossRef]
- Srivastava, S.; et al. Synthesis, quantification, characterization, and signaling properties of glutathionyl conjugates of enals. Methods Enzymol 2010, 474, 297–313. [Google Scholar]
- Way, K.J.; Katai, N.; King, G.L. Protein kinase C and the development of diabetic vascular complications. Diabet Med 2001, 18(12), 945–59. [Google Scholar] [CrossRef] [PubMed]
- Bursell, S.E.; et al. Specific retinal diacylglycerol and protein kinase C beta isoform modulation mimics abnormal retinal hemodynamics in diabetic rats. Invest Ophthalmol Vis Sci 1997, 38(13), 2711–20. [Google Scholar]
- Oishi, N.; et al. Correlation between erythrocyte aldose reductase level and human diabetic retinopathy. Br J Ophthalmol 2002, 86(12), 1363–6. [Google Scholar] [CrossRef] [PubMed]
- Akagi, Y.; et al. Aldose reductase localization in human retinal mural cells. Invest Ophthalmol Vis Sci 1983, 24(11), 1516–9. [Google Scholar] [PubMed]
- Akagi, Y.; et al. Localization of aldose reductase in the human eye. Diabetes 1984, 33(6), 562–6. [Google Scholar] [CrossRef]
- Kador, P.F.; et al. Prevention of pericyte ghost formation in retinal capillaries of galactose-fed dogs by aldose reductase inhibitors. Arch Ophthalmol 1988, 106(8), 1099–102. [Google Scholar] [CrossRef]
- Kador, P.F.; et al. Prevention of retinal vessel changes associated with diabetic retinopathy in galactose-fed dogs by aldose reductase inhibitors. Arch Ophthalmol 1990, 108(9), 1301–9. [Google Scholar] [CrossRef]
- Takahashi, Y.; et al. Diabeteslike preproliferative retinal changes in galactose-fed dogs. Arch Ophthalmol 1992, 110(9), 1295–302. [Google Scholar] [CrossRef]
- Heesom, A.E.; Millward, A.; Demaine, A.G. Susceptibility to diabetic neuropathy in patients with insulin dependent diabetes mellitus is associated with a polymorphism at the 5’ end of the aldose reductase gene. J Neurol Neurosurg Psychiatry 1998, 64(2), 213–6. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.P.; et al. Aldose reductase (AC)(n) microsatellite polymorphism and diabetic microvascular complications in Caucasian Type 1 diabetes mellitus. Diabetes Res Clin Pract 2001, 52(1), 21–7. [Google Scholar] [CrossRef]
- Park, H.K.; et al. (AC)(n) polymorphism of aldose reductase gene and diabetic microvascular complications in type 2 diabetes mellitus. Diabetes Res Clin Pract 2002, 55(2), 151–7. [Google Scholar] [CrossRef]
- Abhary, S.; et al. A systematic meta-analysis of genetic association studies for diabetic retinopathy. Diabetes 2009, 58(9), 2137–47. [Google Scholar] [CrossRef]
- Lee, A.Y.; Chung, S.K.; Chung, S.S. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc Natl Acad Sci U S A 1995, 92(7), 2780–4. [Google Scholar] [CrossRef]
- Ramana, K.V.; et al. Aldose reductase mediates cytotoxic signals of hyperglycemia and TNF-alpha in human lens epithelial cells. FASEB J 2003, 17(2), 315–7. [Google Scholar] [CrossRef] [PubMed]
- Zatechka, D.S., Jr.; et al. Diabetes can alter the signal transduction pathways in the lens of rats. Diabetes 2003, 52(4), 1014–22. [Google Scholar] [CrossRef] [PubMed]
- Yagihashi, S.; et al. Neuropathy in diabetic mice overexpressing human aldose reductase and effects of aldose reductase inhibitor. Brain 2001, 124 Pt 12, 2448–58. [Google Scholar] [CrossRef]
- Song, Z.; et al. Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol Cell Neurosci 2003, 23(4), 638–47. [Google Scholar] [CrossRef]
- Wang, X.; et al. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem J 1998, 333 Pt 2)(Pt 2, 291–300. [Google Scholar] [CrossRef]
- Purves, T.; et al. A role for mitogen-activated protein kinases in the etiology of diabetic neuropathy. FASEB J 2001, 15(13), 2508–14. [Google Scholar] [CrossRef] [PubMed]
- Ho, E.C.; et al. Aldose reductase-deficient mice are protected from delayed motor nerve conduction velocity, increased c-Jun NH2-terminal kinase activation, depletion of reduced glutathione, increased superoxide accumulation, and DNA damage. Diabetes 2006, 55(7), 1946–53. [Google Scholar] [CrossRef]
- Kasajima, H.; et al. Enhanced in situ expression of aldose reductase in peripheral nerve and renal glomeruli in diabetic patients. Virchows Arch 2001, 439(1), 46–54. [Google Scholar] [CrossRef]
- Kapor-Drezgic, J.; et al. Effect of high glucose on mesangial cell protein kinase C-delta and -epsilon is polyol pathway-dependent. J Am Soc Nephrol 1999, 10(6), 1193–203. [Google Scholar] [CrossRef]
- Noh, H.; King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int Suppl 2007, 106, S49–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; et al. Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia 2011, 54(5), 1242–51. [Google Scholar] [CrossRef]
- Sango, K.; et al. Physiological and Pathological Roles of Aldose Reductase in Schwann Cells. Journal of Molecular and Genetic Medicine 2014, 02(s1). [Google Scholar]
- Kobayashi, T.; et al. Localization and physiological implication of aldose reductase and sorbitol dehydrogenase in reproductive tracts and spermatozoa of male rats. J Androl 2002, 23(5), 674–83. [Google Scholar] [CrossRef] [PubMed]
- Steffgen, J.; et al. Osmoregulation of aldose reductase and sorbitol dehydrogenase in cultivated interstitial cells of rat renal inner medulla. Nephrol Dial Transplant 2003, 18(11), 2255–61. [Google Scholar] [CrossRef]
- Pastel, E.; et al. Aldo-Keto Reductases 1B in Endocrinology and Metabolism. Front Pharmacol 2012, 3, 148. [Google Scholar] [CrossRef]
- Cappiello, M.; et al. Basic models for differential inhibition of enzymes. Biochem Biophys Res Commun 2014, 445(3), 556–60. [Google Scholar] [CrossRef]
- Petrash, J.M.; et al. Aldose reductase catalysis and crystallography. Insights from recent advances in enzyme structure and function. Diabetes 1994, 43(8), 955–9. [Google Scholar] [CrossRef]
- Guo, M.; et al. Role of AKR1B10 in inflammatory diseases. Scand J Immunol 2024, 100(2), p. e13390. [Google Scholar] [CrossRef]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Oxidative and reductive metabolism of lipid-peroxidation derived carbonyls. Chem Biol Interact 2015, 234, 261–73. [Google Scholar] [CrossRef]
- Endo, S.; Matsunaga, T.; Nishinaka, T. The Role of AKR1B10 in Physiology and Pathophysiology. Metabolites 2021, 11(6). [Google Scholar] [CrossRef]
- Rondeau, J.M.; et al. Novel NADPH-binding domain revealed by the crystal structure of aldose reductase. Nature 1992, 355(6359), 469–72. [Google Scholar] [CrossRef]
- Borhani, D.W.; Harter, T.M.; Petrash, J.M. The crystal structure of the aldose reductase.NADPH binary complex. J Biol Chem 1992, 267(34), 24841–7. [Google Scholar] [CrossRef]
- Wilson, D.K.; et al. An unlikely sugar substrate site in the 1.65 A structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science 1992, 257(5066), 81–4. [Google Scholar] [CrossRef]
- Biadene, M.; et al. The atomic resolution structure of human aldose reductase reveals that rearrangement of a bound ligand allows the opening of the safety-belt loop. Acta Crystallogr D Biol Crystallogr 2007, 63 Pt 6, 665–72. [Google Scholar] [CrossRef] [PubMed]
- Barski, O.A.; Tipparaju, S.M.; Bhatnagar, A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev 2008, 40(4), 553–624. [Google Scholar] [CrossRef]
- Bohren, K.M.; et al. The structure of Apo R268A human aldose reductase: hinges and latches that control the kinetic mechanism. Biochim Biophys Acta 2005, 1748(2), 201–12. [Google Scholar] [CrossRef] [PubMed]
- Urzhumtsev, A.; et al. A ‘specificity’ pocket inferred from the crystal structures of the complexes of aldose reductase with the pharmaceutically important inhibitors tolrestat and sorbinil. Structure 1997, 5(5), 601–12. [Google Scholar] [CrossRef] [PubMed]
- Sotriffer, C.A.; Kramer, O.; Klebe, G. Probing flexibility and “induced-fit” phenomena in aldose reductase by comparative crystal structure analysis and molecular dynamics simulations. Proteins 2004, 56(1), 52–66. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Kramer, O.; Sotriffer, C. Strategies for the design of inhibitors of aldose reductase, an enzyme showing pronounced induced-fit adaptations. Cell Mol Life Sci 2004, 61(7-8), 783–93. [Google Scholar] [CrossRef] [PubMed]
- El-Kabbani, O.; et al. Ultrahigh resolution drug design. II. Atomic resolution structures of human aldose reductase holoenzyme complexed with Fidarestat and Minalrestat: implications for the binding of cyclic imide inhibitors. Proteins 2004, 55(4), 805–13. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.K.; et al. Refined 1.8 A structure of human aldose reductase complexed with the potent inhibitor zopolrestat. Proc Natl Acad Sci U S A 1993, 90(21), 9847–51. [Google Scholar] [CrossRef]
- Steuber, H.; et al. Merging the binding sites of aldose and aldehyde reductase for detection of inhibitor selectivity-determining features. J Mol Biol 2008, 379(5), 991–1016. [Google Scholar] [CrossRef]
- Steuber, H.; et al. Expect the unexpected or caveat for drug designers: multiple structure determinations using aldose reductase crystals treated under varying soaking and co-crystallisation conditions. J Mol Biol 2006, 363(1), 174–87. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Niu, Y. Inhibitory selectivity to the AKR1B10 and aldose reductase (AR): insight from molecular dynamics simulations and free energy calculations. RSC Adv 2023, 13(38), 26709–26718. [Google Scholar] [CrossRef]
- Ruiz, F.X.; Pares, X.; Farres, J. Perspective on the Structural Basis for Human Aldo-Keto Reductase 1B10 Inhibition. Metabolites 2021, 11(12). [Google Scholar] [CrossRef]
- Tarle, I.; et al. Probing the active site of human aldose reductase. Site-directed mutagenesis of Asp-43, Tyr-48, Lys-77, and His-110. J Biol Chem 1993, 268(34), 25687–93. [Google Scholar] [CrossRef]
- Kanazu, T.; et al. Aldehyde reductase is a major protein associated with 3-deoxyglucosone reductase activity in rat, pig and human livers. Biochem J 1991, 279 Pt 3)(Pt 3, 903–6. [Google Scholar] [CrossRef]
- Oya, T.; et al. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J Biol Chem 1999, 274(26), 18492–502. [Google Scholar] [CrossRef]
- Harrison, D.H.; et al. An anion binding site in human aldose reductase: mechanistic implications for the binding of citrate, cacodylate, and glucose 6-phosphate. Biochemistry 1994, 33(8), 2011–20. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.H.; et al. The alrestatin double-decker: binding of two inhibitor molecules to human aldose reductase reveals a new specificity determinant. Biochemistry 1997, 36(51), 16134–40. [Google Scholar] [CrossRef]
- Ramunno, A.; et al. Progresses in the pursuit of aldose reductase inhibitors: the structure-based lead optimization step. Eur J Med Chem 2012, 51, 216–26. [Google Scholar] [CrossRef]
- Rojas, J.J.; et al. Computational screening identifies selective aldose reductase inhibitors with strong efficacy and limited off target interactions. Sci Rep 2025, 15(1), 28111. [Google Scholar] [CrossRef]
- Gabbay, K.H.; et al. Aldose reductase inhibition: studies with alrestatin. Metabolism 1979, 28 (4 Suppl 1), 471–6. [Google Scholar] [CrossRef]
- Judzewitsch, R.G.; et al. Aldose reductase inhibition improves nerve conduction velocity in diabetic patients. N Engl J Med 1983, 308(3), 119–25. [Google Scholar] [CrossRef]
- Goto, Y.; et al. A placebo-controlled double-blind study of epalrestat (ONO-2235) in patients with diabetic neuropathy. Diabet Med 1993, 10 Suppl 2, 39S–43S. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; et al. Effects of an aldose reductase inhibitor, epalrestat, on diabetic neuropathy. Clinical benefit and indication for the drug assessed from the results of a placebo-controlled double-blind study. Biomed Pharmacother 1995, 49(6), 269–77. [Google Scholar] [CrossRef]
- Hotta, N.; et al. Clinical investigation of epalrestat, an aldose reductase inhibitor, on diabetic neuropathy in Japan: multicenter study. Diabetic Neuropathy Study Group in Japan. J Diabetes Complications 1996, 10(3), 168–72. [Google Scholar] [CrossRef]
- Hotta, N.; et al. Clinical efficacy of fidarestat, a novel aldose reductase inhibitor, for diabetic peripheral neuropathy: a 52-week multicenter placebo-controlled double-blind parallel group study. Diabetes Care 2001, 24(10), 1776–82. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; et al. Fidarestat (SNK-860), a potent aldose reductase inhibitor, normalizes the elevated sorbitol accumulation in erythrocytes of diabetic patients. J Diabetes Complications 2002, 16(2), 133–8. [Google Scholar] [CrossRef] [PubMed]
- Bril, V.; et al. Ranirestat for the management of diabetic sensorimotor polyneuropathy. Diabetes Care 2009, 32(7), 1256–60. [Google Scholar] [CrossRef]
- Perfetti, R.; et al. Safety, Pharmacokinetics, and Pharmacodynamics of the New Aldose Reductase Inhibitor Govorestat (AT-007) After a Single and Multiple Doses in Participants in a Phase 1/2 Study. J Clin Pharmacol 2024, 64(11), 1397–1406. [Google Scholar] [CrossRef]
- Imran, A.; et al. Development and exploration of novel substituted thiosemicarbazones as inhibitors of aldose reductase via in vitro analysis and computational study. Sci Rep 2022, 12(1), p. 5734. [Google Scholar] [CrossRef]
- Zhang, L.; et al. Inhibitor selectivity between aldo-keto reductase superfamily members AKR1B10 and AKR1B1: role of Trp112 (Trp111). FEBS Lett 2013, 587(22), 3681–6. [Google Scholar] [CrossRef]
- Fujii, J.; et al. Pleiotropic Actions of Aldehyde Reductase (AKR1A). Metabolites 2021, 11(6). [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; et al. Discovery of new selective human aldose reductase inhibitors through virtual screening multiple binding pocket conformations. J Chem Inf Model 2013, 53(9), 2409–22. [Google Scholar] [CrossRef]
- Eisenmann, M.; et al. Structure-based optimization of aldose reductase inhibitors originating from virtual screening. ChemMedChem 2009, 4(5), 809–19. [Google Scholar] [CrossRef]
- Bon, M.; et al. Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol Oncol 2022, 16(21), 3761–3777. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; et al. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov 2016, 15(9), 605–619. [Google Scholar] [CrossRef] [PubMed]
- Schuffenhauer, A.; et al. Library design for fragment based screening. Curr Top Med Chem 2005, 5(8), 751–62. [Google Scholar] [CrossRef]
- Jacquemard, C.; Kellenberger, E. A bright future for fragment-based drug discovery: what does it hold? Expert Opin Drug Discov 2019, 14(5), 413–416. [Google Scholar] [CrossRef]
- Kirsch, P.; et al. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24(23). [Google Scholar] [CrossRef]
- Davis, B.J.; Giannetti, A.M. The Synthesis of Biophysical Methods In Support of Robust Fragment-Based Lead Discovery, in Fragment-based Drug Discovery Lessons and Outlook; 2016; pp. 119–138. [Google Scholar]
- Bancet, A.; et al. Fragment Linking Strategies for Structure-Based Drug Design. J Med Chem 2020, 63(20), 11420–11435. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, T.; et al. How to Find a Fragment: Methods for Screening and Validation in Fragment-Based Drug Discovery. ChemMedChem 2024, 19(24), p. e202400342. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, L.; et al. Integrated Strategy for Lead Optimization Based on Fragment Growing: The Diversity-Oriented-Target-Focused-Synthesis Approach. J Med Chem 2018, 61(13), 5719–5732. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; et al. Identification of novel lysine demethylase 5-selective inhibitors by inhibitor-based fragment merging strategy. Bioorg Med Chem 2019, 27(6), 1119–1129. [Google Scholar] [CrossRef]
- Keseru, G.M.; et al. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia. J Med Chem 2016, 59(18), 8189–206. [Google Scholar] [CrossRef]
- Yamaotsu, N.; Hirono, S. In silico fragment-mapping method: a new tool for fragment-based/structure-based drug discovery. J Comput Aided Mol Des 2018, 32(11), 1229–1245. [Google Scholar] [CrossRef]
- Zhu, T.; et al. Hit identification and optimization in virtual screening: practical recommendations based on a critical literature analysis. J Med Chem 2013, 56(17), 6560–72. [Google Scholar] [CrossRef]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med Chem Lett 2010, 1(1), 44–8. [Google Scholar] [CrossRef]
- Murray, J.B.; et al. Off-rate screening (ORS) by surface plasmon resonance. An efficient method to kinetically sample hit to lead chemical space from unpurified reaction products. J Med Chem 2014, 57(7), 2845–50. [Google Scholar] [CrossRef]
- Neumann, T.; et al. SPR-based fragment screening: advantages and applications. Curr Top Med Chem 2007, 7(16), 1630–42. [Google Scholar] [CrossRef]
- Chavanieu, A.; Pugniere, M. Developments in SPR Fragment Screening. Expert Opin Drug Discov 2016, 11(5), 489–99. [Google Scholar] [CrossRef]
- Senisterra, G.; Chau, I.; Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev Technol 2012, 10(2), 128–36. [Google Scholar] [CrossRef]
- Lo, M.C.; et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem 2004, 332(1), 153–9. [Google Scholar] [CrossRef] [PubMed]
- Jerabek-Willemsen, M.; et al. MicroScale Thermophoresis: Interaction analysis and beyond. Journal of Molecular Structure 2014, 1077, 101–113. [Google Scholar] [CrossRef]
- Mueller, A.M.; et al. MicroScale Thermophoresis: A Rapid and Precise Method to Quantify Protein-Nucleic Acid Interactions in Solution. Methods Mol Biol 2017, 1654, 151–164. [Google Scholar] [PubMed]
- Asmari, M.; et al. Thermophoresis for characterizing biomolecular interaction. Methods 2018, 146, 107–119. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem 2017, 61(5), 453–464. [Google Scholar] [PubMed]
- Linke, P.; et al. An Automated Microscale Thermophoresis Screening Approach for Fragment-Based Lead Discovery. J Biomol Screen 2016, 21(4), 414–21. [Google Scholar] [CrossRef]
- Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-based screening by protein crystallography: successes and pitfalls. Int J Mol Sci 2012, 13(10), 12857–79. [Google Scholar] [CrossRef]
- Fragment-Based Drug Discovery and X-Ray Crystallography. In Topics in Current Chemistry; 2012.
- Davies, T.G.; Tickle, I.J. Fragment Screening Using X-Ray Crystallography, in Fragment-Based Drug Discovery and X-Ray Crystallography; Davies, T.G., Hyvönen, M., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp. 33–59. [Google Scholar]
- Muller, I. Guidelines for the successful generation of protein-ligand complex crystals. Acta Crystallogr D Struct Biol 2017, 73 Pt 2, 79–92. [Google Scholar] [CrossRef]
- Cusack, S.; et al. Small is beautiful: protein micro-crystallography. In Nat Struct Biol; 1998; pp. 634–7. [Google Scholar]
- Wolff, A.M.; et al. Comparing serial X-ray crystallography and microcrystal electron diffraction (MicroED) as methods for routine structure determination from small macromolecular crystals. IUCrJ 2020. 7, Pt 2, 306–323. [Google Scholar] [CrossRef]
- Nannenga, B.L.; et al. Structure of catalase determined by MicroED. Elife 2014, 3, p. e03600. [Google Scholar] [CrossRef]
- Yonekura, K.; et al. Electron crystallography of ultrathin 3D protein crystals: atomic model with charges. Proc Natl Acad Sci U S A 2015, 112(11), 3368–73. [Google Scholar] [CrossRef] [PubMed]
- Clabbers, M.T.B.; et al. Protein structure determination by electron diffraction using a single three-dimensional nanocrystal. Acta Crystallogr D Struct Biol 2017, 73 Pt 9, 738–748. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, M.J.; et al. Atomic-resolution structures from fragmented protein crystals with the cryoEM method MicroED. Nat Methods 2017, 14(4), 399–402. [Google Scholar] [CrossRef]
- Xu, H.; et al. A Rare Lysozyme Crystal Form Solved Using Highly Redundant Multiple Electron Diffraction Datasets from Micron-Sized Crystals. Structure 2018, 26(4), 667–675 e3. [Google Scholar] [CrossRef]
- Henderson, R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys 1995, 28(2), 171–93. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; et al. Single particle cryo-EM reconstruction of 52 kDa streptavidin at 3.2 Angstrom resolution. Nat Commun 2019, 10(1), p. 2386. [Google Scholar] [CrossRef]
- Purdy, M.D.; et al. MicroED structures of HIV-1 Gag CTD-SP1 reveal binding interactions with the maturation inhibitor bevirimat. Proc Natl Acad Sci U S A 2018, 115(52), 13258–13263. [Google Scholar] [CrossRef]
- Clabbers, M.T.B.; et al. Visualizing drug binding interactions using microcrystal electron diffraction. Commun Biol 2020, 3(1), 417. [Google Scholar] [CrossRef]
- Clabbers, M.T.B.; Xu, H. Microcrystal electron diffraction in macromolecular and pharmaceutical structure determination. Drug Discov Today Technol 2020, 37, 93–105. [Google Scholar] [CrossRef]
- Shi, D.; et al. Three-dimensional electron crystallography of protein microcrystals. Elife 2013, 2, p. e01345. [Google Scholar] [CrossRef]
- Liu, S.; Gonen, T. MicroED structure of the NaK ion channel reveals a Na(+) partition process into the selectivity filter. Commun Biol 2018, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, C.E. Aldose reductase: model for a new paradigm of enzymic perfection in detoxification catalysts. Biochemistry 1992, 31(42), 10139–45. [Google Scholar] [CrossRef]
- de Souza Neto, L.R.; et al. In silico Strategies to Support Fragment-to-Lead Optimization in Drug Discovery. Front Chem 2020, 8, 93. [Google Scholar] [CrossRef]
- Strecker, C.; et al. Fragment Growing to Design Optimized Inhibitors for Human Blood Group B Galactosyltransferase (GTB). ChemMedChem 2019, 14(14), 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; et al. Impact of linker strain and flexibility in the design of a fragment-based inhibitor. Nat Chem Biol 2009, 5(6), 407–13. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, O.; et al. Compound Design by Fragment-Linking. Mol Inform 2011, 30(4), 298–306. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; et al. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 2002, 45(12), 2615–23. [Google Scholar] [CrossRef]
- Xu, X.; et al. Identification of novel ROS inducer by merging the fragments of piperlongumine and dicoumarol. Bioorg Med Chem Lett 2017, 27(5), 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, P.O.; et al. A fragment merging approach towards the development of small molecule inhibitors of Mycobacterium tuberculosis EthR for use as ethionamide boosters. Org Biomol Chem 2016, 14(7), 2318–26. [Google Scholar] [CrossRef]
- Friberg, A.; et al. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem 2013, 56(1), 15–30. [Google Scholar] [CrossRef]
- Fesik, S.W. Drugging Challenging Cancer Targets Using Fragment-Based Methods. Chem Rev 2025, 125(6), 3586–3594. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.M.; Teague, S.J.; Kleywegt, G.J. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew Chem Int Ed Engl 2003, 42(24), 2718–36. [Google Scholar] [CrossRef]
- Holcomb, J.; et al. Protein crystallization: Eluding the bottleneck of X-ray crystallography. AIMS Biophys 2017, 4(4), 557–575. [Google Scholar] [CrossRef]
- Kascakova, B.; et al. Revealing protein structures: crystallization of protein-ligand complexes - co-crystallization and crystal soaking. FEBS Open Bio 2025, 15(4), 542–550. [Google Scholar] [CrossRef]
- Tremlett, C.J.; et al. Small but mighty: the power of microcrystals in structural biology. IUCrJ 2025, 12 Pt 3, 262–279. [Google Scholar] [CrossRef]
- Oberthur, D. Microcrystals in structural biology: small samples, big insights. IUCrJ 2025, 12 Pt 3, 259–261. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Gonen, T. The cryo-EM method microcrystal electron diffraction (MicroED). Nat Methods 2019, 16(5), 369–379. [Google Scholar] [CrossRef] [PubMed]
- Boudes, M. D. Garriga, and F. Coulibaly, Reflections on the Many Facets of Protein Microcrystallography. Australian Journal of Chemistry 2014, 67(12). [Google Scholar] [CrossRef]
- Fearon, D.; et al. Accelerating Drug Discovery With High-Throughput Crystallographic Fragment Screening and Structural Enablement. Applied Research 2024, 4(1). [Google Scholar] [CrossRef]
- Acehan, D.; et al. Reaching the potential of electron diffraction. Cell Rep Phys Sci 2024, 5(6). [Google Scholar] [CrossRef] [PubMed]
- Bochtler, M. X-rays, electrons, and neutrons as probes of atomic matter. Structure 2024, 32(5), 630–643 e6. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Gonen, T. MicroED: a versatile cryoEM method for structure determination. Emerg Top Life Sci 2018, 2(1), 1–8. [Google Scholar] [PubMed]
- Meyer, A.; et al. Single-drop optimization of protein crystallization. Acta Crystallogr Sect F Struct Biol Cryst Commun 2012, 68 (Pt 8), 994–8. [Google Scholar] [CrossRef]
- Duyvesteyn, H.M.E.; et al. Machining protein microcrystals for structure determination by electron diffraction. Proc Natl Acad Sci U S A 2018, 115(38), 9569–9573. [Google Scholar] [CrossRef]
- Martynowycz, M.W.; et al. Collection of Continuous Rotation MicroED Data from Ion Beam-Milled Crystals of Any Size. Structure 2019, 27(3), 545–548 e2. [Google Scholar] [CrossRef]
- Guha, R. On exploring structure-activity relationships. Methods Mol Biol 2013, 993, 81–94. [Google Scholar] [PubMed]
Figure 1.
The polyol pathway (In conditions of hyperglycemia, as occurring in diabetes, glucose underwent to the polyol pathway to be converted to fructose through the consecutive action of Aldose Reductase (AR) and sorbitol dehydrogenase (SDH).
Figure 1.
The polyol pathway (In conditions of hyperglycemia, as occurring in diabetes, glucose underwent to the polyol pathway to be converted to fructose through the consecutive action of Aldose Reductase (AR) and sorbitol dehydrogenase (SDH).
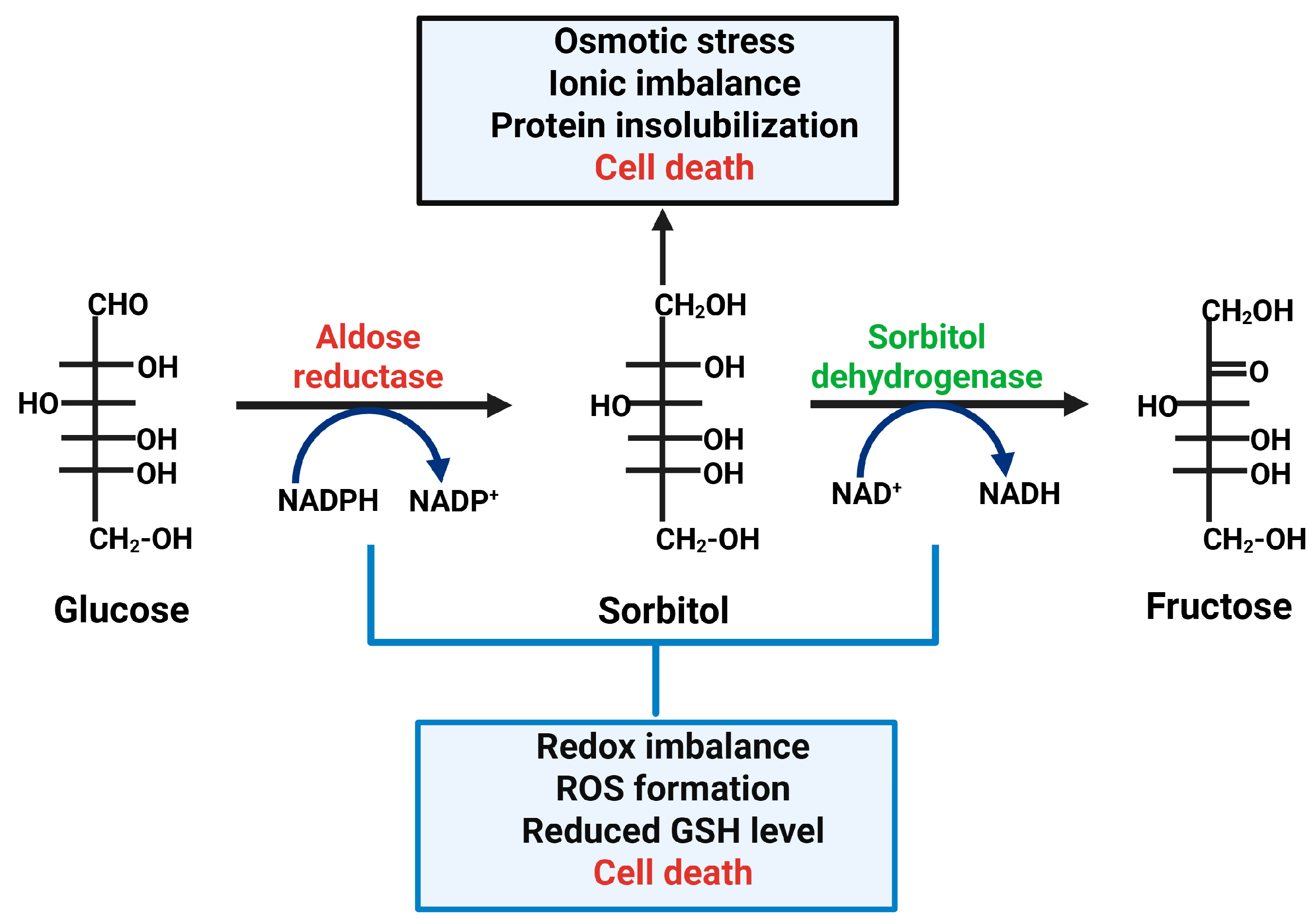
Figure 2.
Schematic view of the binding pocket of Aldose Reductase (AR). The central insert depicts the AR structure, as a cartoon representation (PDB 3S3G).
Figure 2.
Schematic view of the binding pocket of Aldose Reductase (AR). The central insert depicts the AR structure, as a cartoon representation (PDB 3S3G).
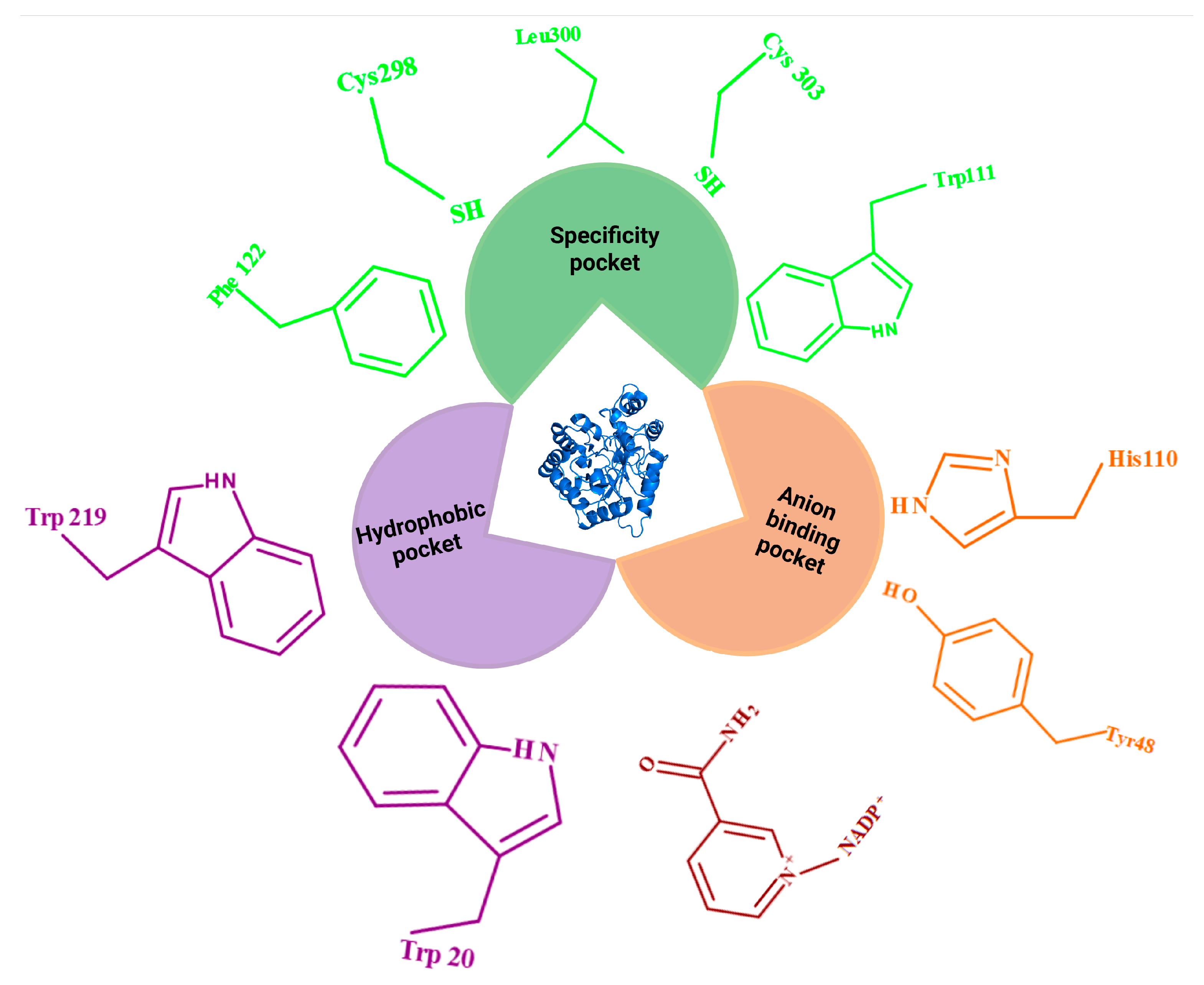
Figure 3.
Workflow of FBDD and structural validation.
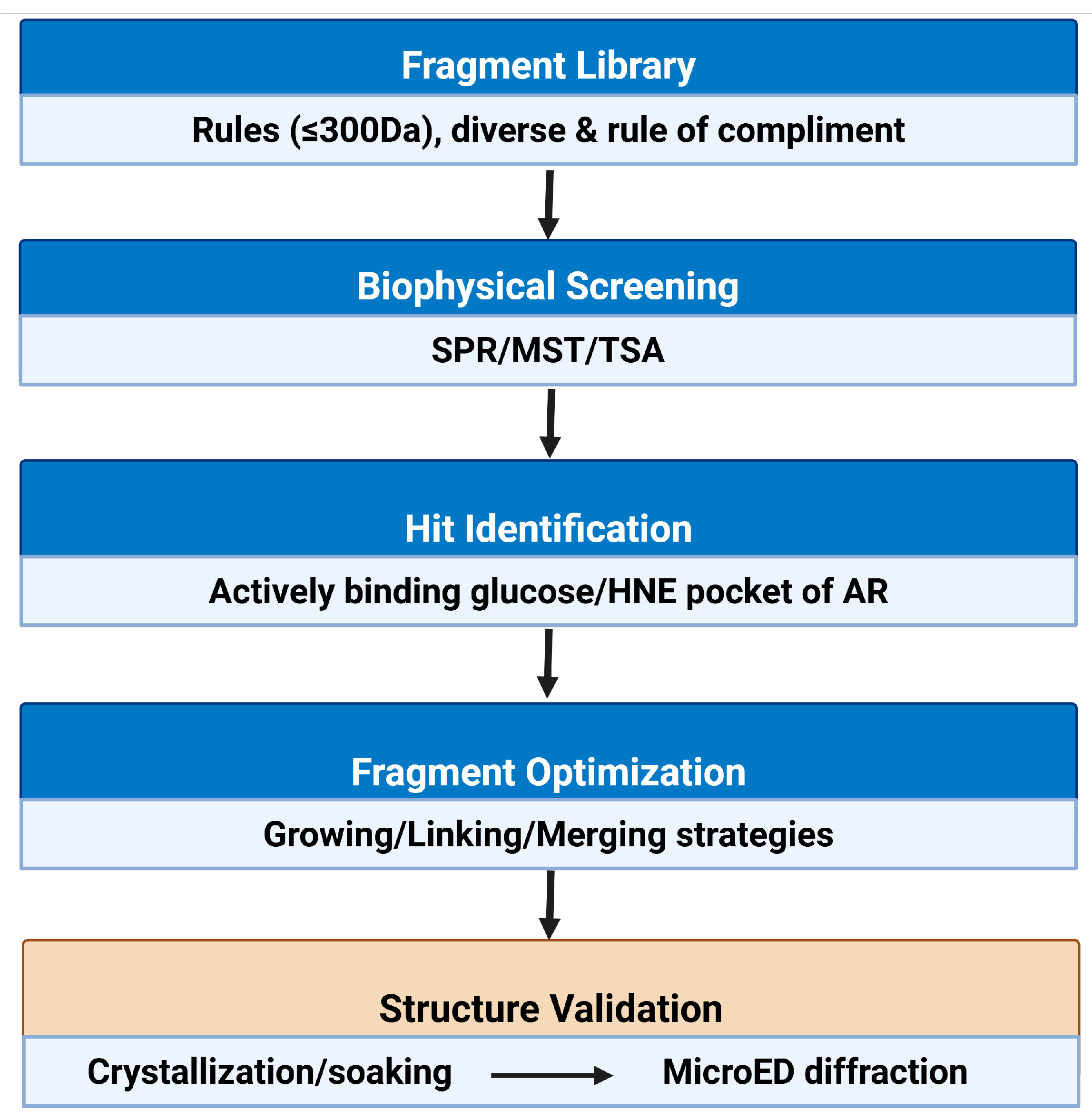
Figure 4.
Schematic representation of fragment optimization to structure validation.
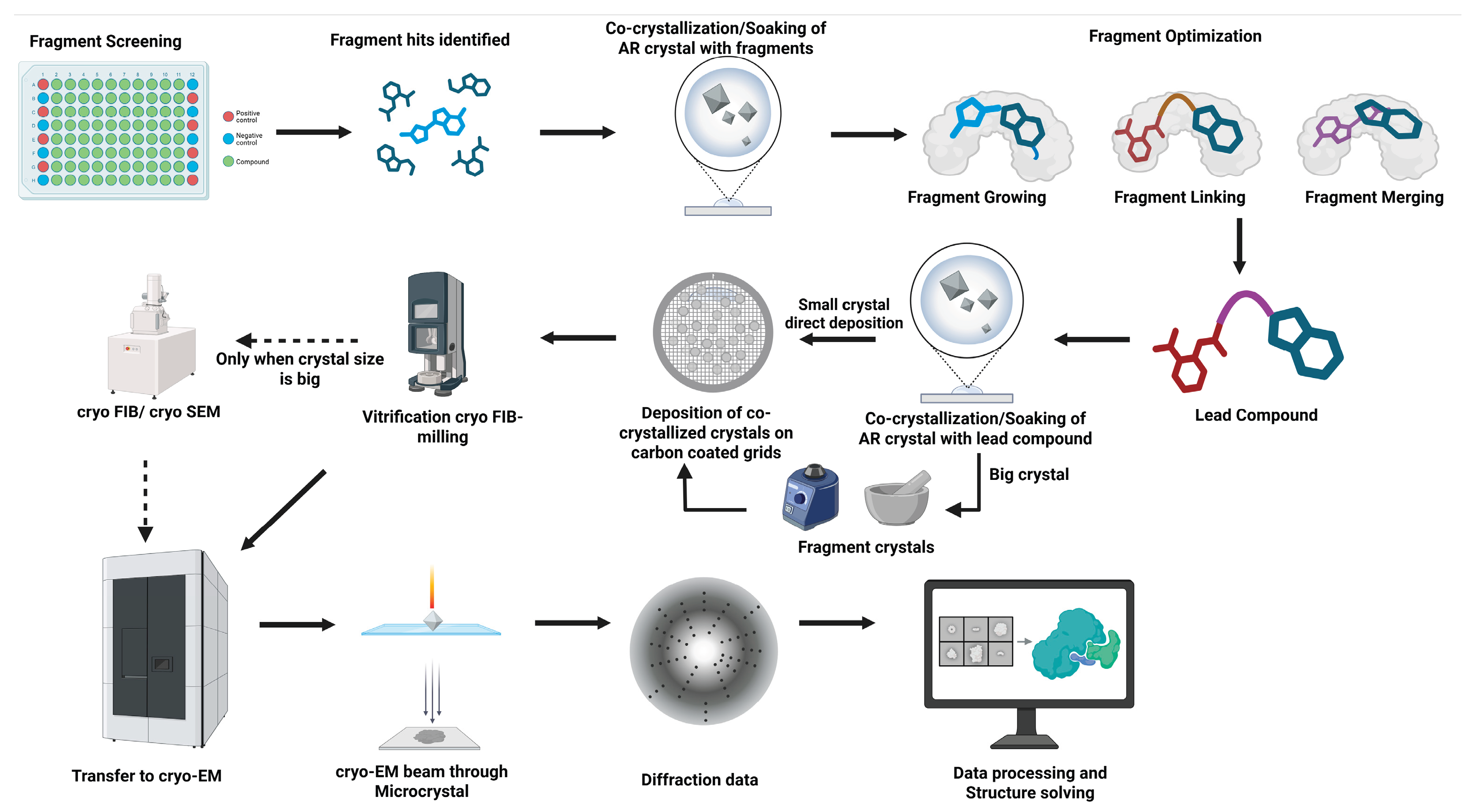
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



