
Submitted:
15 December 2025
Posted:
16 December 2025
You are already at the latest version
Abstract
Background: Prader-Willi syndrome (PWS) is a multisystemic complex imprinting disorder. Prenatal diagnosis of PWS is still a challenge with non-specific ultrasound markers and limitations for diagnosis with non-invasive screening methods. Prenatal suspicion and early postnatal diagnosis are mandatory for promoting healthy growth and development, preventing complications, and providing health care professionals and families with the necessary support and resources for effective management.
Presentation: We report two PWS cases caused by maternal uniparental disomy, who presented with IUGR, specifically reduced fetal abdominal circumference (AC) during the second and early third trimesters against the background of reduced fetal move-ments, normal Doppler indicators and oligohydramnios. They were diagnosed in the early neonatal period with no prenatal suspicion but with similar ultrasound markers of the developing pregnancies, analyzed retrospectively.
Aim: The aim of this study is to emphasise the need to raise awareness among specialists about genetic syndromes such as Prader–Willi syndrome, in order to improve the information provided to couples regarding the limitations of current prenatal screening methods, as well as to ensure that, in cases of prenatal suspicion, appropriate genetic testing can be initiated. A confirmed diagnosis would allow timely and adequate measures to be taken, given the complications of the postnatal period in these patients and their need for specialised care and management
Conclusions: The presence of the aforementioned prenatal characteristics may raise suspicion for PWS. In such cases, invasive diagnostic procedures and methylation testing may be indicated, enabling earlier diagnosis and timely management, which can ultimately improve the quality of life of affected individuals and their families.
Keywords:
Prader–Willi syndrome
; prenatal diagnosis
; intrauterine growth restriction
; reduced fetal movements
; imprinting disorders
; methylation analysis
1. Introduction
PWS was first described by Langdon Down in 1887, but it was not described in the medical literature until 1956, when Prader, Labhart, and Willi reported 9 cases (5 boys and 4 girls, aged 5-23) with similar clinical symptoms [1,2]. It has been recognized nowadays as a genetic imprinting disorder with birth incidence 1/15,000 – 25,000 worldwide (3.1 per 100,000 live births in Europe) which affects males and females equally. There are three main genetic subtypes in PWS: paternal 15q11-q13 deletion (65–75% of cases), maternal uniparental disomy 15 (20–30% of cases), and imprinting defect (1–3%) [3]. DNA methylation analysis is the only technique that will diagnose PWS in all three molecular genetic classes and differentiate PWS from Angelman syndrome. Unfortunately, prenatal detection of PWS is still lacking, because of non-specific criteria from morphology scans and non-invasive testing to indicate invasive procedure with methylation analysis. There is a need to raise awareness among healthcare providers regarding the potential clinical signs that lead to suspicion of underling genetic condition in certain pregnancy complications during the late second and third trimester [2,4,5].
Analyses have identified significant discrepancies in both genotypes and phenotypes in PWS cases resulting from maternal uniparental disomy compared with those with deletions of chromosomal regions, demonstrating less typical facial phenotypes, shorter duration of tube feeding, later onset of hyperphagia, significantly higher birth weight, and milder degree of hypotonia. The frequency of chromosome 15 nondisjunction may increase exponentially with maternal age. Children of older mothers have an increased rate of maternal uniparental disomy of chromosome 15 compared with those born from mothers younger than 35 years at birth [6,7].
PWS is difficult to diagnose prenatally due to the lack of accurate and well-characterized fetal phenotypes that would otherwise provide the basis for further molecular genetic studies. To date, only a few reports of diagnosis of PWS during pregnancy have been published [3,4,5,8]. Most of them describe nonspecific prenatal signs suggestive of PWS. They are incidentally identified as trisomy 15 during invasive diagnosis by chorionic villus sampling or amniocentesis for other established abnormalities during pregnancy. Prenatal diagnosis of cases of paternal translocation of chromosome 15 is more commonly described [8] also increased nuchal translucency and abnormal serum maternal screening in first trimester, which are indicative for an invasive procedure may be associated with PWS, incidentally diagnosed [3].
Reported prenatal findings associated with PWS are non-specific with late onset in second half of pregnancy which include abnormal position of feet and toes [4], polyhydramnios [4,8,9], decreased fetal activity [4,10], hypoplasia of external genitalia [11], breech position [4], diminished fetal movement [4,8], intrauterine growth restriction (IUGR) [8,12], facial dysmorphism [4], and increased head to abdominal circumference and decreased abdominal circumference [4].
We present two cases of postnatal diagnosis of PWS in newborns following normal non-invasive prenatal testing results, including exclusion of PWS deletion type, as well as normal fetal morphology scans. Retrospective analyses of the ultrasound protocols during pregnancies found suspicion for non-classical and non-characteristic signs in both pregnancies, described in the late third trimester.
The purpose is to increase awareness among obstetricians and gynaecologists that rare imprinting syndromes like PWS may not be reliably detected or excluded by current prenatal testing and fetal morphology scans, and parents should be appropriately counselled.
2. Case Presentation
2.1. Case 1
A 43-year-old primiparous woman was admitted for an elective cesarean section (CS) following a high-risk pregnancy. This was her fourth pregnancy achieved through in vitro fertilization (IVF), with a history of three previous pregnancy losses: one spontaneous abortion, one missed abortion, and one termination due to fetal diagnosis of Down syndrome.
Her obstetric history was notable for two prior myomectomies and four hysteroscopic procedures. The current pregnancy progressed uneventfully. Routine fetal morphology assessments and basic non-invasive prenatal testing (NIPT) showed no abnormalities. Non-stress test (NST) maintained normal variability and reactivity throughout, and amniotic liquid remained within physiological limits.
However, both second and third trimester ultrasound examinations consistently reported difficulty visualizing the fetal profile and genitalia due to persistent fetal positioning.
At 35 weeks of gestation, due to the onset of genital bleeding and signs of active labor, the patient underwent cesarean delivery. A live male infant was delivered, with birth weight 2810 grams and birth length 48 cm. While initially appearing full-term and stable, the newborn was found to have bilateral cryptorchidism, without any other overt congenital anomalies (Figure 1 and Figure 2).
On the second day of life, the neonate exhibited progressive muscular hypotonia and feeding difficulties (Figure 3). A pediatric consultation raised clinical suspicion for PWS. The infant was subsequently referred for comprehensive medical-genetic evaluation and counseling, and the diagnosis was confirmed for maternal uniparental disomy.
2.2. Case 2
A multiparous woman was admitted for cesarean section at 36 weeks of gestation. Indications for early operative delivery included a history of previous cesarean section, oligohydramnios, and intrauterine growth restriction (IUGR) noted since the 20th week of gestation. Additionally, non-stress test (NST) revealed reduced to absent variability and reactivity, despite normal Doppler flow measurements.
Following delivery, the newborn (birth weight 2225 g, birth length 46 cm) exhibited signs of respiratory compromise, including acrocyanosis, tachydyspnea, increased oxygen requirements and feeding difficulties. Neuromuscular assessment revealed progressive muscular hypotonia (Figure 4).
Physical examination demonstrated a combination of dysmorphic features including a narrow forehead, almond-shaped eyes, a short nose, downturned corners of the mouth, and a thin upper lip (Figure 5). A working diagnosis of PWS was established, pending confirmatory genetic testing.
In light of the clinical presentation and phenotypic characteristics, both neonates were referred to the Expert center for rare endocrine diseases at “Sveta Marina” University Hospital, Varna, Bulgaria for further diagnostic evaluation and treatment. A confirmed diagnosis of PWS allows timely and adequate measures to be taken, given the complications of the postnatal period in these patients and their need for specialized care and management.
3. Discussion
Although PWS occurs in approximately 1 in 15,000 to 1 in 30,000 live births, the syndrome is underdiagnosed or misdiagnosed in the neonatal period due to its nonspecific early manifestations [13]. This highlights the critical need for heightened clinical suspicion among obstetricians, neonatologists, and pediatricians, particularly in the presence of suggestive features such as severe hypotonia, feeding difficulties, poor weight gain despite low birth weight, and facial dysmorphisms.
The most common ultrasound feature noted in published reports is the presence of fetal growth retardation (usually abdominal circumference below the 5th percentile) and polyhydramnios, which in some cases may be severe enough to require amnioreduction [14]. This is likely due to an impaired swallowing reflex resulting from the associated skeletal muscle hypotonia. The phenotype of PWS includes persistent severe hypotonia, facial dysmorphism with a narrow bifrontal head diameter, almond-shaped eyes, short nose, down-turned corners of the mouth, thin upper lip, smoothed philtrum, lighter skin tone than the parents, acromicry with elongated and pointed fingers. Hypogonadism with micropenis, hypoplastic scrotum, and cryptorchidism in boys, and hypoplastic labia majora in girls [15].
Reduced fetal movements are reported in 4-16% of pregnancies with euploid fetuses, in most cases transient and clinically insignificant [4,15]. However, in some cases this can be a sign of congenital musculoskeletal defects or fetal distress.
Intrauterine growth restriction (IUGR) is most often due to placental insufficiency, especially with an increased AC/HC ratio and abnormal umbilical artery Doppler values. Normal Doppler values exclude placental insufficiency, and IUGR may be an indirect sign of underling genetic disease [5].
Cryptorchidism is present in 4% of healthy full-term male infants and 1–2% of healthy boys during the first year of life. It may be an isolated minor anomaly or part of various genetic and endocrine syndromes. Bilateral cryptorchidism has been reported to occur in almost all infants with PWS [15]. Screening for cryptorchidism is recommended in the presence of other suspicious findings for fetal PWS [16].
A study of 106 families, 47 of whom had a child with PWS, aged up to 10 years was conducted. When analyzing data from ultrasound examinations during pregnancy and history taken from mothers, the authors found that reduced fetal movements, small for gestational age fetuses (SGA), asymmetric retardation (increased HC/AC ratio), and polyhydramnios were found in 88%, 65%, 43% and 34%, respectively. No concomitant morphological defects were found. The authors recommend that prenatal genetic screening for PWS by methylation testing may be recommended in the combination of polyhydramnios, SGA, or asymmetric growth retardation with normal Doppler findings [5].
The early recognition of PWS has substantial implications for patient outcomes. In the early neonatal period, impaired respiratory control in PWS is believed to arise from a combination of hypothalamic dysfunction, immature brainstem respiratory circuits, blunted chemosensitivity and generalized hypotonia. Loss of paternally expressed genes in the 15q11–q13 region—whether due to paternal deletion or UPD—disrupts the development and modulation of central respiratory networks, leading to reduced ventilatory responses to both hypercapnia and hypoxia [17,18]. Clinically, neonates with PWS may present with central apnoeas, hypoventilation and oxygen desaturations [19,20]. Several reports describe newborns requiring supplemental oxygen due to persistent hypoxaemia or central apnoeas [21,22]. Case series show that low-flow oxygen can reduce central apnoeic events and improve oxygenation without worsening hypoventilation in infants with PWS, suggesting that oxygen therapy may serve as a supportive intervention until respiratory control matures, which typically improves over the first months of life [22,23]. Together, this evidence indicates that neonatal respiratory instability in PWS reflects a primary disturbance of central ventilatory regulation with impaired chemoreflexes, for which careful monitoring—and, in selected neonates, supplemental oxygen—may be beneficial. This approach was also applied in our two patients after their referral to an Expert Center (initially in the intensive care unit, and subsequently in the regular ward).
Other supportive care, needed in the neonatal period are targeted nutritional management and early physiotherapy. In many neonates with Prader–Willi syndrome (PWS), severe hypotonia and poor suck–swallow coordination often require temporary nasogastric tube feeding to ensure adequate caloric intake and avoid prolonged failure to thrive [24]. Early, closely supervised tube feeding also supports a smoother transition to oral feeding as oromotor function improves [25]. Given the profound axial hypotonia and delayed motor milestones typical of PWS, early physiotherapy focusing on postural control and antigravity activation is strongly recommended [26]. Structured early motor-training programs have been shown to advance acquisition of sitting and walking, particularly when combined with growth hormone therapy [27]. Early initiation of recombinant human growth hormone (rhGH) therapy, after appropriate clinical assessment, is increasingly recognized as a key component of comprehensive early care. Studies in infants and toddlers demonstrate that rhGH started within the first years of life improves linear growth and body composition and accelerates motor development [25,28]. In the two presented patients, all necessary measures were taken in a timely manner, resulting in favourable outcomes in terms of their physical and neuropsychological development. This further emphasises the importance of raising awareness about rare imprinting syndromes already at the stage of early prenatal diagnostics and allows timely referral to multidisciplinary care teams and genetic counseling services, which are essential for early-intervention model and long-term management.
4. Conclusions
Prenatal diagnosis of Prader–Willi syndrome remains challenging, particularly in cases caused by maternal uniparental disomy, where classical structural anomalies and abnormal non-invasive prenatal screening results are often absent. The two presented cases highlight that subtle and non-specific prenatal findings—such as intrauterine growth restriction with predominant reduction of abdominal circumference, reduced fetal movements, abnormal fetal positioning, altered amniotic fluid volume, and normal Doppler indices—may represent early indicators of an underlying imprinting disorder.
Although prenatal suspicion was not established, retrospective analysis of fetal ultrasound examinations revealed overlapping patterns that could have prompted consideration of Prader–Willi syndrome. Early postnatal recognition enabled timely genetic confirmation and referral to a specialized multidisciplinary center, allowing prompt initiation of respiratory support, nutritional management, physiotherapy, and endocrine follow-up.
Clinical message: In pregnancies complicated by unexplained late-onset growth restriction, reduced fetal activity, and normal placental Doppler findings, clinicians should consider rare genetic imprinting syndromes such as Prader–Willi syndrome, even when routine prenatal screening results are normal. Awareness of these limitations and early use of targeted invasive diagnostics with methylation analysis may reduce diagnostic delay, facilitate early intervention, and ultimately improve long-term outcomes and quality of life for affected children and their families
Author Contributions
All authors made a significant contribution to the work reported. All authors took part in drafting, revising, or critically reviewing this article. All authors gave final approval of the version to be published and agreed on the journal to which this article was submitted. All authors have read and agreed to the published version of the manuscript.
Funding
No external funding supported this research project.
Institutional Review Board Statement
Ethical review and approval were waived for this study, as it is a retrospective case report describing routine clinical practice and anonymized patient data. Written informed consent for publication was obtained from the parents of both patients.
Informed Consent Statement
Written informed consent for publication of clinical data and images was obtained from the parents of both patients.
Data Availability Statement
All relevant data supporting the findings of this case report are contained within this manuscript. Additional details can be obtained from the corresponding author upon reasonable request.
Acknowledgments
The authors extend their deepest thanks to the patients and their families who demonstrated cooperation and bravery during their diagnostic and treatment journey.
Conflicts of Interest
The authors state they have no conflicts of interest.
Abbreviations
| AC | abdominal circumference |
| HC | head circumference |
| IUGR | intrauterine growth restriction |
| NIPT | non-invasive prenatal testing |
| NST | non-stress test |
| PWS | Prader–Willi syndrome |
| rhGH | recombinant human growth hormone |
| SGA | small for gestational age |
| UPD | uniparental disomy |
References
- Hirsch, H.J.; Eldar-Geva, T.; Bennaroch, F.; Pollak, Y.; Gross-Tsur, V. Sexual dichotomy of gonadal function in Prader–Willi syndrome from early infancy through the fourth decade. Hum. Reprod. 2015, 30, 2587–2596. [Google Scholar] [CrossRef]
- Butler, M.G.; Hartin, S.N.; Hossain, W.A.; Manzardo, A.M.; Kimonis, V.; Dykens, E.; Gold, J.A.; Kim, S.J.; Weisensel, N.; Tamura, R.; Miller, J.L.; Driscoll, D.J. Molecular genetic classification in Prader–Willi syndrome: A multisite cohort study. J. Med. Genet. 2019, 56, 149–153. [Google Scholar] [CrossRef]
- Chen, C.P.; Lin, M.H.; Chen, Y.Y.; Chern, S.R.; Wu, P.S.; Chen, S.W.; Wu, F.T.; Town, D.D.; Lee, M.S.; Pan, C.W.; Wang, W. Prenatal diagnosis of a 15q11.2–q14 deletion of paternal origin associated with increased nuchal translucency, mosaicism for de novo multiple unbalanced translocations and Prader–Willi syndrome. Taiwan J. Obstet. Gynecol. 2021, 60, 335–340. [Google Scholar] [CrossRef]
- Bigi, N.; Faure, J.M.; Coubes, C.; Puechberty, J.; Lefort, G.; Sarda, P.; Blanchet, P. Prader–Willi syndrome: Is there a recognizable fetal phenotype? Prenat. Diagn. 2008, 28, 796–799. [Google Scholar] [CrossRef]
- Gross, N.; Rabinowitz, R.; Gross-Tsur, V.; Hirsch, H.J.; Eldar-Geva, T. Prader–Willi syndrome can be diagnosed prenatally. Am. J. Med. Genet. A 2015, 167A, 80–85. [Google Scholar] [CrossRef]
- Fridman, C.; Koiffmann, C.P. Origin of uniparental disomy 15 in patients with Prader–Willi or Angelman syndrome. Am. J. Med. Genet. 2000, 94, 249–253. [Google Scholar] [CrossRef]
- Ye, Y.; Fu, Y.; Zhang, Z.; Ning, H.; Tao, F.; Wang, X.; Fang, Q.; Chen, Z.; Hao, X. Prenatal diagnosis of Prader–Willi syndrome via maternal UPD15 with placental mosaicism: Incidental discovery of fetal DMD carrier status. Front. Genet. 2025, 16, 1675663. [Google Scholar] [CrossRef]
- Srebnik, N.; Gross-Even Zohar, N.; Salama, A.; Sela, H.Y.; Hirsch, H.J.; Gross-Tsur, V.; Eldar-Geva, T. Recognizing the unique prenatal phenotype of Prader–Willi syndrome indicates the need for a diagnostic methylation test. Prenat. Diagn. 2020, 40, 878–884. [Google Scholar] [CrossRef]
- Adam, M.J.; Enderle, I.; Le Bouar, G.; Cabaret-Dufour, A.S.; Tardif, C.; Contin, L.; Arnaud, A.; Proisy, M.; Jaillard, S.; Pasquier, L.; Le Lous, M. Performance of diagnostic ultrasound to identify causes of hydramnios. Prenat. Diagn. 2021, 41, 111–122. [Google Scholar] [CrossRef]
- Insoft, R.M.; Hurvitz, J.; Estrella, E.; Krishnamoorthy, K.S. Prader–Willi syndrome associated with fetal goiter: A case report. Am. J. Perinatol. 1999, 16, 29–31. [Google Scholar] [CrossRef]
- Akiba, Y.; Ono, M.; Shirahashi, M.; Noda, S.; Nishijima, S.; Amagata, T.; Kusano, R.; Fuke, T.; Hayashida, S.; Ikeda, T.; Yakubo, K.; Fukuiya, T. Polyhydramnios associated with Prader–Willi syndrome. J. Obstet. Gynaecol. 2015, 35, 752–753. [Google Scholar] [PubMed]
- Dong, Y.; Liu, S.; Li, J.; Chen, Q.; Luo, J.; Li, C.; Li, H.; Qi, H.; Li, R. Possibility of early diagnosis in a fetus affected by Prader–Willi syndrome with maternal hetero-UPD15: A lesson to be learned. Mol. Med. Rep. 2019, 20, 95–102. [Google Scholar] [CrossRef]
- Naotunna, C.; Lucas-Herald, A.; Donaldson, M.; Shaikh, M. Prader–Willi syndrome: An update on the multidisciplinary approach. Paediatr. Child Health 2025, 35, 118–123. [Google Scholar] [CrossRef]
- Denizot, S.; Boscher, C.; Le Vaillant, C.; et al. Distal arthrogryposis and neonatal hypotonia: An unusual presentation of Prader–Willi syndrome. J. Perinatol. 2004, 24, 733–734. [Google Scholar] [CrossRef]
- Rodprasert, W.; Virtanen, H.E.; Mäkelä, J.A.; Toppari, J. Hypogonadism and cryptorchidism. Front. Endocrinol. 2020, 10, 906. [Google Scholar] [CrossRef]
- McCandless, S.E.; Committee on Genetics. Clinical report—Health supervision for children with Prader–Willi syndrome. Pediatrics 2011, 127, 195–204. [Google Scholar] [CrossRef]
- Gozal, D. Absent peripheral chemosensitivity in Prader–Willi syndrome. J. Appl. Physiol. 1994, 77, 2231–2236. [Google Scholar] [CrossRef]
- Arens, R.; Gozal, D.; Omlin, K.J.; et al. Hypoxic and hypercapnic ventilatory responses in Prader–Willi syndrome. J. Appl. Physiol. 1994, 77, 2224–2230. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, A.C.; Hellström, L.G.; Ritzén, E.M.; et al. Growth hormone treatment increases CO₂ response, ventilation and central inspiratory drive in children with Prader–Willi syndrome. Eur. J. Pediatr. 1999, 158, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.M.; Brouillette, R.T. Sleep and breathing in Prader–Willi syndrome. Pediatr. Pulmonol. 2002, 34, 209–217. [Google Scholar] [CrossRef]
- Urquhart, D.S.; Gulliver, T.; Williams, G.; Harris, M.A.; Blackmore, A.M. Central sleep-disordered breathing and the effect of oxygen therapy in infants with Prader–Willi syndrome. Arch. Dis. Child. 2013, 98, 592–595. [Google Scholar] [CrossRef]
- Cohen, M.; Hamilton, J.; Narang, I. Clinically important central sleep apnea in infants with Prader–Willi syndrome. PLoS ONE 2014, 9, e115011. [Google Scholar] [CrossRef]
- Tan, H.L.; Urquhart, D.S. Respiratory complications in children with Prader–Willi syndrome. Paediatr. Respir. Rev. 2017, 23, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Çizmecioğlu, F.M.; Jones, J.H.; Forsyth Paterson, W.; et al. Neonatal features of Prader–Willi syndrome: The case for making the diagnosis during the first week of life. J. Clin. Res. Pediatr. Endocrinol. 2018, 10, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Carrel, A.L.; Moerchen, V.; Myers, S.E.; Bekx, M.T.; Whitman, B.Y.; Allen, D.B. Growth hormone improves mobility and body composition in infants and toddlers with Prader–Willi syndrome. J. Pediatr. 2004, 145, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Reus, L.; Pelzer, B.J.; Otten, B.J.; Siemensma, E.P.C.; van Alfen-van der Velden, J.A.A.E.M.; Maes-Festen, D.; et al. Growth hormone combined with child-specific motor training improves motor development in infants with Prader–Willi syndrome: A randomized controlled trial. Res. Dev. Disabil. 2013, 34, 3092–3103. [Google Scholar] [CrossRef]
- Festen, D.A.M.; Wevers, M.; Lindgren, A.C.; Böhm, B.; Otten, B.J.; Wit, J.M.; et al. Mental and motor development before and during growth hormone treatment in infants and toddlers with Prader–Willi syndrome. Clin. Endocrinol. (Oxf.) 2008, 68, 919–925. [Google Scholar] [CrossRef]
- Donze, S.H.; Damen, L.; Mahabier, E.F.; Hokken-Koelega, A.C.S. Improved mental and motor development during 3 years of growth hormone treatment in very young children with Prader–Willi syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 3714–3719. [Google Scholar] [CrossRef]
Figure 1.
Facial features of the male neonate (Case 1) on the second day of life, showing increased interorbital distance and mild facial dysmorphism, which raised clinical suspicion of an underlying genetic syndrome.
Figure 1.
Facial features of the male neonate (Case 1) on the second day of life, showing increased interorbital distance and mild facial dysmorphism, which raised clinical suspicion of an underlying genetic syndrome.
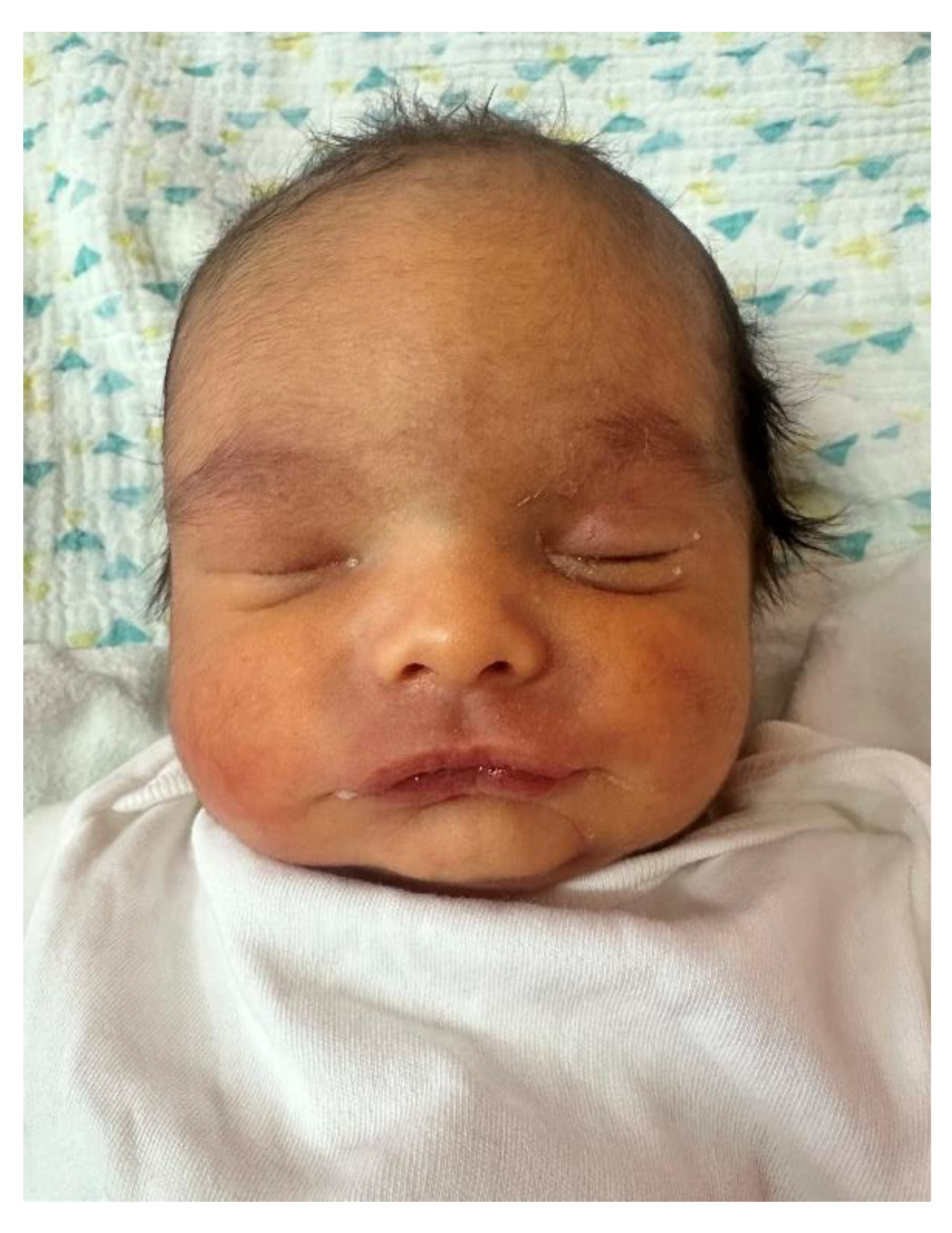
Figure 2.
Lateral facial profile of the male neonate (Case 1) demonstrating a recessed chin and retrognathic appearance observed during postnatal clinical examination.
Figure 2.
Lateral facial profile of the male neonate (Case 1) demonstrating a recessed chin and retrognathic appearance observed during postnatal clinical examination.
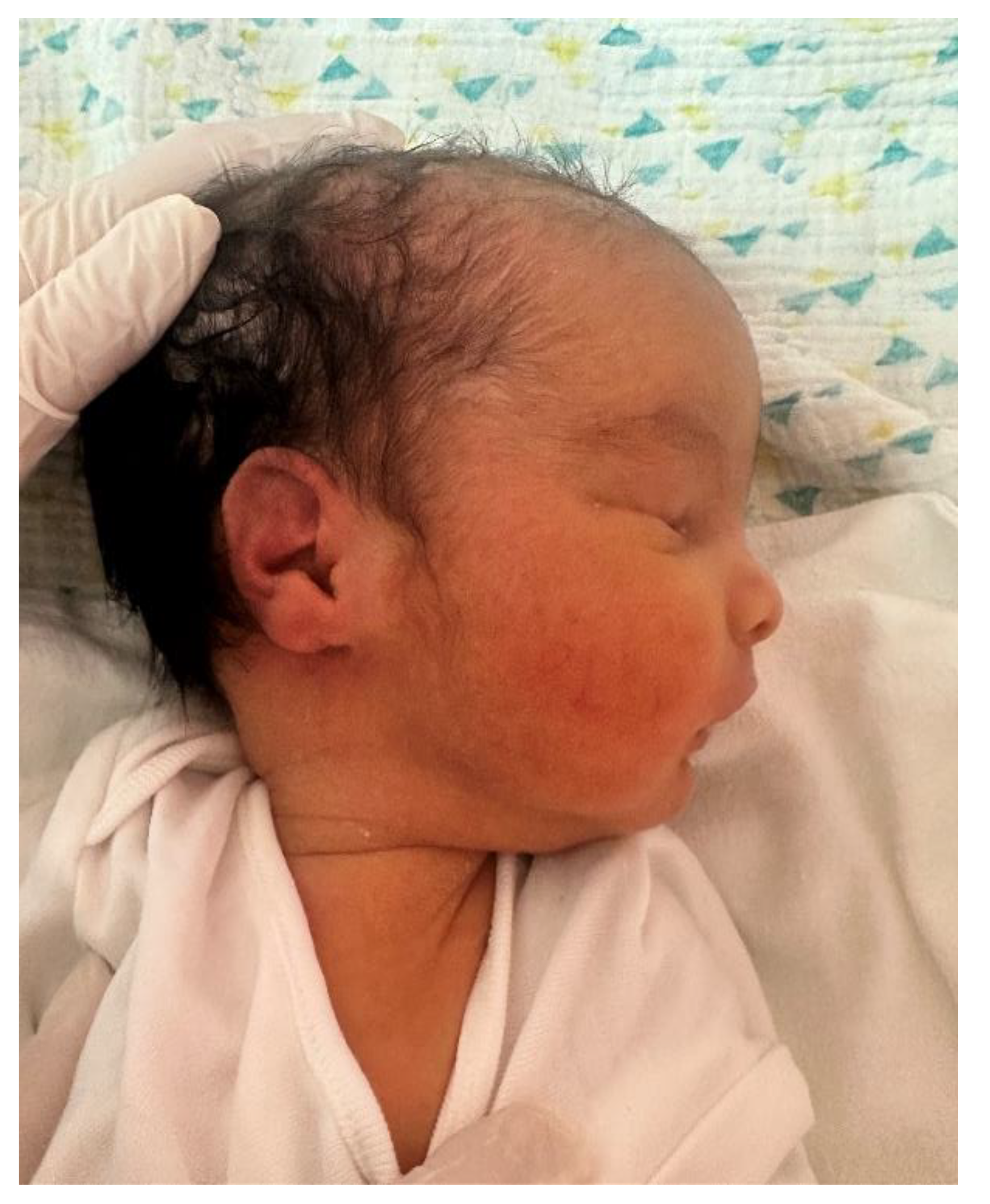
Figure 3.
Generalized muscular hypotonia and bilateral cryptorchidism in the male neonate (Case 1), observed during neonatal physical examination.
Figure 3.
Generalized muscular hypotonia and bilateral cryptorchidism in the male neonate (Case 1), observed during neonatal physical examination.
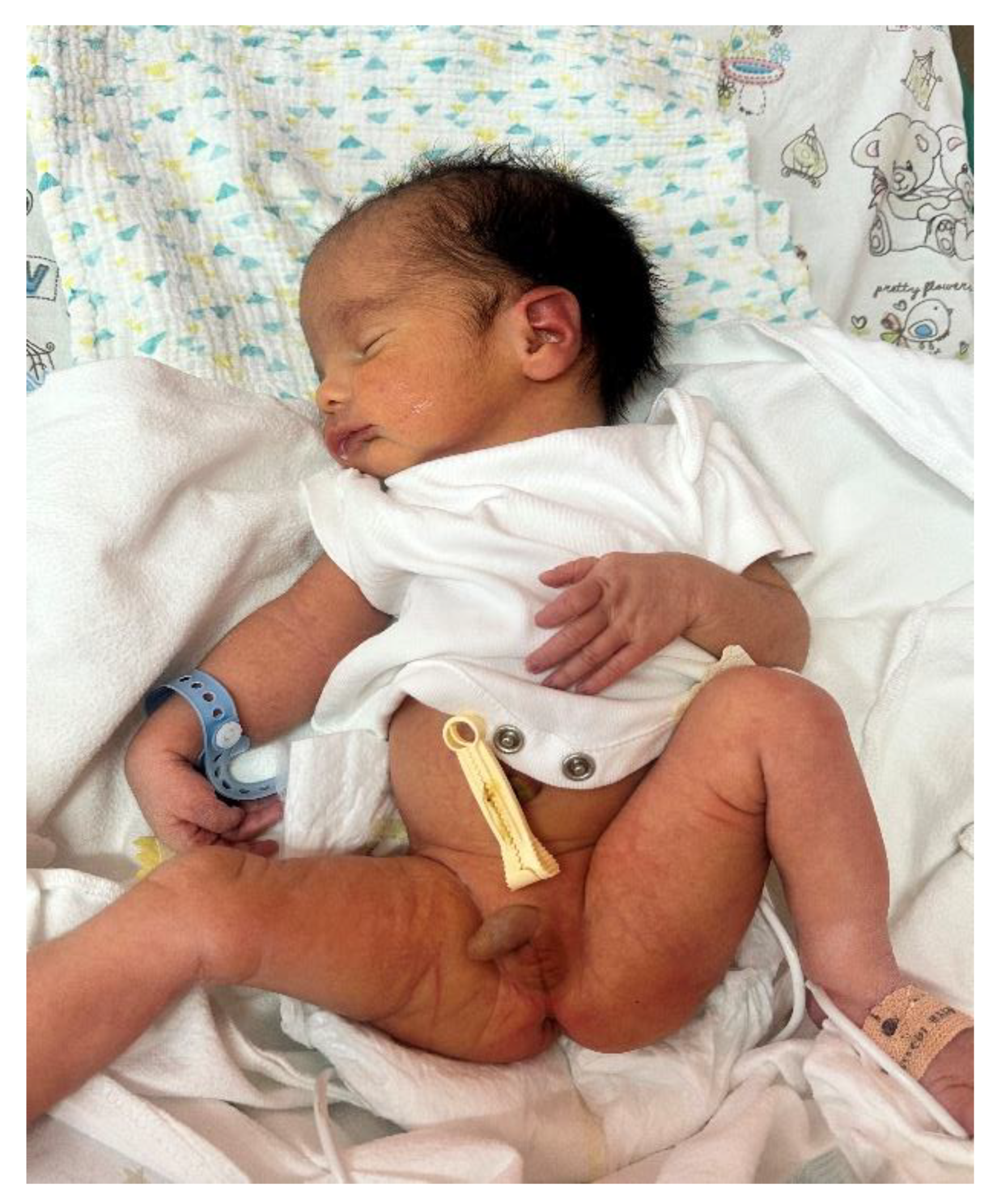
Figure 4.
Marked generalized muscular hypotonia in the neonate (Case 2), evident during early postnatal neurological assessment.
Figure 4.
Marked generalized muscular hypotonia in the neonate (Case 2), evident during early postnatal neurological assessment.
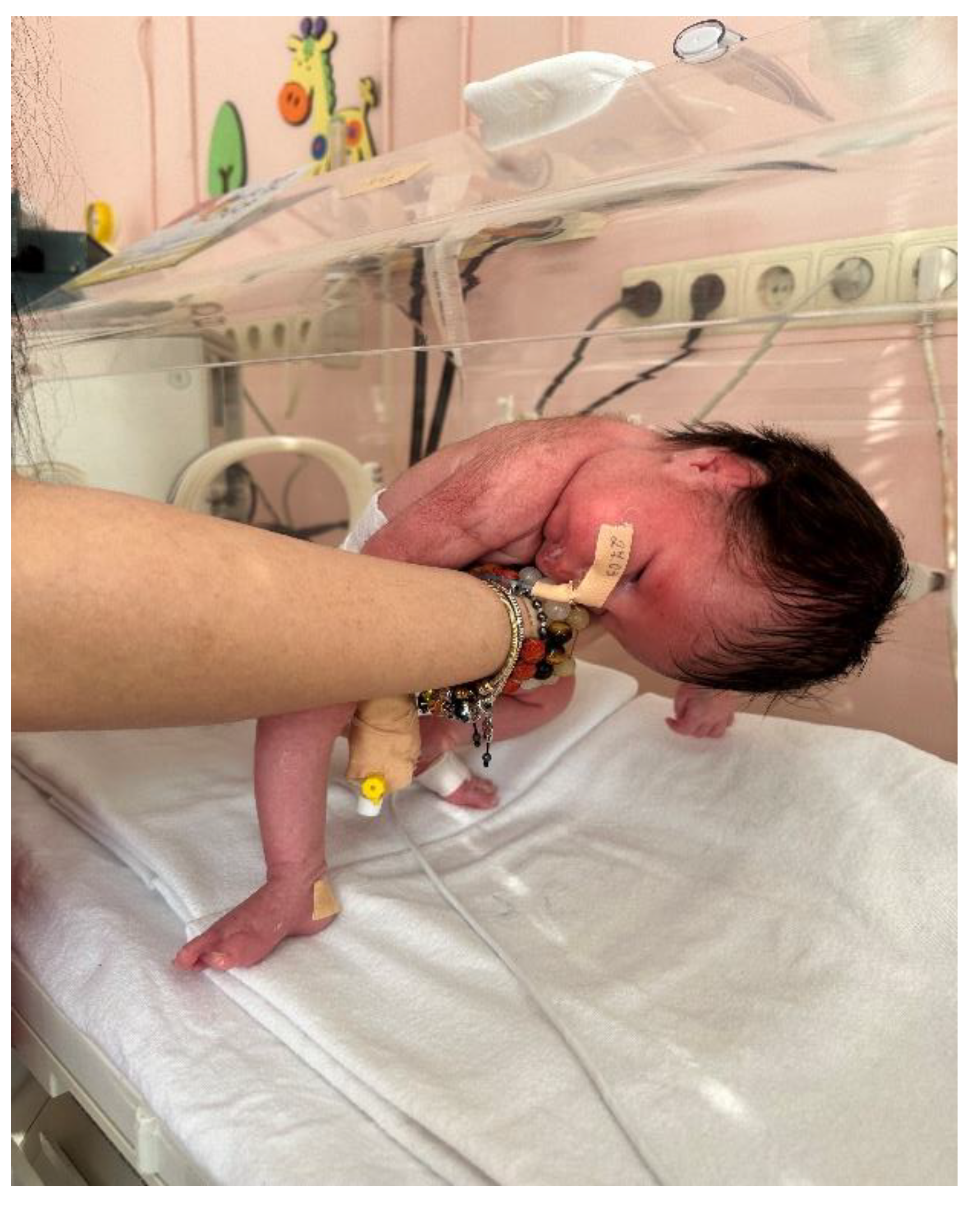
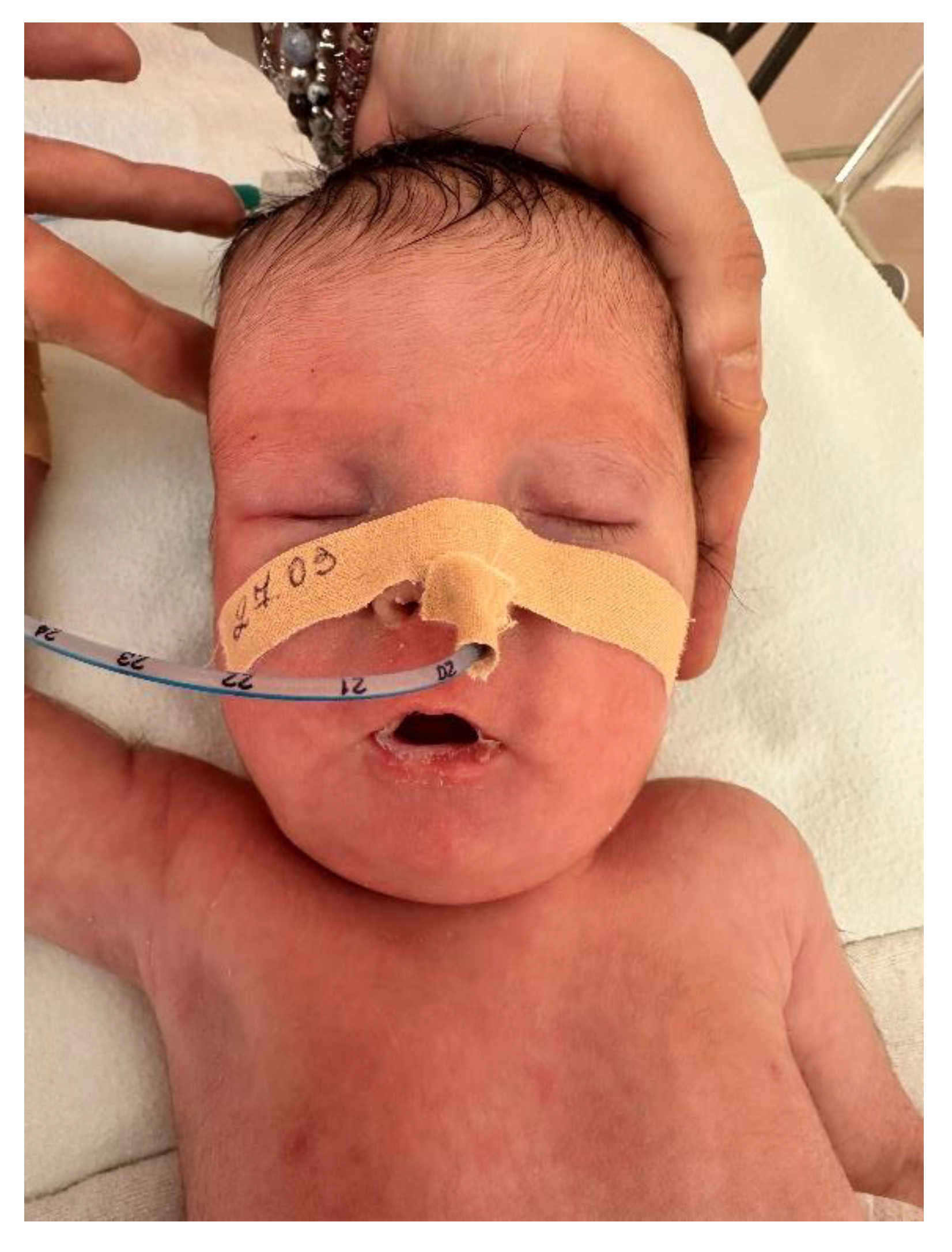
Figure 5.
Facial dysmorphic features in the neonate (Case 2), including a narrow bifrontal diameter, almond-shaped eyes, short nose, downturned corners of the mouth, and a thin upper lip, characteristic of Prader–Willi syndrome.
Figure 5.
Facial dysmorphic features in the neonate (Case 2), including a narrow bifrontal diameter, almond-shaped eyes, short nose, downturned corners of the mouth, and a thin upper lip, characteristic of Prader–Willi syndrome.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



