
Submitted:
02 December 2025
Posted:
10 December 2025
You are already at the latest version
Abstract
The H5N1 subtype of highly pathogenic avian influenza (HPAI) poses a major zoonotic threat due to its high fatality rate and capacity for cross species transmission. In early 2025, Argentina detected a novel triple reassortant A(H5N1) virus in Chaco Province, combining Eurasian, North American, and South American lineage segments. Genomic analyses of subsequent outbreaks in Buenos Aires and Entre Ríos confirmed persistence of this reassortant and additional HA substitutions (T204K, P251S) potentially linked to increased mammalian receptor affinity. Although PB2 sequences lacked canonical mammalian-adaptive markers (E627K, Q591K, D701N), all contained I292M, a mutation associated with human adaptation. Phylogenetic analyses revealed distinct genotypes and increasing divergence. These findings indicate ongoing viral evolution and adaptation within Argentina, emphasizing the urgent need for sustained genomic surveillance, timely data sharing, and integrated One Health strategies to mitigate zoonotic and socioeconomic risks associated with H5N1 spread in South America.
Keywords:
Argentina
; triple reassortant virus
; highly pathogenic avian influenza (HPAI)
; H5N1
; one health
; molecular evolution
1. Introduction
Avian influenza and the H5N1 subtype in particular, poses a significant public health risk due to its zoonotic potential and the high fatality rate among reported human cases. There is also recent evidence of sustained mammal-to-mammal transmission of highly pathogenic avian influenza (HPAI) [1], which increases the risk of the virus acquiring mutations enabling sustained spread among humans; raising concerns for a potential pandemic [2,3]. Moreover, outbreaks in poultry result in substantial economic losses and represent a critical threat to global food security, emphasizing the necessity of sustained active surveillance, rigorous biosecurity measures, and comprehensive preparedness strategies for potential public health emergencies [4].
Since its first detection in South America in late 2022, clade 2.3.4.4b HPAI H5N1 has caused extensive outbreaks in wild birds and poultry across the continent. In Argentina during 2023, several outbreaks of A(H5N1) clade 2.3.4.4b were reported in birds —including wild birds, backyard poultry, and commercial flocks — as well as in marine mammals (South American sea lions) [5].
In early 2025, Argentina reported the first evidence of a novel triple reassortant A(H5N1) virus in the Chaco Province (located in the northeast of the country), with gene segments derived from Eurasian H5N1 (HA, NA and MP), North American low pathogenic avian influenza (LPAI) (NP) and South American LPAI lineages (PA, PB1, PB2 and NS). Such events highlight the dynamic evolution of H5N1 in the Americas [6].
In this study, we present a preliminary genomic analysis of H5N1 viruses detected during subsequent outbreaks in Argentina in 2025 (Figure 1). Between July and September, five additional outbreaks were reported in Buenos Aires and Entre Ríos provinces, in the central region of the country, where the triple-reassortant H5N1 virus was also identified. Importantly, the most recent sequences obtained maintained the reassortant constellation while also showing additional amino acid substitutions.
2. Materials and Methods
Tracheal and/or cloacal swabs were collected from domestic birds during official investigations of four avian influenza outbreaks that occurred in Buenos Aires and Entre Ríos provinces between July and September 2025 as part of the active and passive surveillance activities conducted by SENASA. Samples were submitted to the SENASA Official Laboratory for influenza A detection and H5 subtype confirmation by RT-qPCR following the recommendations of the WOAH Manual of Diagnostic Tests and Vaccines for Terrestrial Animals (2022) and the laboratory’s standard operating procedures.
A total of 23 A(H5N1)-positive samples (22 poultry and 1 duck) were selected for whole-genome sequencing. Viral RNA was extracted from clinical material, reverse-transcribed, and amplified using a multisegment RT-PCR approach targeting all eight influenza genome segments. Sequencing libraries were prepared from the resulting amplicons and sequenced on Illumina platforms. Consensus genomes were assembled using IRMA (v1.0.3) with default parameters.
To assess potential adaptive changes, amino acid substitutions in HA and PB2 were screened for known host-specificity markers and other relevant mutations using the GISAID FluServer tool [7]. In addition, all genomes were analyzed with GenoFLU [8] to evaluate genomic constellations and determine whether sequences corresponded to previously recognized H5N1 genotypes or represented novel reassortant combinations.
3. Results
A phylogenetic tree of HA sequences including viruses from the July–September 2025 outbreaks and the triple-reassortant strain identified in January 2025 in Chaco Province showed that all recent viruses cluster within the same reassortant lineage but with clear diversification, consistent with ongoing local evolution (Figure 1).
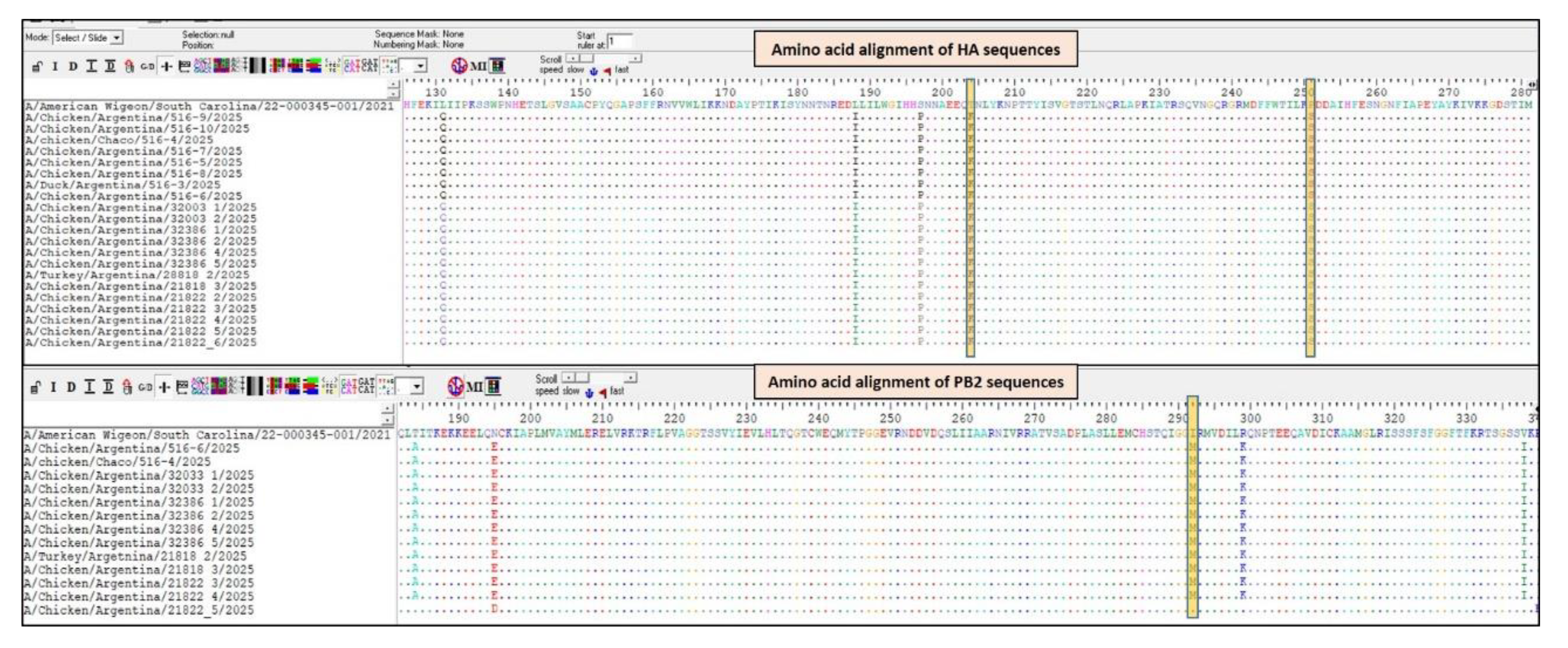
Analysis of the HA gene sequences revealed several amino-acid substitutions (Figure 2). The P251S mutation in the HA of H5N1, located within the 130-loop, a region critical for receptor binding. While this specific mutation has not been widely reported in the literature, its structural location suggests it could influence receptor affinity and potentially affect mammalian adaptation. Mutations in nearby positions, such as S227N (H3 numbering), have been associated with shifts in receptor specificity toward human-type α2,6-linked sialic acids [9].The T204K mutation, is located within the receptor-binding site (RBS) and has also been associated with shifts in receptor specificity. Studies have demonstrated that mutations at position 192 can convert the HA from recognizing avian-type α2,3-linked sialic acids to human type α2,6-linked sialic acids, enhancing the virus’s ability to infect human respiratory epithelial cells [10].
Regarding the PB2 gene, none of the 12 contained the E627K, Q591K or D701N substitution, well-recognized determinants of enhanced polymerase activity in mammalian hosts [1,11,12]. However, they all shared several mutations, including I292M, which, according to the FluServer tool, is associated with a host-specific characteristic involved in adaptation for human-to-human transmission (Figure 2).
GenoFLU analysis showed that the 2025 Argentine viruses could not be assigned to any previously described genotype, supporting the occurrence of reassortment events. Considerable genetic divergence was also observed among the sequences.
4. Discussion
The genomic characterization of A(H5N1) viruses detected during poultry outbreaks in central Argentina in 2025 demonstrates continued circulation and diversification of the triple-reassortant lineage first reported earlier that year. The persistence of this lineage across multiple outbreaks, together with the detection of recurrent HA substitutions linked to human-type receptor binding, suggests ongoing adaptive evolution. Although classical PB2 mammalian-adaptation markers were absent, the consistent presence of I292M highlights the need to further investigate its functional significance in the context of clade 2.3.4.4b viruses.
The inability to classify these viruses into existing genotypes through GenoFLU, along with the observed genomic heterogeneity, reinforces the dynamic nature of H5N1 evolution in the region. These findings underscore the importance of sustained genomic surveillance and rapid data sharing to detect emerging variants and evaluate their potential zoonotic risk. Continued multisectoral coordination within the national surveillance system is essential to support early outbreak detection, risk assessment, and prompt implementation of control measures.Authors should discuss the results and how they can be interpreted from the perspective of previous studies and of the working hypotheses. The findings and their implications should be discussed in the broadest context possible. Future research directions may also be highlighted.
Author Contributions
Conceptualization, E.B. and M.C.A.; methodology, E.B., M.C.A., M.B.G., M.A., L.P., A.Ch., V.M., M.R., M.E.D. and E.M.M, ; software, E.B., M.C.A. and A.B; validation, M.G., N.L. and A.P.; formal analysis, E.B., M.C.A., A.B. and A.P.; investigation, E.B., M.C.A., A.B. and A.P.; resources, M.C.A., M.G. and A.P.; data curation, E.B. and M.C.A.; writing—original draft preparation, E.B.; writing—review and editing, E.B., M.C.A., A.B., M.B.G., M.A., L.P., A.Ch., V.M., M.R., M.E.D., E.M.M, M.G., N.L. and A.P.; visualization, E.B., M.C.A. and A.P.; supervision, A.P.. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The sequences generated in this study will be deposited in the GISAID database and will be available.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| HPAI | Highly Pathogenic Avian Influenza |
| LPAI | Low Pathogenic Avian Influenza |
| RBS | Receptor Binding Site |
References
- Pardo-Roa, C.; Nelson, M.I.; Ariyama, N.; Aguayo, C.; Almonacid, L.I.; Gonzalez-Reiche, A.S.; Muñoz, G.; Ulloa, M.; Ávila, C.; Navarro, C.; et al. Cross-Species and Mammal-to-Mammal Transmission of Clade 2.3.4.4b Highly Pathogenic Avian Influenza A/H5N1 with PB2 Adaptations. Nat Commun 2025, 16, 2232. [Google Scholar] [CrossRef] [PubMed]
- Influenza: A(H5N1). Available online: https://www.who.int/news-room/questions-and-answers/item/influenza-h5n1 (accessed on 17 November 2025).
- Q&A Avian Influenza (Bird Flu) - PAHO/WHO | Pan American Health Organization. Available online: https://www.paho.org/en/topics/avian-influenza/qa-avian-influenza-bird-flu (accessed on 17 November 2025).
- The Spread of H5N1 Highly Pathogenic Avian Influenza Calls for Stepped up Action, FAO Says. Available online: https://www.fao.org/newsroom/detail/the-spread-of-h5n1-highly-pathogenic-avian-influenza-calls-for-stepped-up-action--fao-says/en (accessed on 17 November 2025).
- Artuso, M.C.; Marchione, V.D.; Benedetti, E.; Bonastre, P.; Alvarez, A.M.; Piccini, L.; Ponde, A.; Barrios Benito, E.; Fabeiro, M.; Waisman, K.; et al. Detection and Characterization of Highly Pathogenic Avian Influenza A (H5N1) Clade 2.3.4.4b Virus Circulating in Argentina in 2023. Rev Argent Microbiol 2024. S0325-7541(24)00115-9. [Google Scholar] [CrossRef] [PubMed]
- Vanstreels, R.E.T.; Nelson, M.I.; Artuso, M.C.; Marchione, V.D.; Piccini, L.E.; Benedetti, E.; Crespo-Bellido, A.; Pierdomenico, A.; Wolff, T.; Uhart, M.; et al. Novel Highly Pathogenic Avian Influenza (A)H5N1 Triple Reassortant in Argentina, 2025. bioRxiv 2025, 2025.05.23.655175. [Google Scholar] [CrossRef]
- BII Flusurver - Prepared for the next Wave. Available online: https://flusurver.bii.a-star.edu.sg/ (accessed on 18 November 2025).
- Youk, S.; Torchetti, M.K.; Lantz, K.; Lenoch, J.B.; Killian, M.L.; Leyson, C.; Bevins, S.N.; Dilione, K.; Ip, H.S.; Stallknecht, D.E.; et al. H5N1 Highly Pathogenic Avian Influenza Clade 2.3.4.4b in Wild and Domestic Birds: Introductions into the United States and Reassortments, December 2021–April 2022. Virology 2023, 587, 109860. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Suzuki, Y.; Suzuki, T.; Le, M.Q.; Nidom, C.A.; Sakai-Tagawa, Y.; Muramoto, Y.; Ito, M.; Kiso, M.; Horimoto, T.; et al. Haemagglutinin Mutations Responsible for the Binding of H5N1 Influenza A Viruses to Human-Type Receptors. Nature 2006, 444, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Y.; Wei, C.-J.; Kong, W.-P.; Wu, L.; Xu, L.; Smith, D.F.; Nabel, G.J. Immunization by Avian H5 Influenza Hemagglutinin Mutants with Altered Receptor Binding Specificity. Science 2007, 317, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Uhart, M.M.; Vanstreels, R.E.T.; Nelson, M.I.; Olivera, V.; Campagna, J.; Zavattieri, V.; Lemey, P.; Campagna, C.; Falabella, V.; Rimondi, A. Epidemiological Data of an Influenza A/H5N1 Outbreak in Elephant Seals in Argentina Indicates Mammal-to-Mammal Transmission. Nat Commun 2024, 15, 9516. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, A.; Naguib, M.M.; Nogales, A.; Barre, R.S.; Stewart, J.P.; García-Sastre, A.; Martinez-Sobrido, L. Avian Influenza A (H5N1) Virus in Dairy Cattle: Origin, Evolution, and Cross-Species Transmission. mBio 2024, 15, e0254224. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Maximum-likelihood phylogeny of hemagglutinin (HA) gene sequences of HPAI H5N1 viruses detected in Argentina, 2023–2025. All Argentine sequences are shown in blue; 2025 outbreaks are indicated by colored squares corresponding to distinct geographic events. Viruses from 2025 form a separate clusters relative to those detected in 2023.
Figure 1.
Maximum-likelihood phylogeny of hemagglutinin (HA) gene sequences of HPAI H5N1 viruses detected in Argentina, 2023–2025. All Argentine sequences are shown in blue; 2025 outbreaks are indicated by colored squares corresponding to distinct geographic events. Viruses from 2025 form a separate clusters relative to those detected in 2023.
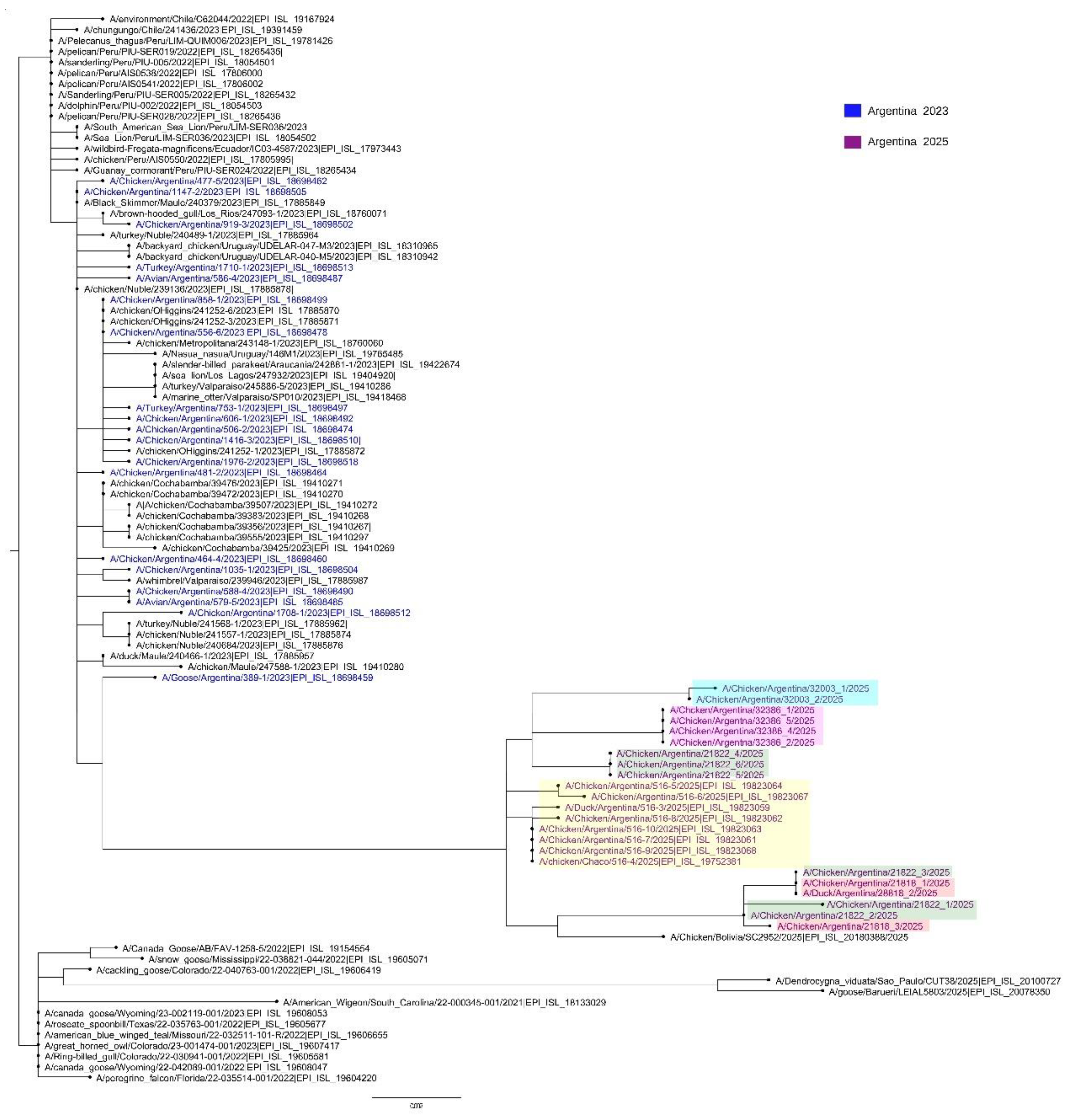
Figure 2.
Amino acid alignments of hemagglutinin (HA) and polymerase basic protein 2 (PB2) genes of highly pathogenic avian influenza A(H5N1) viruses detected in Argentina, 2025, compared with reference sequences A/American_wigeon/South_Carolina/22-000345-001/2021 (HA) and A/Duck/Guangdong/E1/2012(H10N8) (PB2).
Figure 2.
Amino acid alignments of hemagglutinin (HA) and polymerase basic protein 2 (PB2) genes of highly pathogenic avian influenza A(H5N1) viruses detected in Argentina, 2025, compared with reference sequences A/American_wigeon/South_Carolina/22-000345-001/2021 (HA) and A/Duck/Guangdong/E1/2012(H10N8) (PB2).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



