
Submitted:
09 December 2025
Posted:
10 December 2025
You are already at the latest version
Abstract
Tuberculosis (TB) persists as a devastating global health crisis, exacting a disproportionate burden on low-middle-income countries (LMIC), within which the burgeoning pediatric TB population is especially vulnerable to severe TB manifestations. Rifampicin, a cornerstone of the first-line TB regimen, is indispensable; yet its therapeutic potential is substantially constrained by intrinsic poor aqueous solubility, limited permeability, physicochemical instabilities, and deleterious drug-drug interactions with drugs such as isoniazid. Although current research focuses on formulating rifampicin into fixed-dose combinations – some reporting improved outcomes – the variable and often suboptimal rifampicin bioavailability in such formulations remains a critical concern. Consequently, the development of a stable, independent rifampicin oral formulation should be prioritized to ensure effective TB treatment. There are currently no commercially available rifampicin-only liquid preparations, despite the urgent need for palatable, flexible, age-appropriate formulations. This paucity constitutes a profound and persistent gap in pediatric TB, compromising accurate weight-based dosing, treatment adherence, and therapeutic outcomes while contributing to rifampicin resistance. This paper highlights the key characteristics and formulation challenges of rifampicin, with particular emphasis on pediatric TB. It further discusses liquid self-emulsifying drug delivery systems (SEDDSs) as a novel approach to enhance oral rifampicin delivery. These feasible lipid-based delivery systems offer notable promise, as they are capable of maintaining rifampicin in a solubilized and stable state throughout the gastrointestinal tract, which in turn will likely improve bioavailability. The possible development of a stable liquid rifampicin SEDDS represents a paradigm shift in addressing the longstanding neglect of pediatric TB treatment.
Keywords:
lipid-based drug delivery system
; oral administration
; pediatric tuberculosis
; rifampicin
; physicochemical instabilities
; self-emulsifying drug delivery system (SEDDS)
1. Introduction
Tuberculosis (TB) remains a dynamic and devastating global health crisis firmly ingrained in persistent multifaced socioeconomic inequities [1,2,3,4,5,6], including the denial of basic human rights, systemic neglect, and socioeconomic disparities [1,3,7]. In fact, this disease imposes a disproportionate burden on low- and middle-income countries (LMICs), which account for more than 95% of global TB occurrences and morbidity cases [5,8,9], as will be elaborated on further below. Moreover, despite endeavors to eradicate TB [8,10], a disease caused by various strains of Mycobacterium tuberculosis [11,12,13,14], it has reclaimed its grim title as the ‘world’s leading cause of death’ due to a single infectious organism [5,8,15]. Indeed, with shortcomings in currently available pharmacological treatment strategies, significant scientific challenges remain for drug discovery, treatment strategies, refinement and optimization of drug formulations, all warranting further investigation and developments.
The World Health Organization (WHO) estimates that approximately 10.8 million people developed TB in 2023 alone, with a concerning 1.3 million cases occurring in the pediatric population [8]. Throughout history, pediatric TB has been neglected or overlooked, primarily because of the misperception that children are minor contributors to the global epidemic, predominantly given their lower TB infection incidence relative to that of adults, and the assumption that children seldom develop severe forms of TB [16,17]. In fact, children under the age of five years are at higher risk of developing severe or fatal active forms of TB compared to older pediatric patients [4,18,19,20]. Furthermore, without appropriate testing and treatment of exposed children, a silent biological reservoir of TB infection could persist. This could lead to TB cases resurfacing in future, thereby undermining global TB control and eradication efforts [4,16].
Emerging evidence indicates that, similar to adults, pediatric pulmonary TB survivors remain vulnerable to the long-term consequences associated with TB, such as post-tuberculosis lung disease, which causes lasting respiratory impairment [21,22,23,24]. This is especially concerning for pediatric patients due to their longer anticipated remaining lifespan [24]. Moreover, malnutrition, erratic treatment compliance, as well as inaccurate and subtherapeutic rifampicin doses can lead to drug resistance and treatment failure in children suffering from active TB [17,25,26]. Drug-resistant pediatric TB often requires prolonged treatment and increases the susceptibility to lung damage [21,24,27]. As a result, TB can profoundly affect young people, disrupting their education, development, and future economic prospects [28].
Currently, chemotherapy remains the only viable and effective clinical treatment option for TB patients [29]. Anti-TB drugs often exhibit poor therapeutic profiles, including poor bioavailability due to undesirable first-pass metabolism and variable drug absorption [29,30,31,32]. Among these therapeutic limitations associated with effective TB treatment regimens are the toxicity of individual and combined drugs, drug interactions, lengthy treatment regimes, as well as severe adverse drug reactions [29,33,34,35,36] that consequently also contribute to the emergence of multi-drug resistant strains, treatment failure, and poor patient adherence [29].
Rifampicin, a frontier drug in the first-line anti-TB regimen, is often incorporated with isoniazid, pyrazinamide, and ethambutol in fixed-dose combination therapy, such as tablets and oral dispersible tablets [8,37,38]. However, this drug is thermally unstable and exhibits increased degradation when exposed to acidic conditions and/or when co-formulated with isoniazid [30,39,40,41]. Consequently, current pediatric fixed-dose combination therapy often results in suboptimal rifampicin therapeutic levels [25,42]. Moreover, since ample water is required for the effective delivery of pediatric dispersible tablets [37], restricted access to clean and safe water poses yet another notable treatment barrier in resource-limited countries.
Furthermore, pediatric TB treatment regimens are usually extrapolated from adult treatment regimens and are adjusted according to the age and weight of the child, to compensate for the developmental, physiological, and pharmacodynamic differences between children and adults [17,43,44]. From a pharmacological perspective, children are not merely “small adults”. They often display qualitative and quantitative differences in pharmacodynamics and pharmacokinetics, and therefore simple extrapolations from adult data frequently fail to produce child-appropriate formulations or optimal drug dosing [37]. Research has also reported that the majority of commercially available drug products prescribed for pediatric TB patients are not suitable dosage forms for pediatric consumption [17,45,46,47,48,49]. In South Africa, a region with a high TB burden, only two rifampicin-containing dosage forms, namely capsules and vails, are currently registered and commercially available for adult use. Added to that, these may not necessarily be readily available, particularly following a recent discontinuation of the only rifampicin oral suspension [8,50]. Thus, the lack of child-appropriate dosage forms often necessitates reconstitution into extemporaneous formulations [51].
Despite the clinical need for commercially available liquid formulations of rifampicin, such products remain scarce due to the challenging physicochemical properties of rifampicin and the niche market for pediatric use [51]. This exposes a significant gap in pediatric TB treatment. Effective pediatric TB treatment requires age-appropriate formulations carefully designed to ensure efficient and safe drug delivery and pharmacotherapy [52,53].
The persistent challenges with rifampicin prompt a fundamental question: Can the stability of rifampicin in liquid formulations be adequately improved via science and innovation? Addressing this gap is imperative to meet the clinical needs of pediatric TB patients around the world.
2. Pediatric Tuberculosis
As stated above, pediatric TB constitutes a significant yet underprioritized global health concern, where preventable morbidity and mortality among the most fragile age groups persist. In fact, epidemiological data suggest that the global pediatric TB distribution mirrors that of adults [4], with a substantial disease burden in Africa and Southeast Asia, accounting for 80% of global HIV-negative TB-related deaths [8]. It is worth noting that five countries — China, India, Indonesia, Pakistan, and the Philippines — account for more than half of all the TB cases globally [8]. In 2023, the WHO reported that children under 15 years old make up approximately 12% of the estimated total TB incident cases; however, despite this lower incidence, they represent 15% of the projected global TB mortality [8]. These reported statistics on the global pediatric TB burden may not reflect the full picture, as most cases often go unreported or undiagnosed, and as such, the true global numbers may be considerably higher [8,16]. In most high-burden regions, TB deaths are often attributed, without a confirmed diagnosis, to meningitis or pneumonia – common causes of death among children under five years, especially in LMICs [16,17,54,55]. Moreover, diagnosing pediatric TB patients is known to be especially challenging, given the different clinical manifestations, the paucibacillary nature of TB, clinical resemblance with other pediatric diseases [4], age-related and non-specific symptoms, gaps in diagnosis, and limited access to treatment and diagnostic services, especially for children under the age of 15 years [55,56,57]. Pediatric TB screening and diagnostic methods include culture, smear microscopy, nucleic acid amplification assays, tuberculin skin test, and imaging [55,58]. However, these techniques are limited by challenges in obtaining quality specimens, low bacterial loads, and decreased test performance in immunocompromised or young children. As a result, there is no single diagnostic test that can rapidly, accurately, and reliably detect pediatric TB [55,58]. Without accurate estimates of the global pediatric TB burden, resources for treatment, diagnosis, and the demand for new drug development will be inadequately allocated, which in turn diminishes the incentive of pharmaceutical industries to prioritize the development of anti-TB drugs. Collectively, these factors are crucial to advocate for and manage pediatric TB, initiate timely treatment, and prevent fatalities [57,59].
2.1. Pediatric Tuberculosis Transmission and Disease Progression
Mycobacterium tuberculosis, complex intracellular organisms, such as M. canetti, M. bovis BCG, M. bovis, M. africanum (along with other species and strains that are rarely pathogenic to humans), are transmitted through the respiratory route when minute infected droplets are aerosolized from pulmonary or laryngeal TB patients, followed by inhalation into the lungs of close contacts [55,60,61]. The risk of TB infection and progression after a primary M. tuberculosis infection is greatly influenced by the immunologic function and age of the patient [62, 63, 64, 65]. Accordingly, the guideline on clinical investigation of medicinal products in the pediatric population (CHMP/ICH/2711/99) categorizes pediatric age groups based on developmental changes as follows: preterm newborn infants; term newborn infants (0–27 days); infants and toddlers (1 months to 23 months); children (2–11 years), subdivided into pre-school children (2–5 years) and school children (6–11 years), and ultimately adolescents (12–16 or 18 years), with the applicable classification depending on regional definitions [66].
Furthermore, child-to-child TB transmission is rare [60,63,67]. In fact, cavitary forms of TB are less frequent in children, except for those with genetic immune response modifications or HIV [60]. Therefore, TB transmission among pediatrics is often due to close and prolonged exposure to adults with infectious pulmonary TB [4,17]. It is worth noting, however, that pediatric TB follows a bimodal risk profile. The highest transmission risks are observed in children under two years of age and in late adolescence, whereas the risk for children aged between five and ten years is considerably lower [64,67]. Since children under two years depend extensively on adult supervision, they usually contract TB from close household members [65,67], whereas older children, being more independent and socially active, often acquire TB infection from an external source [17,68]. Adults receiving regular and appropriate treatment for pulmonary TB seldom infect children; however, the greater risk lies with those having chronic TB that remains undiagnosed, relapse due to drug resistance, or when inadequately treated [67].
Regarding disease progression, children under two years of age or with impaired immunological response facilitate persistent replication of mycobacteria and their progression to pulmonary disease or its dissemination to other physiological regions [60,65]. Conversely, similar to the risks of contracting TB mentioned above, the risk of disease progression is low in children aged five to ten years, whereas adolescents are more susceptible to disease reactivation [69]. The age-related risk of TB progression is shown in Figure 1 [64].
Moreover, numerous factors contribute to the transition from pediatric TB infection to active disease and the severity thereof, including the virulence of the mycobacterium strain, the burden of mycobacterial exposure, and the time since exposure, during which the first two post-exposure years coincide with the highest risk [55]. In addition, disease progression is influenced by environmental and host risk factors encompassing immunocompromising status (including HIV/AIDS, chronic renal disease, and the use of immunosuppressants such as chemotherapy for malignancy), environmental exposures to indoor air pollutants and tobacco smoke, and socioeconomic conditions, comprising overcrowding living conditions, and malnutrition [55,70,71].
2.2. Clinical Manifestation of Pediatric Tuberculosis
Following exposure, the pathogen is either eliminated or persists within the body; consequently, its persistence may result in a latent or active TB infection [60]. Latent or dormant TB refers to a non-replicating state of the pathogen, occurring without active disease manifestations and symptoms [56] and which may persist for years until reactivation. The risk of a healthy adult with a latent TB infection developing active TB is 5–10% over their lifetime, with a higher likelihood of progression to TB within the first two years after primary infection [69]. Compared to adults, infants with a latent TB infection have a 5–10 times greater probability of developing active TB and are at greater risk of severe disseminated manifestations [4,18,19,20]. Moreover, with proper management, such as prompt initiation of treatment with appropriate drug dosages, pediatric treatment outcomes are generally favorable [55]. This, however, highlights a troubling reality that, despite TB being a preventable and generally curable disease [8], the most vulnerable groups continue to bear the greatest burden of neglect.
Pulmonary TB, a lung-localized infection, remains the most prevalent form in the overall pediatric population, with marginal interindividual variation in age-dependent symptom severity [4]. Approximately 50% of infants exhibit systemic or respiratory symptoms during the primary phase of TB, whereas up to 90% of newly infected older children will remain asymptomatic [72]. Younger children often present with symptoms such as cough, chills, fever, enlarged lymph nodes, weight loss, and stunted growth. Manifestation in adolescents, on the other hand, often resembles adult-type TB and tends to have slightly different signs and symptoms, including chest pain, prolonged cough, night sweats, chills, fever, fatigue, weakness, hemoptysis, anorexia, enlarged lymph nodes, and weight loss [67].
Extrapulmonary:TB accounts for 20–30% of all pediatric TB cases [73]. The clinical presentation varies according to the site of infection, and these sites may include, but are not confined to, the lymph nodes, pleura, abdomen, cutaneous, musculoskeletal system, the cardiac system, meninges, and widespread or miliary dissemination [74]. Notably, the type and severity of extrapulmonary TB in pediatric patients are largely determined by host-pathogen interactions, which often present differently among this diverse age group [17]. Following, several manifestations of pediatric extrapulmonary TB infection are described next.
2.2.1. Indications of Pediatric Extrapulmonary Tuberculosis Infection
The most prevalent type of extrapulmonary TB among children aged 2 to 10 years is TB lymphadenitis [74,75]. Lymph nodes localized in the neck are usually affected; however, lymph node involvement of the axillary, abdominal, supraclavicular, and thoracic regions may also occur [17]. Clinically, it presents as asymmetrical, non-tender, painless lymph node enlargement for more than a month, with or without a discharging sinus, and presents predominantly in the neck [74].
Occasionally, pediatric patients may develop pleural TB, sometimes without pulmonary lesions [74]. Typical clinical presentation in these cases includes reduced breath sounds and dullness on percussion, with or without chest pain [74].
Young children are not often diagnosed with abdominal TB, but this form accounts for about 12% of extrapulmonary TB cases in children under 15 years, with increased susceptibility in children aged five and older [74], representing approximately 1–3% of all TB cases [76]. Abdominal TB typically presents with non-specific symptoms [76,77], but may entail abdominal swelling with abdominal masses or ascites [74]. The most prevalent forms among children are adhesive peritonitis or nodal disease, of either the fibrotic or the wet type, whereas gastrointestinal-involved extrapulmonary TB is more common in adults [77].
Although cutaneous TB accounts for only 1–2% of all TB infections, a considerable number of these cases occur in children aged 10 to 14 years [17]. In younger children, this type of TB often involves systemic organs and lymph nodes, with scrofuloderma (i.e., cutaneous manifestation) as the most prevalent form of this disease [17]. Diagnosing the latter in the pediatric population is challenging due to its variable and non-specific presentation, including both multibacillary and paucibacillary forms [17].
Osteoarticular TB is uncommon in the pediatric population [78], however, infants can still occasionally develop this disease. Most cases occur in children older than five years. Osteoarticular TB symptoms often include unilateral knee or hip effusion and limited movement due to swelling at the ends of long bones. Notably, because of painful joints or limbs and limping, osteoarticular TB is often misattributed to trauma [63,74]. Spinal TB, known as Pott’s spine, accounts for 50% of all osteo-articular TB cases. It primarily affects children aged five years and older; though younger children tend to develop more severe deformities [63,74,78,79]. Spinal TB typically presents with spine deformity, lower limb weakness/inability to walk, or paralysis [63,74,79]. Apart from spinal TB that typically affects young children, skeletal TB usually occurs within the first two decades of life and accounts for 10–20% pediatric extrapulmonary TB cases [80]. As children grow, their transphyseal vessels start to diminish, rendering eighteen-month-old children and younger more prone to develop TB arthritis [81].
Infection of the cardiac system, i.e., pericardial TB, is rare, accounting for less than 4% of the cases [82]. The pericardial tissue is the most frequently infected site, presenting either as a normal TB infection or leading to pericardial effusion and pericarditis, which may subsequently cause hemodynamic effects [74,79]. Typical signs include distant heart sounds, cardiac failure, and difficulty palpating the apex beat [74].
TB meningitis, which is an extrapulmonary TB presentation with central nervous system involvement, is the deadliest and most debilitating form of tuberculosis that disproportionately targets young children, especially children under five with disseminated TB, as well as those with advanced or life-threatening diseases [74]. Despite treatment, mortality rates range from 15% to 40%, and over one-third suffer lasting or permanent neurological defects [4,83]. Typical clinical presentations include headache, neck stiffness, irritability or abnormal behavior, lethargy or reduced consciousness, vomiting (in the absence of diarrhea), convulsions, cranial nerve palsies, and bulging fontanelle [74].
Miliary TB, on the other hand, is a severe, life-threatening form of disseminated TB, resulting from an extensive lymphohematogenous distribution of the pathogen from the primary TB infection site [84]. The typical clinical presentation entails: non-specific, persistent fever, and lethargy [74], and may resemble those of other common pediatric diseases, depending on which specific organs are involved [84]. The most affected groups are usually the terminally ill and children under five with disseminated TB [74].
3. Therapeutic Management of Pediatric Drug-Susceptible Tuberculosis
3.1. Current Treatment of Drug-Susceptible Pediatric Tuberculosis
The goal of pediatric TB treatment is to prevent TB infection from progressing to active TB and to reduce the reservoir for future TB incidents [69]. The principles of TB treatment are mostly consistent between adults and children; however, in pediatric patients, weight-based dosing is required for all drugs due to pharmacokinetic differences within the pediatric population, as well as differences between children and adults [4,43,44,74,85]. The selection of TB regimens for pediatric patients is further influenced by various factors such as the age of the pediatric patient, the severity of the disease, comorbidities, HIV status, antiretroviral therapy, and the affordability and availability of child-friendly formulations [18,69,86].
In general, drug-susceptible pediatric TB, without suspicion or confirmation of drug resistance, is treated with a combination of antitubercular drugs, usually available in fixed-dose combinations, over a typical period of six months consisting of an intensive and a continuation phase. Though, the regimen and duration may be adjusted if drug resistance is present [87,88]. The standard regimen recommended for drug-susceptible TB includes a ‘three/four-drug cocktail’ comprising isoniazid, rifampicin, pyrazinamide, with or without ethambutol [8,86,89].
During the intensive TB treatment phase, the drugs used are predominantly bactericidal, rapidly eliminating substantial amounts of pathogenic bacteria and thereby preventing TB progression, transmission, and the development of drug resistance [88,89]. The drugs involved in the continuation phase are sterilizing agents that eradicate intermittently active or dormant bacilli, which often results in complete bacterial eradication, successful TB treatment, and the prevention of disease relapse [88,89]. Both rifampicin and isoniazid serve as the indispensable cornerstones for both treatment phases, whereas pyrazinamide and ethambutol are usually incorporated depending on various patient-specific factors [17,88].
When pediatric patients transition from the intensive phase to the continuation treatment phase, which typically involves two drugs administered multiple times per week rather than daily, directly observed therapy, if required or available, can be an effective supporting strategy to improve adherence in children [4]. The preferred pediatric pulmonary TB treatment, particularly for patients at a high risk of isoniazid resistance, with extensive pulmonary TB, and/or living in areas of high HIV prevalence, is the four-drug regimen. Ethambutol is included to provide additional protection against the development of resistance in cases of high bacterial load [85].
In contrast, the three-drug treatment regimen, which excludes ethambutol, is preferred for pediatric patients with confirmed or suspected pulmonary TB who reside in regions with low HIV prevalence, where HIV negative status is associated with minimal risk of isoniazid resistance [85]. The WHO Module 5 outlines the current pediatric treatment regimens for both TB infections and drug-susceptible TB, covering presentations from non-severe to severe [86]. A summary of these regimens is provided in Table 1.
3.2. General Classification of First-line Antitubercular Drugs
First-line antitubercular drugs that include isoniazid, rifampicin, and pyrazinamide, with or without ethambutol, are commonly used to treat drug-susceptible TB [8,86]. Their pharmacological properties and use are described below.
Isoniazid, also known as isonicotinic acid hydrazide, is a prodrug that is converted to its active metabolite by the enzyme catalase-peroxidase [91,92]. This drug targets M. tuberculosis by inhibiting catalase-peroxidase, an enzyme that neutralizes oxidative stress, and enoyl-acyl-carrier-protein reductase, which is important for producing mycolic acids [89,93]. This combined action compromises the bacterial cell wall and disrupts the metabolism of lipids, carbohydrates, DNA, and nicotinamide-adenine-dinucleotide [93]. Consequently, this bactericidal drug inhibits cell wall synthesis in M. tuberculosis, thereby eliminating active metabolizing extracellular bacilli [94]. Due to the potent bactericidal activity of isoniazid during the intensive treatment phase, it complements the other first-line companion TB drugs [94]. Isoniazid interferes with the therapeutic and pharmacological effects of vitamin B6 during neurotransmitter biosynthesis; therefore, vitamin B6 supplementation is imperative to prevent isoniazid-induced peripheral neuropathy [17,95]. Although neurological and hepatic toxicity are uncommon in pediatric patients, the risk increases in those receiving high-dose isoniazid therapy, in HIV-positive patients, and in malnourished individuals [96].
Despite the increasing prevalence of rifampicin-resistant TB strains, rifampicin remains a front-line drug in the first-line TB treatment regimen [8,97]. It is a semisynthetic drug that is derived from the rifamycin class, which is part of the ansamycin antibiotic family [92,98]. Rifampicin exerts antimicrobial bactericidal effects by inhibiting DNA-dependent RNA polymerase through binding to its ß-subunit and affecting the mRNA synthesis, preventing RNA transcription and consequently interrupting protein synthesis [89,91,92,94,98]. It is worth mentioning that bacterial RNA polymerase binds rifampicin more readily than mammalian RNA polymerase; however, this selectivity does not preclude rifampicin from causing side effects [97]. The most notable side-effect seen is the orange-red discoloration of tears, sweat, and urine, while hepatic adverse effects are uncommon in pediatric patients [99]. Moreover, rifampicin exhibits bactericidal activity against both extracellular and intracellular M. tuberculosis, aiding its overall bactericidal activity [94,97]. Nonetheless, there are critical gaps associated with rifampicin treatment, including long-term side effects (typically diarrhea, nausea, vomiting, and anorexia); the risk of developing rifampicin resistance when used as monotherapy or as a result of inadequate rifampicin exposure in fixed-dose combination formulations; and CYP450 family metabolic activity induction [98]. Subsequent sections will explore these limitations associated with rifampicin and its treatment in more detail.
Pyrazinamide is another standard drug in the TB treatment regimen and is a vital component in the current “short course” therapy [100]. It exerts potent bactericidal effects against M. tuberculosis bacilli [94,100]. The distinctiveness of pyrazinamide lies in its ability to target semi-dormant bacilli under acidic conditions and to eliminate “persister” bacteria that survive despite unfavorable conditions within the acidic centers of the caseating granulomas [100]. Similar to isoniazid, pyrazinamide is a prodrug that is converted to its active metabolite, pyrazinoic acid, through nicotinamidase, which is the bacterial pyrazinamidase enzyme that subsequently terminates the M. tuberculosis bacilli within the granulomas [89,92]. Nevertheless, the exact mechanism of action by which it exerts its effects remains enigmatic [92]. Common side effects include joint discomfort, gastrointestinal distress, and hepatotoxicity. However, while liver toxicity in adults depends on treatment dosage and duration, it is uncommon in pediatric patients, where it usually causes only minor liver enzyme alterations [17]. Moreover, the hepatotoxic effects of pyrazinamide may be exaggerated when used in combination with isoniazid and rifampicin [94].
Ethambutol is a known bacteriostatic agent that acts as a complementary drug within the first-line group of antitubercular drugs [94]. It inhibits mycobacterial cell wall synthesis by interfering with arabinogalactan synthesis through inhibition of the arabinosyl transferases enzyme [91,93,101] and aids in protecting against drug-resistant mutations [85,94]. Ethambutol monotherapy should be avoided, as it not only results in resistance to itself but also leads to isoniazid tolerance if the latter is consequently implemented [94,102]. However, ethambutol exerts a synergistic effect by improving the activity of other companion antitubercular drugs, such as rifampicin and isoniazid [101,103]. It also increases bacterial sensitivity to isoniazid [101]. Another important reason for incorporating ethambutol is its role in preventing rifampicin resistance, especially in instances where isoniazid monoresistance might be present [104].
Historically, the use of ethambutol in children younger than five years was not recommended and was often considered contraindicated due to a common side-effect associated with this drug, namely optic neuritis [85,105]. Diagnosing this condition in this particular age group is challenging due to their inability to verbalize and comprehend symptoms; however, although delayed diagnosis can result in irreversible blindness, early detection followed by prompt discontinuation of this drug can reverse this adverse effect [85]. Fortunately, when proper dosing guidelines are adhered to, the risk of toxicity in pediatric populations is negligible, as this side effect is both dose- and duration-dependent [85]. Ultimately, its use is determined by careful risk-benefit ratio assessment.
4. Rifampicin: Characteristics and Challenges
4.1. Physicochemical Properties and Rifampicin Instabilities
Rifampicin, presented in Figure 2, is a semi-synthetic derivative of rifamycin B, characterized by the rifamycin ansa structure, which consists of a condensed furan and naphthalene core linked by a substituted aliphatic chain bridging two nonadjacent sites of the naphtohydroquinone ring [106,107]. Interestingly, this chromophore is responsible for the characteristic color of rifampicin [106,108]. This macrocyclic antibiotic presents as an odorless crystalline powder with a brownish-red to reddish-brown color [107,109,110]. Rifampicin (C43H58N4O12, 822,95 g/mol) has a melting point of 183–188°C [39,111].
Rifampicin exerts conformational polymorphism, with Polymorphs I and II varying in both bioavailability and physicochemical properties [107,113,114]. Polymorphic Form I is considered the most stable form of rifampicin, and Form II is the metastable crystalline polymorphic form [107]. Characterization of some commercial rifampicin bulk samples indicated the presence of Form I, Form II, and amorphous states; however, these polymorphic variations confer minimal solubility advantage, rendering these differences both regulatory and clinically insignificant [114]. From a chemical perspective, rifampicin is a hydroquinone derivative containing three phenolic –OH groups that are prone to electrochemical or chemical oxidation [106]. Rifampicin possesses both an acidic p-hydroxy naphthyl ketone and a basic piperazine amine, with pKa values of 1.7 and 7.9, respectively. These confer amphoteric properties, enabling rifampicin to adopt a zwitterionic form depending on the pH of the medium in which it is presented [32,106,107,115].
Rifampicin is a lipophilic drug, with a log P value of 3.85, enabling it to partition into the hydrophobic cell wall of M. tuberculosis and potentially also the caseum [116]. Consequently, exploiting these properties can enhance the therapeutic efficacy of rifampicin in TB treatment [116]. Furthermore, the physicochemical properties of rifampicin contribute to its limitations as an ideal therapeutic agent. For instance, rifampicin exhibits pH-dependent solubility, which is further influenced by solvent types [109,117]. Although rifampicin demonstrates maximum chemical stability at pH 6 [118,119], its solubility increases in acidic environments such as the gastric milieu (pH of 1–3), with a maximum solubility at pH 1–2 [39,41]. This hydrophobic drug [110] is poorly soluble in acetone and in water (pH<6), with an aqueous solubility of 0.10 ± 0.01 mg/mL; it is soluble in methanol and ethyl acetate, and freely soluble in methyl chloride, dimethyl sulfoxide, and chloroform [106,109,117]. Alves et al. reported a significant decrease in rifampicin solubility from 100 mg/mL at pH 2 to 4 mg/mL at pH 5.3, with a further reduction to 2.8 mg/mL at pH 7.5 [120].
Rifampicin absorption predominantly occurs in the duodenum (pH 4–6), despite its relatively lower solubility in this region [120]. According to the Biopharmaceutical Classification System (BCS), rifampicin, previously categorized as a Class II drug, has recently been reclassified as a Class IV drug due to its limited intestinal permeability and poor aqueous solubility [121].
Rifampicin is available in both intravenous and oral formulations; however, the oral route is typically preferred, since intravenous administration is reserved for severe infections and is often associated with immunological reactions and adverse effects, which in turn can compromise patient compliance [110,122,123]. Limited research indicates significant differences in drug disposition between adults and pediatric populations, especially in infants [124,125,126,127,128]. This variation can likely be attributed to Cytochrome P450 (CYP) ontogeny, which begins shortly after birth and matures to adult levels between the ages of 1–10 years [122,124]. Rifampicin is, furthermore, a potent inducer for various enzymes, including permeability-glycoprotein (P-glycoprotein), 5’-diphospho-glucuronosyltransferase (UGTs), and CYP P450 isoenzymes, where CYP3A4 specifically mediates the autoinduction of rifampicin [129,130]. This drug undergoes hepatic deacetylation primarily through CYP3A4 to form its main metabolite, 25-diacetyl rifampicin, with the involvement of CYP2C isoenzymes such as CYP2C8, CYP2C9, CYP2C18, and CYP2C19 [123,125,129]. Figure 2 (a) illustrates the degradation pathway of rifampicin to form 25-diacetyl rifampicin under alkaline conditions [112]. Furthermore, since rifampicin is a potent CYP3A4 inducer, drug-drug interactions merit consideration, as they can result in suboptimal drug levels [122,129]. Therefore, the enzymatic instability of rifampicin further challenges its therapeutic efficacy [112,122,129].
Moreover, numerous challenges, such as efflux pump-mediated extrusion, resistance development, first-pass hepatic metabolism, limited intracellular bacterial accumulation, and peripheral neuropathy are associated with this gold-standard antitubercular drug [110]. The clinical efficacy of this drug is also compromised by adverse dose-dependent effects and inadequate drug penetration as a result of poor vascularization of pathological loci [110]. Rifampicin is subjected to enterohepatic circulation and is predominantly eliminated by means of the urinary and biliary routes [106,123,125]. In pediatrics, aged 3 months to 12 years, the half-life of rifampicin is approximately 2 hours [122].
A persistent challenge that continues to undermine TB treatment is the pronounced chemical instability of rifampicin [106,115,131,132], which is influenced by various factors such as the storage temperature and duration, light exposure, ionic strength of the solution, solvent type, and the pH of the medium [106,118]. In addition, rifampicin exhibits a high propensity for drug-drug interactions, which further compromises its bioavailability [30,32,38,123]. The degradation rate of rifampicin, being an endothermic reaction, accelerates significantly with an increase in storage temperature and prolonged duration, especially above 40°C [118]. Moreover, the degradation of this light-sensitive drug [132] is also accelerated by increased ionic strength in solution [118].
Rifampicin quinone, the primary degradation product of rifampicin, is formed through nonenzymatic autoxidation when in solution [131] – a temperature-dependent reaction that is potentiated under alkaline conditions, Figure 2 (b) [112,131,133]. Oxidation of rifampicin occurs in two steps: first, the hydroquinone functional group reversibly oxidizes to benzoquinone; second, the phenolic ring of rifampicin undergoes irreversible oxidation [133]. Rifampicin quinone is considered an impurity, as its presence in pharmaceutical formulations serves as an indicator of poor quality [109,131,132]. Interestingly, while rifampicin quinone exerts a limited antimicrobial effect, it retains distinct anti-inflammatory and immunosuppressive properties [134]. Under physiologically relevant temperatures and in solution, rifampicin quinone can undergo temperature-dependent chemical conversion to form rifampicin; nonetheless, its presence limits the bioavailability of rifampicin and contributes to the development of antimicrobial resistance [32,131,132,133]. Weinstein and Zaman [135] demonstrated that resistance to rifampicin quinone alarmingly confers cross-resistance to relevant clinical rifamycins, for example, rifampicin, rifapentine, and rifabutin. It is also evident that rifampicin quinone promotes rifampicin resistance in M. smegmatis and Escherichia coli [135]. It is worth mentioning that the other first-line anti-TB drugs can react with rifampicin quinone via their free amino groups; the reaction rate is high in alkaline environments and low in acidic environments, with ethambutol and pyrazinamide exhibiting the highest and lowest reaction rates, respectively [133].
The formulation of an acceptable drug delivery system for rifampicin is further complicated by its susceptibility to degradation, especially under acidic conditions [32,39,115]. Under such conditions, the azo-methine group of rifampicin undergoes reversible acid hydrolysis, resulting in the formation of 1-amino-4-methylpiperazine and 3-formyl-rifamycin SV, Figure 2 (c) [30,39,120]. The bioavailability of rifampicin is further reduced with the formation of 3-formyl-rifamycin SV, a poorly soluble compound that exerts in vitro antimicrobial activity but remains inactive in vivo [30,112,132]. It is worth noting that in vivo degradation is enhanced in fasting subjects with gastric pH values of 1.4–2.1 [12,38,112].
Furthermore, under acidic conditions, 3-formyl-rifamycin SV forms can subsequently react with isoniazid via a second-order reaction to form hydrazone, which, if unstable, can revert to 3-formyl-rifamycin SV and isoniazid through a pseudo-first-order reaction, Figure 2 (d) [12,38,112,136]. Since the second-order reaction occurs more rapidly than both the initial rifampicin and 3-formyl rifampicin reaction and the hydrazone with 3-formyl-rifamycin SV and isoniazid first-order reaction, the overall predominant response is hydrazone formation [12,132]. As shown in Figure 3, hydrazone is formed via a direct reaction between rifampicin and isoniazid, where the amino group of isoniazid nucleophilically attacks the imine group of rifampicin through a tetrahedral mechanism [38]. Consequently, this insoluble metabolite further exacerbates the poor bioavailability of rifampicin [12,30,32,38,132].
Considering the above, it was concluded that rifampicin and isoniazid decomposition in a fixed-dose combination with pyrazinamide could not be attributed solely to the direct transhydrazone formation between rifampicin and isoniazid. Instead, pyrazinamide plays a catalytic role in the decomposition process [38]. Therefore, the presence of pyrazinamide accelerates the decomposition via a base-catalyzed transhydrazone formation, mainly resulting in hydrazone. Pyrazinamide and ethambutol catalyze the decomposition of rifampicin and isoniazid through different mechanisms [38]. Ethambutol (both as the hydrochloride salt and free base), on the other hand, exerts a stronger catalytic effect than pyrazinamide due to its ethylenediamine or 1,2-amino alcohol functional group, which can abstract protons from two isoniazid molecules simultaneously. This results not only in hydrazone formation but also in additional degradation products, such as rifampicin-N-oxide and 25-desacetyl rifampicin, especially under alkaline conditions [38].
Therefore, it is not surprising that fixed-dose combination products comprising rifampicin, isoniazid, pyrazinamide, and/or ethambutol exhibit significantly greater chemical instability compared to rifampicin and isoniazid fixed-dose combinations [38]. Various strategies have been employed to prevent the undesirable interaction between rifampicin and isoniazid, to enhance the stability and, consequently, the bioavailability of rifampicin [137,138,139,140,141]. Although some studies have reported improved outcomes, the variable and poor bioavailability of rifampicin, most pronounced in fixed-dose combination formulations, remains a topic of considerable concern. Hence, the development of a stable, independent rifampicin formulation should be prioritized to ensure consistent bioavailability before it is incorporated into fixed-dose combination products.
4.2. Available Child-Friendly Rifampicin Dosage Forms
Despite the pivotal role of rifampicin in TB management since the 1960s and the alarming statistics of active pediatric TB cases, the majority of TB medicines remain adult-focused; a disquieting reality that underscores the persistent and widespread neglect of pediatric TB [8,12,46,85,98]. This is evident by the prevailing assumption that children are merely ‘small adults’, as pediatric TB treatment regimens are typically extrapolated from those of adults, necessitating adjustments for weight and age [17,43,44,46,48,142]. Such extrapolations can compromise pediatric treatment safety and efficacy, as extemporaneous preparations or the manipulation of adult formulations often result in suboptimal dosage forms with inaccurate weight-based dosing [12,37,45,46,85,142].
According to research, most commercially available drug products prescribed for pediatric TB are not available as child-friendly dosage forms, which contributes to poor adherence and, in turn, exacerbates morbidity and mortality from drug-resistant TB in children [17,45,46,48,49,143]. Notwithstanding these concerns, the development of child-friendly TB dosage forms remains limited. Pharmaceutical companies and research are deterred by this high-risk, small, and less lucrative population, which continues to impede the development of child-friendly medicines [12,17,37,45,144]. It is important to realize that, even when pediatric TB formulations exist, their clinical availability varies considerably across the world [48].
First-line TB drugs are available in water-dispersible fixed-dose combination formulations through the Stop TB Global Drug Facility, enabling more precise dosing and circumventing the need for extemporaneous preparations of adult dosage forms [143,145]. Besides the absence of child-friendly rifampicin-only formulations [145], these water-dispersible fixed-dose combination formulations are not widely available worldwide, especially in countries with low TB rates. [69,144,146].
Furthermore, a study by Wademan et al. revealed that, even with clear instructions on the proper administration of water-dispersible fixed-dose combination tablets, caregivers often modified the medicine using various strategies to improve pediatric TB treatment administration [37]. These strategies included administering the water-dispersible fixed-dose combination formulations in their solid tablet form or mixing them with fluids or food (for example, porridge, yoghurt, or water) to improve their poor palatability [37,45,53]. Such approaches are often employed in resource-constrained countries due to restricted access to safe water [48,144]. However, further research is required to determine the pharmacokinetic impact of these real-world administration practices [37,45]. Pragmatic challenges, including the time-intensive administration process and the need to dissolve the dispersible tablet, also hinder caregivers from integrating this treatment within their daily routines [37]. Furthermore, inadequate reconstitution of dispersible formulations can cause tissue injury (similar to tablets adhering to the esophagus) and prolong drug onset of action [142].
The WHO recommends the administration of fixed-dose combination formulations that combine drugs for both the intensive and continuation phases of TB treatment [86]. However, the available pediatric fixed-dose combinations for the intensive phase contain rifampicin and isoniazid in a 2:1 ratio instead of the recommended pediatric ratio of 1.5:1. As a result, additional isoniazid tablets are often required to achieve recommended weight-based dosing [48,85,147]. Moreover, the currently available child-friendly dispersible fixed-dose combination fails to provide adequate doses, underscoring an urgent need for higher dose formulations [146]. Pediatric patients often require higher dosages of rifampicin to attain serum concentrations extrapolatable to those in adults [85,147]. Additionally, higher rifampicin doses may be critical in severe TB forms, such as miliary TB and TB meningitis [146].
Aside from the advancements in the development of child-friendly dosage forms for drug-susceptible TB, the persistent absence of a rifampicin-only formulation [146] delineates a critical and unmet therapeutic need. Although rifampicin oral suspension is theoretically available globally, its absence in real-world practice is striking [143]. Alarmingly, in South Africa – where the TB burden is among the highest – the only available liquid rifampicin formulation has been withdrawn [50]. Furthermore, previous rifampicin oral suspensions failed to achieve target drug concentrations in the pediatric population at the recommended doses, plausibly attributed to its poor bioavailability [148].
4.3. General Requirements and Considerations for Child-Friendly Dosage Forms
Acceptability can be described as “the overall ability and willingness of the patient to use, and of the caregiver to administer, the medicine as intended” [53]. This paramount requirement ensures optimal adherence to treatment, hence, safeguarding both the safety and efficacy of TB therapy [53]. Enhancing pediatric acceptability, and consequently, adherence to rifampicin treatment, necessitates consideration of characteristics that are both age-appropriate and acceptable across this heterogeneous population [149].
However, non-adherence to pediatric medicines is particularly arduous to circumvent due to multifactorial influences such as socioeconomic status, cultural background, health literacy, family structure, and age [142,143,150]. This represents a multifaceted problem encompassing the collective involvement of interdisciplinary healthcare teams, caregivers, and the patient [150]. Despite the growing availability of child-friendly dosage forms, including dispersible and mini-tablets, oral liquid formulations remain the preferred choice for children under 12 years and their caregivers, owing to their palatability, flexible dosing, ease of administration, and suitability across the pediatric population (including infants) [12,37,142,144,149]. Furthermore, the swallowability of liquid formulations reduces the risk of choking and benefits not only the pediatric population but also the general population, particularly those with swallowing difficulties [12,149].
Pediatric acceptability towards treatment is strongly influenced by key attributes, prioritized as follows: taste, swallowability, ease of administration, mouthfeel/texture, color/appearance, smell, and aftertaste. Notably, pediatric patients are more sensitive to sensory characteristics than adults [37,53,144,149]. Therefore, formulation-related factors, particularly the recalcitrance and organoleptic characteristics of the dosage form, constitute significant contributors to treatment non-adherence in this population [150]. The unpalatability of first-line anti-TB drugs is often cited as the main cause of treatment non-adherence in children. Rifampicin, in particular, is considered the most bitter [37,151]. What further exacerbates the complexity of this problem is the pronounced sensitivity of the pediatric population to excipients intended to enhance palatability, coupled with the frequent exclusion of pediatric patients from clinical trials, rendering the ethical development of child-friendly dosage forms particularly challenging [17,53]. Although the 2013 Guideline on Pharmaceutical Development of Medicines for Pediatric Use (EMA/CHMP/QWP/805880/2012 Rev. 2) issued by the European Medicines Agency emphasized the importance of incorporating pediatric medicine acceptability into clinical studies, achieving adequate pediatric medicine acceptability in practice remains a persistent barrier to safe and efficient treatment [53]. To this day, an urgent need remains to develop appropriate, palatable, child-friendly rifampicin formulations to ensure optimal pediatric TB treatment outcomes [12,17].
5. Can the Therapeutic Efficacy of Rifampicin Liquid Formulations be Improved?
Approximately 70% of currently available drug candidates belong to BCS class II (i.e., low solubility and high permeability), while an additional 20% fall under BCS class IV (low solubility and low permeability) [152,153], rendering most anti-TB drugs sparingly soluble in aqueous media. Nevertheless, to address these persistent formulation challenges associated with poorly soluble drugs, lipid-based formulations have become a leading strategy [154,155,156,157]. The potential of these drug delivery systems to improve the delivery of various anti-TB drugs has been extensively investigated, owing to their favorable biopharmaceutical attributes [29,39,158].
Typically composed of fatty acid esters, lipid-based drug delivery systems constitute a continuum from simple drug-in-oil formulations to advanced delivery systems capable of spontaneously emulsifying in an aqueous environment [159,160]. These novel lipid-based formulations – including emulsions, lipid particulate systems, and vesicular systems, along with their subcategories – offer commercially viable options for developing effective and safe pharmaceuticals for ocular, oral, pulmonary, parenteral, and topical delivery routes of administration [159,161].
Self-emulsifying drug delivery systems (SEDDSs), a subset of these formulations that rely on spontaneous emulsification, have attained broad recognition as a well-established approach for the oral administration of poorly soluble drugs [156,162,163]. The development of rifampicin lipid-based delivery systems, particularly SEDDSs, presents a promising approach to overcome several of the aforementioned challenges [163].
5.1. Self-Emulsifying Drug Delivery Systems
SEDDSs are emulsion concentrates comprising a drug incorporated into a mixture of oil, surfactants, and, in some cases, co-surfactants and/or co-solvents [152,159,163]. In their initial state, SEDDSs are not emulsions; however, upon mild agitation in the aqueous environment of the stomach, spontaneous emulsions are formed [152,161,163,164]. Consequently, these delivery systems provide an effective means of improving the solubilization of poorly aqueous soluble drugs [152,163]. The presence of these minute oil-in-water droplets within the gastrointestinal tract bypasses the dissolution stage typically required for dispersed powders, thereby facilitating improved drug solubilization and absorption [162]. Unlike other lipid-based formulations, SEDDSs demonstrate a promising approach for achieving higher drug-loading capacities [157]. This improvement can be ascribed to the enhanced solubility of poorly water-soluble drugs, particularly those with an intermediate partition coefficient (2 < log P < 4), within the formulation’s co-surfactant and amphiphilic surfactant components [157].
SEDDSs production involves a comparatively straightforward and less intricate formulation process than that of conventional emulsions. This process entails mixing oil, surfactants, and/or co-surfactants, followed by the addition of the drug and vortexing to obtain a clear, homogeneous solution. Alternatively, the drug may first be dissolved in one or more excipients before combining with the remaining components. This approach enables the construction of a pseudoternary phase diagram for the identification of the optimal self-emulsification region. Moreover, the methodologies governing SEDDS development are well-established and comprehensively documented in scientific literature [39,165,166].
The term SEDDS, however, is a broad term encompassing several specific lipid-based drug delivery systems, which are primarily distinguished into three categories according to droplet size. Based on their preparation method and composition, drug-loaded SEDDSs can be formulated as conventional SEDDSs, self-microemulsifying drug delivery systems (SMEDDSs), or self-nanoemulsifying drug delivery systems (SNEDDSs) [155,163,165]. The general properties and composition of these systems are briefly summarized in Table 2; however, these properties represent overall trends and may vary depending on specific formulation parameters. Additionally, lipid-based formulations are categorized within the Lipid-Based Formulation Classification System (LFCS) according to their composition and behavior upon dispersion within an aqueous environment [152,155,167]. LFCS divides lipid-based formulations into four types (I–IV), with conventional SEDDSs, SMEDDSs, and SNEDDSs as type II, type IIIA/IIIB, and type IIIB, respectively [162,167,168].
The successful formulation of a SEDDS of maximal therapeutic efficacy necessitates consideration of several key factors, including the physicochemical properties of both the drug and the excipients, drug-excipient interactions (in vivo and in vitro), and the physiological factors that may impede or enhance bioavailability [156]. Furthermore, formulation design must also account for practical considerations such as the cost of materials, regulatory status, miscibility, solubilization capacity, the physical state of excipients at ambient temperature, and chemical stability [156,159,163]. This systematic and rational approach serves to both expedite the formulation development process and reduce the associated costs [156].
5.1.1. Composition of Self-Emulsifying Drug Delivery Systems
Drugs. As per the BCS, Class II and IV drugs, characterized by poor solubility, are often prime candidates for lipid-based delivery, as these systems can significantly enhance solubility and absorption. However, formulation efficiency is strongly influenced by physicochemical properties such as solubility, partition coefficient, molecular weight, and chemical stability within the gastrointestinal tract [156,162]. Ideally, a drug should possess a log P value of at least 2 [157]. In general, drugs with a log P value greater than 5 exhibit optimal solubility in the oil/lipid, surfactant, and co-surfactant/co-solvent phases, thereby promoting efficient absorption through intestinal epithelial cells and subsequent integration into chylomicrons [157]. Moreover, these highly lipophilic drugs possess enhanced solubility in lipid phases and improved absorption through lymphatic transport, effectively bypassing hepatic first-pass metabolism. Nevertheless, such drugs may also encounter formulation challenges, including precipitation upon dilution [156].
Oils/lipids. Within SEDDSs, the oil phase solubilizes the lipophilic/hydrophobic drug, thereby increasing both drug-loading capacity and oral bioavailability [156]. The hydrophilic-lipophilic balance (HLB), physical properties, and melting characteristics of glycerides depend on the nature of the fatty acid composition, degree of esterification, and their interaction with glycerol to produce mono- or diglycerides [166]. Lipids are commonly classified according to fatty acid chain length into short-chain triglycerides (<5C), medium-chain triglycerides (C6–C12), and long-chain triglycerides (C14–C20) [152,156,161,163,166].
Medium-chain triglycerides, such as coconut-derived glyceryl tricaprylate, are widely used in SEDDSs due to their high oxidative stability and superior solvent capacity [152,156,161,163,166]. Long-chain triglycerides, predominantly found in fixed oils such as vegetable oils, consist mainly of glyceride esters derived from unsaturated long-chain fatty acids. These are generally regarded as safe and readily digestible [156,161]. The substantial hydrophobic domains of both medium- and long-chain triglycerides confer considerable solubilization potential for lipophilic drugs. Upon digestion, short- and medium-chain triglycerides are absorbed through the portal vein, whereas long-chain derived chylomicrons promote lymphatic transport [156,160,168]. The self-emulsification process enhances solubility by reducing interfacial tension between the water and oil interface, and modulating the time and curvature of the interfacial film, by minimizing precipitation [166].
A study by van Deventer et al. [39] demonstrated that although rifampicin exhibited higher solubility in olive oil (0.46 ± 0.12 mg/mL) than in water, its solubility in the olive oil remained lower than what was anticipated. Olive oil is predominantly composed of fatty acid components such palmitic acid (11–12%), oleic acid (about 76–77%), as well as smaller quantities of linoleic, linolenic, palmitoleic, and stearic acids. These components contribute to lowering the pH of the oil to approximately 5.17 at ambient temperature, which further decreases to about 4.49 during storage as the %oleic acid (free oil acidity) increases. As a result, these factors limited rifampicin solubilization and promoted its degradation over 24 hours, emphasizing the importance of selecting an appropriate oil phase in SEDDSs containing rifampicin [39].
Surfactants. Amphiphilic molecules, or surfactants, contain both lipophilic and hydrophilic functional groups, enabling them to reduce interfacial tension at the oil-water interface [156,161]. They are classified as ampholytic, anionic, cationic, or non-ionic, depending on their ionization behavior in aqueous media [161]. In SEDDSs, the drug-loaded oil phase is dispersed into minute droplets, significantly increasing the surface area and, therefore, requiring adequate surfactant concentrations to ensure solubilization [169].
Excessive surfactant concentrations, however, can paradoxically increase droplet size by promoting water infiltration into the oil droplets and disrupting the interfacial film. High surfactant concentrations may also cause gastrointestinal irritation [156]. The required surfactant concentration varies with the oil’s molecular volume, with long-chain triglycerides typically necessitating higher surfactant levels than short-chain di- or monoglycerides. The hydrophilic-lipophilic balance (HBL) determines the emulsion type formed, i.e., oil-in-water, water-in-oil, or bicontinuous emulsions [156].
Nonionic surfactants are predominantly preferred in SEDDSs due to their reduced critical micelle concentrations, low toxicity, and high stability across a wide pH range. Moreover, they do not interact with ionic drugs and are effective in reducing interfacial tension at relatively low concentrations [152,166,169]. Certain surfactants and lipids can also inhibit intestinal efflux transporters such as P-glycoprotein and metabolic enzymes (i.e., CYPs), enhancing drug absorption [168]. Additionally, combinations of oils and/or surfactants, along with their digestion products, can increase intestinal permeability by fluidizing cell membranes and opening tight junctions [168]. Collectively, these properties render surfactants indispensable for the stabilization of fine droplets, improved solubilization, and enhanced bioavailability of lipophilic drugs.
Co-surfactants/co-solvents. Co-surfactants further reduce transient negative interfacial tension, and in turn, they impart interfacial film flexibility, enabling varied curvatures necessary for achieving diverse microemulsion concentrations [170]. Moreover, the addition of co-surfactants simulates higher surfactant amounts, [170], thereby facilitating the formation of finely dispersed droplets. While the film becomes adequately depleted, the positive interfacial tension is restored through the absorption of additional surfactant or adjustment of the surfactant-to-co-surfactant ratio, leading to spontaneous emulsification [171].
Co-solvents, on the other hand, improve the solubilization of incorporated drugs and aid in the dispersion of hydrophilic surfactants within the oil phase, which helps maintain stability and uniformity [168]. Accordingly, co-surfactants and co-solvents act synergistically with surfactants to enhance emulsification, minimize drug precipitation, and augment thermodynamic stability, ensuring uniform drug delivery and optimal absorption [162].
5.2. Optimizing Pediatric Oral Bioavailability of Lipophilic Drugs
The inclusion of poorly aqueous soluble or lipophilic drugs, for example, rifampicin, within a formulation remains an arduous endeavor, since their solubility and absorption in the aqueous environment of the gastrointestinal tract are inherently limited [154,160]. The observed enhancement in the bioavailability of lipophilic drugs when co-administered with fat-rich meals has prompted significant interest in developing oral lipid-based formulations designed to increase drug solubilization in the gastrointestinal tract [164,172]. Poor oral bioavailability may result from multiple interrelated factors, of which adequate solubility within the gastrointestinal lumen is a critical prerequisite, particularly in pediatric patients [154,168]. To fully elucidate the mechanisms and benefits of lipid-based formulations, it is fundamental to grasp the physiological response of the body to lipid intake, while considering the distinct subgroups within the pediatric population [154,155].
After oral administration, the presence of lipid-enriched chyme in the duodenum elicits the secretion of the intestinal peptide hormone cholecystokinin. Cholecystokinin induces simultaneous relaxation of the hepatopancreatic sphincter and contraction of the gallbladder, leading to the release of bile – comprising bile salts, cholesterol, and phospholipids (predominantly phosphatidylcholine) – into the small intestine [172,173]. Concurrently, pancreatic fluids containing lipase and co-lipase are released into the intestines [173]. In addition, pancreatic phospholipase A hydrolyzes endogenous and biliary- or formulation-derived phospholipids to produce free fatty acids and lysophosphatidylcholine, which together contribute to the formation of mixed micelles [173].
The digestion and absorption of endogenous lipids provide an interactive and dynamic conduit for rifampicin delivery and other lipophilic drugs [172]. During lipid digestion, pancreatic lipases convert triglycerides into fatty acids, monoglycerides, and diglycerides [163,172]. These enzymatic processes naturally promote the solubilization of ingested lipids, while gastric lipolysis, albeit to a lesser extent, initiates lipid emulsification. Lipid digestion continues in the small intestine via pancreatic esterases and lipases, during which bile constituents combine with phospholipids to form bile-phospholipid mixed micelles that result in various colloidal structures [160,165,173]. These highly dispersed small colloidal entities aid in the solubilization of ingested poorly water-soluble lipids [173] and facilitate the transport of hydrophobic molecules across the viscous unstirred water layer to the absorptive surface of the intestinal epithelium [172].
Because rifampicin exhibits restricted transport across the viscous unstirred water layer [169], it is particularly well-suited to absorption enhancement through lipid-based formulation strategies. Although the concept of lipid-based formulations was initially inspired by the co-administration of lipophilic drugs together with a fat-rich meal, co-administration thereof, especially in the case of rifampicin, can alter drug-lipoprotein interactions and lymphatic transport, potentially enhancing drug plasma concentrations [163,172].
Lipid-based formulations are designed to exploit these intrinsic lipid digestion pathways, thereby creating an ideal microenvironment in which lipophilic drugs with pronounced lipid affinity can partition into exogenous lipid carriers [165,173]. The digested lipid products, along with the solubilized drug, are absorbed upon reaching the enterocytes of the gastrointestinal wall [160,165]. Bile-phospholipid mixed micelles mediate the physiological effect of exogenous lipid absorption, serving a dual function: they enhance the solubilization of poorly water-soluble drugs and establish a lipid absorption concentration gradient [165]. Incorporating rifampicin into lipid-based formulations will therefore probably improve gastrointestinal absorption through the formation of solubilized colloidal structures. These solubilized colloids will promote enterocyte-based transport, modulate efflux and uptake mechanisms, and facilitate absorption into the intestinal lymphatic system and systemic circulation [165,174]. Studies have indicated that lipid quantities of merely 2 g are adequate to elicit biliary secretion and augment gastrointestinal bile salt concentrations, which in turn improve intraluminal lipid metabolism and solubilization of drugs [175]. Once a lipophilic drug is solubilized, several mechanisms contribute to lipid-mediated drug absorption after oral administration (Figure 4). These include: (a) facilitated transcellular drug absorption via increased membrane fluidity; (b) surfactant-mediated inhibition of CYP P450 enzymes and/or P-glycoprotein, resulting in an increased intracellular drug concentration and extended residence time; (c) paracellular drug transport through the opening of tight junctions; and (d) lipid-induced lipoprotein and chylomicron production, enhancing lymphatic drug uptake [163].
The intestinal lymphatic system provides an alternative pathway for the transportation of highly lipophilic drugs into the systemic circulation [169]. Long-chain triglycerides with solubility exceeding 50 mg/kg and drugs with a log P value greater than 5, demonstrate limited diffusion across blood capillaries and are therefore preferentially absorbed via the lymphatic pathway [169,176]. Within the enterocytes, chylomicrons act as carriers for these drugs, forming drug-chylomicron complexes that are subsequently transported through the mesenteric lymphatic system to the systemic circulation at the junction of the left subclavian and left jugular veins [169]. This pathway not only bypasses first-pass hepatic metabolism, improving oral bioavailability, but also contributes to site-specific targeting of lymph-manifested diseases such as lymph node TB [163,169,177].
Ontogenetic physiological differences in the pediatric population profoundly affect the solubilization and absorption of lipid-based formulations. Unlike adults, young children exhibit reduced lipase activity, lower bile salt secretion, smaller gastric volumes, and distinct gastric mortality patterns [150,154]. For instance, the immature bile and pancreatic secretions may negatively affect the overall bioavailability of lipid drugs [150]. In neonates, insufficient luminal bile salt concentrations may further impair lipophilic drug absorption, necessitating careful dose adjustments. However, bile salt secretion progressively increases with postnatal maturation, gradually improving lipid drug absorption [150,154]. In infants aged six to eight months, the gastric emptying rate is slower due to immature neuroregulation of gastric motility [150]. Importantly, significant knowledge gaps persist regarding gastrointestinal drug disposition in neonates and infants younger than six months [154].
6. Liquid Self-Emulsifying Drug Delivery Systems as a Novel Approach to Enhance Oral Rifampicin Delivery
Rifampicin, a pillar of anti-TB drug therapy, remains constrained by its physicochemical instabilities and drug interactions. These limitations have long hindered the development of reliable liquid formulations and perpetuated pediatric TB treatment gaps, particularly in LMICs, where the TB burden is greatest [8,12,38,39,112,123,131,132,136,148].
Various formulation technologies exist to improve drug solubility and mitigate bioavailability issues. Among these, lipid-based formulations such as SEDDSs offer an economically viable solution and are firmly embedded in oral dosage forms [165]. The relevance of SEDDSs in TB treatment stems from their potential to enhance lipophilic TB drug treatment efficacy through improving the solubility and, consequently, bioavailability while also enabling controlled drug release [29,39,158].
Furthermore, it has been demonstrated that effective solubilization of lipophilic drugs can be achieved with lipid quantities as low as 2 g, thereby enabling SEDDSs to facilitate optimal gastrointestinal absorption regardless of low dietary fat intake, an important consideration in TB management [175]. SEDDSs not only improve the stability of highly lipophilic drug emulsions but also support low-cost, scalable manufacturing, offering particular value in LMIC settings. Moreover, these drug delivery systems can protect drugs such as rifampicin from hydrolysis [156,166,172].
In addition, SEDDSs may inhibit P-glycoprotein-mediated efflux and enhance lymphatic drug transport through stimulation of lipoprotein and chylomicron production, consequently circumventing first-pass hepatic metabolism [163,169]. Beyond these intrinsic pharmacokinetic advantages, liquid SEDDS confer practical clinical benefits that enable weight-based dosing, ease of administration, and improved patient acceptability [12,149]. These advantages are particularly relevant for the growing pediatric TB population, where age-appropriate and suitable rifampicin TB formulations remain scarce yet urgently needed [17,37,46].
Despite reports of improved rifampicin solubility in fixed-dose combination SEDDS formulations containing isoniazid and rifampicin, no oral liquid rifampicin lipid-based formulations – such as SEDDSs – have yet been successfully developed or made commercially available. Evidence from literature indicates that, in separate studies, the solubility, physicochemical characteristics and formulation stability of rifampicin were improved when the drug was incorporated into SEDDSs based formulations [39,178]. Collectively, these findings emphasize the importance of excipient selection in the development of rifampicin SEDDSs [39,178].
In conclusion, the development of oral liquid rifampicin-based SEDDSs provides much more than a minor formulation adjustment. These drug delivery systems offer a strategic and scientific means to overcome the longstanding physicochemical barriers of rifampicin. By maintaining rifampicin in a stable and solubilized state throughout the gastrointestinal tract, liquid SEDDS formulations have the potential to deliver reliable, age-appropriate rifampicin therapy, narrowing established pediatric TB treatment disparities in high TB burden areas.
7. Conclusions
Pediatric TB endures as a underprioritized global health concern, where preventable morbidity and mortality continue to burden this vulnerable population [4]. This is further intestified by the susceptibility of young children to TB progression and disseminated disease [60,64,65], diagnosing challenges [58], and shortcomings in pharmacotherapy [4]. Rifampicin, a cornerstone first-line TB drug, continues to face major challenges, such as a lack of rifampicin child-friendly dosage forms [50,51], physicochemical instabilities [106,112], and drug-drug interactions which further compromise its bioavailability [30,38,123]. These barriers often lead to inadequate dosing, poor treatment adherence, and an increased risk of drug resistance [29,37,90]. As emphasized in this review, there is a critical need for commercially available rifampicin child-friendly formulations to ensure effective and safe drug delivery and pharmacotherapy [53,146]. Liquid rifampicin SEDDSs offer a promising approach as they may enhance drug aqueous solubility, improve stability [162], increase gastro-intestinal absorption [175], and may potentially bypass first-pass hepatic metabolism [163,169]. These lipid-based delivery systems are scalable, feasible, and well suited for weight-based dosing, making them for the pediatric population, particulary those in low-resource settings [12,162,166]. Although no oral liquid rifampicin SEDDS has yet reached the market, advancing this promising approach could finally deliver a stable, child-friendly formulation filling the longstanding void in pediatric TB treatment. As a focused examination of pediatric TB, this review is an imperative approach to emphasize the crucial need for sustained research and innovation to not only change the global trajectory of pediatric TB but also to drive decisive action.
Author Contributions
Conceptualization, J.M.V.; methodology, J.V.M. and K.M.dK.; software, J.M.V. and K.M.dK.; research, K.M.dK.; writing—original draft preparation, K.M.dK.; writing—review and editing, J.M.V., K.M.dK., M.B., and C.B.B.; visualization, J.M.V. and K.M.dK.; supervision, J.M.V., M.B., and C.B.B.; project administration, J.M.V.; funding acquisition, J.M.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research did not receive any dedicated funding from public, commercial, or non-profit agencies.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank the Centre of Excellence for Pharmaceutical Sciences (Pharmacen™), Faculty of Health Sciences, North-West University, South Africa, for the financial contribution to this project. During the preparation of this manuscript, the authors used applications in Microsoft Word, namely Writefull and Grammarly, to assist with improving in-text grammar. The authors have reviewed and edited all outputs and take full responsibility for the content of this publication. No AI was used to create any text, paragraphs, or results. ChemSketch (ACD/Labs), ChatGPT, and Canva (free versions) were used only to assist in the partial development of the figures in this manuscript
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Zumla, A.; Sahu, S.; Ditiu, L.; Singh, U.; Park, Y.-J.; Yeboah-Manu, D.; Osei-Wusu, S.; Asogun, D.; Nyasulu, P.; Tembo, J.; et al. Inequities Underlie the Alarming Resurgence of Tuberculosis as the World's Top Cause of Death From an Infectious Disease - Breaking the Silence and Addressing the Underlying Root Causes. IJID Regions 2025, 14, 100587. [Google Scholar] [CrossRef]
- Gulumbe, B.H.; Abdulrahim, A.; Danlami, M.B. The United Nations' Ambitious Roadmap Against Tuberculosis: Opportunities, Challenges and the Imperative of Equity. Future Science OA 2024, 10, 2418787. [Google Scholar] [CrossRef]
- Bhargava, A.; Bhargava, M.; Pai, M. Tuberculosis: A Biosocial Problem That Requires Biosocial Solutions. Lancet 2024, 403, 2467–2469. [Google Scholar] [CrossRef]
- Tristram, D.; Tobin, E.H. Tuberculosis in Children. Available online: https://www.ncbi.nlm.nih.gov/books/NBK610681/ (accessed on 3 June 2025).
- World Health Organization. Tuberculosis Resurges as Top Infectious Disease Killer. Available online: https://www.who.int/news/item/29-10-2024-tuberculosis-resurges-as-top-infectious-disease-killer (accessed on 24 May 2025).
- Chakaya, J.; Khan, M.; Ntoumi, F.; Aklillu, E.; Fatima, R.; Mwaba, P.; Kapata, N.; Mfinanga, S.; Hasnain, S.E.; Katoto, P.D.M.C.; et al. Global Tuberculosis Report 2020 - Reflections on the Global TB Burden, Treatment and Prevention Efforts. International Journal of Infectious Diseases 2021, 113, S7–S12. [Google Scholar] [CrossRef]
- Grange, J.M.; Zumla, A. The Global Emergency of Tuberculosis: What Is the Cause? Journal of the Royal Society for the Promotion of Health 2002, 122, 78–81. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2024; Geneva, Switzerland, 2024. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2023; Geneva, 2023. [Google Scholar]
- Lönnroth, K.; Migliori, G.B.; Abubakar, I.; D'Ambrosio, L.; de Vries, G.; Diel, R.; Douglas, P.; Falzon, D.; Gaudreau, M.-A.; Goletti, D.; et al. Towards Tuberculosis Elimination: An Action Framework for Low-Incidence Countries. European Respiratory Journal 2015, 45, 928–952. [Google Scholar] [CrossRef] [PubMed]
- Tobin, E.H.; Tristram, D. Tuberculosis Overview. Available online: https://www.ncbi.nlm.nih.gov/books/NBK441916/ (accessed on 4 June 2025).
- Frutos, D.M.; Hidalgo, I.M.; Negre, J.S. Tuberculosis Treatment in Paediatrics: Liquid Pharmaceutical Forms. Rev Enf Emerg 2020, 19, 169–176. [Google Scholar]
- Borrell, S.; Trauner, A.; Brites, D.; Rigouts, L.; Loiseau, C.; Coscolla, M.; Niemann, S.; De Jong, B.; Yeboah-Manu, D.; Kato-Maeda, M.; et al. Reference Set of Mycobacterium tuberculosis Clinical Strains: A Tool for Research and Product Development. PLoS ONE 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the Development of New Tuberculosis Drugs and Treatment Regimens. Nature Reviews Drug Discovery 2013, 12, 388–404. [Google Scholar] [CrossRef]
- Suvvari, T.K. The Persistent Threat of Tuberculosis - Why Ending TB Remains Elusive? Journal of Clinical Tuberculosis and Other Mycobacterial Diseases 2025, 38, 100510. [Google Scholar] [CrossRef] [PubMed]
- Mane, S.S.; Shrotriya, P. Current Epidemiology of Pediatric Tuberculosis. Indian Journal of Pediatrics 2024, 91, 711–716. [Google Scholar] [CrossRef]
- Maphalle, L.N.F.; Michniak-Kohn, B.B.; Ogunrombi, M.O.; Adeleke, O.A. Pediatric Tuberculosis Management: A Global Challenge or Breakthrough? Children 2022, 9. [Google Scholar] [CrossRef]
- Huynh, J.; Abo, Y.-N.; Triasih, R.; Singh, V.; Pukai, G.; Masta, P.; Tsogt, B.; Luu, B.K.; Felisia, F.; Pank, N.; et al. Emerging Evidence to Reduce the Burden of Tuberculosis in Children and Young People. International Journal of Infectious Diseases 2025, 155, N.PAG–N.PAG. [Google Scholar] [CrossRef] [PubMed]
- Getahun, H.; Matteelli, A.; Chaisson, R.E.; Raviglione, M. Latent Mycobacterium tuberculosis Infection. New England Journal of Medicine 2015, 372, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Comstock, G.W.; Livesay, V.T.; Woolpert, S.F. The Prognosis of a Positive Tuberculin Reaction in Childhood and Adolescence. American Journal of Epidemiology 1974, 99, 131–138. [Google Scholar] [CrossRef] [PubMed]
- van der Zalm, M.M.; Jongen, V.W.; Swanepoel, R.; Zimri, K.; Allwood, B.; Palmer, M.; Dunbar, R.; Goussard, P.; Schaaf, H.S.; Hesseling, A.C.; et al. Impaired Lung Function in Adolescents with Pulmonary Tuberculosis During Treatment and Following Treatment Completion. EClinicalMedicine 2024, 67. [Google Scholar] [CrossRef]
- Martinez, L.; Gray, D.M.; Botha, M.; Nel, M.; Chaya, S.; Jacobs, C.; Workman, L.; Nicol, M.P.; Zar, H.J. The Long-Term Impact of Early-Life Tuberculosis Disease on Child Health: A Prospective Birth Cohort Study. American Journal of Respiratory and Critical Care Medicine 2023, 207, 1080–1088. [Google Scholar] [CrossRef]
- Nkereuwem, E.; Agbla, S.; Sallahdeen, A.; Owolabi, O.; Sillah, A.K.; Genekah, M.; Tunkara, A.; Kandeh, S.; Jawara, M.; Saidy, L.; et al. Reduced Lung Function and Health-Related Quality of Life After Treatment for Pulmonary Tuberculosis in Gambian Children: A Cross-Sectional Comparative Study. Thorax 2022, 78, 281–287. [Google Scholar] [CrossRef]
- Nkereuwem, E.; Ageiwaa Owusu, S.; Fabian Edem, V.; Kampmann, B.; Togun, T. Post-Tuberculosis Lung Disease in Children and Adolescents: A Scoping Review of Definitions, Measuring Tools, and Research Gaps. Paediatric Respiratory Reviews 2025, 53, 55–63. [Google Scholar] [CrossRef]
- Blomberg, B.; Spinaci, S. The Rationale for Recommending Fixed-Dose Combination Tablets for Treatment of Tuberculosis. Bulletin of the World Health Organization 2001, 79, 61–68. [Google Scholar]
- Seneadza, N.A.H.; Antwi, S.; Yang, H.; Enimil, A.; Dompreh, A.; Wiesner, L.; Peloquin, C.A.; Lartey, M.; Lauzardo, M.; Kwara, A. Effect of Malnutrition on the Pharmacokinetics of Anti-TB drugs in Ghanaian Children. International Journal of Tuberculosis and Lung Disease 2021, 25, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Mallick, M.; Mrigpuri, P.; Singla, N.; Gupta, A. Sequelae of Pulmonary Multidrug-Resistant Tuberculosis at the Completion of Treatment. Lung India 2018, 35, 4–8. [Google Scholar] [CrossRef]
- Atkins, S.; Heimo, L.; Carter, D.; Closa, M.R.; Vanleeuw, L.; Chenciner, L.; Wambi, P.; Sidney-Annerstedt, K.; Egere, U.; Verkuijl, S.; et al. The Socioeconomic Impact of Tuberculosis on Children and Adolescents: A Scoping Review and Conceptual Framework. BMC Public Health 2022, 22. [Google Scholar] [CrossRef] [PubMed]
- Buya, A.B.; Witika, B.A.; Bapolisi, A.M.; Mwila, C.; Mukubwa, G.K.; Memvanga, P.B.; Makoni, P.A.; Nkanga, C.I. Application of Lipid-Based Nanocarriers for Antitubercular Drug Delivery: A Review. Pharmaceutics 2021, 13. [Google Scholar] [CrossRef]
- .
- Sadaphal, P.; Chakraborty, K.; Jassim-AlMossawi, H.; Pillay, Y.; Roscigno, G.; Kaul, A.; Kak, N.; Matji, R.; Mvusi, L.; DeStefano, A. Rifampicin Bioavailability in Fixed-Dose Combinations for Tuberculosis Treatment: Evidence and Policy Actions. Journal of Lung Health and Diseases 2019. [Google Scholar] [CrossRef]
- Angiolini, L.; Agnes, M.; Cohen, B.; Yannakopoulou, K.; Douhal, A. Formation, Characterization and pH Dependence of Rifampicin: Heptakis(2,6-di-O-methyl)-β-Cyclodextrin Complexes. International Journal of Pharmaceutics 2017, 531, 668–675. [Google Scholar] [CrossRef]
- Mereškevičienė, R.; Danila, E. The Adverse Effects of Tuberculosis Treatment: A Comprehensive Literature Review. Medicina 2025, 61. [Google Scholar] [CrossRef]
- Thakur, A. Advancements in Tuberculosis Treatment: From Epidemiology to Innovative Therapies. International Journal of Science and Research (IJSR) 2024, 13. [Google Scholar] [CrossRef]
- Varshney, K.; Patel, H.; Kamal, S. Trends in Tuberculosis Mortality Across India: Improvements Despite the COVID-19 Pandemic. Cureus 2023, 15, e38313. [Google Scholar] [CrossRef]
- Alemu, A.; Bitew, Z.W.; Diriba, G.; Gumi, B. Risk Factors Associated with Drug-Resistant Tuberculosis in Ethiopia: A Systematic Review and Meta-analysis. Transboundary and Emerging Diseases 2022, 69, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Wademan, D.T.; Busakwe, L.; Nicholson, T.J.; van der Zalm, M.; Palmer, M.; Workman, J.; Turkova, A.; Crook, A.M.; Thomason, M.J.; Gibb, D.M.; et al. Acceptability of a First-Line Anti-Tuberculosis Formulation for Children: Qualitative Data from the SHINE Trial. The International Journal of Tuberculosis and Lung Disease 2019, 23, 1263–1268. [Google Scholar] [CrossRef]
- Bhutani, H.; Singh, S.; Jindal, K.C.; Chakraborti, A.K. Mechanistic Explanation to the Catalysis by Pyrazinamide and Ethambutol of Reaction Between Rifampicin and Isoniazid in Anti-TB FDCs. Journal of Pharmaceutical and Biomedical Analysis 2005, 39, 892–899. [Google Scholar] [CrossRef]
- van Deventer, M.; Haynes, R.K.; Brits, M.; Viljoen, J.M. Formulation of Topical Drug Delivery Systems Containing a Fixed-Dose Isoniazid–Rifampicin Combination Using the Self-Emulsification Mechanism. Pharmaceutics 2025, 17, 680. [Google Scholar] [CrossRef]
- Iftikhar, S.; Sarwar, M.R. Potential Disadvantages Associated with Treatment of Active Tuberculosis Using Fixed-Dose Combination: A Review of Literature. Journal of Basic and Clinical Pharmacy 2017, 8, S131–136. [Google Scholar]
- Mariappan, T.T.; Singh, S. Regional Gastrointestinal Permeability of Rifampicin and Isoniazid (Alone and Their Combination) in the Rat. International Journal of Tuberculosis and Lung Disease 2003, 7, 797–803. [Google Scholar]
- Denti, P.; Wasmann, R.E.; van Rie, A.; Winckler, J.; Bekker, A.; Rabie, H.; Hesseling, A.C.; van der Laan, L.E.; Gonzalez-Martinez, C.; Zar, H.J.; et al. Optimizing Dosing and Fixed-Dose Combinations of Rifampicin, Isoniazid, and Pyrazinamide in Pediatric Patients with Tuberculosis: A Prospective Population Pharmacokinetic Study. Clinical Infectious Diseases 2022, 75, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Tsiligiannis, A.; Sfouni, M.; Nalda-Molina, R.; Dokoumetzidis, A. Optimization of a Paediatric Fixed Dose Combination Mini-tablet and Dosing Regimen for the First Line Treatment of Tuberculosis. European Journal of Pharmaceutical Sciences 2019, 138, 105016. [Google Scholar] [CrossRef]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nature Reviews Disease Primers 2016, 2. [Google Scholar] [CrossRef]
- Matawo, N.; Adeleke, O.A.; Wesley-Smith, J. Optimal Design, Characterization and Preliminary Safety Evaluation of an Edible Orodispersible Formulation for Pediatric Tuberculosis Pharmacotherapy. International Journal of Molecular Sciences 2020, 21, 5714–5714. [Google Scholar] [CrossRef] [PubMed]
- Adeleke, O.A.; Hayeshi, R.K.; Davids, H. Development and Evaluation of a Reconstitutable Dry Suspension Containing Isoniazid for Flexible Pediatric Dosing. Pharmaceutics 2020, 12, 286. [Google Scholar] [CrossRef]
- Sosnik, A.; Seremeta, K.P.; Imperiale, J.C.; Chiappetta, D.A. Novel Formulation and Drug Delivery Strategies for the Treatment of Pediatric Poverty-Related Diseases. Expert Opinion on Drug Delivery 2012, 9, 303–323. [Google Scholar] [CrossRef]
- Craig, S.R.; Adams, L.V.; Spielberg, S.P.; Campbell, B. Pediatric Therapeutics and Medicine Administration in Resource-Poor Settings: A Review of Barriers and an Agenda for Interdisciplinary Approaches to Improving Outcomes. Social Science and Medicine 2009, 69, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Nahata, M.C. Safety of “Inert" Additives or Excipients in Paediatric Medicines. Archives of Disease in Childhood Fetal and Neonatal Edition 2009, 94, F392–F393. [Google Scholar] [CrossRef] [PubMed]
- South African Health Products Regulatory Authority. Registered Health Products Database. Available online: https://medapps.sahpra.org.za:6006 (accessed on 11 July 2025).
- Salim, S.; Murshid, M.; Gazzali, A. Stability of Extemporaneous Rifampicin Prepared with X-temp® Oral Suspension System. Journal of Pharmacy 2021, 1, 54–62. [Google Scholar] [CrossRef]
- van Riet-Nales, D.A.; Schobben, A.F.A.M.; Vromans, H.; Egberts, T.C.G.; Rademaker, C.M.A. Safe and Effective Pharmacotherapy in Infants and Preschool Children: Importance of Formulation Aspects. Archives of Disease in Childhood 2016, 101, 662–669. [Google Scholar] [CrossRef]
- European Medicines Agency. Guideline on Pharmaceutical Development of Medicines for Paediatric Use. Available online: https://www.tga.gov.au/sites/default/files/2024-07/Guidelineonpharmaceuticaldevelopmentofmedicinesforpaediatric%20use%20.pdf (accessed on 28 June 2025).
- Mahtab, S.; Blau, D.M.; Madewell, Z.J.; Ogbuanu, I.; Ojulong, J.; Lako, S.; Legesse, H.; Bangura, J.S.; Bassat, Q.; Mandomando, I.; et al. Post-Mortem Investigation of Deaths Due to Pneumonia in Children Aged 1-59 Months in Sub-Saharan Africa and South Asia From 2016 to 2022: An Observational Study. The Lancet Child and Adolescent Health 2024, 8, 201–213. [Google Scholar]
- Thomas, T.A. Tuberculosis in Children. Pediatric Clinics of North America 2017, 64, 893–909. [Google Scholar] [CrossRef]
- Russo, D.O.; Jimenez, A.L.L.; Diniz, L.M.O.; Cardoso, C.A. Missed Opportunities in the Prevention and Diagnosis of Pediatric Tuberculosis: A Scoping Review. Jornal de Pediatria 2024, 100, 343–349. [Google Scholar] [CrossRef]
- Plata-Casas, L.; González-Támara, L.; Cala-Vitery, F. Tuberculosis Mortality in Children under Fifteen Years of Age: Epidemiological Situation in Colombia, 2010-2018. Tropical Medicine and Infectious Disease 2022, 7. [Google Scholar] [CrossRef]
- Basu, S.; Chakraborty, S. A Comprehensive Review of the Diagnostics for Pediatric Tuberculosis Based on Assay Time, Ease of Operation, and Performance. Microorganisms 2025, 13. [Google Scholar] [CrossRef]
- Dodd, P.J.; Yuen, C.M.; Sismanidis, C.; Seddon, J.A.; Jenkins, H.E. The Global Burden of Tuberculosis Mortality in Children: A Mathematical Modelling Study. Lancet Global Health 2017, 5, e898–e906. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, U.; Pintus, R.; Savasta, S.; Fanos, V. Pulmonary Tuberculosis in Children: A Forgotten Disease? Microorganisms 2023, 11. [Google Scholar] [CrossRef]
- Fennelly, K.P.; Martyny, J.W.; Fulton, K.E.; Orme, I.M.; Cave, D.M.; Heifets, L.B. Cough-Generated Aerosols of Mycobacterium Tuberculosis: A New Method to Study Infectiousness. American Journal of Respiratory and Critical Care Medicine 2004, 169, 604–609. [Google Scholar] [CrossRef]
- Vanden Driessche, K.; Persson, A.; Marais, B.J.; Fink, P.J.; Urdahl, K.B. Immune Vulnerability of Infants to Tuberculosis. Clinical and Developmental Immunology 2013, 2013, 781320. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.P.; Schaaf, H.S.; Nuttall, J.; Marais, B.J.; Brink, A. Childhood Tuberculosis Guidelines of the Southern African Society for Paediatric Infectious Diseases. In Proceedings of the Southern African Journal of Epidemiology and Infection, Johannesburg, South Africa, 2009/01/01/2009, 2009; pp. 57–68. [Google Scholar]
- Marais, B.J.; Gie, R.P.; Schaaf, H.S.; Hesseling, A.C.; Obihara, C.C.; Nelson, L.J.; Enarson, D.A.; Donald, P.R.; Beyers, N. The Clinical Epidemiology of Childhood Pulmonary Tuberculosis: A Critical Review of Literature from the Pre-Chemotherapy Era. International Journal of Tuberculosis and Lung Disease 2004, 8, 278–285. [Google Scholar]
- Newton, S.M.; Brent, A.J.; Anderson, S.; Whittaker, E.; Kampmann, B. Paediatric Tuberculosis. The Lancet Infectious Diseases 2008, 8, 498–510. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Reflection Paper: Formulations of Choice for the Paediatric Population. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-formulations-choice-paediatric-population_en.pdf (accessed on 17 July 2025).
- Lamb, G.S.; Starke, J.R. Tuberculosis in Infants and Children. Microbiology Spectrum 2017, 5. [Google Scholar] [CrossRef]
- Laycock, K.M.; Enane, L.A.; Steenhoff, A.P. Tuberculosis in Adolescents and Young Adults: Emerging Data on TB Transmission and Prevention among Vulnerable Young People. Tropical Medicine and Infectious Disease 2021, 6. [Google Scholar] [CrossRef]
- Nolt, D.; Starke, J.R. Tuberculosis Infection in Children and Adolescents: Testing and Treatment. Pediatrics 2021, 148, 1–23. [Google Scholar] [CrossRef]
- Siddalingaiah, N.; Chawla, K.; Nagaraja, S.B.; Hazra, D. Risk Factors for the Development of Tuberculosis among the Pediatric Population: A Systematic Review and Meta-Analysis. European Journal of Pediatrics 2023, 182, 3007–3019. [Google Scholar] [CrossRef]
- Lancella, L.; Vecchio, A.L.; Chiappini, E.; Tadolini, M.; Cirillo, D.; Tortoli, E.; de Martino, M.; Guarino, A.; Principi, N.; Villani, A.; et al. How to Manage Children Who Have Come into Contact with Patients Affected by Tuberculosis. Journal of Clinical Tuberculosis and Other Mycobacterial Diseases 2015, 1, 1–12. [Google Scholar] [CrossRef]
- Castillo-Barrientos, H.d.; Centeno-Luque, G.; Untiveros-Tello, A.; Simms, B.; Lecca, L.; Nelson, A.K.; Lastimoso, C.; Shin, S. Clinical Presentation of Children with Pulmonary Tuberculosis: 25 Years of Experience in Lima, Peru. International Journal of Tuberculosis and Lung Disease 2014, 18, 1066–1073. [Google Scholar] [CrossRef]
- World Health Organization. WHO Roadmap for Childhood Tuberculosis: Towards Zero Deaths; Geneva, Switzerland, 2013. [Google Scholar]
- Graham, S.M.; Oliwa, J.N. Diagnosis and Management of Tuberculosis in Children and Adolescents: A Desk Guide for Primary Health Care Workers; Paris, France, 2023. [Google Scholar]
- Esposito, S.; Tagliabue, C.; Bosis, S. Tuberculosis in Children. Mediterranean Journal of Hematology and Infectious Diseases 2013, 5, e2013064. [Google Scholar] [CrossRef]
- Sartoris, G.; Seddon, J.A.; Rabie, H.; Nel, E.D.; Schaaf, H.S. Abdominal Tuberculosis in Children: Challenges, Uncertainty, and Confusion. Journal of the Pediatric Infectious Diseases Society 2020, 9, 218–227. [Google Scholar] [CrossRef]
- Tinsa, F.; Essaddam, L.; Fitouri, Z.; Brini, I.; Douira, W.; Ben Becher, S.; Boussetta, K.; Bousnina, S. Abdominal Tuberculosis in Children. Journal of Pediatric Gastroenterology and Nutrition 2010, 50, 634–638. [Google Scholar] [CrossRef]
- Agarwal, A. Paediatric Osteoarticular Tuberculosis: A Review. Journal of Clinical Orthopaedics and Trauma 2020, 11, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.K.; Somvanshi, D.S. Spinal Tuberculosis: A Review. The Journal of Spinal Cord Medicine 2011, 34, 440–454. [Google Scholar] [CrossRef] [PubMed]
- Teo, H.E.L.; Peh, W.C.G. Skeletal Tuberculosis in Children. Pediatric Radiology 2004, 34, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Pattamapaspong, N.; Muttarak, M.; Sivasomboon, C. Tuberculosis Arthritis and Tenosynovitis. Seminars in Musculoskeletal Radiology 2011, 15, 459–469. [Google Scholar] [CrossRef]
- Imazio, M.; Gaita, F.; LeWinter, M. Evaluation and Treatment of Pericarditis: A Systematic Review. JAMA: Journal of the American Medical Association 2015, 314, 1498–1506. [Google Scholar] [CrossRef]
- Ramírez-Lapausa, M.; Menéndez-Saldaña, A.; Noguerado-Asensio, A. [Extrapulmonary tuberculosis]. Revista Espanola De Sanidad Penitenciaria 2015, 17, 3–11. [Google Scholar] [CrossRef]
- Sharma, S.K.; Mohan, A. Miliary Tuberculosis. Microbiology Spectrum 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.M. Treatment of Paediatric TB: Revised WHO Guidelines. Paediatric Respiratory Reviews 2011, 12, 22–26. [Google Scholar] [CrossRef]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis Module 5: Management of Tuberculosis in Children and Adolescents. Available online: https://iris.who.int/bitstream/handle/10665/352522/9789240046764-eng.pdf?sequence=1 (accessed on 7 August 2025).
- Nahid, P.; Mase, S.R.; Migliori, G.B.; Sotgiu, G.; Bothamley, G.H.; Brozek, J.L.; Cattamanchi, A.; Cegielski, J.P.; Chen, L.; Daley, C.L.; et al. Treatment of Drug-Resistant Tuberculosis. An Official ATS/CDC/ERS/IDSA Clinical Practice Guideline. American Journal of Respiratory and Critical Care Medicine 2019, 200, e93–e142. [Google Scholar] [CrossRef]
- Schaaf, H.S.; Garcia-Prats, A.J.; Donald, P.R. Antituberculosis Drugs in Children. Clinical Pharmacology and Therapeutics 2015, 98, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. International Journal of Molecular Sciences 2023, 24, 5202. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Tuberculosis in Children. Available online: https://www.cdc.gov/tb/about/children.html (accessed on 4 August 2025).
- Bhanu, M.L.S.; Manasa P Kumari; Begum, T.M. Anti-Tuberculosis Drugs and Mechanisms of Action: Review. International Journal of Infectious Diseases and Research 2023, 4, 1-7.
- Chauhan, A.; Kumar, M.; Kumar, A.; Kanchan, K. Comprehensive Review on Mechanism of Action, Resistance and Evolution of Antimycobacterial Drugs. Life Sciences 2021, 274, N.PAG-N.PAG. [CrossRef]
- Omoteso, O.A.; Fadaka, A.O.; Walker, R.B.; Khamanga, S.M. Innovative Strategies for Combating Multidrug-Resistant Tuberculosis: Advances in Drug Delivery Systems and Treatment. Microorganisms 2025, 13. [CrossRef]
- Britton, P.; Perez-Velez, C.M.; Marais, B.J. Diagnosis, Treatment and Prevention of Tuberculosis in Children. New South Wales Public Health Bulletin 2013, 24, 15-21. [CrossRef]
- Schellack, N.; Yotsombut, K.; Sabet, A.; Nafach, J.; Hiew, F.L.; Kulkantrakorn, K. Expert Consensus on Vitamin B6 Therapeutic Use for Patients: Guidance on Safe Dosage, Duration and Clinical Management. Drug, Healthcare and Patient Safety 2025, 17, 97-108. [CrossRef]
- Tostmann, A.; Boeree, M.J.; Aarnoutse, R.E.; de Lange, W.C.M.; van der Ven, A.J.A.M.; Dekhuijzen, R. Antituberculosis Drug-Induced Hepatotoxicity: Concise Up-To-Date Review. Journal of Gastroenterology and Hepatology 2008, 23, 192-202. [CrossRef]
- Adams, R.A.; Leon, G.; Miller, N.M.; Reyes, S.P.; Thantrong, C.H.; Thokkadam, A.M.; Lemma, A.S.; Sivaloganathan, D.M.; Wan, X.; Brynildsen, M.P. Rifamycin Antibiotics and the Mechanisms of Their Failure. The Journal of Antibiotics 2021, 74, 786-798. [CrossRef]
- Brucoli, F.; D McHugh, T. Rifamycins: Do Not Throw the Baby Out With the Bathwater. Is Rifampicin Still an Effective Anti-Tuberculosis Drug? Future Medicinal Chemistry 2021, 13, 2129-2131. [CrossRef]
- Hoagland, D.; Zhao, Y.; Lee, R.E. Advances in Drug Discovery and Development for Pediatric Tuberculosis. Mini Rev Med Chem 2016, 16, 481-497. [CrossRef]
- Zhang, N.; Savic, R.M.; Boeree, M.J.; Peloquin, C.A.; Weiner, M.; Heinrich, N.; Bliven-Sizemore, E.; Phillips, P.P.J.; Hoelscher, M.; Whitworth, W.; et al. Optimising Pyrazinamide for The Treatment of Tuberculosis. European Respiratory Journal 2021, 58. [CrossRef]
- Chen, Z.; Yu, L.; Lihua, H.; Min, Y.; Zheng-Guo, H. Molecular Mechanism of The Synergistic Activity of Ethambutol and Isoniazid Against Mycobacterium Tuberculosis. Journal of Biological Chemistry 2018, 293, 16741-16750. [CrossRef]
- Srivastava, S.; Musuka, S.; Sherman, C.; Meek, C.; Leff, R.; Gumbo, T. Efflux-Pump-Derived Multiple Drug Resistance to Ethambutol Monotherapy in Mycobacterium tuberculosis and the Pharmacokinetics and Pharmacodynamics of Ethambutol. Journal of Infectious Diseases 2010, 201, 1225-1231. [CrossRef]
- Jnawali, H.N.; Ryoo, S. First– and Second–Line Drugs and Drug Resistance. In Tuberculosis - Current Issues in Diagnosis and Management; InTech: 2013.
- Castro, N.D.; Mechaï, F.; Bachelet, D.; Canestri, A.; Joly, V.; Vandenhende, M.; Boutoille, D.; Kerjouan, M.; Veziris, N.; Molina, J.M.; et al. Treatment With a Three-Drug Regimen for Pulmonary Tuberculosis Based on Rapid Molecular Detection of Isoniazid Resistance: A Noninferiority Randomized Trial (FAST-TB). Open Forum Infectious Diseases 2022, 9, 1-8. [CrossRef]
- Levy, M.; Rigaudière, F.; de Lauzanne, A.; Koehl, B.; Melki, I.; Lorrot, M.; Faye, A. Ethambutol-related Impaired Visual Function in Childrens Less than 5 Years of Age Treated for a Mycobacterial Infection. The Pediatric Infectious Disease Journal 2015, 34, 346-350. [CrossRef]
- Noor, H.; David, I.G.; Jinga, M.L.; Popa, D.E.; Buleandra, M.; Iorgulescu, E.E.; Ciobanu, A.M. State of the Art on Developments of (Bio)Sensors and Analytical Methods for Rifamycin Antibiotics Determination. Sensors 2023, 23, 976. [CrossRef]
- De Pinho Pessoa Nogueira, L.; De Oliveira, Y.S.; De C. Fonseca, J.; Costa, W.S.; Raffin, F.N.; Ellena, J.; Ayala, A.P. Crystalline Structure of the Marketed Form of Rifampicin: A Case of Conformational and Charge Transfer Polymorphism. Journal of Molecular Structure 2018, 1155, 260-266. [CrossRef]
- Zaborniak, I.; Macior, A.; Chmielarz, P. Stimuli-Responsive Rifampicin-Based Macromolecules. Materials (Basel, Switzerland) 2020, 13. [CrossRef]
- British Pharmacopoeia. Rifampicin. Available online: https://www.pharmacopoeia.com/bp-2025/monographs/rifampicin.html?date=2025-04-01&text=Rifampicin (accessed on 13 March 2025).
- Hussain, A.; Shakeel, F.; Singh, S.K.; Alsarra, I.A.; Faruk, A.; Alanazi, F.K.; Peter Christoper, G.V. Solidified SNEDDS for the Oral Delivery of Rifampicin: Evaluation, Proof of Concept, In Vivo Kinetics, and In Silico GastroPlusTM Simulation. International Journal of Pharmaceutics 2019, 566, 203-217. [CrossRef]
- Abbas, M.H.; Taha, D.N. Determination of Rifampicin in Pure form and Pharmaceutical Preparations by Using Merging Zone-Continuous Flow Injection Analysis. International Journal of Pharmaceutical Quality Assurance 2019, 10, 303–310. [CrossRef]
- Karaźniewicz-Łada, M. A Review on Recent Advances in the Stability Study of Anti-Mycobacterial Drugs. Polimery W Medycynie 2024, 54, 135-142. [CrossRef]
- Henwood, S.Q.; Liebenberg, W.; Tiedt, L.R.; Lötter, A.P.; de Villiers, M.M. Characterization of the Solubility and Dissolution Properties of Several New Rifampicin Polymorphs, Solvates, and Hydrates. Drug Development and Industrial Pharmacy 2001, 27, 1017-1030. [CrossRef]
- Agrawal, S.; Ashokraj, Y.; Bharatam, P.V.; Pillai, O.; Panchagnula, R. Solid-State Characterization of Rifampicin Samples and Its Biopharmaceutic Relevance. European Journal of Pharmaceutical Sciences 2004, 22, 127-144. [CrossRef]
- Arca, H.Ç.; Mosquera-Giraldo, L.I.; Pereira, J.M.; Sriranganathan, N.; Taylor, L.S.; Edgar, K.J. Rifampin Stability and Solution Concentration Enhancement Through Amorphous Solid Dispersion in Cellulose ω-Carboxyalkanoate Matrices. Journal of Pharmaceutical Sciences 2018, 107, 127-138. [CrossRef]
- Piccaro, G.; Poce, G.; Biava, M.; Giannoni, F.; Fattorini, L. Activity of Lipophilic and Hydrophilic Drugs Against Dormant and Replicating Mycobacterium Tuberculosis. Journal of Antibiotics 2015, 68, 711-714.
- O’Neil, M.J. The Merck Index - An Encyclopedia of Chemicals, Drugs, and Biologicals. 2001, 1474.
- Ibrahim, S.A.; Kassem, A.A.; Attia, I.A.; Mohamed, S.I. Studies on Some Factors Affecting the Stability of Rifampicin in Solution. Bulletin of Pharmaceutical Sciences Assiut University 1979, 2, 11-25. [CrossRef]
- He, D.; Deng, P.; Yang, L.; Tan, Q.; Liu, J.; Yang, M.; Zhang, J. Molecular Encapsulation of Rifampicin as an Inclusion Complex of Hydroxypropyl-β-Cyclodextrin: Design; Characterization and In Vitro Dissolution. Colloids and Surfaces B: Biointerfaces 2013, 103, 580-585. [CrossRef]
- Alves, R.; Reis, T.V.d.S.; Silva, L.C.C.d.; Storpírtis, S.; Mercuri, L.P.; Matos, J.d.R. Thermal Behavior and Decomposition Kinetics of Rifampicin Polymorphs Under Isothermal and Non-Isothermal Conditions. Brazilian Journal of Pharmaceutical Sciences 2010, 46, 343-351.
- Mariappan, T.T.; Singh, S. Positioning of Rifampicin in the Biopharmaceutics Classification System (BCS). Clinical Research and Regulatory Affairs 2006, 23, 1–10. [Google Scholar] [CrossRef]
- Vander Schaaf, M.; Luth, K.; Townsend, D.M.; Chessman, K.H.; Mills, C.M.; Garner, S.S.; Peterson, Y.K. CYP3A4 Drug Metabolism Considerations in Pediatric Pharmacotherapy. Medicinal Chemistry Research 2024, 33, 2221–2235. [Google Scholar] [CrossRef]
- Beloor Suresh, A.; Rosani, A.; Wadhwa, R. Rifampin. Available online: https://research.ebsco.com/linkprocessor/plink?id=ac1553a5-ec8b-30e2-9a43-8cc3ed3110f5 (accessed on 26 February 2025).
- Kearns, G.L.; Abdel-Rahman, S.M.; Alander, S.W.; Blowey, D.L.; Leeder, J.S.; Kauffman, R.E. Developmental Pharmacology – Drug Disposition, Action, and Therapy in Infants and Children. New England Journal of Medicine 2003, 349, 1157–1167. [Google Scholar] [CrossRef]
- Smith, P.B.; Cotten, C.M.; Hudak, M.L.; Sullivan, J.E.; Poindexter, B.B.; Cohen-Wolkowiez, M.; Boakye-Agyeman, F.; Lewandowski, A.; Anand, R.; Benjamin, D.K., Jr.; et al. Rifampin Pharmacokinetics and Safety in Preterm and Term Infants. Antimicrobial Agents and Chemotherapy 2019, 63. [Google Scholar] [CrossRef]
- Cohen-Wolkowiez, M.; Benjamin, D.K., Jr.; Fowler, V.G., Jr.; Wade, K.C.; Alexander, B.D.; Worley, G.; Goldstein, R.F.; Smith, P.B. Mortality and Neurodevelopmental Outcome after Staphylococcus Aureus Bacteremia in Infants. The Pediatric Infectious Disease Journal 2007, 26, 1159–1161. [Google Scholar] [CrossRef]
- Pullen, J.; Stolk, L.M.L.; Degraeuwe, P.L.J.; van Tiel, F.H.; Neef, C.; Zimmermann, L.J.I. Pharmacokinetics of Intravenous Rifampicin (Rifampin) in Neonates. Therapeutic Drug Monitoring 2006, 28, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Nahata, M.C.; Fan-Havard, P.; Barson, W.J.; Bartkowski, H.M.; Kosnik, E.J. Pharmacokinetics, Cerebrospinal Fluid Concentration, and Safety of Intravenous Rifampin in Pediatric Patients Undergoing Shunt Placements. European Journal of Clinical Pharmacology 1990, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Nassr, N.; Huennemeyer, A.; Herzog, R.; von Richter, O.; Hermann, R.; Koch, M.; Duffy, K.; Zech, K.; Lahu, G. Effects of Rifampicin on the Pharmacokinetics of Roflumilast and Roflumilast N-Oxide in Healthy Subjects. British Journal of Clinical Pharmacology 2009, 68, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Raymond, K. Roles of Rifampicin in Drug-Drug Interactions: Underlying Molecular Mechanisms Involving the Nuclear Pregnane X Receptor. Annals of Clinical Microbiology and Antimicrobials 2006, 5, 3. [Google Scholar] [CrossRef]
- Sutradhar, I.; Zaman, M.H. Evaluation of the Effect of Temperature on the Stability and Antimicrobial Activity of Rifampicin Quinone. Journal of Pharmaceutical and Biomedical Analysis 2021, 197, N.PAG–N.PAG. [Google Scholar] [CrossRef]
- Karaźniewicz-Łada, M.; Kosicka-Noworzyń, K.; Rao, P.; Modi, N.; Xie, Y.L.; Heysell, S.K.; Kagan, L. New Approach to Rifampicin Stability and First-Line Anti-Tubercular Drug Pharmacokinetics by UPLC-MS/MS. Journal of Pharmaceutical and Biomedical Analysis 2023, 235, N.PAG–N.PAG. [Google Scholar] [CrossRef]
- Zivari-Moshfegh, F.; Javanmardi, F.; Nematollahi, D. A Comprehensive Electrochemical Study on Anti-Tuberculosis Drug Rifampicin. Investigating Reactions of Rifampicin-Quinone with Other Anti-Tuberculosis Drugs, Isoniazid, Pyrazinamide and Ethambutol. Electrochimica Acta 2023, 457, N.PAG–N.PAG. [Google Scholar] [CrossRef]
- Acuna, L.; Corbalán, N.S.; Raisman-Vozari, R. Rifampicin Quinone Pretreatment Improves Neuronal Survival by Modulating Microglia Inflammation Induced by α-Synuclein. Neural Regeneration Research 2020, 15, 1473–1474. [Google Scholar] [CrossRef]
- Weinstein, Z.B.; Zaman, M.H. Evolution of Rifampin Resistance in Escherichia coli and Mycobacterium smegmatis Due to Substandard Drugs. Antimicrobial Agents and Chemotherapy 2018, 63. [Google Scholar] [CrossRef]
- Singh, S.; Mariappan, T.T.; Sharda, N.; Kumar, S.; Chakraborti, A. The Reason for an Increase in Decomposition of Rifampicin in the Presence of Isoniazid under Acid Conditions. Pharmacy and Pharmacology Communications 2010, 6, 405–410. [Google Scholar] [CrossRef]
- Singh, H.; Bhandari, R.; Kaur, I.P. Encapsulation of Rifampicin in a Solid Lipid Nanoparticulate System to Limit Its Degradation and Interaction with Isoniazid at Acidic pH. International Journal of Pharmaceutics 2013, 446, 106–111. [Google Scholar] [CrossRef]
- Pund, S.; Joshi, A.; Vasu, K.; Nivsarkar, M.; Shishoo, C. Gastroretentive Delivery of Rifampicin: In Vitro Mucoadhesion and In Vivo Gamma Scintigraphy. International Journal of Pharmaceutics 2011, 411, 106–112. [Google Scholar] [CrossRef]
- Gohel, M.; Sarvaiya, K. A Novel Solid Dosage Form of Rifampicin and Isoniazid with Improved Functionality. AAPS PharmSciTech 2007, 8, E133–E139. [Google Scholar] [CrossRef]
- Bhise, S.B.; More, A.B.; Malayandi, R. Formulation and In Vitro Evaluation of Rifampicin Loaded Porous Microspheres. Scientia Pharmaceutica 2010, 78, 291–302. [Google Scholar] [CrossRef]
- Anita, G.S.; Lata, S.M.; Tejraj, M.A. Novel pH-Sensitive Hydrogels Prepared from the Blends of Poly(vinyl alcohol) with Acrylic Acid-graft-Guar Gum Matrixes for Isoniazid Delivery. Industrial and Engineering Chemistry Research 2010, 49, 7323–7329. [Google Scholar] [CrossRef]
- Batchelor, H.K.; Marriott, J.F. Formulations for Children: Problems and Solutions. British Journal of Clinical Pharmacology 2015, 79, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.E.; Millar, B.C. Readability of Patient-Facing Information of Antibiotics Used in the WHO Short 6-Month and 9-Month All Oral Treatment for Drug-Resistant Tuberculosis. Lung (0341-2040) 2024, 202, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Orubua, E.S.F.; Tuleua, C. Medicines for Children: Flexible Solid Oral Formulations. Bulletin of the World Health Organization 2017, 95, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Stop TB Partnership Global Drug Facility. Global Drug Facility September 2025 Medicines Catalog. Available online: https://www.stoptb.org/sites/default/files/documents/2025.09.02_gdf_medicines_catalog.pdf (accessed on 3 September 2025).
- Schaaf, H.S.; Wademan, D.T.; van der Laan, L.E. Global Child-Friendly Anti-TB Medicines - Where Do We Stand? IJTLD Open 2025, 2, 501–504. [Google Scholar] [CrossRef]
- Principi, N.; Galli, L.; Lancella, L.; Tadolini, M.; Migliori, G.; Villani, A.; Esposito, S. Recommendations Concerning the First-Line Treatment of Children with Tuberculosis. Pediatric Drugs 2016, 18, 13–23. [Google Scholar] [CrossRef] [PubMed]
- McIlleron, H.; Hundt, H.; Smythe, W.; Bekker, A.; Winckler, J.; van der Laan, L.; Smith, P.; Zar, H.J.; Hesseling, A.C.; Maartens, G.; et al. Bioavailability of Two Licensed Paediatric Rifampicin Suspensions: Implications for Quality Control Programmes. The International Journal of Tuberculosis and Lung Disease 2016, 20, 915–919. [Google Scholar] [CrossRef]
- Alessandrini, E.; Brako, F.; Scarpa, M.; Lupo, M.; Bonifazi, D.; Pignataro, V.; Cavallo, M.; Cullufe, O.; Enache, C.; Nafria, B.; et al. Children's Preferences for Oral Dosage Forms and Their Involvement in Formulation Research via EPTRI (European Paediatric Translational Research Infrastructure). Pharmaceutics 2021, 13, 730. [Google Scholar] [CrossRef]
- Domingues, C.; Jarak, I.; Veiga, F.; Dourado, M.; Figueiras, A. Pediatric Drug Development: Reviewing Challenges and Opportunities by Tracking Innovative Therapies. Pharmaceutics 2023, 15, 2431. [Google Scholar] [CrossRef]
- Keating, A.V.; Soto, J.; Forbes, C.; Zhao, M.; Craig, D.Q.M.; Tuleu, C. Multi-Methodological Quantitative Taste Assessment of Anti-Tuberculosis Drugs to Support the Development of Palatable Paediatric Dosage Forms. Pharmaceutics 2020, 12, 369–369. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Partheniadis, I. Self-Emulsifying Granules and Pellets: Composition and Formation Mechanisms for Instant or Controlled Release. Pharmaceutics 2017, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, L.; Hirvonen, J. Drug Nanocrystals – Versatile Option for Formulation of Poorly Soluble Materials. International Journal of Pharmaceutics 2018, 537, 73–83. [Google Scholar] [CrossRef]
- Salunke, S.; O'Brien, F.; Cheng Thiam Tan, D.; Harris, D.; Math, M.; Ariën, T.; Klein, S.; Timpe, C. Oral Drug Delivery Strategies for Development of Poorly Water Soluble Drugs in Paediatric Patient Population. Advanced Drug Delivery Reviews 2022, 190. [Google Scholar] [CrossRef]
- Holm, R.; Kuentz, M.; Ilie-Spiridon, A.-R.; Griffin, B.T. Lipid Based Formulations as Supersaturating Oral Delivery Systems: From Current to Future Industrial Applications. European Journal of Pharmaceutical Sciences 2023, 189, 106556. [Google Scholar] [CrossRef] [PubMed]
- Gurram, A.K.; Deshpande, P.B.; Kar, S.S.; Nayak, U.Y.; Udupa, N.; Reddy, M.S. Role of Components in the Formation of Self-microemulsifying Drug Delivery Systems. Indian Journal of Pharmaceutical Sciences 2015, 77, 249–257. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid Formulations for Oral Administration of Drugs: Non-Emulsifying, Self-Emulsifying and ‘Self-Microemulsifying' Drug Delivery Systems. European Journal of Pharmaceutical Sciences 2000, 11 Suppl 2, S93–S98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Virmani, T.; Kumar, G.; Deshmukh, R.; Sharma, A.; Duarte, S.; Brandão, P.; Fonte, P. Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies. Pharmaceuticals 2023, 16. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, H.; Bala, R.; Arora, S. Lipid-Based Drug Delivery Systems. Journal of Pharmaceutics 2014, 801820. [Google Scholar] [CrossRef]
- Mu, H.; Holm, R.; Müllertz, A. Lipid-Based Formulations for Oral Administration of Poorly Water-Soluble Drugs. International Journal of Pharmaceutics 2013, 453, 215–224. [Google Scholar] [CrossRef]
- Rani, E.R.; Radha, G.V. Insights into Novel Excipients of Self-Emulsifying Drug Delivery Systems and Their Significance: An Updated Review. Critical Reviews in Therapeutic Drug Carrier Systems 2021, 38, 27–74. [Google Scholar] [CrossRef]
- Uttreja, P.; Karnik, I.; Adel Ali Youssef, A.; Narala, N.; Elkanayati, R.M.; Baisa, S.; Alshammari, N.D.; Banda, S.; Vemula, S.K.; Repka, M.A. Self-Emulsifying Drug Delivery Systems (SEDDS): Transition from Liquid to Solid-A Comprehensive Review of Formulation, Characterization, Applications, and Future Trends. Pharmaceutics 2025, 17. [Google Scholar] [CrossRef]
- Kalepu, S.; Manthina, M.; Padavala, V. Oral Lipid-Based Drug Delivery Systems – An Overview. Acta Pharmaceutica Sinica B 2013, 3, 361–372. [Google Scholar] [CrossRef]
- Xu, Y.; Michalowski, C.B.; Beloqui, A. Advances in Lipid Carriers for Drug Delivery to the Gastrointestinal Tract. Current Opinion in Colloid and Interface Science 2021, 52. [Google Scholar] [CrossRef]
- Seo, E.B.; du Plessis, L.H.; Viljoen, J.M. Solidification of Self-Emulsifying Drug Delivery Systems as a Novel Approach to the Management of Uncomplicated Malaria. Pharmaceuticals 2022, 15, 120. [Google Scholar] [CrossRef]
- Salawi, A. Self-Emulsifying Drug Delivery Systems: A Novel Approach to Deliver Drugs. Drug Delivery 2022, 29, 1811–1823. [Google Scholar] [CrossRef]
- Pouton, C.W. Formulation of Poorly Water-Soluble Drugs for Oral Administration: Physicochemical and Physiological Issues and the Lipid Formulation Classification System. European Journal of Pharmaceutical Sciences 2006, 29, 278–287. [Google Scholar] [CrossRef]
- Cerpnjak, K.; Zvonar, A.; Gašperlin, M.; Vrečer, F. Lipid-Based Systems as a Promising Approach for Enhancing the Bioavailability of Poorly Water-Soluble Drugs. Acta Pharmaceutica 2013, 63, 427–445. [Google Scholar] [CrossRef] [PubMed]
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975–975. [Google Scholar] [CrossRef] [PubMed]
- Kohli, K.; Chopra, S.; Dhar, D.; Arora, S.; Khar, R.K. Self-Emulsifying Drug Delivery Systems: An Approach to Enhance Oral Bioavailability. Drug Discovery Today 2010, 15, 958–965. [Google Scholar] [CrossRef]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-Emulsifying Oral Lipid Drug Delivery Systems: Advances and Challenges. AAPS PharmSciTech 2019, 20, 129. [Google Scholar] [CrossRef]
- Feeney, O.M.; Crum, M.F.; McEvoy, C.L.; Trevaskis, N.L.; Williams, H.D.; Pouton, C.W.; Charman, W.N.; Bergström, C.A.S.; Porter, C.J.H. 50 Years of Oral Lipid-Based Formulations: Provenance, Progress and Future Perspectives. Advanced Drug Delivery Reviews 2016, 101, 167–194. [Google Scholar] [CrossRef]
- Porter, C.J.H.; Trevaskis, N.L.; Charman, W.N. Lipids and Lipid-Based Formulations: Optimizing the Oral Delivery of Lipophilic Drugs. Nature Reviews Drug Discovery 2007, 6, 231–248. [Google Scholar] [CrossRef]
- Pramod, K.; Peeyush, K.; Rajeev, K.; Nitish, K.; Rakesh, K. An Overview on Lipid Based Formulation for Oral Drug Delivery. Drug Invention Today 2010, 2, 390–395. [Google Scholar]
- Kossena, G.A.; Charman, W.N.; Wilson, C.G.; O'Mahony, B.; Lindsay, B.; Hempenstall, J.M.; Davison, C.L.; Crowley, P.J.; Porter, C.J.H. Low Dose Lipid Formulations: Effects on Gastric Emptying and Biliary Secretion. Pharmaceutical Research 2007, 24, 2084–2096. [Google Scholar] [CrossRef]
- Charman, W.N.; Stella, V.J. Estimating the Maximal Potential for Intestinal Lymphatic Transport of Lipophilic Drug Molecules. International Journal of Pharmaceutics 1986, 34, 175–178. [Google Scholar] [CrossRef]
- Singh, I.; Swami, R.; Khan, W.; Sistla, R. Delivery Systems for Lymphatic Targetting. Focal Controlled Drug. Delivery 2013, 429–458. [Google Scholar] [CrossRef]
- Arimurni, D. Optimization of Self-Nano Emulsifying Drug Delivery System of Rifampicin for Nebulization Using Cinnamon Oil as Oil Phase. Pharmaciana 2024, 14, 365–369. [Google Scholar] [CrossRef]
Figure 1.
Age-related risk of TB development after initial infection prior to the chemotherapy era. Adapted from [55,64].
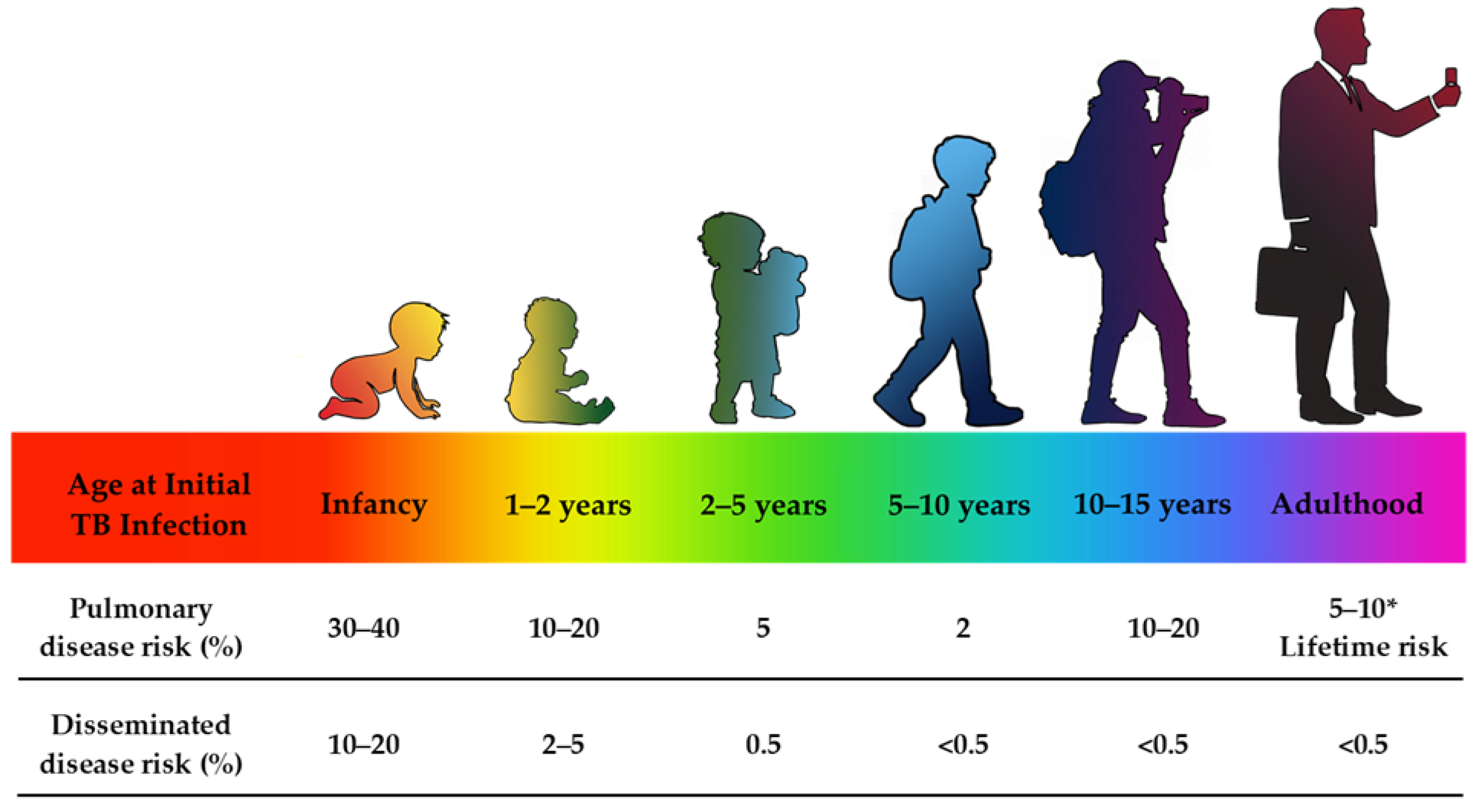
Figure 2.
Degradation pathways of rifampicin: (a) 24-diacetyl rifampicin; (b) rifampicin quinone; (c) 3-formyl rifamycin SV; (d) 3-formyl-rifamycin-isonicotinylhydrazone. Adapted from [112].
Figure 2.
Degradation pathways of rifampicin: (a) 24-diacetyl rifampicin; (b) rifampicin quinone; (c) 3-formyl rifamycin SV; (d) 3-formyl-rifamycin-isonicotinylhydrazone. Adapted from [112].
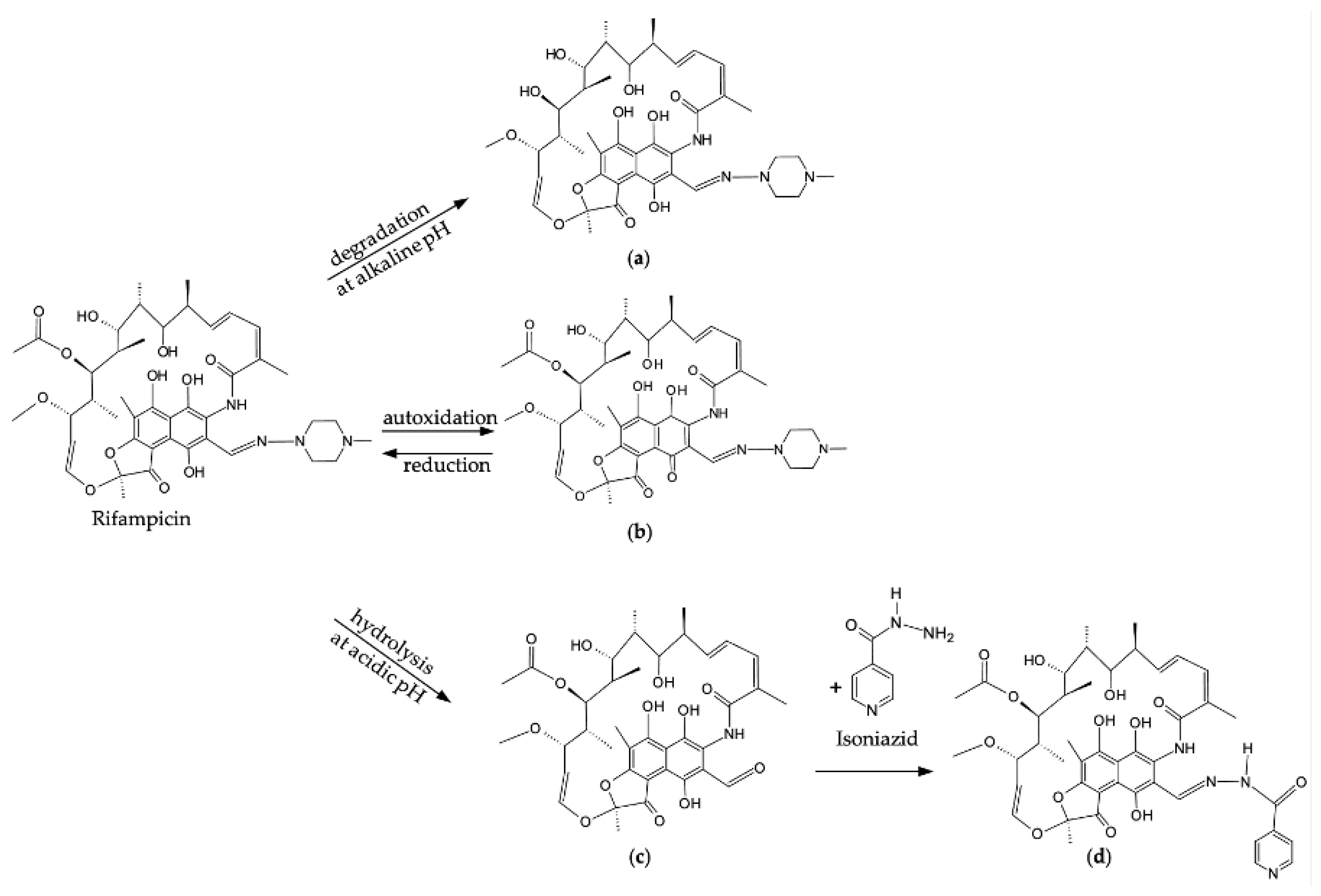
Figure 3.
The interaction between rifampicin and isoniazid (also known as isonicotinylhydrazide) results in the formation of hydrazone. (I) first intermediate; (II) second intermediate. Adapted from [30,38].
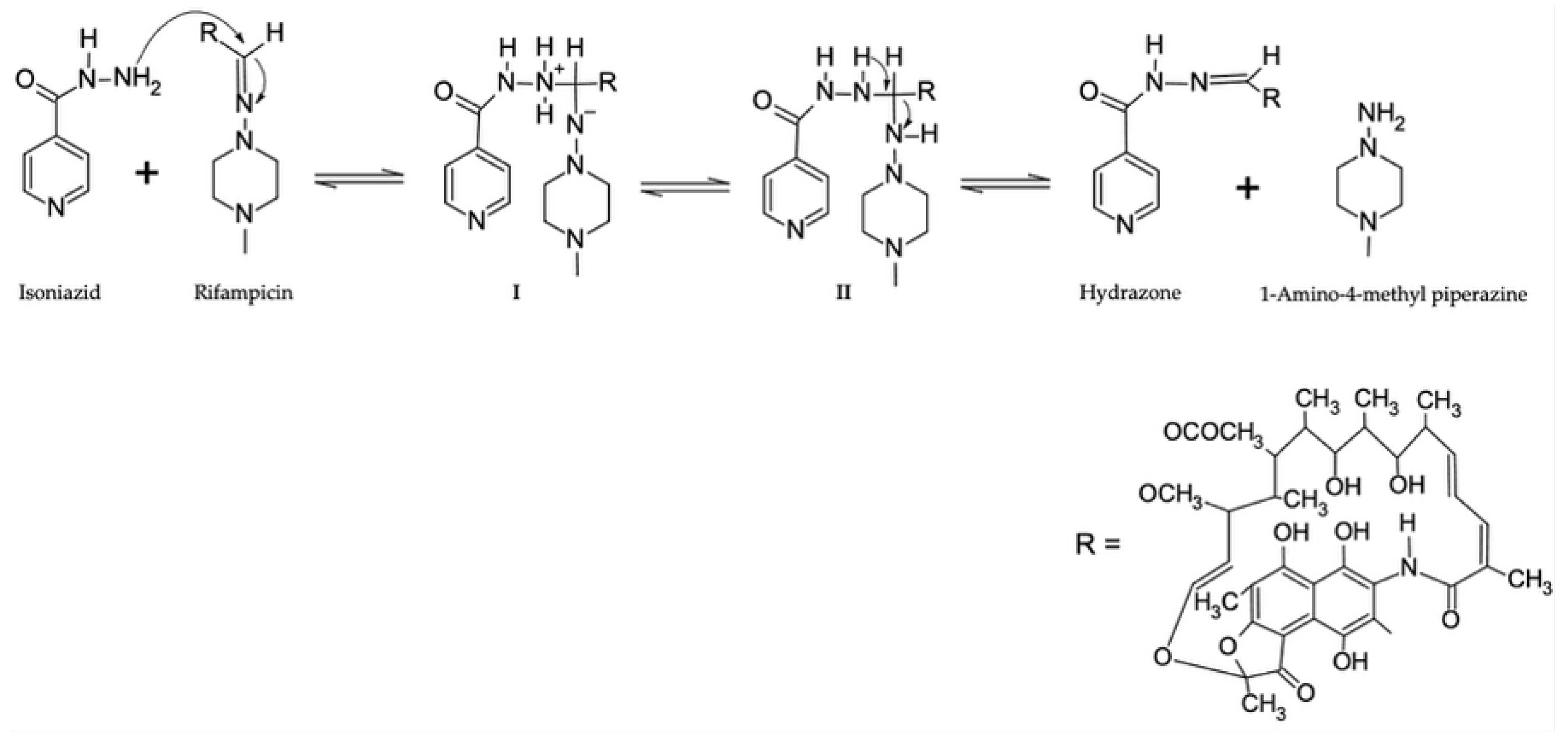
Figure 4.
Mechanism by which lipid-based formulations such as SEDDSs facilitate intestinal drug transport.
Figure 4.
Mechanism by which lipid-based formulations such as SEDDSs facilitate intestinal drug transport.
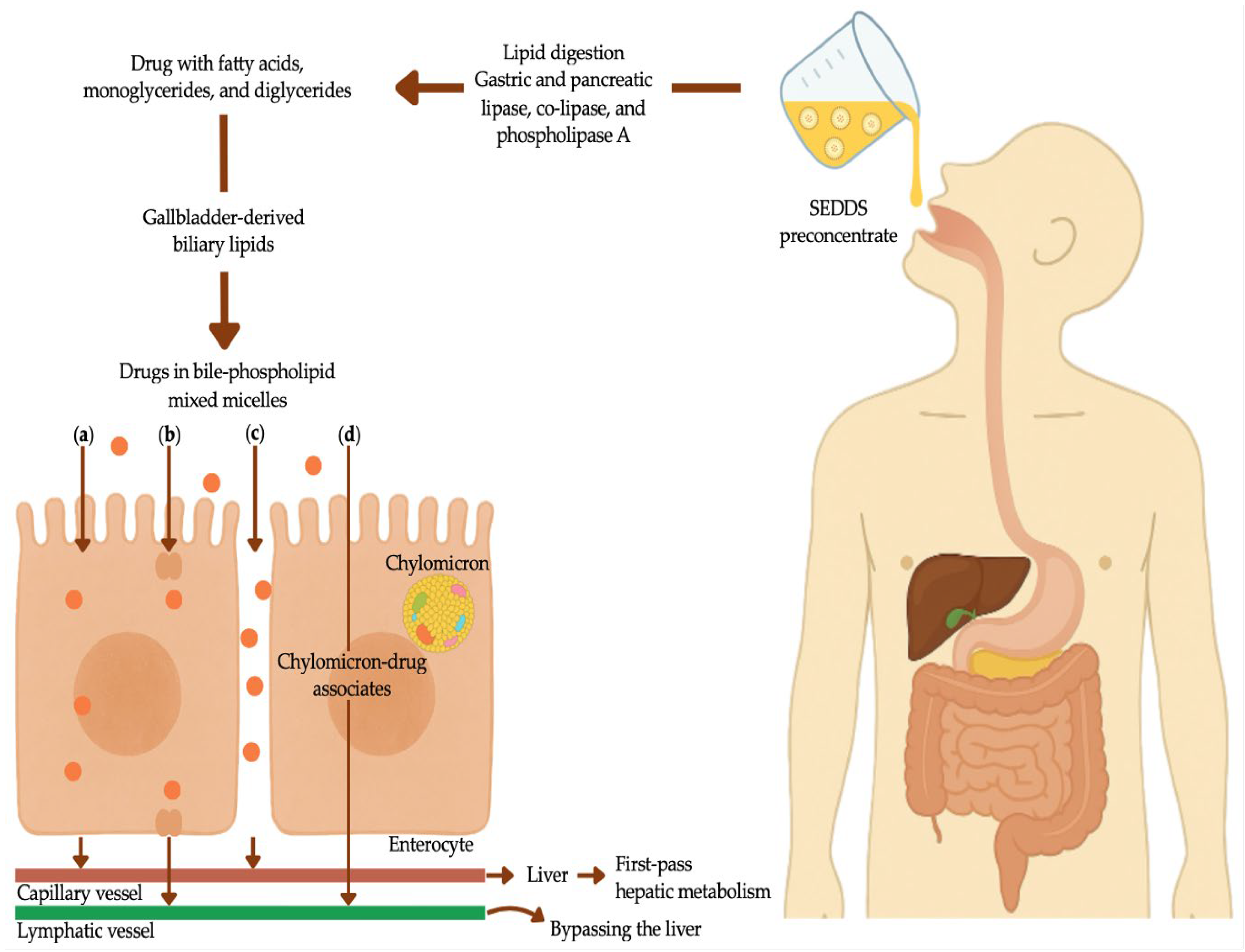

Table 1.
A summary of pediatric TB treatment regimens for TB infection, as well as drug-susceptible TB, including non-severe and severe TB [63,69,86].
| Type of TB | Regimen | Drugs | Duration (months) | Recommended Regimen | Key Consideration |
|---|---|---|---|---|---|
| Tuberculosis Infection | 3HR | Isoniazid (H) Rifampicin (R) |
3 | All pediatric patients. Preferred for HIV-negative children <25 kg. | Use dispersible fixed dose combinations if available. Vitamin B6 supplements for selected patients.* |
| 4R | Rifampicin (R) | 4 | All pediatric patients. | No pediatric-friendly formulation is readily available: In adolescents and older children, a rifampicin tablet may be used; compounding or tablet crushing may be necessary for younger children. Use if suspected or resistance to isoniazid. | |
| 6H or 9H | Isoniazid (H) | 6 or 9 | All pediatric patients. Preferred for HIV-positive children on anti-retroviral therapy. | Use if rifampicin is contraindicated or not available. Vitamin B6 supplements for selected patients.* | |
| 3HP | Isoniazid (H) Rifapentine (P) |
3 | ≥2 years. This regimen is not recommended for HIV-positive children. | Administer with fatty food. Vitamin B6 supplements for selected patients.* | |
| Non-severe drug- susceptible TB |
2HRZE/2HR | Isoniazid (H) Rifampicin (R) Pyrazinamide (P) ±Ethambutol (E) |
4 | Pediatric patients ≥3 months to <16 years with non-severe peripheral lymph node or pulmonary TB. | Meets WHO non-severe TB criteria (uncomplicated pleural effusion; non-cavitary, paucibacillary disease. Confined to one lung lobe without miliary pattern; intrathoracic lymph node TB without airway obstruction). Ethambutol optional. Exclude rifampicin resistance. Child-friendly formulations available. |
| Severe drug-susceptible TB | 2HRZE/4HR | Isoniazid (H) Rifampicin (R) Pyrazinamide (P) ±Ethambutol (E) |
6 | All pediatric patients with severe TB. | Include severe pulmonary TB (cavitary, extensive, and miliary TB) and all forms of extrapulmonary TB, except osteoarticular TB and TB meningitis. Ethambutol should always be included in the intensive phase. Child-friendly dispersible formulations available. |
| Osteoarticular TB and TB meningitis | 2HRZE/10HR | Isoniazid (H) Rifampicin (R) Pyrazinamide (P) ±Ethambutol (E) |
12 | All pediatric patients with osteoarticular TB or TB meningitis. | For osteoarticular TB, consider surgical intervention if needed and focus on adherence. For TB meningitis, consider high rifampicin dosages to achieve optimal central nervous system penetration. Add corticosteroids. Initiate treatment in hospital. Monitor for neurological and hepatotoxicity changes. Child-friendly formulations available |
* The selected patients for Vitamin B6 supplementation include infants who are exclusively breastfed, children, and adolescents with milk- or meat-deficient diets, malnourished pediatric patients, and symptomatic HIV-positive children.Completing the prescribed TB regimen is crucial, as interruption or incorrect use can result in disease recurrence, drug resistance, as well as prolonged, more costly, or even failed treatment [90]. Moreover, determining the optimal TB antibiotic dose to achieve effective systemic drug levels – thereby minimizing toxicity while maximizing the therapeutic efficacy – within the pediatric population is often a formidable challenge due to low drug serum concentration and poor patient treatment adherence [17,37]. This frequently leads to unsuccessful treatment and the emergence of drug-resistant strains, which are considerably more challenging to manage [17]. Furthermore, TB management during this stage of disease is more arduous due to the more extended treatment period, higher costs compared to first-line treatment, and the necessity for injectable drugs; all of which contribute to poor patient adherence and increased severity of side effects [17].
| Property | SEDDSs | SMEDDSs | SNEDDSs |
|---|---|---|---|
| Average droplet size | >300 nm | 100–200 nm | <100 nm |
| Appearance | Cloudy/turbid | Clear to translucent | Optically clear |
| Bioavailability | Moderate | Enhanced | Superior |
| Classification as per LFCS | Type II | Type IIIA/IIIB | Type IIIB |
| Concentration of oil | 40–80% | 40–80%/<20% | <20% |
| Concentration of surfactant |
30–40% | 40–80% | 40–80% |
| HBL of surfactants | <10 | 10–12 | >12 |
| Oil types | Long-chain triglycerides |
Medium-chain triglycerides |
Medium- and short-chain triglycerides |
| Solubilizing capacity | High | High | High |
| Stability | Thermodynamically unstable | Thermodynamically stable | Kinetically stable |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



