Submitted:
25 November 2025
Posted:
09 December 2025
You are already at the latest version
Abstract
Epithelial, endothelial and many connective tissue cells are normally attached to the extracellular matrix (ECM). These cells rely on the ECM for structural support, signaling, and regulation of their behavior. When these cells lose this attachment or are in an inappropriate location, they soon die by a mechanism called anoikis (homelessness). Anoikis, is a programmed cell death of apoptotic nature, however, in certain cases it can be overcome and detached cells can survive in the absence of the correct signals from the ECM. This is the case for malignant cells, where anoikis resistance is a prerequisite for invasion and metastasis. Without anoikis resistance (anchorage-independency), tumors would be unable to abandon their normal sites and invade neighboring tissues and metastasize at distant locations. Anoikis is the natural barrier against cancer progression. Therefore, overcoming anoikis is a major step in cellular transformation. Cancer cells have developed many successful strategies to bypass anoikis. The main mechanism, albeit not the only one, involves hyper-activating survival pathways and over-expressing anti-apoptotic molecules. There is a strong and intertwined association between epithelial mesenchymal transition and anoikis resistance that is discussed in depth. A better understanding of anoikis resistance mechanisms has led to the research and development of pharmaceuticals that can counteract them.
Keywords:
Anoikis
; Anoikis resistance
; Cancer
; Apoptosis
; Epithelial mesenchymal transition
; Anoikis resistance in pancreatic cancer
1. Introduction
Normal tissues have a delicate dynamic balance between cell proliferation and cell death that form part of an essential homeostasis of multicellular organisms. Anoikis (which means homelessness in Greek) contributes to this homeostasis. Anoikis is defined as a programmed cell death, that is induced upon cell detachment from the extracellular matrix (ECM). Anoikis is also known as cell-detachment-induced apoptosis. Sakamoto and Kyprianou [1] described this phenomenon clearly and succinctly: cells gain freedom and meet death. In laymen’s terms it could be restated as a form of apoptosis that occurs when cells are detached from the extracellular matrix. Those cells that lose their established permanent contacts with the extracellular matrix are bound to die. The words "established permanent" differentiate cells that have permanent contact with their ECM such as:
epithelial, endothelial, osteoblasts, chondrocytes, and even some connective tissue cells, from those cells that can have occasional, but not usually firmly established contact, such as circulating leucocytes, dendritic cells, and macrophages. Therefore, we can differentiate attachment-dependent cells like epithelial cells and others from attachment-independent cells such as white blood cells. Attachment-dependent cells “need” to be attached to the ECM to survive and thrive while attachment-independent cells do not. Solid tumors are characterized by the transformation of attachment-dependent into attachment-independent cells. This transformation involves different pathways and molecules that will be analyzed in this review.
Normal attachment-dependent cells have permanent crosstalk with the ECM to which they are attached. This crosstalk is essential for their survival because it influences cell shape, migration, proliferation, and differentiation [2]. The ECM is not just a scaffold - it’s a dynamic environment that communicates with cells to regulate their fate and function. When the cell-ECM communication is interrupted, the cells die. This is called anoikis. On the other hand, cancer cells manage to survive after the interruption of the crosstalk, meaning that they became attachment-independent. Attachment-independent growth is a hallmark of transformation in cancer biology, often used as a test for tumorigenicity in vitro [3].
3.1.- Resistance to anoikis, a very important hallmark of cancer, consists of the cancer cell’s survival despite its detachment from the ECM. Resistance to anoikis (AR), or cell survival despite detachment, is the loss of the natural barrier that prevents invasion and metastasis. Among the many changes that circulating cancer cells develop, one of the most essential is anoikis resistance. Cancer cells that resist anoikis can survive in suspension, circulate through the bloodstream, and seed metastases [4]. While anoikis leads to the death of circulating tumor cells and inhibits colonization, AR promotes distant organ colonization and metastatic potential [5]. Furthermore, AR generally correlates with poor prognosis. This has therapeutic implications, because targeting AR mechanisms may:
- Prevent metastasis
- Sensitize circulating cancer cells to apoptosis
- Improve outcomes in advanced cancers
3.2.- Cell-matrix interactions have profound effects on phenotype and gene expression and govern several aspects of cell growth, differentiation, and motility [6]. Attachment-dependent cells can survive for a very short time when they lose their attachment and finally die. This means that by losing their attachment they are no longer viable. On the other hand, cancer cells can survive when they are detached from their ECM. Multiple mechanisms are developed in cancer cells to achieve an attachment-independent state. Developing this independence is an event that precedes mobility, invasion and metastasis.
It would be impossible for the millions of cancer cells found in most tumors to circulate without acquiring attachment-independent status. This is a pre-condition for invasion and metastasis [7].
Crosstalk between the extracellular matrix (ECM) and malignant cells plays a pivotal role in promoting anoikis resistance, enabling cancer cells to survive detachment and metastasize. This interaction reprograms signaling and metabolic pathways that normally trigger cell death upon loss of adhesion.
2. Historical Background
In 1968, Stoker et al. [8] found that “Many cell types will grow when attached to a rigid surface but not in suspension, a phenomenon termed „anchorage dependence”. Further: “The state of inhibited cells in suspension culture was examined by dispersing them in a methyl cellulose gel, in vessels lined with agar. In this system aggregation is prevented and the cells may be recovered quantitatively. Normal, as well as transformed, cells increase in size, and a proportion synthetize DNA during the first 24 hours in suspension culture. Growth and DNA synthesis in normal cells then virtually cease, while transformed cells continue to grow into colonies.”
Based on these previous experiments, anoikis was described for the first time in 1994 by Frisch and Francis [9]. They also coined the name. Furthermore, in the same article they showed that transformed cells were resistant to anoikis, and that there were attachment-dependent cells such as the epithelial cells, and attachment-independent cells like fibroblasts. In that single article they established the pillars on which the anoikis building was erected. Importantly, they clearly showed the essential relationship between the ECM and cells and that apoptosis could be regulated by the ECM. The origin of the Frisch and Francis studies stemmed from their idea that “cell motility or transformation might require matrix-independent survival”
3. Anoikis Concept
Under physiological conditions, anoikis maintains appropriate cell numbers balance by triggering apoptosis in cells with inadequate cell-to-ECM and cell-to-cell interactions [10]. A good example of anoikis is the death of intestinal epithelial cells that are detached and shed into the lumen and undergo apoptosis [11]. Anoikis is found in many normal epithelial cells that permanently or periodically renew their portfolio. This includes mammary glands [12,13], thyroid cells, intestinal mucosa, keratinocytes and other cell types [14]. Anoikis is also present in normal endothelial cells and osteoclasts [15].
Many publications maintain that anoikis is a mechanism for prevention of adherent-independent cell growth and attachment to an inappropriate matrix, thus avoiding colonization of distant organs. This would mean that the cell commits this suicide because it "knows" it is dangerous to allow a cell to circulate out of control. We do not agree with this concept. It is hard to believe that individual cells are conscious of the danger they represent. We believe that what drives anoikis is not the cell’s “knowledge”. What drives it is the loss of stimulatory and regulatory signals received from the extracellular matrix and fellow epithelial cells. Anoikis occurs "because of something" rather than "for something". This does not mean that anoikis is purposeless.
Anoikis is an evolutionary trait developed in a Darwinian fashion in multicellular organisms. Its development confers selective advantages such as preventing cell growth in unusual sites, invasion among tissues, epithelial mesenchymal transition, and metastasis. It would thus be selected evolutionarily. At a certain point, evolution from a unicellular to a multicellular organism requires the ability to grow and differentiate only when the cell is in the correct place in a tissue. Otherwise, by the process of anoikis, a cell removes itself through apoptosis. However, we could not find any publications on the phylogenetic evolution of anoikis. Detection of "being in the right place" means that the cell has developed sensing mechanisms that inform it where it is. And these sensing mechanisms have the ability to trigger death pathways if the cell’s place is not the correct one.
Anoikis also means that cells have intercommunication with the matrix and with other cells in order to recognize their place. In this regard, Gilmore [16] wrote "Anoikis, therefore, should not be considered as an experimental system in vitro, but the mechanism by which cells in vivo use ECM-derived signals to maintain tissue integrity". Here, we must add that cells need to recognize their correct extracellular site as well as being with their neighbor cells.
To understand anoikis it is necessary to study the main interaction point between the ECM and the cell: that is focal adhesion (see below). Focal adhesion is the origin of the signaling that keeps the cell alive while attached to the ECM and initiates apoptosis when detached.
In summary: Attachment-dependent cells die when detached, which is due to anoikis; cancer cells do not die when detached, they have resistance to anoikis. Attached normal cells permanently receive signals from the ECM, mainly through integrins and detachment interrupts these signals triggering anoikis. When cells become detached, the cell cycle is interrupted and a caspase-dependent cell death occurs resulting in anoikis. Focal adhesions are hubs that permit cell-to-matrix adhesion and at the same time transmit signals from the ECM to the cell and vice versa. ECM detachment in tumors precede abnormal dissemination that leads to metastasis.
4. Focal Adhesions (FAs)
Focal adhesions are mechanical linkages between the cell and the ECM. These adhesions represent a biochemical signaling and an adhesion hub where there is a large concentration of adhesive molecules such as integrins. Focal adhesions are very dynamic and are made up of more than 100 different proteins that are in a constant state of change. Focal adhesions form following the cell’s attached to the ECM through integrins, anchoring actin filaments and microtubules (MTs) attached to the cell’s membrane [17]. Two kinases, FAK (focal adhesion kinase) and SRC (sarcoma oncogene, also known as Proto-oncogene tyrosine-protein kinase Src) and a protein Paxillin are key elements that connect integrins to the actin cytoskeleton through talin and vinculin, two other important proteins.
Some proteins of focal adhesions associate more permanently, while others disassociate continuously. This generates signals that are transmitted to the rest of the cell by the ECM-integrin-cell axis. Many cell functions such as motility, invasion, cell cycle, apoptosis or resistance to apoptosis are thus influenced by the protein trafficking and modifications at the focal adhesion. The large number of different proteins forming these structures may suggest that not all the focal adhesions have the same function. However, there is a lack of information in this respect. The dynamic and permanent changes in the proteins of focal adhesions create the conditions for cell adhesion, survival, growth, differentiation, and motility of normal cells as well as invasion, metastasis and resistance to anoikis, in cancer cells.
4.1. Focal Adhesions in Cancer
Tumor cell migration and invasion:
FAs facilitate cell motility by coordinating cytoskeleton remodeling and ECM degradation.
Cancer cells exploit FA dynamics to invade surrounding tissues and enter circulation.
Anoikis resistance:
Normally, detachment from the ECM triggers anoikis (a form of apoptosis).
In normal cells, FA signaling ceases after detachment. In cancer cells, FA signaling persists even after detachment, allowing cells to survive and metastasize [18].
Survival and proliferation:
FAs activate key pathways like FAK, PI3K/AKT, and MAPK/ERK, promoting cell survival and growth.
Mechanotransduction:
FAs help cancer cells adapt to mechanical stress in the tumor microenvironment, enhancing their invasive potential.
4.2. Key Molecules Involved in Focal Adhesions
FAK (Focal Adhesion Kinase):
Central to FA signaling; over-expressed in many cancers.
Drives survival, migration, and drug resistance.
Integrins:
Integrins are transmembrane receptors that initiate FA formation and influence tumor cell resistance to apoptosis. Altered integrins expression contributes to metastasis and therapy resistance [19]. Changes in integrins profiles enable tumor cells to evade apoptosis, migrate through tissues, and adapt to hostile microenvironments, including during treatment. For example, integrins αvβ3 and α5β1 integrins are associated with poor prognosis and metastatic spread in melanoma, glioblastoma, and colorectal cancer, and integrin β1 is linked to resistance in breast, lung, and pancreatic cancers [20,21,22,23]. Integrins such as β1, αvβ3, and α5β1 bind ECM components and activate survival pathways like FAK/Src, PI3K/AKT, and MAPK/ERK. This signaling cascade inhibits pro-apoptotic factors and promotes cytoskeleton stability, allowing cells to resist detachment-induced apoptosis [24,25].
Adaptor proteins:
Talin, vinculin, paxillin, vimentin, and Src coordinate FA assembly and signal transduction. They are called adaptor proteins because they connect the cytoskeleton to integrins. Although they are called adaptor proteins their functions seem to go beyond adaptation. For example, talin participates in integrin activation [26,27]. Talin over-expression in tumors correlates with resistance to anoikis and metastasis. Furthermore, talin homodimers can bind four integrins establishing a cross-link of integrins that are essential for clustering [28].
Figure 1 shows a simplified view of the main players in the focal adhesion structure. The few proteins shown in the drawing are the most important and best known.
The first step in cell adhesion to the ECM consists of the contact between fibers (fibronectin, collagens, elastin, and laminins) in the ECM with the extracellular segment of integrin dimers. This step activates integrins and they undergo a conformational change (Figure 2). Next, activated integrins trigger a key protein in the intracellular segment: FAK. This activation causes FAK autophosphorylation at Tyr397. As mentioned above, talin also plays a role in integrin activation.
In a second step, the phosphorylated FAK recruits and activates another kinase: SRC. SRC continually adds phosphate groups to the many proteins it services. In this case SRC also adds further phosphates to FAK permitting full FAK activation (Figure 2 (4)). Now the highly phosphorylated FAK initiates signaling to downstream effectors.
5. The Main Players in Anoikis
5.1. Integrins
Integrins are known to inhibit apoptosis in attached cells by transmitting anti-apoptotic survival signals initiated from the ECM [35]. They transmit both outside-in and inside-out signals that regulate survival, proliferation, and migration [36]. Integrins are transmembrane adhesive proteins that transmit signals between cells and the extracellular matrix, probably in a bidirectional fashion [37]. Integrins are heterodimers formed by an α and a β subunit that are associated through non-covalent bonding [38]. The integrin family is formed by 24 different heterodimers. Their extracellular ligands make it possible to classify them into four groups:
- RGD-binding integrins: RGD receptors (Arg-Gly-Asp (RGD) attachment site), constitute a major recognition system for cell adhesion [39,40]. Several integrins recognize and bind to the RGD motif, a key tripeptide sequence found in many extracellular matrix (ECM) proteins like fibronectin, vitronectin, and fibrinogen. These RGD-binding integrins play crucial roles in cell adhesion, migration, and signaling. Importantly, RGD-binding integrins like αvβ3 and αvβ5 are over-expressed in tumors and promote angiogenesis, invasion, and metastasis [41];
-
leukocyte-specific receptors are a specialized subset of integrins that mediate immune cell adhesion, migration, and signaling. They are essential for immune surveillance, inflammation, and host defense [44]. These integrins are primarily expressed on white blood cells and are often referred to as β2 integrins or CD18 family;∙ collagen receptors that regulate proliferation, migration and adhesion [45].
Importantly, integrins suppress anoikis [46]. Thus, while the epithelial cell is anchored to the matrix there is no anoikis. This means that integrin’s signaling to the cell prevents anoikis-induced apoptosis. But as soon as the cell is detached this inhibition disappears and the cell dies. If the cell is malignant, it does not die because it develops resistance to anoikis.
Integrins have a double function: they anchor the cell to the matrix (adhesive function) and transmit signals between matrix and cell, and vice versa (communication function). The precise molecular mechanism of how integrins participate in anoikis and resistance to anoikis is far from clear. What is clear is that anoikis is mediated by membrane adhesion signaling molecules such as integrins. Interestingly, integrins can also inhibit anoikis in some detached cells [47].
5.1.1 Integrins as Sensors
Integrins act as biochemical and biomechanical sensors, enabling cells to detect and respond to changes in their extracellular environment. This sensing function is crucial for regulating adhesion, migration, survival, and differentiation—especially in cancer and immune responses.
Their sensing capabilities operate through two main modes:
Biochemical Sensing
Ligand recognition: Integrins bind to ECM proteins like fibronectin, collagen, and laminin via specific motifs (e.g., RGD).
Biomechanical Sensing [51]
There are two important modifications within the ECM that play an essential role in the cancer cell’s fate: stiffness (rigidity) and degradation [52]. ECM stiffness is frequently found in cancer and plays a role in migration, invasion and metastasis [53,54]. Importantly, stiffness is detected by cancer cells through their integrins-mediated biomechanical sensing abilities which elicit intracellular changes that reinforce the malignant phenotype, including resistance to apoptosis. The involved mechanisms are:
Force transmission: Integrins connect the ECM to the actin cytoskeleton, allowing cells to sense mechanical tension.
Mechanotransduction [55]: Integrins convert mechanical stimuli into biochemical signals, influencing cell fate and behavior. Integrins can detect matrix stiffness leading cells to adjust their adhesion and migration based on ECM rigidity, which is usually altered in cancer.
The integrin properties mentioned above activate various signaling pathways, such as:
the focal adhesion kinase (FAK) pathway,
the Src kinase pathway, and
the PI3K/Akt pathway.
However, integrin signaling is not limited to these pathways and can also interact with other signaling molecules, such as growth factor receptors and GPCRs (G protein-coupled receptors), to modulate their activity.
Integrin antagonists are being developed to block aberrant signaling in cancer. RGD-based drug delivery systems exploit integrin sensing to target tumors with high αvβ3 expression. In this regard, there are now drugs, such as cilengitide, TDI4161, MK0469 that target αvβ3 integrins (which are discussed below).
5.2. FAK
FAK is a cytoplasmic protein tyrosine kinase that is activated by phosphorylation, when integrins bind to their extracellular ligands such as collagen or fibronectin. Integrins do not have kinase activity, and they do not phosphorylate FAK. However, they induce FAK’s autophosphorylation. This activation triggers a cascade of signaling events that regulate cell behavior including cell survival, migration, and proliferation [56,57]. FAK has several functional domains that interact with other proteins at the focal adhesion and can execute various biological processes [58]. FAK has a FERM domain at the N-terminus, a central catalytic kinase domain and a C-terminal focal adhesion- targeting domain (FAT). (Lower right panel in Figure 1).
FAK acts as a signaling hub, interacting with various proteins such as Src kinases, paxillin and p130Cas, transmitting downstream signals regulating:
FAK is often over-expressed and hyperactive in various types of cancer, such as exocrine pancreas [64,65], non-small cell lung [66], small cell lung [67], ovarian [68], gastric [69], colorectal [70], head and neck squamous cell [71], breast [72,73], thyroid [74,75,76], hepatocellular carcinoma [77], clear cell renal cell carcinoma [78], glioblastoma [79,80], and melanoma [81,82]. FAK inhibitors are subject of active research [83]. Defactinib is one of the most promising drugs in this regard (see below).
Hypothetically, we can say that cells require permanent and specific signaling from the correct ECM in order to stay alive. Normal cells seem to be continuously monitoring their microenvironment. If this signaling is absent or is incorrect, the cell undergoes apoptosis with a different triggering mechanism, and this triggers anoikis (detachment-induced apoptosis). Cancer cells can block or modify the vital need for this signaling, and this is called resistance to anoikis. This resistance means that the cancer cell can survive despite the loss of ECM signaling. Once a cancer cell reaches this status, the road for epithelial mesenchymal transition, invasion and metastasis is wide open. This explains the importance of anoikis and the role of resistance to anoikis [84,85,86,87,88,89].
It should be noted that not every cell can undergo anoikis. Only attachment-dependent cells are susceptible. When they are detached from the epithelium, attachment-dependent normal cells will die. On the other hand, when cancer cells are detached from the epithelium they resist anoikis and survive. This means that cancer cells develop survival mechanisms, despite the lack of stimulation and signaling that comes from the extracellular matrix and from fellow epithelial cells. Anoikis resistance is a mechanism to resist apoptosis. It is mainly found in cancer cells and is a necessary step in the progression towards motility, invasion, and metastasis. When anoikis resistance is absent, the cancer cell is unable to progress towards metastasis. This is why anoikis has become an important issue in cancer research.
5.3. Integrin Linked Kinase (ILK)
ILK is a kinase with multifunctional characteristics. It regulates integrin and growth factor receptor signaling [90]. It acts as a signal transduction protein and scaffolding structure. It is usually located at the cell membrane and is a component of focal adhesions. ILK has been found to play a role in cancer progression, and it is a therapeutic target [91]. Despite its name, it is not clear if ILK is really a kinase [92]. ILK has been found to bind the intracellular portion of integrins and its signaling suppresses anoikis [93,94]. Notwithstanding its multiple pro-tumoral functions, it is not clear if it represents another link between integrin and focal adhesions with anti-apoptotic proteins.
5.4. SRC
SRC was identified in the cell in 1976 as the equivalent counterpart of the transforming gene of the avian Rous sarcoma virus, v-src, discovered by Peyton Rous in 1911. His initial observations describe it as a filterable agent, because viruses were not known at that time [95]. This was the first viral oncogene to be discovered and gave rise to the concepts of proto-oncogenes, oncogenes and tumor suppressor genes.
The SRC protein is a tyrosine kinase signaling protein that specializes in messages that control the growth of cells. It is located inside cells next to the cell membrane. Its main function consists of passing on signals from various protein receptors to intracellular proteins related to growth, survival, cell division, and motility. SRC adds phosphate groups to FAK, one of its “clients”, . This further increases FAK activity [96. 97]. SRC is activated by phosphorylation of Tyr530 and it can phosphorylate several Tyr residues of FAK [98]. (Figure 3).
SRC-mediated phosphorylation of FAK is necessary to couple actin to focal adhesion, and adhesion dynamics to survival signaling [99]. SRC has three domains: SH3, SH2, and kinase (SH1). There is also a SH4 domain in some members of the SRC family. SH2 and SH3 are the protein binding regions, while SH1 is the tyrosine kinase region that has an auto-phosphorylation site required for activation.
5.5. p130Cas
p130Cas also known as breast cancer anti-estrogen resistance 1 (BCAR1), is an adaptor protein that interacts with several proteins at the intracellular portion of focal adhesion. It has a regulatory role in migration and apoptosis [100]. In addition to its interaction with FAK and SRC [101,102], it also interacts with caspase 3 [103], and growth factor receptors [104].
5.6. Paxillin
6. Molecular Mechanisms of Anoikis
Integrins regulate cell viability and other intracellular features by integrating extracellular conditions with specific intracellular pathways [108]. Integrins also regulate the activity of many cell proteins, such as growth factor receptors, intracellular kinases such as FAK (Focal Adhesion Kinase) and SRC (proto-oncogene tyrosine-protein kinase Src), the cytoskeleton, and other proteins [109,110]. Matrix proteins and/or ligands can activate the heterodimeric integrins to induce intracellular events such as cytoskeletal actin polymerization or signaling towards intracellular pathways. These pathways and intracellular organization are essential for cell viability while attached to the appropriate matrix. Figure 4.
In 2018, the Nomenclature Committee on Cell Death [116] defined anoikis as a specific form of intrinsic apoptosis triggered by integrin-dependent anchorage deficiency. (Intrinsic apoptosis is activated by internal signals and involves the mitochondria, while extrinsic apoptosis is apoptosis triggered by external signals such as binding to death receptors on the cell surface [117]).
Anoikis is a triggering mechanism that ends in extrinsic apoptosis and secondarily leads to intrinsic apoptosis. There is evidence showing that the development of resistance to anoikis favors the development of cancers [118], epithelial mesenchymal transition [119], and metastasis [120].
Integrin signaling is not the only pathway that prevents anoikis. Growth factor receptors also play a role [121,122,123,124]. Below we discuss mechanisms of anoikis suppression. Figure 5.
Anoikis uses both the intrinsic and extrinsic apoptosis pathways. Therefore, it is not different from other death pathways. What differs from classic apoptosis is the trigger mechanism. Anoikis can use different pathways leading to cell death, but they all end up triggering apoptosis, one way or another. We can consider anoikis as a special form of apoptosis triggered by epithelial or endothelial cell detachment from the matrix. Since it is integrins that connect the cell with its matrix, these transmembrane proteins are the key players in anoikis and are probably also important in resistance to anoikis.
7. Specificity of the Surface to Which the Cell Is Attached
Meredith et al. [128] showed that some types of cells need to be attached to specific surfaces to rescue them from anoikis. The title of the paper by Meredith et al. fully depicts the relationship between ECM and cell: "The Extracellular Matrix as a Cell Survival Factor".
8. Specificity of Integrins
9. Relationship Between Cell Detachment and the Apoptosis Pathway
In non-malignant cells, cell surface death receptors are activated by death ligands. These receptors recruit death proteins that form the DISC (Death induced signaling complex) which in turn cleaves caspase 8, which is the initiator caspase. Cleaved caspase 8 in turn cleaves the executor caspases leading to cell breakdown. FLIP (FLICE-inhibitory protein) is the main inhibitor of the death receptors pathway by preventing caspase 8 activation.
When the cell loses contact with the ECM, death receptors and death receptor ligands are up-regulated, and FLIP is down-regulated [133]. This leads to caspase 8 activation. However, anoikis can occur even without participation of the death receptor: unligated β integrins tails can recruit caspase-8 to the membrane without FADD (Fas-associated protein with death domain) intervention. There, caspase 8 is activated in a death receptor-independent manner [134]. Figure 6.
Matrix attachment seems to protect cells from Fas receptor-induced apoptosis (Fas is a death receptor on the cell surface), and the opposite occurs with matrix detachment. The interruption of the integrin signaling that follows the Integrin-FAK-PI3K-AKT pathway seems to be what increases the activity of the death receptors and decreases FLIP expression [136]. Figure 7.
Activation of caspase 8 is the central event of extrinsic apoptosis. This, in turn, can initiate the intrinsic apoptosis through cleavage of BID (BH3-interacting domain death agonist) releasing t-BID (truncated BID) which unleashes mitochondrial outer membrane permeabilization with cytochrome C release. This is the key in intrinsic apoptosis.
Figure 8 shows how extrinsic apoptosis initiated by caspases 8 activation continues through the intrinsic pathway by cleavage of BID to t-BID (truncated BID). Caspase 8 can cleave BID and thus increase the apoptotic activity reinforced by the intrinsic pathway. However, it has been shown that full length BID can achieve similar results without being cleaved to t-BID [145].
10. Resistance to Anoikis
Resistance to anoikis is essential for tumor development. The malignant cell needs to detach and separate from the ECM during the epithelial mesenchymal transition process conferring migratory, invasive, and metastatic abilities. This progression can only take place when the risk of anoikis is neutralized. Therefore, malignant cells need to develop one or more mechanisms to prevent anoikis. Anoikis resistance (AR) is a possible therapeutic target which is under intense investigation. Unfortunately, the exact molecular players and their sequence in anoikis resistance are only partially known. For efficient AR, both apoptotic pathways (intrinsic and extrinsic) need to be inhibited. Evidence shows that cancer cells can efficiently abrogate both.
Let’s examine the known facts.
A) Intrinsic apoptotic pathway
Apoptosis or anti-apoptosis is the result of the balance between pro- and anti- apoptotic proteins. Pro- and anti- apoptotic proteins can heterodimerize and it is believed that they can interfere with each other’s functions. Anti-apoptotic proteins can bind BH3 domains of the apoptotic proteins inhibiting their effects [146,147]. Therefore, the increased expression of anti-apoptotic proteins will result in resistance to apoptosis [148].
► There is evidence that the expression of the anti-apoptotic members of the Bcl2 family induces increased anoikis resistance [149].
► RAS activation prevents down-regulation of antiapoptotic proteins during detachment [150].
► SRC activation induces antiapoptotic proteins expression and resistance to anoikis [151].
► According to Woods et al. [152] the antiapoptotic protein Mcl1 is targeted for proteasomal degradation, and this along with up-regulation of BIM are the initiators of anoikis. Mcl1 ubiquitination and degradation does not occur in malignant cells.
B) Extrinsic apoptotic pathway
► FLIP is the natural inhibitor of the extrinsic pathway. It is a protein with remarkable similarities to caspase 8 and with a higher bind affinity for DISC, thus replacing caspase 8 binding on DISC. This prevents caspase 8 activation at the DISC [153]. FLIP over-expression has been clearly identified as one of the main causes of AR [154,155,156,157].
While normal cells down-regulate FLIP expression after detachment, malignant cells do not.
► Majwi et al. [158] have shown that inhibiting FLIP at a post-transcriptional level, induces anoikis in AR cells when they are detached but not while they are attached.
11. Drivers of Anoikis Resistance
AR is the result of multifactorial changes. Although there is no direct experimental proof, it seems that not one, but many alterations are necessary to induce AR. An indirect proof of this concept is the variety of multiple genetic signatures found in AR in different tumors. These different AR factors are interrelated at some point forming what we may call an AR network. Parts of this network are:
11.1. Major Drivers of Anoikis Resistance
11.1.1. Altered Integrins Expression
Cancer cells down-regulate integrins that promote apoptosis upon detachment (e.g., α5β1) and up-regulate those that support survival (e.g., αvβ3, α6β4). The integrin switch is linked to EMT, which enhances migratory and invasive capabilities while reducing dependence on ECM attachment. The integrin switch has been identified as a cause of AR in melanoma and other tumors. When melanoma invades the dermis, it switches from the normal αvβ1 integrin to αvβ3, which facilitates the acquisition of anoikis resistance [159].
11.1.2. Activation of survival pathways such as:
FAK-SRC pathway.- FAK and Src kinases are often activated by switched integrins, promoting cell survival and motility.
11.1.3. Metabolic Reprogramming
Malignant tumors rewire their metabolic landscape by adopting a high glycolytic flux phenotype in which glycolysis and oxidative phosphorylation metabolism coexist even in presence of adequate amount of oxygen (Warburg effect). This shift supports ATP production (through oxidative phosphorylation) and biosynthesis (through glycolysis that provides building blocks) under stress, helping cells survive in suspension [170]. Metabolic reprogramming enables cancer cells to resist anoikis by adapting their energy production, redox balance, and biosynthetic pathways to survive without extracellular matrix (ECM) attachment. Enhanced glycolysis, glutaminolysis, increased fatty acid oxidation, increased pentose phosphate pathway, and mitochondrial oxidative phosphorylation support anoikis resistance [171].
Proteomic studies have shown distinct metabolic profiles between adherent and suspended cancer cells, revealing up-regulation of survival-promoting pathways [170]. However, the molecular mechanisms involved have not been clearly established in all cases.
ENOX2 (Ecto-NOX disulfide-thiol exchanger 2) is a cell surface protein involved in redox regulation and cell growth control. It has enzymatic activity and acts as a hydroquinone/NADH oxidase and a protein disulfide-thiol exchanger. ENOX2 is frequently over-expressed in malignant cells [172]. During ECM detachment, ENOX2 supports antioxidant defenses, preventing oxidative stress-induced apoptosis. Although the role of ENOX2 in anoikis resistance has not been fully proved, we believe that by enabling survival in suspension, ENOX2 facilitates the dissemination of cancer cells through the bloodstream. Cells with elevated ENOX2 expression show higher metastatic potential supporting this contention [173].
Enolase 2 (ENO2).- ENO2 over-expression helps detached cells resist anoikis by maintaining the redox balance. Figure 13. Glycolysis and glycolytic enzymes are increased in detached cells [174,175]. By enhancing glycolysis, ENO2 ensures energy supply in anchorage-independent conditions. ENO2 expression correlates with increased migration and invasion, suggesting its role in facilitating metastasis through anoikis resistance. Over-expression of ENO2 as a cause of AR was initially found in anaplastic thyroid cancers [176], but recently it has also been found in other tumors [177]. Figure 9.
11.1.4. Autophagy
Cells recycle components to survive nutrient deprivation during detachment. It is a protective autophagy but under certain circumstances it can lead to cell death [178,179,180]. Although there is no clear line that separates the autophagy cell death from protective autophagy, it is presumed that in AR the role is protective, at least at the beginning.
Yu et al. [181] found that the over-expression of the cell migration-inducing protein (CEMIP) in detaching cells resulted in protective autophagy by inducing the dissociation of the B-cell lymphoma-2 (Bcl-2)/Beclin1 complex. The Bcl-2/Beclin1 complex regulates the balance between autophagy and apoptosis, playing a critical role in cancer cell survival and resistance to therapy. The Bcl-2/Beclin1 complex forms when Bcl-2 binds to Beclin1 via its BH3 domain, inhibiting Beclin1’s ability to promote autophagy [182]. Figure 10.
Autophagy begins when Beclin 1 is released. Bcl-2 also inhibits apoptosis by sequestering pro-apoptotic proteins like Bax and Bak.The balanced dual role of Bcl-2 in suppressing both autophagy and apoptosis make it a central survival factor in cancer.
11.1.5. Cytoskeleton Reorganization
Cancer cells evade anoikis through dynamic changes in their cytoskeleton. These changes affect cell mechanics, signaling, and interactions with the microenvironment [183]. Cytoskeleton reorganization includes:
- actin remodeling that supports anchorage-independent growth and facilitates migration through tissues;
- microtubule stabilization that maintains intracellular transport and polarity in detached cells and promotes the formation of survival-promoting structures like giant unilamellar vacuoles which buffer mechanical stress.
- during epithelial mesenchymal transition (EMT) intermediate filaments such as vimentin are up-regulated contributing to structural integrity and resistance to mechanical stress;
- activation of survival pathways such as the Hippo pathway, particularly YAP/TAZ transcription factors, which promote cell survival and proliferation in detached conditions. Cell detachment activates the Hippo pathway kinases Lats1/2 and leads to YAP phosphorylation and inhibition. This detachment-induced YAP inactivation is essential for anoikis in non-malignant cells, whereas in cancer cells the deregulation of the Hippo pathway inhibits anoikis. Furthermore, knockdown of YAP and TAZ restores anoikis [184].
11.1.6. Epithelial Mesenchymal Transition (EMT)
EMT is a biological process where epithelial cells lose their polarity and adhesion properties, acquiring mesenchymal traits like motility and invasiveness. This transformation is closely linked to anoikis resistance in cancer progression [185].
Down-regulation of E-cadherin in EMT disrupts epithelial junctions, allowing cells to detach. This detachment normally triggers anoikis, but EMT suppresses apoptotic signaling [186]. EMT activates the PI3K/Akt, MAPK/ERK, and NF-κB pathways, which inhibit apoptosis and promote survival in detached conditions. These pathways also support anchorage-independent growth and therapy resistance. We suggest that anoikis resistance development is a necessary and integral part of the EMT process. Both phenomena are so intertwined and interdependent that it is not easy to establish a clear dividing line. For example, EMT involves remodeling of actin and intermediate filaments (e.g. vimentin), enhancing cell motility and structural integrity during detachment. Another example is EMT increasing the population of cancer stem-like cells, which are inherently resistant to anoikis and contribute to recurrence and metastasis. E-cadherin and ankyrin-G are lost coordinately during EMT, conferring anoikis resistance. Finally, targeting EMT regulators or restoring epithelial traits may sensitize tumors to anoikis and reduce metastasis.
11.2. Other Drivers of Anoikis Resistance
11.2.1. Extracellular Acidity
It is a well-known fact that extracellular acidity, a constant hallmark of cancer [187], is an important facilitator of metastasis. What is less known is that one of the mechanisms involved in this facilitation is an increased resistance to anoikis. The exact molecular steps that lead from extracellular acidity to increased anoikis resistance are not well known. On a speculative basis, but based on some experimental evidence, we may assume that extracellular acidity increases anoikis resistance by stimulating autophagy [188]. mTORC1 and NF-kB signaling seem to have a role in resistance to anoikis in an acidic extracellular matrix [189].
11.2.2. V-ATPase Pump up-Regulation
Up-regulation of these proton exporters seems to promote anoikis resistance [190]. We believe that this is due to their contribution to extracellular acidity.
11.2.3. Nitric Oxide (NO)
Chanvorachote et al. [191] found that NO can impair the apoptotic function of lung carcinoma cells after detachment by inhibiting the ubiquitin-proteasomal degradation of Caveolin-1.
11.2.4. Reactive Oxygen Species (ROS) and Growth Factor Receptors
NADPH oxidase 4 (NOX4) expression and ROS generation are up-regulated in some tumors such as gastric cancer, leading to anoikis resistance by inducing EGFR activation [192].
11.2.5. EWS/FLI Oncogenic Protein
Ewing sarcoma is an aggressive pediatric bone and soft tissue cancer characterized at the molecular level by the presence of a chromosomal translocation: t(11;22)(q24;q12). The encoded chimeric oncoprotein from this translocation is known as EWS/FLI, which is the result of the fusion of the amino-terminal domain of EWS with the carboxyl-terminus of FLI [193]. This protein has been shown to play a key role in anoikis resistance, because when the protein is down-regulated, anoikis follows [194]. This protein is not found in other tumors.
11.2.6. Oncoviruses and Anoikis Resistance
11.2.7. Mir141-Sp1 Axis
11.2.8. NHE1 (Sodium Hydrogen Exchanger 1)
NHE1 plays a role in anoikis resistance, although this has not been firmly proven. Pedersen [198] offered a hypothesis of the mechanism: “…it is well-established that NHE1 is an important effector downstream from integrin activation and necessary for integrin-dependent cell spreading. It is, thus, tempting to speculate that the loss of anchorage-dependence seen in many cancer cells could be related to the elevated NHE1 activity and/or altered NHE1-dependent signaling in these cells, circumventing the requirement for cell adhesion for survival”.
11.2.9. FER Kinase (Feline Sarcoma Related Kinase)
FER kinase is a non-receptor cytoplasmic tyrosine kinase [205] that controls migration and metastasis of invasive human breast cancer cell lines by regulating α6- and β1-integrin-dependent adhesion [206]. FER boosts integrin signaling, enabling cells to survive without ECM attachment. FER kinase activity has also been identified in AR in other tumors such as ovarian, lung and prostate cancers [207,208,209,210]. FER kinase can activate PI3K/Akt and MAPK/ERK pathways, suppressing apoptosis and supporting anchorage-independent growth.
11.2.10. Epigenetic Factors
DNA methylation and histone modifications alter gene expression to favor survival. These changes can silence pro-apoptotic genes and activate anti-apoptotic ones [211].
11.2.11. Loss of E cadherin
Loss of E cadherin is a characteristic feature of EMT. Cells with lost expression of E cadherin show a higher rate of metastasis and increased resistance to anoikis [212].
12. Targeting Anoikis Resistance
Pharmacological targeting of resistance to anoikis is a promising therapeutic objective [213]. While no drugs have yet been approved, several are currently in late-stage development. There are also approved drugs that can be repurposed to target AR. Drugs targeting anoikis resistance aim to re-sensitize cancer cells to anoikis. By overcoming anoikis resistance, these drugs can potentially inhibit cancer cell survival, metastasis and recurrence.
12.1. V-ATPase Pump Inhibitors
It has been shown that archazolid, a V-ATPase inhibitor, reduced FLIP expression, increased caspase 8 activity, reduced active integrin-β1, and increased the proapoptotic protein BIM in several types of malignant cells in vitro. This was the initial effect of archazolid, however, later it could increase anoikis resistance. In vivo, it reduced the number of metastases in mouse breast cancer [214]. Archazolid-treated endothelial cells increased tumor cell adhesion mediated by β1-integrins expressed on the tumor cells [215]. Archazolid induces a pro-adhesive phenotype that seems to counteract AR.
12.2. Microtubule Destabilizing Agents
These drugs interfere with the cytoskeleton, disrupting focal adhesions and inducing anoikis [219]. Microtubule-disrupting agents induced paxillin phosphorylation and activated anoikis through the loss of focal adhesion structures. ILK over-expression rescued the antimicrotubule-mediated loss of cell viability.
12.3. Anoikis Resistance in Pancreatic Cancer
Anoikis resistance in pancreatic cancer is a crucial factor in its progression and metastasis, allowing cancer cells to survive detachment from the extracellular matrix. Several intertwined and overlapping mechanisms contribute to this resistance. However, one mechanism is considered the main promoter of anoikis resistance in pancreatic cancer: over-expression and activation of the transcription factor STAT3. STAT3 activation is achieved by increased phosphorylation at Tyr705, leading to enhanced expression of anti-apoptotic proteins like Bcl-2 and Mcl-1, and increased cancer cells' migratory, invasive, and metastatic potential. Inhibiting STAT3 reduces anoikis resistance and tumor formation in pancreatic cancer models, highlighting its vital role in cancer progression and metastasis.
The following AR drivers have been identified in PDAC:
12.3.1. The PI3K/AKT Pathway Activation
This pathway promotes cell survival, proliferation, angiogenesis, and resistance to apoptosis. We believe that the activation of this pathway represents the central core of AR in PDAC (pancreatic ductal adenocarcinoma) [220] as shown in Figure 12. PI3K/AKT is often hyperactivated in PDAC due to mutations in KRAS, PTEN loss, or over-expression of growth factor receptors.
Chronic inflammation, a risk factor for PDAC, is triggered by multiple mechanisms and is important in its development. Cytokines, chemokines, and growth factors present in the PDAC desmoplastic environment activate the PI3K/AKT pathway [221]. This is a clearly pro-survival pathway that induces the expression of the anti-apoptotic proteins of the BCL2 family. This starts with PI3K (a lipid kinase) which phosphorylates PIP2 to generate PIP3. This recruits AKT to the plasma membrane where it is activated by PDK1 and PDK2. Activated AKT phosphorylates multiple downstream targets that inhibit apoptosis [222,223]. For example, AKT phosphorylates and inactivates BAD, a pro-apoptotic Bcl-2 family member. It also inhibits caspase-9, a key initiator of the intrinsic apoptotic pathway [224]. Furthermore, AKT enhances the activity of transcription factors like NF-κB, which up-regulate anti-apoptotic genes such as Bcl-2 and Bcl-xL [225]. Blocking the PI3K/AKT pathway led to a decreased metastatic burden in cell lines and mouse models by lowering the survival rate of circulating cancer cells [226].
The PI3K/AKT pathway can be activated by many receptors and proteins:
Growth Factors and Cytokines Produced by CAFs
Cancer associated fibroblasts (CAFs) of the PDAC desmoplastic stroma produce growth factors such as TGF-β, HGF, FGF, PDGF, and IGF-1 [227,228,229,230], which bind to their receptors which in turn initiate signaling pathways that can activate PI3K/AKT. TGF-β is a potent activator of the PI3K/AKT pathway via TRAF6-dependent mechanisms, especially in cancer and fibrotic conditions [231,232]. FGF binding its cognate receptors can also activate the PI3K/AKT pathway [233]. The same situation happens with IGF1 [234], and PDGF [235].
STAT3 Activation
Signal transducer and activator of transcription 3 (STAT3) plays a critical role in conferring anoikis resistance to pancreatic cancer cells. Cells that resist anoikis show increased expression and phosphorylation of STAT3 at Tyr 705, which also enhances their migratory and invasive characteristics. Interleukin-6 (IL-6) treatment can enhance anoikis resistance by phosphorylating STAT3, while STAT3 inhibitors like the natural alkaloid piperlongumine can induce anoikis and prevent tumor formation and metastasis in pancreatic cancer models [236]. STAT3 can be activated by IL-6 as upstream effector. IL6 can simultaneously activate both the JAK/STAT3 and PI3K/AKT pathways. This dual activation is crucial for inflammatory and survival responses in cells [237]. Furthermore, CAFs produce IL-6 in pancreatic cancer [238]. In PTEN-deficient cancer cells, inhibition of the PI3K/AKT pathway can lead to feedback activation of STAT3, which then limits the efficacy of PI3K-targeted therapies. STAT3 can upregulate genes that encode components or regulators of the PI3K/AKT pathway, thereby indirectly enhancing its activity. In cells transformed by the cancer associated p110α-H1047R mutant PI3K, STAT3 phosphorylation is increased and this contributes to oncogenic transformation. Blocking STAT3 impairs PI3K-driven tumorigenesis. Probably for this reason, many authors consider STAT3 as the main driver of AR [239,240].
MUC1 (mucin 1)
MUC1 is a transmembrane glycoprotein often over-expressed and abnormally glycosylated in many cancers, and especially in PDAC. Its cytoplasmic tail interacts with key signaling molecules, including components of the PI3K/AKT pathway. The cytoplasmic tail of MUC1 can bind to PI3K, facilitating its activation and subsequent phosphorylation of AKT. Furthermore, MUC1 enhances the stability and activity of growth factor receptors, amplifying PI3K/AKT signaling cascades [241]. Targeting tumor-associated MUC1 with agents like TAB004 has shown promise in reducing colony-forming potential and triggering anoikis in pancreatic ductal adenocarcinoma (PDA) cells, suggesting its potential to curb tumor relapse, prevent metastasis, and enhance chemotherapy efficacy [242]. Furthermore, MUC1 over-expression can induce TGF-β signaling in PDAC [243].
HMGA1
Over-expression of HMGA1 (High Mobility Group A1) promotes anoikis resistance in pancreatic adenocarcinoma cells through a PI3-K/Akt-dependent mechanism, leading to increased Akt phosphorylation and kinase activity, and reduced caspase 3 activation [244].
PTEN Inactivation: Virus-Induced Protein APOBEC3G
APOBEC3G can induce AR by inactivating PTEN and subsequently activating Akt kinase [245].
PAXILLIN
MicroRNA-137 (miR-137) is down-regulated in pancreatic cancer and promotes anoikis by modulating the AKT signaling pathway, with paxillin (PXN) identified as a target of miR-137 that promotes AKT activation [246].
FOXM1 (Forkhead Box M1)Regulation
FOXM1 is a transcription factor, that has been found to be differently regulated in pancreatic cancer cells, and its expression is influenced by platelets. Manipulating FOXM1 expression affects platelet-mediated anoikis resistance, suggesting it as a potential therapeutic target [247]. FOXM1 plays a pivotal role in regulating cell cycle progression, DNA repair, and tumorigenesis. Its interaction with the PI3K/AKT signaling pathway is especially significant in cancer biology. FOXM1 can transcriptionally upregulate genes like RacGAP1, which in turn activates the PI3K/AKT pathway [248]. This leads to enhanced cell proliferation, migration, and invasion, particularly in cervical cancer cells. The PI3K/AKT pathway can also regulate FOXM1 expression. AKT activation promotes FOXM1 nuclear localization and transcriptional activity, creating a positive feedback loop. FOXM1 and FOXO3 influences sensitivity to AKT inhibitors. FOXM1 over-expression can reduce responsiveness to these therapies [249].
12.3.2. ERK/BCL-2 Pathway
ERK signaling activation increases BCL-2 expression, which helps pancreatic cancer cells resist apoptosis[250]. This pathway seems to be independent of the PI3K/AKT pathway. The ERK-BCL2 pathway links extracellular signals to anti-apoptotic responses, promoting cell survival and contributing to cancer progression. ERK activation is triggered by growth factors, cytokines, or oncogenic signals via the RAS/RAF/MEK cascade. ERK1/2 signaling promotes cell survival by activating pro-survival BCL2 proteins and by repressing pro-death proteins [51].
Figure 12 summarizes the intertwining and overlapping factors that play a role in AR in PDAC.
12.3.3. STAT3 as Independent Driver of AR
Figure 12 and the preceding text illustrate STAT3 AR effects mediated by the PI3K/AKT pathway.
However, STAT3 has important independent effects. STAT3 can directly promote the expression of anti-apoptotic and survival genes. In some pancreatic cancer models STAT3-driven survival is found even when PI3K/AKT is not chronically activated. PI3K/AKT signaling can enhance STAT3 activity reinforcing survival signals.
STAT3 activation induces V-ATPase pump expression and activity, aiding cancer cells manage intracellular pH and ROS levels, further promoting resistance to anoikis [252]. In addition, STAT3 also positively regulates the expression of another proton extruder: NHE3 (sodium-hydrogen exchanger 3) through interaction with Sp1, Sp3 (specificity protein 1and 3). Sp1, Sp3, and STAT3 bind cooperatively to the NHE3 promoter [253]. The connection between NHE1 and pancreatic cancer is not well understood. In gastric cancer, STAT3 has been found to translocate to the nucleus after activation and binds to promoter regions of target genes, including SLC9A1, which encodes NHE1. In gastric cancer cells resistant to 5-FU NHE1 is up-regulated. STAT3 activation leads to this increased NHE1 expression, which helps maintain intracellular pH and supports cell survival under stress [254].
STAT3 can cooperate with HIF-1α to enhance carbonic anhydrase IX (CAIX) gene transcription, especially in a hypoxic tumor microenvironments [255]. CAIX helps cancer cells survive acidic stress by regulating intracellular and extracellular pH, a process enhanced by STAT3 signaling.
In melanoma and pancreatic cancer cells, silencing STAT3 leads to a 70-80% reduction in anoikis resistance. It also reduces migration and invasion. STAT3 also influences the expression of EMT-related proteins and signaling pathways that promote cell survival in suspension, contributing to aggressive cancer phenotypes through anoikis resistance [256,257,258]. STAT3 has been found to be a genetic modifier of TGF- beta-induced EMT in KRAS mutant pancreatic cancer cells. D’Amico et al. [259] showed that STAT3 activation conferred increased KRAS-dependency to PDAC cells.
PDAC is a particularly hypoxic tumor where the hypoxia inducible factors (HIFs) play an important pro-tumoral role. Adaptation to hypoxia is a necessary and essential role for pancreatic cancer cell survival. STAT3 and HIF-1α cooperate to regulate gene expression under hypoxic conditions, enhancing tumor survival, angiogenesis, and metabolic adaptation. STAT3 enhances HIF-1α’s transcriptional activity by stabilizing its expression and facilitating its nuclear localization [260,261,262]. This interaction forms part of an autocrine loop where STAT3 and HIF1α mutually regulate each other to sustain cancer cell survival, growth, and adaptation under hypoxic stress [263].
12.3.4. Genetic Signature of Anoikis Resistance in PDAC
Anoikis-Related Gene Signature: A multi-omics study identified seven prognostic genes (MET, DYNLL2, CDK1, TNFSF10, PIP5K1C, MSLN, GKN1) linked to pancreatic adenocarcinoma prognosis and anoikis. Their related proteins, such as EGFR and MMP2, also significantly impact prognosis. These genes were found to be highly expressed in pancreatic cancer cell lines compared to normal pancreatic cells [264].
Mutations: Mutations in KRAS, P53, and CDKN2A have a significant impact on the prognosis of pancreatic adenocarcinoma, highlighting their role in the tumor microenvironment and potentially anoikis resistance. For instance, constitutively active KRAS persistently stimulates downstream signaling cascades such as PI3K/AKT and RAF/MEK/ERK, resulting in the inhibition of apoptotic pathways. KRAS-mutant cells shift toward glycolysis and glutamine metabolism, supporting survival in anchorage-independent conditions.
Integrin and ephrin receptor families were up-regulated in all PDAC samples, regardless of clinical outcome, indicating that interactions between pancreatic cancer cells and the desmoplastic reaction play a key role in tumor development and progression [265].
Figure 12 shows that AR in PDAC is a multifactorial phenomenon, as it is also in other tumors. One or other factors may be more important in specific tumors at a specific moment, but probably one factor alone would not be enough to support AR. This issue has not been experimentally proven as yet. In pancreatic cancer, it is evident that different AR players yield to create an AR phenotype. We must also underline that in PDAC the desmoplastic environment is a contributor to AR.
Cross-talk and redundancy of AR molecules and pathways contribute to reinforce the malignant cell survival, and inhibiting one pathway can be compensated by others [266].
12.4. Targeting Signaling Pathways
12.4.1. Curcumol
Curcumol is a bioactive hemiketal sesquiterpenoid obtained from the essential oil of the rhizomes of Curcuma rhizoma. It shows a good absorption and tissue distribution after oral administration to rats. Curcumol’s anti-cancer activity can be increased by chemical manipulation [267]. Curcumol is a potent inducer of apoptosis in numerous cancer cells. It targets key signaling pathways such as MAPK/ERK, PI3K/Akt and NF-κB. It also inhibits cell survival in triple negative breast cancer (TNBC) by targeting AR through the BIM pathway [268], and it can activate p73 and PUMA [269]. Furthermore, it enhances the sensitivity of doxorubicin in TNBC [270]. Additionally, it has shown pleiotropic anti-cancer effects in many other tumors [271,272].
12.4.2. Fucoxanthinol
12.4.3. Tunicamycin
Tunicamycin causes endoplasmic reticulum stress and unfolded protein response due to protein aggregation that leads to anoikis [277].
12.4.4. Thapsigargin
12.4.5. Dasatinib
Dasatinib is an FDA approved tyrosine kinase inhibitor used for the treatment of chronic myeloid leukemia and other hematologic cancers with the Philadelphia chromosome. It inhibits the activity of BCR-ABL kinase and SRC family kinases along with other specific oncogenic kinases including c-KIT, ephrin receptor (EPH) kinases and the PDGFß receptor. Importantly, it can induce anoikis through SRC inhibition [280,281,282,283].
12.4.6. Celecoxib
Celecoxib is a COX2-selective inhibitor that triggers anoikis in colon carcinoma by targeting protein Crk-associated substrate 130 kDa (p130Cas) thus deregulating focal adhesions. Celecoxib induces the proteolytic cleavage of p130Cas and the nuclear translocation of the 31 kDa generated fragment leading to apoptosis [284]. This effect is independent of COX2 inhibition. Celecoxib induces anoikis in osteosarcoma by inhibiting the PI3K/Akt pathway [285]. Similar effects have been found in canine breast cancer cells [286].
12.4.7. Gefitinib
12.4.8. MEK Inhibitors
These agents block the MAP kinase pathway at the level of MEK, thereby preventing ERK phosphorylation. It has been shown that MEK inhibitors can restore anoikis sensitivity in human breast cancer cells [289] through increased BIM [290]. Selumetinib (AZD6244) is a MEK1 and MEK2 inhibitor that has been found useful for the treatment of neurofibromas [291]. It has also been tested in melanoma metastasis [292].
12.4.9. Disulfiram
this is used for chronic alcohol-dependence and it is a drug that has shown many anti-tumor properties. Among them, disulfiram can induce copper-dependent cell death. Importantly, it induces anoikis in TNBC cells [293].
12.4.10. Metformin
Metformin can counteract anoikis resistance in medullary thyroid carcinoma probably through down-regulation of mTOR/6SK and phosphorylated ERK [294].
12.5. Integrin Inhibitors
12.5.1. Cilengitide
Cilengitide is a selective αv integrin inhibitor that has been tested against glioblastoma [295]. The integrin αvβ3 is involved in angiogenesis, cell migration, and proliferation. It is expressed at low levels in normal cells and over-expressed in glioblastoma, melanoma, breast, prostate, and pancreatic cancer cells [296]. By targeting integrin αvβ3, cilengitide down-regulates the FAK-SRC-AKT pathway in endothelial cells acting as an angiogenic drug. The mechanism of action seems to be complex. In a first step, the integrin inhibition produces anchorage loss of the endothelial cells and this in turn induces anoikis [297,298,299,300].
However, the clinical results with cilengitide in glioblastoma have been disappointing with no overall survival improvement. For this reason, there has been no further development of the drug [301].
12.5.2. JSM6427
JSM6427 is an α1β3 inhibitor that showed decreased growth in glioblastoma cells in vitro and in vivo [302].
12.5.3. ILKAS
ILKAS is an ILK anti-sense oligonucleotide that has been used in investigative studies to treat glioblastoma [303].
12.5.4. TDI4161
TDI4161 is a small molecule that has been developed for the treatment of osteoporosis that specifically targets αvβ3. It has not been fully tested in cancer [304].
12.6. FAK Inhibitors
12.6.1. Doxycycline
This antibiotic has been found to induce growth inhibition and apoptosis in some tumors. One of the mechanisms involved in these effects is inhibition of FAK phosphorylation [305].
A group of specific FAK inhibitors has been developed. Their use as monotherapy has shown poor results. On the other hand, results have improved substantially when they are used together with MEK inhibitors.
12.6.2. Defactinib
Defactinib is a FAK inhibitor that has undergone encouraging clinical trials in association with the MEK inhibitor avutometinib for the treatment of low-grade serous ovarian cancer. Table 1.
12.7. Many Over-the-Counter Drugs and Nutraceuticals Have Been Shown to Counteract AR. Among Them Are:
12.7.1. Alpha-Mangostin
Alpha-mangostin is a member of the class of xanthones, isolated from the stems of Cratoxylum cochinchinense. It has antioxidant, antimicrobial and antitumor effects. Mangostin was found to induce anoikis in anoikis resistant hepatocarcinoma cells [311].
12.7.2. Aspirin
Aspirin increased sensitivity of breast cancer cells to anoikis decreasing the number of distant metastases. It also decreased the number of circulating tumor cells in vitro. Thromboxane A2 is increased upon detachment from the ECM and aspirin decreased thromboxane A2 through COX1 inhibition [312,313,314].
12.7.3. Berberine [315]
The natural pentacyclic triterpenoids betulinic acid (BA) and betulin are found in many trees and plants. White birch tree (Betula alba) bark is particularly rich in BA. Many therapeutic effects of these compounds have been described in AIDS, malaria and cancer, which are probably the most important. BA has clear anti-cancer effects in some tumors, particularly melanoma. However, these benefits do not apply to all tumors. Probably, they induce apoptosis in ectoderm originated malignancies such as melanoma and neuroblastoma. However, betulinic acid was also found to induce apoptosis and anoikis in other tumors such as prostate, colon, breast cancers, among others. BA was found to be an effective inducer of apoptosis in malignant cells [316,317,318,319,320], while preserving normal cells [321].
BA in melanoma. In 1995, Pischa et al. [322] found that BA was a selective inhibitor of human melanoma in vivo. Multiple studies have shown that its primary action is to induce apoptosis [323,324,325,326,327,328,329,330,331,332]. Furthermore, it has been found that BA has additive or synergistic effects with chemotherapeutic drugs against melanoma [333,334], and other non-chemotherapeutic drugs such as digoxin [335]. However, one publication did find that BA induced increased expression of the anti-apoptotic protein Mcl1 [336]. This seems to contradict all the above findings, and we have no plausible explanation for this result.
12.7.4. Apigenin
Apigenin is a natural component of celery. It has been found to sensitize breast cancer cells to anoikis by a mechanism similar to aspirin [337].
12.8. Targeting Integrin-EGFR Interaction
Doxazosin
Doxazosin is used to treat arterial hypertension and benign prostatic hyperplasia. Doxasosin derivatives have been found to exert important anti-tumor effects in prostate cancer and importantly, it was found to reverse anoikis resistance in vitro [338].
12.9. Digoxin and Its Derivatives
Digoxin has important effects promoting apoptosis and reducing anoikis resistance in circulating [339] and non-circulating cancer cells [340,341]. This effect is achieved by targeting the alpha subunit of the Na+/K+ ATPase pump. It also sensitizes cancer cells to anoikis by stimulating the proteasomal degradation of the anti-apoptotic protein Mcl1 [342].
12.10. Targeting STAT3
Several STAT3 inhibitors with potential therapeutic applications have been identified. Most STAT3 inhibitors work by preventing its dimerization through binding to the SH2 domain, blocking downstream signaling pathways involved in tumor progression and immune suppression. Several are under evaluation for clinical development, with some showing promising in vitro and animal models experimental results. None has achieved FDA approval.
12.10.1. N4
Of particular interest in pancreatic cancer is a small molecule inhibitor called N4 that has shown strong antitumor activity by directly targeting STAT3 activation [343]. This inhibitor binds specifically to the SH2 domain of STAT3, suppressing tumor growth, metastasis, and remodeling the tumor microenvironment. It has been tested in vitro and in animal models. Human testing is not available as yet.
12.10.2. WB436B
WB436B selectively binds to STAT3 inhibiting Tyr705 phosphorylation. WB436B suppresses pancreatic cancer growth and metastasis in vivo and prolonged survival of mice bearing the tumor [344].
12.10.3. C188-9 (TTI-101)
It is an inhibitor evaluated in clinical trials for various cancers, designed to suppress STAT3 activity by targeting its SH2 domain. There are preclinical tests in breast cancer showing increased reduction of cancer cell viability which was superior to that reached with other traditional chemotherapy drugs [345].
12.10.4. OPB-111077
It is a high-affinity inhibitor that inhibits STAT3 activity and mitochondrial function, undergoing clinical evaluation [346,347]. A phase I clinical trial [348] in patients with hepatocellular carcinoma showed limited preliminary benefits. There are also other phase I trials with some encouraging results in acute myeloid leukemia in combination with conventional chemotherapeutics [349].
12.10.5. Stattic
Is an earlier-discovered small molecule inhibitor targeting the STAT3-SH2 domain and inhibiting STAT3 activit. It is used mainly in research and preclinical studies.
12.10.6. YY002
It is a highly selective STAT3 inhibitor with favorable pharmacokinetics showing promising in vitro and in vivo effects. It is potentially superior to some compounds in clinical development [350].
12.10.7. Ibuprofen
Ibuprofen has been shown to inhibit the STAT3 pathway, but it acts indirectly rather than as a direct STAT3 inhibitor. Ibuprofen reduces both the total STAT3 protein and its phosphorylated form (p-STAT3 Y705), particularly under hypoxic conditions in cancer cells, leading to decreased STAT3 activity. This inhibition contributes to reduced cancer cell viability, migration, and inflammation-related gene expression. The mechanism involves ibuprofen's effect on histone modifications (H3 methylation and acetylation) [351] and suppression of related inflammatory and stemness genes via COX2-dependent pathways. It also downregulates other transcription factors like NFκB, further impairing STAT3-mediated pathways. However, binding assays indicate ibuprofen does not directly bind to STAT3 but modulates its expression and activation levels through upstream regulatory mechanisms. This means that while ibuprofen impacts STAT3 activity, it is not a classical direct STAT3 inhibitor like small molecules designed to bind STAT3's SH2 domain (e.g., N4 or Stattic). Rather it exerts modulatory effects on STAT3 expression and phosphorylation through upstream pathways like COX2 and epigenetic regulation [352].
12.10.8. Other Non-Steroidal Anti-Inflammatory Drugs (NSAIDs)
12.10.9. Targeting Sp1 Transcription Factor
Some NSAIDs such as tolfenamic acid and celecoxib have been shown to down-regulate Sp1 transcription factor.
13. Discussion
High turnover epithelial tissues, such as the gastrointestinal mucosa, permanently shed a huge number of cells. These cells need to be eliminated because they would be able to colonize other tissues generating eventual neoplastic growth if they have the appropiate mutations. Since the number of shed cells is in the order of billions, the risk of finding a few of them with adequate mutations to develop a cancer, is very high. Fortunately, multicellular organisms have developed anoikis as a mechanism that induces the programmed destruction of these cells. Anoikis begins shortly after the cell detaches from the extracellular matrix and is shed. However, malignant cells have also developed a mechanism to resist anoikis.
Epithelial mesenchymal transition (EMT) is an early step in transformation. During this process, cells acquire mesenchymal characteristics including morphology and biochemistry. Functional changes include migration and invasion abilities, and for this, anoikis resistance is of capital importance, otherwise transformed cells would die as a consequence of anoikis-induced apoptosis [358]. Therefore, the moment for the development of anoikis resistance installation is the EMT or close to it. Part of the essential function of EMT in cancer cells is to transform an epithelial attachment-dependent cell into a mesenchymal-like attachment-independent one.
During EMT, cells down-regulate epithelial markers and up-regulate mesenchymal genes, enabling detachment, migration, and resistance to anoikis. Key traits include:
- (1)
- CDH1 (E-cadherin gene), EPCAM, and occludin down-regulation thus facilitating detachment.
- (2)
- Up-regulating VIM (vimentin gene, supporting cytoskeleton reorganization), CDH2 (N-cadherin that replaces E-cadherin), SNAI1 (Snail, that represses E-cadherin), TWIST1/2 (promote mesenchymal gene expression and stemness), and ZEB1(increases migratory abilities).
Vimentin supports anoikis resistance by boosting survival signaling and promoting cell clustering in detached cells; both are essential for metastasis. Vimentin binds to the internal pool of β1 integrins protecting them from lysosomal degradation. Cell detachment induces vimentin phosphorylation at amino acid Ser38 residue which induces the release of β1 integrins and their translocation to the cell surface. The increased surface expression of β1, in turn, induces cell clustering and activates the PI3K/AKT survival pathway, thereby evading anoikis [359,360]. Vimentin has also other AR actions such as:
interacting with the 14-3-3 protein preventing its availability for the pro-apoptotic cascade [361];
up-regulating pro-survival molecules like NF-kB [362];
activating the Ras/Raf/Erk proliferative pathway;
14-3-3 interaction with vimentin enhances cell motility, particularly during EMT and cancer metastasis. Vimentin’s reorganization, supported by 14-3-3, enables cells to adapt to new environments and resist anoikis [365,366].
Smit et al. [367] described a twist-snail-TrkB axis which represents a clear relation between two EMT-related genes like twist and snail with an AR-related gene like TrkB (see below). Figure 13.
Netrin-1, up-regulated during EMT, supports survival signaling and prevents apoptosis by binding to its receptor UNC5H [370]. NP137 is a monoclonal antibody against netrin-1, and inhibits EMT increasing epithelial cell populations, reducing metastasis, down-regulating anoikis resistance, and enhancing chemotherapy response [371].
The transcription factor Sp1 (specificity protein 1), binds GC-rich promoter regions and regulates genes involved in cell survival, proliferation, and stress response. It is another link between EMT and AR. Sp1 promotes resistance to anoikis and drives epithelial-mesenchymal transition (EMT) by regulating key survival and migration-related genes, contributing to cancer progression and metastasis [372]. Sp1:
- Activates survival pathways: Sp1 up-regulates components of the PI3K/Akt, MAPK, and JAK/STAT pathways, which suppress apoptosis triggered by ECM detachment [373].
- Supports anchorage-independent growth: By maintaining survival signals, Sp1 enables cancer cells to thrive in suspension, a key step in metastasis.
- Sp1 can increase intracellular pH: Intracellular alkalinity is a handicap for the apoptotic process. Sp1 can alkalinize the cell by increasing proton export through the promotion of NHE1, NHE2, and NHE3 [376].
- Sp1represses epithelial markers: such as E-cadherin, weakening cell-cell adhesion.
- Activates mesenchymal genes: It promotes expression of vimentin, fibronectin, and N-cadherin, facilitating cytoskeletal remodeling and migration. Sp1 directly regulates the transcription of the vimentin gene by binding to its promoter [377].
- Cooperates with EMT transcription factors: Sp1 interacts with Snail, ZEB1, and Twist, amplifying EMT signaling and enhancing resistance to anoikis [378].
All the data mentioned above hint towards a unified integral idea of EMT and AR as part of the same indivisible process.
During AR implementation, the cell over-expresses a group of genes and pathways such as:
13.1. GENES
- MUC1: A big transmembrane glycoprotein, is usually found over-expressed in epithelial cancers [384] and particularly in pancreatic cancer. It disrupts cell adhesion and promotes survival signaling, helping cells evade anoikis. MUC1 glycosylation stimulates apoptosis and chemotherapy resistance to drugs such as bortezomib, trastuzumab and tamoxifen among others [385,386]. It also promotes multidrug resistance genes. Under stressful conditions, MUC1 is cleaved into two molecules, MUC1-N and MUC1-C which create inward pro-survival signals. The role of MUC1 in cancer goes well beyond anoikis resistance but will not be discussed here (for a review see Chen et al. [387], Lan et al. [388], and Qing et al. [389].
- KL (Klotho): The KL gene was identified in 1997 as an anti-aging gene and was initially believed to be a tumor suppressor gene/protein [390]. It is now evident that KL is a controversial gene/protein. Most articles describe KL as a tumor suppressor [391,392] but it also shows pro-tumoral effects that “increases cellular migration, anchorage-independent growth, and anoikis resistance in hepatoma cells” [393,394].
- MNX1 (motor neuron and pancreas homeobox 1): Is a pro-tumoral transcription factor linked to oncogenic transformation and anoikis resistance through metabolic and proliferative pathways. MNX1 was found to play an important role in developing anoikis resistance in glioblastoma. MNX1 was up-regulated in highly malignant glioma cell lines. MNX1 allowed malignant cells to bypass anoikis while reducing fibronectin adhesion [395]. MNX1- induced anoikis resistance was mediated by the activation of tyrosine kinase receptor B (TrkB) which is a downstream effector. It also promotes proliferation by up-regulating cyclin E [396], and CCDC34 (Coiled-coil domain-containing 34) [397]. In addition to glioblastoma, MNX1 was identified as an anoikis resistance gene in many tumors such as renal cell carcinoma [398], colon cancer [399], and acute myeloid leukemia [400].
- MMP3 and TIMP1 have been recognized as anoikis resistance genes in laryngeal squamous cell carcinoma [401].
- ADCY10 (Adenylate Cyclase 10): Is involved in cAMP signaling, which can modulate survival pathways under stress. It has been identified as an AR gene signature in lung adenocarcinoma [404].
- TrkB (tropomyosin receptor kinase B) is a key suppressor of anoikis, enabling cancer cells to survive detachment and promoting metastasis. TrkB is a receptor tyrosine kinase that binds brain-derived neurotrophic factor (BDNF). It plays a critical role in neuronal survival and development but is hijacked by cancer cells to evade anoikis. TrkB activation blocks apoptotic signals triggered by loss of cell adhesion, allowing cells to survive detachment [405]. It also activates PI3K/AKT and MAPK/ERK signaling cascades promoting proliferation and survival [406], and it promotes epithelial mesenchymal transition increasing growth and metastatic potential [407]. TrkB activity is increased in tumors such as neuroblastoma, breast, lung, pancreatic, gastric, colorectal, ovarian and cervical cancers [408,409,410,411]. TrkB inhibitors have been developed:
- Experimental selective TrkB Inhibitors
ANA-12: A non-competitive TrkB antagonist used in preclinical studies. It inhibits BDNF-induced TrkB activation and downstream signaling.
GNF-5837: A small molecule that selectively inhibits TrkA, TrkB, and TrkC, with potent activity against TrkB in vitro.
FDA approved pan-TrkB inhibitors
Larotrectinib (Vitrakvi): Highly selective for TRK proteins; approved for solid tumors with NTRK fusions.
Entrectinib (Rozlytrek): Inhibits TRK, ROS1, and ALK; approved for NTRK-fusion positive cancers.
TrkB inhibitors have shown promise in preclinical models by restoring anoikis sensitivity and reducing metastasis.
13.2. PATHWAYS
- PI3K/AKT and MAPK/ERK signaling: These pathways are frequently activated in anoikis-resistant cells, promoting survival and proliferation.
- Metabolic reprogramming: Cancer cells adapt their metabolism (e.g., increased glycolysis) to survive without matrix attachment.
- EMT (Epithelial-Mesenchymal Transition): EMT-related genes are often up-regulated, enhancing motility and resistance to cell death.
The list of AR- related genes and pathways is incomplete. We have only discussed the most important. Furthermore, different tumors show different AR genetic signatures.
For example: the following anoikis resistance genes have been identified in prostate cancer: EGF, MYC, PLK1, EZH2, AFP, NOX4, BMP6, and MMP11 [412]. Expression of PLK1 (polo like kinase 1) was the most predictive gene regarding survival and tumor evolution. PLK1 is a cell cycle regulator, that is activated by NF-kB during cancer cell detachment; it prevents β-catenin degradation [413] and activates STAT3 [414]. PLK1 induces epithelial mesenchymal transition and cell motility [415]. PLK1 inhibitors such as volasertib are under development [416].
In lung adenocarcinoma an anoikis score was developed based on four genes: TLE1, GLI2, PLK1, and BAK1 [417].
A predictive model for cutaneous melanoma was developed utilizing three anoikis-related genes: FASLG, IGF1, and PIK3R2. Additional AR-related genes have been identified, though they were not included in predictive models for melanoma. These genes are DAPK2, BIRC5, PIK3CG, MMP9, PRKCQ, HAVCR2, SPIB, CDC25C, LCK, SPTA1, and IKZF3.
Jiao et al. [418] evaluated a risk model for breast cancer based on 10 prognostic AR related genes: CD24, PTK6, NAT1, BRCA2, IKZF3, HMGA1, EDA2R, SPIB, BST2, NTRK3).
The reason for mentioning these AR gene signatures is to show their heterogeneity in different tumors. Therefore, although anoikis resistance is a universal phenomenon in cell transformation, the way to achieve it shows that there are many different genetic mechanisms.
13.3. The Anoikis Resistance Environment
Cancer cells are characterized by extracellular acidity and intracellular alkalinity. This inversion of the normal pH gradient is the ideal condition for AR because apoptosis-related enzymes require an acidic cytoplasm. Therefore, intracellular alkalinity is a barrier to apoptosis.
Intracellular acidification can trigger apoptosis by activating specific proteases and disrupting cellular homeostasis [419,420,421,422]. This process plays a critical role in programmed cell death across various biological contexts. We believe that alkalinization of the intracellular pH plays an important role in achieving anoikis resistance.
Tumor’s intracellular pH research has been mainly based on NHE1 effects and their inhibition, however, there is evidence showing that the isoform NHE7 can be an important player in anchorage-independent growth [423]. NHE7 is a member of the NHE gene family (there are 9 members) that the regulates the luminal pH of Golgi apparatus and endosomes.
14. Conclusions
Normal cells die upon detachment from their ECM through an apoptotic mechanism that usually starts as extrinsic apoptosis and finally involves the intrinsic apoptotic pathway as well. This is known as anoikis. Cell detachment from the ECM is a major event in cells life because it inevitably leads to death. On the other hand, cancer cells develop resistance to anoikis which permits invasion and metastasis. To survive ECM detachment, cancer cells initiate multiple mechanisms that prevent their death. Without anoikis resistance there would be no metastasis.
If we have to define AR in a very simplified manner, we will say that anoikis resistance is controlled by constitutive activation of FAK. Thus, FAK should be considered a primary target for inhibiting AR. Though targeting anoikis resistance is a promising therapeutic avenue which is under intensive research, it is a difficult target. Although many drugs have been shown to induce anoikis in tests, no truly effective pharmaceutical has been developed to date. Defactinib, a specific FAK inhibitor, showed encouraging results in low-grade serous ovarian cancer when administered with MEK inhibitors such as avutometinib. However, many drugs other have failed initial promise, with cilengitide being the best example. Unfortunately, there is no ideal drug to prevent anoikis resistance as yet. In this paper we have reviewed the strong relationship between EMT and AR in some detail and we believe that they both are part of one integrated and indivisible process, and this relationship could be exploiting in developing treatments for AR.
Declarations
Both authors participated equally in the workup of this manuscript.
Funding
No financial support of any kind has been received for this article.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
There are no conflicts of interest.
References
- Sakamoto, S.; Kyprianou, N. Targeting anoikis resistance in prostate cancer metastasis. Molecular aspects of medicine 2010, 31, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Talbott, G.A.; Juliano, R.L. Integrin-mediated signaling events in human endothelial cells. Molecular biology of the cell 1998, 9, 1969–1980. [Google Scholar] [CrossRef]
- Giverso, C.; Jankowiak, G.; Preziosi, L.; Schmeiser, C. The influence of nucleus mechanics in modelling adhesion-independent cell migration in structured and confined environments. Bulletin of Mathematical Biology 2023, 85, 88. [Google Scholar] [CrossRef]
- Joussaume, A.; Karayan-Tapon, L.; Benzakour, O.; Dkhissi, F. A comparative study of anoikis resistance assays for tumor cells. Biomedical Journal of Scientific & Technical Research 2020, 29. [Google Scholar] [CrossRef]
- Khan, S.U.; Fatima, K.; Malik, F. Understanding the cell survival mechanism of anoikis-resistant cancer cells during different steps of metastasis. Clinical & experimental metastasis 2022, 39, 715–726. [Google Scholar]
- Adams, J.C.; Watt, F.M. Regulation of development and differentiation by the extracellular matrix. Development 1993, 117, 1183–1198. [Google Scholar] [CrossRef]
- Tan, K.; Goldstein, D.; Crowe, P.; Yang, J.L. Uncovering a key to the process of metastasis in human cancers: a review of critical regulators of anoikis. Journal of cancer research and clinical oncology 2013, 139, 1795–1805. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.; O'Neill, C.; Berryman, S.; Waxman, V. Anchorage and growth regulation in normal and virus-transformed cells. International Journal of Cancer 1968, 3, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell-matrix interactions induces apoptosis. The Journal of cell biology 1994, 124, 619–626. [Google Scholar] [CrossRef]
- Chiarugi, P.; Giannoni, E. Anoikis: a necessary death program for anchorage-dependent cells. Biochemical pharmacology 2008, 76, 1352–1364. [Google Scholar] [CrossRef]
- Strater, J.; Wedding, U.; Barth, T.F.; Koretz, K.; Elsing, C.; Moller, P. Rapid onset of apoptosis in vitro follows disruption of beta 1-integrin/matrix interactions in human colonic crypt cells. Gastroenterology 1996, 110, 1776–1784. [Google Scholar] [CrossRef]
- Wen, H.C.; Avivar-Valderas, A.; Sosa, M.S.; Girnius, N.; Farias, E.F.; Davis, R.J.; Aguirre-Ghiso, J.A. p38α signaling induces anoikis and lumen formation during mammary morphogenesis. Science signaling 2011, 4, ra34–ra34. [Google Scholar] [CrossRef]
- Strange, R.; Metcalfe, T.; Thackray, L.; Dang, M. Apoptosis in normal and neoplastic mammary gland development. Microscopy research and technique 2001, 52, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Vachon, P.H. Integrin signaling, cell survival, and anoikis: distinctions, differences, and differentiation. Journal of signal transduction 2011, 2011, 738137. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Kobayashi, Y.; Sakai, E.; Shibata, M.; Kato, Y. Cell adhesion is a prerequisite for osteoclast survival. Biochemical and Biophysical Research Communications 2000, 270, 550–556. [Google Scholar] [CrossRef]
- Gilmore; Anoikis, A. Cell Death Differ 2005, 12 Suppl 2, 1473–1477. [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: in command and control of cell motility. Nature reviews Molecular cell biology 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Liu, B.; Wu, Q.; Xuan, Z.; Zheng, Z.; Du, Y.; Sui, X.; Wu, H.; Zhang, Z.; Zhang, Z.; Zhong, M.; et al. Mechanisms Involved in Focal Adhesion Signaling Regulating Tumor Anoikis Resistance. Cancer Science 2025, 116, 2640–2648. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Wang, J.; Li, W.; Hao, X.; Hang, Q. Roles of integrins in gastrointestinal cancer metastasis. Frontiers in Molecular Biosciences 2021, 8, 708779. [Google Scholar] [CrossRef]
- Cooper, C.R.; Chay, C.H.; Pienta, K.J. The role of αvβ3 in prostate cancer progression. Neoplasia 2002, 4, 191–194. [Google Scholar] [CrossRef]
- Sloan, E.K.; Pouliot, N.; Stanley, K.L.; Chia, J.; Moseley, J.M.; Hards, D.K.; Anderson, R.L. Tumor-specific expression of αvβ3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Research 2006, 8, R20. [Google Scholar] [CrossRef]
- Mierke, C.T. The integrin alphav beta3 increases cellular stiffness and cytoskeletal remodeling dynamics to facilitate cancer cell invasion. New Journal of Physics 2013, 15, 015003. [Google Scholar] [CrossRef]
- Hou, J.; Yan, D.; Liu, Y.; Huang, P.; Cui, H. The roles of integrin α5β1 in human cancer. OncoTargets and therapy 2020, 13329–13344. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.K. Regulation of apoptosis by integrin receptors. Journal of pediatric hematology/oncology 1997, 19, 541–545. [Google Scholar] [CrossRef]
- Lee, J.W.; Juliano, R.L. α5β1 integrin protects intestinal epithelial cells from apoptosis through a phosphatidylinositol 3-kinase and protein kinase B–dependent pathway. Molecular biology of the cell 2000, 11, 1973–1987. [Google Scholar] [CrossRef]
- Desiniotis, A.; Kyprianou, N. Significance of talin in cancer progression and metastasis. International review of cell and molecular biology 2011, 289, 117–147. [Google Scholar]
- Calderwood, D.A.; Yan, B.; de Pereda, J.M.; Alvarez, B.G.; Fujioka, Y.; Liddington, R.C.; Ginsberg, M.H. The phosphotyrosine binding-like domain of talin activates integrins. Journal of Biological Chemistry 2002, 277, 21749–21758. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Legate, K.R.; Zent, R.; Fässler, R. The tail of integrins, talin, and kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the Focal Adhesion Kinase, ppl25FAK, Directs SH2-Dependent Binding of pp60 src. Molecular and cellular biology 1994, 14, 1680–1688. [Google Scholar]
- Tapial Martínez, P.; López Navajas, P.; Lietha, D. FAK structure and regulation by membrane interactions and force in focal adhesions. Biomolecules 2020, 10, 179. [Google Scholar] [CrossRef]
- Owen, J.D.; Ruest, P.J.; Fry, D.W.; Hanks, S.K. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto-and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Molecular and cellular biology 1999, 19, 4806–4818. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Frame, M.C. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Current opinion in cell biology 2005, 17, 542–547. [Google Scholar] [CrossRef]
- Katoh, K. Signal transduction mechanisms of focal adhesions: Src and FAK-mediated cell response. Frontiers in Bioscience-Landmark 2024, 29, 392. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.C.; Cary, L.A.; Jamieson, J.S.; Cooper, J.A.; Turner, C.E. Src and FAK kinases cooperate to phosphorylate paxillin kinase linker, stimulate its focal adhesion localization, and regulate cell spreading and protrusiveness. Molecular biology of the cell 2005, 16, 4316–4328. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. science 1999, 285, 1028–1033. [Google Scholar] [CrossRef]
- Shen, B.; Delaney, M.K.; Du, X. Inside-out, outside-in, and inside–outside-in: G protein signaling in integrin-mediated cell adhesion, spreading, and retraction. Current opinion in cell biology 2012, 24, 600–606. [Google Scholar] [CrossRef]
- Li, S.; Sampson, C.; Liu, C.; Piao, H.L.; Liu, H.X. Integrin signaling in cancer: bidirectional mechanisms and therapeutic opportunities. Cell Communication and Signaling 2023, 21, 266. [Google Scholar] [CrossRef]
- Berman, A.E.; Kozlova, N.I.; Morozevich, G.E. Integrins: structure and signaling. Biochemistry (Moscow) 2003, 68, 1284–1299. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annual review of cell and. [CrossRef]
- developmental biology 1996, 12, 697–715.
- Akiyama, S.K. Integrins in cell adhesion and signaling. Human cell 1996, 9, 181–186. [Google Scholar]
- Ludwig, B.S.; Kessler, H.; Kossatz, S.; Reuning, U. RGD-binding integrins revisited: how recently discovered functions and novel synthetic ligands (re-) shape an ever-evolving field. Cancers 2021, 13, 1711. [Google Scholar] [CrossRef]
- Wewer, U.M.; Taraboletti, G.; Sobel, M.E.; Albrechtsen, R.; Liotta, L.A. Role of laminin receptor in tumor cell migration. Cancer research 1987, 47, 5691–5698. [Google Scholar]
- Banerjee, S.; Lo, W.-C.; Majumder, P.; Roy, D.; Ghorai, M.; Shaikh, N.K.; Kant, N.; Shekhawat, M.S.; Gadekar, V.S.; Ghosh, S.; et al. Multiple roles for basement membrane proteins in cancer progression and EMT. European journal of cell biology 2022, 101, 151220. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Patzak, I.; Willenbrock, F. The insider's guide to leukocyte integrin signalling and function. Nature Reviews Immunology 2011, 11, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B. Transmembrane collagen receptors. Annual review of cell and developmental biology 2011, 27, 265–290. [Google Scholar] [CrossRef] [PubMed]
- Alanko, J.; Mai, A.; Jacquemet, G.; Schauer, K.; Kaukonen, R.; Saari, M.; Goud, B.; Ivaska, J. Integrin endosomal signalling suppresses anoikis. Nature cell biology 2015, 17, 1412–1421. [Google Scholar] [CrossRef]
- Maubant, S.; Saint-Dizier, D.; Boutillon, M.; Perron-Sierra, F.; Casara, P.J.; Hickman, J.A.; Tucker, G.C.; Van Obberghen-Schilling, E. Blockade of αvβ3 and αvβ5 integrins by RGD mimetics induces anoikis and not integrin-mediated death in human endothelial cells. Blood 2006, 108, 3035–3044. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: bidirectional, allosteric signaling machines. cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Welf, E.S.; Naik, U.P.; Ogunnaike, B.A. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophysical journal 2012, 103, 1379–1389. [Google Scholar] [CrossRef]
- Changede, R.; Sheetz, M. Integrin and cadherin clusters: A robust way to organize adhesions for cell mechanics. BioEssays 2017, 39, 1–12. [Google Scholar] [CrossRef]
- Kechagia, J.Z.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nature reviews Molecular cell biology 2019, 20, 457–473. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. Journal of cellular biochemistry 2019, 120, 2782–2790. [Google Scholar]
- Wullkopf, L.; West, A.K. V.; Leijnse, N.; Cox, T.R.; Madsen, C.D.; Oddershede, L.B.; Erler, J.T. Cancer cells’ ability to mechanically adjust to extracellular matrix stiffness correlates with their invasive potential. Molecular biology of the cell 2018, 29, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Gkretsi, V.; Stylianopoulos, T. Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Frontiers in oncology 2018, 8, 145. [Google Scholar] [CrossRef]
- Katoh, K. Integrin and Its Associated Proteins as a Mediator for Mechano-Signal Transduction. Biomolecules 2025, 15, 166. [Google Scholar] [CrossRef]
- Hanks, S.K.; Ryzhova, L.; Shin, N.Y.; Brábek, J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci 2003, 8, d982–996. [Google Scholar] [CrossRef]
- Mierke, C.T.; Fischer, T.; Puder, S.; Kunschmann, T.; Soetje, B.; Ziegler, W.H. Focal adhesion kinase activity is required for actomyosin contractility-based invasion of cells into dense 3D matrices. Scientific reports 2017, 7, 42780. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.H.; Zhen, Y.Y.; Tsai, Y.C.; Chuang, C.H.; Hsiao, M.; Huang, M.S.; Yang, C.J. FAK in cancer: from mechanisms to therapeutic strategies. International journal of molecular sciences 2022, 23, 1726. [Google Scholar] [CrossRef] [PubMed]
- Tomar, A.; Schlaepfer, D.D. Focal adhesion kinase: switching between GAPs and GEFs in the regulation of cell motility. Current opinion in cell biology 2009, 21, 676–683. [Google Scholar] [CrossRef]
- Genna, A.; Lapetina, S.; Lukic, N.; Twafra, S.; Meirson, T.; Sharma, V.P.; Condeelis, J.S.; Gil-Henn, H. Pyk2 and FAK differentially regulate invadopodia formation and function in breast cancer cells. Journal of Cell Biology 2018, 217, 375–395. [Google Scholar] [CrossRef]
- Mousson, A.; Legrand, M.; Steffan, T.; Vauchelles, R.; Carl, P.; Gies, J.-P.; Lehmann, M.; Zuber, G.; De Mey, J.; Dujardin, D.; et al. Inhibiting FAK–paxillin interaction reduces migration and invadopodia-mediated matrix degradation in metastatic melanoma cells. Cancers 2021, 13, 1871. [Google Scholar] [CrossRef]
- Xuefeng, X.; Hou, M.-X.; Yang, Z.-W.; Agudamu, A.; Wang, F.; Su, X.-L.; Li, X.; Shi, L.; Terigele, T.; Bao, L.-L.; et al. Epithelial–mesenchymal transition and metastasis of colon cancer cells induced by the FAK pathway in cancer-associated fibroblasts. Journal of International Medical Research 2020, 48, 0300060520931242. [Google Scholar] [CrossRef] [PubMed]
- Tilghman, R.W.; Parsons, J.T. Focal adhesion kinase as a regulator of cell tension in the progression of cancer. In Seminars in cancer biology; Academic Press, February 2008; Vol. 18, No. 1, pp. 45–52. [Google Scholar]
- Sawai, H.; Okada, Y.; Funahashi, H.; Matsuo, Y.; Takahashi, H.; Takeyama, H.; Manabe, T. Activation of focal adhesion kinase enhances the adhesion and invasion of pancreatic cancer cells via extracellular signal-regulated kinase-1/2 signaling pathway activation. Molecular cancer 2005, 4, 37. [Google Scholar] [CrossRef] [PubMed]
- Duxbury, M.S.; Ito, H.; Zinner, M.J.; Ashley, S.W.; Whang, E.E. Focal adhesion kinase gene silencing promotes anoikis and suppresses metastasis of human pancreatic adenocarcinoma cells. Surgery 2004, 135, 555–562. [Google Scholar] [CrossRef]
- Carelli, S.; Zadra, G.; Vaira, V.; Falleni, M.; Bottiglieri, L.; Nosotti, M.; Di Giulio, A.M.; Gorio, A.; Bosari, S. Up-regulation of focal adhesion kinase in non-small cell lung cancer. Lung cancer 2006, 53, 263–271. [Google Scholar] [CrossRef]
- Ocak, S.; Chen, H.; Callison, C.; Gonzalez, A.L.; Massion, P.P. Expression of focal adhesion kinase in small-cell lung carcinoma. Cancer 2012, 118, 1293–1301. [Google Scholar] [CrossRef]
- Sood, A.K.; Coffin, J.E.; Schneider, G.B.; Fletcher, M.S.; DeYoung, B.R.; Gruman, L.M.; Gershenson, D.M.; Schaller, M.D.; Hendrix, M.J. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. The American journal of pathology 2004, 165, 1087–1095. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, B.L.; Yoon, J.; Kim, J.; Kim, M.A.; Yang, H.K.; Kim, W.H. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Human pathology 2010, 41, 1664–1673. [Google Scholar] [CrossRef]
- Lark, A.L.; Livasy, C.A.; Calvo, B.; Caskey, L.; Moore, D.T.; Yang, X.; Cance, W.G. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clinical Cancer Research 2003, 9, 215–222. [Google Scholar]
- Zhang, Y.; Sun, X. Role of focal adhesion kinase in head and neck squamous cell carcinoma and its therapeutic prospect. OncoTargets and therapy 2020, 10207–10220. [Google Scholar] [CrossRef]
- Rigiracciolo, D.C.; Cirillo, F.; Talia, M.; Muglia, L.; Gutkind, J.S.; Maggiolini, M.; Lappano, R. Focal adhesion kinase fine tunes multifaced signals toward breast cancer progression. Cancers 2021, 13, 645. [Google Scholar] [CrossRef]
- Yom, C.K.; Noh, D.Y.; Kim, W.H.; Kim, H.S. Clinical significance of high focal adhesion kinase gene copy number and overexpression in invasive breast cancer. Breast cancer research and treatment 2011, 128, 647–655. [Google Scholar] [CrossRef]
- Šelemetjev, S.; Bartolome, A.; Išić Denčić, T.; Đorić, I.; Paunović, I.; Tatić, S.; Cvejić, D. Overexpression of epidermal growth factor receptor and its downstream effector, focal adhesion kinase, correlates with papillary thyroid carcinoma progression. International journal of experimental pathology 2018, 99, 87–94. [Google Scholar] [CrossRef]
- Ignjatović, V.B.; Janković Miljuš, J.R.; Rončević, J.V.; Tatić, S.B.; Išić Denčić, T.M.; Đorić, I.Đ.; Šelemetjev, S.A. Focal adhesion kinase splicing and protein activation in papillary thyroid carcinoma progression. Histochemistry and Cell Biology 2022, 157, 183–194. [Google Scholar] [CrossRef]
- Kim, L.T.; Fleming, J.B.; Lopez-Guzman, C.; Nwariaku, F. Focal adhesions and associated proteins in medullary thyroid carcinoma cells. Journal of Surgical Research 2003, 111, 177–184. [Google Scholar] [CrossRef]
- Fujii, T.; Koshikawa, K.; Nomoto, S.; Okochi, O.; Kaneko, T.; Inoue, S.; Yatabe, Y.; Takeda, S.; Nakao, A. Focal adhesion kinase is overexpressed in hepatocellular carcinoma and can be served as an independent prognostic factor. Journal of hepatology 2004, 41, 104–111. [Google Scholar] [CrossRef]
- Ghosh, A.P.; Willey, C.D.; Anderson, J.C.; Welaya, K.; Chen, D.; Mehta, A.; Ghatalia, P.; Madan, A.; Naik, G.; Sudarshan, S.; et al. Kinomic profiling identifies focal adhesion kinase 1 as a therapeutic target in advanced clear cell renal cell carcinoma. Oncotarget 2017, 8, 29220. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Sun, X.; You, Y.; Liu, N.; Fu, Z. Expression of focal adhesion kinase and phosphorylated focal adhesion kinase in human gliomas is associated with unfavorable overall survival. Translational Research 2010, 156, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.-J.; LaFortune, T.; Honda, T.; Ohmori, O.; Hatakeyama, S.; Meyer, T.; Jackson, D.; de Groot, J.; Yung, W.A. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Molecular cancer therapeutics 2007, 6, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Kahana, O.; Micksche, M.; Witz, I.P.; Yron, I. The focal adhesion kinase (P125FAK) is constitutively active in human malignant melanoma. Oncogene 2002, 21, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Kircher, D.A.; Trombetti, K.A.; Silvis, M.R.; Parkman, G.L.; Fischer, G.M.; Angel, S.N.; Stehn, C.M.; Strain, S.C.; Grossmann, A.H.; Duffy, K.L.; et al. AKT1E17K activates focal adhesion kinase and promotes melanoma brain metastasis. Molecular Cancer Research 2019, 17, 1787–1800. [Google Scholar] [CrossRef]
- Hu, H.H.; Wang, S.Q.; Shang, H.L.; Lv, H.F.; Chen, B.B.; Gao, S.G.; Chen, X.B. Roles and inhibitors of FAK in cancer: current advances and future directions. Front Pharmacol 2024, 15, 1274209. [Google Scholar] [CrossRef]
- Simpson, C.D.; Anyiwe, K.; Schimmer, A.D. Anoikis resistance and tumor metastasis. Cancer letters 2008, 272, 177–185. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 2013, 1833, 3481–3498. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Srivastava, S.K. STAT3 induces anoikis resistance, promotes cell invasion and metastatic potential in pancreatic cancer cells. Carcinogenesis 2015, 36, 142–150. [Google Scholar] [CrossRef]
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis–pathways to anchorage-independent growth in cancer. Journal of cell science 2011, 124, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Brakebusch, C.; Bouvard, D.; Stanchi, F.; Sakai, T.; Fässler, R. Integrins in invasive growth. The Journal of clinical investigation 2002, 109, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Nagaprashantha, L.D.; Vatsyayan, R.; Lelsani, P.C.R.; Awasthi, S.; Singhal, S.S. The sensors and regulators of cell–matrix surveillance in anoikis resistance of tumors. International Journal of Cancer 2011, 128, 743–752. [Google Scholar] [CrossRef]
- Dedhar, S.; Williams, B.; Hannigan, G. Integrin-linked kinase (ILK): a regulator of integrin and growth-factor signalling. Trends in cell biology 1999, 9, 319–323. [Google Scholar] [CrossRef]
- Górska, A.; Mazur, A.J. Integrin-linked kinase (ILK): the known vs. the unknown and perspectives. Cellular and Molecular Life Sciences 2022, 79, 100. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Gupta, S.; Chen, K.; Wu, C.; Qin, J. The pseudoactive site of ILK is essential for its binding to α-Parvin and localization to focal adhesions. Molecular cell 2009, 36, 819–830. [Google Scholar] [CrossRef]
- Attwell, S.; Roskelley, C.; Dedhar, S. The integrin-linked kinase (ILK) suppresses anoikis. Oncogene 2000, 19, 3811–3815. [Google Scholar] [CrossRef] [PubMed]
- Benoit, D.S.; Tripodi, M.C.; Blanchette, J.O.; Langer, S.J.; Leinwand, L.A.; Anseth, K.S. Integrin-linked kinase production prevents anoikis in human mesenchymal stem cells. Journal of Biomedical Materials Research Part A: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2007, 81, 259–268. [Google Scholar] [CrossRef]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene (s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef]
- Downloaded from https://pdb101.rcsb.org/motm/43 Accessed February 2024.
- Abram, C.L.; Courtneidge, S.A. Src family tyrosine kinases and growth factor signaling. Experimental cell research 2000, 254, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacological research 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Westhoff, M.A.; Serrels, B.; Fincham, V.J.; Frame, M.C.; Carragher, N.O. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Molecular and cellular biology 2004, 24, 8113–8133. [Google Scholar] [CrossRef]
- Barrett, A.; Pellet-Many, C.; Zachary, I.C.; Evans, I.M.; Frankel, P. p130Cas: a key signalling node in health and disease. Cellular signalling 2013, 25, 766–777. [Google Scholar] [CrossRef]
- Harte, M.T.; Hildebrand, J.D.; Burnham, M.R.; Bouton, A.H.; Parsons, J.T. p130Cas, a substrate associated with v-Src and v-Crk, localizes to focal adhesions and binds to focal adhesion kinase. Journal of Biological Chemistry 1996, 271, 13649–13655. [Google Scholar] [CrossRef]
- Defilippi, P.; Di Stefano, P.; Cabodi, S. p130Cas: a versatile scaffold in signaling networks. Trends in cell biology 2006, 16, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kook, S.; Shim, S.R.; Choi, S.J.; Ahnn, J.; Kim, J.I.; Eom, S.H.; Jung, Y.K.; Paik, S.G.; Song, W.K. Caspase-mediated cleavage of p130cas in etoposide-induced apoptotic Rat-1 cells. Molecular biology of the cell 2000, 11, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Ojaniemi, M.; Vuori, K. Epidermal growth factor modulates tyrosine phosphorylation of p130Cas: involvement of phosphatidylinositol 3′-kinase and actin cytoskeleton. Journal of Biological Chemistry 1997, 272, 25993–25998. [Google Scholar] [CrossRef] [PubMed]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: a crossroad in pathological cell migration. Journal of hematology & oncology 2017, 10, 50. [Google Scholar]
- Panera, N.; Crudele, A.; Romito, I.; Gnani, D.; Alisi, A. Focal adhesion kinase: insight into molecular roles and functions in hepatocellular carcinoma. International journal of molecular sciences 2017, 18, 99. [Google Scholar] [CrossRef]
- Indovina, P.; Forte, I.M.; Pentimalli, F.; Giordano, A. Targeting SRC family kinases in mesothelioma: Time to upgrade. Cancers 2020, 12, 1866. [Google Scholar] [CrossRef]
- Giancotti, F.G. Complexity and specificity of integrin signalling. Nature cell biology 2000, 2, E13–E14. [Google Scholar] [CrossRef]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: an emerging hallmark in health and diseases. The Journal of pathology 2012, 226, 380–393. [Google Scholar] [CrossRef]
- Stupack, D.G.; Cheresh, D.A. Get a ligand, get a life: integrins, signaling and cell survival. Journal of cell science 2002, 115, 3729–3738. [Google Scholar] [CrossRef]
- Bouchard, V.; Demers, M.; Thibodeau, S.; Laquerre, V.; Fujita, N.; Tsuruo, T.; Beaulieu, J.; Gauthier, R.; Vézina, A.; Villeneuve, L.; et al. Fak/Src signaling in human intestinal epithelial cell survival and anoikis: differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. Journal of cellular physiology 2007, 212, 717–728. [Google Scholar] [CrossRef]
- Moreno-Layseca, P.; Streuli, C.H. Signalling pathways linking integrins with cell cycle progression. Matrix Biology 2014, 34, 144–153. [Google Scholar] [CrossRef]
- Jo, M.H.; Li, J.; Jaumouillé, V.; Hao, Y.; Coppola, J.; Yan, J.; Waterman, C.M.; Springer, T.A.; Ha, T. Single-molecule characterization of subtype-specific β1 integrin mechanics. Nature communications 2022, 13, 7471. [Google Scholar] [CrossRef] [PubMed]
- Khwaja, A.; Lehmann, K.; Marte, B.M.; Downward, J. Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. Journal of Biological Chemistry 1998, 273, 18793–18801. [Google Scholar] [CrossRef] [PubMed]
- Stracke, M.L.; Soroush, M.; Liotta, L.A.; Schiffmann, E. Cytoskeletal agents inhibit motility and adherence of human tumor cells. Kidney international 1993, 43, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death & Differentiation 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ashkenazi, A. Targeting the extrinsic apoptotic pathway in cancer: lessons learned and future directions. The Journal of clinical investigation 2015, 125, 487–489. [Google Scholar] [CrossRef]
- Derouet, M.; Wu, X.; May, L.; Yoo, B.H.; Sasazuki, T.; Shirasawa, S.; Rake, J.; Rosen, K.V. Acquisition of anoikis resistance promotes the emergence of oncogenic K-ras mutations in colorectal cancer cells and stimulates their tumorigenicity in vivo. Neoplasia 2007, 9, 536–545. [Google Scholar] [CrossRef]
- Cao, Z.; Livas, T.; Kyprianou, N. Anoikis and EMT: lethal" liaisons" during cancer progression. Critical Reviews™ in Oncogenesis 2016, 21. [Google Scholar] [CrossRef]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis resistance: an essential prerequisite for tumor metastasis. International journal of cell biology 2012, 2012, 306879. [Google Scholar] [CrossRef]
- Luey, B.C.; May, F.E. Insulin-like growth factors are essential to prevent anoikis in oestrogen-responsive breast cancer cells: importance of the type I IGF receptor and PI3-kinase/Akt pathway. Molecular cancer 2016, 15, 1–15. [Google Scholar] [CrossRef]
- Singh, A.B.; Sugimoto, K.; Harris, R.C. Juxtacrine activation of epidermal growth factor (EGF) receptor by membrane-anchored heparin-binding EGF-like growth factor protects epithelial cells from anoikis while maintaining an epithelial phenotype. Journal of Biological Chemistry 2007, 282, 32890–32901. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Sung, J.Y.; Park, E.-K.; Kho, S.; Koo, K.H.; Park, S.-Y.; Goh, S.-H.; Jeon, Y.K.; Oh, S.; Park, B.-K.; et al. Regulation of anoikis resistance by NADPH oxidase 4 and epidermal growth factor receptor. British journal of cancer 2017, 116, 370–381. [Google Scholar] [CrossRef]
- Valentinis, B.; Reiss, K.; Baserga, R. Insulin-like growth factor-I-mediated survival from Anoikis: Role of cell aggregation and focal adhesion kinase. Journal of cellular physiology 1998, 176, 648–657. [Google Scholar] [CrossRef]
- Adeshakin, F.O.; Adeshakin, A.O.; Afolabi, L.O.; Yan, D.; Zhang, G.; Wan, X. Mechanisms for modulating anoikis resistance in cancer and the relevance of metabolic reprogramming. Frontiers in Oncology 2021, 11, 626577. [Google Scholar] [CrossRef]
- Okayama, H. Cell cycle control by anchorage signaling. Cellular Signalling 2012, 24, 1599–1609. [Google Scholar] [CrossRef]
- Dai, Y.; Zhang, X.; Ou, Y.; Zou, L.; Zhang, D.; Yang, Q.; Qin, Y.; Du, X.; Li, W.; Yuan, Z.; et al. Anoikis resistance––protagonists of breast cancer cells survive and metastasize after ECM detachment. Cell Communication and Signaling 2023, 21, 190. [Google Scholar] [CrossRef]
- Meredith Jr, J.E.; Fazeli, B.; Schwartz, M.A. The extracellular matrix as a cell survival factor. Molecular biology of the cell 1993, 4, 953–961. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, V.; Frisch, S.M.; Juliano, R.L. Expression of the integrin α5 subunit in HT29 colon carcinoma cells suppresses apoptosis triggered by serum deprivation. Experimental cell research 1996, 224, 208–213. [Google Scholar] [CrossRef]
- Brooks, P.C.; Montgomery, A.M.; Rosenfeld, M.; Reisfeld, R.A.; Hu, T.; Klier, G.; Cheresh, D.A. Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell 1994, 79, 1157–1164. [Google Scholar] [CrossRef]
- An, T.; Zhang, Z.; Li, Y.; Yi, J.; Zhang, W.; Chen, D.; Ao, J.; Xiao, Z.-X.; Yi, Y. Integrin β1-mediated cell–cell adhesion augments metformin-induced anoikis. International Journal of Molecular Sciences 2019, 20, 1161. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, M.; Sahin, E.; Johannsen, M.; Somasundaram, R.; Manski, D.; Riecken, E.O.; Schuppan, D. Soluble collagen VI drives serum-starved fibroblasts through S phase and prevents apoptosis via down-regulation of Bax. Journal of Biological Chemistry 1999, 274, 34361–34368. [Google Scholar] [CrossRef]
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: a role for c-flip and implications for anoikis. The Journal of cell biology 2001, 152, 633–644. [Google Scholar] [CrossRef]
- Stupack, D.G.; Puente, X.S.; Boutsaboualoy, S.; Storgard, C.M.; Cheresh, D.A. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. Journal of Cell Biology 2001, 155, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Laguinge, L.M.; Samara, R.N.; Wang, W.; El-Deiry, W.S.; Corner, G.; Augenlicht, L.; Jessup, J.M. DR5 receptor mediates anoikis in human colorectal carcinoma cell lines. Cancer Research 2008, 68, 909–917. [Google Scholar] [CrossRef]
- Goel, H.L.; Li, J.; Kogan, S.; Languino, L.R. Integrins in prostate cancer progression. Endocrine-related cancer 2008, 15, 657. [Google Scholar] [CrossRef]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef]
- Tang, D.; Okada, H.; Ruland, J.; Liu, L.; Stambolic, V.; Mak, T.W.; Ingram, A.J. Akt is activated in response to an apoptotic signal. Journal of Biological Chemistry 2001, 276, 30461–30466. [Google Scholar] [CrossRef] [PubMed]
- Madrid, L.V.; Wang, C.Y.; Guttridge, D.C.; Schottelius, A.J.; Baldwin Jr, A.S.; Mayo, M.W. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-κB. Molecular and cellular biology 2000, 20, 1626–1638. [Google Scholar] [CrossRef]
- de Sousa Mesquita, A.P.; de Araújo Lopes, S.; Pernambuco Filho, P.C.A.; Nader, H.B.; Lopes, C.C. Acquisition of anoikis resistance promotes alterations in the Ras/ERK and PI3K/Akt signaling pathways and matrix remodeling in endothelial cells. Apoptosis 2017, 22, 1116–1137. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Zhao, H.; Han, Z.C. Signalling mechanisms of anoikis. Histology and histopathology 19, 973–983.
- Horowitz, J.C.; Rogers, D.S.; Sharma, V.; Vittal, R.; White, E.S.; Cui, Z.; Thannickal, V.J. Combinatorial activation of FAK and AKT by transforming growth factor-β1 confers an anoikis-resistant phenotype to myofibroblasts. Cellular signalling 2007, 19, 761–771. [Google Scholar] [CrossRef]
- Liu, G.; Meng, X.; Jin, Y.; Bai, J.; Zhao, Y.; Cui, X.; Chen, F.; Fu, S. Inhibitory role of focal adhesion kinase on anoikis in the lung cancer cell A549. Cell biology international 2008, 32, 663–670. [Google Scholar] [CrossRef]
- Chen, I.H.; Shih, H.C.; Hsieh, P.W.; Chang, F.R.; Wu, Y.C.; Wu, C.C. HPW-RX40 restores anoikis sensitivity of human breast cancer cells by inhibiting integrin/FAK signaling. Toxicology and applied pharmacology 2015, 289, 330–340. [Google Scholar] [CrossRef]
- Valentijn, A.J.; Gilmore, A.P. Translocation of full-length Bid to mitochondria during anoikis. Journal of Biological Chemistry 2004, 279, 32848–32857. [Google Scholar] [CrossRef] [PubMed]
- Chota, A.; George, B.P.; Abrahamse, H. Interactions of multidomain pro-apoptotic and anti-apoptotic proteins in cancer cell death. Oncotarget 2021, 12, 1615. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: function and dysfunction of its modulators and targeted therapeutic strategies. Aging (albany NY) 2016, 8, 603. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y. Cell death regulation by the Bcl-2 protein family in the mitochondria. Journal of cellular physiology 2003, 195, 158–167. [Google Scholar] [CrossRef]
- Frisch, S.M.; Vuori, K.; Kelaita, D.; Sicks, S. A role for Jun-N-terminal kinase in anoikis; suppression by bcl-2 and crmA. The Journal of cell biology 1996, 135, 1377–1382. [Google Scholar] [CrossRef]
- Rosen, K.; Rak, J.; Leung, T.; Dean, N.M.; Kerbel, R.S.; Filmus, J. Activated ras Prevents Downregulation of Bcl-XL Triggered by Detachment from the Extracellular MatrixA Mechanism of ras-Induced Resistance to Anoikis in Intestinal Epithelial Cells. Journal of Cell Biology 2000, 149, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Coll, M.L.; Rosen, K.; Ladeda, V.; Filmus, J. Increased Bcl-xL expression mediates v-Src-induced resistance to anoikis in intestinal epithelial cells. Oncogene 2002, 21, 2908–2913. [Google Scholar] [CrossRef]
- Woods, N.T.; Yamaguchi, H.; Lee, F.Y.; Bhalla, K.N.; Wang, H.G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer research 2007, 67, 10744–10752. [Google Scholar] [CrossRef]
- Irmler, M.; Thome, M.; Hahne, M.; Schneider, P.; Hofmann, K.; Steiner, V.; Bodmer, J.-L.; Schröter, M.; Burns, K.; Mattmann, C.; et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388, 190–195. [Google Scholar] [CrossRef]
- Rippo, M.R.; Moretti, S.; Vescovi, S.; Tomasetti, M.; Orecchia, S.; Amici, G.; Catalano, A.; Procopio, A. FLIP overexpression inhibits death receptor-induced apoptosis in malignant mesothelial cells. Oncogene 2004, 23, 7753–7760. [Google Scholar] [CrossRef]
- Dolcet, X.; Llobet, D.; Pallares, J.; Rue, M.; Comella, J.X.; Matias-Guiu, X. FLIP is frequently expressed in endometrial carcinoma and has a role in resistance to TRAIL-induced apoptosis. Laboratory investigation 2005, 85, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. c-FLIP, a master anti-apoptotic regulator. Experimental oncology 2012, 34, 176. [Google Scholar] [PubMed]
- Mathas, S.; Lietz, A.; Anagnostopoulos, I.; Hummel, F.; Wiesner, B.; Janz, M.; Jundt, F.; Hirsch, B.; Jöhrens-Leder, K.; Vornlocher, H.-P.; et al. c-FLIP mediates resistance of Hodgkin/Reed-Sternberg cells to death receptor–induced apoptosis. The Journal of experimental medicine 2004, 199, 1041–1052. [Google Scholar] [CrossRef]
- Mawji, I.A.; Simpson, C.D.; Hurren, R.; Gronda, M.; Williams, M.A.; Filmus, J.; Jonkman, J.; Da Costa, R.S.; Wilson, B.C.; Thomas, M.P.; et al. Critical Role for Fas-Associated Death Domain–Like Interleukin-1–Converting Enzyme–Like Inhibitory Protein in Anoikis Resistance and Distant Tumor Formation. Journal of the National Cancer Institute 2007, 99, 811–822. [Google Scholar] [CrossRef]
- Montgomery, A.M.; Reisfeld, R.A.; Cheresh, D.A. Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc Nat Acad Sci. 1994, 91, 8856–60. [Google Scholar] [CrossRef] [PubMed]
- Bello, L.; Francolini, M.; Marthyn, P.; Zhang, J.; Carroll, R.S.; Nikas, D.C.; Strasser, J.F.; Villani, R.; Cheresh, D.A.; Black, P.M. αvβ3 and αvβ5 integrin expression in glioma periphery. Neurosurgery 2001, 49, 380–390. [Google Scholar]
- Felding-Habermann, B.; O'Toole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin activation controls metastasis in human breast cancer. Proceedings of the National Academy of Sciences 2001, 98, 1853–1858. [Google Scholar] [CrossRef]
- Dolinschek, R.; Hingerl, J.; Benge, A.; Zafiu, C.; Schüren, E.; Ehmoser, E.K.; Lössner, D.; Reuning, U. Constitutive activation of integrin αvβ3 contributes to anoikis resistance of ovarian cancer cells. Molecular Oncology 2021, 15, 503–522. [Google Scholar] [CrossRef]
- Bates, R.C.; Bellovin, D.I.; Brown, C.; Maynard, E.; Wu, B.; Kawakatsu, H.; Sheppard, D.; Oettgen, P.; Mercurio, A.M. Transcriptional activation of integrin β6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. The Journal of clinical investigation 2005, 115, 339–347. [Google Scholar] [CrossRef]
- Jones, J.; Watt, F.M.; Speight, P.M. Changes in the expression of αv integrins in oral squamous cell carcinomas. Journal of oral pathology & medicine 1997, 26, 63–68. [Google Scholar]
- Valencia-Expósito, A.; Gómez-Lamarca, M.J.; Widmann, T.J.; Martín-Bermudo, M.D. Integrins cooperate with the EGFR/Ras pathway to preserve epithelia survival and architecture in development and oncogenesis. Frontiers in Cell and Developmental Biology 2022, 10, 892691. [Google Scholar] [CrossRef] [PubMed]
- Braunholz, D.; Saki, M.; Niehr, F.; Öztürk, M.; Puértolas, B.B.; Konschak, R.; Budach, V.; Tinhofer, I. Spheroid culture of head and neck cancer cells reveals an important role of EGFR signalling in anchorage independent survival. PLoS One 2016, 11, e0163149. [Google Scholar] [CrossRef] [PubMed]
- Jost, M.; Huggett, T.M.; Kari, C.; Rodeck, U. Matrix-independent survival of human keratinocytes through an EGF receptor/MAPK-kinase-dependent pathway. Molecular biology of the cell 2001, 12, 1519–1527. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Montero, C.M.; Wygant, J.N.; McIntyre, B.W. PI3-K/Akt-mediated anoikis resistance of human osteosarcoma cells requires Src activation. European Journal of Cancer 2006, 42, 1491–1500. [Google Scholar] [CrossRef]
- Juan, Z.; Dake, C.; Tanaka, K.; Shuixiang, H. EGFL7 as a novel therapeutic candidate regulates cell invasion and anoikis in colorectal cancer through PI3K/AKT signaling pathway. International Journal of Clinical Oncology 2021, 26, 1099–1108. [Google Scholar] [CrossRef]
- He, C.; He, J. Metabolic reprogramming and signaling adaptations in anoikis resistance: mechanisms and therapeutic targets. Molecular and Cellular Biochemistry 2025, 1–28. [Google Scholar] [CrossRef]
- Peppicelli, S.; Kersikla, T.; Menegazzi, G.; Andreucci, E.; Ruzzolini, J.; Nediani, C.; Bianchini, F.; Calorini, L. The critical role of glutamine and fatty acids in the metabolic reprogramming of anoikis-resistant melanoma cells. Frontiers in Pharmacology 2024, 15, 1422281. [Google Scholar] [CrossRef]
- Tang, X.; Tian, Z.; Chueh, P.J.; Chen, S.; Morré, D.M.; Morré, D.J. Alternative splicing as the basis for specific localization of tNOX, a unique hydroquinone (NADH) oxidase, to the cancer cell surface. Biochemistry 2007, 46, 12337–12346. [Google Scholar] [CrossRef]
- Morré, DJ; Morré, D.M. Cancer Therapeutic Applications of ENOX2 Proteins. ECTO-NOX Proteins: Growth, Cancer, and Aging, 345-417.
- Shiraishi, T.; Verdone, J.E.; Huang, J.; Kahlert, U.D.; Hernandez, J.R.; Torga, G.; Zarif, J.C.; Epstein, T.; Gatenby, R.; McCartney, A.; et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 2014, 6, 130. [Google Scholar] [CrossRef]
- Mason, J.A.; Hagel, K.R.; Hawk, M.A.; Schafer, Z.T. Metabolism during ECM detachment: achilles heel of cancer cells? Trends in cancer 2017, 3, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ji, X.; Wang, Y. ENO2 promotes anoikis resistance in anaplastic thyroid cancer by maintaining redox homeostasis. Gland Surgery 2024, 13, 209. [Google Scholar] [CrossRef]
- Chen, W.-J.; Yang, W.; Gong, M.; He, Y.; Xu, D.; Chen, J.-X.; Chen, W.-J.; Li, W.-Y.; Wang, Y.-Q.; Dong, K.-Q.; et al. ENO2 affects the EMT process of renal cell carcinoma and participates in the regulation of the immune microenvironment. Oncology Reports 2022, 49, 33. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Z.; Yan, X.; Li, X.; Liu, Z.; Ma, Z. Integration of autophagy and anoikis resistance in solid tumors. The Anatomical Record 2013, 296, 1501–1508. [Google Scholar] [CrossRef]
- Chen, J.L.; David, J.; Cook-Spaeth, D.; Casey, S.; Cohen, D.; Selvendiran, K.; Bekaii-Saab, T.; Hays, J.L. Autophagy induction results in enhanced anoikis resistance in models of peritoneal disease. Molecular Cancer Research 2017, 15, 26–34. [Google Scholar] [CrossRef]
- Gulia, S.; Chandra, P.; Das, A. The prognosis of cancer depends on the interplay of autophagy, apoptosis, and anoikis within the tumor microenvironment. Cell Biochemistry and Biophysics 2023, 81, 621–658. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, B.; Li, X.; Lu, D.; Yang, L.; Chen, L.; Li, Y.; Cheng, L.; Lv, F.; Zhang, P.; et al. ATF4/CEMIP/PKCα promotes anoikis resistance by enhancing protective autophagy in prostate cancer cells. Cell death & disease 2022, 13, 46. [Google Scholar]
- Palabiyik, A.A. The role of Bcl-2 in controlling the transition between autophagy and apoptosis. Molecular Medicine Reports 2025, 32, 172. [Google Scholar] [CrossRef]
- Brokatzky, D.; Mostowy, S. Rearranging to resist cell death. eLife 2024, 13, e104942. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes & development 2012, 26, 54–68. [Google Scholar]
- Zhou, J.; Yang, S.; Zhu, D.; Li, H.; Miao, X.; Gu, M.; Xu, W.; Zhang, Y.; Tang, W.; Shen, R.; et al. The crosstalk between anoikis and epithelial-mesenchymal transition and their synergistic roles in predicting prognosis in colon adenocarcinoma. Frontiers in Oncology 2023, 13, 1184215. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Schaller, M.; Cieply, B. Mechanisms that link the oncogenic epithelial–mesenchymal transition to suppression of anoikis. Journal of cell science 2013, 126, 21–29. [Google Scholar] [CrossRef]
- Koltai, T.; Fliegel, L.; Reshkin, S.J., Baltazar, F., Cardone, R.A., Alfarouk, K.O., Afonso, J. pH deregulation as the eleventh hallmark of cancer. Book. Publisher: Elsevier.
- Wang, S.; Lv, Y.; Zhou, Y.; Ling, J.; Wang, H.; Gu, D.; Wang, C.; Qin, W.; Zheng, X.; Jin, H. Acidic extracellular pH induces autophagy to promote anoikis resistance of hepatocellular carcinoma cells via downregulation of miR-3663-3p. Journal of Cancer 2021, 12, 3418. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Ruzzolini, J.; Bianchini, F.; Andreucci, E.; Nediani, C.; Laurenzana, A.; Margheri, F.; Fibbi, G.; Calorini, L. Anoikis Resistance as a Further Trait of Acidic-Adapted Melanoma Cells. Journal of Oncology 2019, 2019, 8340926. [Google Scholar] [CrossRef]
- Adeshakin, F.O.; Adeshakin, A.O.; Liu, Z.; Lu, X.; Cheng, J.; Zhang, P.; Yan, D.; Zhang, G.; Wan, X. Upregulation of V-ATPase by STAT3 activation promotes anoikis resistance and tumor metastasis. Journal of Cancer 2021, 12, 4819. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Nimmannit, U.; Lu, Y.; Talbott, S.; Jiang, B.H.; Rojanasakul, Y. Nitric oxide regulates lung carcinoma cell anoikis through inhibition of ubiquitin-proteasomal degradation of caveolin-1. Journal of Biological Chemistry 2009, 284, 28476–28484. [Google Scholar] [CrossRef]
- Du, S.; Miao, J.; Zhu, Z.; Xu, E.; Shi, L.; Ai, S.; Wang, F.; Kang, X.; Chen, H.; Lu, X.; et al. NADPH oxidase 4 regulates anoikis resistance of gastric cancer cells through the generation of reactive oxygen species and the induction of EGFR. Cell death & disease 2018, 9, 948. [Google Scholar]
- Grünewald, T.G.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Dirksen, U. Ewing sarcoma. Nature reviews Disease primers 2018, 4, 5. [Google Scholar] [CrossRef]
- Zhang, H.; Hughes, C.S.; Delaidelli, A.; Huang, Y.Z.; Shyp, T.; Yang, X.; Sorensen, P.H. Abstract PR002: Identification of metabolic adaptation mechanisms that drive anoikis suppression and metastasis in Ewing sarcoma. Cancer Research 2023, 83. [Google Scholar] [CrossRef]
- Amjad, Z.S.; Shojaeian, A.; Nahand, J.S.; Bayat, M.; Taghizadieh, M.; Rostamian, M.; Babaei, F.; Moghoofei, M. Oncoviruses: induction of cancer development and metastasis by increasing anoikis resistance. Heliyon 2023, 9. [Google Scholar] [CrossRef]
- Kakavandi, E.; Shahbahrami, R.; Goudarzi, H.; Eslami, G.; Faghihloo, E. Anoikis resistance and oncoviruses. Journal of cellular biochemistry 2018, 119, 2484–2491. [Google Scholar] [CrossRef]
- Mak, C.S.L.; Yung, M.M.H.; Hui, L.M.N.; Leung, L.L.; Liang, R.; Chen, K.; Liu, S.S.; Qin, Y.; Leung, T.H.Y.; Lee, K.-F.; et al. MicroRNA-141 enhances anoikis resistance in metastatic progression of ovarian cancer through targeting KLF12/Sp1/survivin axis. Molecular cancer 2017, 16, 11. [Google Scholar] [CrossRef]
- Pedersen, S.F. The Na+/H+ exchanger NHE1 in stress-induced signal transduction: implications for cell proliferation and cell death. Pflügers Archiv 2006, 452, 249–259. [Google Scholar] [CrossRef]
- Park, H.J.; Lyons, J.C.; Ohtsubo, T.; Song, C.W. Acidic environment causes apoptosis by increasing caspase activity. British journal of cancer 1999, 80, 1892–1897. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Nordberg, J.; Skowronski, E.; Babior, B.M. Apoptosis induced in Jurkat cells by several agents is preceded by intracellular acidification. Proceedings of the National Academy of Sciences 1996, 93, 654–658. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Reed, J.C. Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nature cell biology 2000, 2, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Segal, M.S.; Beem, E. Effect of pH, ionic charge, and osmolality on cytochrome c-mediated caspase-3 activity. American Journal of Physiology-Cell Physiology 2001, 281, C1196–C1204. [Google Scholar] [CrossRef] [PubMed]
- Slepkov, E.; Fliegel, L. Structure and function of the NHE1 isoform of the Na+/H+ exchanger. Biochemistry and cell biology 2002, 80, 499–508. [Google Scholar] [CrossRef]
- Hu, Y.; Lou, J.; Jin, Z.; Yang, X.; Shan, W.; Du, Q.; Liao, Q.; Xu, J.; Xie, R. Advances in research on the regulatory mechanism of NHE1 in tumors. Oncology Letters 2021, 21, 273. [Google Scholar] [CrossRef] [PubMed]
- Pawson, T.; Letwin, K.; Lee, T.; Hao, Q.L.; Heisterkamp, N.; Groffen, J. The FER gene is evolutionarily conserved and encodes a widely expressed member of the FPS/FES protein-tyrosine kinase family. Molecular and cellular biology 1989, 9, 5722–5725. [Google Scholar]
- AIvanova, I.; Vermeulen, J.F.; Ercan, C.; Houthuijzen, J.M.; ASaig, F.; Vlug, E.J.; van der Wall, E.; van Diest, P.J.; Vooijs, M.; Derksen, P.W.B. FER kinase promotes breast cancer metastasis by regulating α6-and β1-integrin-dependent cell adhesion and anoikis resistance. Oncogene 2013, 32, 5582–5592. [Google Scholar] [CrossRef]
- Ahn, J.; Truesdell, P.; Meens, J.; Kadish, C.; Yang, X.; Boag, A.H.; Craig, A.W. Fer protein-tyrosine kinase promotes lung adenocarcinoma cell invasion and tumor metastasis. Molecular Cancer Research 2013, 11, 952–963. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Rocha, J.; Zouanat, F.Z.; Hamel, L.; Scarlata, E.; Aprikian, A.G.; Chevalier, S. The Fer tyrosine kinase cooperates with interleukin-6 to activate signal transducer and activator of transcription 3 and promote human prostate cancer cell growth. Molecular Cancer Research 2009, 7, 142–155. [Google Scholar] [CrossRef]
- Fan, G.; Zhang, S.; Gao, Y.; Greer, P.A.; Tonks, N.K. HGF-independent regulation of MET and GAB1 by nonreceptor tyrosine kinase FER potentiates metastasis in ovarian cancer. Genes & development 2016, 30, 1542–1557. [Google Scholar]
- Zhang, Y.; Xiong, X.; Zhu, Q.; Zhang, J.; Chen, S.; Wang, Y.; Cao, J.; Chen, L.; Hou, L.; Zhao, X.; et al. FER-mediated phosphorylation and PIK3R2 recruitment on IRS4 promotes AKT activation and tumorigenesis in ovarian cancer cells. Elife 2022, 11, e76183. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.; Bhowmik, A.D.; Pillai, M.S.G.; Robbins, N.; Dwivedi, S.K.D.; Rao, G. Anoikis resistance in cancer: mechanisms, therapeutic strategies, potential targets, and models for enhanced Understanding. Cancer Letters 2025, 624, 217750. [Google Scholar] [CrossRef] [PubMed]
- Derksen, P.W.; Liu, X.; Saridin, F.; van der Gulden, H.; Zevenhoven, J.; Evers, B.; van Beijnum, J.R.; Griffioen, A.W.; Vink, J.; Krimpenfort, P.; et al. Somatic inactivation of E-cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer cell 2006, 10, 437–449. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, S.; Fleishman, J.S.; Chen, J.; Tang, H.; Chen, Z.-S.; Chen, W.; Ding, M. Targeting anoikis resistance as a strategy for cancer therapy. Drug Resistance Updates 2024, 101099. [Google Scholar] [CrossRef]
- Schempp, C.M.; von Schwarzenberg, K.; Schreiner, L.; Kubisch, R.; Müller, R.; Wagner, E.; Vollmar, A.M. V-ATPase inhibition regulates anoikis resistance and metastasis of cancer cells. Molecular cancer therapeutics 2014, 13, 926–937. [Google Scholar] [CrossRef]
- Luong, B.; Schwenk, R.; Bräutigam, J.; Müller, R.; Menche, D.; Bischoff, I.; Fürst, R. The vacuolar-type ATPase inhibitor archazolid increases tumor cell adhesion to endothelial cells by accumulating extracellular collagen. PLoS one 2018, 13, e0203053. [Google Scholar] [CrossRef]
- Michel, V.; Licon-Munoz, Y.; Trujillo, K.; Bisoffi, M.; Parra, K.J. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. International journal of cancer 2013, 132, E1–E10. [Google Scholar] [CrossRef]
- Cotter, K.; Capecci, J.; Sennoune, S.; Huss, M.; Maier, M.; Martinez-Zaguilan, R.; Forgac, M. Activity of plasma membrane V-ATPases is critical for the invasion of MDA-MB231 breast cancer cells. Journal of Biological Chemistry 2015, 290, 3680–3692. [Google Scholar] [CrossRef]
- Ohta, T.; Tajima, H.; Yachie, A.; Yokoyama, K.; Elnemr, A.; Fushida, S.; Kitagawa, H.; Kayahara, M.; Nishimura, G.; Miwa, K.; et al. Activated lansoprazole inhibits cancer cell adhesion to extracellular matrix components. International journal of oncology 1999, 15, 33–42. [Google Scholar] [CrossRef]
- Deschesnes, R.G.; Patenaude, A.; Rousseau, J.L.C.; Fortin, J.S.; Ricard, C.; Côté, M.-F.; Huot, J.; C-Gaudreault, R.; Petitclerc, E. Microtubule-destabilizing agents induce focal adhesion structure disorganization and anoikis in cancer cells. The Journal of pharmacology and experimental therapeutics 2007, 320, 853–864. [Google Scholar] [CrossRef]
- Momin, S.; Nagaraju, G.P. PIK3-AKT and Its Role in Pancreatic Cancer. In Role of Tyrosine Kinases in Gastrointestinal Malignancies; Springer Singapore: Singapore, 2018; pp. 57–61. [Google Scholar]
- Stanciu, S.; Ionita-Radu, F.; Stefani, C.; Miricescu, D.; Stanescu-Spinu, I.-I.; Greabu, M.; Totan, A.R.; Jinga, M. Targeting PI3K/AKT/mTOR signaling pathway in pancreatic cancer: from molecular to clinical aspects. International journal of molecular sciences 2022, 23, 10132. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.A.; Ito, H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Pungsrinont, T.; Kallenbach, J.; Baniahmad, A. Role of PI3K-AKT-mTOR pathway as a pro-survival signaling and resistance-mediating mechanism to therapy of prostate cancer. International journal of molecular sciences 2021, 22, 11088. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell death & disease 2020, 11, 797. [Google Scholar]
- Rascio, F.; Spadaccino, F.; Rocchetti, M.T.; Castellano, G.; Stallone, G.; Netti, G.S.; Ranieri, E. The pathogenic role of PI3K/AKT pathway in cancer onset and drug resistance: an updated review. Cancers 2021, 13, 3949. [Google Scholar] [CrossRef]
- Mehra, S.; Deshpande, N.; Nagathihalli, N. Targeting PI3K pathway in pancreatic ductal adenocarcinoma: rationale and progress. Cancers 2021, 13, 4434. [Google Scholar] [CrossRef]
- Manoukian, P.; Bijlsma, M.; Van Laarhoven, H. The cellular origins of cancer-associated fibroblasts and their opposing contributions to pancreatic cancer growth. Frontiers in Cell and Developmental Biology 2021, 9, 743907. [Google Scholar] [CrossRef]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef]
- Heinemann, V.; Reni, M.; Ychou, M.; Richel, D.J.; Macarulla, T.; Ducreux, M. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer Treat. Rev. 2014, 40, 118–128. [Google Scholar] [CrossRef]
- Gorchs, L.; Kaipe, H. Interactions between cancer-associated fibroblasts and T cells in the pancreatic tumor microenvironment and the role of chemokines. Cancers 2021, 13, 2995. [Google Scholar] [CrossRef]
- Hamidi, A.; Song, J.; Thakur, N.; Itoh, S.; Marcusson, A.; Bergh, A.; Heldin, C.-H.; Landström, M. TGF-β promotes PI3K-AKT signaling and prostate cancer cell migration through the TRAF6-mediated ubiquitylation of p85α. Science signaling 2017, 10, eaal4186. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Landström, M. TGFβ activates PI3K-AKT signaling via TRAF6. Oncotarget 2017, 8, 99205. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The fibroblast growth factor signaling pathway. In Wiley Interdisciplinary Reviews: Developmental Biology; 2015; Volume 4, pp. 215–266. [Google Scholar]
- Zhang, R.; Lian, Y.; Xie, K.; Cai, Y.; Pan, Y.; Zhu, Y. Ropivacaine suppresses tumor biological characteristics of human hepatocellular carcinoma via inhibiting IGF-1R/PI3K/AKT/mTOR signaling axis. Bioengineered 2021, 12, 9162–9173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. The Journal of clinical investigation 2007, 117, 730–738. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Srivastava, S.K. Inhibition of STAT-3 by piperlongumine induces anoikis, prevents tumor formation in pancreatic cancer in vitro and in vivo. Cancer Research 2014, 74, 1233–1233. [Google Scholar] [CrossRef]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U.U. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Communication and Signaling 2018, 16, 55. [Google Scholar] [CrossRef]
- Guan, J.; Zhang, H.; Wen, Z.; Gu, Y.; Cheng, Y.; Sun, Y.; Zhang, T.; Jia, C.; Lu, Z.; Chen, J. Retinoic acid inhibits pancreatic cancer cell migration and EMT through the downregulation of IL-6 in cancer associated fibroblast cells. Cancer letters 2014, 345, 132–139. [Google Scholar] [CrossRef]
- An, H.; Kim, J.Y.; Oh, E.; Lee, N.; Cho, Y.; Seo, J.H. Salinomycin promotes anoikis and decreases the CD44+/CD24-stem-like population via inhibition of STAT3 activation in MDA-MB-231 cells. PloS one 2015, 10, e0141919. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, H.; Duan, C.; Liu, D.; Qian, L.; Yang, Z.; Guo, L.; Song, L.; Yu, M.; Hu, M.; et al. Deficiency of Erbin induces resistance of cervical cancer cells to anoikis in a STAT3-dependent manner. Oncogenesis 2013, 2, e52–e52. [Google Scholar] [CrossRef]
- Palollathil, A.; Dagamajalu, S.; Ahmed, M.; Vijayakumar, M.; Prasad, T.S. K.; Raju, R. The network map of mucin 1 mediated signaling in cancer progression and immune modulation. Discover Oncology 2025, 16, 1–15. [Google Scholar] [CrossRef]
- Bose, M.; Sanders, A.; De, C.; Zhou, R.; Lala, P.; Shwartz, S.; Mitra, B.; Brouwer, C.; Mukherjee, P. Targeting tumor-associated MUC1 overcomes anoikis-resistance in pancreatic cancer. Translational Research 2023, 253, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Bose, M.; Grover, P.; Sanders, A.J.; Zhou, R.; Ahmad, M.; Shwartz, S.; Lala, P.; Nath, S.; Yazdanifar, M.; Brouwer, C.; et al. Overexpression of MUC1 induces non-canonical TGF-β signaling in pancreatic ductal adenocarcinoma. Frontiers in cell and developmental biology 2022, 10, 821875. [Google Scholar] [CrossRef]
- Liau, S.S.; Jazag, A.; Ito, K.; Whang, E.E. Overexpression of HMGA1 promotes anoikis resistance and constitutive Akt activation in pancreatic adenocarcinoma cells. British journal of cancer 2007, 96, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Pan, T.-H.; Xu, S.; Jia, L.-T.; Zhu, L.-L.; Mao, J.-S.; Zhu, Y.-L.; Cai, J.-T. The virus-induced protein APOBEC3G inhibits anoikis by activation of Akt kinase in pancreatic cancer cells. Scientific Reports 2015, 5, 12230. [Google Scholar] [CrossRef]
- Li, L.; He, Z.; Zhu, C.; Chen, S.; Yang, Z.; Xu, J.; Bi, N.; Yu, C.; Sun, C. MiR-137 promotes anoikis through modulating the AKT signaling pathways in Pancreatic Cancer. J Cancer 2020, 11, 6277–6285. [Google Scholar] [CrossRef]
- Ernesti, A.; Heydel, B.; Blümke, J.; Gutschner, T.; Hämmerle, M. FOXM1 regulates platelet-induced anoikis resistance in pancreatic cancer cells. bioRxiv 2024, 2024–10. [Google Scholar] [CrossRef]
- Yao, H.; Li, J.; Zhou, D.; Pan, X.; Chu, Y.; Yin, J. FOXM1 transcriptional regulation of RacGAP1 activates the PI3K/AKT signaling pathway to promote the proliferation, migration, and invasion of cervical cancer cells. International Journal of Clinical Oncology 2024, 29, 333–344. [Google Scholar] [CrossRef]
- Cutano, V.; Chia, M.L.; Wigmore, E.M.; Hopcroft, L.; Williamson, S.C.; Christie, A.L.; Willis, B.; Kerr, J.; Ashforth, J.; Fox, R.; et al. The interplay between FOXO3 and FOXM1 influences sensitivity to AKT inhibition in PIK3CA and PIK3CA/PTEN altered estrogen receptor positive breast cancer. NPJ Breast Cancer 2025, 11, 36. [Google Scholar] [CrossRef]
- Galante, J.M.; Mortenson, M.M.; Bowles, T.L.; Virudachalam, S.; Bold, R.J. ERK/BCL-2 pathway in the resistance of pancreatic cancer to anoikis. Journal of Surgical Research 2009, 152, 18–25. [Google Scholar] [CrossRef]
- Cook, S.J.; Stuart, K.; Gilley, R.; Sale, M.J. Control of cell death and mitochondrial fission by ERK 1/2 MAP kinase signalling. The FEBS journal 2017, 284, 4177–4195. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Palmfeldt, J.; Lin, L.; Colaço, A.; Clemmensen, K.K.B.; Huang, J.; Xu, F.; Liu, X.; Maeda, K.; Luo, Y.; et al. STAT3 associates with vacuolar H+-ATPase and regulates cytosolic and lysosomal pH. Cell Research 2018, 28, 996–1012. [Google Scholar] [CrossRef] [PubMed]
- Su, H.W.; Wang, S.W.; Ghishan, F.K.; Kiela, P.R.; Tang, M.J. Cell confluency-induced Stat3 activation regulates NHE3 expression by recruiting Sp1 and Sp3 to the proximal NHE3 promoter region during epithelial dome formation. American Journal of Physiology-Cell Physiology 2009, 296, C13–C24. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Luan, S.; Yao, Y.; Qin, T.; Xu, X.; Shen, Z.; Yao, R.; Yue, L. NHE1 mediates 5-Fu resistance in gastric cancer via STAT3 signaling pathway. OncoTargets and therapy 2020, 8521–8532. [Google Scholar] [CrossRef]
- Grossmannova, K.; Belvoncikova, P.; Puzderova, B.; Simko, V.; Csaderova, L.; Pastorek, J.; Barathova, M. Carbonic anhydrase IX downregulation linked to disruption of HIF-1, NFκB and STAT3 pathways as a new mechanism of ibuprofen anti-cancer effect. PLoS One 2025, 20, e0323635. [Google Scholar] [CrossRef]
- Huang, C.; Yang, G.; Jiang, T.; Zhu, G.; Li, H.; Qiu, Z. The effects and mechanisms of blockage of STAT3 signaling pathway on IL-6 inducing EMT in human pancreatic cancer cells in vitro. Neoplasma 2011, 58, 396. [Google Scholar] [CrossRef]
- Guo, H.; Hu, Z.; Yang, X.; Yuan, Z.; Gao, Y.; Chen, J.; Xie, L.; Chen, C.; Guo, Y.; Bai, Y. STAT3 inhibition enhances gemcitabine sensitivity in pancreatic cancer by suppressing EMT, immune escape and inducing oxidative stress damage. International immunopharmacology 2023, 123, 110709. [Google Scholar] [CrossRef]
- Li, B.; Huang, C. Regulation of EMT by STAT3 in gastrointestinal cancer. International Journal of Oncology 2017, 50, 753–767. [Google Scholar] [CrossRef]
- D'Amico, S.; Kirillov, V.; Petrenko, O.; Reich, N.C. STAT3 is a genetic modifier of TGF-beta induced EMT in KRAS mutant pancreatic cancer. Elife 2024, 13, RP92559. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, A.; Betto, R.M.; Diamante, L.; Tesoriere, A.; Ghirardo, R.; Cioccarelli, C.; Argenton, F. STAT3 and HIF1α cooperatively mediate the transcriptional and physiological responses to hypoxia. Cell death discovery 2023, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Grillo, M.; Palmer, C.; Holmes, N.; Sang, F.; Larner, A.C.; Bhosale, R.; Shaw, P.E. Stat3 oxidation-dependent regulation of gene expression impacts on developmental processes and involves cooperation with Hif-1α. Plos one 2020, 15, e0244255. [Google Scholar] [CrossRef]
- Carlsson, R.; Özen, I.; Barbariga, M.; Gaceb, A.; Roth, M.; Paul, G. STAT3 precedes HIF1α transcriptional responses to oxygen and oxygen and glucose deprivation in human brain pericytes. PLoS One 2018, 13, e0194146. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.L.; Cassone, M.; Otvos, L., Jr.; Vogiatzi, P. HIF-1α and STAT3 client proteins interacting with the cancer chaperone Hsp90: Therapeutic considerations. Cancer biology & therapy 2008, 7, 10–14. [Google Scholar]
- Zhang, J.; Li, X.; Lu, Y.; Wang, G.; Ma, Y. Anoikis-related gene signature for prognostication of pancreatic adenocarcinoma: a multi-omics exploration and verification study. Cancers 2023, 15, 3146. [Google Scholar] [CrossRef]
- Van den Broeck, A.; Vankelecom, H.; Van Eijsden, R.; Govaere, O.; Topal, B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. Journal of Experimental & Clinical Cancer Research 2012, 31, 68. [Google Scholar] [CrossRef]
- Wang, J.; Lv, X.; Guo, X.; Dong, Y.; Peng, P.; Huang, F.; Wang, P.; Zhang, H.; Zhou, J.; Wang, Y.; et al. Feedback activation of STAT3 limits the response to PI3K/AKT/mTOR inhibitors in PTEN-deficient cancer cells. Oncogenesis 2021, 10, 8. [Google Scholar] [CrossRef]
- Zhai, S.; Wang, R.; Wang, J.; Xu, X.; Niu, L.; Guo, M.; Zhang, Y.; Shi, Y.; Tang, X. Curcumol: a review of its pharmacology, pharmacokinetics, drug delivery systems, structure–activity relationships, and potential applications. Inflammopharmacology 2024, 32, 1659–1704. [Google Scholar] [CrossRef]
- Li, C.-L.; Huang, C.-W.; Ko, C.-J.; Fang, S.-Y.; Ou-Yang, F.; Pan, M.-R.; Luo, C.-W.; Hou, M.-F. Curcumol suppresses triple-negative breast cancer metastasis by attenuating anoikis resistance via inhibition of skp2-mediated transcriptional addiction. Anticancer Research 2020, 40, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, A.; Liao, G.; Yang, F.; Yang, J.; Chen, X.; Jiang, X. Curcumol triggers apoptosis of p53 mutant triple-negative human breast cancer MDA-MB 231 cells via activation of p73 and PUMA. Oncology letters 2017, 14, 1080–1088. [Google Scholar] [CrossRef]
- Zeng, C.; Fan, D.; Xu, Y.; Li, X.; Yuan, J.; Yang, Q.; Zhou, X.; Lu, J.; Zhang, C.; Han, J.; et al. Curcumol enhances the sensitivity of doxorubicin in triple-negative breast cancer via regulating the miR-181b-2-3p-ABCC3 axis. Biochemical Pharmacology 2020, 174, 113795. [Google Scholar] [CrossRef]
- Sheng, W.; Xu, W.; Ding, J.; Li, L.; You, X.; Wu, Y.; He, Q. Curcumol inhibits the malignant progression of prostate cancer and regulates the PDK1/AKT/mTOR pathway by targeting miR-9. Oncology Reports 2021, 46, 1–11. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; Tao, Y.; Li, X.; Qin, J.; Bai, Z.; Chi, B.; Yan, W.; Chen, X. Curcumol inhibits colorectal cancer proliferation by targeting miR-21 and modulated PTEN/PI3K/Akt pathways. Life sciences 2019, 221, 354–361. [Google Scholar] [CrossRef]
- Terasaki, M.; Maeda, H.; Miyashita, K.; Mutoh, M. Induction of anoikis in human colorectal cancer cells by fucoxanthinol. Nutrition and Cancer 2017, 69, 1043–1052. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M.; Inoue, T.; Murase, W.; Kubota, A.; Kojima, H.; Kojoma, M.; Ohta, T.; Maeda, H.; Miyashita, K.; Mutoh, M.; et al. Fucoxanthinol induces apoptosis in a pancreatic intraepithelial neoplasia cell line. Cancer Genomics & Proteomics 2021, 18, 133–146. [Google Scholar] [CrossRef]
- Shukla, M.; Varalakshmi, K.N. Apoptosis induction in cancer cell lines by the carotenoid Fucoxanthinol from Pseudomonas stutzeri JGI 52. Indian journal of pharmacology 2018, 50, 116–122. [Google Scholar] [PubMed]
- Konishi, I.; Hosokawa, M.; Sashima, T.; Kobayashi, H.; Miyashita, K. Halocynthiaxanthin and fucoxanthinol isolated from Halocynthia roretzi induce apoptosis in human leukemia, breast and colon cancer cells. Comparative Biochemistry and Physiology Part C: Toxicology & Pharmacology 2006, 142(1-2), 53–59. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.H.; Wang, Y.; Lee, S.J.; Jin, M.H.; Sun, H.N.; Kwon, T. Regulation of anoikis by extrinsic death receptor pathways. Cell Communication and Signaling 2023, 21, 227. [Google Scholar] [CrossRef]
- Bondar, V.M.; McConkey, D.J. Anoikis is regulated by BCL-2-independent pathways in human prostate carcinoma cells. The Prostate 2002, 51, 42–49. [Google Scholar] [CrossRef]
- Xia, S.; Wu, J.; Zhou, W.; Zhang, M.; Zhao, K.; Tian, D.; Liu, J.; Liao, J. HRC promotes anoikis resistance and metastasis by suppressing endoplasmic reticulum stress in hepatocellular carcinoma. International Journal of Medical Sciences 2021, 18, 3112. [Google Scholar] [CrossRef]
- Eguchi, R.; Fujita, Y.; Tabata, C.; Ogawa, H.; Wakabayashi, I.; Nakano, T.; Fujimori, Y. Inhibition of Src family kinases overcomes anoikis resistance induced by spheroid formation and facilitates cisplatin-induced apoptosis in human mesothelioma cells. Oncology Reports 2015, 34, 2305–2310. [Google Scholar] [CrossRef]
- Li, Y.J.; He, Y.F.; Han, X.H.; Hu, B. Dasatinib suppresses invasion and induces apoptosis in nasopharyngeal carcinoma. International journal of clinical and experimental pathology 2015, 8, 7818. [Google Scholar]
- Liang, W.; Kujawski, M.; Wu, J.; Lu, J.; Herrmann, A.; Loera, S.; Yen, Y.; Lee, F.; Yu, H.; Wen, W.; et al. Antitumor activity of targeting SRC kinases in endothelial and myeloid cell compartments of the tumor microenvironment. Clinical Cancer Research 2010, 16, 924–935. [Google Scholar] [CrossRef]
- Heilmann, T.; Rumpf, A.-L.; Roscher, M.; Tietgen, M.; Will, O.; Gerle, M.; Damm, T.; Borzikowsky, C.; Maass, N.; Glüer, C.-C.; et al. Dasatinib prevents skeletal metastasis of osteotropic MDA-MB-231 cells in a xenograft mouse model. Archives of Gynecology and Obstetrics 2020, 301, 1493–1502. [Google Scholar] [CrossRef]
- Casanova, I.; Parreño, M.; Farré, L.; Guerrero, S.; Céspedes, M.V.; Pavon, M.A.; Sancho, F.J.; Marcuello, E.; Trias, M.; Mangues, R. Celecoxib induces anoikis in human colon carcinoma cells associated with the deregulation of focal adhesions and nuclear translocation of p130Cas. International journal of cancer 2006, 118, 2381–2389. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yang, Z.; Tao, H. Cyclooxygenase-2 inhibitors induce anoikis in osteosarcoma via PI3K/Akt pathway. Medical hypotheses 2012, 79, 98–100. [Google Scholar] [CrossRef] [PubMed]
- Tamura, D.; Saito, T.; Murata, K.; Kawashima, M.; Asano, R. Celecoxib exerts antitumor effects in canine mammary tumor cells via COX-2-independent mechanisms. international journal of oncology 2015, 46, 1393–1404. [Google Scholar] [CrossRef]
- Jung, B.C.; Woo, S.H.; Kim, S.H.; Kim, Y.S. Gefitinib induces anoikis in cervical cancer cells. BMB reports 2024, 57, 104. [Google Scholar] [CrossRef]
- Jung, B.C.; Woo, S.H.; Kim, S.H.; Kim, Y.S. Parkin enhances gefitinib-induced anoikis in HeLa cervical cancer cells. Anticancer Research 2024, 44, 1853–1862. [Google Scholar] [CrossRef]
- Fukazawa, H.; Noguchi, K.; Murakami, Y.; Uehara, Y. Mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK) inhibitors restore anoikis sensitivity in human breast cancer cell lines with a constitutively activated extracellular-regulated kinase (ERK) pathway. Molecular Cancer Therapeutics 2002, 1, 303–309. [Google Scholar]
- Fukazawa, H.; Noguchi, K.; Masumi, A.; Murakami, Y.; Uehara, Y. BimEL is an important determinant for induction of anoikis sensitivity by mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitors. Molecular cancer therapeutics 2004, 3, 1281–1288. [Google Scholar] [CrossRef]
- Coyne, G.H.O.; Gross, A.M.; Dombi, E.; Tibery, C.; Carbonell, A.; Takebe, N.; Derdak, J.; Pichard, D.; Srivastava, A.K.; Herrick, W.; et al. Phase II trial of the MEK 1/2 inhibitor selumetinib (AZD6244, ARRY-142886 Hydrogen Sulfate) in adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PN). 2020. [Google Scholar] [CrossRef]
- Neuendorf, H.M.; Simmons, J.L.; Boyle, G.M. Therapeutic targeting of anoikis resistance in cutaneous melanoma metastasis. Frontiers in Cell and Developmental Biology 2023, 11, 1183328. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, N.; Kim, Y.-J.; Cho, Y.; An, H.; Oh, E.; Cho, T.-M.; Sung, D.; Seo, J.H. Disulfiram induces anoikis and suppresses lung colonization in triple-negative breast cancer via calpain activation. Cancer letters 2017, 386, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Klubo-Gwiezdzinska, J.; Jensen, K.; Costello, J.; Patel, A.; Hoperia, V.; Bauer, A.; Burman, K.D.; Wartofsky, L.; Vasko, V. Metformin inhibits growth and decreases resistance to anoikis in medullary thyroid cancer cells. Endocrine-related cancer 2012, 19, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Fink, K.L.; Mikkelsen, T.; Cloughesy, T.F.; O'Neill, A.; Plotkin, S.; Glantz, M.; Ravin, P.; Raizer, J.J.; Rich, K.M.; et al. Randomized phase II study of cilengitide, an integrin-targeting arginine-glycine-aspartic acid peptide, in recurrent glioblastoma multiforme. Journal of clinical oncology 2008, 26, 5610–5617. [Google Scholar] [CrossRef]
- Chinot, O.L. Cilengitide in glioblastoma: when did it fail? The Lancet Oncology 2014, 15, 1044–1045. [Google Scholar] [CrossRef] [PubMed]
- Leblond, P.; Dewitte, A.; Le Tinier, F.; Bal-Mahieu, C.; Baroncini, M.; Sarrazin, T.; Lartigau, E.; Lansiaux, A.; Meignan, S. Cilengitide targets pediatric glioma and neuroblastoma cells through cell detachment and anoikis induction. Anti-cancer drugs 2013, 24, 818–825. [Google Scholar] [CrossRef]
- Meignan, S.; Leblond, P.; Dewitte, A.; Wattez, N.; Lartigau, E.; Lansiaux, A. Cilengitide targets more efficiently pediatric than adult glioma cells in vitro through cell detachment and anoikis induction. Cancer Research 2011, 71, 3266–3266. [Google Scholar] [CrossRef]
- Cheng, N.C.; van Zandwijk, N.; Reid, G. Cilengitide inhibits attachment and invasion of malignant pleural mesothelioma cells through antagonism of integrins αvβ3 and αvβ5. PLoS one 2014, 9, e90374. [Google Scholar] [CrossRef] [PubMed]
- Alghisi, G.C.; Ponsonnet, L.; Rüegg, C. The integrin antagonist cilengitide activates αVβ3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PloS one 2009, 4, e4449. [Google Scholar] [CrossRef]
- Stupp, R; Hegi, ME; Gorlia T; et al; for the European Organisation for Research and Treatment of Cancer (EORTC); the Canadian Brain Tumor Consortium; and the CENTRIC study team Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol published online. 2014. [Google Scholar] [CrossRef]
- Färber, K.; Synowitz, M.; Zahn, G.; Vossmeyer, D.; Stragies, R.; van Rooijen, N.; Kettenmann, H. An α5β1 integrin inhibitor attenuates glioma growth. Molecular and Cellular Neuroscience 2008, 39, 579–585. [Google Scholar] [CrossRef]
- Edwards, L.A.; Thiessen, B.; Dragowska, W.H.; Daynard, T.; Bally, M.B.; Dedhar, S. Inhibition of ILK in PTEN-mutant human glioblastomas inhibits PKB/Akt activation, induces apoptosis, and delays tumor growth. Oncogene 2005, 24, 3596–3605. [Google Scholar] [CrossRef]
- Li, J.; Fukase, Y.; Shang, Y.; Zou, W.; Muñoz-Félix, J.M.; Buitrago, L.; van Agthoven, J.; Zhang, Y.; Hara, R.; Tanaka, Y.; et al. Novel pure αVβ3 integrin antagonists that do not induce receptor extension, prime the receptor, or enhance angiogenesis at low concentrations. ACS Pharmacology & Translational Science 2019, 2, 387–401. [Google Scholar] [CrossRef]
- Sun, T.; Zhao, N.; Ni, C.-S.; Zhao, X.-L.; Zhang, W.-Z.; Su, X.; Zhang, D.-F.; Gu, Q.; Sun, B.-C. Doxycycline inhibits the adhesion and migration of melanoma cells by inhibiting the expression and phosphorylation of focal adhesion kinase (FAK). Cancer letters 2009, 285, 141–150. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Lockhart, A.C.; Tan, B.R.; Suresh, R.; Lim, K.H.; Ratner, L.; DeNardo, D.G. Phase I study of defactinib combined with pembrolizumab and gemcitabine in patients with advanced cancer. 2018. [Google Scholar]
- Patel, M.R.; Infante, J.R.; Moore, K.N.; Keegan, M.; Poli, A.; Padval, M.; Burris, H.A. Phase 1/1b study of the FAK inhibitor defactinib (VS-6063) in combination with weekly paclitaxel for advanced ovarian cancer. 2014. [Google Scholar] [CrossRef]
- Gerber, D.E.; Camidge, D.R.; Morgensztern, D.; Cetnar, J.; Kelly, R.J.; Ramalingam, S.S.; Burns, T.F. Phase 2 study of the focal adhesion kinase inhibitor defactinib (VS-6063) in previously treated advanced KRAS mutant non-small cell lung cancer. Lung Cancer 2020, 139, 60–67. [Google Scholar] [CrossRef]
- Fennell, D.A.; Baas, P.; Taylor, P.; Nowak, A.K.; Gilligan, D.; Nakano, T.; Pachter, J.A.; Weaver, D.T.; Scherpereel, A.; Pavlakis, N.; et al. Maintenance defactinib versus placebo after first-line chemotherapy in patients with merlin-stratified pleural mesothelioma: COMMAND—a double-blind, randomized, phase II study. Journal of Clinical Oncology 2019, 37, 790–798. [Google Scholar] [CrossRef]
- Banerjee, S.N.; Monk, B.J.; Van Nieuwenhuysen, E.; Moore, K.N.; Oaknin, A.; Fabbro, M.; Colombo, N.; O'MAlley, D.M.; Coleman, R.L.; Oza, A.M.; et al. ENGOT-ov60/GOG-3052/RAMP 201: A phase 2 study of VS-6766 (RAF/MEK clamp) alone and in combination with defactinib (FAK inhibitor) in recurrent low-grade serous ovarian cancer (LGSOC). 2022, TPS5615–TPS5615. [Google Scholar] [CrossRef]
- Wang, Y.; Fleishman, J.S.; Wang, J.; Chen, J.; Zhao, L.; Ding, M. Pharmacologically inducing anoikis offers novel therapeutic opportunities in hepatocellular carcinoma. Biomedicine & Pharmacotherapy 2024, 176, 116878. [Google Scholar] [CrossRef]
- Xu, R.; Yan, Y.; Zheng, X.; Zhang, H.; Chen, W.; Li, H.; Dong, Z. Aspirin suppresses breast cancer metastasis to lung by targeting anoikis resistance. Carcinogenesis 2022, 43, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, S. (2016). Aspirin affects early phases of metastasis through the inhibition of COX-1-thromboxane A2 axis (Doctoral dissertation, University of Oxford). Downloaded from https://ora.ox.ac.uk/objects/uuid:5a12480c-6b8a-4f46-b38a-4b59122e9280 Accessed June 2025.
- Yang, J.; Yamashita-Kanemaru, Y.; Morris, B.I.; Contursi, A.; Trajkovski, D.; Xu, J.; Patrascan, I.; Benson, J.; Evans, A.C.; Roychoudhuri, R.; et al. Aspirin prevents metastasis by limiting platelet TXA2 suppression of T cell immunity. Nature 2025, 1–10. [Google Scholar] [CrossRef]
- Kim, J.B.; Yu, J.H.; Ko, E.; Lee, K.W.; Song, A.K.; Park, S.Y.; Noh, D.Y. The alkaloid Berberine inhibits the growth of Anoikis-resistant MCF-7 and MDA-MB-231 breast cancer cell lines by inducing cell cycle arrest. Phytomedicine 2010, 17, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic acid for cancer treatment and prevention. International journal of molecular sciences 2008, 9, 1096–1107. [Google Scholar] [CrossRef]
- Fulda, S.; Scaffidi, C.; Susin, S.A.; Krammer, P.H.; Kroemer, G.; Peter, M.E.; Debatin, K.M. Activation of mitochondria and release of mitochondrial apoptogenic factors by betulinic acid. J. Biol. Chem. 1998, 273, 33942–33948. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Grimmel, C.; Wagenknecht, B.; Dichgans, J.; Weller, M. Betulinic acid-induced apoptosis in glioma cells: A sequential requirement for new protein synthesis, formation of reactive oxygen species, and caspase processing. J. Pharmacol. Exp. Ther. 1999, 289, 1306–1312. [Google Scholar] [CrossRef]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nunez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53- independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Fulda, S.; Debatin, K.M. Betulinic acid induces apoptosis through a direct effect on mitochondria in neuroectodermal tumors. Med. Pediatr. Oncol. 2000, 35, 616–618. [Google Scholar] [CrossRef]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Pisha, E.; Chai, H.; Lee, I.-S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M.; et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nature medicine 1995, 1, 1046–1051. [Google Scholar] [CrossRef]
- Liu, W.K.; Ho, J.C.; Cheung, F.W.; Liu, B.P.; Ye, W.C.; Che, C.T. Apoptotic activity of betulinic acid derivatives on murine melanoma B16 cell line. European Journal of Pharmacology 2004, 498, 71–78. [Google Scholar] [CrossRef]
- Tan, Y.; Yu, R.; Pezzuto, J.M. Betulinic acid-induced programmed cell death in human melanoma cells involves mitogen-activated protein kinase activation. Clinical Cancer Research 2003, 9, 2866–2875. [Google Scholar]
- Jeong, H.J.; Chai, H.B.; Park, S.Y.; Kim, D.S. Preparation of amino acid conjugates of betulinic acid with activity against human melanoma. Bioorganic & medicinal chemistry letters 1999, 9, 1201–1204. [Google Scholar]
- Coricovac, D.; Dehelean, C.A.; Pinzaru, I.; Mioc, A.; Aburel, O.-M.; Macasoi, I.; Draghici, G.A.; Petean, C.; Soica, C.; Boruga, M.; et al. Assessment of betulinic acid cytotoxicity and mitochondrial metabolism impairment in a human melanoma cell line. International Journal of Molecular Sciences 2021, 22, 4870. [Google Scholar] [CrossRef]
- Weber, L.A.; Meißner, J.; Delarocque, J.; Kalbitz, J.; Feige, K.; Kietzmann, M.; Michaelis, A.; Paschke, R.; Michael, J.; Pratscher, B.; et al. Betulinic acid shows anticancer activity against equine melanoma cells and permeates isolated equine skin in vitro. BMC veterinary research 2020, 16, 44. [Google Scholar] [CrossRef]
- Ghiulai, R.; Mioc, A.; Racoviceanu, R.; Mioc, M.; Milan, A.; Prodea, A.; Semenescu, A.; Dehelean, C.; Tudoran, L.B.; Avram, Ș.; et al. The anti-melanoma effect of betulinic acid functionalized gold nanoparticles: a mechanistic in vitro approach. Pharmaceuticals 2022, 15, 1362. [Google Scholar] [CrossRef] [PubMed]
- Liebscher, G.; Vanchangiri, K.; Mueller, T.; Feige, K.; Cavalleri, J.M.; Paschke, R. In vitro anticancer activity of Betulinic acid and derivatives thereof on equine melanoma cell lines from grey horses and invivo safety assessment of the compound NVX-207 in two horses. Chemico-biological interactions 2016, 246, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Gheorgheosu, D.; Jung, M.; Ören, B.; Schmid, T.; Dehelean, C.; Muntean, D.; Brüne, B. Betulinic acid suppresses NGAL-induced epithelial-to-mesenchymal transition in melanoma. Biological Chemistry 2013, 394, 773–781. [Google Scholar] [CrossRef]
- Drąg-Zalesińska, M.; Drąg, M.; Poręba, M.; Borska, S.; Kulbacka, J.; Saczko, J. Anticancer properties of ester derivatives of betulin in human metastatic melanoma cells (Me-45). Cancer Cell International 2017, 17, 4. [Google Scholar] [CrossRef]
- Bratu, L.M.; Marcovici, I.; Macasoi, I.O.A.N.A.; Manea, A.; Niculescu, B.; Olaru, F.; Coricovac, D. In vitro insights regarding the role of melanin in melanoma cells’ response to betulinic acid treatment. Farmacia 2022, 70. [Google Scholar]
- Wróblewska-Łuczka, P.; Cabaj, J.; Bąk, W.; Bargieł, J.; Grabarska, A.; Góralczyk, A.; Łuszczki, J.J. Additive interactions between betulinic acid and two taxanes in in vitro tests against four human malignant melanoma cell lines. International Journal of Molecular Sciences 2022, 23, 9641. [Google Scholar] [CrossRef]
- Sawada, N.; Kataoka, K.; Kondo, K.; Arimochi, H.; Fujino, H.; Takahashi, Y.; Miyoshi, T.; Kuwahara, T.; Monden, Y.; Ohnishi, Y. Betulinic acid augments the inhibitory effects of vincristine on growth and lung metastasis of B16F10 melanoma cells in mice. British Journal of Cancer 2004, 90, 1672–1678. [Google Scholar] [CrossRef]
- Rednic, R.; Macasoi, I.; Pinzaru, I.; Dehelean, C.A.; Tomescu, M.C.; Susan, M.; Feier, H. Pharmaco-toxicological assessment of the combined cytotoxic effects of digoxin and betulinic acid in melanoma cells. Life 2022, 12, 1855. [Google Scholar] [CrossRef]
- Selzer, E.; Thallinger, C.; Hoeller, C.; Oberkleiner, P.; Wacheck, V.; Pehamberger, H.; Jansen, B. Betulinic acid-induced Mcl-1 expression in human melanoma—mode of action and functional significance. Molecular Medicine 2002, 8, 877–884. [Google Scholar] [CrossRef]
- Xu, R.; Yao, Z.; Zhang, H.; Li, H.; Chen, W. Apigenin is an anoikis sensitizer with strong anti-metastatic properties in experimental breast cancer. Food Science and Human Wellness 2024, 13, 2221–2233. [Google Scholar] [CrossRef]
- Keledjian, K.; Kyprianou, N. Anoikis induction by quinazoline based α1-adrenoceptor antagonists in prostate cancer cells: antagonistic effect of bcl-2. The Journal of urology 2003, 169, 1150–1156. [Google Scholar] [CrossRef]
- Numata, Y.; Fujii, T.; Toda, C.; Okumura, T.; Manabe, T.; Takeda, N.; Shimizu, T.; Tabuchi, Y.; Fujii, T.; Sakai, H. Digoxin promotes anoikis of circulating cancer cells by targeting Na+/K+-ATPase α3-isoform. Cell Death & Disease 2025, 16, 373. [Google Scholar] [CrossRef]
- Rocha, S.C.; Pessoa, M.T.C.; Neves, L.D.R.; Alves, S.L.G.; Silva, L.M.; Santos, H.L.; Oliveira, S.M.F.; Taranto, A.G.; Comar, M.; Gomes, I.V.; et al. 21-Benzylidene digoxin: a proapoptotic cardenolide of cancer cells that up-regulates Na, K-ATPase and epithelial tight junctions. PLoS One 2014, 9, e108776. [Google Scholar] [CrossRef]
- Simpson, C.D.; Mawji, I.A.; Anyiwe, K.; Williams, M.A.; Wang, X.; Venugopal, A.L.; Gronda, M.; Hurren, R.; Cheng, S.; Serra, S.; et al. D. Inhibition of the sodium potassium adenosine triphosphatase pump sensitizes cancer cells to anoikis and prevents distant tumor formation. Cancer research 2009, 69, 2739–2747. [Google Scholar] [CrossRef]
- Pongrakhananon, V.; Stueckle, T.A.; Wang, H.Y.L.; O’Doherty, G.A.; Dinu, C.Z.; Chanvorachote, P.; Rojanasakul, Y. Monosaccharide digitoxin derivative sensitize human non-small cell lung cancer cells to anoikis through Mcl-1 proteasomal degradation. Biochemical pharmacology 2014, 88, 23–35. [Google Scholar] [CrossRef]
- Chen, H.; Bian, A.; Yang, L.-F.; Yin, X.; Wang, J.; Ti, C.; Miao, Y.; Peng, S.; Xu, S.; Liu, M.; et al. Targeting STAT3 by a small molecule suppresses pancreatic cancer progression. Oncogene 2021, 40, 1440–1457. [Google Scholar] [CrossRef]
- Chen, H.; Zhou, W.; Bian, A.; Zhang, Q.; Miao, Y.; Yin, X.; Ye, J.; Xu, S.; Ti, C.; Sun, Z.; et al. Selectively targeting STAT3 using a small molecule inhibitor is a potential therapeutic strategy for pancreatic cancer. Clinical Cancer Research 2023, 29, 815–830. [Google Scholar] [CrossRef]
- Zheng, R.; Guan, T.; Hong, C.; Yao, Y.; Fang, Y.; Huang, W.; Chen, C.; Zeng, H.; Huang, J.; Lin, H.; et al. C188-9 reduces patient-specific primary breast cancer cells proliferation at the low, clinic-relevant concentration. Journal of Translational Medicine 2024, 22, 784. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.; Flaherty, K.; Shapiro, G.I.; Berlin, J.; Witzig, T.; Habermann, T.; Bullock, A.; Rock, E.; Elekes, A.; Lin, C.; et al. A first-in-human phase I study of OPB-111077, a small-molecule STAT3 and oxidative phosphorylation inhibitor, in patients with advanced cancers. The Oncologist 2018, 23, 658–e72. [Google Scholar] [CrossRef] [PubMed]
- Takaki, E.O.; Ohi, N. Synergistic antitumor effect of a novel first-in-class small molecule OPB-111077 that inhibits mitochondrial oxidative phosphorylation in combination with alkylating agent. 2024. [Google Scholar] [CrossRef]
- Yoo, C.; Kang, J.; Lim, H.Y.; Kim, J.H.; Lee, M.-A.; Lee, K.-H.; Kim, T.-Y.; Ryoo, B.-Y. Phase I dose-finding study of OPB-111077, a novel STAT3 inhibitor, in patients with advanced hepatocellular carcinoma. Cancer Research and Treatment: Official Journal of Korean Cancer Association 2019, 51, 510–518. [Google Scholar] [CrossRef]
- Santoni, M.; Miccini, F.; Cimadamore, A.; Piva, F.; Massari, F.; Cheng, L.; Lopez-Beltran, A.; Montironi, R.; Battelli, N. An update on investigational therapies that target STAT3 for the treatment of cancer. Expert opinion on investigational drugs 2021, 30, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Bian, A.; Zhou, W.; Miao, Y.; Ye, J.; Li, J.; He, P.; Zhang, Q.; Sun, Y.; Sun, Z.; et al. Discovery of the highly selective and potent STAT3 inhibitor for pancreatic cancer treatment. ACS Central Science 2024, 10, 579–594. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Zhang, X.; Du, R.; Gao, W.; Wang, J.; Bao, Y.; Yang, W.; Luo, N.; Li, J. Ibuprofen mediates histone modification to diminish cancer cell stemness properties via a COX2-dependent manner. British Journal of Cancer 2020, 123, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Pingali, P.; Wu, Y.J.; Boothello, R.; Sharon, C.; Li, H.; Sistla, S.; Sankaranarayanan, N.V.; Desai, U.R.; Le, A.T.; Doebele, R.C.; et al. High dose acetaminophen inhibits STAT3 and has free radical independent anti-cancer stem cell activity. Neoplasia 2021, 23, 348–359. [Google Scholar] [CrossRef]
- Cheng, L.; Hu, Z.; Gu, J.; Li, Q.; Liu, J.; Liu, M.; Li, J.; Bi, X. Exploring COX-independent pathways: A novel approach for meloxicam and other NSAIDs in cancer and cardiovascular disease treatment. Pharmaceuticals 2024, 17, 1488. [Google Scholar] [CrossRef]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Baer-Dubowska, W. Unlocking the potential: Novel NSAIDs hybrids unleash chemopreventive power toward liver cancer cells through Nrf2, NF-κB, and MAPK signaling pathways. Molecules 2023, 28, 5759. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, B.; Brecher, P. Selective inhibition of STAT3 phosphorylation by sodium salicylate in cardiac fibroblasts. Biochemical pharmacology 2002, 63, 1197–1207. [Google Scholar] [CrossRef]
- Vaish, V.; Sanyal, S.N. Chemopreventive effects of NSAIDs on cytokines and transcription factors during the early stages of colorectal cancer. Pharmacological Reports 2011, 63, 1210–1221. [Google Scholar] [CrossRef]
- Reed, S.; Li, H.; Li, C.; Lin, J. Celecoxib inhibits STAT3 phosphorylation and suppresses cell migration and colony forming ability in rhabdomyosarcoma cells. Biochemical and biophysical research communications 2011, 407, 450–455. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell death & disease 2013, 4, e915–e915. [Google Scholar]
- Jang, J.; Park, H.J.; Seong, W.; Kim, J.; Kim, C. Vimentin-mediated buffering of internal integrin β1 pool increases survival of cells from anoikis. BMC biology 2024, 22, 139. [Google Scholar] [CrossRef]
- Usman, S.; Waseem, N.H.; Nguyen, T.K.N.; Mohsin, S.; Jamal, A.; Teh, M.T.; Waseem, A. Vimentin is at the heart of epithelial mesenchymal transition (EMT) mediated metastasis. Cancers 2021, 13, 4985. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Luo, Z.J.; Avruch, J. Calyculin A-induced vimentin phosphorylation sequesters 14-3-3 and displaces other 14-3-3 partners in vivo. Journal of Biological Chemistry 2000, 275, 29772–29778. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the NLRP3 inflammasome. Nature communications 2015, 6, 6574. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, K.; Wang, T.; Qiu, X.; Zhang, X.; He, T.; Chen, L.; Chen, J.; Cui, X.; Wu, H. Vimentin inhibits neuronal apoptosis after spinal cord injury by enhancing autophagy. CNS Neuroscience & Therapeutics 2025, 31, e70200. [Google Scholar] [CrossRef]
- Mohanasundaram, P.; Coelho-Rato, L.S.; Modi, M.K.; Urbanska, M.; Lautenschläger, F.; Cheng, F.; Eriksson, J.E. Cytoskeletal vimentin regulates cell size and autophagy through mTORC1 signaling. PLoS Biology 2022, 20, e3001737. [Google Scholar] [CrossRef]
- Yasuda, H.; Fukusumi, Y.; Zhang, Y.; Kawachi, H. 14-3-3 Proteins stabilize actin and vimentin filaments to maintain processes in renal glomerular podocyte. The FASEB Journal 2023, 37, e23168. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Gupta, V.S.; Kaplun, L.; Balan, V. 14-3-3 proteins as potential oncogenes. In Seminars in cancer biology; Academic Press, June 2006; Vol. 16, No. 3, pp. 203–213. [Google Scholar]
- Smit, M.A.; Geiger, T.R.; Song, J.Y.; Gitelman, I.; Peeper, D.S. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Molecular and cellular biology 2009, 29, 3722–3737. [Google Scholar] [CrossRef]
- Song, J.; Liu, Y.; Liu, F.; Zhang, L.; Li, G.; Yuan, C.; Yu, C.; Lu, X.; Liu, Q.; Chen, X.; et al. The 14-3-3σ protein promotes HCC anoikis resistance by inhibiting EGFR degradation and thereby activating the EGFR-dependent ERK1/2 signaling pathway. Theranostics 2021, 11, 996. [Google Scholar] [CrossRef]
- Smit, M.A.; Peeper, D.S. Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene 2011, 30, 3735–3744. [Google Scholar] [CrossRef]
- Llambi, F.; Causeret, F.; Bloch-Gallego, E.; Mehlen, P. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. The EMBO journal. 2001, 20, 2715–2722. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. Targeting EMT in cancer through netrin-1. Nat. Rev. Drug Discov 2023, 22, 785. [Google Scholar] [CrossRef]
- Ivanenko, K.A.; Prassolov, V.S.; Khabusheva, E.R. Transcription factor Sp1 in the expression of genes encoding components of MAPK, JAK/STAT, and PI3K/Akt signaling pathways. Molecular Biology 2022, 56, 756–769. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.-X.; Zhou, S.-Y.; Wang, S.-X.; Zheng, K.; Xu, D.-D.; Liu, Y.-T.; Wang, X.-Y.; Wang, X.; Yan, H.-Z.; et al. Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Molecular cancer 2015, 14, 56. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Li, W.; Zhang, H.; Zhao, C.; Li, Y.; Wang, Z.; Chen, C. Sp1 upregulates survivin expression in adenocarcinoma of lung cell line A549. The Anatomical Record: Advances in Integrative Anatomy and Evolutionary Biology 2011, 294, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.-M.; Li, L.; Zhu, P.; Dai, Y.-J.; Zhao, T.-T.; Liao, L.-Y.; Chen, G.G.; Liu, Z.-M. Up-regulation of Hsp27 by ERα/Sp1 facilitates proliferation and confers resistance to apoptosis in human papillary thyroid cancer cells. Molecular and cellular endocrinology 2016, 431, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Hua, P.; Xu, H.; Uno, J.K.; Lipko, M.A.; Dong, J.; Kiela, P.R.; Ghishan, F.K. Sp1 and Sp3 mediate NHE2 gene transcription in the intestinal epithelial cells. American Journal of Physiology-Gastrointestinal and Liver Physiology 2007, 293, G146–G153. [Google Scholar] [CrossRef]
- Zhang, X.; Diab, I.H.; Zehner, Z.E. ZBP-89 represses vimentin gene transcription by interacting with the transcriptional activator, Sp1. Nucleic acids research 2003, 31, 2900–2914. [Google Scholar] [CrossRef]
- Tomecka, P.; Kunachowicz, D.; Górczyńska, J.; Gebuza, M.; Kuźnicki, J.; Skinderowicz, K.; Choromańska, A. Factors determining epithelial-mesenchymal transition in cancer progression. International Journal of Molecular Sciences 2024, 25, 8972. [Google Scholar] [CrossRef]
- Sarmadhikari, D.; Asthana, S. Structural insights into Beclin 1 interactions with it's regulators for autophagy modulation. Computational and Structural Biotechnology Journal 2025, 27, 3005–3035. [Google Scholar] [CrossRef]
- Huang, C.; Chen, Y.; Lai, B.; Chen, Y.X.; Xu, C.Y.; Liu, Y.F. Overexpression of SP1 restores autophagy to alleviate acute renal injury induced by ischemia-reperfusion through the miR-205/PTEN/Akt pathway. Journal of Inflammation 2021, 18, 7. [Google Scholar] [CrossRef]
- Zutter, M.M.; Ryan, E.E.; Painter, A.D. Binding of phosphorylated Sp1 protein to tandem Sp1 binding sites regulates α2 integrin gene core promoter activity. Blood, The Journal of the American Society of Hematology 1997, 90, 678–689. [Google Scholar]
- Gingras, M.E.; Masson-Gadais, B.; Zaniolo, K.; Leclerc, S.; Drouin, R.; Germain, L.; Guerin, S.L. Differential binding of the transcription factors Sp1, AP-1, and NFI to the promoter of the human α5 integrin gene dictates its transcriptional activity. Investigative ophthalmology & visual science 2009, 50, 57–67. [Google Scholar]
- Nam, E.H.; Lee, Y.; Park, Y.K.; Lee, J.W.; Kim, S. ZEB2 upregulates integrin α5 expression through cooperation with Sp1 to induce invasion during epithelial–mesenchymal transition of human cancer cells. Carcinogenesis 2012, 33, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Piyush, T.; Rhodes, J.M.; Yu, L.G. MUC1 O-glycosylation contributes to anoikis resistance in epithelial cancer cells. Cell death discovery 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Farahmand, L.; Merikhian, P.; Jalili, N.; Darvishi, B.; Majidzadeh-A, K. Significant role of MUC1 in development of resistance to currently existing anti-cancer therapeutic agents. Current Cancer Drug Targets 2018, 18, 737–748. [Google Scholar] [CrossRef]
- Supruniuk, K.; Radziejewska, I. MUC1 is an oncoprotein with a significant role in apoptosis. International Journal of Oncology 2021, 59, 68. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, Z.; Zhang, S.; Zhu, P.; Ko, J.K. S.; Yung, K.K.L. MUC1: structure, function, and clinic application in epithelial cancers. International journal of molecular sciences 2021, 22, 6567. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Ni, W.; Tai, G. Expression of MUC1 in different tumours and its clinical significance. Molecular and clinical oncology 2022, 17, 161. [Google Scholar] [CrossRef]
- Qing, L.; Li, Q.; Dong, Z. MUC1: An emerging target in cancer treatment and diagnosis. Bulletin du Cancer 2022, 109, 1202–1216. [Google Scholar] [CrossRef]
- Chen, B.; Wang, X.; Zhao, W.; Wu, J. Klotho inhibits growth and promotes apoptosis in human lung cancer cell line A549. Journal of Experimental & Clinical Cancer Research 2010, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, K.I.; Glinka, Y.Y.; Perfilova, V.N.; Nesterova, A.A.; Kaplanov, K.D. Antiaging klotho protein as a prospective novel tumor suppressor. Annals of the Russian academy of medical sciences 2023, 78, 24–44. [Google Scholar] [CrossRef]
- Dai, D.; Wang, Q.; Li, X.; Liu, J.; Ma, X.; Xu, W. Klotho inhibits human follicular thyroid cancer cell growth and promotes apoptosis through regulation of the expression of stanniocalcin-1. Oncology reports 2016, 35, 552–558. [Google Scholar] [CrossRef]
- Chen, L.; Liu, H.; Liu, J.; Zhu, Y.; Xu, L.; He, H.; Zhang, H.; Wang, S.; Wu, Q.; Liu, W.; et al. Klotho endows hepatoma cells with resistance to anoikis via VEGFR2/PAK1 activation in hepatocellular carcinoma. PLoS One 2013, 8, e58413. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X. Klotho: a novel biomarker for cancer. Journal of cancer research and clinical oncology 2015, 141, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, S.; Zhao, D.; Yan, J.; Chen, J.; Yang, C.; Zheng, G. MNX1 reduces sensitivity to anoikis by activating TrkB in human glioma cells. Molecular medicine reports 2018, 18, 3271–3279. [Google Scholar] [CrossRef]
- Xiao, L.; Hong, L.; Zheng, W. Motor neuron and pancreas homeobox 1 (MNX1) is involved in promoting squamous cervical cancer proliferation via regulating cyclin E. Medical science monitor: international medical journal of experimental and clinical research 2019, 25, 6304. [Google Scholar] [CrossRef]
- Wu, J.; Yue, C.; Xu, W.; Li, H.; Zhu, J.; Li, L. MNX1 facilitates the malignant progress of lung adenocarcinoma through transcriptionally upregulating CCDC34. Oncology Letters 2023, 26, 325. [Google Scholar] [CrossRef]
- Zhou, L.; Lu, H.; Fu, B.; Fu, J. Anoikis-related genes predicts prognosis and therapeutic response in renal cell carcinoma. Annals of Medicine 2025, 57, 2548042. [Google Scholar] [CrossRef]
- Zhong, G.; Zhou, M.; Wang, Z.; Zhu, W. Prognostic potential of MNX1-AS1 in chemotherapy of colorectal carcinoma. Cellular and Molecular Biology 2023, 69, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Kelekçi, S. Epigenetic compound screening in AML with MNX1 overexpression (Doctoral dissertation).Downloaded from https://archiv.ub.uni-heidelberg.de/volltextserver/35795/ Novembre 2025.
- Wang, C.; Gao, G.; Che, Q.; Zheng, S.; Yang, Y.; Li, T.; Liu, M. Deciphering the value of anoikis-related genes in prognosis, immune microenvironment, and drug sensitivity of laryngeal squamous cell carcinoma. Pathology-Research and Practice 2025, 268, 155849. [Google Scholar] [CrossRef]
- He, Z.; Gu, Y.; Yang, H.; Fu, Q.; Zhao, M.; Xie, Y.; Liu, Y.; Du, W. Identification and verification of a novel anoikis-related gene signature with prognostic significance in clear cell renal cell carcinoma. Journal of Cancer Research and Clinical Oncology 2023, 149, 11661–11678. [Google Scholar] [CrossRef]
- Liu, C.L.; Pan, H.W.; Torng, P.L.; Fan, M.H.; Mao, T.L. SRPX and HMCN1 regulate cancer-associated fibroblasts to promote the invasiveness of ovarian carcinoma. Oncology reports 2019, 42, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, S.; Teng, M.; Qing, Y.; Dong, X.; Chen, L.; Ai, K. A novel anoikis-related prognostic signature associated with prognosis and immune infiltration landscape in lung adenocarcinoma. The journal of gene medicine 2024, 26, e3610. [Google Scholar] [CrossRef]
- Geiger, T.R.; Peeper, D.S. The neurotrophic receptor TrkB in anoikis resistance and metastasis: a perspective. Cancer research 2005, 65, 7033–7036. [Google Scholar] [CrossRef]
- Yuan, Y.; Ye, H.Q.; Ren, Q.C. Proliferative role of BDNF/TrkB signaling is associated with anoikis resistance in cervical cancer. Oncology reports 2018, 40, 621–634. [Google Scholar] [CrossRef]
- Geiger, T.R.; Peeper, D.S. Critical role for TrkB kinase function in anoikis suppression, tumorigenesis, and metastasis. Cancer research 2007, 67, 6221–6229. [Google Scholar] [CrossRef]
- Oyama, Y.; Nagao, S.; NA, L.; Yanai, K.; Umebayashi, M.; Nakamura, K.; Nagai, S.; Fujimura, A.; Yamasaki, A.; Nakayama, K.; et al. TrkB/BDNF signaling could be a new therapeutic target for pancreatic cancer. Anticancer research 2021, 41, 4047–4052. [Google Scholar] [CrossRef]
- Johnson, M.D.; Stone, B.; Thibodeau, B.J.; Baschnagel, A.M.; Galoforo, S.; EFortier, L.; Ketelsen, B.; Ahmed, S.; Kelley, Z.; Hana, A.; et al. The significance of Trk receptors in pancreatic cancer. Tumor Biology 2017, 39, 1010428317692256. [Google Scholar] [CrossRef]
- Odate, S.; Nakamura, K.; Onishi, H.; Kojima, M.; Uchiyama, A.; Nakano, K.; Kato, M.; Tanaka, M.; Katano, M. TrkB/BDNF signaling pathway is a potential therapeutic target for pulmonary large cell neuroendocrine carcinoma. Lung Cancer 2013, 79, 205–214. [Google Scholar] [CrossRef]
- Tanaka, K.; Shimura, T.; Kitajima, T.; Kondo, S.; Ide, S.; Okugawa, Y.; Saigusa, S.; Toiyama, Y.; Inoue, Y.; Araki, T.; et al. Tropomyosin-related receptor kinase B at the invasive front and tumour cell dedifferentiation in gastric cancer. British journal of cancer 2014, 110, 2923–2934. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, Z.; Tang, Z.; Hu, J.; Zhou, Y.; Ge, J.; Dong, J.; Xu, S. An anoikis-related gene signature for prediction of the prognosis in prostate cancer. Frontiers in oncology 2023, 13, 1169425. [Google Scholar] [CrossRef]
- Lin, D.-C.; Zhang, Y.; Pan, Q.-J.; Yang, H.; Shi, Z.-Z.; Xie, Z.-H.; Wang, B.-S.; Hao, J.-J.; Zhang, T.-T.; Xu, X.; et al. PLK1 Is transcriptionally activated by NF-κB during cell detachment and enhances anoikis resistance through inhibiting β-catenin degradation in esophageal squamous cell carcinoma. Clinical Cancer Research 2011, 17, 4285–4295. [Google Scholar] [CrossRef]
- Zhang, Y.; Du, X.; Wang, C.; Lin, D.; Ruan, X.; Feng, Y.; Huo, Y.; Peng, H.; Cui, J.; Zhang, T.; et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology 2012, 142, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ivanov, A.I.; Fisher, P.B.; Fu, Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife 2016, 5, e10734. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Chu, Q.S.; Stephenson, J.; Johnson, B.E.; Govindan, R.; Bonomi, P.; Eaton, K.; Fritsch, H.; Munzert, G.; Socinski, M. An open label phase II trial of the Plk1 inhibitor BI 2536, in patients with sensitive relapse small cell lung cancer (SCLC). Journal of Clinical Oncology 2009, 27, 8108–8108. [Google Scholar] [CrossRef]
- Mo, G.; Long, X.; Hu, Z.; Tang, Y.; Zhou, Z. Anoikis-related gene signatures can aid prognosis of lung adenocarcinoma. Advances in Clinical and Experimental Medicine 2024, 33, 751–761. [Google Scholar] [CrossRef]
- Jiao, Y.; Ji, F.; Hou, L.; Zhang, J. A novel gene signature associated with anoikis predicts prognosis and unveils immune infiltration in breast cancer patients. Discover Oncology 2025, 16, 1–17. [Google Scholar] [CrossRef]
- Furlong, I.J.; Ascaso, R.; Rivas, A.L.; Collins, M.K. Intracellular acidification induces apoptosis by stimulating ICE-like protease activity. Journal of cell science 1997, 110, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: origins and roles. Cell Death & Differentiation 2004, 11, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A. Cell acidification in apoptosis. Apoptosis 1996, 1, 40–48. [Google Scholar] [CrossRef]
- Su, Y.; Ren, H.; Tang, M.; Zheng, Y.; Zhang, B.; Wang, C.; Hou, X.; Niu, Z.; Wang, Z.; Gao, X.; et al. Role and dynamics of vacuolar pH during cell-in-cell mediated death. Cell death & disease 2021, 12, 119. [Google Scholar]
- Numata, M.; Onishi, I.; Lin, P.J.; Numata, Y.; Austin, P.; Cipollone, J.; Roberge, M.; Roskelley, C.D. Organellar (Na+, K+)/H+ exchanger NHE7 regulates cell adhesion, invasion and anchorage-independent growth of breast cancer MDA-MB-231 cells. Oncology reports 2012, 27, 311–317. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The figure shows the main proteins that interact at the focal adhesion site, where there is a concentration of integrin heterodimers within the membrane that are attached to extracellular matrix proteins such as fibronectin, collagens, elastin, and laminins. The main attachment of integrins is to fibronectin. In addition to its adhesion function, this attachment triggers signaling to intracellular proteins. The main signaling protein is FAK which has many binding partners as shown in the right panel. Autophosphorylation of FAK at Tyr397 creates a binding site for the SRC kinase family. The recruitment of SRC further leads to full FAK activation through phosphorylation of other tyrosine residues [29].
Figure 1.
The figure shows the main proteins that interact at the focal adhesion site, where there is a concentration of integrin heterodimers within the membrane that are attached to extracellular matrix proteins such as fibronectin, collagens, elastin, and laminins. The main attachment of integrins is to fibronectin. In addition to its adhesion function, this attachment triggers signaling to intracellular proteins. The main signaling protein is FAK which has many binding partners as shown in the right panel. Autophosphorylation of FAK at Tyr397 creates a binding site for the SRC kinase family. The recruitment of SRC further leads to full FAK activation through phosphorylation of other tyrosine residues [29].
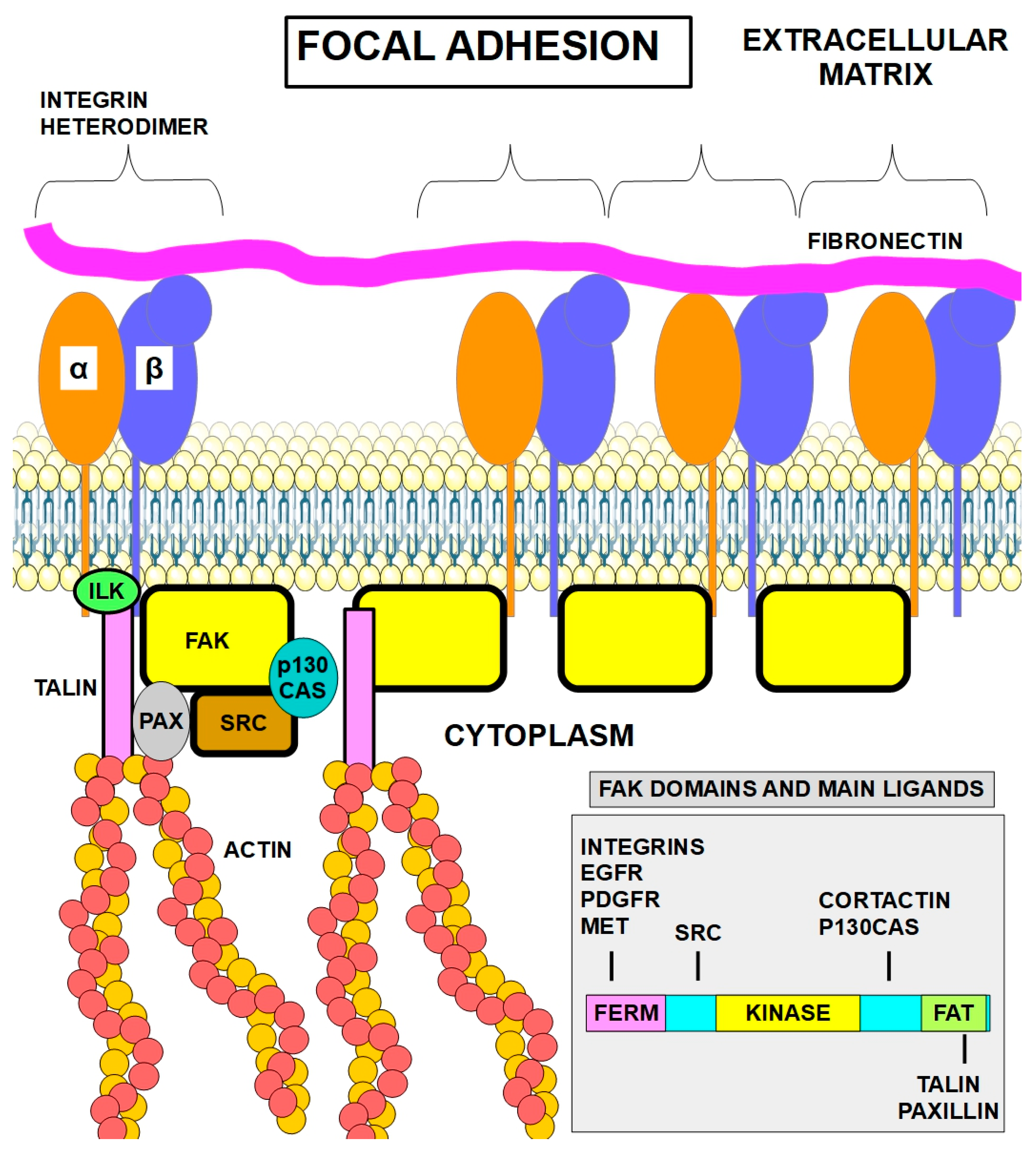
Figure 2.
Integrin and FAK activation [30,31,32,33,34] initiated by fibronectin contact with the extracellular portion of the integrin dimer (1) and (2). When the integrin is not in contact with fibronectin, FAK is inactive (1). Contact between fibronectin and integrin (2) causes autophosphorylation of FAK. Phosphorylation of FAK at amino acid Tyr347 recruits SCR (3) which further phosphorylates FAK (4) exerting downstream effects (signaling) (5).
Figure 2.
Integrin and FAK activation [30,31,32,33,34] initiated by fibronectin contact with the extracellular portion of the integrin dimer (1) and (2). When the integrin is not in contact with fibronectin, FAK is inactive (1). Contact between fibronectin and integrin (2) causes autophosphorylation of FAK. Phosphorylation of FAK at amino acid Tyr347 recruits SCR (3) which further phosphorylates FAK (4) exerting downstream effects (signaling) (5).
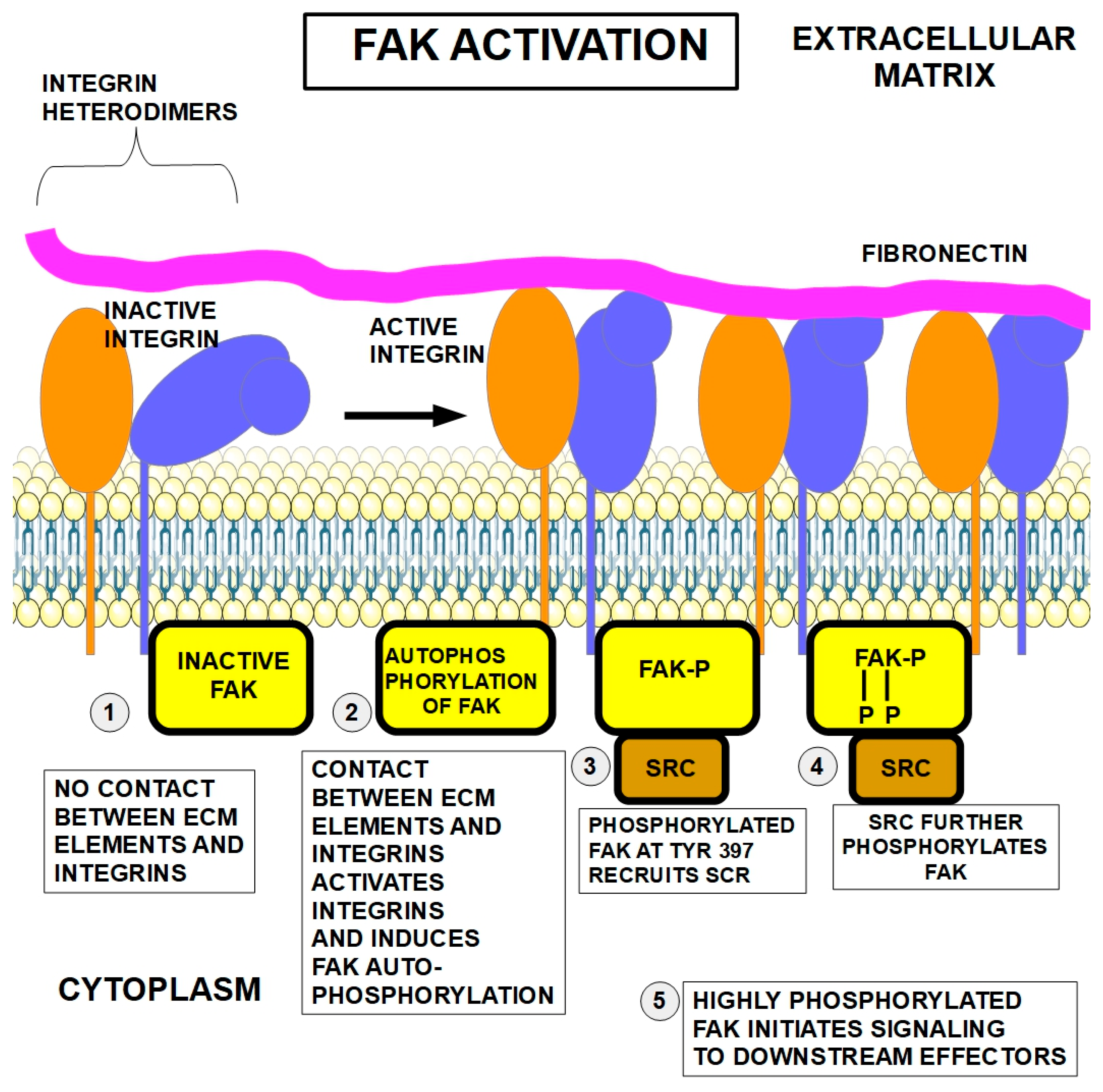
Figure 3.
FAK activation [106], and SRC structure [107]. Upper panel, dephosphorylated FAK is inactive. FAK has a N-terminal FERM (For Ezrin, Radixin and Moesin) domain, a kinase domain and a FAT (focal adhesion targeting) domain. Middle panel, FAK autophosphorylation at Tyr397 induces a conformational change and activates the protein. Lower panel, full activation of FAK at multiple sites is achieved through SRC phosphorylation at several additional sites.
Figure 3.
FAK activation [106], and SRC structure [107]. Upper panel, dephosphorylated FAK is inactive. FAK has a N-terminal FERM (For Ezrin, Radixin and Moesin) domain, a kinase domain and a FAT (focal adhesion targeting) domain. Middle panel, FAK autophosphorylation at Tyr397 induces a conformational change and activates the protein. Lower panel, full activation of FAK at multiple sites is achieved through SRC phosphorylation at several additional sites.
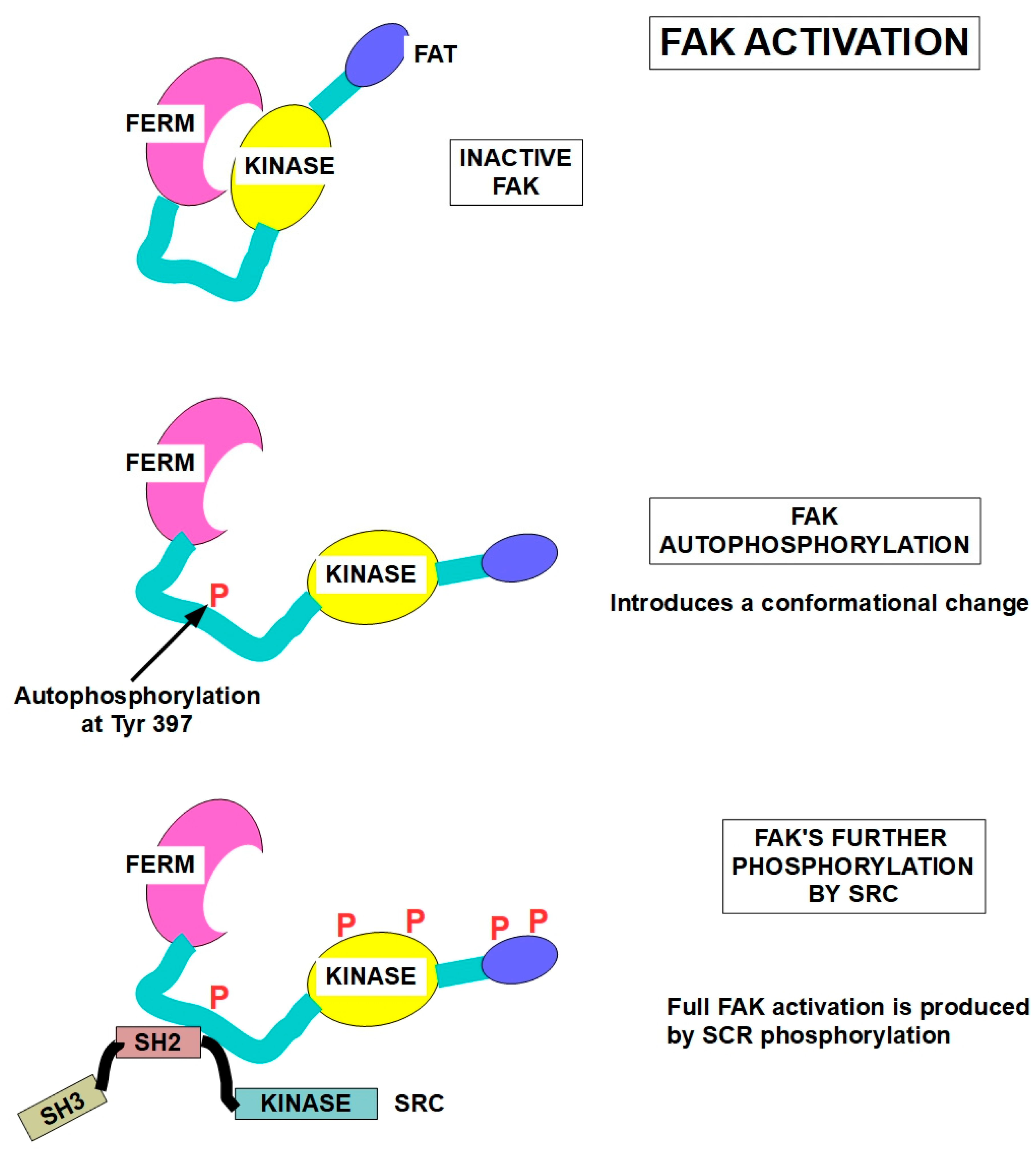
Figure 4.
Focal adhesions at the cell membrane concentrate integrins that act mechanically to bind the actin cytoskeleton to the ECM. Importantly, focal adhesions are also signaling hubs that permit crosstalk between the cell and the ECM. The heterodimer formed by integrins α and β is activated by the matrix proteins. Integrins transmit this activation to various intracellular pathways [111,112]. This keeps the cell "alive" and prevents anoikis [113]. While the cell is attached in its right place, it is the integrins and additionally growth factor receptor signaling that maintains cell viability. The FAK-SRC complex activates several signaling pathways, such as the pro-survival PI3K/Akt pathway (right panel) [114]. Microtubule-deregulating agents can disrupt the focal adhesions leading to loss of adhesion and anoikis [115].
Figure 4.
Focal adhesions at the cell membrane concentrate integrins that act mechanically to bind the actin cytoskeleton to the ECM. Importantly, focal adhesions are also signaling hubs that permit crosstalk between the cell and the ECM. The heterodimer formed by integrins α and β is activated by the matrix proteins. Integrins transmit this activation to various intracellular pathways [111,112]. This keeps the cell "alive" and prevents anoikis [113]. While the cell is attached in its right place, it is the integrins and additionally growth factor receptor signaling that maintains cell viability. The FAK-SRC complex activates several signaling pathways, such as the pro-survival PI3K/Akt pathway (right panel) [114]. Microtubule-deregulating agents can disrupt the focal adhesions leading to loss of adhesion and anoikis [115].
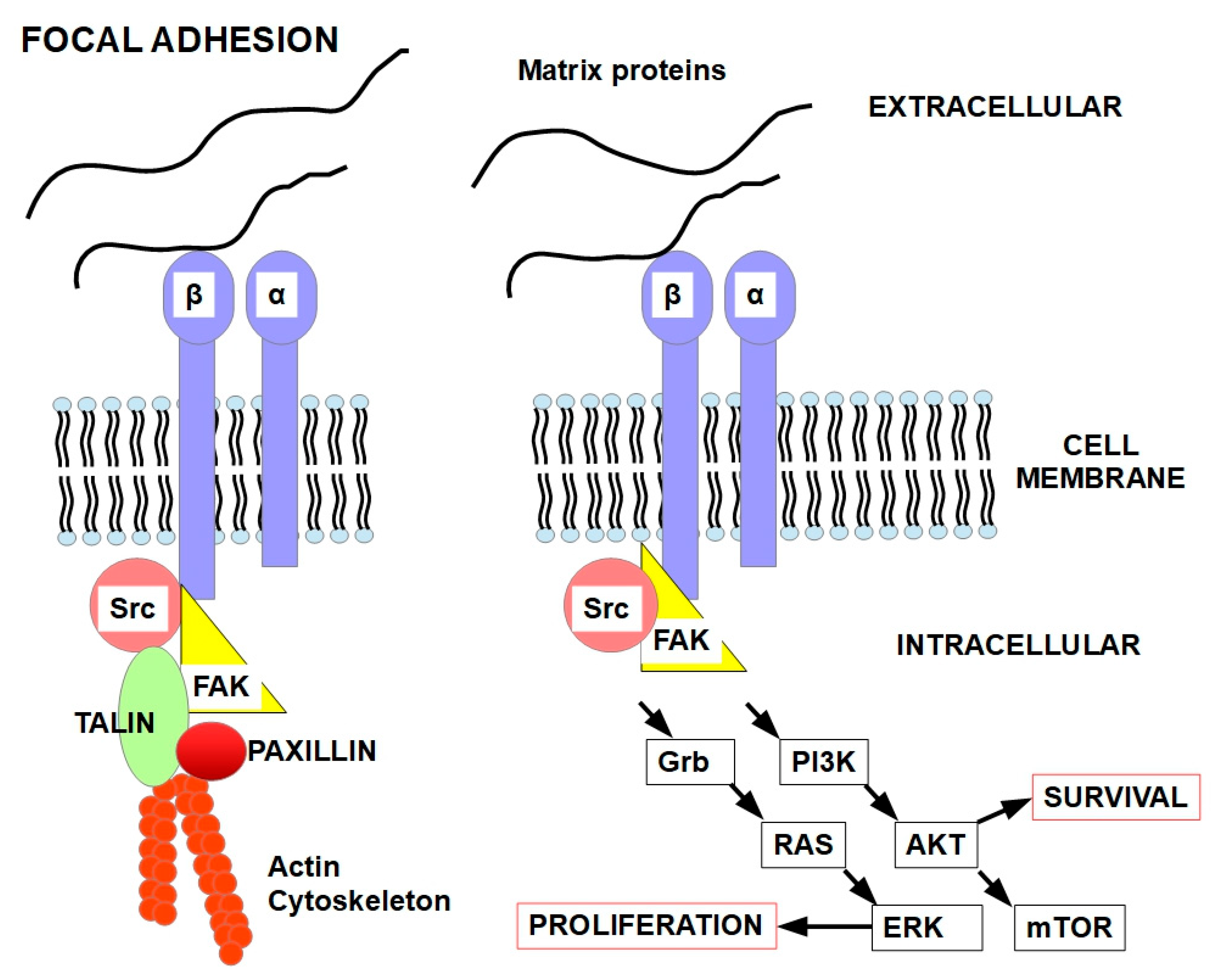
Figure 5.
- ECM detachment in tumors precedes the abnormal dissemination of cells that leads to metastasis [125]. The drawing shows the destiny of a cell when anoikis is functional (left lower panel) and when it is not (right lower panel). Under normal conditions, epithelial cells’ attachment to the matrix keeps them viable (left upper side), otherwise they die [126,127]. This drawing shows that, in addition to the genetic regulation of the cell itself, the microenvironment plays an important role in the cell’s growth, differentiation, and fate.
Figure 5.
- ECM detachment in tumors precedes the abnormal dissemination of cells that leads to metastasis [125]. The drawing shows the destiny of a cell when anoikis is functional (left lower panel) and when it is not (right lower panel). Under normal conditions, epithelial cells’ attachment to the matrix keeps them viable (left upper side), otherwise they die [126,127]. This drawing shows that, in addition to the genetic regulation of the cell itself, the microenvironment plays an important role in the cell’s growth, differentiation, and fate.
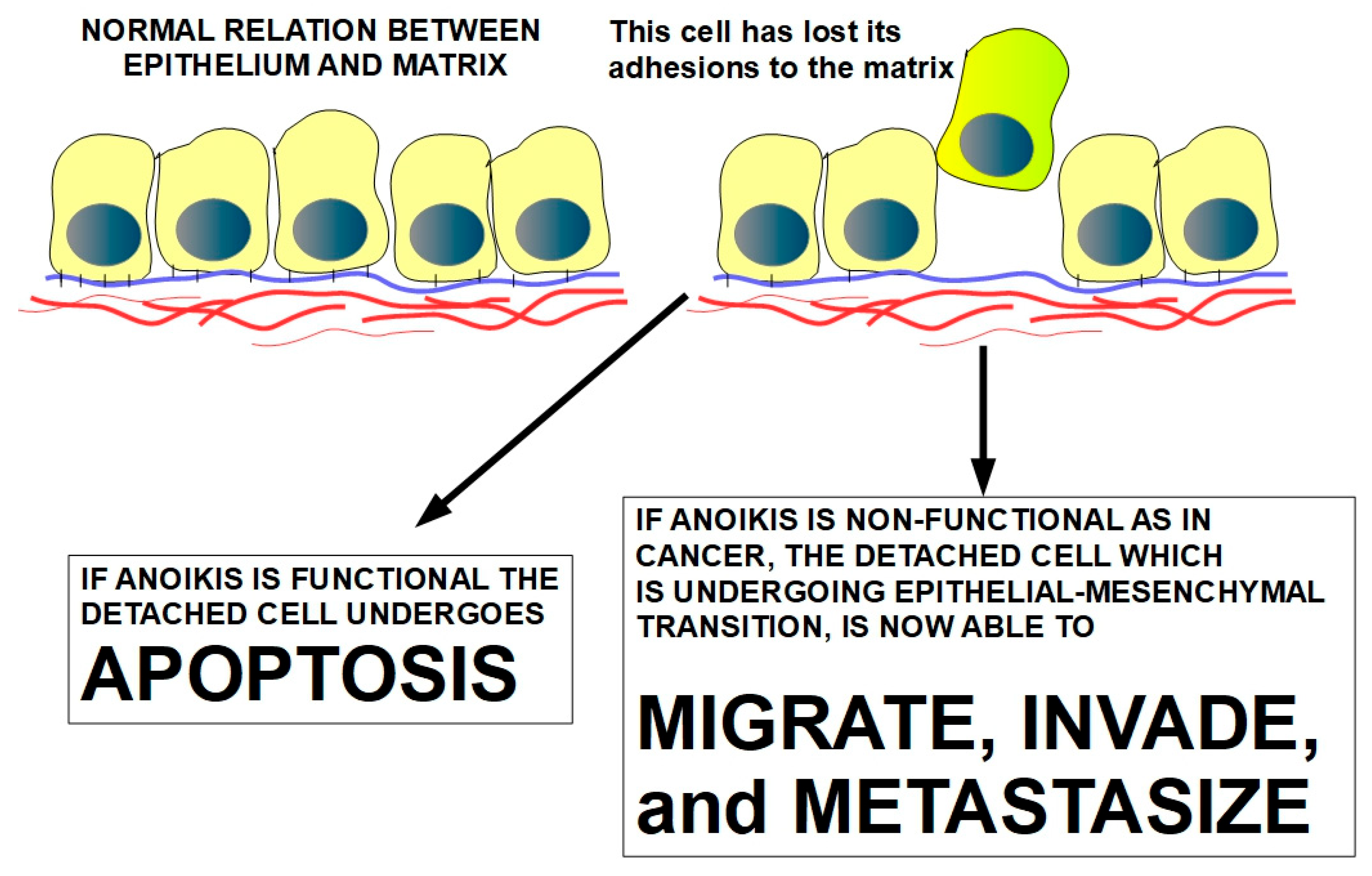
Figure 6.
Apoptosis the extrinsic pathway. The usual extrinsic pathway is shown, where external ligand binding to the death receptor (2) is necessary to activate the pathway through DISC formation (3). DISC: Death inducing signaling complex Procaspase 8 is initially activated at DISC to form active caspase 8 (4) which is an initiator caspase. It activates executioner caspases like 3, 6, and 7 (5). Once these are activated, they will degrade the cell (6). FLIP (FLICE-like inhibitory protein) is a key regulator of procaspase 8 activation. In anoikis, the lack of integrin signaling leads to an increased number of death receptors and decreased FLIP leads to caspase 8 activation without the need for an extracellular ligand. The extrinsic pathways seem to predominate as the main pathway in anoikis. Experiments have shown that cell death is prevented when caspase-3 and caspase-8 are inhibited. Inhibiting caspase 9 does not achieve the same result [135].
Figure 6.
Apoptosis the extrinsic pathway. The usual extrinsic pathway is shown, where external ligand binding to the death receptor (2) is necessary to activate the pathway through DISC formation (3). DISC: Death inducing signaling complex Procaspase 8 is initially activated at DISC to form active caspase 8 (4) which is an initiator caspase. It activates executioner caspases like 3, 6, and 7 (5). Once these are activated, they will degrade the cell (6). FLIP (FLICE-like inhibitory protein) is a key regulator of procaspase 8 activation. In anoikis, the lack of integrin signaling leads to an increased number of death receptors and decreased FLIP leads to caspase 8 activation without the need for an extracellular ligand. The extrinsic pathways seem to predominate as the main pathway in anoikis. Experiments have shown that cell death is prevented when caspase-3 and caspase-8 are inhibited. Inhibiting caspase 9 does not achieve the same result [135].
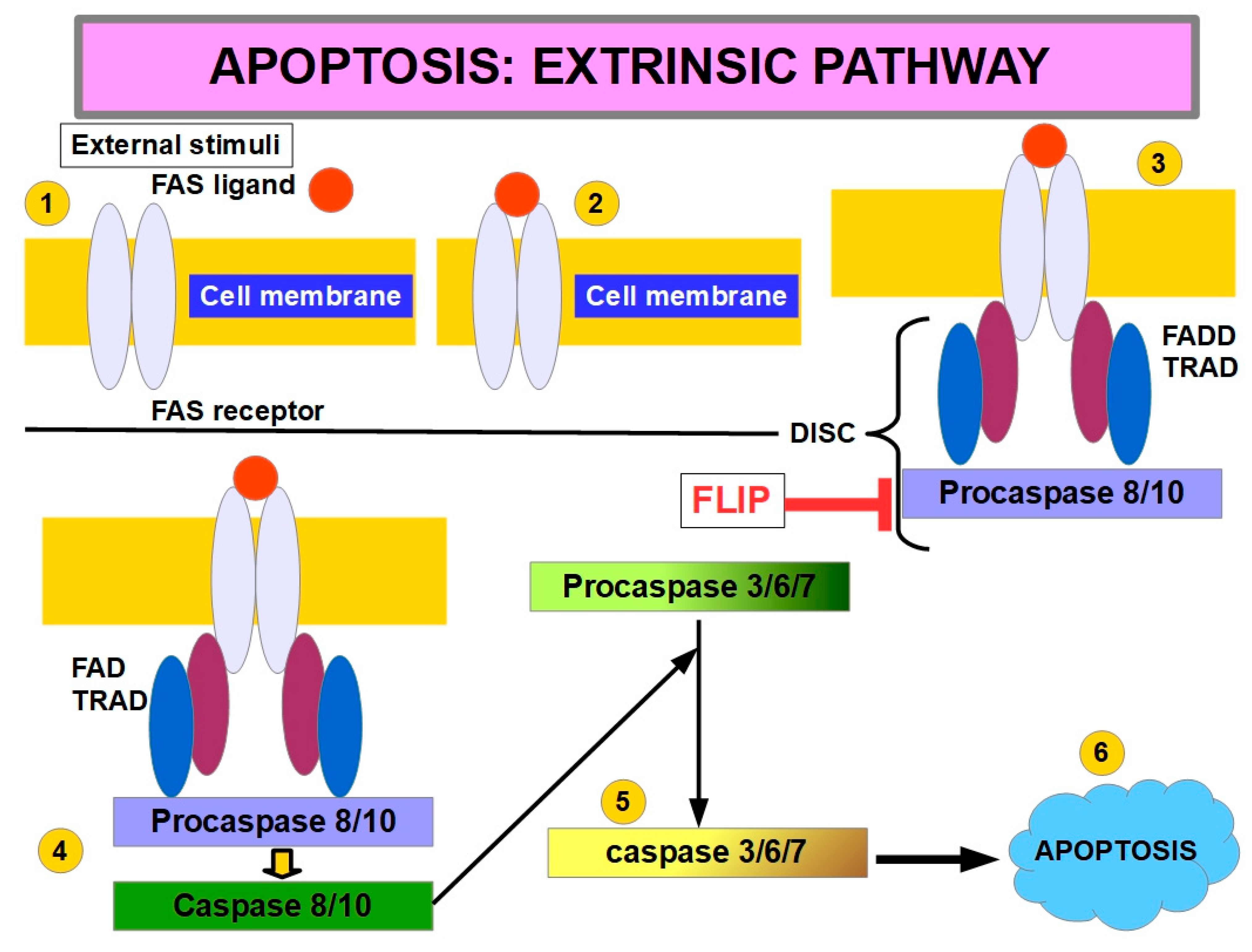
Figure 7.
Role of cell detachment in interruption of the apoptosis-repressing pathway [137,138,139,140]. Integrins continuously regulate cell viability through their interaction with the ECM. Integrins are mechanosensors of forces arising from the matrix and through their linked pathways they convert these stimuli to chemically transduced signals [141]. Activating FAK (focal adhesive kinase) and AKT can prevent anoikis [142,143,144].
Figure 7.
Role of cell detachment in interruption of the apoptosis-repressing pathway [137,138,139,140]. Integrins continuously regulate cell viability through their interaction with the ECM. Integrins are mechanosensors of forces arising from the matrix and through their linked pathways they convert these stimuli to chemically transduced signals [141]. Activating FAK (focal adhesive kinase) and AKT can prevent anoikis [142,143,144].
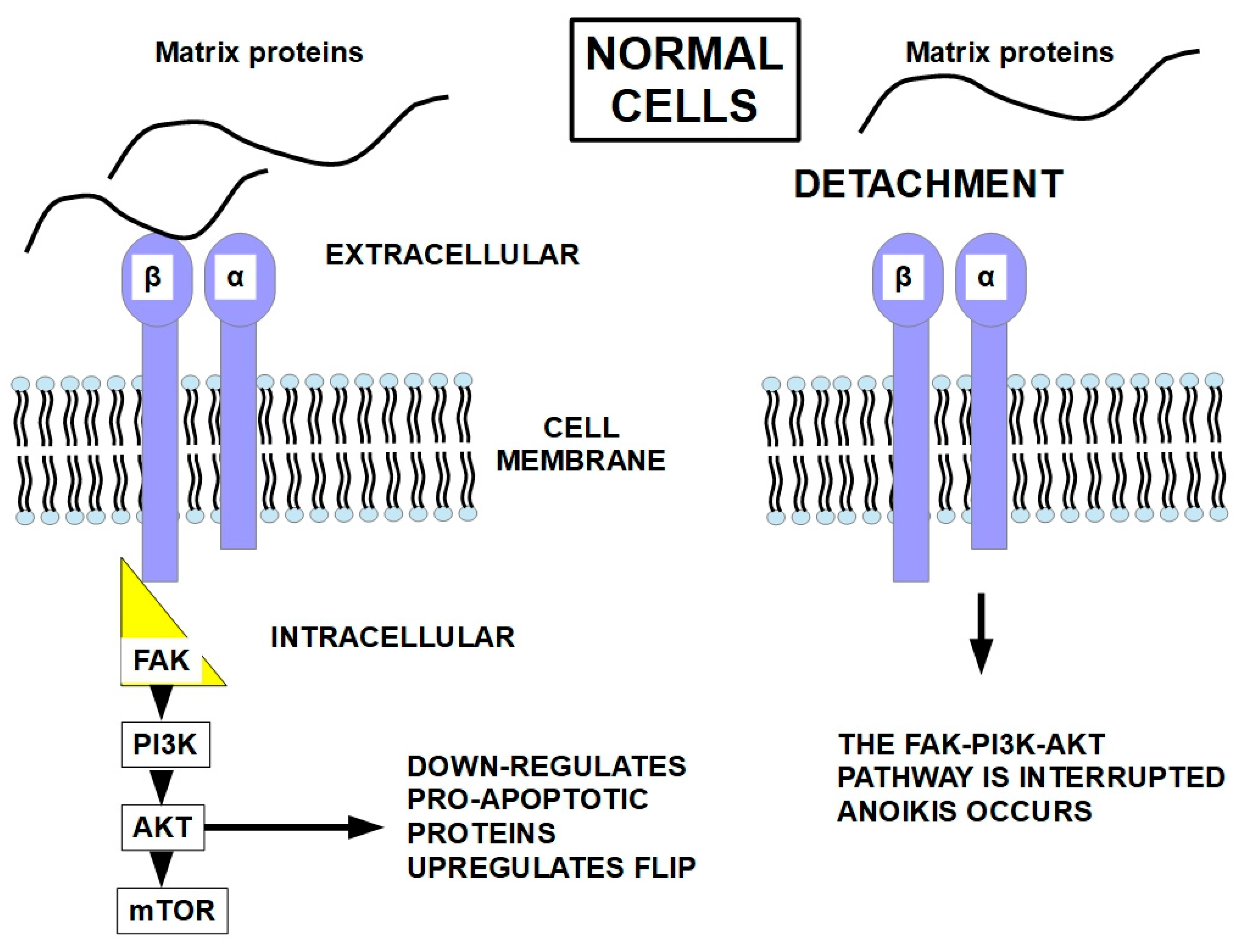
Figure 8.
Truncated BID (t-BID) is the connecting protein between extrinsic and intrinsic apoptosis. Truncated BID is generated by the action of Caspase 8 (or Caspase 10). It can then translocate to the mitochondria where it promotes outer membrane permeability leading to Cytochrome C release and activation of downstream caspases. This triggers apoptosome assembly which activates caspases that lead to apoptosis.
Figure 8.
Truncated BID (t-BID) is the connecting protein between extrinsic and intrinsic apoptosis. Truncated BID is generated by the action of Caspase 8 (or Caspase 10). It can then translocate to the mitochondria where it promotes outer membrane permeability leading to Cytochrome C release and activation of downstream caspases. This triggers apoptosome assembly which activates caspases that lead to apoptosis.
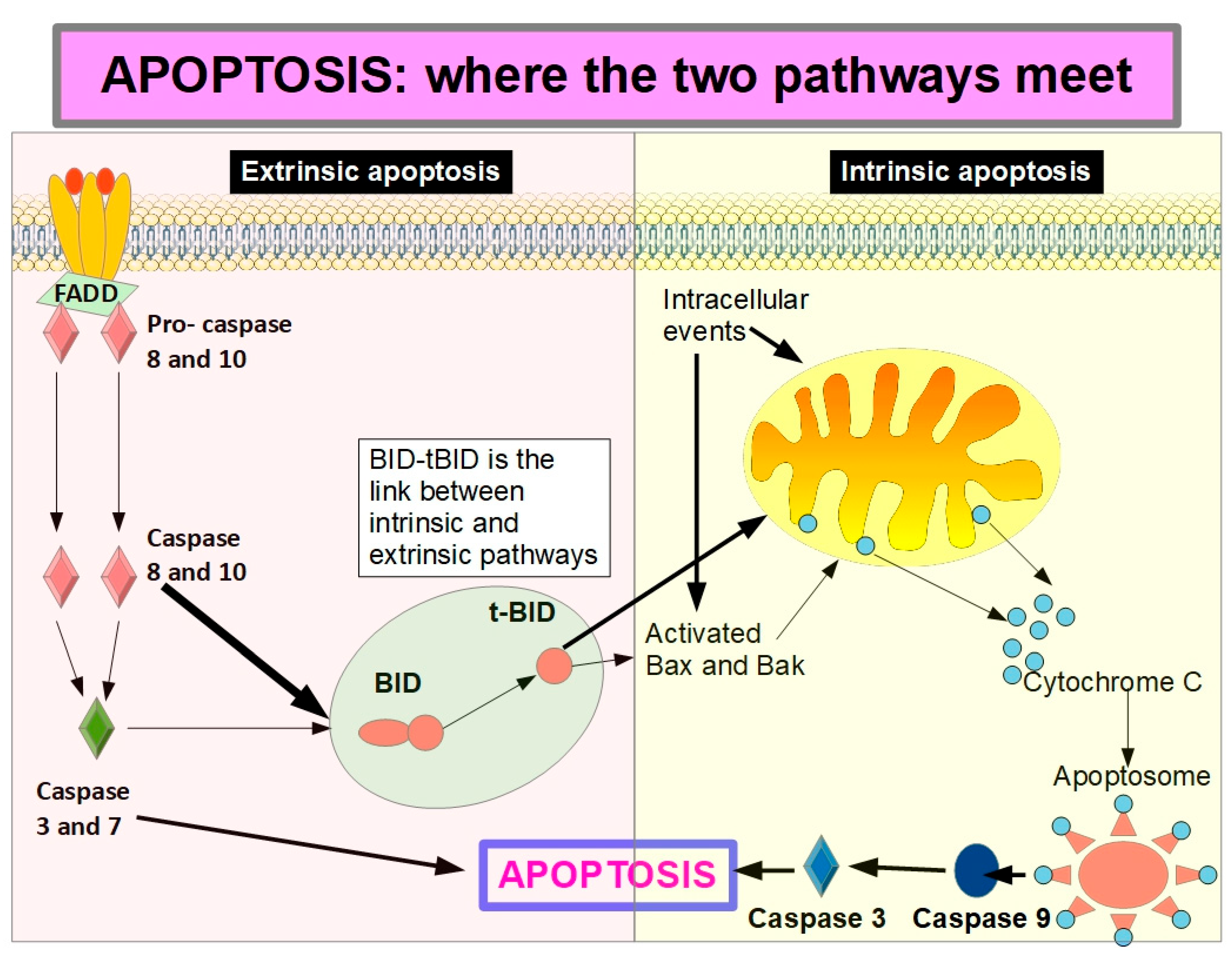
Figure 9.
- The ENO2 pathway increases detached cell survival by improving redox homeostasis.
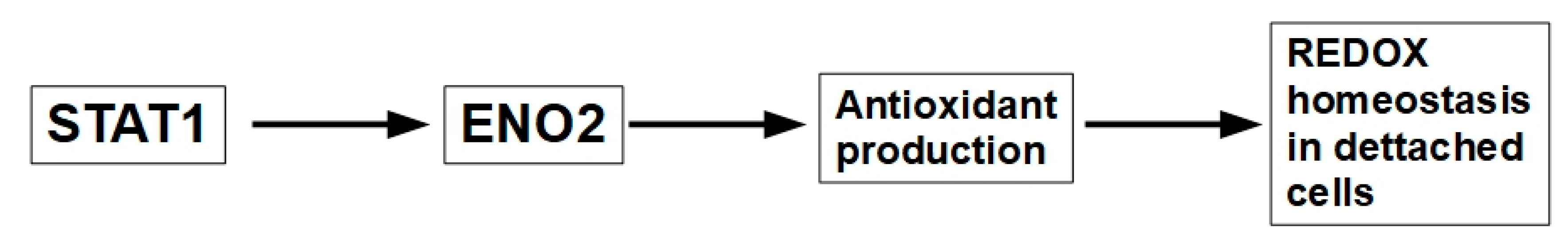
Figure 10.
The steps of autophagy. Beclin1, a key component of autophagy initiation, is released from the BCL2-Beclin complex by over-expression of CEMIP permitting the formation of the PI3K complex that induces autophagosome formation. Autophagosomes can then cause autophagy, or self-eating of the cell.
Figure 10.
The steps of autophagy. Beclin1, a key component of autophagy initiation, is released from the BCL2-Beclin complex by over-expression of CEMIP permitting the formation of the PI3K complex that induces autophagosome formation. Autophagosomes can then cause autophagy, or self-eating of the cell.
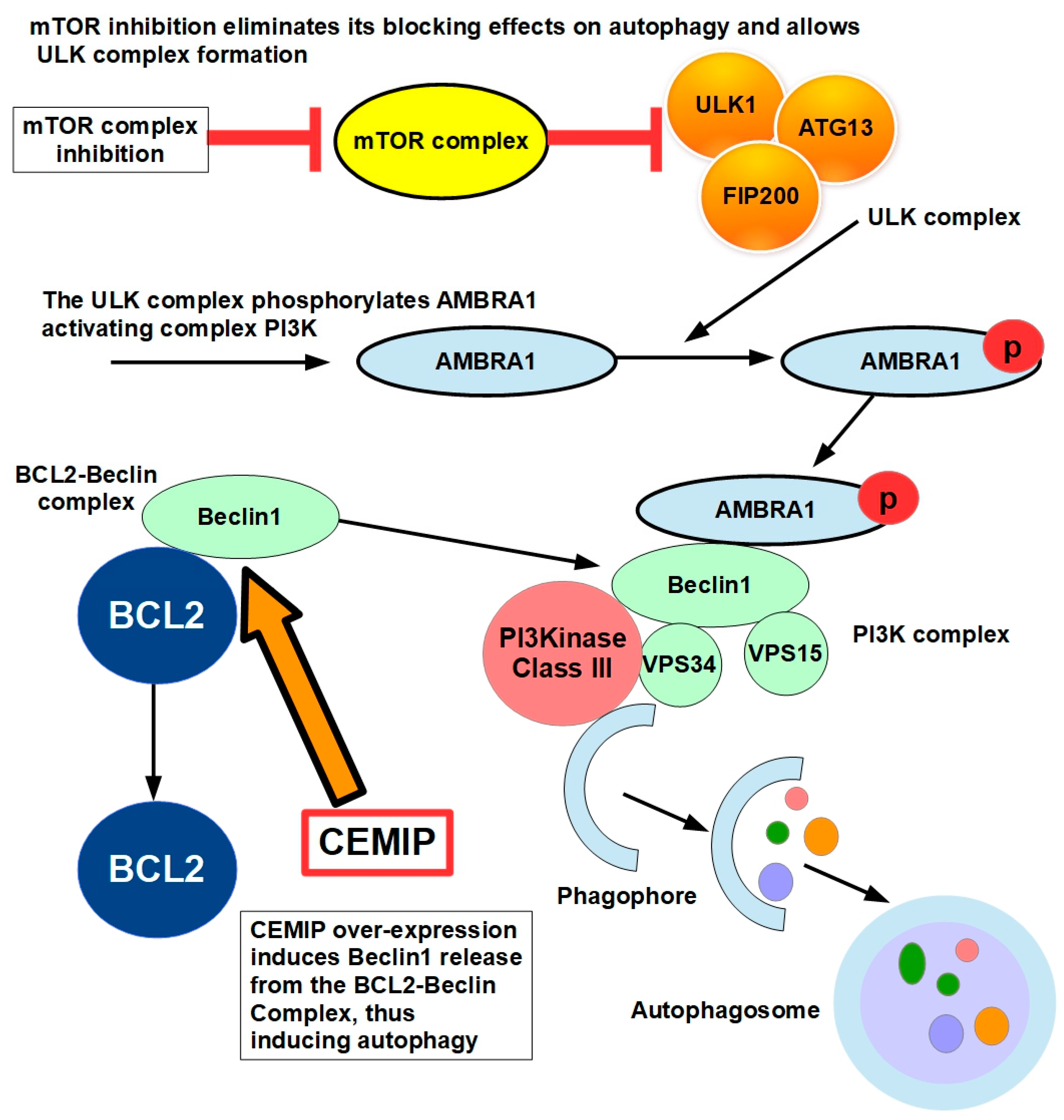
Figure 11.
The role of MiR141 and Sp1 in anoikis resistance. KLF12 expression and activity is a barrier to anoikis resistance.
Figure 11.
The role of MiR141 and Sp1 in anoikis resistance. KLF12 expression and activity is a barrier to anoikis resistance.
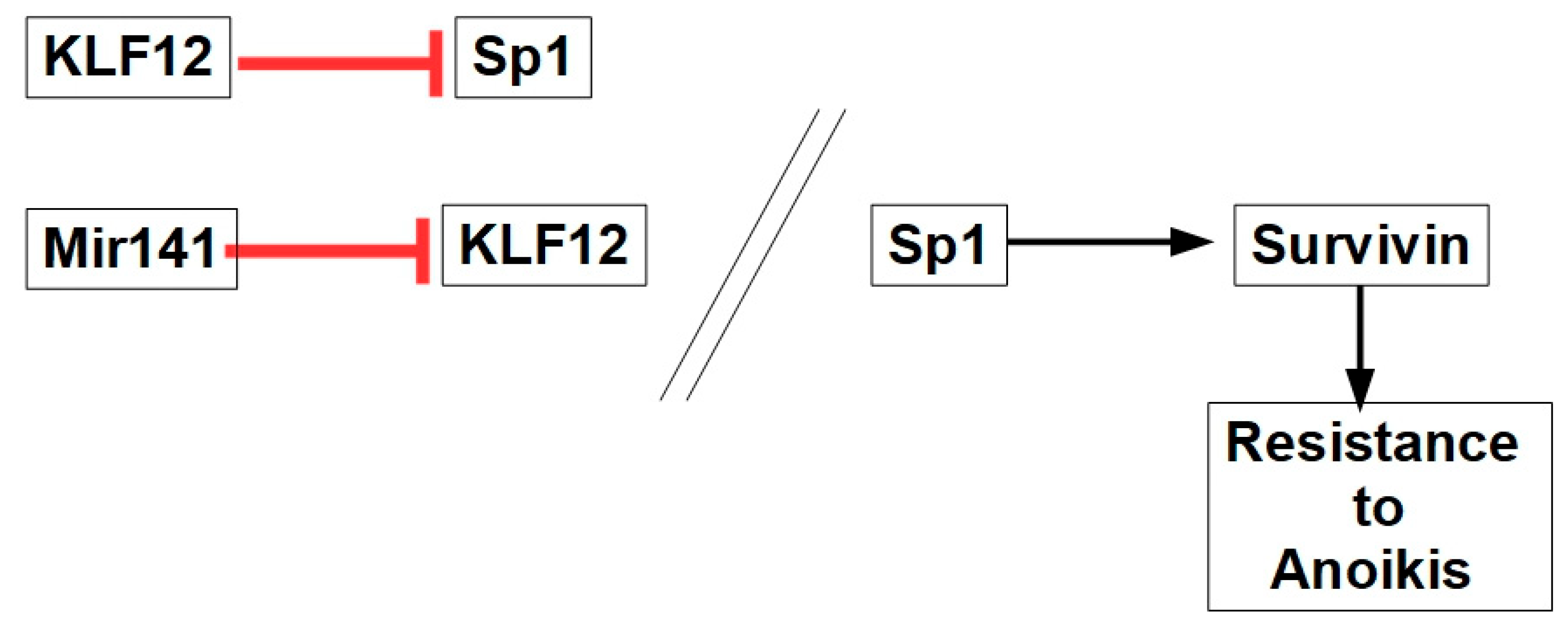
Figure 12.
Factors involved in mediating anoikis resistance in PDAC. STAT3 is considered the main driver of anoikis resistance. However, its effects on AR are mediated through the PI3K/AKT pathway. This is also the case of most of the AR players with the exception of ERK.
Figure 12.
Factors involved in mediating anoikis resistance in PDAC. STAT3 is considered the main driver of anoikis resistance. However, its effects on AR are mediated through the PI3K/AKT pathway. This is also the case of most of the AR players with the exception of ERK.
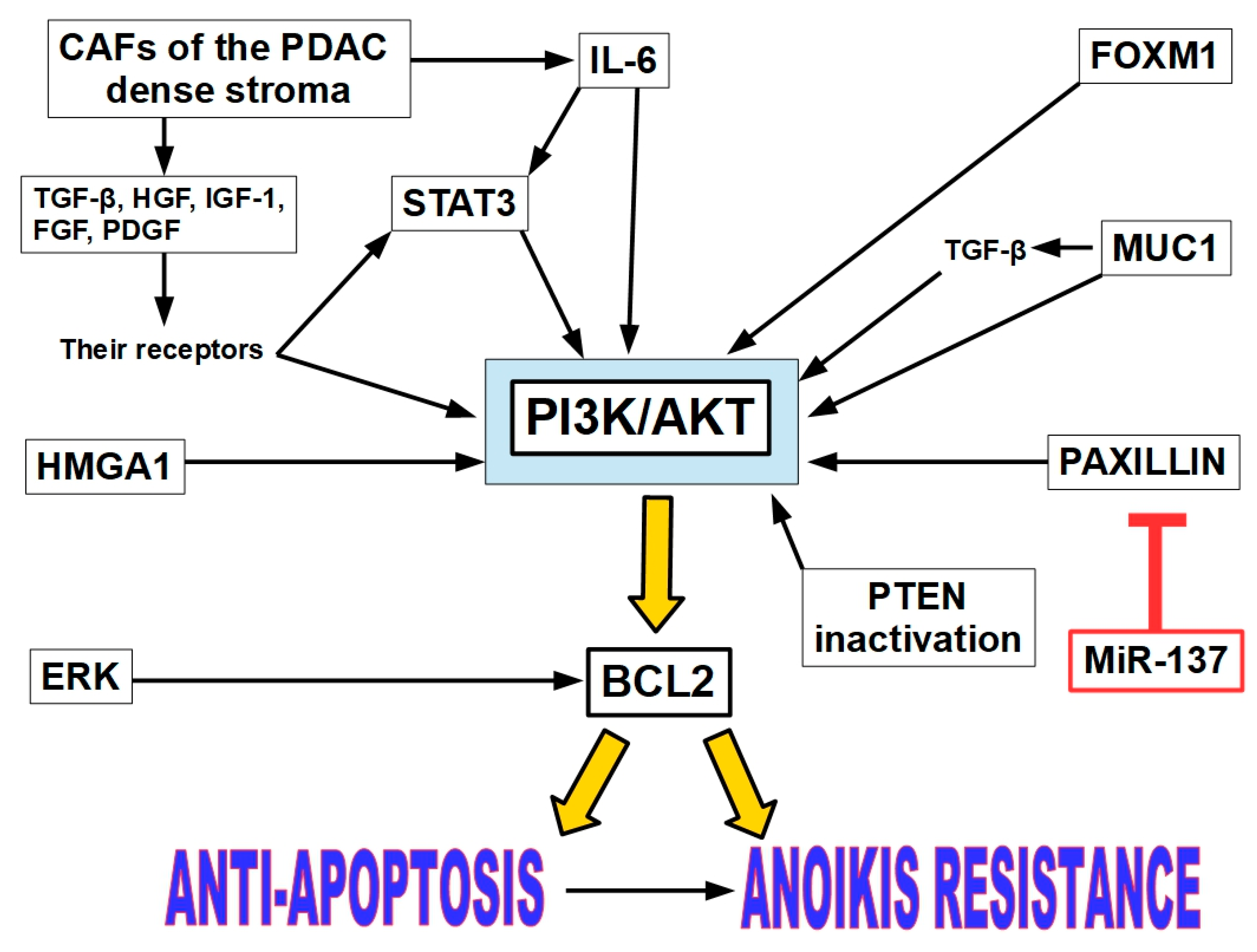
Figure 13.
A snapshot showing the multiple relations between EMT and AR. *[368]; **[369]. Targeting EMT-related proteins restores anoikis sensitivity. ZEB1 suppression restores E-cadherin expression and sensitizes cells to anoikis. Snail and Twist inhibitors reverse EMT, promoting cell-cell adhesion and apoptosis upon detachment.
Figure 13.
A snapshot showing the multiple relations between EMT and AR. *[368]; **[369]. Targeting EMT-related proteins restores anoikis sensitivity. ZEB1 suppression restores E-cadherin expression and sensitizes cells to anoikis. Snail and Twist inhibitors reverse EMT, promoting cell-cell adhesion and apoptosis upon detachment.
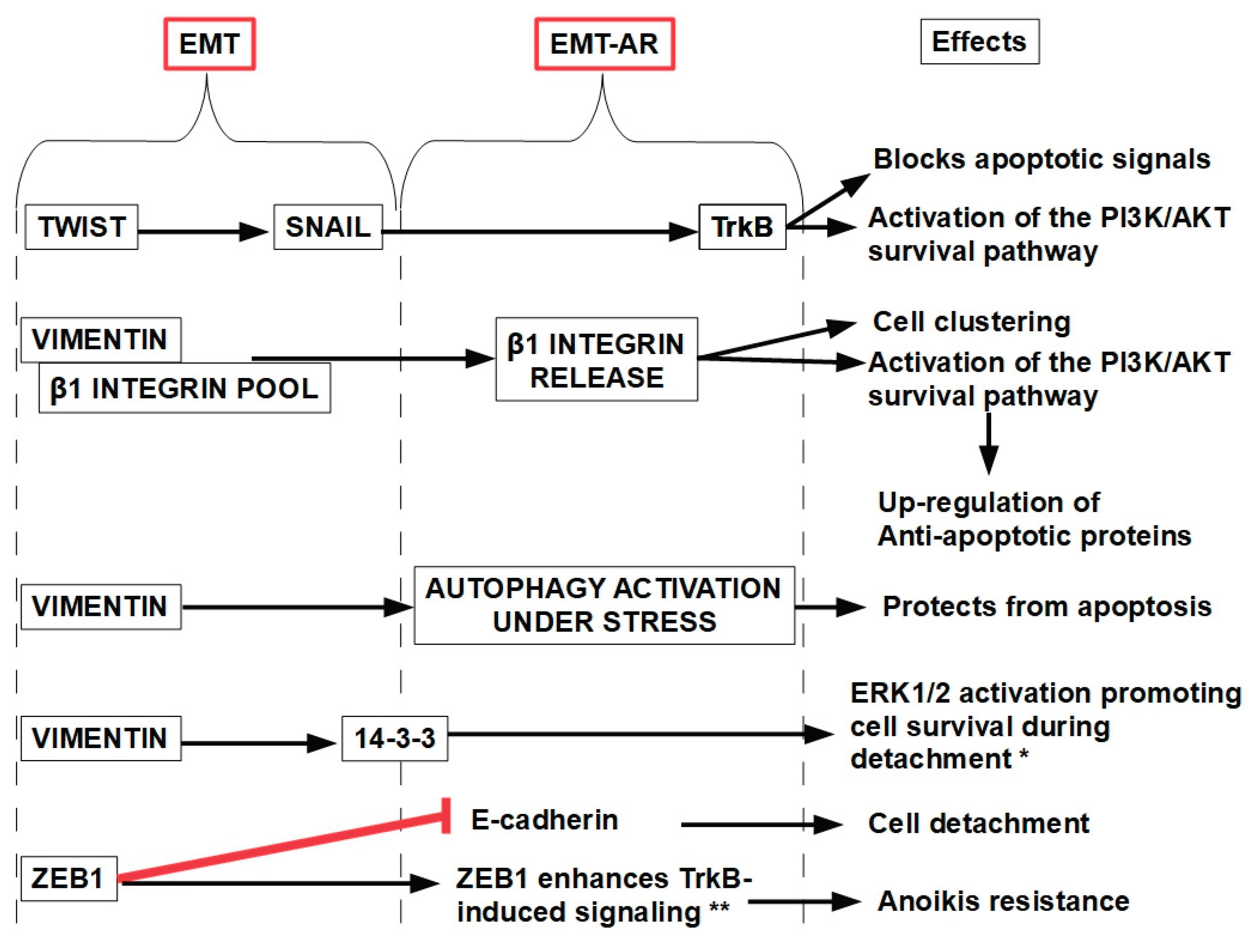

Table 1.
Clinical Trials associating defactinib with other drug(s).
| Pathology | Trial data | Associated drug | Stage/results |
| Diffuse type gastric cancer | NCT06487221 | Enrolling patients | |
| Pancreatic ductal adenocarcinoma | [306] | Pembrolizumab and gemcitabine |
Phase I. 15 patients. 7% partial response, 53% stable disease, 40% disease progression. |
| Refractory ovarian cancer | NCT01778803 [307] |
Paclitaxel | Phase I/Ib. 18 patients. 1 complete response, 1 partial response, 1 stable disease. |
| Kras mutant non-small cell lung cancer | [308] | Defactinib monotherapy | Phase II. 55 patients. One patient with partial response |
| Pleural Mesothelioma |
[309] | Defactinib monotherapy | Phase II. 173 patients treated with 171 controls. There were no overall survival benefits. |
| Low-grade serous ovarian cancer | [310] | Defactinib with avutometinib |
Response of 45% with the association and 10% with avutometinib monotherapy. Tumor shrinkage was observed in the vast majority of patients on the combination and monotherapy arms, 86% and 90% respectively. |
FDA breakthrough therapy designation was given to avutometinib + defactinib in recurrent low-grade serous ovarian cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



