Submitted:
05 December 2025
Posted:
08 December 2025
You are already at the latest version
Abstract
Epilepsy is a neurological disorder characterized by a long-lasting predisposition to recurrently generate unprovoked seizures. Epilepsy affected over 70 million people worldwide, with approximately one third suffering from pharmacoreisitant seizures. Currently, the clinic antiseizure drugs have not the efficacy in alteration of epilepto-genesis. Adenosine, as an endogenous anticonvulsant, inhibits the development of ep-ilepsy via interaction with the molecular epileptogenic network on several levels: i) Activation of A1 receptor inhibits glutamate release via presynaptic inhibition, and hyperpolarize the synaptic potentials in post-synaptic neurons. ii) A2A receptor on as-trocyte interacts with glutamate transporter GLT-1, controlling glial glutamate homeo-stasis. iii) Activation of A3 receptor inhibits GABA transporter type 1 -mediated GABA uptake. iv) Adenosine kinase (ADK) is highlighted as a pathological hallmark of epi-lepsy. The short isoform adenosine kinase (ADK-S) in the cytoplasm of astrocytes, controls the extracellular levels of adenosine and hence adenosine receptor-mediated mechanisms. The long isoform of adenosine kinase (ADK-L) in the nucleus of astrocytes and a subset of neurons, modulates adenosine receptor independent epigenetic mech-anisms. In the present review, we focus on some of the most recent and exciting pro-gresses including mechanisms, evaluation biomarkers for epilepsy as well as therapy aim to modify the progression of epilepsy.
Keywords:
epilepsy
; adenosine
; adenosine receptor
; adenosine kinase
; epileptogenesis
1. Introduction
Epilepsy is a chronic neurological disorder characterized by recurrent abnormal synchronous neuronal discharges, affecting approximately 70 million people worldwide. Approximately 36% of these patients are insensitive to existing antiseizure medications (ASMs), leading to pharmacoresistant epilepsy [1,2]. Epileptic seizures in pharmacoresistant epilepsy cause brain neuronal damage [3,4,5], resulting in intellectual and psychiatric disorders, severely impairing patients' quality of life [6]. Currently, ASMs primarily target neurons, mainly by modulating voltage-gated ion channels, augmenting gamma-aminobutyric acid (GABA)-ergic inhibition or dampening excitatory neurotransmitter glutamate. However, these ASMs fail to decrease significantly the percentage of patients with pharmacoresistant epilepsy or intervene in the disease progression process effectively (i.e., epileptogenesis or disease modification) [7,8,9], and often come with severe systemic adverse effects. The crucial role of astrocyte dysfunction in the initiation and maintenance of epilepsy has been highlighted in recent years. Metabolic pathways offer novel directions for drug development [10]. Particularly, the imbalance of glia-derived adenosine homeostasis is regarded as one of the key factors leading to neuronal hyperexcitability [9,11,12].
Adenosine is regarded as an endogenous anticonvulsant and neuroprotective regulator of the brain. Adenosine is maintained by the enzymes adenosine kinase (ADK) and adenosine deaminase (ADA), in conjunction with ectonucleotidase (CD39, CD73) and equilibrative nucleoside transporters (ENTs). Adenosine homeostasis in the brain is primarily governed by metabolic clearance through ADK levels under physiological conditions, due to the high affinity of ADK for adenosine [13]. Increasing evidence has demonstrated that ADK is a marker of epileptogenesis and a target for its prevention [13,14,15,16,17,18,19,20,21,22,23,24,25]. There are two alternatively spliced forms of ADK: ADK-long (ADK-L), which is located in the nucleus, and ADK-short (ADK-S), which is located in the cytoplasm [26]. The cytoplasmic form of ADK-S regulates both intra- and extracellular adenosine levels; therefore, intracellular ADK-S controls the activation of adenosine receptors, thereby modulating neuronal excitability. In contrast, ADK-L is primarily located in the nucleus and is essential for sustaining methylation processes, such as DNA methylation [27,28,29], which are involved in controlling epileptogenesis[13].
Adenosine activates G protein-coupled receptors with four subtypes: A1 receptors(A1Rs), A2A receptors(A2ARs), A2B receptors (A2BRs) and A3 receptors(A3Rs) [30]. Adenosine in the brain primarily regulates excitatory neurotransmission by activating G protein-coupled inhibitory A1Rs and excitatory A2Ars [30,31,32,33]. Imbalance of adenosine receptor activation (decreased A1R signaling and/or increased A2AR signaling) can aggravate the development and progression of epilepsy. A3Rs are regarded as a promising target for the development of antiseizure medications via A3Rs-mediated action upon GABA transporter type 1(GAT-1) and GABAergic currents [34]. Currently, little is known about the contribution of A2BRs in epilepsy. Extensive evidence from experimental epilepsy models and research on human specimens demonstrates that aberrant adenosine signaling is a common pathologic feature in epilepsy [15,16,17,18,19,20,21,35]. Therefore, the adenosine system might therefore be a therapeutic target for the treatment of epilepsy, including adenosine receptor agonists, adenosine kinase inhibitors, and gene therapy [23,24,36,37,38,39,40,41,42,43,44].
The present review is intended not to offer a comprehensive overview of all potential mechanisms, but to focus on the role of adenosine receptors (A1Rs, A2ARs, A3Rs), adenosine kinase in epilepsy and propose perspectives on potential therapeutic targets.
2. Adenosine A1 Receptors (A1Rs) in Epilepsy
A1Rs belong to the G protein-coupled receptor (GPCR) family and mediates signal transduction through Gi/o proteins, leading to inhibition of cyclic adenosine monophosphate synthesis [45]. A1Rs are enriched in the central nervous system, where they are expressed in the cerebral cortex, hippocampus, cerebellum, thalamus, and brainstem, present at both pre- and postsynaptic sites in neurons, and also found in astrocytes, microglia, and oligodendrocytes. Adenosine has been proved to be a major endogenous anticonvulsant acting via A1Rs. In the brain, adenosine modulates neuronal activity via: i) decreasing presynaptic release of various neurotransmitters, and the most dramatic inhibitory actions are on the glutamatergic system through presynaptic A1Rs [46]; ii) activating potassium ion channels, leading to hyperpolarization of postsynaptic neurons and promoting NMDA receptor inhibition through postsynaptic A1Rs [47]. Genetic deletion of A1Rs induces spontaneous electrographic seizures [15,16,37] and leads to lethal status epilepticus in mice models of epilepsy [48]. Downregulation of A1Rs in human temporal lobe epilepsy may contribute to the human epileptic condition [21,49]. Pharmacological block of adenosine A1Rs could transiently provoke seizures in epileptic animals treated with focal adenosine augmentation [36]. Deep brain stimulation (DBS) markedly attenuated spontaneous recurrent seizures (SRSs) via an increase in extracellular adenosine [38], while pharmacological block of adenosine A1Rs reversed the effect of DBS on interictal epileptic discharges [23]. Pharmacological activation of A1R has been proved to mediate a strong anticonvulsant action in human neocortical slices from patients with temporal lobe epilepsy [50], as well as prevent the spread of seizures in mouse epileptic models. Of importance, in patients after a severe traumatic brain injury, variants in the A1Rs gene were associated with the development of posttraumatic seizures, which indicated that deficiency in A1R signaling might be associated with posttraumatic epileptogenesis [51]. The development of A1 receptor-targeting drugs faces challenges such as cardiovascular side effects (bradycardia, atrioventricular block, diminished atrial contractility), sedation as well as insufficient receptor selectivity [52].
3. A2A Receptors (A2ARs) in Epilepsy
A2ARs are coupled to stimulatory Gs proteins, facilitating cAMP production [45]. A2ARs are predominantly localized in the striatum, olfactory tubercle, and to a lesser extent in the cortex and hippocampus, with expression in neurons, astrocytes, microglia, and oligodendrocytes. A2ARs are pivotal in synaptic plasticity, counteract the A1Rs-mediated inhibition of synaptic transmission, increase expression in neurons and glial cells upon injury induced by seizures [20,35], and act as a STOP signal of the immune-inflammatory system [53]. Adenosine enhances seizure thresholds and discontinues seizures via activation of inhibitory A1Rs, while stimulatory A2ARs may promote synaptic transmission within a globally inhibited network. Therefore, alterations in crosstalk between A1Rs and A2ARs signaling might be linked to seizure generation: prolonged activation of high-affinity A1Rs can lead to their internalization and a consequent increase in A2ARs density [54]. A distinct synaptic adenosine pool is largely under the control of the adenosine-producing enzyme CD73 and allows the control of A2ARs activation at the single-synapse level. Extracellular CD73-mediated formation of adenosine provides an important source for the activation of A2ARs. The concomitant increase in CD73 and A2ARs immunoreactivity has been demonstrated in specimens from patients with epilepsy associated with Rasmussen encephalitis [35], focal cortical dysplasia [20], as well as patients with temporal lobe epilepsy [55].
Astroglial glutamate transporter-1 (GLT-1), the transporter responsible for the majority of glutamate clearance in the central nervous system, is essential for preventing seizures [56]. GLT-1 is under the control of A2ARs, which regulate astrocytic glutamate clearance through their physical associations with GLT-1[57]. Increased A2ARs expression coupled with diminished GLT-1 density in reactive astrocytes within the epileptic focus in patients with Rasmussen encephalitis [35] and focal cortical dysplasia [20] further supported A2ARs-mediated modulation of glutamate uptake. Currently, most research using both selective antagonists and genetic manipulations of A2ARs has consistently substantiated proconvulsant effects of endogenously activated A2ARs. Both selective antagonists and genetic ablation of A2ARs markedly reduced both the incidence and severity of seizures, supporting a proconvulsant role of A2ARs via modulation of excitatory neurotransmission [58,59]. A2ARs expression was found to be enhanced in epileptic WAG/Rij rats, a genetic model of human absence epilepsy [60]. Of importance, the dual modulation of A2ARs and GLT-1 represents a synergistic approach to attenuate brain hyperexcitability, with therapeutic potential for mitigating TBI-related persistent hyperexcitability. Therefore, targeting A2ARs and glial glutamate transporter GLT-1 synergistically might be a novel disease-modifying approach [61]. A2ARs, the primary modulators of excitatory transmission and neuronal excitability, exhibit increased expression in epilepsy. However, the causality of these changes—whether they drive epilepsy or are secondary adaptations—remains unresolved [62].
4. A3 Receptors (A3ARs) in Epilepsy
Similar to A1Rs, A3ARs belong to the G protein-coupled receptor (GPCR) family and mediates signal transduction through Gi/o proteins, leading to inhibition of cyclic adenosine monophosphate synthesis. A3Rs are widely expressed at hippocampal synapses, both pre- and postsynaptically, with expression in neurons [63], astrocytes [64], and microglia [65]. The role of the A3 receptor in epilepsy, through the regulation of the GAT-1-mediated GABA transport mechanism, provides new targets and strategies for modulating the balance of central inhibitory neurotransmitters. Its activation inhibits GAT-1 function, increases GABA concentration in the synaptic cleft, thereby enhancing inhibitory signals, reducing neuronal excitability, and alleviating the frequency and intensity of epileptic seizures [34]. Compared to A1Rs, A3Rs are a more ideal target for epilepsy therapy. i) without side effects on cardiac function: as a non-ubiquitous adenosine receptor, A3Rs are a putative target for novel antiseizure medications. Adenosine has been proved to be a major endogenous anticonvulsant acting via A1Rs. However, the widespread distribution of A1Rs throughout the body raises the concern that A1Rs agonists may lead to bradycardia. ii) minimal sedative effects: A3Rs antagonist has selective actions in epileptic tissue (rodent and human), sparing non-epileptic tissue [66]. A3Rs antagonist selectively affects epileptic tissue while preserving excitatory transmission in healthy tissue [34]. iii) upregulation of A3Rs versus downregulation of A1Rs in epileptic focus: A decrease in A1Rs [21,67] while an increase in A3Rs [34] in epileptic tissue have been demonstrated in patients and the majority of studies in animal models, which indicated that activation of A3Rs might have more antiseizure potential. A3Rs agonists, as a potentially safe new class of antiseizure medications, are still in the early stages of development from bench to bed for epilepsy, with key issues including blood-brain barrier permeability, long-term safety, and efficacy stability requiring further attention.
5. ADK-S in Epilepsy: Modulating Extracellular/Synaptic Adenosine Levels and Inhibiting Seizures in an Adenosine Receptor-Dependent Way
On the one hand, ADK-S modulates extracellular/synaptic adenosine. ADK-S is primarily located in the cytoplasm, regulating extracellular/synaptic adenosine levels, thereby modulating the activation of adenosine receptors [68]. The primary source of synaptic adenosine is ATP, which is released through vesicular transport [69] or via hemichannels in astrocytes [70]. After entering the synaptic cleft, ATP is degraded into adenosine by a series of extracellular nucleotidases (CD39 and CD73) [71]. Astrocytic membranes contain equilibrative nucleoside transporters (ENTs), which rapidly balance extracellular and cytoplasmic adenosine levels [72]. Intracellular adenosine levels are largely controlled by ADK-S, which phosphorylates adenosine into AMP. Due to the lack of a classical transporter-regulated reuptake system for adenosine and its high affinity for adenosine, cytoplasmic astrocytic ADK-S is considered to be a metabolic reuptake system for adenosine, causing an inward net flux through ENTs and maintaining a low extracellular adenosine level [13]. Extracellular adenosine plays an important role in epilepsy by modulating neuronal excitability via G protein-coupled adenosine receptors. On the other hand, ADK-S modulates cytoplasmic adenosine concentrations and influences the transmethylation process. Adenosine generated through the transmethylation pathway may represent a significant source of intracellular adenosine, in addition to ATP catabolism via AMP [73]. DNA methylation is catalyzed by DNA methyltransferases (DNMTs), which transfer a methyl group from S-adenosylmethionine (SAM) to the fifth carbon atom of cytosine, forming 5-methylcytosine (5-mC). Concurrently, SAM is converted to S-adenosylhomocysteine (SAH), which is then hydrolyzed by SAH hydrolase (SAHH) to produce adenosine and homocysteine (HCY) (Figure 1). In addition, the adenosine produced via the transmethylation pathway is predominantly salvaged by ADK-S within the cell [74]. Of note, the thermodynamic equilibrium of the SAH hydrolysis reaction favors SAH formation. Consequently, this reaction—which is essential for regulating methyl group flux through the transmethylation pathway—can only proceed when adenosine levels are kept low [75] and adenosine is effectively removed by ADK [76]. DNA methylation is one of the most important types of epigenetic modification. Studies on brain tissue from patients with temporal lobe epilepsy (TLE) and TLE animal models suggest that DNA methylation is involved in the pathophysiological process of TLE and plays a critical role in the pathogenesis of epilepsy [40,77,78,79,80]. Whether adenosine levels and ADK can regulate the activity of these cytosolic methyltransferases remains unclear and warrants further investigation [29].
Adenosine is an endogenous neuromodulator released during seizures and implicated in seizure arrest, postictal refractoriness, and suppression of epileptogenesis[81]. Therefore, ADK-S inhibition is a potential strategy to boost extracellular adenosine for epilepsy control. This concept is supported by multiple lines of evidence: i) Animal Models: ADK-overexpressing transgenic mice show increased sensitivity to brain injury and seizures, whereas mice with reduced forebrain ADK are resistant to pharmacologically induced epileptogenesis[16]; ii) Viral Induction: Wild-type mice with virus-induced ADK overexpression in hippocampal CA3 astrocytes develop electrographic seizures[82]; iii) Genetic Intervention: Intrahippocampal implantation of stem cells with biallelic ADK gene disruption (Adk−/−) prevents kainate-induced epileptogenesis in a limbic mouse model[16]; iv) Human Studies: ADK overexpression is observed in human temporal lobe epilepsy tissue[5,21], focal cortical dysplasia tissue[18,20], Rasmussen encephalitis tissue[17,19], Sturge-Weber Syndrome tissue[83] and glial tumor tissue as well as peritumoral regions infiltrated by glia[84], suggesting that reduced adenosine may contribute to epileptogenesis in these conditions; Additionally, basal adenosine levels are lower in the epileptic human hippocampus compared to controls[81], further supporting ADK's role in epileptogenesis; v) Dietary Therapy: A ketogenic diet suppresses seizures in ADK-overexpressing transgenic mice and A1R+/− mice by inhibiting ADK expression, thereby increasing ambient adenosine and activating A1Rs[37]. The suppression of seizures induced by the diet was reversed by either glucose administration (metabolic reversal) or an A1Rs antagonist (pharmacological reversal). Notably, in A1Rs-deficient mice, the diet failed to influence spontaneous seizures. Together, these genetic and pharmacological manipulations of the adenosine system provide strong evidence that the diet’s anticonvulsant effect is specifically mediated by A1Rs activation, aligning with the proposed role of adenosine signaling in epilepsy control; vi) Neuromodulation Therapy: DBS of the anterior nucleus of the thalamus can reduce the frequency of spontaneous recurrent seizures (SRS) and attenuate seizure progression in epileptic rats via inhibition of ADK [23,85]. DBS significantly elevated hippocampal adenosine levels via in vivo measurements of adenosine using fiber photometry [38] or microdialysis [86], which might be derived from the downregulation of ADK. A1Rs antagonists reversed the effect of DBS on interictal epileptic discharges in epileptic rats [23]. In vitro experiments demonstrated that the DBS-induced reduction in neuronal excitability was completely blocked in animals pretreated with A1Rs antagonists, whereas it was markedly enhanced by A1Rs agonists. The findings indicate that DBS can reduce SRS in epileptic rats via inhibition of ADK-S, the subsequent increase of extracellular/synaptic adenosine levels and activation of A1Rs, which are consistent with the proposed role of adenosine signaling in epilepsy control. Therefore, ADK-S plays a critical role in maintaining adenosine receptor-mediated signal transduction, and changes in its activity directly affect the availability of extracellular/synaptic adenosine and the extent of adenosine receptor activation. The characteristic that inhibition of ADK-S can effectively elevate extracellular/synaptic adenosine levels makes it a potential therapeutic target for seizure suppression.
6. ADK-L in Epilepsy: Modulating Adenosine Receptor-Independent Epigenetic Mechanisms
Distinct from its cytoplasmic ADK-S counterpart, ADK-L is specifically localized in the nucleus [26]. Within the nucleus, ADK-L regulates adenosine metabolism to control DNA methylation through transmethylation reactions driven by DNA methyltransferases (DNMTs) (Figure 1). The dynamic regulation of DNA methylation is directly mediated by the adenosine-responsive transmethylation pathway. Hippocampal infusion of adenosine, the end product in the transmethylation pathway, induces global DNA hypomethylation, while infusion of SAM, the primary methyl donor for transmethylation reactions, causes hypermethylation [40]. Pharmacological ADK inhibition (5-iodotubercidin, 5-ITU) or genetic ADK reduction leads to brain DNA hypomethylation. Notably, 5-ITU-dependent hypomethylation persisted in mice lacking the adenosine A1Rs, demonstrating that receptor activation-the primary mechanism underlying the anticonvulsant effects of adenosine [48]-is dispensable for triggering ADO-induced hypomethylation. Of importance, increased levels of both ADK-L and ADK-S isoforms contribute to elevated global DNA methylation (400% versus 50%), with the nuclear isoform ADK-L playing a more dominant role in regulating DNA methylation status [40]. These findings establish ADK-L as an epigenetic regulator independent of adenosine receptor signaling. On the one hand, extensive evidence suggests that upregulation of DNA methylating enzymes and DNA hypermethylation play a role in the pathogenesis of human and experimental epilepsy, especially as DNA methylation makes a contribution to epileptogenesis [77,78,80,87,88,89]. On the other hand, ADK-L plays a chief role in regulating DNA methylation status [40,90], and the antiepileptogenic effects of adenosine are mediated by its ability to regulate epigenetic mechanisms. Therefore, blockade of ADK-L can modify the disease course of epilepsy or prevent epileptogenesis through an epigenetic mechanism. Recently, novel ADK-L inhibitors developed through structure-based approaches (MRS4380/MRS4203) offer enhanced potency over traditional nucleoside-based inhibitors [91]. These compounds demonstrate concentration-dependent DNA hypomethylation effects in cancer cells [90], highlighting their potential as epigenetic modulators. Future development requires optimizing nuclear entry, ADK-L selectivity, and the ability to modulate DNA methylation, as well as achieving sustained antiepileptic effects through refined structure-based design.
Clinical therapies for pharmacoresistant epilepsy - such as ketogenic diet, DBS, and vagus nerve stimulation (VNS) - highlight the clinical utility of ADK-L modulation for therapeutic benefits. i) Dietary Therapy: Epilepsy rodent models reveal persistent disease-modifying effects of ketogenic diet therapy, driven by epigenetic mechanisms [77,92,93]. Likewise, a transient use of ketogenic diet therapy in children with epilepsy can yield lasting seizure freedom after discontinuation of the diet [94,95]. Furthermore, ketogenic diet therapy increases adenosine levels [37,92] while decreasing adenosine kinase expression [37]. These findings suggest that the sustained therapeutic benefits of ketogenic diet therapy after discontinuation may be attributed to epigenetic changes triggered by elevated adenosine and ADK-L modulation. ii) DBS therapy: DBS disrupts the kindling process, with seizure suppression persisting even one week after stimulation cessation. These results indicate that ANT-DBS may offer therapeutic advantages in slowing epilepsy progression, disease-modifying benefits or potential antiepileptogenic effect [85]. In addition, DBS therapy increases adenosine levels [38,86] while decreasing adenosine kinase expression [37]. Elevated ADK-L expression during epileptogenesis promotes DNA hypermethylation, a mechanism independently demonstrated to contribute to epileptogenesis. These results demonstrate that the therapeutic benefits of DBS for disease modification are due to elevated adenosine and ADK-L modulation. iii) VNS therapy: VNS is applied in pharmacoresistant epilepsy cases where resective surgery is not feasible. It results in a more than 50% reduction in seizure frequency in 50%–60% of patients, with 6%–11% attaining complete seizure control [96]. Optimized stimulation parameters (higher duty cycle and output current) were associated with better seizure control and substantial subjective improvements in quality of life [97]. Experimental studies indicate that VNS has antiepileptogenic properties [98]. Evidence from diverse studies suggests that neurostimulation activates adenosine release [38,86,99]. Genetic variations in the Adk gene have been identified as a reliable biomarker for predicting the effectiveness of VNS treatment. All patients with homozygosity for the minor allele in specific Adk single nucleotide polymorphisms (SNPs)—specifically homozygous rs11001109 (AA) and rs946185 (AA) —achieved > 50% seizure reduction, and 40% of these patients attained complete seizure freedom [22]. In addition, VNS inhibits DNA methylation in a rat model of pilocarpine-induced temporal lobe epilepsy [80]. These discoveries underscore a pivotal role of ADK-L in VNS therapy and reinforce the idea that the enzyme plays an intricate role in the pathogenesis of epilepsy.
7. Future Perspective
The adenosine system, as a core component of the endogenous antiepileptic network, regulates neuronal excitability through various receptors and metabolic pathways. Via interacting with the molecular epileptogenic network on several levels, adenosine has been shown inhibition of the development of epilepsy. From molecular mechanisms to models and clinical translation, substantial evidence supports the critical value of adenosine-related targets in epilepsy treatment. Intervention strategies such as global ADK inhibitors, A1Rs agonists and A2ARs antagonists demonstrate promising antiepileptic potential. However, overcoming peripheral side effects and achieving precise intervention through targeted delivery remain major challenges in clinical translation.
A3ARs agonists represent a promising novel class of antiseizure medications with an improved safety profile, currently undergoing early-stage development from preclinical research to clinical application for epilepsy treatment. However, their therapeutic potential hinges on addressing critical challenges, including optimizing blood-brain barrier permeability to ensure sufficient central nervous system exposure, establishing long-term safety profiles through comprehensive preclinical and clinical evaluation, and ensuring sustained efficacy stability during chronic administration.
Novel inhibitors with preferential inhibition of ADK-L over ADK-S are particularly promising. The enduring disease-modifying effects of adenosine are rooted in an epigenetic and receptor-independent mechanism regulated by ADK-L, which orchestrates DNA methylation to sustain long-term therapeutic outcomes, and avoids the cardiovascular and sedative side effects of adenosine which arise from excessive activation of adenosine receptors (linked to ADK-S).
Author Contributions
Writing—original draft preparation, X.Z. and T.L.; writing—review and editing, J.D. and T.L.; visualization, T.L. and J.D.; supervision, Z.X., T.L. and J.D.; funding acquisition, T.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| GAT-1 | GABA transporter type 1 |
| ADK | adenosine kinase |
| ADK-S | short isoform of adenosine kinase |
| ADK-L | long isoform of adenosine kinase |
| ASMs | antiseizure medications |
| GABA | gamma-aminobutyric acid |
| ADA | adenosine deaminase |
| ENTs | equilibrative nucleoside transporters |
| A1Rs | A1 receptors |
| A2ARs | A2A receptors |
| A2BRs | A2B receptors |
| A3Rs | A3 receptors |
| GPCR | G protein-coupled receptor |
| DBS | deep brain stimulation |
| SRSs | spontaneous recurrent seizures |
| GLT-1 | glutamate transporter-1 |
| SAM | S-adenosylmethionine |
| 5-mC | 5-methylcytosine |
| SAH | S-adenosylhomocysteine |
| SAHH | SAH hydrolase |
| HCY | homocysteine |
| TLE | temporal lobe epilepsy |
| DNMTs | DNA methyltransferases |
| 5-ITU | 5-iodotubercidin |
| VNS | vagus nerve stimulation |
| SNPs | single nucleotide polymorphisms |
| h-ch | hemichannels |
References
- Kwan, P.; Schachter, S.C.; Brodie, M.J. Drug-resistant epilepsy. N Engl J Med 2011, 365, 919–926. [Google Scholar] [PubMed]
- Chen, Z.; Brodie, M.J.; Liew, D.; Kwan, P. Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol 2018, 75, 279–286. [Google Scholar]
- Li, T.; Fan, Y.; Luo, Y.; Xiao, B.; Lu, C. In vivo delivery of a XIAP (BIR3-RING) fusion protein containing the protein transduction domain protects against neuronal death induced by seizures. Exp Neurol 2006, 197, 301–308. [Google Scholar]
- Li, T.; Lu, C.; Xia, Z.; Xiao, B.; Luo, Y. Inhibition of caspase-8 attenuates neuronal death induced by limbic seizures in a cytochrome c-dependent and Smac/DIABLO-independent way. Brain Res 2006, 1098, 204–211. [Google Scholar]
- Aronica, E.; Zurolo, E.; Iyer, A.; de Groot, M.; Anink, J.; Carbonell, C.; van Vliet, E.A.; Baayen, J.C.; Boison, D.; Gorter, J.A. Upregulation of adenosine kinase in astrocytes in experimental and human temporal lobe epilepsy. Epilepsia 2011, 52, 1645–1655. [Google Scholar] [CrossRef]
- Li, T. Epilepsy and Associated Comorbidities. In Neuropsychiatry (Lond); 2017. [Google Scholar]
- Klein, P.; Kaminski, R.M.; Koepp, M.; Loscher, W. New epilepsy therapies in development. Nat Rev Drug Discov 2024, 23, 682–708. [Google Scholar] [PubMed]
- Terman, S.W.; Kirkpatrick, L.; Akiyama, L.F.; Baajour, W.; Atilgan, D.; Dorotan, M.K.C.; Choi, H.W.; French, J.A. Current state of the epilepsy drug and device pipeline. Epilepsia 2024, 65, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Ravizza, T.; Bedner, P.; Aronica, E.; Steinhauser, C.; Boison, D. Astrocytes in the initiation and progression of epilepsy. Nat Rev Neurol 2022, 18, 707–722. [Google Scholar]
- Xiong, Z.; Gao, R.; Wang, R.; Li, T. Proteome-Wide Mendelian Randomization Reveals Novel Protein Targets for Epilepsy. Curr Neuropharmacol 2025. [Google Scholar]
- Boison, D. Adenosine dysfunction in epilepsy. Glia 2012, 60, 1234–1243. [Google Scholar]
- Boison, D.; Steinhauser, C. Epilepsy and astrocyte energy metabolism. Glia 2018, 66, 1235–1243. [Google Scholar] [CrossRef]
- Boison, D. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev 2013, 65, 906–943. [Google Scholar]
- Boison, D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol 2008, 84, 249–262. [Google Scholar] [PubMed]
- Li, T.; Lytle, N.; Lan, J.Q.; Sandau, U.S.; Boison, D. Local disruption of glial adenosine homeostasis in mice associates with focal electrographic seizures: a first step in epileptogenesis? Glia 2012, 60, 83–95. [Google Scholar] [PubMed]
- Li, T.; Ren, G.; Lusardi, T.; Wilz, A.; Lan, J.Q.; Iwasato, T.; Itohara, S.; Simon, R.P.; Boison, D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest 2008, 118, 571–582. [Google Scholar]
- Luan, G.; Gao, Q.; Guan, Y.; Zhai, F.; Zhou, J.; Liu, C.; Chen, Y.; Yao, K.; Qi, X.; Li, T. Upregulation of adenosine kinase in Rasmussen encephalitis. J Neuropathol Exp Neurol 2013, 72, 1000–1008. [Google Scholar] [PubMed]
- Luan, G.; Gao, Q.; Zhai, F.; Zhou, J.; Liu, C.; Chen, Y.; Li, T. Adenosine kinase expression in cortical dysplasia with balloon cells: analysis of developmental lineage of cell types. J Neuropathol Exp Neurol 2015, 74, 132–147. [Google Scholar]
- Luan, G.; Wang, X.; Gao, Q.; Guan, Y.; Wang, J.; Deng, J.; Zhai, F.; Chen, Y.; Li, T. Upregulation of Neuronal Adenosine A1 Receptor in Human Rasmussen Encephalitis. J Neuropathol Exp Neurol 2017, 76, 720–731. [Google Scholar]
- Guo, M.; Zhang, J.; Wang, J.; Wang, X.; Gao, Q.; Tang, C.; Deng, J.; Xiong, Z.; Kong, X.; Guan, Y.; Zhou, J.; Boison, D.; Luan, G.; Li, T. Aberrant adenosine signaling in patients with focal cortical dysplasia. Mol Neurobiol 2023, 60, 4396–4417. [Google Scholar]
- Guo, M.; Wang, J.; Xiong, Z.; Wang, X.; Yang, Y.; Zhang, Y.; Tang, C.; Zhang, J.; Guan, Y.; Chen, F.; Yao, K.; Teng, P.; Zhou, J.; Zhai, F.; Boison, D.; Luan, G.; Li, T. Ectopic expression of neuronal adenosine kinase, a biomarker in mesial temporal lobe epilepsy without hippocampal sclerosis. Neuropathol Appl Neurobiol 2023, 49, e12926. [Google Scholar]
- Zhang, Y.; Wang, X.; Tang, C.; Guan, Y.; Chen, F.; Gao, Q.; Wang, J.; Zhou, J.; Zhai, F.; Boison, D.; Luan, G.; Li, T. Genetic variations of adenosine kinase as predictable biomarkers of efficacy of vagus nerve stimulation in patients with pharmacoresistant epilepsy. J Neurosurg 2022, 136, 726–735. [Google Scholar] [PubMed]
- Xie, P.; Liu, S.; Huang, Q.; Xiong, Z.; Deng, J.; Tang, C.; Xu, K.; Zhang, B.; He, B.; Wang, X.; Liu, Z.; Wang, J.; Zhou, J.; Guan, Y.; Luan, G.; Li, T.; Zhai, F. Deep brain stimulation suppresses epileptic seizures in rats via inhibition of adenosine kinase and activation of adenosine A1 receptors. CNS Neurosci Ther 2023, 29, 2597–2607. [Google Scholar]
- Guo, M.; Li, T. Adenosine Dysfunction in Epilepsy and Associated Comorbidities. Curr Drug Targets 2022, 23, 344–357. [Google Scholar]
- Chen, F.; He, X.; Li, T. Adenosine Kinase, a Common Pathologic Biomarker for Human Pharmacoresistant Epilepsy. Neuropsychiatry 2018, 08. [Google Scholar]
- Cui, X.A.; Singh, B.; Park, J.; Gupta, R.S. Subcellular localization of adenosine kinase in mammalian cells: The long isoform of AdK is localized in the nucleus. Biochem Biophys Res Commun 2009, 388, 46–50. [Google Scholar]
- Peter-Okaka, U.; Boison, D. Adenosine Kinase: An Epigenetic Modulator and Drug Target. J Inherit Metab Dis 2025, 48, e70033. [Google Scholar]
- Murugan, M.; Fedele, D.; Millner, D.; Alharfoush, E.; Vegunta, G.; Boison, D. Adenosine kinase: An epigenetic modulator in development and disease. Neurochem Int 2021, 147, 105054. [Google Scholar] [PubMed]
- Murugan, M.; Boison, D. Adenosine Kinase: Cytoplasmic and Nuclear Isoforms. In Jasper's Basic Mechanisms of the Epilepsies, 5th ed.; Oxford University Press: New York, 2024; p. 555-568 555-68. [Google Scholar]
- Boison, D. Adenosinergic signaling in epilepsy. Neuropharmacology 2016, 104, 131–139. [Google Scholar] [CrossRef]
- Boison, D.; Chen, J.F.; Fredholm, B.B. Adenosine signaling and function in glial cells. Cell Death Differ 2010, 17, 1071–1082. [Google Scholar] [PubMed]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and brain function. Int Rev Neurobiol 2005, 63, 191–270. [Google Scholar]
- Matos, M.; Augusto, E.; Agostinho, P.; Cunha, R.A.; Chen, J.F. Antagonistic interaction between adenosine A2A receptors and Na+/K+-ATPase-alpha2 controlling glutamate uptake in astrocytes. J Neurosci 2013, 33, 18492–18502. [Google Scholar] [CrossRef]
- Ghosh, A.; Ribeiro-Rodrigues, L.; Ruffolo, G.; Alfano, V.; Domingos, C.; Rei, N.; Tosh, D.K.; Rombo, D.M.; Morais, T.P.; Valente, C.A.; Xapelli, S.; Bordadagua, B.; Rainha-Campos, A.; Bentes, C.; Aronica, E.; Diogenes, M.J.; Vaz, S.H.; Ribeiro, J.A.; Palma, E.; Jacobson, K.A.; Sebastiao, A.M. Selective modulation of epileptic tissue by an adenosine A(3) receptor-activating drug. Br J Pharmacol 2024, 181, 5041–5061. [Google Scholar] [CrossRef]
- He, X.; Chen, F.; Zhang, Y.; Gao, Q.; Guan, Y.; Wang, J.; Zhou, J.; Zhai, F.; Boison, D.; Luan, G.; Li, T. Upregulation of adenosine A2A receptor and downregulation of GLT1 is associated with neuronal cell death in Rasmussen's encephalitis. Brain Pathol 2019. [Google Scholar] [CrossRef]
- Li, T.; Steinbeck, J.A.; Lusardi, T.; Koch, P.; Lan, J.Q.; Wilz, A.; Segschneider, M.; Simon, R.P.; Brustle, O.; Boison, D. Suppression of kindling epileptogenesis by adenosine releasing stem cell-derived brain implants. Brain 2007, 130 Pt 5, 1276–1288. [Google Scholar] [CrossRef]
- Masino, S.A.; Li, T.; Theofilas, P.; Sandau, U.S.; Ruskin, D.N.; Fredholm, B.B.; Geiger, J.D.; Aronica, E.; Boison, D. A ketogenic diet suppresses seizures in mice through adenosine A(1) receptors. J Clin Invest 2011, 121, 2679–2683. [Google Scholar] [CrossRef]
- Xiong, Z.; Deng, J.; Xie, P.; Tang, C.; Wang, J.; Deng, Q.; Yang, Y.; Zhang, J.; Guo, M.; Wang, X.; Guan, Y.; Luan, G.; Zhou, J.; Li, T. Deep Brain Stimulation Inhibits Epileptic Seizures via Increase of Adenosine Release and Inhibition of ENT1, CD39, and CD73 Expression. Mol Neurobiol 2025, 62, 1800–1812. [Google Scholar] [CrossRef] [PubMed]
- Wilz, A.; Pritchard, E.M.; Li, T.; Lan, J.Q.; Kaplan, D.L.; Boison, D. Silk polymer-based adenosine release: therapeutic potential for epilepsy. Biomaterials 2008, 29, 3609–3616. [Google Scholar] [CrossRef] [PubMed]
- Williams-Karnesky, R.L.; Sandau, U.S.; Lusardi, T.A.; Lytle, N.K.; Farrell, J.M.; Pritchard, E.M.; Kaplan, D.L.; Boison, D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest 2013, 123, 3552–3563. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, T. Role of Adenosine Kinase Inhibitor in Adenosine Augmentation Therapy for Epilepsy: A potential novel drug for epilepsy. Curr Drug Targets 2019. [Google Scholar]
- Guo, M.; Xie, P.; Liu, S.; Luan, G.; Li, T. Epilepsy and Autism Spectrum Disorder (ASD): The Underlying Mechanisms and Therapy Targets Related to Adenosine. Curr Neuropharmacol 2023, 21, 54–66. [Google Scholar]
- Chen, F.; He, X.; Luan, G.; Li, T. Role of DNA Methylation and Adenosine in Ketogenic Diet for Pharmacoresistant Epilepsy: Focus on Epileptogenesis and Associated Comorbidities. Front Neurol 2019, 10, 119. [Google Scholar] [PubMed]
- Boison, D. Adenosine augmentation therapies (AATs) for epilepsy: prospect of cell and gene therapies. Epilepsy Res 2009, 85, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Fredholm, B.B.; IJzerman, A.P.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors-an update. Pharmacol Rev 2011, 63, 1–34. [Google Scholar]
- Reppert, S.M.; Weaver, D.R.; Stehle, J.H.; Rivkees, S.A. Molecular cloning and characterization of a rat A1-adenosine receptor that is widely expressed in brain and spinal cord. Mol Endocrinol 1991, 5, 1037–1048. [Google Scholar] [CrossRef]
- Wardas, J. Neuroprotective role of adenosine in the CNS. Pol J Pharmacol 2002, 54, 313–326. [Google Scholar]
- Fedele, D.E.; Li, T.; Lan, J.Q.; Fredholm, B.B.; Boison, D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol 2006, 200, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Glass, M.; Faull, R.L.; Bullock, J.Y.; Jansen, K.; Mee, E.W.; Walker, E.B.; Synek, B.J.; Dragunow, M. Loss of A1 adenosine receptors in human temporal lobe epilepsy. Brain Res 1996, 710, 56–68. [Google Scholar] [CrossRef]
- Klaft, Z.J.; Hollnagel, J.O.; Salar, S.; Caliskan, G.; Schulz, S.B.; Schneider, U.C.; Horn, P.; Koch, A.; Holtkamp, M.; Gabriel, S.; Gerevich, Z.; Heinemann, U. Adenosine A1 receptor-mediated suppression of carbamazepine-resistant seizure-like events in human neocortical slices. Epilepsia 2016, 57, 746–756. [Google Scholar] [CrossRef]
- Wagner, A.K.; Miller, M.A.; Scanlon, J.; Ren, D.; Kochanek, P.M.; Conley, Y.P. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res 2010, 90, 259–272. [Google Scholar]
- Hack, S.P.; Christie, M.J. Adaptations in adenosine signaling in drug dependence: therapeutic implications. Crit Rev Neurobiol 2003, 15, 235–274. [Google Scholar] [CrossRef]
- Gomes, C.V.; Kaster, M.P.; Tome, A.R.; Agostinho, P.M.; Cunha, R.A. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta 2011, 1808, 1380–1399. [Google Scholar] [CrossRef]
- Stockwell, J.; Jakova, E.; Cayabyab, F.S. Adenosine A1 and A2A Receptors in the Brain: Current Research and Their Role in Neurodegeneration. Molecules 2017, 22. [Google Scholar] [CrossRef]
- Barros-Barbosa, A.R.; Ferreirinha, F.; Oliveira, A.; Mendes, M.; Lobo, M.G.; Santos, A.; Rangel, R.; Pelletier, J.; Sevigny, J.; Cordeiro, J.M.; Correia-de-Sa, P. Adenosine A(2A) receptor and ecto-5'-nucleotidase/CD73 are upregulated in hippocampal astrocytes of human patients with mesial temporal lobe epilepsy (MTLE). Purinergic Signal 2016, 12, 719–734. [Google Scholar] [CrossRef]
- Kong, Q.; Takahashi, K.; Schulte, D.; Stouffer, N.; Lin, Y.; Lin, C.L. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis 2012, 47, 145–154. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Santos-Rodrigues, A.D.; Schwarzschild, M.A.; Chen, J.F.; Cunha, R.A.; Agostinho, P. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 2012, 60, 702–716. [Google Scholar] [CrossRef] [PubMed]
- Canas, P.M.; Porciuncula, L.O.; Simoes, A.P.; Augusto, E.; Silva, H.B.; Machado, N.J.; Goncalves, N.; Alfaro, T.M.; Goncalves, F.Q.; Araujo, I.M.; Real, J.I.; Coelho, J.E.; Andrade, G.M.; Almeida, R.D.; Chen, J.F.; Kofalvi, A.; Agostinho, P.; Cunha, R.A. Neuronal Adenosine A2A Receptors Are Critical Mediators of Neurodegeneration Triggered by Convulsions. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, M.; Ledent, C.; Parmentier, M.; Daoust, M.; Costentin, J.; Vaugeois, J. Absence of the adenosine A(2A) receptor or its chronic blockade decrease ethanol withdrawal-induced seizures in mice. Neuropharmacology 2001, 40, 424–432. [Google Scholar] [CrossRef]
- D'Alimonte, I.; D'Auro, M.; Citraro, R.; Biagioni, F.; Jiang, S.; Nargi, E.; Buccella, S.; Di Iorio, P.; Giuliani, P.; Ballerini, P.; Caciagli, F.; Russo, E.; De Sarro, G.; Ciccarelli, R. Altered distribution and function of A2A adenosine receptors in the brain of WAG/Rij rats with genetic absence epilepsy, before and after appearance of the disease. Eur J Neurosci 2009, 30, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.; Gerbatin, R.; Kalmeijer, R.; Fedele, D.; Engel, T.; Boison, D. Adenosine A(2A) receptor and glial glutamate transporter GLT-1 are synergistic targets to reduce brain hyperexcitability after traumatic brain injury in mice. Neuropharmacology 2025, 278, 110599. [Google Scholar] [CrossRef]
- Tescarollo, F.C.; Rombo, D.M.; DeLiberto, L.K.; Fedele, D.E.; Alharfoush, E.; Tome, A.R.; Cunha, R.A.; Sebastiao, A.M.; Boison, D. Role of Adenosine in Epilepsy and Seizures. J Caffeine Adenosine Res 2020, 10, 45–60. [Google Scholar] [CrossRef]
- Lopes, L.V.; Rebola, N.; Pinheiro, P.C.; Richardson, P.J.; Oliveira, C.R.; Cunha, R.A. Adenosine A3 receptors are located in neurons of the rat hippocampus. Neuroreport 2003, 14, 1645–1648. [Google Scholar]
- Bjorklund, O.; Shang, M.; Tonazzini, I.; Dare, E.; Fredholm, B.B. Adenosine A1 and A3 receptors protect astrocytes from hypoxic damage. Eur J Pharmacol 2008, 596, 6–13. [Google Scholar] [PubMed]
- Hammarberg, C.; Schulte, G.; Fredholm, B.B. Evidence for functional adenosine A3 receptors in microglia cells. J Neurochem 2003, 86, 1051–1054. [Google Scholar]
- Tosh, D.K.; Paoletta, S.; Deflorian, F.; Phan, K.; Moss, S.M.; Gao, Z.G.; Jiang, X.; Jacobson, K.A. Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking and potent anticonvulsant activity. J Med Chem 2012, 55, 8075–8090. [Google Scholar] [PubMed]
- Spanoghe, J.; Larsen, L.E.; Craey, E.; Manzella, S.; Van Dycke, A.; Boon, P.; Raedt, R. The Signaling Pathways Involved in the Anticonvulsive Effects of the Adenosine A(1) Receptor. Int J Mol Sci 2020, 22. [Google Scholar]
- Boison, D. Adenosine and seizure termination: endogenous mechanisms. Epilepsy Curr 2013, 13, 35–37. [Google Scholar]
- Pascual, O.; Casper, K.B.; Kubera, C.; Zhang, J.; Revilla-Sanchez, R.; Sul, J.Y.; Takano, H.; Moss, S.J.; McCarthy, K.; Haydon, P.G. Astrocytic purinergic signaling coordinates synaptic networks. Science 2005, 310, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J Neurosci 2008, 28, 4702–4711. [Google Scholar]
- Zimmermann, H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol 2000, 362, 299–309. [Google Scholar]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med 2013, 34, 529–547. [Google Scholar]
- Headrick, J.P.; Willis, R.J. 5'-Nucleotidase activity and adenosine formation in stimulated, hypoxic and underperfused rat heart. Biochem J 1989, 261, 541–550. [Google Scholar] [PubMed]
- Lloyd, H.G.; Deussen, A.; Wuppermann, H.; Schrader, J. The transmethylation pathway as a source for adenosine in the isolated guinea-pig heart. Biochem J 1988, 252, 489–494. [Google Scholar]
- Hoffman, D.R.; Cornatzer, W.E.; Duerre, J.A. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can J Biochem 1979, 57, 56–65. [Google Scholar]
- Boison, D.; Scheurer, L.; Zumsteg, V.; Rulicke, T.; Litynski, P.; Fowler, B.; Brandner, S.; Mohler, H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA 2002, 99, 6985–6990. [Google Scholar]
- Kobow, K.; Kaspi, A.; Harikrishnan, K.N.; Kiese, K.; Ziemann, M.; Khurana, I.; Fritzsche, I.; Hauke, J.; Hahnen, E.; Coras, R.; Muhlebner, A.; El-Osta, A.; Blumcke, I. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta Neuropathol 2013, 126, 741–756. [Google Scholar]
- Miller-Delaney, S.F.; Bryan, K.; Das, S.; McKiernan, R.C.; Bray, I.M.; Reynolds, J.P.; Gwinn, R.; Stallings, R.L.; Henshall, D.C. Differential DNA methylation profiles of coding and non-coding genes define hippocampal sclerosis in human temporal lobe epilepsy. Brain 2015, 138 Pt 3, 616–631. [Google Scholar]
- Kobow, K.; Ziemann, M.; Kaipananickal, H.; Khurana, I.; Muhlebner, A.; Feucht, M.; Hainfellner, J.A.; Czech, T.; Aronica, E.; Pieper, T.; Holthausen, H.; Kudernatsch, M.; Hamer, H.; Kasper, B.S.; Rossler, K.; Conti, V.; Guerrini, R.; Coras, R.; Blumcke, I.; El-Osta, A.; Kaspi, A. Genomic DNA methylation distinguishes subtypes of human focal cortical dysplasia. Epilepsia 2019, 60, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Zhang, J.; Deng, Q.; Wang, M.; Li, T. Vagus Nerve Stimulation Inhibits DNA and RNA Methylation in a Rat Model of Pilocarpine-Induced Temporal Lobe Epilepsy. CNS Neurosci Ther 2025, 31, e70484. [Google Scholar] [PubMed]
- During, M.J.; Spencer, D.D. Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 1992, 32, 618–624. [Google Scholar]
- Theofilas, P.; Brar, S.; Stewart, K.A.; Shen, H.Y.; Sandau, U.S.; Poulsen, D.; Boison, D. Adenosine kinase as a target for therapeutic antisense strategies in epilepsy. Epilepsia 2011, 52, 589–601. [Google Scholar] [CrossRef]
- Luan, G.; Wang, X.; Chen, F.; Gao, Q.; Zhou, J.; Guan, Y.; Wang, J.; Zhai, F.; Chen, Y.; Li, T. Overexpression of Adenosine Kinase in Patients with Epilepsy Associated With Sturge-Weber Syndrome. Neuropsychiatry 2018, 08. [Google Scholar]
- de Groot, M.; Iyer, A.; Zurolo, E.; Anink, J.; Heimans, J.J.; Boison, D.; Reijneveld, J.C.; Aronica, E. Overexpression of ADK in human astrocytic tumors and peritumoral tissue is related to tumor-associated epilepsy. Epilepsia 2012, 53, 58–66. [Google Scholar]
- Gimenes, C.; Motta Pollo, M.L.; Diaz, E.; Hargreaves, E.L.; Boison, D.; Covolan, L. Deep brain stimulation of the anterior thalamus attenuates PTZ kindling with concomitant reduction of adenosine kinase expression in rats. Brain Stimul 2022, 15, 892–901. [Google Scholar]
- Miranda, M.F.; Hamani, C.; de Almeida, A.C.; Amorim, B.O.; Macedo, C.E.; Fernandes, M.J.; Nobrega, J.N.; Aarao, M.C.; Madureira, A.P.; Rodrigues, A.M.; Andersen, M.L.; Tufik, S.; Mello, L.E.; Covolan, L. Role of adenosine in the antiepileptic effects of deep brain stimulation. Front Cell Neurosci 2014, 8, 312. [Google Scholar] [CrossRef]
- Qureshi, I.A.; Mehler, M.F. Epigenetic mechanisms underlying human epileptic disorders and the process of epileptogenesis. Neurobiol Dis 2010, 39, 53–60. [Google Scholar]
- Younus, I.; Reddy, D.S. Epigenetic interventions for epileptogenesis: A new frontier for curing epilepsy. Pharmacol Ther 2017, 177, 108–122. [Google Scholar]
- Kobow, K.; Blumcke, I. The emerging role of DNA methylation in epileptogenesis. Epilepsia 2012, 53 (Suppl. 9), 11–20. [Google Scholar] [CrossRef]
- Wahba, A.E.; Fedele, D.; Gebril, H.; AlHarfoush, E.; Toti, K.S.; Jacobson, K.A.; Boison, D. Adenosine Kinase Expression Determines DNA Methylation in Cancer Cell Lines. ACS Pharmacol Transl Sci 2021, 4, 680–686. [Google Scholar]
- Toti, K.S.; Osborne, D.; Ciancetta, A.; Boison, D.; Jacobson, K.A. South (S)- and North (N)-Methanocarba-7-Deazaadenosine Analogues as Inhibitors of Human Adenosine Kinase. J Med Chem 2016, 59, 6860–6877. [Google Scholar] [PubMed]
- Lusardi, T.A.; Akula, K.K.; Coffman, S.Q.; Ruskin, D.N.; Masino, S.A.; Boison, D. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 2015, 99, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; He, X.; Luan, G.; Li, T. Role of DNA Methylation and Adenosine in Ketogenic Diet for Pharmacoresistant Epilepsy: Focus on Epileptogenesis and Associated Comorbidities. Front Neurol 2019, 10, 119. [Google Scholar]
- Taub, K.S.; Kessler, S.K.; Bergqvist, A.G. Risk of seizure recurrence after achieving initial seizure freedom on the ketogenic diet. Epilepsia 2014, 55, 579–583. [Google Scholar] [CrossRef]
- Schoeler, N.E.; Ridout, D.; Neal, E.G.; Becirovic, M.; Whiteley, V.J.; Meskell, R.; Lightfoot, K.; Mills, N.; Ives, T.; Bara, V.; Cameron, E.; Thomas, P.; Wilford, E.; Fox, R.; Fabe, J.; Leong, J.Y.; Tan-Smith, C. Maintenance of response to ketogenic diet therapy for drug-resistant epilepsy post diet discontinuation: A multi-centre case note review. Seizure 2024, 121, 78–84. [Google Scholar]
- Elliott, R.E.; Rodgers, S.D.; Bassani, L.; Morsi, A.; Geller, E.B.; Carlson, C.; Devinsky, O.; Doyle, W.K. Vagus nerve stimulation for children with treatment-resistant epilepsy: a consecutive series of 141 cases. J Neurosurg Pediatr 2011, 7, 491–500. [Google Scholar]
- Bansal, L.; Jaafar, F.; Kapoor, A.; Abdelmoity, L.; Madkoor, A.; Kaufman, C.; Abdelmoity, A. Transforming pediatric epilepsy care: Real-world insights from a leading single-center study on vagus nerve treatment outcomes. Epilepsia 2025. [Google Scholar]
- Fernandez-Guardiola, A.; Martinez, A.; Valdes-Cruz, A.; Magdaleno-Madrigal, V.M.; Martinez, D.; Fernandez-Mas, R. Vagus nerve prolonged stimulation in cats: effects on epileptogenesis (amygdala electrical kindling): behavioral and electrographic changes. Epilepsia 1999, 40, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Bekar, L.; Libionka, W.; Tian, G.F.; Xu, Q.; Torres, A.; Wang, X.; Lovatt, D.; Williams, E.; Takano, T.; Schnermann, J.; Bakos, R.; Nedergaard, M. Adenosine is crucial for deep brain stimulation-mediated attenuation of tremor. Nat Med 2008, 14, 75–80. [Google Scholar] [PubMed]
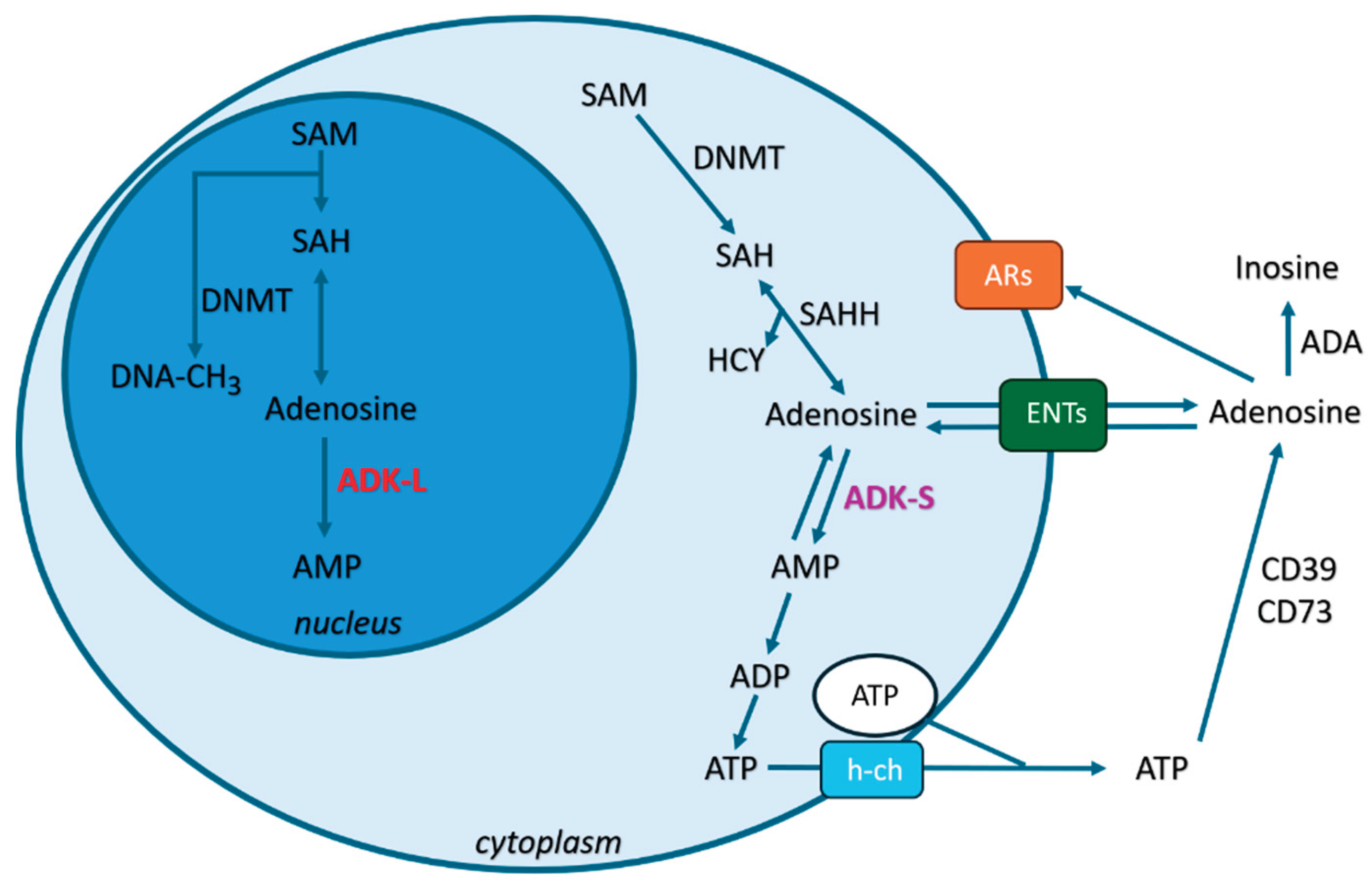
Figure 1.
Figure 1. The role of adenosine in epilepsy: subcellular regulation and mechanisms in epilepsy. Adenosine is localized in extracellular, cytoplasmic, and nuclear compartments. i) Extracellular adenosine is maintained by ATP degradation via ectonucleotidases (CD39/CD73), ADA catabolism, and equilibrative nucleoside transporters (ENTs). Extracellular adenosine mainly activates adenosine receptors (ARs: A1Rs/A2ARs/A3Rs), modulating neuronal excitability. ii) Cytoplasmic adenosine arises from transmethylation reactions and is regulated by ADK-S, with metabolism by ADA and ENTs playing key roles. Cytoplasmic adenosine regulates transmethylation by DNMT (DNA methylation), influencing neurotransmitter balance. iii) Nuclear adenosine, generated by transmethylation, is controlled by ADK-L. Nuclear adenosine controls epigenetic mechanisms via DNMT, potentially affecting gene expression in epilepsy. This compartmentalized regulation highlights adenosine's dual role in both suppressing seizures (via A1Rs activation) and contributing to neuroplasticity (via epigenetic modulation).
Figure 1.
Figure 1. The role of adenosine in epilepsy: subcellular regulation and mechanisms in epilepsy. Adenosine is localized in extracellular, cytoplasmic, and nuclear compartments. i) Extracellular adenosine is maintained by ATP degradation via ectonucleotidases (CD39/CD73), ADA catabolism, and equilibrative nucleoside transporters (ENTs). Extracellular adenosine mainly activates adenosine receptors (ARs: A1Rs/A2ARs/A3Rs), modulating neuronal excitability. ii) Cytoplasmic adenosine arises from transmethylation reactions and is regulated by ADK-S, with metabolism by ADA and ENTs playing key roles. Cytoplasmic adenosine regulates transmethylation by DNMT (DNA methylation), influencing neurotransmitter balance. iii) Nuclear adenosine, generated by transmethylation, is controlled by ADK-L. Nuclear adenosine controls epigenetic mechanisms via DNMT, potentially affecting gene expression in epilepsy. This compartmentalized regulation highlights adenosine's dual role in both suppressing seizures (via A1Rs activation) and contributing to neuroplasticity (via epigenetic modulation).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



