Submitted:
04 December 2025
Posted:
05 December 2025
You are already at the latest version
Abstract
The Cladia aggregata group of lichen-forming fungi comprises multiple species that are difficult to differentiate based on phenotypic characters. The Cladia aggregata group has a wide distribution across several continents, but is most diverse in Australasia. We aimed to delimit the species complex further, investigate the relatedness of the lineages, and examine their distributional ranges and phenotypic traits. We used Restriction Site Associated DNA Sequencing (RAD-seq) to compare thousands of loci across 91 individuals from the Americas, Asia, and Australasia. All Asian samples formed a distinct, monophyletic clade in all phylogenetic trees, while the American samples divided into two clades: one comprising South American samples and another comprising Caribbean samples—the latter representing C. aggregata sensu stricto, as the type specimen was collected in Jamaica. Further population-genomic analyses support the conclusion that the Asian samples are genetically distinct and are here described as a new species. The new species, Cladia asiatica, accommodates the Asian samples previously included in C. aggregata. Our analysis highlights the potential of next-generation sequencing to reveal hidden diversity and resolve the phylogeny of this species complex and lichen-forming fungi in general.
Keywords:
next-generation sequencing
; systemics
; phylogenetics
; molecular variation
; biogeography
1. Introduction
Taxonomy has three primary roles in science: assigning organisms to recognized species, organizing species into higher categories such as genus and family, and delimiting species. Species delimitation is the designation of species when individuals do not adhere to current species categories [1]. Accurate species delimitations are necessary for ecological and conservation research [3]; however, estimates suggest that a substantial proportion of the world’s species remain hidden, with potentially 86% of terrestrial species and 91% of marine species yet to be described [2].
Among these yet-undescribed organisms are many lichens, which are stable symbiotic associations between a fungal partner (the mycobiont) and a photosynthetic partner (the photobiont) [3]. Traditionally, species delimitation of lichen-forming fungi has relied on morphological and chemical traits. While this approach is helpful for some taxa, it can be challenging for those with few distinctive traits or high phenotypic plasticity [1]. Many cryptic species of lichen-forming fungi likely remain hidden within phenotype-based species descriptions, reflecting substantial unexplored or undiscovered diversity [4].
Next-generation sequencing has successfully delimited species complexes that were previously difficult to distinguish through multi-locus, morphological, or chemical approaches. This technology allows an in-depth exploration of many single-nucleotide polymorphisms (SNPs) across the entire genome [5]. In particular, restriction site-associated DNA sequencing (RAD-seq) has improved our understanding of the number of lichen-forming fungus species, their phylogenetic relationships, and their geographic distribution [6,7,8,9,10,11,12].
The Cladia aggregata group includes fruticose lichens belonging to the family Cladoniaceae [13], which exhibit substantial morphological variation. Different taxonomic approaches have been used to interpret or classify this variation. While Filson [14] applied a broad species concept, subsequent authors separated certain taxa from C. aggregata s. lat. based on morphological and chemical traits [15,16]. The type specimen was collected in Jamaica in 1788 by the Swedish botanist Olof Peter Swartz [17]. The species has a wide distribution, primarily in the Southern Hemisphere [18].
Recently, the C. aggregata species complex was studied using multi-gene analyses, leading to the formal description of additional species within the complex [19,20]. Currently, twelve species are recognized in the group, but distinct geographical patterns were found in the remaining clades under C. aggregata sensu stricto [19]. We have now revisited this C. aggregata group, with special emphasis on Asian populations and their relationships to Neotropical populations from which the type material is derived. We employed RAD-seq data, which have been successfully used to disentangle phylogenetic relationships in taxonomically difficult groups, such as the neuropogonoid species in Usnea [8] [10] and species pairs in the genus Usnea [21].
We addressed the following research questions by using RAD-seq on the C. aggregata species complex: (1) Are there more species within the complex than previously identified? (2) How are these species related to previously described species? (3) What is the geographic range of these species? and (4) Which phenotypic traits correspond to genetic variation within the complex? We hypothesized that the Asian and Australian populations represent a separate species from the neotropical populations. The wide geographic range of the species complex suggests that gene flow between geographically separated populations may be limited.
2. Materials and Methods
2.1. NGS sequencing and initial data processing
A total of 95 specimens of C. aggregata were sequenced for this project from the Neotropics, Australasia, East Asia, and Thailand to represent its wide geographic range (Supplementary Materials Table S3). We initially included a sample of Pulchrocladia ferdinandii and P. retipora as outgroups in this study and sequenced them alongside all C. aggregata samples. However, after reconstructing an initial phylogenetic tree and a subsequent morphological examination, we found that three samples identified as C. neocaledonica (C1011219F, C2035641F, and C2035148F) were a more suitable outgroup, and the two initial outgroup samples were removed. Further, after sequencing and data processing, a sample (C1011230F) was dropped due to low data coverage (<200 loci), and another (RAMK40318) was dropped due to a high estimated error rate (.00648). Consequently, our revised, final dataset contained 91 samples.
DNA extractions and the construction of the RAD-seq library were done at the University of Wisconsin-Madison Biotechnology Center. DNA was extracted using the DNeasy mericon Food Kit, and the RAD-seq library was prepared using the restriction enzyme ApeKI before sequencing on an Illumina Novaseq 6000 (Illumina, San Diego, CA, USA) following a procedure similar to [22]. The RAD-seq data were received as gzipped fastq files.
The RAD-seq data were processed using ipyrad v0.9.90 using the servers of the Grainger Bioinformatics Center at the Field Museum as previously described in Otero et al. (2023) [10]. We set the ipyrad parameter files for paired GBS data, the assembly method to reference, and the ploidy to 1 (haploid). We kept the minimum sample coverage of 4. The reference sequence was the genome of Cladonia squamosa from the Wellcome Sanger Institute, which was downloaded from GenBank (GCA_947623385.1). The reference sequence was used to filter reads from the lichen-forming fungus from the rest of the metagenome. Sequences that could not be mapped to the reference genome were discarded by ipyrad.
2.2. Phylogenetics
RAxML (Randomized Axelerated Maximum Likelihood) is a maximum likelihood method for reconstructing phylogenetic trees. We used RAxML v8.2.12 [23]. We initially made a tree with 95 samples prior to removing two samples for poor data quality and the two outgroup samples, because three samples later identified as C. neocaledonia functioned as a better outgroup. The input data was the phylip file of 91 samples created by ipyrad. The phylip or .phy file contains all the loci from the data concatenated into a supermatrix with missing data filled in as Ns [24]. 1,000 bootstraps were run for each analysis using the GTRCATX substitution model. See Supplementary Materials Figure S2 for the RAxML tree. Additionally, we used IQ-TREE v2 [25] as another method to calculate maximum-likelihood trees using the same input file. We used the model checker [26], which assigns BIC values to multiple different nucleotide substitution models to choose the best model for the specific data entered. The ultrafast bootstrap [27] was used to reduce the computation time. We used the best-fit model based on BIC values (TPM3u+F+R5) and ran 1,000 bootstraps. The output trees from both maximum-likelihood methods were visualized using FigTree [28]. The trees were rooted at the midpoint, then the node order was set to “increasing” for both trees.
SVDquartets [29] is a coalescent-based method implemented in the PAUP v4.0a package [30]. SVDquartets calculates several quartet trees, each using 4 SNPs, before using those trees to make an overall species tree. We used PAUP to convert the file of unlinked SNPs to a Nexus format for our input file. We used CASTER-site v1.20.2.5 (Coalescence-aware Alignment-based Species Tree EstimatoR) [31] to assign bootstrap values to the SVD Quartets tree. In CASTER-site, we used the newick-formatted output tree from SVDquartets as a constraint tree and the phylip file from ipyrad as a loci matrix. Due to the speed of CASTER-site, we set the argument for the number of placements (-r) and the number of rounds of subsampling (-s) to 10,000. We used FigTree [28] to visualize the results. The branches were converted to a cladogram, the node order set to “increasing”, and the tree was manually rooted with the outgroup. We used different software, each using different models to calculate maximum likelihood trees and a coalescent-based method to calculate another tree, so that we had multiple lines of evidence to further support our conclusions of the relationships between C. aggregata complex populations.
2.3. Statistics
Statistical analyses were conducted on a reduced dataset comprising only samples from the American and Asian clades. Other samples were excluded because they formed small clades, showed inconsistent topologies across phylogenetic methods, or had low bootstrap support. We used the R package poppr v2.9.7 [32] to run an AMOVA (Analysis of Molecular Variance). The AMOVA was used to calculate the genomic variation within and between clades. We also used the ade4 package v1.7.23 [33] to compare the variation between our assigned groups and randomly assigned groups. We used the randtest command for this comparison, which uses a Monte Carlo test to simulate randomly assigned groups of samples and ran 999 iterations. Our input file was a genind object generated with the adegenet v2.1.11 package [34]. The genind object was made from a vcf file recoded using VCFtools v0.1.16 [35] so that sites found in less than 50% of the data and those with a minor allele frequency below 5% were excluded.
2.4. Population Genetics
All population-genomic analyses were performed using the same reduced dataset described above. A Principal Component Analysis (PCA) was performed to summarize genetic variation between samples using the same genind object as used before. We used the dudi.pca function in ade4 [33] to calculate the PCA. values were replaced with the mean allele frequencies. We used ggplot2 v3.5.2 [36] to visualize the plot.
We used fineRADstructure to group samples into estimated populations based on co-ancestry or relatedness among individuals. FineRADstructure can show shared ancestry in more detail than similar methods, including the level of shared ancestry between populations rather than just the number of separate populations [37]. The ipyrad alleles file was converted to a fineRADstructure file using the finerad_input.py script in the fineRADstructure tools. The minimum number of samples per locus was set to four. RADpainter was used to calculate a co-ancestry matrix for the data. We used fineSTRUCTURE to group individuals into populations using an MCMC clustering algorithm. The number of sample iterations (-y) was set to 100000, the number of burn-in iterations (-x) was set to 100000, and the thin interval (-z) was set to 1000. The method (-m) was set to the default, which performs the full range of moves to obtain the best posterior state. To visualize the tree with estimated population sizes, we used the R packages ape [38] and XML [39].
FastSTRUCTURE [40] was used to measure admixture between populations. The dataset was initially cleaned with VCFtools v0.1.16 [35] as described in the Statistics section. We used PLINK v1.9 [41] to convert the newly created vcf files into plink files [42]. The sample assignment was studied across different population sizes (K values). Then we ran fastSTRUCTURE for K = 1-10. The R package POPHELPER [43] was used to visualize results grouped by population and to view Fast Structure results for several K values simultaneously.
2.5. Phenotypical Characteristics
We performed high-performance thin-layer chromatography (HPTLC) on all samples. All 95 samples, including the outgroup, were soaked in acetone and chromatographed using solvent B and C [44]. We used the traits described in [15] to measure morphological differences among the samples in this study. These traits include the shape of apices, perforation of samples, sample color, branch pattern, and others. We followed a study that assigned a numerical value to each possible option for every trait, for example, samples with abundant perforations are assigned 1, and those with little to no perforations are assigned 0 for that trait [19]. Assigning numerical values to each trait enabled us to create a matrix of morphological traits, allowing us to quantify the physical differences between samples.
2.6. Biogeography
RASP v4.4 [44] is a software that can apply several methods of estimating the geographic distribution of ancestral nodes in phylogenies. The treefile from IQ-TREE 2 was provided as a consensus tree. RASP requires a states file, and we provided a text file in the format recommended for BioGeoBears [45]. We limited the number of states a node could occupy to two areas for our analysis. We ran BBM (Bayesian Binary MCMC) on the consensus tree [46,47]. The number of cycles was 50,000 with 10 chains. The frequencies of samples and discard samples were both set to 100, and the temperature was set to 0.1. The model for the state frequencies was a fixed Jukes-Cantor model, and a gamma distribution with a shape parameter from 0.001 to 100 was selected for among-site rate variation.
3. Results
3.1. RAD Sequencing
We recovered 5,720 loci from the complete set of 95 samples after filtering, yielding an average of 1,480.48 loci per sample. The average number of raw reads per sample was 7,068,030.81, while the average number of reads mapped to the reference genome per sample was 187,914.98. Therefore, the percentage of total raw unfiltered reads mapped to the reference is 2.7%. After processing the data with ipyrad, the resulting matrix contained 46,731 total SNPs with 81% missing sites. The average error estimate per sample was .0017 ( Table 1 )
We removed four samples from our final analysis. We removed C1011230F, due to low coverage (<200 loci), and RAMK40318, because it had the highest error estimate (.00648). We then identified samples in our data that were used in prior studies of the C. aggregata complex [20] as well as records on GenBank and the BOLD (Barcode Of Life Data Systems) database. We identified five samples that had already been identified using single-locus markers. After additional morphological inspection, it was determined that the three samples were Cladia neocaledonica. We replaced the intended outgroup (Pulchrocladia retipora and P. ferdinandii) with the three samples, as they functioned as a more closely related outgroup to the rest of our samples. See Supplementary Materials Table S1 for information on the samples already identified as belonging to other species in the C. aggregata complex. We did not drop any of the previously identified samples. After these four samples, the final dataset consisted of 88 ingroup samples and three outgroup samples.
3.2. Phylogenetic Analyses
The maximum likelihood phylogenetic trees generated with IQ-TREE v2 (Figure 1) and RAxML (Figure S2) showed consistent overall topologies. Both analyses strongly supported three major clades: a Caribbean clade (100/100), a South American clade (100/100), and an Asian clade (100/100). The internal topology of the Asian clade differed between the two analyses. In both trees, the taxa from Thailand formed a moderately supported subclade (86/100). The remaining Asian samples (from China, Taiwan, and Japan) were divided into two weakly supported, paraphyletic groups. The Chinese specimen 66707 KUN-L was placed within one of these clades in the RAxML tree, but appeared as sister to all other Asian samples in the IQ-TREE v2 analysis. In both trees, two taxa identified as C. terebrata from Australia and New Zealand were recovered as the sister group of the Asian clade. These four lineages (Asia, South America, Caribbean, and C. terebrata) were in turn sister to a group consisting mainly of Australasian taxa, together with one C. inflata sample from southern Argentina and one Taiwanese sample. The two analyses differed in how these taxa were arranged: in the IQ-TREE v2 reconstruction, they formed an unsupported clade, whereas in RAxML, they were paraphyletic, with two samples identified as C. inflata and one as C. blanchonii forming a moderately supported sister clade (85) to all remaining taxa. In both analyses, the well-supported New Caledonian clade was resolved as the earliest-branching lineage.
See Figure S1 in the Supplementary Materials section for the RAxML tree with all samples, including samples that were later dropped. In the 95-sample tree the Thailand and East Asian clades are combined into one poorly supported Asian clade. The C. terebrata samples are sister to an Australasian clade rather than the Asian samples and the American samples are also sister to another Australasian clade in the 95-sample tree, although both these differences from the 91-sample RAxML tree have low support. The two samples in the ingroup that were dropped have long branches relative to the rest of the tree.
The coalescent-based tree (Figure S3 also showed strong support for the major clades recovered in the Maximum Likelihood analyses, including the Asian, South American, Caribbean, C. terebrata, and New Caledonian clades. However, it differed in several key aspects. In this analysis, both the Thailand and East Asian subclades within the Asian lineage received high support, whereas their relationships were weakly resolved in the ML trees. The C. terebrata clade was recovered as the earliest-branching lineage, sister to all other taxa. One C. inflata sample clustered with the C. blanchonii specimen as sister to the Asian clade, thereby occupying the position held by the C. terebrata clade in the Maximum Likelihood analyses. The remaining C. inflata sample grouped with the Australasian and New Caledonian taxa, which together formed a paraphyletic assemblage.
3.3. Statistics
The AMOVA analysis revealed that most of the variation in the dataset is due to differences between groups rather than within groups (Table 2). The samples were selected and grouped into four main groups based on a coalescent-based phylogeny: Thailand, East Asia, South America, and the Caribbean. The Monte Carlo test indicated considerable variation between groups. The average variation between random simulated groups was far lower than the variation between our actual groups that correspond to clades of the phylogeny. The p-value for our Monte Carlo test was well below the 0.05 threshold, indicating a significant result.
3.4. Population Genetics
The PCA confirms the separation of samples from Asia and the Americas. These four groups contained 77 samples, and the filtered dataset included 1,542 loci and 3,084 alleles. That is, following the removal of sites with less than 50% coverage and those with a MAF below 5% as described in the methods. PC 1 explains 41.5% of the variation in the data and separates the Asian and American samples, while the Asian samples build a more distinct cluster. PC 2 explains 8.4% of the variation and separates East Asia from Thailand, as well as South America from the Caribbean samples, though less pronouncedly (Figure 2A).
Using fineRADstructure, we assessed patterns of co-ancestry among individuals to evaluate genetic structure within and among major clades. The resulting co-ancestry matrix resolved distinct clusters corresponding to Thailand, East Asia, South America, and the Caribbean. Samples within each of these four groups showed higher relatedness to one another than to samples from other regions. The Caribbean and South American groups exhibited stronger within-group co-ancestry than the East Asian and Thai groups. Co-ancestry between the Asian and American samples was low, indicating limited recent gene flow between regions. Although a separation between East Asia and Thailand, and between South America and the Caribbean, was detected, these divisions were comparatively weak.
Population patterns and gene flow indicated by co-ancestry were also observed with fastSRUCTURE. As with other population-genomics analyses, fastSTRUCTURE results clearly separated samples from Asia and the Americas. K = 3 through 6 also separated Thailand from East Asia, although only K = 5 showed no admixture between East Asia and Thailand, while K =5 also separated the Caribbean and South America.

3.5. Phenotypic Characteristics
The chemistry of our samples did not aid in further species delimitation because there was not enough variation for us to use for sample comparison between different populations or localities. Most samples produced the same chemotype 1. Most of the samples producing compounds different from chemotype 1 were identified as belonging to species other than C. aggregata sensu stricto. A few of the samples that were still identified as belonging to C. aggregata sensu stricto and produced different compounds were all from the same locality in Bolivia. Chemotype 1 is characterized by barbatic and 4-O-demethylbarbatic acid. Two samples were within chemotype 2 (stictic acid complex). One of these was previously identified as C. blacnchonii [20], and the other was more recently identified as C. inflata. Moreover, chemotype 3 (fumarprotocetraric, succinprotocetraric, and protocetraric acid) was only found in one Australian sample from the outgroup. Chemotype 4 (barbatic, thamnolic, and 4-O-demethylbarbatic acid) was only present in three Bolivian samples. Chemotype 5 (barbatic, 4-O-demethylbarbatic, and fumarprotocetraric acid) is only present in one sample from Bolivia, one sample in the outgroup from New Caledonia, which was previously identified as C. neocaledonica [20], and one sample from Thailand (RAMK40318), which was excluded from this study. Both samples of the previous outgroup are in chemotype six (Atranorin), but they were excluded from this study. Chemotype 7 (fumarprotocetraric acid) is only present in one sample from New Zealand in the outgroup and another sample from New Zealand (C1011230F), which was dropped from our analysis. See supplementary table S2 for the major compounds of each chemotype and the geographic distribution of the samples within each chemotype. In addition, the C. aggregata complex exhibits extensive intraspecific morphological variation, much of which is non-diagnostic and thus unsuitable for reliable species delimitation.
3.6. Biogeography
Range reconstruction suggests that the ancestral populations of C. aggregata originated in Australasia (Figure 4). The BBM results indicate that dispersal to Asia and the Americas was more recent compared to ancestral splits within Australasian populations. BBM also estimates that the common ancestor of both the Thai and East Asian populations was most likely from East Asia. However, the common ancestor of the American populations is less clear and could have originated from South America, the Caribbean, or Australasia, before dispersing to both areas independently.
Figure 3.
Bayesian Binary MCMC (BBM) analysis on the consensus tree calculated in IQ-TREE V2. Each node has a corresponding pie chart showing the likelihood of that ancestor occupying a given distribution. The corresponding colors for each region are explained in the legend at the top left of the figure.
Figure 3.
Bayesian Binary MCMC (BBM) analysis on the consensus tree calculated in IQ-TREE V2. Each node has a corresponding pie chart showing the likelihood of that ancestor occupying a given distribution. The corresponding colors for each region are explained in the legend at the top left of the figure.
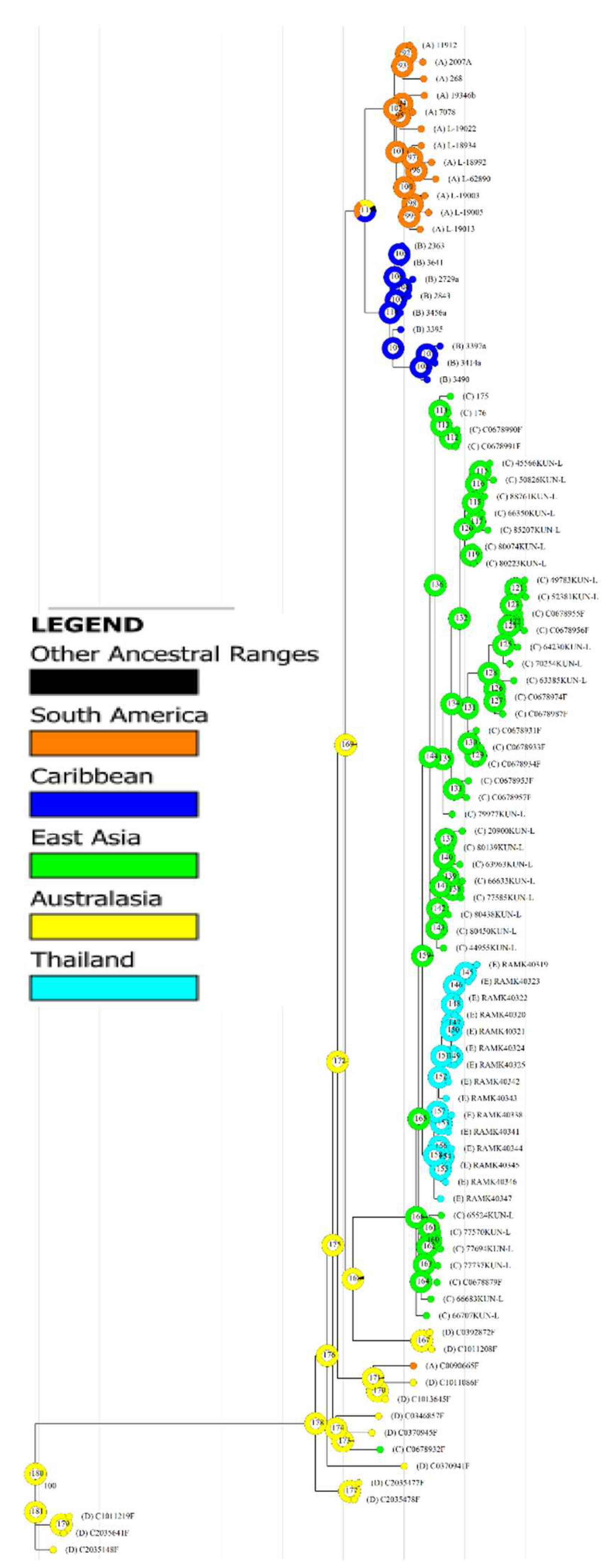
Figure 4.
Habit of Cladia asiatica sp. nov. (Holotype). Cladia asiatica de Abreu, Barcenas, Pengsungnoen, Wang, Huang, Lumbsch, & Grewe MycoBank No. 861532
Figure 4.
Habit of Cladia asiatica sp. nov. (Holotype). Cladia asiatica de Abreu, Barcenas, Pengsungnoen, Wang, Huang, Lumbsch, & Grewe MycoBank No. 861532

3.7. Taxonomy
Characterized by slightly inflated and tapered sterile pseudopodetia, with glossy, pale yellow to green or greenish brown or brownish surface, mostly dichotomously branched toward the apex. Perforations mostly abundant. Generative pseudopodetia robust, inflated, and racemosely branched. Perforations abundant. Conidiomata on terminal branches of sterile pseudopodetia, conidia up to 5 μm long, rod-shaped. Apothecia not seen. Secondary chemistry: barbatic and 4-O-demethylbarbatic acid.
Type: Thailand, Loei Province, Phu Luang Wildlife Sanctuary, Khok Nok Kraba Ranger Station, Tha Sala, Phu Ruea, 17°16'39.4"N 101°31'33.3"E, on moss, 1555 M, June 2008, Kawinnat Buaruang (holotype RAMK 40342, isotype F).
Phylogenetic notes: Strongly supported as a monophyletic lineage in a maximum likelihood tree made using IQ-TREE2 v2, where the species is sister to samples of Cladia terebrata (Figure 1)
Reference sequence: GenBank No. GCA_947623385.1
Distribution and habitat: This species occurs in high altitudes of montane forests in East Asia and has been found in China, Taiwan, Thailand, and Japan. It occurs on mossy soil and rocky outcrops.
Etymology: The new species is named after the geographic range it inhabits.
Additional specimens examined (paratypes): Taiwan, Ilan, Taiping Shan National Forest, Cuifong lake circle Trail, 1840 M, August 2023, J.P. Huang, 175 China, Yunnan Province, Dali Ci., Ma'er Mt., 26°15′21.67″N 100°06′32.21″E, on soil, 3502 M, June 2018, 王立松(LS Wang),王欣宇(XY Wang), 张雁云(YY Zhang),银安城(AC Yin),姜龙芊(LQ Jiang), KUN-L 18-59051
4. Discussion
4.1. Species Delimitation
The results supported our hypotheses that RAD-seq data would reveal additional species within the Cladia aggregata complex and that geographic distance among continents limits gene flow between populations. Unlike previous studies, our analysis identified distinct Asian and American species within the Cladia aggregata complex, as evidenced by restricted gene flow between the two well-supported, reciprocally monophyletic groups. While earlier studies relied on the analysis of a limited number of molecular markers, the large volume of genomic data generated through next-generation sequencing in this study enabled more precise species delimitation within this group of closely related lichen-forming fungi. Most striking was the separation of most Asian specimens in a monophyletic clade, sister to C. terebrata and distinct from C. aggregata sensu stricto. Due to its clear delimitation, we propose that the Asia samples constitute a new species, designated as Cladia asiatica.
As the amount and resolution of data used for taxonomic inference have increased from morphology to single and multilocus phylogenies, and now to genome-scale datasets, our understanding of species boundaries within the Cladia aggregata complex has become progressively more detailed. Early morphology-based studies, which also relied on chemical traits, treated the complex very broadly, recognizing it as only one, highly variable species [14] or delimiting eight species within the complex [15]. Later, multi-locus molecular studies using phylogenetic approaches expanded the diversity to twelve species, most of which were restricted to Australasia [19,20]. Only a few species included samples from outside the region: C. cryptica from Malaysia, C. inflata from Chile, and C. gorgonea, which occurs in both Australasia and La Réunion Island. The remaining Asian and American samples were still placed within C. aggregata [20]. Our study extends this work by combining a broader taxon sampling, particularly from China, Taiwan, and Japan, with a genome-scale dataset, providing the resolution necessary to accurately reconstruct evolutionary relationships within the C. aggregata complex across Asia and the Americas. The expanded dataset reveals limited gene flow between continents, likely due to geographic distance, and supports the notion that Asian and American populations represent distinct species.
Although some analyses suggest further structure between taxa from Thailand and the remaining Asian samples, or between South American taxa and Caribbean taxa, we refrain from formally delimiting additional species. The Thai samples form a distinct monophyletic group in some analyses but cluster within the broader Asian clade in others. They also exhibit partial separation in the PCA and in some fastSTRUCTURE K values, yet most analyses reveal admixture between Thailand and East Asia. Given this inconsistent signal and potential gene flow, we do not consider the Thai population to be a distinct species. Similarly, while South American and Caribbean samples form separate clades in some trees, the low sample size from the Caribbean and the high genetic similarity between the two regions argue against further species delimitation. Both fastSTRUCTURE and fineRADstructure indicate greater relatedness between these populations than between Asian and American samples, consistent with either incomplete lineage sorting or limited gene flow. Although these populations represent distinct genetic clusters, the level of divergence does not justify recognizing them as separate species.
Our results indicate that all samples from East Asia and Thailand represent a distinct species, differing from Cladia aggregata sensu stricto. Jamaican specimens analyzed in this study form a well-supported clade that we interpret as representing C. aggregata sensu stricto, consistent with the type specimen originally described by Olof Peter Swartz from Jamaica [17]. Any sample clustering with this lineage, or not sufficiently differentiated from it, should continue to be recognized as C. aggregata. In contrast, all Asian samples form a strongly supported monophyletic clade across multiple phylogenetic methods. While population genetics methods and statistical tests also confirm a high degree of genetic divergence between samples from Asia and those from the Caribbean, including the Jamaican samples, indicating limited or no gene flow.
4.2. Biogeography
In the Bayesian Binary MCMC tree, most ancestral nodes were assigned to Australasia, which aligns with previous research on the genus Cladia, suggesting that the genus first evolved in Australia [52]. The common ancestor of the Asian populations is probably from East Asia. There is some uncertainty about the common ancestor of both the Caribbean and South American populations, and we cannot definitively determine whether this common ancestor occupied South America, the Caribbean, Australasia, or a combination of those regions.
Gene flow between Australasia and the Southern tip of South America explains the connection of a sample from Argentina (C0090665F) with Australasian samples. A similar pattern was observed in a study on Pseudocyphellaria glabra using RAD-seq data, in which some samples from Chile and samples from New Zealand were closely interpreted as resulting from long-distance dispersal via migratory birds or wind currents carrying spores west to east [11]. An earlier study on the circumarctic lichen-forming fungus Porpidia flavicunda found that the disjunct populations shared haplotypes and lacked fixed nucleotide polymorphisms corresponding to their geographical locations, likely due to long-distance dispersal [48]. The odd placement of a single Taiwan sample (C0678932F) as sister to a New Zealand sample and apart from the larger Asian clade was further investigated by ITS barcoding and confirmed not to be an error. Therefore, further sampling is necessary to verify whether this sample represents a second species in Taiwan.
Further sampling is also required across Australasia to comprehensively assess the diversity of the Cladia aggregata complex in the region. Previous studies have documented considerable morphological and chemical variation within New Zealand and Australia, and our results suggest the presence of distinct populations and potentially distinct species within Australasia. But a broader and more geographically balanced sampling is needed for any definitive conclusions. RAD-seq of more evenly and extensively sampled Australasia populations could reveal the true biodiversity of C. aggregata in that region.
5. Conclusion
Our phylogenomic and population genetic analyses based on RAD-seq data clarified species boundaries within the C. aggregata species complex and revealed a previously unknown species from East Asia and Thailand, here described as Cladia asiatica. The genome-scale data clearly distinguished Asian from American samples, a separation not seen in earlier multilocus studies. Population genetics results showed little to no evidence of admixture or co-ancestry, likely due to limited gene flow across the continents. Biogeographic patterns derived from this larger dataset align with previous findings on the genus Cladia, indicating a diversification event that occurred before the more recent dispersals to Asia and the Americas. Overall, this work highlights the importance of genome-scale data for accurate species delimitation and the identification of previously overlooked diversity within the C. aggregata complex.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/doi/s1, Table S1: Sample Identified as other species in the C. aggregata species complex.; Table S2: The main compounds of each chemotype in the dataset and the geographic distribution of the samples within those chemotypes.; Table S3: Collection Data and herbaria accession numbers for all sequenced individuals.; Figure S1: RAxML tree with all 95 samples.; Figure S2: A phylogenetic, maximum likelihood tree made with RAxML from the 91 sample dataset.; Figure S3: A coalescent-based tree made with SVDquartets in PAUP.
Author Contributions
Author Contributions: Conceptualization, H.T.L., F.G. and J.D.; methodology, all authors; software, J.D.; validation, F.G., A.B.-P., and H.T.L.; formal analysis, J.D.; investigation, all authors; resources, F.G., H.T.L., V.P., J-P.H., and X.W.; data curation, J.D.; writing—original draft preparation, J.D.; writing— review and editing, all authors; funding acquisition, F.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Grainger Foundation and the National Natural Science Foundation of China, grant number 32570250.
Data Availability Statement
The RAD sequence data used in this study were deposited in the NCBI Sequence Read Archive (SRA) through the accession number PRJNA1337906. Accession numbers for RADseq raw sequences are listed in Table S3. All the scripts that were used in this study can be found on GitHub at: https://github.com/jddeabreu/Cladia_aggregata
Acknowledgments
The first author would like to thank Drs. John Dean and Windsor Aguirre (DePaul University, Chicago) for their advice on his thesis and helpful guidance. We are grateful to our colleagues at the Field Museum, especially Meredith Doellman, Yukun Sun, Matt Nelsen, Todd Widhelm, and the Pritzker lab team for their support. Funding was provided by the Grainger Bioinformatics Center at the Field Museum.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rannala, B.; Yang, Z. Species Delimitation. In Phylogenetics in the Genomic Era; No commercial publisher, 2020; p. pp.5.5:1--5.5:18.
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.B.; Worm, B. How Many Species Are There on Earth and in the Ocean? PLOS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef] [PubMed]
- Honegger, R.; Edwards, D.; Axe, L. The Earliest Records of Internally Stratified Cyanobacterial and Algal Lichens from the Lower Devonian of the Welsh Borderland. New Phytol. 2013, 197, 264–275. [Google Scholar] [CrossRef]
- Lumbsch, H.T.; Leavitt, S.D. Goodbye Morphology? A Paradigm Shift in the Delimitation of Species in Lichenized Fungi. Fungal Divers. 2011, 50, 59–72. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLOS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Barcenas-Peña, A.; Divakar, P.K.; Crespo, A.; Nuñez-Zapata, J.; Lumbsch, H.T.; Grewe, F. Reference-Based Restriction-Site-Associated DNA Sequencing Data Are Useful for Species Delineation in a Recently Diverged Asexually Reproducing Species Complex (Parmeliaceae, Ascomycota). J. Fungi 2023, 9, 1180. [Google Scholar] [CrossRef]
- Grewe, F.; Huang, J.-P.; Leavitt, S.D.; Lumbsch, H.T. Reference-Based RADseq Resolves Robust Relationships among Closely Related Species of Lichen-Forming Fungi Using Metagenomic DNA. Sci. Rep. 2017, 7, 9884. [Google Scholar] [CrossRef] [PubMed]
- Grewe, F.; Lagostina, E.; Wu, H.; Printzen, C.; Lumbsch, H.T. Population Genomic Analyses of RAD Sequences Resolves the Phylogenetic Relationship of the Lichen-Forming Fungal Species Usnea Antarctica and Usnea Aurantiacoatra. MycoKeys 2018, 43, 91–113. [Google Scholar] [CrossRef]
- Jorna, J.; Linde, J.B.; Searle, P.C.; Jackson, A.C.; Nielsen, M.-E.; Nate, M.S.; Saxton, N.A.; Grewe, F.; Herrera-Campos, M. de L.A.; Spjut, R.W.; et al. Species Boundaries in the Messy Middle-A Genome-Scale Validation of Species Delimitation in a Recently Diverged Lineage of Coastal Fog Desert Lichen Fungi. Ecol. Evol. 2021, 11, 18615–18632. [Google Scholar] [CrossRef]
- Otero, A.; Barcenas-Peña, A.; Lumbsch, H.T.; Grewe, F. Reference-Based RADseq Unravels the Evolutionary History of Polar Species in ‘the Crux Lichenologorum’ Genus Usnea (Parmeliaceae, Ascomycota). J. Fungi 2023, 9, 99. [Google Scholar] [CrossRef]
- Widhelm, T.J.; Grewe, F.; Huang, J.-P.; Ramanauskas, K.; Mason-Gamer, R.; Lumbsch, H.T. Using RADseq to Understand the Circum-Antarctic Distribution of a Lichenized Fungus, Pseudocyphellaria Glabra. J. Biogeogr. 2021, 48, 78–90. [Google Scholar] [CrossRef]
- Widhelm, T.J.; Rao, A.; Grewe, F.; Lumbsch, H.T. High-Throughput Sequencing Confirms the Boundary between Traditionally Considered Species Pairs in a Group of Lichenized Fungi (Peltigeraceae, Pseudocyphellaria). Bot. J. Linn. Soc. 2023, 201, 471–482. [Google Scholar] [CrossRef]
- Stenroos, S.; Pino-Bodas, R.; Hyvönen, J.; Lumbsch, H.T.; Ahti, T. Phylogeny of the Family Cladoniaceae (Lecanoromycetes, Ascomycota) Based on Sequences of Multiple Loci. Cladistics 2019, 35, 351–384. [Google Scholar] [CrossRef]
- Filson, R.B. A REVISION OF THE LICHEN GENUS CLAD/A NYL. Hattori Bot. Lab. 1981, 49, 1–75. [Google Scholar]
- Kantvilas, G.; Kantvilas, G.; Elix, J.A. Studies on the Lichen Genus Cladia Nyl. in Tasmania: The C. Aggregata Complex. Muelleria Aust. J. Bot. 1999, 12, 135–162. [Google Scholar] [CrossRef]
- Kantvilas, G.; Elix, J. A New Species of Cladia (Lichenized Ascomycotina) from Tasmania. Mycotaxon 1987, 29, 199–205. [Google Scholar] [CrossRef]
- Swartz, O.; Swartz, O.; Nordstrom, A.J.; Swederus, M. Nova Genera & Species Plantarum, Seu, Prodromus Descriptionum Vegetabilium :Maximam Partem Incognitorum Quæ Sub Itinere in Indiam Occidentalem Annis 1783-87; in bibliopoliis Acad. M. Swederi: Holmiae [Stockholm], Upsaliæ & Aboæ, 1788. [Google Scholar]
- Ahti, T. Cladoniaceae; Organization for Flora Neotropica: New York Botanical Garden, 2000; Vol. 78; ISBN 978-0-89327-431-3.
- Parnmen, S.; Rangsiruji, A.; Mongkolsuk, P.; Boonpragob, K.; Nutakki, A.; Lumbsch, H.T. Using Phylogenetic and Coalescent Methods to Understand the Species Diversity in the Cladia Aggregata Complex (Ascomycota, Lecanorales). PLOS ONE 2012, 7, e52245. [Google Scholar] [CrossRef]
- Parnmen, S.; Leavitt, S.; Rangsiruji, A.; Lumbsch, T. Identification of Species in the Cladia Aggregata Group Using DNA Barcoding (Ascomycota: Lecanorales). Phytotaxa 2013, 115, 1. [Google Scholar] [CrossRef]
- Otero, A.; Lumbsch, H.T.; Degtjarenko, P.; Grewe, F. Speciation Continuum in Non-Model Organisms: Revisiting the Species-Pair Concept in Lichens. Mol. Phylogenet. Evol. 2025, 108407. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLOS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.; Overcast, I. Output Formats — Ipyrad Documentation. Available online: https://ipyrad.readthedocs.io/en/latest/output_formats.html (accessed on 29 October 2024).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- FigTree. Available online: https://tree.bio.ed.ac.uk/software/figtree/ (accessed on 16 May 2025).
- Chifman, J.; Kubatko, L. Quartet Inference from SNP Data Under the Coalescent Model. Bioinformatics 2014, 30, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods) 2003.
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A General Species Delimitation Method with Applications to Phylogenetic Placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R Package for Genetic Analysis of Populations with Clonal, Partially Clonal, and/or Sexual Reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A.-B. The Ade4 Package: Implementing the Duality Diagram for Ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New Tools for the Analysis of Genome-Wide SNP Data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag New York, 2016; ISBN 978-3-319-24277-4.
- Malinsky, M.; Trucchi, E.; Lawson, D.J.; Falush, D. RADpainter and fineRADstructure: Population Inference from RADseq Data. Mol. Biol. Evol. 2018, 35, 1284–1290. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Lang, D.T.; Kalibera, T. XML: Tools for Parsing and Generating XML Within R and S-Plus 2025.
- Raj, A.; Stephens, M.; Pritchard, J.K. fastSTRUCTURE: Variational Inference of Population Structure in Large SNP Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef]
- Purcell, S.; Chang, C. PLINK 1. 9 2024.
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Francis, R.M. Pophelper: An R Package and Web App to Analyse and Visualize Population Structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef]
- Yu, Y.; Blair, C.; He, X. RASP 4: Ancestral State Reconstruction Tool for Multiple Genes and Characters. Mol. Biol. Evol. 2020, 37, 604–606. [Google Scholar] [CrossRef]
- Matzke, N.J. Probabilistic Historical Biogeography: New Models for Founder-Event Speciation, Imperfect Detection, and Fossils Allow Improved Accuracy and Model-Testing. Front. Biogeogr. 2013, 5. [Google Scholar] [CrossRef]
- Ali, S.S.; Yu, Y.; Pfosser, M.; Wetschnig, W. Inferences of Biogeographical Histories within Subfamily Hyacinthoideae Using S-DIVA and Bayesian Binary MCMC Analysis Implemented in RASP (Reconstruct Ancestral State in Phylogenies). Ann. Bot. 2012, 109, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian Phylogenetic Inference under Mixed Models. Bioinforma. Oxf. Engl. 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Buschbom, J. Migration between Continents: Geographical Structure and Long-Distance Gene Flow in Porpidia Flavicunda (Lichen-Forming Ascomycota). Mol. Ecol. 2007, 16, 1835–1846. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A phylogenetic, Maximum Likelihood tree calculated with IQ-TREE v2. Each sample ID is represented in the following format: the collection or accession number that was used in the original ipyrad analysis as a sample ID, followed by the species and information found in Parnmen et al. (2013) [20] when applicable, then the country where the sample was collected, and finally the chemotype. Bootstrap values above 75 are shown next to their respective branches
Figure 1.
A phylogenetic, Maximum Likelihood tree calculated with IQ-TREE v2. Each sample ID is represented in the following format: the collection or accession number that was used in the original ipyrad analysis as a sample ID, followed by the species and information found in Parnmen et al. (2013) [20] when applicable, then the country where the sample was collected, and finally the chemotype. Bootstrap values above 75 are shown next to their respective branches
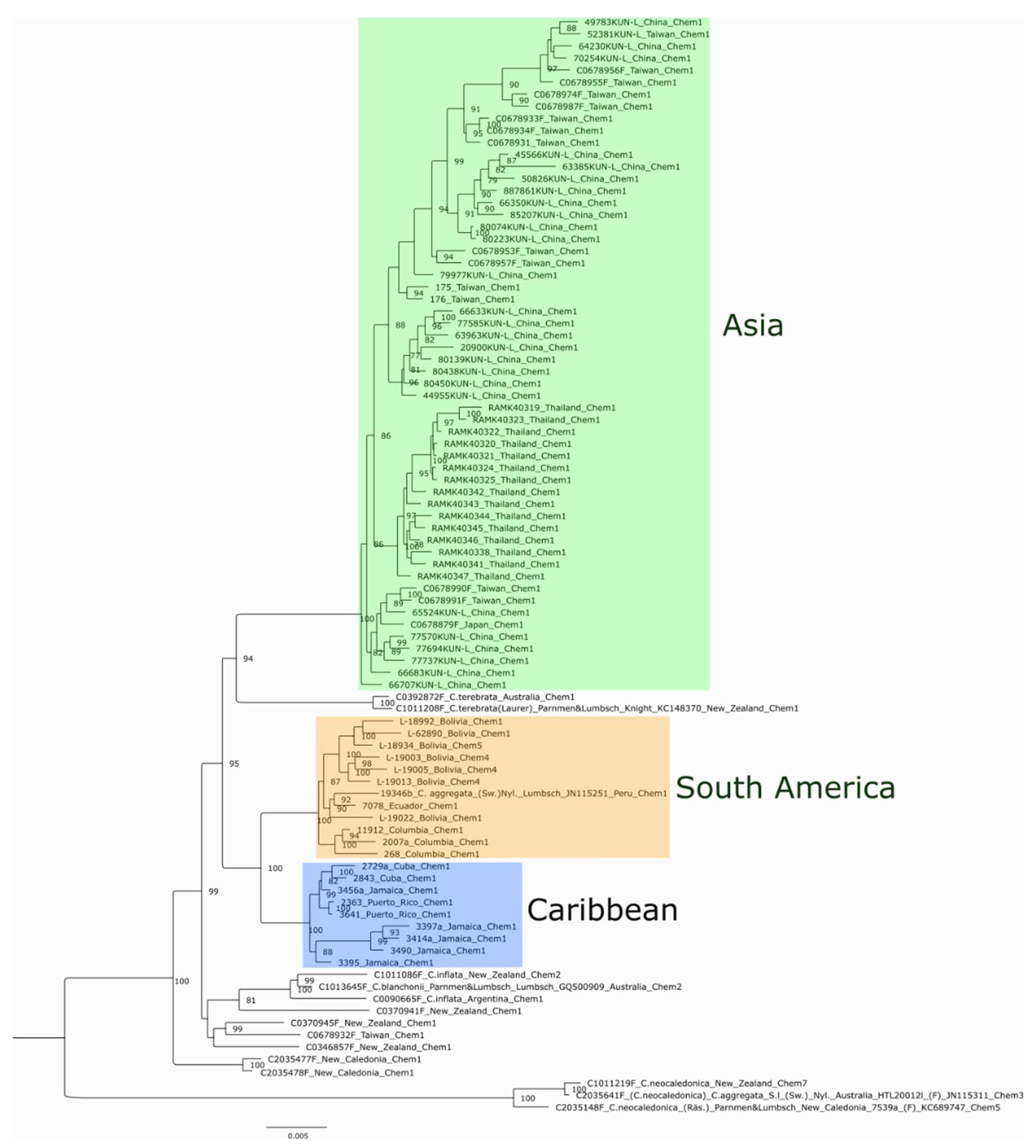
Figure 2.
Population structure of Cladia aggregata samples from Asia and the Americas. (A) Principal component analysis (PCA) of Asian and American samples, with PC1 (x-axis) explaining 41.5% and PC2 (y-axis) explaining 8.4% of the total genetic variation. (B) Co-ancestry heatmap generated with fineRADstructure, showing relatedness among individuals. The dendrogram above the plot represents the hierarchical clustering of samples inferred by fineRADstructure. (C) fastSTRUCTURE results for K = 2–6. Each horizontal bar represents an individual sample, with colors indicating inferred genomic ancestry components. Results are grouped by K value (y-axis) and population (x-axis).
Figure 2.
Population structure of Cladia aggregata samples from Asia and the Americas. (A) Principal component analysis (PCA) of Asian and American samples, with PC1 (x-axis) explaining 41.5% and PC2 (y-axis) explaining 8.4% of the total genetic variation. (B) Co-ancestry heatmap generated with fineRADstructure, showing relatedness among individuals. The dendrogram above the plot represents the hierarchical clustering of samples inferred by fineRADstructure. (C) fastSTRUCTURE results for K = 2–6. Each horizontal bar represents an individual sample, with colors indicating inferred genomic ancestry components. Results are grouped by K value (y-axis) and population (x-axis).
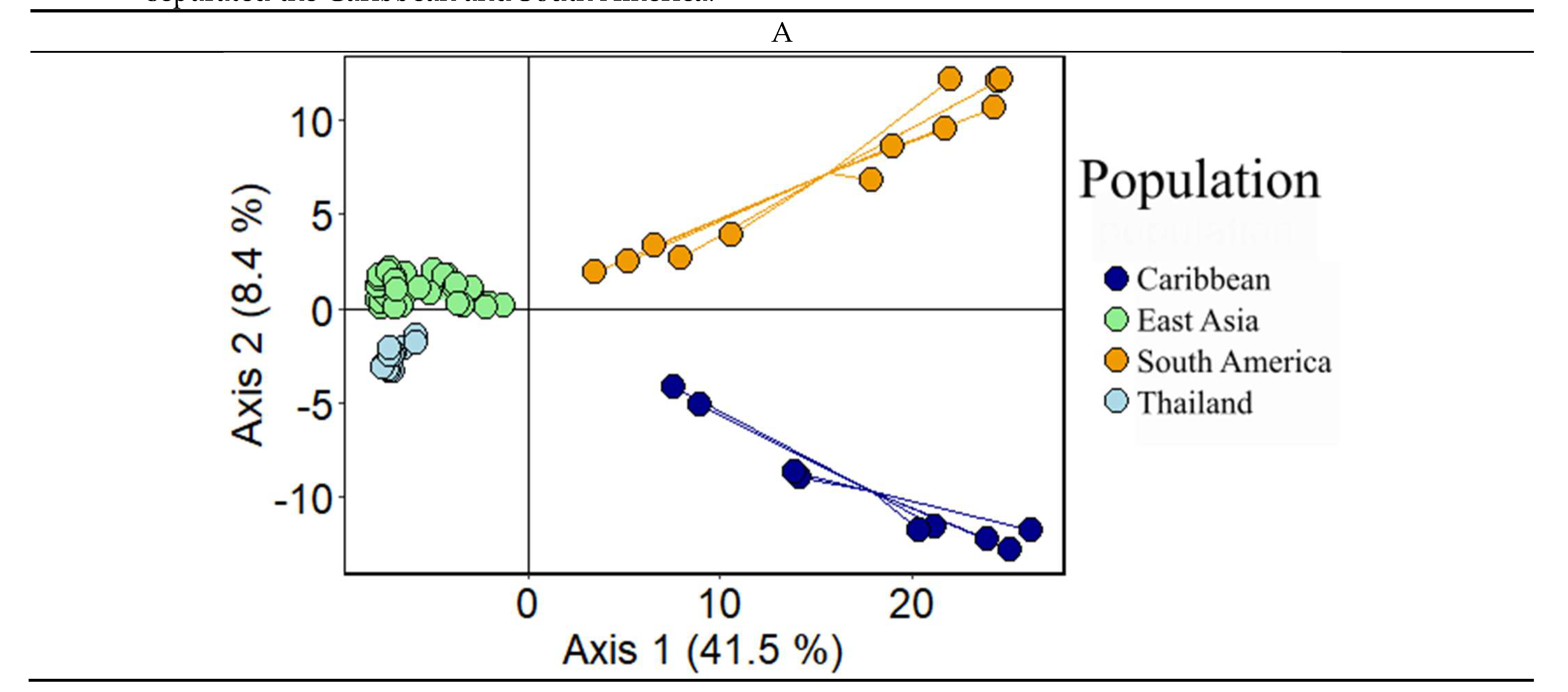
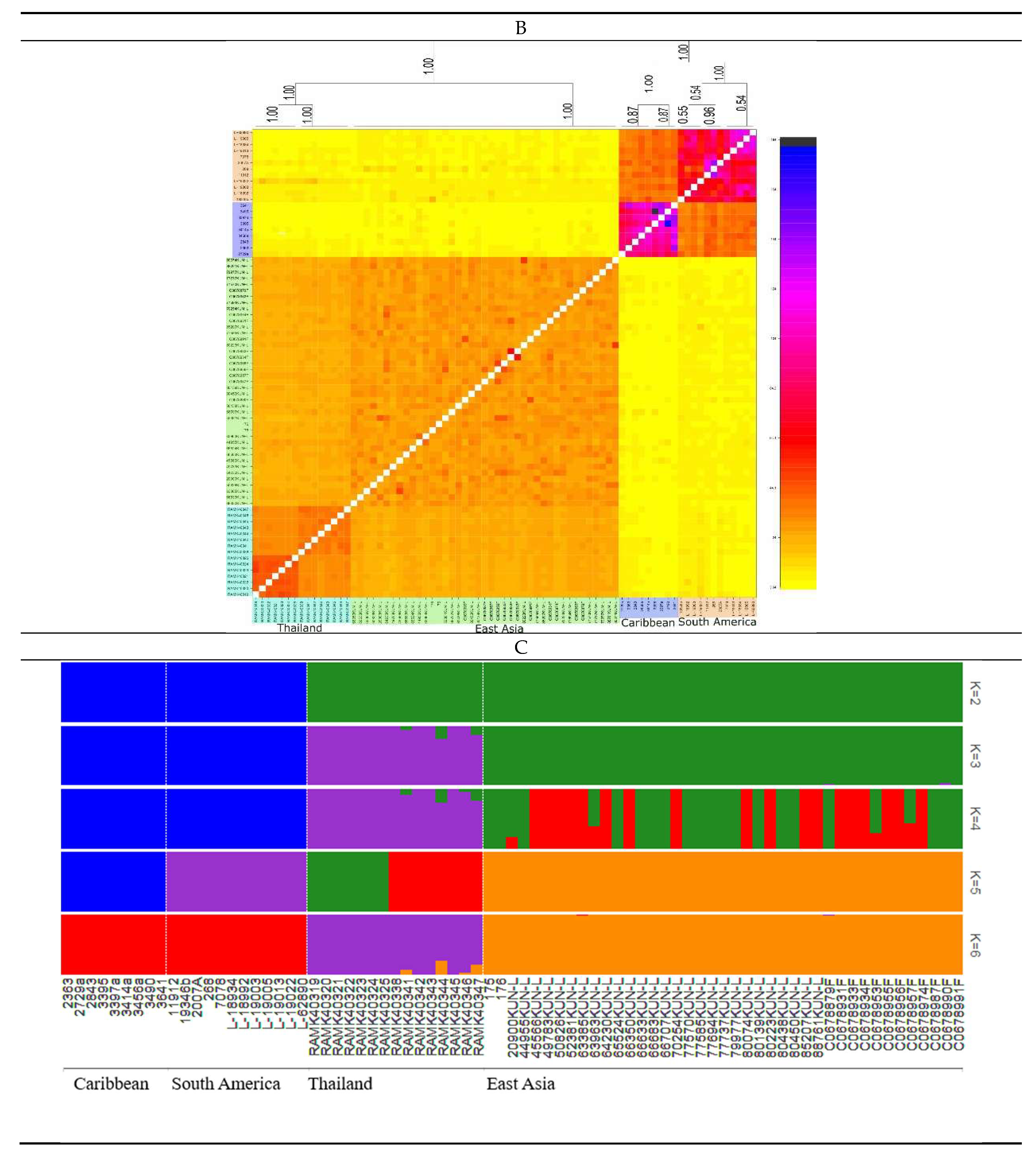

Table 1.
Results of ipyrad Assembly and Analysis of RAD-seq Data.
| Number of Filtered Loci | 5,720 |
| Number of SNPs | 46,731 |
| Percentage of Missing Data | snps matrix size: 81.00% missing sites. sequence matrix size: 87.95% missing sites. |
| Average Number of Loci Per Sample | 1,480.48 |
| Average Number of Raw Reads | 7,068,030.81 |
| Average Number of Mapped Reads | 187,914.98 |
| Average Error Estimate | 0.0017 |
Table 2.
Analysis of Molecular Variance (AMOVA) and Monte Carlo Test for Samples of C. aggregata from East Asia, Thailand, South America, and the Caribbean.
Table 2.
Analysis of Molecular Variance (AMOVA) and Monte Carlo Test for Samples of C. aggregata from East Asia, Thailand, South America, and the Caribbean.
| Variation Between Groups | 544.9762 | 79.19% |
| Variation Within Groups | 143.2486 | 20.81% |
| Total Variation | 688.2249 | 100% |
| Phi-samples-total | 0.7918578 | |
| Monte-Carlo Test P Value | 0.001 | |
| Average Variance of Simulated Groups | 170.36 | |
| Observed Variance Between Actual Groups | 544.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



