
Submitted:
03 December 2025
Posted:
05 December 2025
You are already at the latest version
Abstract
Terpyridines are well-known ligands in coordination chemistry due to their conforma-tional flexibility and strong metal-binding properties; therefore, they serve as attractive platforms for the design and synthesis of new functional derivatives. This study focused on the synthesis and comprehensive investigation of a new class of bis-oxazolo[5,4-b]pyridine derivatives, designed based on their structural similarity to terpyridines. Four novel compounds 4a–d were synthesized by cyclization of amide de-rivatives of 3-aminopyridin-2(1H)-ones using pyridine-2,6-dicarboxylic acid and its di-chloride as key acidic components. Their structures and purity were confirmed by melt-ing point analysis, high-resolution mass spectrometry, and detailed 1H NMR spectros-copy. Photophysical studies in chloroform, dichloromethane, and acetonitrile revealed that compounds 4a–c exhibit intense blue to deep blue fluorescence (л_max 323–347 nm) with high quantum yields (φi ≈ 0.32–0.84), attributed to р–π* transitions within the con-jugated ring system. These findings suggest their potential as air-stable phosphors for organic electronics. Computational modeling of 4a–c molecules provided insight into their electronic structures, conformational stability, and predicted optical behavior. The most stable conformers (4a–II, 4b–II, 4c–II′) showed decreasing HOMO–LUMO gaps and photoactivity from 4a to 4c, with 4a–II emerging as the most promising fluorophore due to its high symmetry and consistent emission. Overall, this study lays the foundation for future studies of bis(oxazolo[5,4-b]pyridine) derivatives in coordination chemistry and optoelectronic materials development.
Keywords:
3-aminopyridin-2(1H)-ones
; oxazolo[5
; 4-b]pyridines
; intramolecular heterocyclization
; structural analogs of terpyridine
; UV spectroscopy
; optical properties
; quantum exit
; Stokes shift
; in silico
; molecular docking
; computational study
1. Introduction
One of the key challenges in modern organic and coordination chemistry is creating new, highly effective fluorophores and ligands with useful properties that can be applied in industry, everyday technology, and daily life. Many organic fluorophores are successfully used as biosensors, biomarkers, and dyes for labeling cells [1,2,3,4,5]. They are also widely used in organic electronics, including LEDs, transistors, batteries, solar cells, and other devices [6,7,8].
Finding such materials is especially important not only for electronics and optics, but also for medicine. This search is a major focus in chemistry and related fields. Today, most active research on new useful materials is based on ligands that contain coordinating parts from different types of organic compounds, mainly heterocycles like macrocycles, crown ethers, terpyridines, porphyrins, phthalocyanines, and others.
To evaluate how well fluorophores work, scientists look at properties like fluorescence quantum yield, Stokes shift, molar extinction coefficient, and absorption/emission peaks [1]. These values, measured using UV–Vis spectroscopy, help choose the right fluorophores for tasks like bioimaging. However, only a small number of known or new fluorophores have ideal properties, which limits their wider use.
Designing and testing new fluorophores with better light-related properties is still a difficult and time-consuming process, even with general guidelines for making new dyes. That’s why finding and improving simple methods to make better dyes remains an important goal.
It’s also well known that organic ligands often play a key role in controlling how metal ions react [9]. Since organometallic chemistry studies how metal ions interact with organic ligands, sometimes a single “special” ligand can make a big difference in coordination chemistry [10,11,12,13]. Nitrogen-containing ligands are especially important and are widely used in coordination compounds [14,15,16]. These ligands can be either monodentate or multidentate, depending on how many coordination sites they have.
Among nitrogen-based ligands, 2,2’:6’,2″-terpyridine 1 (tpy) has gained a lot of attention over the past 20 years [17,18,19]. Terpyridine derivatives and their metal complexes are used in making fluorescent sensors and biological labels, in photocatalysis, in solar cell sensitization, in molecular logic devices, organic LEDs, and in catalysis.
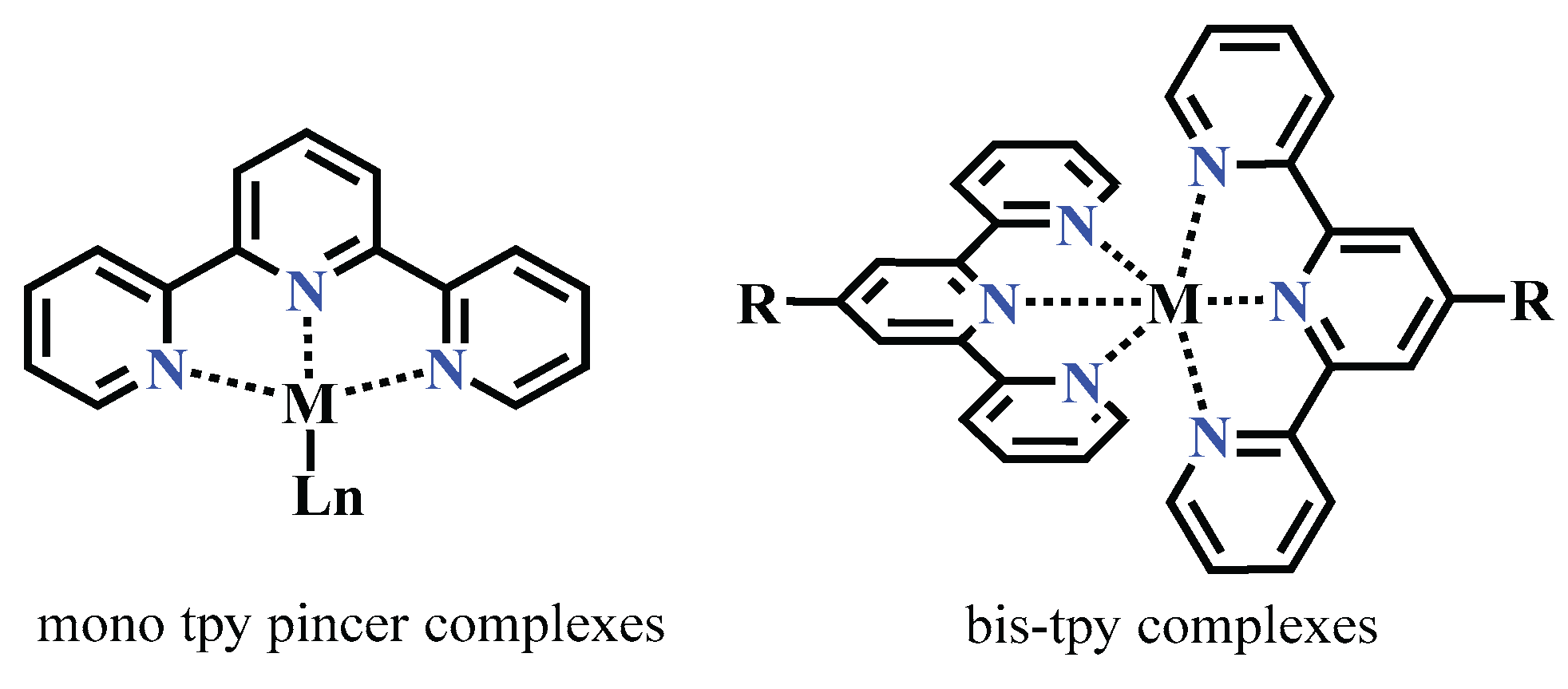
The uniqueness of terpyridine 1 and its metal complexes is based on three key features.
First, terpyridine 1 contains three electron-deficient pyridine rings, which makes it not only a strong σ-donor but also an excellent π-acceptor. With three nitrogen atoms as coordination sites, it acts as a tridentate NNN-type chelating ligand, capable of forming stable complexes with various metal cations in a nearly planar geometry. The lone pairs on the nitrogen atoms allow terpyridine to coordinate not only with transition, heavy, and colored metal ions, but also with lanthanide ions.
Second, depending on the metal-to-ligand ratio, two types of coordination are possible: mono-chelate complexes of the type {M(tpy)} and bis-chelate complexes of the type {M(tpy)₂} (Scheme 1).
As can be seen in Scheme 1, a trans–trans configuration relative to the central ring is typically adopted by the two outer pyridine rings in terpyridine before coordination with metal cations. This arrangement helps minimize repulsion between the lone pairs of electrons on the nitrogen atoms.
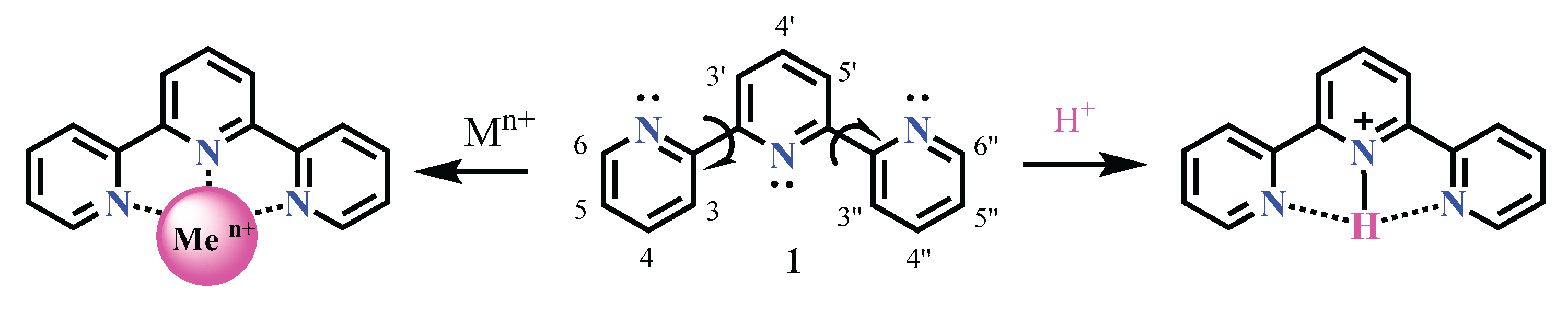
Thirdly, upon coordination with a metal ion or under simple acidification, the terpyridine ligand undergoes a conformational change to a cis–cis geometry [20,21,22], in which all three pyridine rings become ideally coplanar. This coplanarity allows for better conjugation between the aromatic rings and the metal center, enhancing electronic communication and stabilizing the complex (Scheme 2).
Based on its three nitrogen coordination sites and flexible sigma bonds between the pyridine rings, 2,2’:6’,2″-terpyridine 1 can act as a tridentate chelating ligand, capable of stabilizing cations of various transition metals. This property makes tpy a promising ligand for supporting low-valent metal centers, including nickel, iron, cobalt, manganese, and others.
Another important feature of the terpyridine ligand is its ability to promote single-electron transfer processes, allowing metal-centered sites in the complex to participate in catalytic reactions. For example, terpyridine–nickel complexes are known to catalyze various C–C bond-forming cross-coupling reactions [23,24,25,26].
In recent years, it has been demonstrated that tpy–Mn, tpy–Fe, and tpy–Co complexes can be successfully used for the hydrofunctionalization of multiple C–C bonds and in cross-coupling reactions [27,28,29]. These advances have generated considerable interest in further exploring the potential catalytic activity of such unique terpyridine-based complexes.
For example, it is well known that low-valent nickel complexes (Ni(0) and Ni(I)) exhibit good reactivity in oxidative addition reactions to carbon–halogen bonds [30]. However, one of the main challenges is stabilizing the highly reactive Ni center to ensure efficient catalytic conversion.
In addition to the excellent properties that terpyridine-based metal complexes have demonstrated in organometallic chemistry, terpyridine also attracts great interest in the design and construction of complex supramolecular frameworks through hierarchical self-assembly. Over the past decades, many researchers have focused on developing various and often intricate supramolecular organic architectures that can mimic biological systems and may be useful in chemical sensing, molecular imaging, drug delivery, metal extraction, and other applications [31].
However, building such large supramolecular structures and controlling their self-assembly processes effectively remains a major challenge. In this context, terpyridine–metal complexes have proven especially helpful, as they allow researchers to guide self-assembly through non-covalent interactions between the coordinated transition metal and the pre-organized ligand.
As a result, several research groups have successfully synthesized impressive and representative structures, such as the “Star of David,” “Nock’s Snowflake,” and the “Sierpinski Triangle” [32,33].
Thus, the brief overview presented above clearly shows that the range of potential and practical applications of 2,2’:6’,2″-terpyridine derivatives and their metal complexes is remarkably broad. Substituted terpyridines are commonly used in research and applications, so finding new ligands based on 2,2’:6’,2″-terpyridine and related structures is a timely and important task.
However, there are still some unresolved issues in the use of terpyridines and their derivatives, such as low overall synthetic yields and poor solubility of terpyridine-based ligands.
For this reason, discovering new terpyridine-like structures that keep or even improve its coordination abilities is a pressing and important goal in modern organic and especially coordination chemistry. This is particularly true in today’s fast-moving scientific environment, where there is a constant demand for new, practically useful materials for broad applications in areas such as organic electronics, molecular science, medicine, and drug discovery [34,35].
The main goal of this study was to identify and comprehensively examine a series of structurally unique and previously undescribed bis-derivatives of oxazolo[5,4-b]pyridines. These compounds were selected as close structural analogues of terpyridines, which are well known for their versatility and effectiveness as ligands in coordination chemistry.
2. Materials and Methods
2.1. Material and Instrumentation
The Materials and Methods should be described with sufficient details to allow others to replicate and build on the published results. Please note that the publication of your manuscript implicates that you must make all materials, data, computer code, and protocols associated with the publication available to readers. Please disclose at the submission stage any restrictions on the availability of materials or information. New methods and protocols should be described in detail while well-established methods can be briefly described and appropriately cited.
1H and 13C NMR spectra were recorded on Bruker AVANCE 500 (500 and 126 MHz, respectively), Bruker Avance NEO (400 and 101 MHz, respectively) and Magritek spinsolve 80 carbon ultra (81 and 20 MHz, respectively) instruments using CDCl3; residual solvent signals (7.26 and 77.2 ppm for 1H and 13C nuclei) served as an internal standard.
Chromatographic mass spectrometric studies were carried out on a Trace GC Ultra chromatograph with a DSQ II mass-selective detector in electron ionization mode (70 eV) on a Thermo TR-5 MS quartz capillary column 15 m long, with an internal diameter of 0.25 mm, and a stationary phase film thickness of 0.25 μm. The splitless input mode was used. The carrier gas flow rate was 20 ml/min. The carrier gas (helium) flow rate was 1 ml/min. The following temperature parameters were set: evaporator temperature 200 °C, transition chamber temperature 200 °C, ion source temperature 200 °C. The temperature of the column thermostat was changed according to the following program: 20 °C per minute - in the temperature range from 15 (delay 5 min) to 220 °C; 15 °C per minute - in the temperature range up to 290 °C. The total analysis time was 30 minutes. The volume of the injected sample was 1 µl. Chromatograms were recorded in TIC mode. The mass scanning range was 30 - 500 a.u.m.
Melting points were determined on a Stuart SMP10 apparatus. Elemental analysis was performed on a Carlo Erba 1106CHN instrument. The reaction progress and product purity were monitored by TLC on Sorbfil plates and visualized using iodine vapor or UV light.
UV-visible spectra were recorded in the range of 200-600 nm in a quartz cell with an optical path length of 1 cm using a Shimadzu UV-2600 spectrophotometer. Absorption spectrum of the studied compounds were recorded in the range of 300-700 nm at room temperature using a Shimadzu RF 5301 PC spectrofluorimeter.
Fluorescence spectra were recorded using an RF-5301 PC spectrofluorimeter (Shimadzu). Quinine sulfate in 0.1 M H₂SO₄ was used as the internal standard for compounds 4c and 4d, while tryptophan at a concentration of 10⁻⁶ mol/L was used for compounds 4a and 4b. The pH of the tryptophan solution was adjusted to 6.1 using a phosphate buffer.
The relative quantum yield was calculated using the following formula:
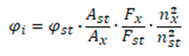
where φi - is the fluorescence quantum yield of the studied compound (4a-d);
φst - is the fluorescence quantum yield of the standard used (quinine sulfate or tryptophan);
Ast, Ax - is the absorbances at excitation wavelength for the standard sample (st) and the studied compound (4a-d);
Fst, Fx - is the areas of fluorescence spectrum with spectral correction for the standard sample (st) and the studied compound (4a-d);
nst, nx - is the refractive index of the standard sample (st) and the studied compound (4a-d).
Synthesis and Spectral Analysis of Synthesized Compounds
3-Aminopyridine-2(1H)-ones 1а-d were prepared according to a similar literature procedure [37].
Synthesis of Diacyl Derivatives of Pyridine-2,6-dicarboxylic Acid
General Procedure for Compounds 3a–d
A mixture of 1.0 mmol of 3-aminopyridin-2(1H)-one (1a–d) and 2.0 mmol of triethylamine (TEA) was dissolved in 10 mL of dichloromethane. While stirring and cooling, 0.5 mmol of pyridine-2,6-dicarboxylic acid dichloride dissolved in 3 mL of dichloromethane was added dropwise. The reaction mixture was stirred at room temperature for 20–25 hours. After completion, the mixture was washed with distilled water and extracted with dichloromethane. The combined organic layers were dried over Na₂SO₄, and the solvent was removed. The residue was recrystallized from a hexane–isopropanol mixture (1:2).
N²,N⁶-Bis(4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)pyridine-2,6-dicarboxamide3aYield: 232 mg (57%), colorless microcrystalline powder, mp 306–307 °C. ¹H NMR (500 MHz, DMSO-d₆) δ (J, Hz): 2.02 (s, 6H, 2CH₃), 2.16 (s, 6H, 2CH₃), 5.97 (s, 2H, H-5,5′), 8.21–8.29 (m, 3H, H-3,4,5 Py), 10.43 (s, 2H, 2NHCO), 11.73 (s, 2H, 2NHCO). 13C NMR (125 MHz, DMSO-d₆) δ: 18.2, 106.6, 122.1, 124.8, 139.6, 142.4, 147.6, 148.7, 160.4, 162.1. Found: C 61.72; H 5.02; N 17.70. Calculated for C₂₁H₂₁N₅O₄: C 61.91; H 5.20; N 17.19.
N²,N⁶-Bis(6-methyl-2-oxo-4-phenyl-1,2-dihydropyridin-3-yl)pyridine-2,6-dicarboxamide 3b Yield: 306 mg (62%), light gray microcrystalline powder, mp 238–239 °C. 1H NMR (400 MHz, DMSO-d₆) δ: 2.25 (s, 6H), 6.10 (s, 2H), 7.28 (br. t, J=3.1, 6H, Ph), 7.49–7.52 (m, 4H, Ph), 8.09 (br. s, 3H, H-3,4,5 Py), 10.34 (s, 2H), 11.96 (s, 2H). 13C NMR (100 MHz, DMSO-d₆) δ: 18.4, 105.6, 120.9, 124.4, 127.6, 128.1, 128.3, 137.3, 139.6, 143.7, 148.4, 149.9, 160.9, 162.7. Found: C 70.28; H 4.50; N 13.48. Calculated for C₃₁H₂₅N₅O₄: C 70.05; H 4.74; N 13.18.
N²,N⁶-Bis(6-methyl-2-oxo-4-(thiophen-2-yl)-1,2-dihydropyridin-3-yl)pyridine-2,6-dicarboxamide 3c Yield: 315 mg (58%), light gray microcrystalline powder, mp 255–256 °C. 1H NMR (500 MHz, DMSO-d₆) δ: 2.24 (s, 6H), 6.54 (s, 2H), 7.10 (dd, J=4.9, 4.0, 2H), 7.67 (d, J=4.9, 2H), 7.74 (d, J=3.1, 2H), 8.23–8.29 (m, 3H, H-3,4,5 Py), 10.62 (s, 2H), 11.78 (s, 2H). 13C NMR (125 MHz, DMSO-d₆) δ: 18.5, 102.3, 119.2, 124.6, 127.0, 128.9, 130.4, 139.6, 141.3, 143.2, 148.7, 160.8, 163.2. Found: C 59.80; H 3.67; N 13.03. Calculated for C₂₇H₂₁N₅O₄S₂: C 59.66; H 3.89; N 12.88.
N²,N⁶-Bis(6-methyl-4-(5-methylfuran-2-yl)-2-oxo-1,2-dihydropyridin-3-yl)pyridine-2,6-dicarboxamide 3d Yield: 318 mg (59%), light gray microcrystalline powder, mp 258–260 °C. 1H NMR (80 MHz, DMSO-d₆) δ: 2.25 (br. s, 12H), 6.54 (d, J=3.1, 2H), 6.42 (s, 2H), 6.92 (d, J=3.1, 2H), 8.28 (br. s, 3H, H-3,4,5 Py), 10.56 (br. s, 2H), 11.75 (br. s, 2H). 13C NMR (20 MHz, DMSO-d₆) δ: 13.3, 18.6, 100.1, 109.0, 114.8, 117.5, 124.8, 137.2, 139.8, 143.7, 146.8, 148.9, 153.7, 160.8, 162.5. Found: C 64.89; H 4.94; N 13.23. Calculated for C₂₉H₂₅N₅O₆: C 64.56; H 4.67; N 12.98.
Synthesis of 2,5-Bis(oxazolo[5,4-b]pyridine) Derivatives
General Procedure for Compounds 4a–d
A mixture of 1.0 mmol of N²,N⁵-bis(2-oxo-1,2-dihydropyridin-3-yl) pyridine-2,5-dicarboxamide (3a–d) and 5.0 mmol of phosphorus oxychloride was heated at 90–100 °C under reflux with a calcium chloride drying tube for 5 hours. Excess phosphorus oxychloride was removed under vacuum. The reaction mixture was treated with cold water, and the resulting precipitate was filtered, washed with distilled water, and air-dried. The product was recrystallized from a mixture of isopropanol and dichloromethane (2:3).
2-(6-(5,7-Dimethyl-3a,4-dihydrooxazolo[5,4-b]pyridin-2-yl)pyridin-2-yl)-5,7-dimethyloxazolo[5,4-b]pyridine 4a Yield: 304 mg (82%), colorless crystals, mp 259–260 °C. 1H NMR (500 MHz, DMSO-d₆), δ, (J, Hz): 2.03 (s, 6H, 2 CH₃), 2.16 (s, 6H, 2 CH₃), 7.30 (s, 2H), 8.32 (t, J=2.7, 1H, H-4 of Py), 8.53 (d, J=7.8, 2H, H-3,5 of Py). High-resolution mass spectrum (ESI-QTOF): found m/z [M+H]⁺ 372.1453; calculated for C₂₁H₁₈N₅O₂⁺: 372.1455.
5-Methyl-2-(6-(5-methyl-7-phenyl-3a,4-dihydrooxazolo[5,4-b]pyridin-2-yl)pyridin-2-yl)-7-phenyloxazolo[5,4-b]pyridine 4b Yield: 342 mg (69%), white microcrystalline powder, mp 288–289 °C. ¹H NMR (400 MHz, DMSO-d₆), δ, (J, Hz): 2.70 (s, 6H, 2CH₃), 7.57 (t, J = 7.1, 2H, H-4,4′ of Ph), 7.64 (t, J = 7.3, 4H, H-3,5,3′,5′ of Ph), 7.80 (s, 2H, H-6,6′), 8.30 (d, J = 7.8, 4H, H-2,6,2′,6′ of Ph), 8.34–8.36 (m, 1H, H-4 of Py), 8.60 (d, J = 7.8, 2H, H-3,5 of Py). Found: C 75.42; H 3.98; N 14.46. Calculated for C₃₁H₂₁N₅O₂: C 75.14; H 4.27; N 14.13.
2,6-Bis(5-methyl-7-(thiophen-2-yl)-3a,4-dihydrooxazolo[5,4-b]pyridin-2-yl)pyridine 4c Yield: 368 mg (72%), light gray microcrystalline powder, mp 363–365 °C. ¹H NMR (80 MHz, DMSO-d₆) δ (J, Hz): 2.65 (br. s, 6H, 2CH₃), 7.35 (br. s, 2H, H-4′,4″ of thiophene), 7.74 (s, 2H, H-6,6′), 7.93 (d, J = 4.0, 2H, H-3′,3″ of thiophene), 8.25 (d, J=4.5, 2H, H-5′,5″ of thiophene), 8.30–8.61 (m, 3H, H-3,4,5 Py). Found: C 63.75; H 4.38; N 13.47. Calculated for C₂₇H₂₁N₅O₂S₂: C 63.39; H 4.14; N 13.69.
2,6-Bis(5-methyl-7-(5-methylfuran-2-yl)-3a,4-dihydrooxazolo[5,4-b]pyridin-2-yl)pyridine 4d Yield: 431 mg (85%), white microcrystalline powder, mp 296–298 °C. ¹H NMR (80 MHz, DMSO-d₆) δ (J, Hz): 2.41 (s, 6H, 2 CH3); 2.62 (s, 6H, 2 CH3); 6.26 (d, 2H, J=2.5 Hz, Н-4’,4’’ Fu); 7.47 (s, 2Н, Н-6,6’); 7.63 (d, 2Н, J=2.9, Н-3’,3’’ Fu); 8.16 (t, 1Н, J=7.7, H-4 Py); 8.53 (d, J=7.4, 2Н, H-3,5 Py). Found: C 68.92; H 5.18; N 14.06. Calculated for C₂₉H₂₅N₅O₄: C 68.63; H 4.97; N 13.80.
2.3. Conformational Analysis and Electronic Structure of the New 2,6-Bis(oxazolo[5,4-b]pyridin-2-yl)pyridines
Geometry optimizations and frequency calculations (OPT+FREQ) were performed using DFT with the ωB97XD functional [38] and 6-311++G(d,p) basis set [39] in Gaussian 16 [40]. Molecular structures were built and visualized in GaussView 6.0 [41].
The ωB97XD functional was chosen for its reliable treatment of dispersion forces, conformational flexibility, and π-delocalization—key features for modeling heteroaromatic systems like 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridines. Its long-range correction and empirical dispersion terms ensure accurate representation of noncovalent and stacking interactions [42].
Optimizations were first performed in vacuum to assess intrinsic conformational preferences. Solvent effects were then evaluated via geometry optimizations CPCM calculations [43] in dichloromethane and acetonitrile, representing specroscopic medium.
All optimized geometries were confirmed as true minima by the absence of imaginary frequencies [44]. Thermochemical data, dipole moments, and electronic descriptors were extracted from the output files [45].
Frontier molecular orbitals (HOMO and LUMO) were visualized to assess electron distribution, energy gaps, and donor–acceptor potential. Orbital localization was analyzed to identify reactive centers and evaluate π-conjugation. Molecular electrostatic potential (MEP) maps were generated on the electron density isosurface (0.001 a.u.) to visualize charge distribution [46]. Color-coded gradients highlight nucleophilic and electrophilic regions, aiding interpretation of coordination and supramolecular behavior [47].
2.4. DFT-Based Simulation of Electronic Absorption and Luminescence Properties
UV–vis absorption and emission spectra were simulated using time-dependent density functional theory (TD DFT) as implemented in Gaussian 16 [48]. All calculations were performed with the long-range corrected ωB97XD functional and the 6-311++G(d,p) basis set, incorporating solvent effects via the conductor-like polarizable continuum model (CPCM) with acetonitrile as the solvent.
Initial geometry optimizations of the ground state (S₀) were carried out using DFT at the ωB97XD/6-311++G(d,p) level with CPCM solvation. Vertical excitation energies and oscillator strengths were then computed via TD DFT using the keyword TD=(NStates=30, Root=1) to simulate the UV–vis absorption spectrum. The resulting transitions from S₀ to excited singlet states (Sn) were analyzed to identify key absorption bands.
To simulate luminescence, the geometry of the first excited state (S₁) was optimized. Following excited-state optimization, vertical transitions from the relaxed excited state back to the ground state (S₁ → S₀) were computed using TD DFT. These transitions represent the emission processes and were used to construct theoretical luminescence spectra.
Excitation energies and oscillator strengths were extracted from Gaussian output files. Spectral profiles were visualized using MS Excel software.
3. Results and Discussion
3.1. Chemistry
Our previous research has shown that 4-aryl-(hetaryl-)substituted 3-aminopyridin-2(1H)-ones are largely unexplored in the literature [34] and can serve as excellent synthons for the development of efficient luminophores [37]. Even the simplest derivative, 4-phenyl-3-aminopyridin-2(1H)-one, demonstrated a remarkably high fluorescence quantum yield of 76% [49]. The presence of an amino acid-like fragment in the 3-aminopyridone structure also makes these compounds valuable building blocks for the synthesis of potential peptidomimetics [45,46,47,48,49,50,51,52] and more complex biologically active heterocyclic systems [53,54,55].
The main strategy and goal of this study was to synthesize structural analogues of terpyridines using our previously developed cyclization methods for amide derivatives of 3-aminopyridin-2(1H)-ones [56,57,58,59]. For this purpose, pyridine-2,6-dicarboxylic acid and its dichloride derivative were selected as the acid components.
We have previously established that 4-aryl-substituted 3-aminopyridin-2(1H)-ones 2a–d undergo acylation with pyridine-2,6-dicarboxylic acid dichloride in the presence of triethylamine, forming the corresponding diamides 3a–d. Further heating of these diamides with phosphorus oxychloride leads to cyclization products of 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridines 4a–d with the yields of around 80% (Scheme 3).
A comparative analysis of the synthesized 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridines 4a–d showed that they are structurally very similar to terpyridines. However, unlike terpyridine, these compounds contain three heteroatoms (one oxygen and two nitrogen atoms) in each peripheral condensed oxazolopyridine ring system. This structural feature may lead to improved coordination and luminescent properties due to the increased number of conjugated bonds, the presence of additional electron-accepting oxygen and nitrogen atoms, and the potential for new conformations resulting from rotation around the C(2) and C(6) bonds of the central pyridine core.
3.2. Photophysical Properties of Compounds
The synthesized 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridines (4a–d) show strong fluorescence in the blue and deep-blue region when exposed to UV light (Figure 1). This property is especially important for potential applications in organic electronics, where there is a known shortage of air-stable organic luminophores emitting in the blue range.
In addition, these compounds may serve as promising chelating agents and fluorescent markers for various metal cations, including rare-earth elements, with the potential to enhance luminescence upon complex formation.
Spectral properties, including λmax, Stokes shift, and quantum yield of the synthesized compounds 4a–d in chloroform, dichloromethane and acetonitrile medium were measured since they are of interest as potential luminophores. As a first step, we recorded the UV–Vis absorption spectra of the bis(oxazolopyridine) derivatives 4a–d in three solvents: chloroform, dichloromethane, and acetonitrile. The measurements were carried out at a concentration of 2×10⁻⁵ M for compounds 4a, 4c, and 4d, and 1×10⁻⁵ M for compound 4b. The spectral characteristics of the compounds 4a–d are summarized in Table 1. The absorption spectra of compounds 4a–d are shown in Figure 2.
It was found that all synthesized 2,2′-bis(oxazolo[5,4-b]pyridine) derivatives show absorption maxima in the range of 323–357 nm. This is attributed to π–π* electronic transitions within the conjugated bis(oxazolo[5,4-b]pyridine) ring system. Spectral analysis revealed that compounds 4c and 4d display strong absorption bands in the longer wavelength region: λabsmax= 351 nm (chloroform), 350 nm (dichloromethane), and 347 nm (acetonitrile) for compound 4c; λabsmax = 343 nm (chloroform), 357 nm (dichloromethane), and 354 nm (acetonitrile) for compound 4d. These values are shifted compared to compound 4a, which shows λabsmax = 329 nm (chloroform), 327 nm (dichloromethane), and 323 nm (acetonitrile). The methyl group at the 4-position of the pyridine ring in 4a does not participate in conjugation with the π-system, which explains the shorter wavelength absorption. The bathochromic shift observed in compounds 4c and 4d is may be due to the presence of heteroatoms (sulfur and oxygen) with lone electron pairs that engage in additional n–π conjugation with the pyridine ring. The difference in absorption maxima between the thiophene and furan rings is related to the higher polarizability of the sulfur atom compared to oxygen. Compound 4b also shows a bathochromic shift relative to 4a, which is caused by π–π conjugation between the phenyl group and the pyridine ring.
Given that the synthesized compounds 4a–d contain two conjugated oxazolo[5,4-b]pyridine ring systems and are of significant interest as potential luminophores, we aimed to determine their fluorescence quantum yields. This parameter, defined as the ratio of emitted photons to absorbed photons, indicates how efficiently a substance converts absorbed light energy into fluorescent emission.
To measure the quantum yields of compounds 4a–d, fluorescence spectra were recorded in chloroform, dichloromethane, and acetonitrile solutions at concentrations of 2×10⁻⁵ M (for compounds 4a, 4c, and 4d) and 1×10⁻⁵ M (for compound 4b). The excitation wavelengths were selected to match the absorption maxima of each compound (Figure 3). The emission maxima of the studied compounds fall within the range of 323 to 357 nm, corresponding to blue and deep-blue photoluminescence (Table 1).
Thus, we confirmed the photoluminescent properties of the newly synthesized 2,2′-bis(oxazolo[5,4-b]pyridine) derivatives 4a-d and found that they exhibit high fluorescence quantum yields (ц ≈ 0.32-0.84) compared to well-known standards — quinine sulfate (ц ≈ 0.55) and tryptophan (ц ≈ 0.15). These results suggest that the new compounds based on 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridines 4a–d are promising both as potential ligands for metal complexation and as efficient luminophores.
3.3. Conformational Analysis and Electronic Structure of the New 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridines
The analysis of the photophysical properties of compounds 4a–d showed that structural modifications within the 2,2′-bis(oxazolo[5,4-b]pyridine) framework influence their spectral characteristics, fluorescence efficiency, and solvent sensitivity. Compounds 4a–c exhibited shorter-wavelength fluorescence (λemmax ≈ 358–414 nm) in the blue and deep-blue region, while compound 4d displayed a more red-shifted emission (λemmax ≈ 444–474 nm), extending into the blue-green region. Since our focus was on blue and deep-blue luminophores, we selected compounds 4a–c for further computational modeling (Table 2) in order to gain deeper insight into their electronic structure, assess the stability of possible conformations, and simulate their absorption and fluorescence spectra. The calculations were carried out using density functional theory (DFT) with the ωB97XD functional [38] and the 6-311++G(d,p) basis set [39] in the Gaussian 16 software package [40]. Molecular structures were built and visualized using GaussView 6.0 [41].
Table 2 summarizes the structures and IUPAC names of the synthesized 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridine derivatives 4a–c. As can be seen in Table 2, these compounds share a common terpyridine-like scaffold, in which the central pyridine ring is symmetrically substituted at positions 2 and 6 with fused oxazolo[5,4-b]pyridine moieties bearing various substituents at positions 5 and 7 of the oxazole ring. At the same time compound 4a (2,6-bis(5,7-dimethyloxazolo[5,4-b]pyridin-2-yl)pyridine) features methyl groups at both positions 5 and 7 of each oxazolopyridine unit, enhancing symmetry and electron-donating character. Compound 4b (2,6-bis(5-methyl-7-phenyloxazolo[5,4-b]pyridin-2-yl)pyridine) contains a phenyl substituent at position 7, introducing extended π-conjugation and potential for intermolecular interactions via aromatic stacking. Compound 4c (2,6-bis(5-methyl-7-(thiophen-2-yl)oxazolo[5,4-b]pyridin-2-yl)pyridine) incorporates a thiophene ring at position 7, which may influence electronic properties through sulfur-based heteroaromatic conjugation and increase polarizability. These structural variations are designed to modulate the electronic properties of the terpyridine analogues, with potential applications in luminescent materials and metal complexation.
Also, structural formulas of compounds 4a–c presented in Table 1 clearly reveal the presence of rotational degrees of freedom associated with the substituents at the 2 and 6 positions of the central pyridine ring. In addition, compound 4c possesses an extra rotational degree of freedom associated with the thiophene substituents at the 7-position of the oxazolo[5,4-b]pyridine rings. Conformations can differ in energy, dipole moment, and HOMO–LUMO gap, which is important for photophysical and complexing properties.
Based on these structural features, rotational transformation Scheme 1, Scheme 2 and Scheme 3 were developed to systematically explore the conformational landscape of compounds 4a–c. These schemes account for the torsional flexibility of the substituents at the 2 and 6 positions of the central pyridine ring, as well as the additional rotational freedom introduced by the thiophene groups in compound 4c. Transitions between forms occur due to rotation around the C(2)–C and C(6)–C bonds of the central pyridine ring.
Scheme 4 includes three conformations of 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridine, designated 4a–I, 4a–II, and 4a–III, connected by double-headed arrows, indicating the possibility of interconversion between them.
Conformation 4a-I in Scheme 4 represents the most symmetrical structure, in which both oxazolo[5,4-b]pyridine moieties are oriented in the same plane as the central pyridine core. This molecular configuration promotes maximum conjugation of the π-systems, which can enhance electron delocalization and luminescence properties. In conformation 4a-II, both peripheral moieties are rotated relative to the central core, resulting in pronounced conformational deformation. This form may be less stable in vacuum, but is potentially active under complexation conditions, where the spatial accessibility of donor atoms plays a key role. In conformation 4a-III, one of the peripheral moieties is rotated relative to the plane of the central pyridine. This leads to a partial disruption of conjugation and can affect the electron density and charge distribution. This form can be stabilized in certain solvents or upon coordination with metals.
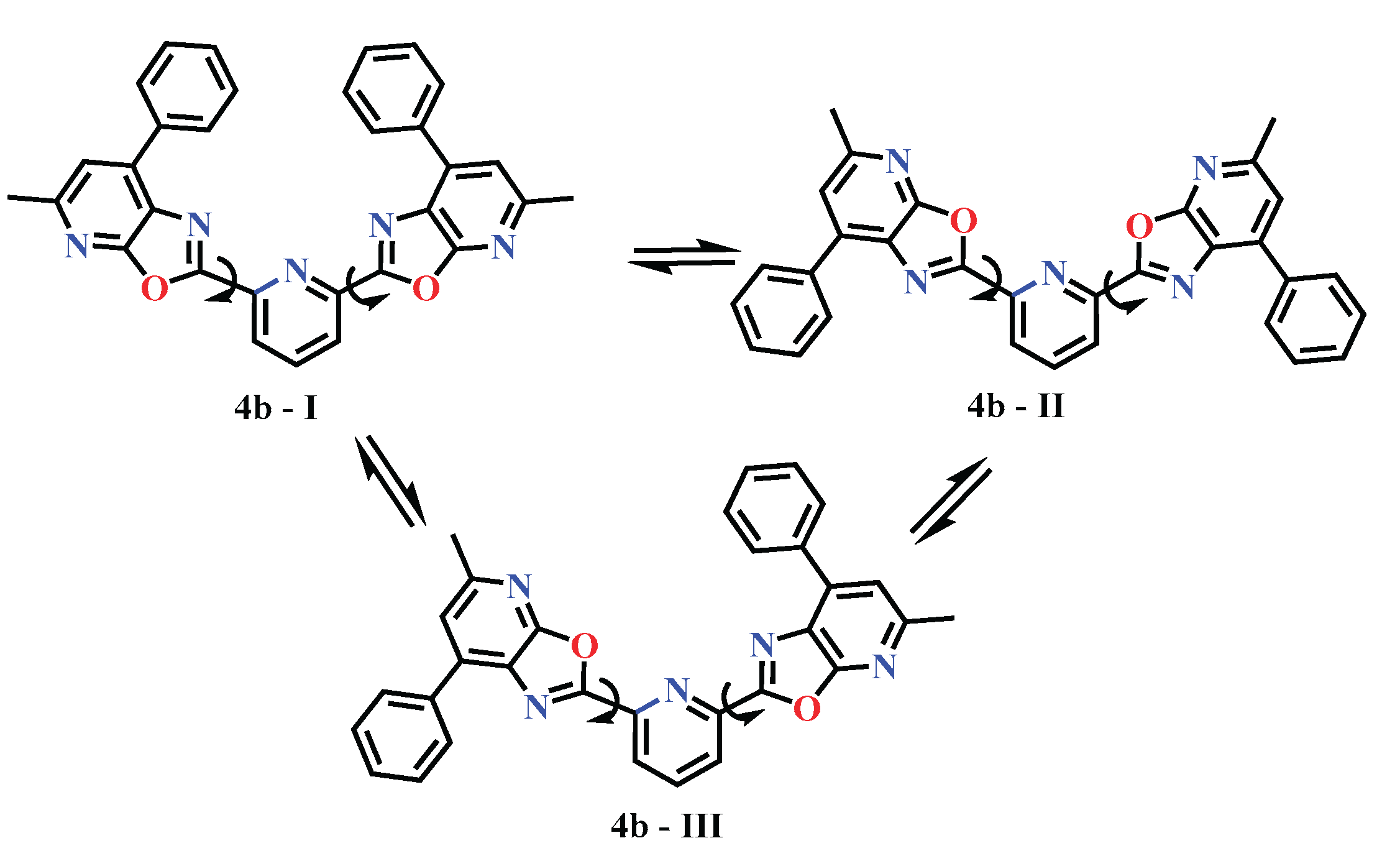
Scheme 5 shows three conformations of 2,6-bis(5-methyl-7-phenyloxazolo[5,4-b]pyridin-2-yl)pyridine, designated 4b-I, 4b-II, and 4b-III, connected by double-headed arrows, indicating the potential for interconversion between them.
Сonformation 4b-I in Scheme 5 represents the most symmetrical conformation, in which both peripheral fragments (oxazolo[5,4-b]pyridine with a phenyl substituent) are in a conjugated plane with the central pyridine. This configuration promotes maximum π-conjugation and may be the most stable in the gas phase or in a non-polar environment. In conformation 4b-II, both peripheral fragments are rotated, which leads to a pronounced conformational deformation. This form may be less stable, but is potentially active under complexation conditions or when interacting with biomolecules, where the spatial accessibility of the phenyl groups plays a role. In conformation 4b-III, one of the peripheral fragments is rotated relative to the plane of the central core, which leads to a partial disruption of conjugation. This may reflect the influence of steric factors or interaction with the solvent. This form may be intermediate in energy.
Scheme 6 includes seven conformations of 2,6-bis(5-methyl-7-(thiophen-2-yl)oxazolo[5,4-b]pyridin-2-yl)pyridine: 4c–I, 4c–II, 4c–III, as well as their mirror or alternative versions — 4c–I′, 4c–II′, 4c–III′ and 4c–III″.
Scheme 6 depicts 4c–I / I′ as symmetrical forms in which both peripheral fragments are in a conjugated plane with the central pyridine. The difference between I and I′ is due to the orientation of the thiophenes. Conformations 4c–II / II′ are forms with partial rotation of one of the peripheral fragments, which leads to disruption of conjugation and a change in electron density. Conformations 4c–III / III′ / III″ are forms with pronounced conformational deformation, where both peripheral fragments are rotated. These forms may be less stable but are important for analyzing steric accessibility and potential complexation.
The thermodynamic stability of the conformations of compounds 4a–c in different media was evaluated based on their total electronic energy (Etotal) and Gibbs free energy (G), calculated using DFT level of theory with the ωB97XD functional and the 6-311++G(d,p) basis set in the Gaussian 16 software package (Table 3).
The data presented in Table 3 show that each compound has a preferred conformation with the lowest Gibbs free energy in a given solvent. Specifically, the most stable conformer is 4a–II for compound 4a, 4b–II for compound 4b, and 4c–II′ for compound 4c. The differences in Gibbs free energy between alternative conformers are less than 3 kJ/mol, suggesting that multiple conformations may coexist thermodynamically in solution. The largest energy variations were observed for compound 4c, where several conformers have similar energy values. This may be due to the increased flexibility of the molecular framework and the presence of multiple local minima on the potential energy surface. The effect of the solvent (CH₂Cl₂ and CH₃CN) on the energy parameters was moderate: the relative stability order of the conformers remained unchanged, although the absolute Gibbs energy values showed slight shifts, reflecting specific interactions between the molecule and the surrounding medium.
Figure 4 shows the optimized geometries of the most stable conformers 4a–II (Table S1), 4b–II (Table S2), and 4c–II′ (Table S3), obtained by DFT geometry optimization (щB97XD/6-311++G(d,p)) in acetonitrile.
The structures shown in Figure 4 highlight key features of spatial organization that influence the photophysical properties and thermodynamic stability of the compounds. Conformation 4a–II is compact and symmetric, with a planar arrangement of oxazole fragments. This promotes effective conjugation and minimizes steric interactions between substituents, which explains its lowest Gibbs free energy among the 4a conformers. Conformation 4b–II has an extended molecular geometry with a well-developed π-conjugated system. The substituents are positioned to avoid steric clashes while maintaining electronic delocalization. This geometry supports high molar extinction and moderate fluorescence quantum yield. Conformation 4c–II′ is the most branched and spatially crowded structure. The presence of additional donor–acceptor fragments causes molecular bending and leads to an increased Stokes shift. All three conformers show a favorable balance between electronic conjugation and spatial stability, as confirmed by their minimal Gibbs energy values.
To better understand how spatial arrangement affects the electronic properties of these compounds, the next step was to compare the optimized geometries of conformers 4a–II, 4b–II, and 4c–II′ with the distribution of their frontier molecular orbitals (HOMO and LUMO). These orbitals were calculated based on the optimized geometries using DFT (ωB97XD/6-311++G(d,p)) in Gaussian 16, and visualized in GaussView 6.0 (Figure 5).
The diagrams shown in Figure 5 illustrate the spatial distribution of the frontier molecular orbitals, revealing characteristic electronic features of compounds 4a–c. In all structures, the HOMO is mainly localized on the aromatic and heterocyclic fragments, indicating their donor nature and involvement in electronic transitions. The LUMO is distributed over the р-conjugated system, including acceptor regions, which confirms the dominance of р→π* transitions. Conformation 4c–II′ is particularly notable for its more pronounced spatial separation between HOMO and LUMO, which may contribute to an increased Stokes shift and enhanced fluorescence efficiency.
Table 4 presents the calculated HOMO and LUMO energy values for the most stable conformations 4a–II, 4b–II and 4c–II′, along with their corresponding energy gaps.
As shown in Table 4, the energy gap between the HOMO and LUMO orbitals decreases consistently from conformation 4a–II to 4c–II′, reflecting differences in the electronic structure and photophysical behavior of the compounds. The largest gap is observed for conformation 4a–II (ΔE = 7.76 eV), indicating high ground-state stability and a tendency toward shorter-wavelength absorption. Conformation 4b–II shows an intermediate gap (ΔE = 7.63 eV), which may suggest easier excitation and a shift toward longer wavelengths. The smallest gap is found in conformation 4c–II′ (ДE = 7.31 eV), indicating a higher tendency for electronic excitation, which may enhance fluorescence and increase the Stokes shift.
Overall, the decreasing ДE from 4a–II to 4c–II′ suggests increasing photoactivity and potential efficiency of these compounds as fluorophores. The narrowest gap observed for 4c–II′ is consistent with the previously noted spatial separation of the HOMO and LUMO orbitals, which may support more efficient charge transfer in the excited state and stronger fluorescence.
To further explore the distribution of electron density and identify potential reactive sites in the molecules, a molecular electrostatic potential (MEP) analysis was performed for the most stable conformations 4a–II, 4b–II and 4c–II′ (Figure 6).
As shown in Figure 6, the red regions with the most negative molecular electrostatic potential (MEP) in conformations 4a–II, 4b–II and 4c–II′ are mainly located on oxygen and nitrogen atoms, as well as on р-conjugated fragments. These areas have high electron density and can act as nucleophilic sites, interacting with electrophiles such as protons, metal ions, or positively charged molecules. The blue regions on the MEP maps, which indicate areas of positive electrostatic potential, are mostly found on hydrogen atoms—especially those involved in hydrogen bonding or located near electronegative atoms. These regions are electron-deficient and can serve as electrophilic sites, susceptible to attack by nucleophiles such as anions or electron-donating groups. Among the studied structures, conformation 4c–II′ shows the most pronounced spatial separation between donor and acceptor regions.
3.4. DFT-Based Simulation of Electronic Absorption and Luminescence Properties
To elucidate the electronic transitions and photophysical behavior of the studied conformations, we performed TD DFT calculations of UV absorption spectra and emission luminescence spectra for 4a–II 4b–II and 4c–II′ conformations, aiming to establish structure–property relationships relevant to optoelectronic applications. The calculated absorption and emission spectra are presented in Figure 7.
The calculated UV–vis absorption spectra of the 4a–II, 4b–II and 4c–II′ conformations exhibit distinct profiles, reflecting differences in their electronic structures and transition energies (Figure 7a). All spectra show pronounced absorption bands in the 290–320 nm range, characteristic of π→π* or n→π* electronic transitions and corresponding to experimental peaks in the range of 323–354 nm. The observed difference of 25–40 nm between the experimental and calculated absorption maxima is expected since TD-DFT reliably predicts spectral trends, but often shows absolute deviations of 0.3–0.5 eV (approximately 25–40 nm), especially for π→π* transitions [60,61].
The 4a–II conformation displays the most blue-shifted absorption maximum, located near 290 nm, indicating the largest HOMO–LUMO energy gap among the three compounds. The 4b–II conformation peaks slightly later, around 300 nm, with comparable intensity and a narrower band shape, suggesting efficient electronic excitation with minimal structural distortion. In contrast, the 4c–II′ shows the most red-shifted and intense absorption band, centered near 320 nm, accompanied by a broader spectral profile. This behavior implies a reduced excitation energy and potentially greater charge delocalization or π-conjugation within the molecular framework. The observed bathochromic shifts and variations in peak intensity correlate with differences in molecular geometry, electronic distribution, and substituent effects.
The simulated emission spectra of the three compounds reveal distinct photophysical behaviors, as illustrated in Figure 7b. The 4a–II conformation exhibits the most intense and narrow luminescence band, with a maximum at approximately 350.5 nm, indicating the largest energy gap between the first excited singlet state (S₁) and the ground state (S₀) and corresponding to the experimental peak in the 370 nm range. In contrast, the 4b–II and 4c–II′ conformations display progressively broader and red-shifted emission profiles, with maxima near 380 (experimental peak near 380 nm) and 420 nm (experimental peak near 430 nm), respectively. These spectral shifts suggest a reduction in the S₁–S₀ energy gap, likely due to conformational relaxation, substituent effects, or enhanced charge delocalization. The observed trend — 4a–II > 4b–II > 4c–II′ in both peak intensity and energy—correlates with experiment and reflect differences in electronic structure.
Overall, conformation 4a–II shows the most efficient luminescence, which is likely due to its high molecular symmetry and minimal energy loss from conformational changes. The weaker luminescence observed for conformations 4b–II and 4c–II′ may result from greater conformational flexibility, intermolecular interactions, or differences in electronic structure. Conformation 4a–II can be considered a promising candidate for use as a fluorophore or in OLED materials, while 4b–II and 4c–II′ are of interest for studying how molecular structure affects spectral properties.
4. Conclusions
The aim of this study was to investigate a new group of structurally diverse bis-oxazolo[5,4-b]pyridine derivatives, selected for their close similarity to terpyridines, well-known ligands in coordination chemistry due to their flexible structure and strong binding capacity. Four new 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridine derivatives 4a-d were synthesized using our established approach to cyclization of amide derivatives of 3-aminopyridin-2(1H)-ones, with pyridine-2,6-dicarboxylic acid and its dichloride serving as key acidic components. The newly synthesized compounds were fully characterized by melting point analysis, high-resolution mass spectrometry and detailed ¹H NMR spectroscopy to confirm their structure and purity. Сompounds 4a–d were further studied for their photophysical properties; their spectral properties, including λmax, Stokes shift, and quantum yield in chloroform, dichloromethane and acetonitrile medium were measured. It was found that compounds 4a–c exhibited strong blue or deep blue fluorescence under UV light in the range of 323–347 nm attributed to π–π* electronic transitions within the conjugated bis(oxazolo[5,4-b]pyridine) ring system and high fluorescence quantum yields (ц ≈ 0.32-0.84), highlighting their potential as air-stable phosphors for use in organic electronics. Therefore, compounds 4a–c were further investigated using computational modeling to better understand their electronic structure, evaluate the stability of different conformations, and predict their absorption and fluorescence behavior. The most stable conformations 4a–II, 4b–II and 4c–II′ were characterized by HOMO–LUMO energy levels, energy gaps and molecular electrostatic potential (MEP) maps, showing a gradual decrease in gap values and photoactivity from 4a to 4c. Among the studied compounds, conformation 4a–II proved to be the most promising fluorophore both experimentally and theoretically, due to its high molecular symmetry and stable blue emission. Future research can focus on expanding the structural diversity of bis(oxazolo[5,4-b]pyridine) derivatives and studying their coordination behavior with metal ions to develop new functional materials for optoelectronic applications.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org; S2–S23, 1H, 13C NMR spectra and Mass spectra.
Author Contributions
Conceptualization, methodology, resources, supervision, I.V.K. and I.A.P.; software, I.A.P.; investigation, validation, formal analysis, data curation, I.V.P., S.V.V. and A.S.R.; writing—original draft preparation, writing—review and editing, visualization, I.V.P., I.V.K. and I.A.P.; project administration, funding acquisition, I.V.P. All authors have read and agreed to the published version of the manuscript.
Funding
The study was supported by a grant from the Russian Science Foundation (No. 24-23-00472, «Development of methods for the preparation and practical application of new derivatives of bisoxazolo[5,4-b]pyridine with potential luminescent, complexing and catalytic activity», https://rscf.ru/en/project/24-23-00472/ ).
Data Availability Statement
Data are available from the corresponding author upon reasonable request. No publicly archived datasets were generated or analyzed in this study.
Acknowledgments
Spectrophotometric studies were carried out using the equipment of the Center for Collective Use “Rational Nature Management and Physicochemical Research” of University of Tyumen. NMR studies were conducted using equipment purchased under the “Priority 2030” program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lavis, L.D.; Raines, R.T. Bright Ideas for Chemical Biology. ACS Chem. Biol. 2008, 3, 142.
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in Neurobiology: Probing Their Chemistry and Biology with Molecular Imaging. Chem. Rev. 2008, 108, 1517.
- Gonçalves, M.S.T. Fluorescent Labeling of Biomolecules with Organic Probes. Chem. Rev. 2009, 109, 190.
- Urano, Y.; Asanuma, D.; Hama, Y.; Koyama, Y.; Barrett, T.; Kamiya, M.; Nagano, T.; Watanabe, T.; Hasegawa, A.; Choyke, P.L. Selective Molecular Imaging of Viable Cancer Cells with pH-Activatable Fluorescence Probes. Nat. Med. 2008, 15, 104.
- Schäferling, M. The Art of Fluorescence Imaging with Chemical Sensors. Angew. Chem. Int. Ed. 2012, 51, 3532.
- Köhler, A.; Wilson, J.S.; Friend, R.H. Fluorescence and Phosphorescence in Organic Materials. Adv. Eng. Mater. 2002, 4, 453.
- O’Regan, B.; Grätzel, M. A Low-Cost, High-Efficiency Solar Cell Based on Dye-Sensitized Colloidal TiO2 Films. Nature 1991, 353, 737.
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595.
- Hartwig, J.F. Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books: Sausalito, CA, USA, 2010; 1128 pp.
- Liu, Z.; Sadler, P.J. Organoiridium complexes: anticancer agents and catalysts. Acc. Chem. Res. 2014, 47, 1174.
- Luca, O.R.; Crabtree, R.H. Redox-active ligands in catalysis. Chem. Soc. Rev. 2013, 42, 1440.
- Peris, E.; Crabtree, R.H. Recent homogeneous catalytic applications of chelate and pincer N-heterocyclic carbenes. Coord. Chem. Rev. 2004, 248, 2239.
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010, 110, 1746.
- Chi, Y.; Chou, P.T. Transition-metal phosphors with cyclometalating ligands: fundamentals and applications. Chem. Soc. Rev. 2010, 39, 638.
- Deiters, A.; Martin, S.F. Synthesis of oxygen-and nitrogen-containing heterocycles by ring-closing metathesis. Chem. Rev. 2004, 104, 2199.
- Fache, F.; Schulz, E.; Tommasino, M.L.; Lemaire, M. Nitrogen-containing ligands for asymmetric homogeneous and heterogeneous catalysis. Chem. Rev. 2000, 100, 2159.
- Wei, C.; He, Y.; Shi, X.; Song, Z. Terpyridine-metal complexes: Applications in catalysis and supramolecular chemistry. Coord. Chem. Rev. 2019, 385, 1.
- Chelucci, G.; Thummel, R.P. Chiral 2,2‘-bipyridines, 1,10-phenanthrolines, and 2,2‘:6‘,2‘‘-terpyridines: Syntheses and applications in asymmetric homogeneous catalysis. Chem. Rev. 2002, 102, 3129.
- Winter, A.; Newkome, G.R.; Schubert U.S., Catalytic applications of terpyridines and their transition metal complexes. ChemCatChem 2011, 3, 1384.
- Shabunina, O.V.; Starnovskaya, E.S.; Shaitz, Ya.K.; Kopchuk, D.S.; Sadieva, L.K.; Kim, G.A.; Taniya, O.S.; Nikonov, I.L.; Santra, S.; Zyryanov, G.V.; Charushin, V.N. Asymmetrically substituted 5,5′′-diaryl-2,2′:6′,2′′-terpyridines as efficient fluorescence “turn-on” probes for Zn2+ in food/cosmetic samples and human urine. J. Photochem. Photobiol. A Chem. 2021, 408, 113101.
- Armspach, D.; Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Zehnder, M. Carbaborane-functionalised 2,2′:6′,2″-terpyridine ligands for metallosupramolecular chemistry: Syntheses, complex formation, and the crystal and molecular structures of 4′-(ortho-carboranyl)-2,2′:6′,2″-terpyridine and 4′-(ortho-carboranylpropoxy)-2,2′:6′,2″-terpyridine. J. Organomet. Chem. 1998, 550, 193.
- Constable, E.C. 2,2′:6′,2″-Terpyridines: From chemical obscurity to common supramolecular motifs. Chem. Soc. Rev. 2007, 36, 246.
- Garcia-Dominguez, A.; Mueller, S.; Nevado, C.; Nickel-catalyzed intermolecular carbosulfonylation of alkynes via sulfonyl radicals. Angew. Chem. Int. Ed. 2017, 56, 9949.
- Hie, L.; Baker, E.L.; Anthony, S.M.; Desrosiers, J.N.; Senanayake, C.; Garg, N.K. Nickel-catalyzed esterification of aliphatic amides. Angew. Chem. Int. Ed. 2016, 55, 15129.
- Huihui, K.M.M.; Shrestha, R.; Weixs, D.J. Nickel-catalyzed reductive conjugate addition of primary alkyl bromides to enones to form silyl enol ethers. Org. Lett. 2017, 19, 340.
- Joshi-Pangu, A.; Ganesh, M.; Biscoe, M.R. Nickel-catalyzed Negishi cross-coupling reactions of secondary alkylzinc halides and aryl iodides. Org. Lett. 2011, 13, 1218.
- Kamata, K.; Suzuki, A.; Nakai, Y.; Nakazawa, H. Catalytic hydrosilylation of alkenes by iron complexes containing terpyridine derivatives as ancillary ligands. Organometallics 2012, 31, 3825.
- Tondreau, A.M.; Atienza, C.C.H.; Darmon, J.M.; Milsmann, C.; Hoyt, H.M.; Weller, K.J.; Nye, S.A.; Lewis, K.M.; Boyer, J.; Delis, J.G.P.; Lobkovsky, E.; Chirik, P.J. Synthesis, electronic structure, and alkene hydrosilylation activity of terpyridine and bis(imino)pyridine iron dialkyl complexes. Organometallics 2012, 31, 4886.
- Duong, H.A.; Wu, W.Q.; Teo, Y.Y. Cobalt-catalyzed cross-coupling reactions of arylboronic esters and aryl halides. Organometallics 2017, 36, 4363.
- Kurahashi, T.; Matsubara, S. Nickel-catalyzed reactions directed toward the formation of heterocycles. Acc. Chem. Res. 2015, 48, 1703.
- Kolesnichenko, I.V.; Anslyn, E.V. Practical applications of supramolecular chemistry. Chem. Soc. Rev. 2017, 46, 2385.
- Hwang, S.H.; Moorefield, C.N.; Fronczek, F.R.; Lukoyanova, O.; Echegoyen, L.; Newkome, G.R. Construction of triangular metallomacrocycles:[M 3 (1,2-bis(2,2′∶ 6′,2″-terpyridin-4-yl-ethynyl)benzene)3][M= Ru (ii), Fe (ii), 2Ru (ii) Fe (ii)]. Chem. Commun. 2005, 713.
- Sarkar, R.; Guo, K.; Moorefield, C.N.; Saunders, M.J.; Wesdemiotis, C.; Newkome, G.R. One-Step Multicomponent Self-Assembly of a First-Generation Sierpiński Triangle: From Fractal Design to Chemical Reality. Angew. Chem. Int. Ed. 2014, 53, 12182.
- Kulakov I.V.; Matsukevich M.V.; Shulgau Z.T.; Sergazy S.; Seilkhanov T.M.; Puzari A.; Fisyuk A.S. Synthesis and antiradical activity of 4-aryl(hetaryl)-substituted 3-aminopyridin-2(1H)-ones. Chem. Heterocycl. Compd. 2015, 51, 991.
- Mohamed, A. A.; Nassr, A. A.; Sadeek, S. A.; Rashid, N. G.; Abd El-Hamid, S. M. First Report on Several NO-Donor Sets and Bidentate Schiff Base and Its Metal Complexes: Characterization and Antimicrobial Investigation. Compounds 2023, 3 (3), 376–389.
- Moiseev, R. V.; Morrison, P. W. J.; Steele, F.; Khutoryanskiy, V. V. Penetration Enhancers in Ocular Drug Delivery. Pharmaceutics 2019, 11 (7), 321.
- Shatsauskas, A.L.; Abramov, A.A.; Chernenko, S.A.; Kostyuchenko, A.S.; Fisyuk, A.S. Synthesis and Photophysical Properties of 3-Amino-4-arylpyridin-2 (1Н)-ones. Synthesis 2020, 52, 227.
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10 (44), 6615.
- Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72 (1), 650–654.
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford CT, 2016.
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 6; Semichem Inc.: Shawnee Mission, KS, 2016. Available online: http://gaussian.com.
- Huber, R. G.; Margreiter, M. A.; Fuchs, J. E.; von Grafenstein, S.; Tautermann, C. S.; Liedl, K. R.; Fox, T. Heteroaromatic π-Stacking Energy Landscapes. J. Chem. Inf. Model. 2014, 54 (5), 1371–1379.
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102 (11), 1995–2001.
- Hehre, W. J.; Radom, L.; Schleyer, P. v. R.; Pople, J. A. Ab Initio Molecular Orbital Theory; Wiley: New York, 1986; pp 235–240.
- Young, D. C. Computational Chemistry: A Practical Guide for Applying Techniques to Real-World Problems; Wiley-Interscience: New York, 2001; pp 104–110.
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102 (11), 1995–2001.
- Rakhimzhanova, A. S.; Muzaparov, R. A.; Pustolaikina, I. A.; Kurmanova, A. F.; Nikolskiy, S. N.; Kapishnikova, D. D.; Stalinskaya, A. L.; Kulakov, I. V. Series of Novel Integrastatins Analogues: In Silico Study of Physicochemical and Bioactivity Parameters. Eurasian J. Chem. 2024, 29 (4 (116)), 44–60.
- Stratmann, R. E.; Scuseria, G. E.; Frisch, M. J. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 1998, 109 (19), 8218–8224.
- Shatsauskas, A.L.; Shatalin, Y.V.; Shubina, V.S.; Chernenko, S.A.; Kostyuchenko, A.S.; Fisyuk, A.S.5-Ethyl-5,6-dihydrobenzo[c][1,7]naphthyridin-4(3H)-ones – A new class of fluorescent dyes. Dyes and Pigments 2022, 204, 110388.
- Shatsauskas, A.; Shatalin, Yu.; Shubina, V.; Zablodtskii, Yu.; Chernenko, S.; Samsonenko, A.; Kostyuchenko, A.; Fisyuk, A. Synthesis and application of new 3-amino-2-pyridone based luminescent dyes for ELISA. Dyes and Pigments 2021, 187, 109072.
- Åberg, V.; Sellstedt, M.; Hedenström, M.; Pinkner, J. S.; Hultgren, S. Design, synthesis and evaluation of peptidomimetics based on substituted bicyclic 2-pyridones—Targeting virulence of uropathogenic E. coli. J. Bioorg. Med. Chem. 2006, 14, 7563.
- Verissimo, E.; Berry, N.; Gibbons, P.; Cristiano, M. L. S.; Rosenthal, P. J.; Gut, J.; Ward, S. A.; O’Neill, P. M. Design and synthesis of novel 2-pyridone peptidomimetic falcipain 2/3 inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 4210.
- Zhu, S.; Hudson, T.H.; Kyle, D.E.; Lin, A.J. Synthesis and in vitro studies of novel pyrimidinyl peptidomimetics as potential antimalarial therapeutic agents. J. Med. Chem., 2002, 45, 3491.
- Palamarchuk, I.V.; Shulgau, Z.T.; Kharitonova, M.A.; Kulakov, I.V. Synthesis and neurotropic activity of new 3-(arylmethyl)aminopyridine-2(1H)-one. Chem. Pap. 2021, 75, 4729.
- Kulakov, I.V.; Shatsauskas, A.L.; Matsukevich, M.V.; Palamarchuk, I.V.; Seilkhanov, T.M.; Gatilov, Yu.V.; Fisyuk, A.S. A New Approach to the Synthesis of Benzo[c][1,7] naphthyridin-4(3H)-ones. Synthesis 2017, 49, 3700.
- Kulakov, I.V.; Matsukevich, M.V.; Levin, M.L.; Palamarchuk, I.V.; Seilkhanov, T.M.; Fisyuk, A.S. Synthesis of the First Representatives of Thieno[3,2-c][1,7]naphthyridine Derivatives Based on 3-Amino-6-methyl-4-(2-thienyl)pyridin-2(1H)-one. Synlett 2018, 29, 1741.
- Palamarchuk, I.V.; Matsukevich, M.V.; Kulakov, I.V.; Seilkhanov, T.M.; Fisyuk, A.S. Synthesis of N-substituted 2-aminomethyl-5-methyl-7-phenyloxazolo[5,4-b]pyridines. Chem. Heterocycl. Compd. 2019, 55, 788.
- Shatsauskas, A.L.; Abramov, A.A.; Saibulina, E.R.; Palamarchuk, I.V.; Kulakov, I.V.; Fisyuk, A.S. Synthesis of 3-amino-6-methyl-4-phenylpyridin-2(1H)-one and its derivatives. Chem. Heterocycl. Compd. 2017, 53, 186.
- Palamarchuk, I.V.; Kulakov, I.V. A New Method for Obtaining Carboxylic Derivatives of Oxazolo[5,4-b]pyridine Based on 3-Aminopyridine-2(1H)-ones. Eurasian J. Chem. 2024, 29, 32.
- Laurent, A. D.; Jacquemin, D. TD-DFT Benchmarks: A Review. Int. J. Quantum Chem. 2013, 113 (17), 2019–2039.
- Casida, M. E. Time-Dependent Density-Functional Theory for Molecules and Molecular Solids. J. Mol. Struct. THEOCHEM 2009, 914 (1–3), 3–18.
Scheme 1.
Basic coordination mode of terpyridine ligands.
Scheme 2.
Formation of a metal cation complex and protonation of the central nitrogen atom in terpyridine.
Scheme 2.
Formation of a metal cation complex and protonation of the central nitrogen atom in terpyridine.
Scheme 3.
Synthesis of 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridine derivatives 4a-d.
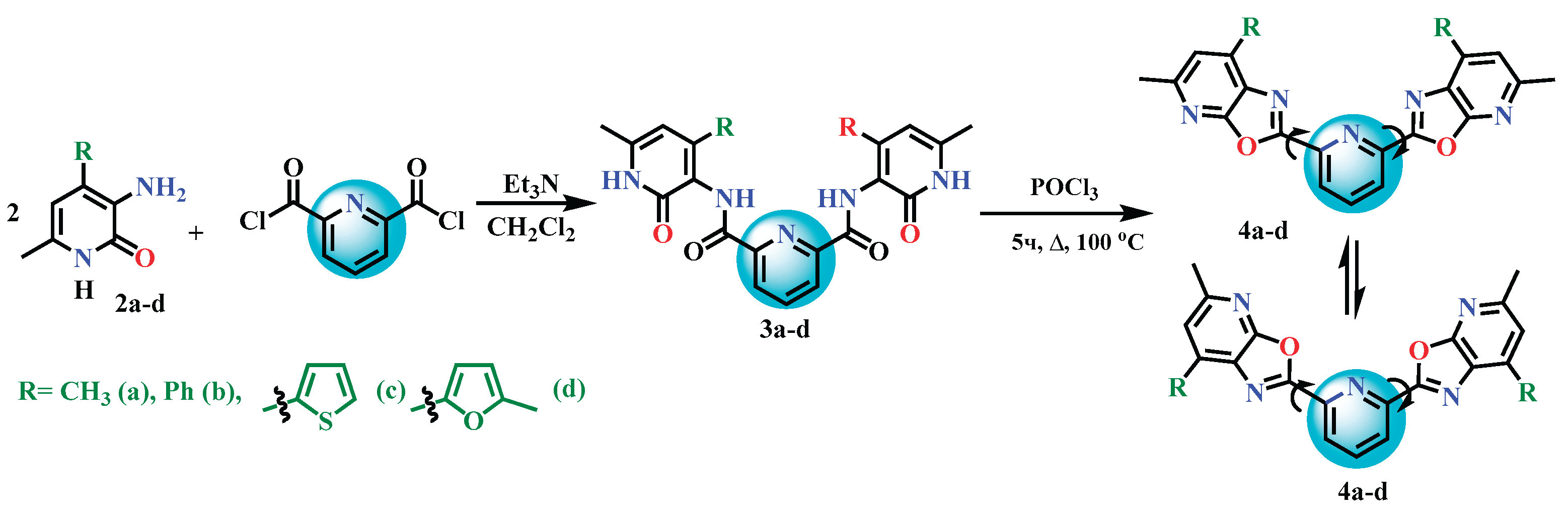
Figure 1.
Observed luminescence of 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridine samples under UV irradiation.
Figure 1.
Observed luminescence of 2,6-bis(oxazolo[5,4-b]pyridin-2-yl)pyridine samples under UV irradiation.

Figure 2.
Normalized absorption spectra of bisoxazolo[5,4-b]pyridine derivatives 4a-d in chloroform (A), dichloromethane (B), acetonitrile (C), general spectrum (D).
Figure 2.
Normalized absorption spectra of bisoxazolo[5,4-b]pyridine derivatives 4a-d in chloroform (A), dichloromethane (B), acetonitrile (C), general spectrum (D).
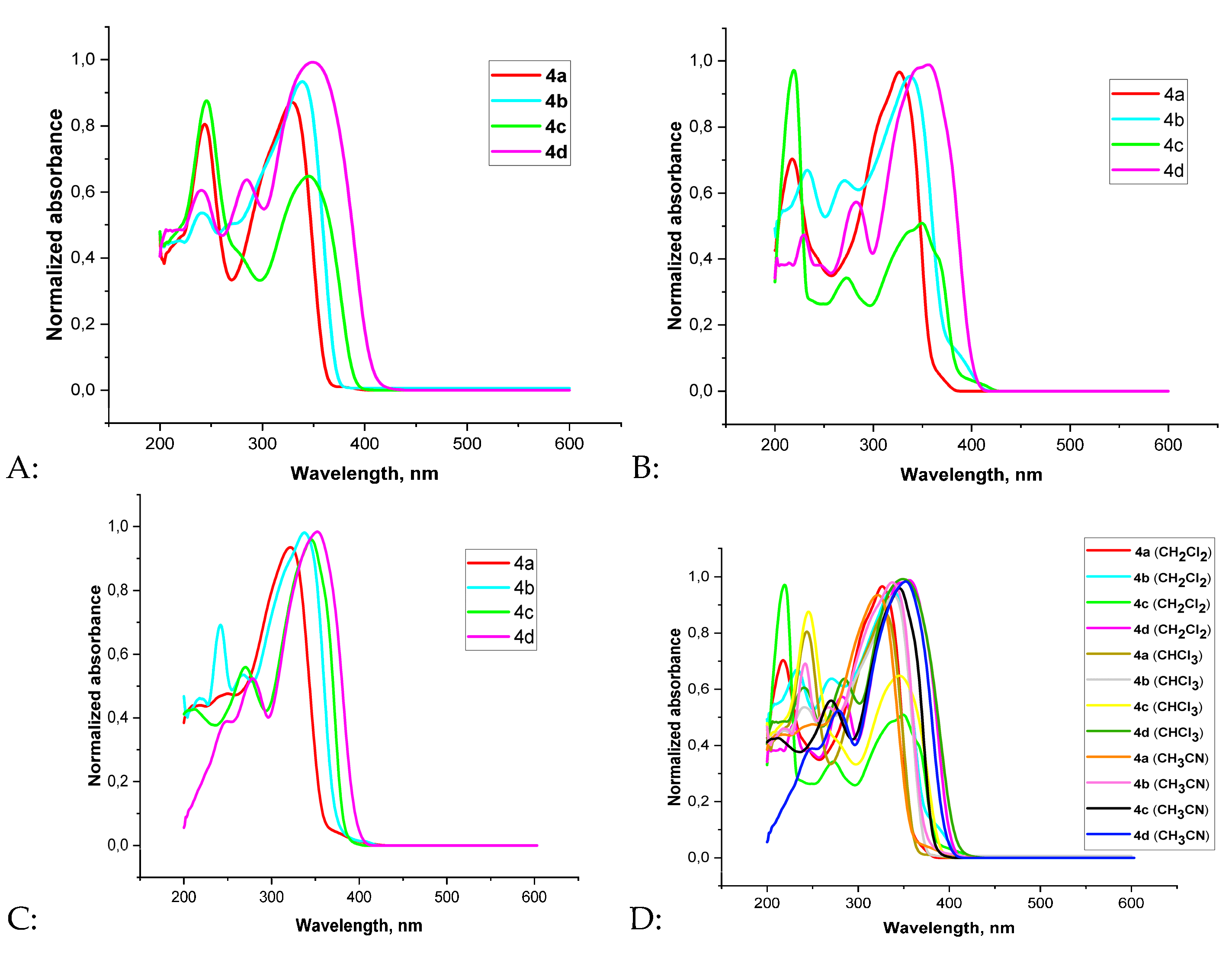
Figure 3.
Normalized fluorescence spectra of bisoxazolo[5,4-b]pyridine derivatives 4a-d in chloroform (A), dichloromethane (B), acetonitrile) (C), general spectrum (D).
Figure 3.
Normalized fluorescence spectra of bisoxazolo[5,4-b]pyridine derivatives 4a-d in chloroform (A), dichloromethane (B), acetonitrile) (C), general spectrum (D).

Scheme 4.
Conformational landscape of compound 4a.

Scheme 5.
Conformational landscape of compound 4b.
Scheme 6.
Conformational landscape of compound 4c.
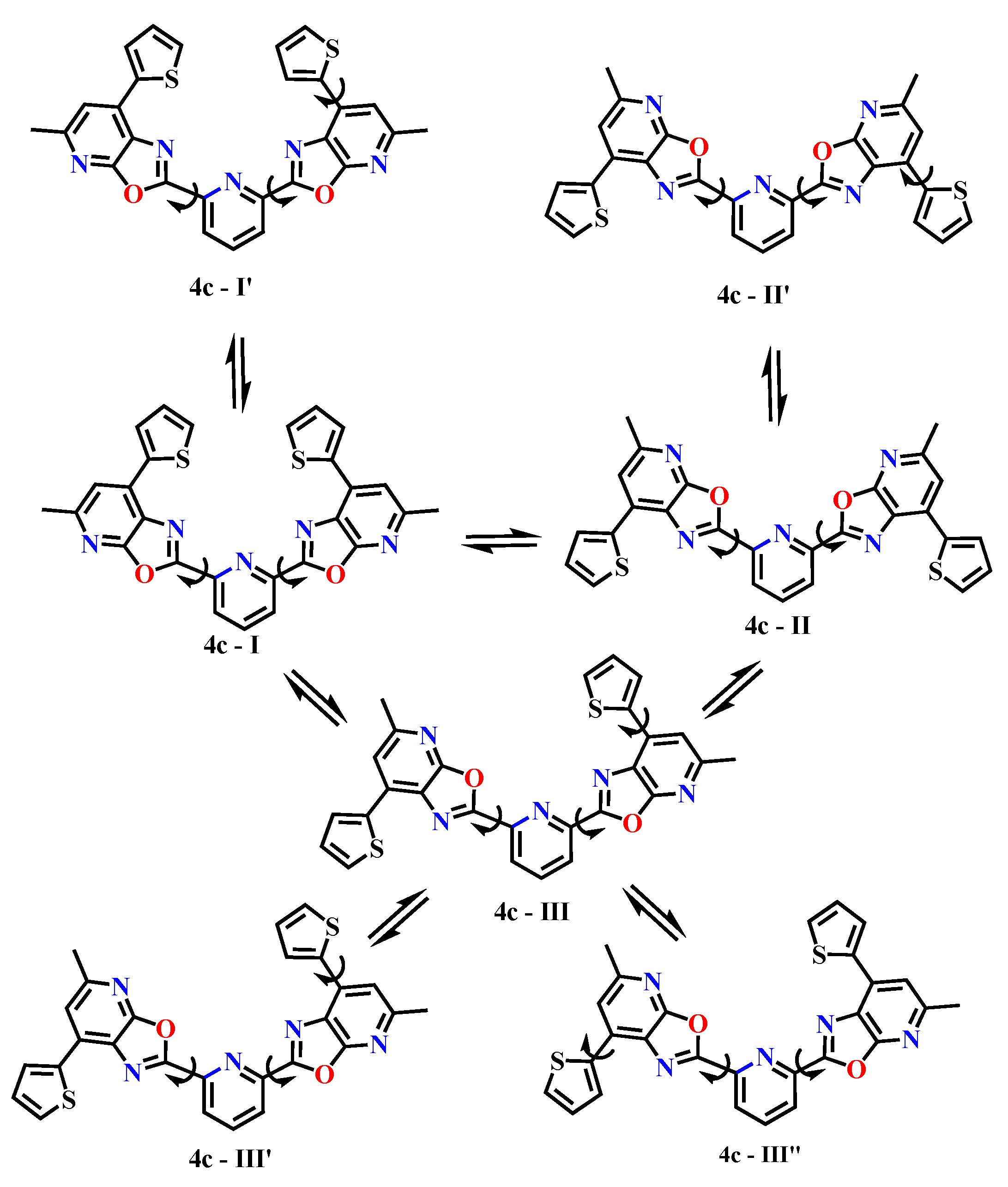
Figure 4.
Optimized geometries of the most stable conformations of compounds 4a–c calculated by DFT (ωB97XD/6-311++G(d,p)) in acetonitrile (CH₃CN).
Figure 4.
Optimized geometries of the most stable conformations of compounds 4a–c calculated by DFT (ωB97XD/6-311++G(d,p)) in acetonitrile (CH₃CN).
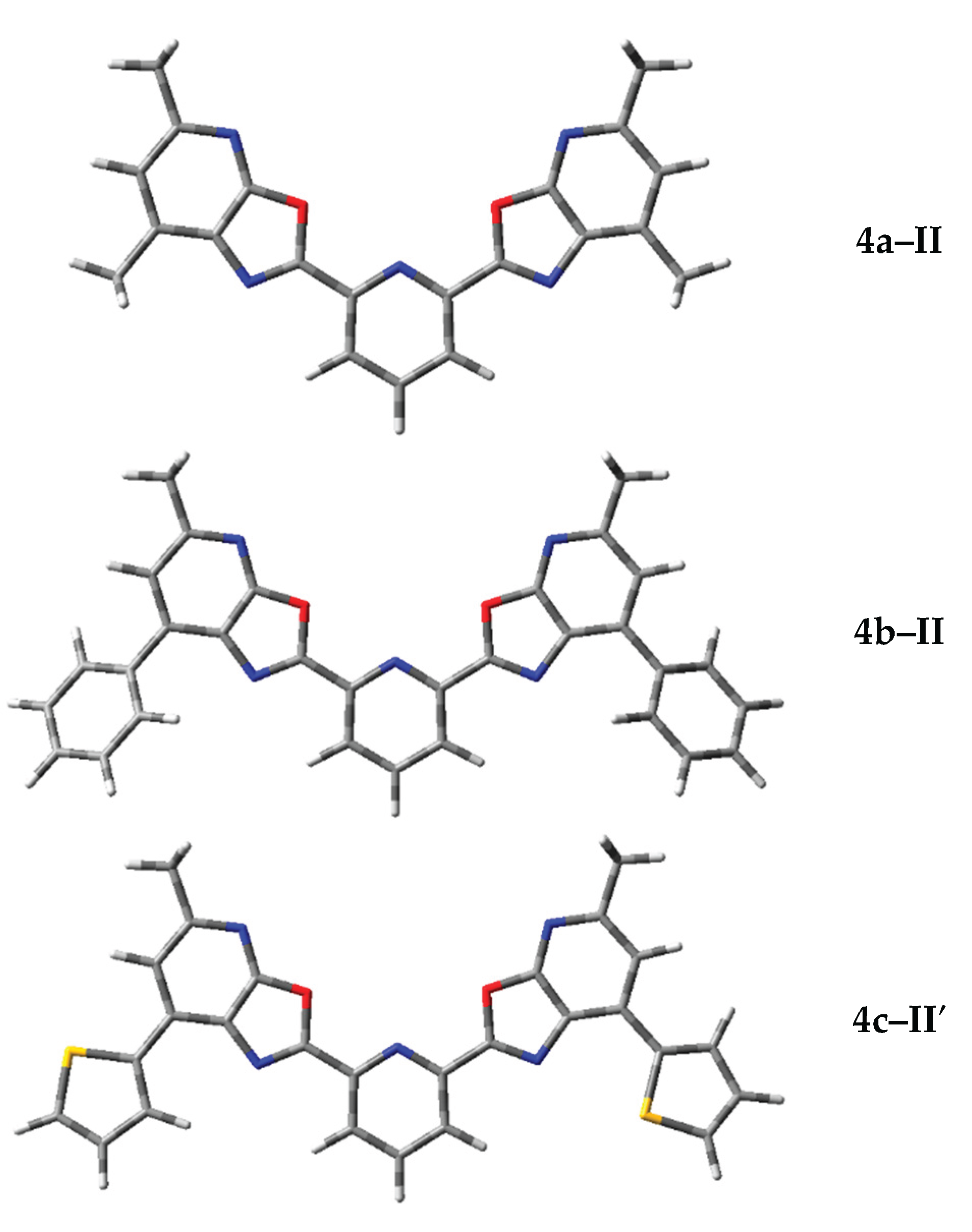
Figure 5.
Frontier Molecular Orbitals (HOMO and LUMO) visualization for the most stable conformations 4a–II, 4b–II and 4c–II′, calculated by DFT (ωB97XD/6-311++G(d,p)) in acetonitrile (CH₃CN).
Figure 5.
Frontier Molecular Orbitals (HOMO and LUMO) visualization for the most stable conformations 4a–II, 4b–II and 4c–II′, calculated by DFT (ωB97XD/6-311++G(d,p)) in acetonitrile (CH₃CN).
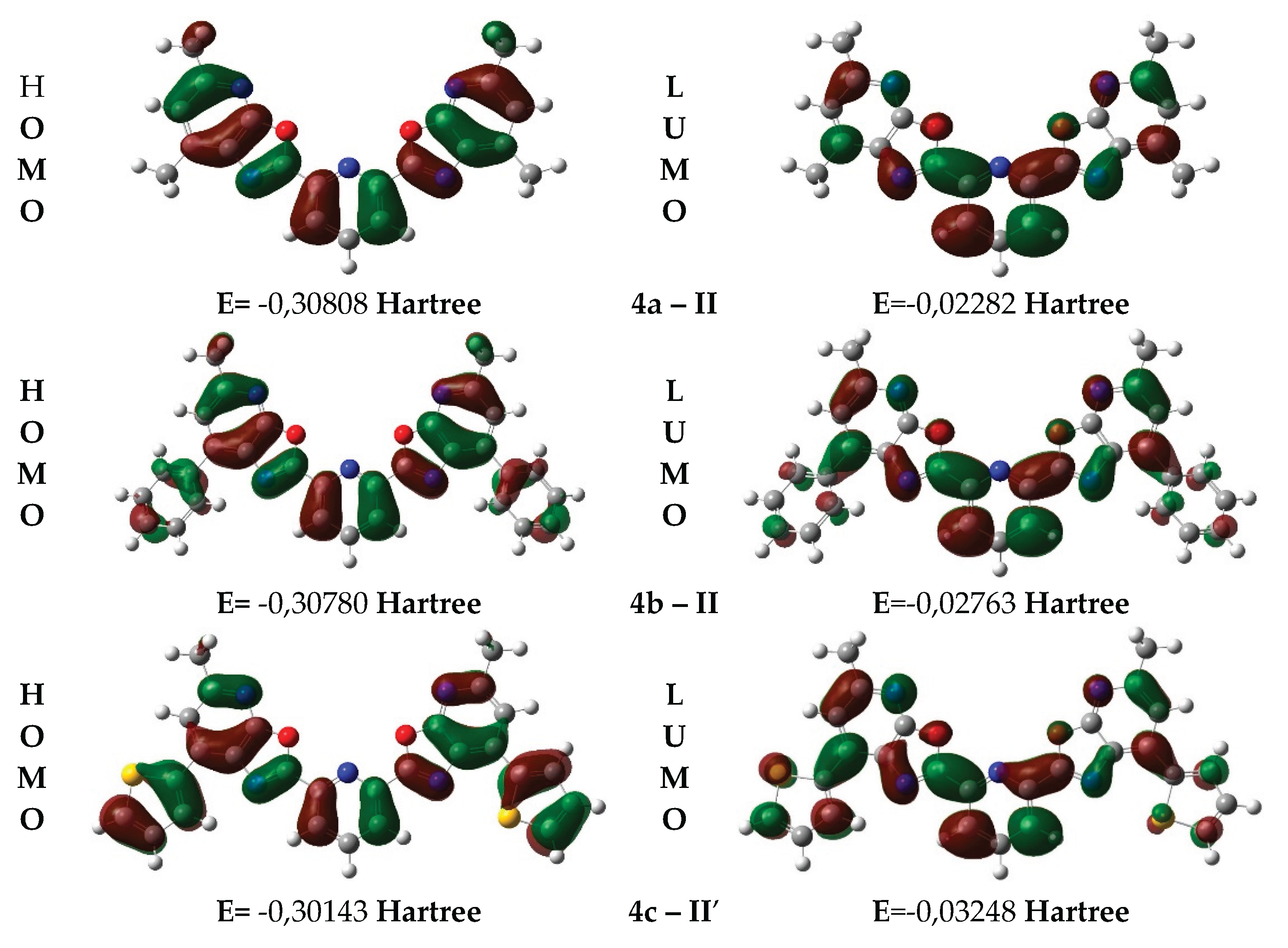
Figure 6.
Molecular Electrostatic Potential (MEP) maps for the most stable conformations 4a–II, 4b–II and 4c–II′ calculated by DFT (ωB97XD/6-311++G(d,p)) in acetonitrile (CH₃CN) MEP color scale: red areas indicate regions with the most negative potential; blue areas show regions with positive potential; green and yellow zones represent neutral charge distribution.
Figure 6.
Molecular Electrostatic Potential (MEP) maps for the most stable conformations 4a–II, 4b–II and 4c–II′ calculated by DFT (ωB97XD/6-311++G(d,p)) in acetonitrile (CH₃CN) MEP color scale: red areas indicate regions with the most negative potential; blue areas show regions with positive potential; green and yellow zones represent neutral charge distribution.

Figure 7.
TD DFT calculated UV–vis a) absorption and b) emission spectra for the 4a–II, 4b–II and 4c–II′ optimized conformation.
Figure 7.
TD DFT calculated UV–vis a) absorption and b) emission spectra for the 4a–II, 4b–II and 4c–II′ optimized conformation.
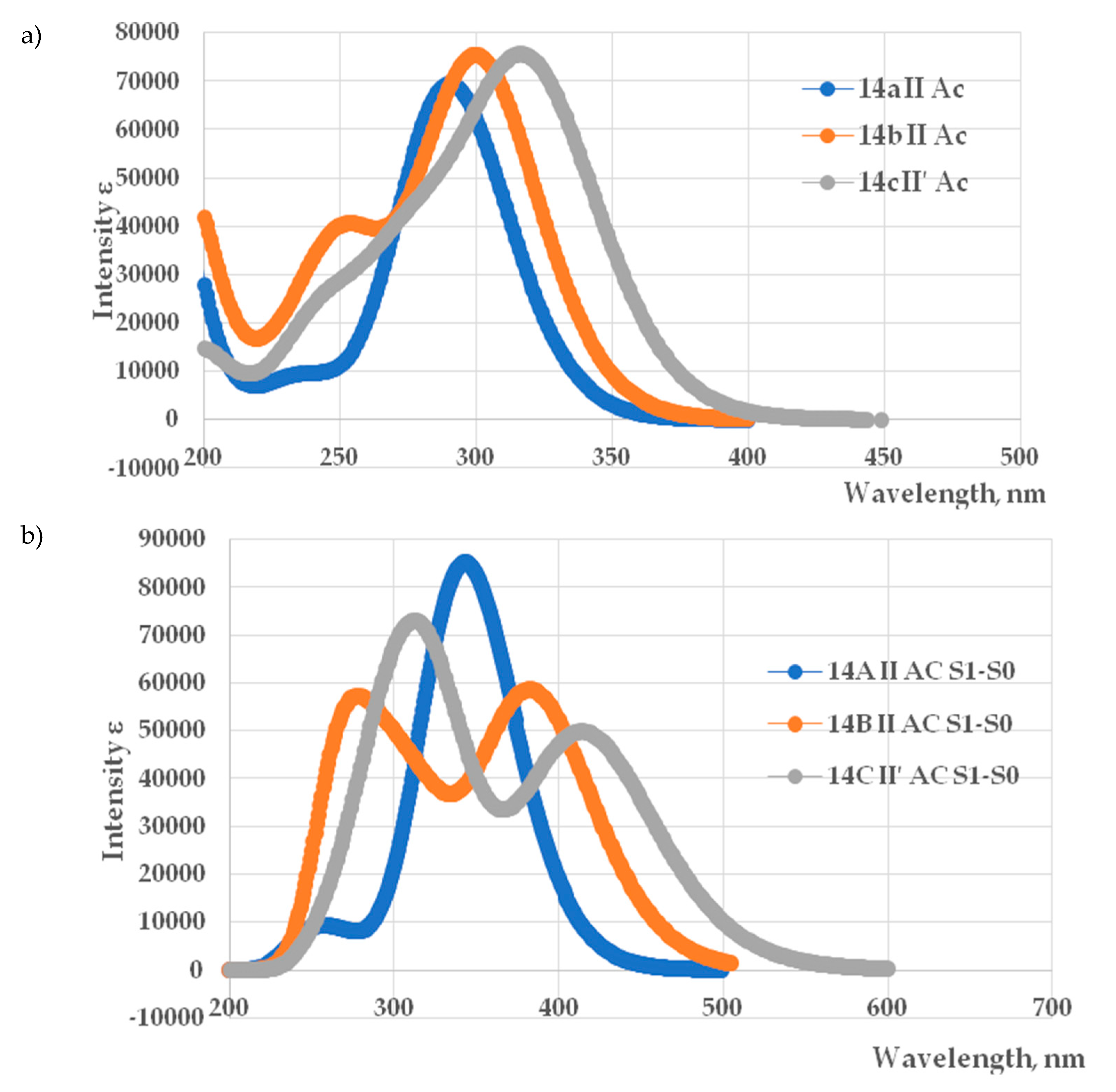

Table 1.
Photophysical properties of compounds 4a-d.
| Compound | UV–Vis | ||||||||
| λabsmax, [nm] | ε, [l/cm*mol] | ||||||||
| CHCl3 | CH2Cl2 | CH3CN | CHCl3 | CH2Cl2 | CH3CN | ||||
| 4а | 329 | 327 | 323 | 4200 | 3750 | 4950 | |||
| 4b | 338 | 337 | 336 | 5100 | 5000 | 7500 | |||
| 4c | 351 | 350 | 347 | 5000 | 4500 | 11150 | |||
| 4d | 343 | 357 | 354 | 4700 | 4100 | 4450 | |||
| Compound | Photoluminescence | ||||||||
| λemmax [nm] | Stokes shift, [nm] | Quantum yield (Φfl)а | |||||||
| CHCl3 | CH2Cl2 | CH3CN | CHCl3 | CH2Cl2 | CH3CN | CHCl3 | CH2Cl2 | CH3CN | |
| 4а | 358; 372 | 357, 372 | 368 | 43 | 45 | 45 | 0.50 | 0.84 | 0.71 |
| 4b | 368; 386 | 367; 384 | 365; 382 | 48 | 47 | 46 | 0.63 | 0.63 | 0.44 |
| 4c | 391; 414 | 415 | 431 | 63 | 65 | 84 | 0.40 | 0.38 | 0.32 |
| 4d | 444 | 454 | 474 | 105 | 97 | 120 | 0.47 | 0.73 | 0.74 |
Table 2.
Compounds 4a–c.
| No | Structural formula | IUPAC name |
| 4а | |
2,6-bis(5,7-dimethyloxazolo[5,4-b]pyridin-2-yl)pyridine |
| 4b |  |
2,6-bis(5-methyl-7-phenyloxazolo[5,4-b]pyridin-2-yl)pyridine |
| 4c |  |
2,6-bis(5-methyl-7-(thiophen-2-yl)oxazolo[5,4-b]pyridin-2-yl)pyridine |
Table 3.
Total Electronic Energy (Etotal) and Gibbs Free Energy (G) of various conformations of compounds 4a–c in vacuum and in solvents (CH₂Cl₂, CH₃CN).
Table 3.
Total Electronic Energy (Etotal) and Gibbs Free Energy (G) of various conformations of compounds 4a–c in vacuum and in solvents (CH₂Cl₂, CH₃CN).
| No |
Confor- mation |
Etotal, Hartree | G, Hartree | ||||
| vacuum | CH2Cl2 | CH3CN | vacuum | CH2Cl2 | CH3CN | ||
| Compound 4a | |||||||
| 1. | 4а - I | -1234.601128 | -1234.619395 | -1234.621546 | -1234.312490 | -1234.329376 | -1234.331575 |
| 2. | 4а - II | -1234.605119 | -1234.621583 | -1234.623556 | -1234.313758 | -1234.330327 | -1234.332393 |
| 3. | 4а - III | -1234.603304 | -1234.620573 | -1234.622619 | -1234.313169 | -1234.329507 | -1234.331598 |
| Compound 4b | |||||||
| 4. | 4b - I | -1618.025748 | -1618.047279 | -1618.049795 | -1617.631494 | -1617.656502 | -1617.658902 |
| 5. | 4b - II | -1618.029901 | -1618.048890 | -1618.051111 | -1617.638304 | -1617.657294 | -1617.659742 |
| 6. | 4b - III | -1618.028236 | -1618.047834 | -1618.050160 | -1617.637415 | -1617.656927 | -1617.659101 |
| Compound 4c | |||||||
| 7. | 4c - I | -2259.571275 | -2259.592992 | -2259.595664 | -2259.248044 | -2259.269901 | -2259.269709 |
| 8. | 4c - I’ | -2259.572917 | -2259.592245 | -2259.596036 | -2259.247327 | -2259.269201 | -2259.273103 |
| 9. | 4c – II | -2259.575259 | -2259.594544 | -2259.596348 | -2259.251650 | -2259.271053 | -2259.272130 |
| 10. | 4c - II’ | -2259.576532 | -2259.595037 | -2259.597195 | -2259.253505 | -2259.271921 | -2259.274708 |
| 11. | 4c - III | -2259.573293 | -2259.593485 | -2259.595891 | -2259.250371 | -2259.270751 | -2259.273289 |
| 12. | 4c - III’ | -2259.575792 | -2259.594097 | -2259.596231 | -2259.252692 | -2259.271836 | -2259.274293 |
| 13. | 4c - III’’ | -2259.574576 | -2259.593995 | -2259.596304 | -2259.252483 | -2259.271385 | -2259.274164 |
Table 4.
Energy characteristics of HOMO and LUMO orbitals for the most stable conformations 4a–II, 4b–II and 4c–II′.
Table 4.
Energy characteristics of HOMO and LUMO orbitals for the most stable conformations 4a–II, 4b–II and 4c–II′.
| Conformation | E(HOMO), Hartree | E(LUMO), Hartree | ДE (LUMO–HOMO), Hartree | ДE (LUMO–HOMO), eV |
| 4a–II | –0.3080 | -0.02282 | 0.28526 | 7.76 |
| 4b–II | -0.30780 | -0.02763 | 0.28017 | 7.63 |
| 4c–II′ | -0.30143 | -0.03248 | 0.26895 | 7.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



