
Submitted:
30 November 2025
Posted:
02 December 2025
You are already at the latest version
Abstract
Brain-derived growth factor, BDNF, has critical roles in a wide variety of neuronal aspects, including cell survival, differentiation, and synaptic function after their maturation. TrkB, a high affinity receptor for BDNF, is a major contributor in these neuronal aspects, and its functions are exerted via stimulating intracellular signaling pathways including the mitogen-activated protein kinase (MAPK) pathways. Especially, extracellular regulated kinase 1/2 (ERK 1/2), a major serine-threonine kinase and belonging to MAPK family, also works as a downstream molecule after activation of BDNF/TrkB system. Interestingly, growing evidence has demonstrated that ERK1/2 signaling exerts positive or negative influence on neurons in both healthy and pathological conditions in the central nervous system (CNS). Indeed, activation of ERK 1/2 stimulated by the BDNF/TrkB system is involved in regulation of synaptic plasticity. On the other hand, over-activation of ERK1/2 signaling under the pathological conditions is closely related to the neurodegeneration. In this review, we show how ERK1/2 signaling affects neuronal fate, including cell survival or cell death, in the CNS. Moreover, we discuss the involvement of overactivation of ERK signaling in the neurodegeneration observed in Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington’s disease (HD).
Keywords:
ERK1/2
; MAPK
; BDNF
; Alzheimer's disease
; neurodegeneration
; Parkinson's disease
1. Introduction
It has been well-established that decreased levels of BDNF in the brain is involved in the pathogenesis of neurodegenerative diseases including AD, PD, and HD because BDNF and its related molecules are essential for various neural events including cell proliferation, survival, synaptic function, and neurogenesis via activating the intracellular signaling such as MAPK signaling pathway [1]. In addition to BDNF, nerve growth factor (NGF), which was found as a neurotrophin firstly, also promotes neuronal cell survival in the peripheral nervous system (PNS) and CNS neurons [1]. Furthermore, neurotrophin-3 (NT3) is also a member of neurotrophin, and NT4/5 has been also identified. As a specific high affinity receptor, it has been established that TrkA receptor is a specific one for NGF, and TrkC is for NT-3 although TrkB is for both NT4/5 and BDNF. Trk receptors are the family of tyrosine kinase receptors, and trigger intracellular signaling pathways such as PLCγ, PI3K/Akt, and MAPK, affecting cell fate decision in the PNS and CNS. In addition to Trk receptors, a low-affinity p75NTR is also involved in neurotrophin actions because the receptor binds to all neurotrophins [1,2].
ERK/MAPKs signaling contributes to various cellular programs such as cell proliferation, cell differentiation, and cell survival maintenance. Although, as the family of MAPKs, ERKs, p38MAPK, and JNKs have been extensively studied, ERKs was firstly identified [3]. ERKs are one of Serine/Threonine (Ser/Thr) protein kinases and are stimulated by a variety of extracellular signals including BDNF/TrkB system or oxidate stress. ERKs, one of the most investigated kinases, are involved in synaptic regulation, learning and memory function [4,5]. For example, in the cultured neurons, BDNF induced upregulation of synaptic proteins including synapsin I, and glutamate receptor subunits (NR2A, NR2B, GluR1), via activating ERK1/2, suggesting positive effects of ERK1/2 on the synapse formation [6]. In contrast, 17beta-estradiol protected cultured neurons against oxidative stress via reducing activation of ERK1/2 [7]. Therefore, ERKs have both positive and negative influences on neurons.
ERK1/2 was reported to be involved in regulation of neural gene expression, neurotransmission, changes of synapses, suggesting that ERK1/2 pathway has a role in the learning and memory function [4]. Interestingly, an abnormal activation (hyperactivation) of ERK1/2 can contribute to disease progression (see [8]). In the CNS, ERK1/2 is one of major contributors in the brain development, and is regulating synaptic functions, maintaining healthy memory formation while the kinases can cause an enhancement of cell death induction and neuroinflammation (see [8]). Oxidative stress, neuroinflammation, and apoptosis and ferroptosis are suggested to affect the progression of traumatic brain injury, and hyperactivated ERK1/2 potentiates oxidative stress, inflammation, and neuronal cell death [9]. Importantly, evidence has demonstrated that a variety of stress in the CNS neurons, such as oxidative stress and neuroinflammatory, contribute to the AD pathophysiology. Remarkedly, it has been well-known that subfamilies of MAPKs (p38MAPKs, JNKs, and ERKs) are associated with an infinitude of physiological events in non-neuronal cells [10]. Since ERK1/2 signaling is important for cell division, it is possible that inhibitors for ERKs had been considered as anticancer drugs [10]. In addition, cell stress and inflammatory cytokines elicit activation of p38MAPKs, suggesting critical contribution of the p38MAPK signaling in the regulation of autoimmunity. Furthermore, the prohibition of JNK signaling is effective to ameliorate rheumatoid arthritis [10].
Because evidence suggests hyperphosphorylation state of various MAPKs, especially ERK1/2, in a variety of pathogenesis conditions reflects the neurodegeneration, a detailed understanding of the MAPK signaling dynamics in the CNS neurons is required. In this review, we show recent studies on positive influence of ERK 1/2, (for example, stimulated form by the BDNF/TrkB system) and on over-activation of ERK1/2 under the pathological conditions, which is suggested to be closely related to the neurodegeneration (see Figure 1). Furthermore, we discuss the status of ERK1/2 signaling when applying drug candidate in the model for AD, PD, and HD.
2. Relationship Among ERK Signaling and BDNF/TrkB System in Neurons
As mentioned above, it has been suggested that downregulation of the neurotrophins and/or their Trks have roles in the AD pathogenesis. Interestingly, using cell lines, cultured cortical neurons, and animals, Fernandez et al. (2023) examined effects of ACD856, which is a novel allosteric modulator for Trks [11]. It was revealed that ACD856 enhanced neurite outgrowth induced by NGF in PC12 cells. In primary cortical neurons, ACD856 upregulated levels of pERK1/2, and an Aβ-caused decrease in SNAP25-positive neurites was restored by ACD856 treatment [11]. Further, it was shown that the administration of ACD856 upregulated levels of BDNF protein in the brains of aged animals (21 months old mice) [11]. Using rats, Kim et al. (2014) showed beneficial effects of monoclonal antibody 29D7, an agonist for TrkB, against perinatal hypoxic ischemia (H-I) [12]. After an intracerebroventricular (icv) administration of 29D7, marked activation of ERK1/2 and Akt in neurons was confirmed. Importantly, when rats received 29D7 administration and subjected to H-I with unilateral carotid artery ligation and to 8% oxygen hypoxia exposure, an activation of caspase-3 (as a hallmark of apoptosis) caused by H-I injury was significantly suppressed by 29D7 [12]. In addition to BDNF/TrkB system, to improve a low bioavailability and its BBB impermeability when using NGF/TrkA system, effects of ENT-A013, which is one of NGF mimetics and selective activator of TrkA, was examined and confirmed its neuroprotective and anti-Aβ function as a potential therapeutic candidate against AD [13]. After injury and/or disease, the transplantation of neural stem cells (NSCs) is expected to induce neural repair, thus, Chen et al. (2017) investigated whether NSCs, which was genetically modified to overexpress BDNF (BDNF/NSCs), is effective in synaptogenesis in the rat model of traumatic brain injury (TBI) [14]. As expected, the transplantation of BDNF/NSCs transplantation significantly upregulated TrkB gene and increased the levels of pTrkB in the lesions caused by TBI. In addition to increased PSD95, levels of Ras and pERK1/2 were also elevated in the BDNF/NSCs transplantation groups compared with those in the naive NSCs ones [14]. These studies demonstrate that ERK1/2 signaling stimulated by BDNF/TrkB system has a positive effect in maintenance of CNS neurons (see Figure 1).
3. Natural Compounds and ERK-Signaling in AD Models
Growing evidence has demonstrated beneficial effects of natural compounds mediating ERK1/2 signaling in AD models. Choi et al. (2025) recently reported that paeoniflorin (PF) exerted an inhibitory effect against amyloidogenesis and neuroinflammation [15]. The amyloidogenesis and neuroinflammation observed in BV-2 microglial cells treated with lipopolysaccharide (LPS) were improved by application of PF. Furthermore, using amnesic mouse model, they found that deficits in memory function of animals were improved by PF [15]. In the hippocampus and cerebral cortex of the animal, although increased pp38MAPK, pJNK, and pERK1/2 caused by LPS were reduced by PF, upregulated pCREB and BDNF, and decreased APP were induced after PF application [15]. Phylloporia ribis (PRG), one of traditional Chinese medicines, has been used to improve weakness and memory loss in aged persons. Indeed, recent study showed that PRG exerted neuroprotective effects in vitro AD model produced by Aβ25-35 exposure [16]. PC12 cells exposed to Aβ25-35 exhibited increased mitochondrial stress and subsequent apoptosis, which were all reversed by PRG, with upregulation of pERK, pCREB and BDNF proteins, suggesting that PRG restored an inhibition of the ERK pathway [16]. Furthermore, Cornuside, an extract obtained from Corni Fructus, improved the decreased learning and memory in D-galactose (D-Gal)-treated mice [17]. Cornuside upregulated BDNF, and downregulated the RAGE, Iba1(for microglial), and GFAP (for astrocyte) in the hippocampus and cortex of the D-Gal-treated animals. Remarkedly, in addition to pERK1/2, phosphorylation level of JNK and p38 MAPK were significantly increased by D-Gal administration although only pERK1/2 was dramatically decreased by Cornuside treatment, suggesting a major contribution of overactivation of ERK1/2 in neuron damage caused by D-Gal [17]. Using Aβ-treated rats, Kuedo et al. (2022) showed beneficial effects of ethanolic extract of white shrimp (Litopenaous vannamei) shells (EESS) against a memory impairment [18]. Interestingly, treatment with EESS alone failed to improve, however, the EESS-loaded liposome significantly improved memory ability in Aβ-treated animals [18]. The EESS-loaded liposome increased protein levels of BDNF, TrkB, GAP-43, and PSD-95 as well as pERK1/2 in the cortex and hippocampus of Aβ-treated animals [18]. Cycloastragenol (CAG), which is a triterpenoid saponins and an ingredient in Astragalus membranaceus (Fisch.) Bunge, is known as one of Chinese medicinal herbs [19]. An increasing body of evidence has demonstrated that CAG has various pharmacological effects, including anti-oxidative properties, anti-inflammatory, and especially telomerase activation [19]. Ikram et al. (2021) examined the effects of CAG against Aβ-induced AD-like phenotypes in animals [20]. It was revealed that stereotaxic injection of Aβ caused oxidative stress and decreased expression of BDNF and pTrkB. As expected, decreased expression of NeuN, a neuron marker, in the cortex and hippocampus of Aβ-injected mice was observed. When CAG was given in Aβ-injected mice, BDNF, pTrkB and NeuN were significantly reversed [20]. Interestingly, the upregulation of pJNK, pp38MAPK, and pERK1/2 induced by Aβ injection were significantly reduced by CAG treatment [20]. Using cultured hippocampal neurons, Munni et al. (2023) examined effects of Garlic (Allium sativum L.) on neurite outgrowth and synapse development [21]. They compared effects of the ethanol extracts of unprocessed (white garlic extract, WGE) and processed (black garlic extract, BGE) garlic on the hippocampal synaptogenesis. Neurite outgrowth was enhanced by both WGE and BGE without cytotoxicity although the more robust effects by WGE was found [21]. Significant upregulation of both NR2A and NR2B was induced by WGE and BGE, but the increased level of NR2A by BGE was higher. The analysis with gas chromatography and mass spectrometry revealed that both extracts contained linalool, which has significant neurite outgrowth effect in the cultured neurons. By using network pharmacology, they demonstrated that effect of linalool was exerted via regulating signaling pathways including GSK3β and ERK1/2 [21]. Interestingly, an enhancement of BDNF/TrkB system with flavonoids (small molecules activating TrkB signaling) has been intensively studied to treat brain diseases including AD (See [2]). Beneficial effects of a variety of flavonoids against the progression of AD have been demonstrated. Tang et al. (2025) recently reported influence of isoliquiritigenin, which is a flavonoid obtained from the root of liquorice, in various aspects of AD model [22]. Treatment with isoliquiritigenin significantly restored the spatial memory capacity of mice receiving streptozotocin injection. Levels of pTau and reactive oxygen species in the cortex and hippocampus of the AD model mice caused by streptozotocin were improved by isoliquiritigenin. Furthermore, isoliquiritigenin restored synaptic impairment and loss of neurons and decreased the levels of mTOR and ERK activity [22]. Recent study also showed that glycosaminoglycan has protective effects against neurotoxicity caused by fluoride exposure [23]. Chondroitin sulfate (CS, one of glycosaminoglycans), improved the deficits in learning and memory of rats received a fluoride exposure [23]. Decreased hippocampal synaptophysin (a synaptic marker) caused by fluoride was reversed by CS. Although upregulation of metalloproteinase-9 (MMP-9), total ERK1/2, and pERK1/2 in vivo and in vitro (using SH-SY5Y cells) after fluoride exposure occurred, CS significantly reversed these fluoride-dependent negative effects, suggesting that CS rescued impaired synaptic functions by fluorosis exposure through repressing the ERK1/2-MMP-9 pathway [23].
These studies suggest the upregulation of BDNF/TrkB system after treatment with a variety of natural compounds is considered as strong methods to inhibit AD pathogenesis. However, activation status of ERK1/2 does not always correspond to that in the BDNF/TrkB system. Among compounds, actions of flavonoids in the disease models seem to be similar with those by BDNF/TrkB system (See [2]).
4. Chemicals and ERK-Signaling in AD Models
Joodi et al. (2025) reported that cabergoline (CAB, a dopamine receptor agonist) improved deficits in spatial and recognition memory using ovariectomized rats injected with D-Gal [24]. The concentrations of β-secretase, Aβ42, and pTau in the hippocampal of the model rats were decreased by CAB administration although pCREB and BDNF was increased by CAB. Importantly, upregulation of glutamate transporter-1 protein, which promoted uptake of glutamate contributing Ca2+ overload and consequently reduced the phosphorylated forms of p38MAPK and ERK1/2, was observed after CAB treatment [24]. A recent study has shown that (2R,6R)-hydroxynorketamine (HNK, a ketamine metabolite) reversed decreased protein synthesis and synaptic plasticity in the hippocampus of AD mouse models [25]. Hippocampal LTP and memory deficits caused by Aβ oligomers infusion was restored by HNK [25]. In addition, ERK1/2, mTOR, and p70S6 kinase 1 (S6K1)/ribosomal protein S6 signaling pathways were activated by treatment with HNK. HNK also rescued synaptic and memory defects and corrected transcriptional alterations in the hippocampus of the APP/PS1 AD mice [25]. Because chemotherapy may contribute to the long-term cognitive deficits in breast cancer survivors, Salas-Ramirez et al. (2015) studied possible mechanisms underlying cognitive impairments when chemotherapeutic agents were used to treat the cancer. Ovariectomized (OVX) and intact female rats were treated with saline or a combination of doxorubicin and cyclophosphamide. Significant impaired working and spatial memory by chemotherapy was observed. Interestingly, no difference in the impaired memory among OVX and intact animals was confirmed [26]. On the other hand, an increased pERK1/2 and pAkt as well as PSD95 expression in OVX female rodents after the chemotherapy was induced although OVX females displayed higher level of BDNF compared with that in intact ones, independent of chemotherapy [26]. Senescence accelerated mouse-prone 8 (SAMP8) mice exhibit significant age-related deficits in learning and memory function in compliance with early onset of senescence [27,28]. It has been demonstrated that SAMP8 mice are useful to investigate the mechanism under the senescence acceleration, the onset of AD, and other cognitive disorders. Recently, because the majority of AD cases are sporadic due to unknown genetic causes, SAMP8 mice, which are displaying Aβ deposits, hyperphosphorylated Tau, inflammation, and neuronal cell death, can be useful for clarifying features of sporadic AD [27]. Lian et al. (2021) showed therapeutic effect of DL0410, a novel acetylcholinesterase inhibitor, using SAMP8 mice. It was revealed that DL0410 administration significantly improves the cognitive deficits in SAMP8 mice [29]. DL0410 increased synaptic proteins including PSD95 and synaptophysin in the mouse brain. Interestingly, DL0410 administration upregulated BDNF and TrkB, and the neurotrophic effect was mediated via the ERK1/2 and PI3K/Akt/GSK3β pathways [29]. Zhang et al. (2020) examined effects of DL0410 against lipopolysaccharides (LPS)-induced neuroinflammation and hydrogen peroxide-induced oxidative stress as both neuroinflammation and oxidative stress are involved in AD [30]. They found that DL0410 reduced both inflammatory responses and ROS production in BV2 cells treated with LPS [30]. DL0410 restored downregulated pCREB, pTrkB, and pERK1/2 and expression of BDNF [30].
When using AD models, the status of pERK1/2 often differs from that in the BDNF/TrkB system (see Figure 1). Further interventions concerning the relationship among upregulation of BDNF (or status of pTrkB) and ERK1/2 singling are required.
5. Inhibitors of ERK Signaling and AD Models
Recently, Hassan et al. (2025) examined the effects of limettin (5,7-dimethoxycoumarin) on AD-like pathology using mouse model [31]. In their system, sporadic AD (SAD) animal model was established using an injection of streptozotocin (STZ, a compound derived from Streptomyces achromogeneshas), which induces activation of microglial cells causing neuroinflammation and oxidative [32,33]. The treatment with limettin and PD98059 (a specific inhibitor for MEK) reversed deficits in cognitive and memory performance of the SAD animals [31]. Furthermore, limettin and PD98059 decreased persistent activation of pERK1/2 in the hippocampus of the SAD mice. Importantly, the enhancement of pCREB (Ser133) and BDNF expressions was observed in SAD mice receiving PD98059 and limettin [31]. In addition to ERK1/2 pathway, p38 MAPK signaling is also involved in the pathophysiology of AD since Aβ peptide and/or tauopathies stimulate activation of the p38 MAPK [34]. For example, significant improvement in associative and spatial memory deficit observed in APP/PS1 Tg AD mice was achieved by treatment with MW01-18-150SRM, a selective inhibitor of p38αMAPK [35]. Using APP/PS1 knock-in mice, it was also revealed that spatial memory deficit in the AD animals was ameliorated by the p38αMAPK inhibitor [35].
6. Neurotoxicity of Aβ and ERK Signaling
Previously, Morroni et al. (2016) showed the early memory deficits caused by soluble beta-amyloid oligomers (AβO). They also reported cognitive decline in mice received intracerebroventricular injection of AβO was associated with elevated caspase-9 activation and oxidative stress, and decreased immunoreactivity of synaptophysin in the hippocampus of the animals [36]. In their system, AβO injection caused an increased ERK1/2 activity [36]. It was reported that expression levels of serine/arginine repetitive matrix 2 (SRRM2) and polyglutamine binding protein 1 (PQBP1, the splicing regulatory protein relating an intellectual disability) were slightly lower in the postmortem cerebral cortex in AD patients, and that ERK1/2 had a role for phosphorylation of SRRM2 at Ser1068 [37]. Interestingly, before Aβ aggregation in the AD model mice, the subcellular localization shift (from the nucleus to the cytoplasm) of SRRM2 occurred when SRRM2 was phosphorylated [37]. It was revealed that the SRRM2 localization shift decreased PQBP1 impairing synapses. suggesting ERK signaling and splicing-related proteins play a role in the early-phase pathologies of AD [37]. As mentioned above, it has been demonstrated that dysregulation of ERK1/2 is involved in cognitive impairments and neuropathogenesis of AD. A recent report showed that E-twenty-six (ETS)-like protein 1 (ELK1), a member of transcription factor, which is activated by ERK1/2, has a role in AD pathogenesis. Yi et al. (2025) reported that ELK1 knockdown or inhibiting its activation via interfering peptide decreased amyloidogenic processing of APP, consequently reducing generation of Aβ in APP23/PS45 double-transgenic AD animal [38]. Interestingly, the suppression of ELK1 expression or activation improved synaptic and memory impairments observed in the AD animal, suggesting possible contribution of ERK1/2 to AD pathogenesis through regulating ELK1 function [38]. Using the AlCl3-induced AD rat model, Salama et al. (2025) examined effects of betanin (BET)-loaded liposomal nanocarriers (LPN, to enhance the brain penetration and bioavailability) on AD-like behaviors [39]. In addition to less cortical and hippocampal degeneration, an improvement in spatial and learning memory was achieved by BET-LPN treatment compared with oral BET treatment. Furthermore, BET-LPN treatment reduced the levels of ERK1/2 and MEK1/2 with more effectiveness compared with traditional oral BET [39]. Wu et al. (2025) investigated influence of long-term cervical lymphadenectomy (cLE) in AD-like tauopathy [40]. They found that male mice which received cLE displayed significant impairment in cognitive function and anxiety-depressive behaviors. Significant impairment in brain wastes (including A and pTau drainage) activated ERK1/2, leading to reduced autophagy, were caused by CLE [40]. To investigate possible impact of prenatal stress (PS) on brain maturation and cognitive function in offspring, Trojan et al. (2023) used male C57BL/6 J (WT) and the knock-in (KI) APPNL-F/NL-F mice carrying Swedish and Beyreuther/Iberian mutations, and pregnant animals were exposed to daily restraint stress (E12.5–E18.5) [41]. They found an increased levels of ApoE and Aβ42/Aβ40 in the frontal cortex and the hippocampus of male offspring of the KI animals [41]. Furthermore, a changed insulin signaling, an interference in ERK1/2- and mTOR-signaling, and pro-inflammatory (IL-6, IL-23, and TNF-α) status were observed in the KI mice [41]. Remarkably, recent study using single-cell (scRNA-seq) and single-nucleus (snRNA-seq) RNA sequencing demonstrated cellular and molecular alterations when mice received distal middle cerebral artery occlusion (dMCAO) [42]. In this experiment, animals were assigned to control, sham, 3day-dMCAO, and 14day-dMCAO. Cell populations, including neurons (both glutamatergic and GABAergic ones), astrocytes, oligodendrocytes, microglia, endothelial cells, fibroblast-like cells, and pericytes were identified by snRNA-seq [42]. Importantly, neuronal death, autophagy, and cAMP biosynthesis pathways were shown by Gene ontology analysis. Increased levels of syngap1, Rock1 and Ikbkb in the glutamatergic populations suggested a contribution to vulnerability to ischemic injury. Dissociation of SynGAP1 from PSD-95 after ischemia may increase pERK1/2 levels, while ischemic preconditioning reduces the dissociation and inhibits ERK1/2 overactivation [42].
Under pathological conditions, it has been demonstrated that hyperactivation of ERK1/2 (or sustained activation of ERK1/2) is recognized as one of major contributors to potentiate the progression of AD pathophysiology. Using pathological system in which ERK1/2 is a major contributor, further investigation concerning drug candidates, which increase both BDNF- and ERK1/2-signaling, may be interesting.
7. Role of ERK Signaling in Parkinson’s Disease
PD affects more than 10 million individuals worldwide and is clinically defined by motor symptoms such as bradykinesia, rigidity, and resting tremor, as well as non-motor manifestations including cognitive impairment and mood disturbances [43]. The pathological hallmarks of PD include degeneration of nigrostriatal dopaminergic neurons and intracellular accumulation of α-synuclein aggregates, known as Lewy bodies. Despite substantial advances in symptomatic therapy, including dopamine replacement strategies, no current treatment effectively halts or reverses neurodegeneration [43].
As mentioned above, neurotrophic factors have emerged as critical regulators of neuronal integrity in both the developing and adult brain. Among them, BDNF has received particular attention due to its high expression in dopaminergic circuits and its ability to promote neuronal survival through activation of TrkB [44]. Downstream of TrkB, the ERK/MAPK pathway transduces extracellular cues into transcriptional programs that support neuronal plasticity, differentiation, and resistance to oxidative or proteotoxic stress. Understanding how ERK signaling mediates BDNF’s neuroprotective effects may reveal novel therapeutic strategies for PD.
BDNF supports the survival of dopaminergic neurons in the substantia nigra and enhances the functional activity of striatal medium spiny neurons, the primary target neurons receiving input from the substantia nigra. It has been also shown that BDNF facilitates both the synthesis and uptake of dopamine [44].
Multiple lines of evidence indicate that BDNF signaling is disrupted in PD. For example, postmortem studies reveal reduced BDNF and TrkB expression in PD brains, particularly in the substantia nigra and striatum [45,46,47]. Another study identified a nigrostriatal phenotype in aged haploinsufficient TrkB mutant mice (TrkB+/−) that mirrors a preclinical stage of PD [48]. This phenotype is characterized by a reduction of tyrosine hydroxylase (TH)-positive neurons in the substantia nigra pars compacta (SNpc), accompanied by a loss of striatal TH-positive fibers and extensive α-synuclein accumulation within dopaminergic cell bodies [48]. The downregulation of this trophic axis may exacerbate neuronal vulnerability to mitochondrial dysfunction and oxidative damage, both of which are central to PD pathogenesis [2]. Thus, the impairment of BDNF signaling represents not only a consequence but also a contributing factor to dopaminergic degeneration.
Experimental models using α-synuclein fibrils or overexpression demonstrate attenuated ERK activation. A recent study reported that aberrant phosphorylation of α-synuclein at Ser129 (pS129) inhibited activation of the BDNF/ERK signaling pathway in PD model rats [49]. Pharmacological activation of ERK1/2 alleviated the pathological effects of pS129, restored BDNF expression in the medial prefrontal cortex (mPFC), and reduced pS129 levels in both the substantia nigra and mPFC [49]. Another study demonstrated that transfection with constitutively active MEK-1 induced ERK phosphorylation in cells overexpressing α-synuclein, which consequently enhanced cell viability, thereby reinforcing the neuroprotective function of ERK1/2 signaling [50].
However, the role of ERK1/2 in PD is complex and context-dependent: while transient activation exerts protective effects, sustained or aberrant activation, often induced by neurotoxins like MPP⁺ or 6-hydroxydopamine (6-OHDA), can lead to neuronal stress and apoptosis [51]. For example, activated ERK1/2 has been observed in Lewy bodies, the pathological hallmarks of PD composed of aggregated α-synuclein within degenerated neurons [52]. Another experimental PD model also showed that exposure to 6-OHDA induced sustained ERK1/2 activation, whereas treatment with PD98059, a MEK inhibitor, mitigated cell death [53]. Using a human iPSC-based PD model, a recent study demonstrated, through transcriptomic profiling and subsequent biochemical validation, that the ERK1/2 pathway is activated in cortical neurons derived from PD patients [54]. They further showed that inhibition of this signaling cascade reduced neuronal cell death [54]. Therefore, understanding the temporal and spatial dynamics of ERK1/2 signaling is crucial for exploiting its neuroprotective potential.
Collectively, the BDNF-ERK1/2 signaling axis is a central mediator of neuronal survival, synaptic integrity, and adaptive plasticity in the nigrostriatal system. Its impairment in PD contributes significantly to dopaminergic neurodegeneration. Re-establishing this pathway, either by augmenting BDNF availability or selectively targeting ERK1/2 signaling, holds great promise for neuroprotective therapy. Future studies should focus on elucidating the precise regulatory mechanisms governing ERK1/2 activation in dopaminergic neurons and developing targeted delivery systems capable of restoring trophic signaling in the diseased brain. A better understanding of the temporal dynamics and context-specific roles of ERK1/2 signaling will be essential for translating these insights into effective, disease-modifying treatments for PD.
8. Role of ERK Signaling in Huntington’s Disease
HD represents one of the most devastating hereditary neurodegenerative diseases, resulting from a CAG repeat expansion in the huntingtin (HTT) gene that produces a mutant huntingtin protein (mHTT) with an elongated polyglutamine tract [55]. The disease is marked by selective degeneration of medium spiny neurons (MSNs) in the striatum, leading to characteristic motor impairments such as chorea and dystonia, accompanied by cognitive and psychiatric disturbances [55]. Despite decades of investigation, the precise molecular mechanisms connecting mHTT expression to neuronal death remain incompletely understood. Among various cellular pathways affected, neurotrophic signaling mediated by BDNF and its downstream effector ERK1/2 has received considerable attention due to its essential role in neuronal survival and plasticity.
BDNF is synthesized predominantly in cortical neurons and transported along corticostriatal projections to reach the striatum, where it supports the viability and function of MSNs [56]. Mutant huntingtin disrupts this process at multiple levels. Transcriptionally, mHTT sequesters the repressor-element 1-silencing transcription/neuron-restrictive silencer factor (REST/NRSF), a transcriptional repressor, inappropriately within the nucleus, repressing BDNF promoter activity [57]. Consequently, cortical neurons in HD exhibit markedly reduced BDNF mRNA and protein levels, which directly correlates with disease progression and striatal degeneration severity [57].
Axonal transport of BDNF-containing vesicles is similarly compromised. Wild-type huntingtin interacts with huntingtin-associated protein 1 (HAP1) and the dynein motor complex to facilitate efficient microtubule-based transport [57]. The polyglutamine-expanded form of huntingtin, however, has been shown to influence its capacity to interact with motor complex subunits and to transport intracellular cargoes, such as BDNF [58]. The resultant depletion of BDNF in the striatum deprives MSNs of essential trophic support, thereby reducing TrkB receptor activation and subsequent ERK1/2 phosphorylation. This early disruption in neurotrophic delivery constitutes one of the earliest detectable molecular events preceding overt neurodegeneration in HD.
Beyond the suppression of BDNF expression and availability by mHtt, TrkB receptor signaling itself is also disrupted by mHtt. BDNF-induced activation of the MEK/ERK pathway was found to be significantly diminished in striatal STHdhQ111 cells, while the activation of other TrkB downstream signaling cascades, such as Akt and PLCγ, remained unaffected [59]. Furthermore, the study demonstrated that attenuated BDNF- ERK1/2 pathway enhances the susceptibility of the mutant huntingtin striatal cells to oxidative stress, and that pharmacological stimulation of the MAPK pathway using phorbol 12-myristate 13-acetate (PMA) effectively protects against oxidative stress–induced cell death, underscoring the critical role of ERK1/2 signaling in mediating BDNF-dependent neuroprotection in STHdhQ111 striatal cells [59].
Postmortem analyses of HD brains, along with studies in transgenic mouse models, have demonstrated diminished ERK1/2-dependent gene expression, characterized by reduced pCREB and lower expression of pro-survival genes [60,61,62]. These findings emphasize the progressive loss of neuroprotective ERK1/2 signaling during disease development.
Given the critical role of the BDNF–ERK signaling pathway in maintaining neuronal viability and synaptic integrity, strategies aimed at restoring or augmenting this pathway have emerged as promising therapeutic directions for HD. The progressive impairment of BDNF production, TrkB receptor activation, and ERK1/2-dependent transcriptional regulation creates a cascade of neurodegenerative processes that can potentially be attenuated or reversed through molecular interventions. Several therapeutic approaches—ranging from neurotrophin supplementation to small-molecule enhancement of ERK1/2 signaling—have been explored in preclinical models of HD.
One of the most direct strategies for rescuing ERK1/2 signaling in HD involves restoring BDNF levels in the corticostriatal circuit. Viral vector-mediated delivery of BDNF has been among the most extensively studied approaches. For example, adeno-associated virus (AAV) to express BDNF in striatal neurons have demonstrated substantial neuroprotective effects in transgenic HD rat model [63].
Another strategy involves pharmacological modulation of TrkB receptors to activate the downstream ERK1/2 cascade. Small-molecule TrkB agonists such as 7,8-dihydroxyflavone (7,8-DHF) have received considerable attention for their neuroprotective potential. 7,8-DHF crosses the blood–brain barrier efficiently and mimics BDNF by binding to the extracellular domain of TrkB, thereby triggering receptor dimerization and phosphorylation. In HD models, administration of 7,8-DHF stimulates both the PI3K/Akt and MAPK signaling pathways, promoting neuronal survival and plasticity while improving synaptic function [64,65]. Another promising small-molecule modulator of the ERK1/2 pathway is fisetin, a naturally occurring plant polyphenol. Fisetin was shown to increase ERK phosphorylation and enhance cell survival in PC12 cells expressing mHTT, while concurrently suppressing JNK and caspase-3 activation. Pharmacological inhibition of MEK abrogated both ERK1/2 activation and the neuroprotective effect of fisetin against mHTT-induced cell death, highlighting the pivotal role of ERK1/2 pathway activation in fisetin-mediated cellular rescue [66].
Taken together, therapeutic strategies targeting the BDNF–ERK pathway offer a biologically grounded and mechanistically rational approach to counteract neurodegeneration in HD. Whether through exogenous BDNF delivery, pharmacological activation of TrkB, fine-tuned modulation of ERK1/2 activity, or epigenetic reactivation of neurotrophic genes, the restoration of this signaling axis holds promise for re-establishing neuronal resilience and delaying disease progression. The success of future interventions will depend on achieving precise control over the magnitude and timing of ERK1/2 activation—transforming this fundamental survival pathway into a clinically effective target for HD therapy.
9. Conclusions
The BDNF–ERK signaling axis emerges as a central determinant of neuronal survival, synaptic integrity, and adaptive plasticity, positioning it at the nexus of mechanisms underlying major neurodegenerative diseases. In AD, deficits in BDNF expression and subsequent attenuation of ERK1/2 activation contribute to synaptic weakening, impaired long-term potentiation, and cognitive decline. In PD, compromised BDNF signaling within the nigrostriatal system exacerbates dopaminergic neuronal vulnerability, increasing susceptibility to oxidative stress, mitochondrial dysfunction, and α-synuclein pathology. HD similarly highlights the consequences of BDNF–ERK disruption, where impaired transcription and axonal transport of BDNF, coupled with defective ERK1/2 signaling, drive striatal neuron degeneration and motor deficits (Figure 2). Across these conditions, the convergence on the BDNF–ERK axis underscores its role as a unifying molecular pathway in neurodegeneration, despite disease-specific variations in upstream triggers and downstream outcomes.
Therapeutically, restoring or enhancing BDNF–ERK signaling represents a compelling strategy. Preclinical evidence suggests that interventions capable of increasing BDNF availability, activating TrkB receptors, or directly modulating ERK1/2 activity can ameliorate synaptic deficits, reduce neuronal loss, and improve functional outcomes (Figure 2). Nonetheless, translating these findings into clinical therapies remains challenging. Achieving spatiotemporally precise activation of ERK1/2 without eliciting off-target effects, maladaptive plasticity, or oncogenic risks requires sophisticated delivery systems and a nuanced understanding of the signaling dynamics in different neuronal populations.
Future research should focus on several key directions. First, it is critical to delineate disease-specific mechanisms of BDNF–ERK dysregulation, including the impact of protein aggregates, genetic mutations, and environmental stressors on pathway integrity. Second, integrating multi-omics approaches—encompassing transcriptomics, proteomics, and phosphoproteomics—may reveal novel regulatory nodes and biomarkers predictive of pathway dysfunction and therapeutic responsiveness. Third, combinatorial strategies that simultaneously target BDNF–ERK signaling and intersecting pathways, such as PI3K/Akt, JNK, and p38MAPK, may offer synergistic neuroprotection, particularly in multifactorial conditions like AD and PD. Additionally, emerging technologies, including gene therapy, engineered neurotrophic mimetics, and nanocarrier-based delivery, hold promise for achieving sustained, targeted modulation of this pathway in specific neuronal populations.
Finally, longitudinal and translational studies are needed to evaluate the efficacy and safety of BDNF–ERK-targeted interventions in human patients. Understanding the temporal window during which pathway restoration is most effective, as well as the long-term effects on synaptic function and cognitive or motor outcomes, will be essential for clinical application. By integrating mechanistic insights with advanced therapeutic approaches, targeting the BDNF–ERK axis offers a realistic avenue not only for slowing neurodegeneration but also for potentially restoring neuronal function and improving quality of life in patients with Alzheimer’s, Parkinson’s, and Huntington’s disease.
Author Contributions
TN and RK wrote and edited the paper. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the Grant-in-Aid for Scientific Research (C) (JSPS KA-KENHI 24K09645) (TN) of the Ministry of Education, Culture, Sports, Science, and Technology of Japan, and from the Takeda Science Foundation (TN). This study was also supported by Japanese Society of Inherited Metabolic Disease/Sanofi LSD Research Grant (RK), Kawano Masanori Memorial Public Interest Incorporated Foundation for Promotion of Pediatrics (RK), and the Grant-in-Aid for Scientific Research (JSPS KAKENHI 23K07772) (RK).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or reported in this review article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol Histopathol 2010, 25, 237–258. [Google Scholar] [CrossRef]
- Numakawa, T.; Kajihara, R. The Role of Brain-Derived Neurotrophic Factor as an Essential Mediator in Neuronal Functions and the Therapeutic Potential of Its Mimetics for Neuroprotection in Neurologic and Psychiatric Disorders. Molecules 2025, 30. [Google Scholar] [CrossRef]
- Boulton, T.G.; Cobb, M.H. Identification of multiple extracellular signal-regulated kinases (ERKs) with antipeptide antibodies. Cell Regul 1991, 2, 357–371. [Google Scholar] [CrossRef]
- Ojea Ramos, S.; Feld, M.; Fustiñana, M.S. Contributions of extracellular-signal regulated kinase 1/2 activity to the memory trace. Front Mol Neurosci 2022, 15, 988790. [Google Scholar] [CrossRef]
- Sweatt, J.D. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 2004, 14, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Niyaz, M.; Kudo, M.; Kunugi, H. Glucocorticoid prevents brain-derived neurotrophic factor-mediated maturation of synaptic function in developing hippocampal neurons through reduction in the activity of mitogen-activated protein kinase. Mol Endocrinol 2008, 22, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, Y.; Matsumoto, T.; Yokomaku, D.; Taguchi, T.; Niki, E.; Hatanaka, H.; Kunugi, H.; Numakawa, T. 17beta-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology 2007, 148, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (Review). Int J Mol Med 2017, 39, 1338–1346. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.; Hou, L.; Wei, G.; He, C.; Li, H.; Liu, L. Advances in ERK Signaling Pathway in Traumatic Brain Injury: Mechanisms and Therapeutic Potential. Neurochem Res 2025, 50, 191. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef]
- Parrado Fernandez, C.; Juric, S.; Backlund, M.; Dahlström, M.; Madjid, N.; Lidell, V.; Rasti, A.; Sandin, J.; Nordvall, G.; Forsell, P. Neuroprotective and Disease-Modifying Effects of the Triazinetrione ACD856, a Positive Allosteric Modulator of Trk-Receptors for the Treatment of Cognitive Dysfunction in Alzheimer's Disease. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.S.; Cho, S.; Nelson, J.W.; Zipfel, G.J.; Han, B.H. TrkB agonist antibody pretreatment enhances neuronal survival and long-term sensory motor function following hypoxic ischemic injury in neonatal rats. PLoS One 2014, 9, e88962. [Google Scholar] [CrossRef] [PubMed]
- Rogdakis, T.; Charou, D.; Latorrata, A.; Papadimitriou, E.; Tsengenes, A.; Athanasiou, C.; Papadopoulou, M.; Chalikiopoulou, C.; Katsila, T.; Ramos, I.; et al. Development and Biological Characterization of a Novel Selective TrkA Agonist with Neuroprotective Properties against Amyloid Toxicity. Biomedicines 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wu, Y.; Wang, Y.; Zhu, J.; Chu, H.; Kong, L.; Yin, L.; Ma, H. Brain-Derived Neurotrophic Factor Increases Synaptic Protein Levels via the MAPK/Erk Signaling Pathway and Nrf2/Trx Axis Following the Transplantation of Neural Stem Cells in a Rat Model of Traumatic Brain Injury. Neurochem Res 2017, 42, 3073–3083. [Google Scholar] [CrossRef]
- Choi, J.W.; Im, J.H.; Balakrishnan, R. Paeoniflorin exercise-mimetic potential regulates the Nrf2/HO-1/BDNF/CREB and APP/BACE-1/NF-κB/MAPK signaling pathways to reduce cognitive impairments and neuroinflammation in amnesic mouse model. Biomed Pharmacother 2025, 189, 118299. [Google Scholar] [CrossRef] [PubMed]
- Bian, Z.; Cao, C.; Ding, J.; Ding, L.; Yu, S.; Zhang, C.; Liu, Q.; Zhu, L.; Li, J.; Zhang, Y.; et al. Neuroprotective effects of PRG on Aβ(25-35)-induced cytotoxicity through activation of the ERK1/2 signaling pathway. J Ethnopharmacol 2023, 313, 116550. [Google Scholar] [CrossRef]
- Yu, L.; Che, R.; Zhang, W.; Xu, J.; Lian, W.; He, J.; Tu, S.; Bai, X.; He, X. Cornuside, by regulating the AGEs-RAGE-IκBα-ERK1/2 signaling pathway, ameliorates cognitive impairment associated with brain aging. Phytother Res 2023, 37, 2419–2436. [Google Scholar] [CrossRef]
- Kuedo, Z.; Chotphruethipong, L.; Raju, N.; Reudhabibadh, R.; Benjakul, S.; Chonpathompikunlert, P.; Klaypradit, W.; Hutamekalin, P. Oral Administration of Ethanolic Extract of Shrimp Shells-Loaded Liposome Protects against Aβ-Induced Memory Impairment in Rats. Foods 2022, 11. [Google Scholar] [CrossRef]
- Yu, Y.; Zhou, L.; Yang, Y.; Liu, Y. Cycloastragenol: An exciting novel candidate for age-associated diseases. Exp Ther Med 2018, 16, 2175–2182. [Google Scholar] [CrossRef]
- Ikram, M.; Jo, M.H.; Choe, K.; Khan, A.; Ahmad, S.; Saeed, K.; Kim, M.W.; Kim, M.O. Cycloastragenol, a Triterpenoid Saponin, Regulates Oxidative Stress, Neurotrophic Dysfunctions, Neuroinflammation and Apoptotic Cell Death in Neurodegenerative Conditions. Cells 2021, 10. [Google Scholar] [CrossRef]
- Munni, Y.A.; Dash, R.; Choi, H.J.; Mitra, S.; Hannan, M.A.; Mazumder, K.; Timalsina, B.; Moon, I.S. Differential Effects of the Processed and Unprocessed Garlic (Allium sativum L.) Ethanol Extracts on Neuritogenesis and Synaptogenesis in Rat Primary Hippocampal Neurons. Int J Mol Sci. [CrossRef]
- Tang, Z.; Sha, T.; Wang, Y.; Xiao, Y.; Ding, Y.; Ni, R.; Qi, X. Isoliquiritigenin attenuated cognitive impairment, cerebral tau phosphorylation and oxidative stress in a streptozotocin-induced mouse model of Alzheimer's disease. Life Sci 2025, 376, 123759. [Google Scholar] [CrossRef]
- Ai, F.; Wang, S.; Ye, L.; Wan, W.; Zhou, X.; Liu, M.; Mo, K.; Lu, Y.; Wei, N.; Guan, Z.; et al. Chondroitin sulfate protects against synaptic impairment caused by fluorosis through the Erk1/2-MMP-9 signaling pathway. Sci Rep 2025, 15, 29760. [Google Scholar] [CrossRef]
- Joodi, S.A.; Khattab, M.M.; Ibrahim, W.W. Repurposing of cabergoline to improve cognitive decline in D-galactose-injected ovariectomized rats: Modulation of AKT/mTOR, GLT-1/P38-MAPK, and ERK1/2 signaling pathways. Toxicol Appl Pharmacol 2025, 500, 117391. [Google Scholar] [CrossRef]
- Ribeiro, F.C.; Cozachenco, D.; Argyrousi, E.K.; Staniszewski, A.; Wiebe, S.; Calixtro, J.D.; Soares-Neto, R.; Al-Chami, A.; Sayegh, F.E.; Bermudez, S.; et al. The ketamine metabolite (2R,6R)-hydroxynorketamine rescues hippocampal mRNA translation, synaptic plasticity and memory in mouse models of Alzheimer's disease. Alzheimers Dement 2024, 20, 5398–5410. [Google Scholar] [CrossRef]
- Salas-Ramirez, K.Y.; Bagnall, C.; Frias, L.; Abdali, S.A.; Ahles, T.A.; Hubbard, K. Doxorubicin and cyclophosphamide induce cognitive dysfunction and activate the ERK and AKT signaling pathways. Behav Brain Res 2015, 292, 133–141. [Google Scholar] [CrossRef]
- Ong, J.; Sasaki, K.; Ferdousi, F.; Suresh, M.; Isoda, H.; Szele, F.G. Senescence accelerated mouse-prone 8: a model of neuroinflammation and aging with features of sporadic Alzheimer's disease. Stem Cells 2025, 43. [Google Scholar] [CrossRef]
- Akiguchi, I.; Pallàs, M.; Budka, H.; Akiyama, H.; Ueno, M.; Han, J.; Yagi, H.; Nishikawa, T.; Chiba, Y.; Sugiyama, H.; et al. SAMP8 mice as a neuropathological model of accelerated brain aging and dementia: Toshio Takeda's legacy and future directions. Neuropathology 2017, 37, 293–305. [Google Scholar] [CrossRef]
- Lian, W.W.; Zhou, W.; Zhang, B.Y.; Jia, H.; Xu, L.J.; Liu, A.L.; Du, G.H. DL0410 ameliorates cognitive disorder in SAMP8 mice by promoting mitochondrial dynamics and the NMDAR-CREB-BDNF pathway. Acta Pharmacol Sin 2021, 42, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhao, J.; Wang, Z.; Xu, L.; Liu, A.; Du, G. DL0410 attenuates oxidative stress and neuroinflammation via BDNF/TrkB/ERK/CREB and Nrf2/HO-1 activation. Int Immunopharmacol 2020, 86, 106729. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.M.; Elsayed, N.S.; Assaf, N.; Budzyńska, B.; Skalicka-Wożniak, K.; Ibrahim, S.M. Limettin and PD98059 Mitigated Alzheimer's Disease Like Pathology Induced by Streptozotocin in Mouse Model: Role of p-ERK1/2/p-GSK-3β/p-CREB/BDNF Pathway. J Neuroimmune Pharmacol 2025, 20, 55. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feng, P.; Peng, A.; Qiu, X.; Lai, W.; Zhang, L.; Li, W. Protective effects of isoquercitrin on streptozotocin-induced neurotoxicity. J Cell Mol Med 2020, 24, 10458–10467. [Google Scholar] [CrossRef]
- Grieb, P. Intracerebroventricular Streptozotocin Injections as a Model of Alzheimer's Disease: in Search of a Relevant Mechanism. Mol Neurobiol 2016, 53, 1741–1752. [Google Scholar] [CrossRef]
- Falcicchia, C.; Tozzi, F.; Arancio, O.; Watterson, D.M.; Origlia, N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Roy, S.M.; Grum-Tokars, V.L.; Schavocky, J.P.; Saeed, F.; Staniszewski, A.; Teich, A.F.; Arancio, O.; Bachstetter, A.D.; Webster, S.J.; Van Eldik, L.J.; et al. Targeting human central nervous system protein kinases: An isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer's disease mouse models. ACS Chem Neurosci 2015, 6, 666–680. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Tarozzi, A.; Rimondini, R.; Hrelia, P. Early effects of Aβ1-42 oligomers injection in mice: Involvement of PI3K/Akt/GSK3 and MAPK/ERK1/2 pathways. Behav Brain Res 2016, 314, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Kondo, K.; Chen, X.; Homma, H.; Tagawa, K.; Kerever, A.; Aoki, S.; Saito, T.; Saido, T.; Muramatsu, S.I.; et al. The intellectual disability gene PQBP1 rescues Alzheimer's disease pathology. Mol Psychiatry 2018, 23, 2090–2110. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Li, J.; He, Y.; Wang, J.; Wang, M.; Guo, S.; Luo, M.; Wu, B.; Xu, M.; Tian, Q.; et al. ELK1 inhibition alleviates amyloid pathology and memory decline by promoting the SYVN1-mediated ubiquitination and degradation of PS1 in Alzheimer's disease. Exp Mol Med 2025, 57, 1032–1046. [Google Scholar] [CrossRef]
- Salama, R.M.; Yehia, R.; Elmongy, N.F.; Sallam, A.A.; Abd-Elgalil, M.M.; Schaalan, M.F.; Abdel-Mottaleb, M.M.A.; Bazan, L.S. Evaluation of betanin-loaded liposomal nanocarriers in AlCl(3)-induced Alzheimer's disease in rats: Impact on cognitive function, neurodegeneration, and TREM2/ADAM10 pathways. Arch Pharm (Weinheim) 2025, 358, e2400641. [Google Scholar] [CrossRef]
- Wu, C.; Yuan, J.; Tian, Y.; Wang, Y.; He, X.; Zhao, K.; Huang, J.; Jiang, R. Tauopathy after long-term cervical lymphadenectomy. Alzheimers Dement 2025, 21, e70136. [Google Scholar] [CrossRef]
- Trojan, E.; Curzytek, K.; Cieślik, P.; Wierońska, J.M.; Graff, J.; Lasoń, W.; Saito, T.; Saido, T.C.; Basta-Kaim, A. Prenatal stress aggravates age-dependent cognitive decline, insulin signaling dysfunction, and the pro-inflammatory response in the APP(NL-F/NL-F) mouse model of Alzheimer's disease. Neurobiol Dis 2023, 184, 106219. [Google Scholar] [CrossRef]
- Ikegami, T.; Sasaki, T.; Shimbo, T.; Kitayama, T.; Yamamoto, Y.; Ouchi, Y.; Yamazaki, S.; Sugiyama, S.; Nishiyama, K.; Gon, Y.; et al. Characterizing stroke-related cellular changes in the surviving neurons of mouse ischemic stroke. Neurochem Int 2025, 191, 106086. [Google Scholar] [CrossRef]
- Tanaka, M. Parkinson's Disease: Bridging Gaps, Building Biomarkers, and Reimagining Clinical Translation. Cells 2025, 14. [Google Scholar] [CrossRef]
- Ali, N.H.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Alexiou, A.; Papadakis, M.; AlAseeri, A.A.; Alruwaili, M.; Saad, H.M.; Batiha, G.E. BDNF/TrkB activators in Parkinson's disease: A new therapeutic strategy. J Cell Mol Med 2024, 28, e18368. [Google Scholar] [CrossRef]
- Mogi, M.; Togari, A.; Kondo, T.; Mizuno, Y.; Komure, O.; Kuno, S.; Ichinose, H.; Nagatsu, T. Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson's disease. Neurosci Lett 1999, 270, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Howells, D.W.; Porritt, M.J.; Wong, J.Y.; Batchelor, P.E.; Kalnins, R.; Hughes, A.J.; Donnan, G.A. Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol 2000, 166, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol 2005, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- von Bohlen und Halbach, O.; Minichiello, L.; Unsicker, K. Haploinsufficiency for trkB and trkC receptors induces cell loss and accumulation of alpha-synuclein in the substantia nigra. Faseb j 2005, 19, 1740–1742. [Google Scholar] [CrossRef]
- Ma, Z.; Xu, Y.; Lian, P.; Wu, Y.; Liu, K.; Zhang, Z.; Tang, Z.; Yang, X.; Cao, X. Alpha-synuclein Fibrils Inhibit Activation of the BDNF/ERK Signaling Loop in the mPFC to Induce Parkinson's Disease-like Alterations with Depression. Neurosci Bull 2025, 41, 951–969. [Google Scholar] [CrossRef]
- Iwata, A.; Maruyama, M.; Kanazawa, I.; Nukina, N. alpha-Synuclein affects the MAPK pathway and accelerates cell death. J Biol Chem 2001, 276, 45320–45329. [Google Scholar] [CrossRef]
- Colucci-D'Amato, L.; Perrone-Capano, C.; di Porzio, U. Chronic activation of ERK and neurodegenerative diseases. Bioessays 2003, 25, 1085–1095. [Google Scholar] [CrossRef]
- Zhu, J.H.; Kulich, S.M.; Oury, T.D.; Chu, C.T. Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol 2002, 161, 2087–2098. [Google Scholar] [CrossRef] [PubMed]
- Kulich, S.M.; Chu, C.T. Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson's disease. J Neurochem 2001, 77, 1058–1066. [Google Scholar] [CrossRef]
- Suzuki, H.; Egawa, N.; Imamura, K.; Kondo, T.; Enami, T.; Tsukita, K.; Suga, M.; Yada, Y.; Shibukawa, R.; Takahashi, R.; et al. Mutant α-synuclein causes death of human cortical neurons via ERK1/2 and JNK activation. Mol Brain 2024, 17, 14. [Google Scholar] [CrossRef]
- Donaldson, J.; Hensman Moss, D.; Ciosi, M.; Usdin, K.; Balmus, G.; Monckton, D.G.; Tabrizi, S.J. Huntington disease: somatic expansion, pathobiology and therapeutics. Nat Rev Neurol 2025. [Google Scholar] [CrossRef]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J Neurosci 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Speidell, A.; Bin Abid, N.; Yano, H. Brain-Derived Neurotrophic Factor Dysregulation as an Essential Pathological Feature in Huntington's Disease: Mechanisms and Potential Therapeutics. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Li, X.J.; Li, S.H.; Sharp, A.H.; Nucifora, F.C., Jr.; Schilling, G.; Lanahan, A.; Worley, P.; Snyder, S.H.; Ross, C.A. A huntingtin-associated protein enriched in brain with implications for pathology. Nature 1995, 378, 398–402. [Google Scholar] [CrossRef]
- Ginés, S.; Paoletti, P.; Alberch, J. Impaired TrkB-mediated ERK1/2 activation in huntington disease knock-in striatal cells involves reduced p52/p46 Shc expression. J Biol Chem 2010, 285, 21537–21548. [Google Scholar] [CrossRef]
- Bodai, L.; Marsh, J.L. A novel target for Huntington's disease: ERK at the crossroads of signaling. The ERK signaling pathway is implicated in Huntington's disease and its upregulation ameliorates pathology. Bioessays. [CrossRef]
- Choi, Y.S.; Lee, B.; Cho, H.Y.; Reyes, I.B.; Pu, X.A.; Saido, T.C.; Hoyt, K.R.; Obrietan, K. CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington's disease. Neurobiol Dis 2009, 36, 259–268. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Hennessey, T.; Johri, A.; Tiwari, S.K.; Mishra, D.; Agarwal, S.; Kim, Y.S.; Beal, M.F. Transducer of regulated CREB-binding proteins (TORCs) transcription and function is impaired in Huntington's disease. Hum Mol Genet 2012, 21, 3474–3488. [Google Scholar] [CrossRef] [PubMed]
- Connor, B.; Sun, Y.; von Hieber, D.; Tang, S.K.; Jones, K.S.; Maucksch, C. AAV1/2-mediated BDNF gene therapy in a transgenic rat model of Huntington's disease. Gene Ther 2016, 23, 283–295. [Google Scholar] [CrossRef] [PubMed]
- García-Díaz Barriga, G.; Giralt, A.; Anglada-Huguet, M.; Gaja-Capdevila, N.; Orlandi, J.G.; Soriano, J.; Canals, J.M.; Alberch, J. 7,8-dihydroxyflavone ameliorates cognitive and motor deficits in a Huntington's disease mouse model through specific activation of the PLCγ1 pathway. Hum Mol Genet 2017, 26, 3144–3160. [Google Scholar] [CrossRef]
- Ahmed, S.; Kwatra, M.; Gawali, B.; Panda, S.R.; Naidu, V.G.M. Potential role of TrkB agonist in neuronal survival by promoting CREB/BDNF and PI3K/Akt signaling in vitro and in vivo model of 3-nitropropionic acid (3-NP)-induced neuronal death. Apoptosis 2021, 26, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Bodai, L.; Gerard, P.E.; Purcell, J.M.; Marsh, J.L. ERK activation by the polyphenols fisetin and resveratrol provides neuroprotection in multiple models of Huntington's disease. Hum Mol Genet 2011, 20, 261–270. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Roles of ERK1/2 and BDNF/TrkB in the CNS neurons. Extracellular regulated kinase 1/2 (ERK 1/2), which is well-studied serine-threonine kinases, are stimulated by a variety of extracellular stimuli, and their hyperactivation is known to be involved in the enhancement of pathological conditions including Aβ plaques production, resulting in neurodegeneration. On the other hand, an activation of ERK1/2 pathway occurs as one of downstream signaling of the BDNF/TrkB and has a role in the maintenance of CNS neurons. Hyperactivation of ERK1/2 signaling caused by various extracellular stimuli (not by the BDNF/TrkB system) has been suggested to induce synapse loss and/or neuronal cell death confirmed in neurodegenerative diseases such as AD, PD, and HD.
Figure 1.
Roles of ERK1/2 and BDNF/TrkB in the CNS neurons. Extracellular regulated kinase 1/2 (ERK 1/2), which is well-studied serine-threonine kinases, are stimulated by a variety of extracellular stimuli, and their hyperactivation is known to be involved in the enhancement of pathological conditions including Aβ plaques production, resulting in neurodegeneration. On the other hand, an activation of ERK1/2 pathway occurs as one of downstream signaling of the BDNF/TrkB and has a role in the maintenance of CNS neurons. Hyperactivation of ERK1/2 signaling caused by various extracellular stimuli (not by the BDNF/TrkB system) has been suggested to induce synapse loss and/or neuronal cell death confirmed in neurodegenerative diseases such as AD, PD, and HD.
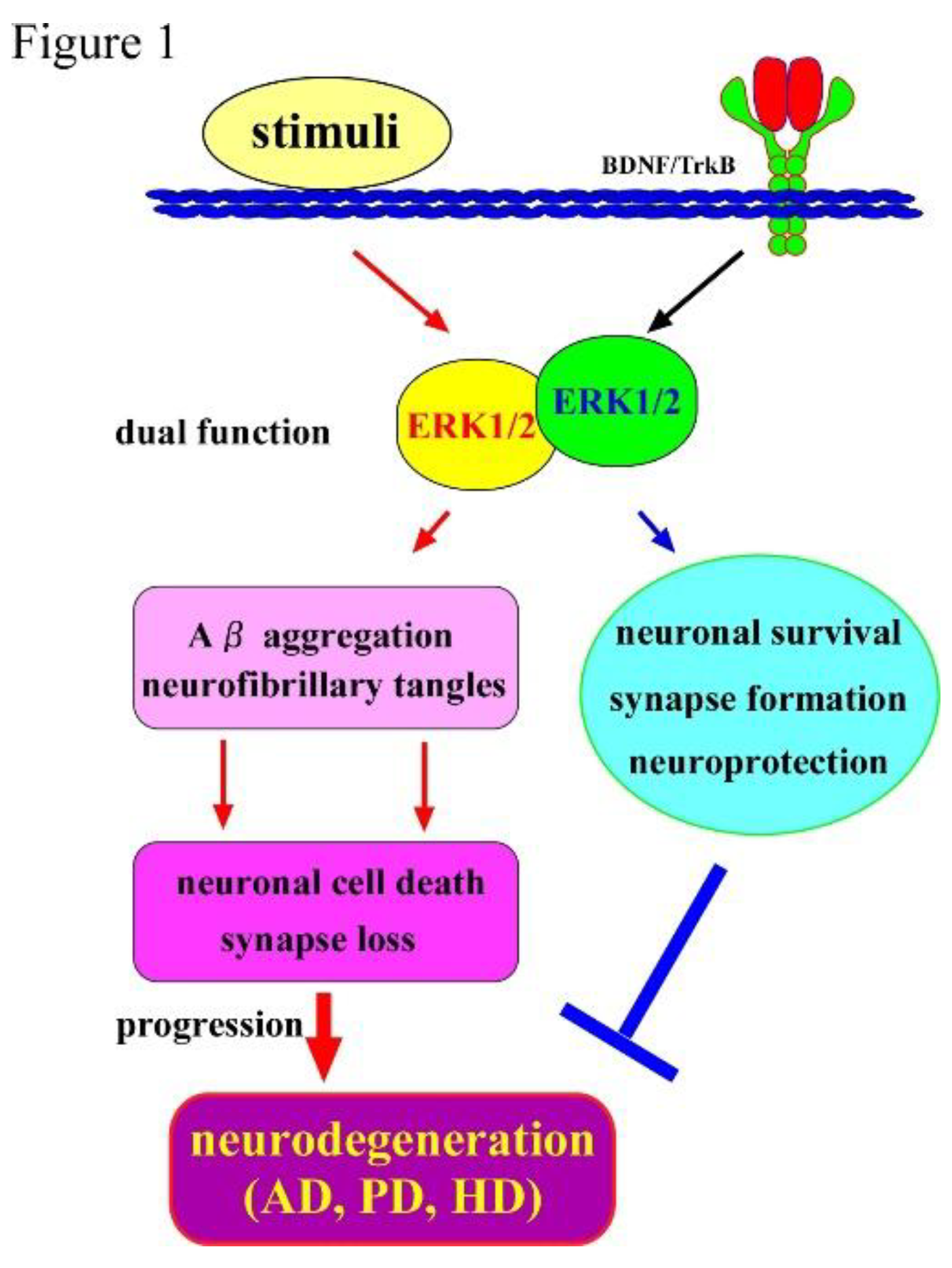
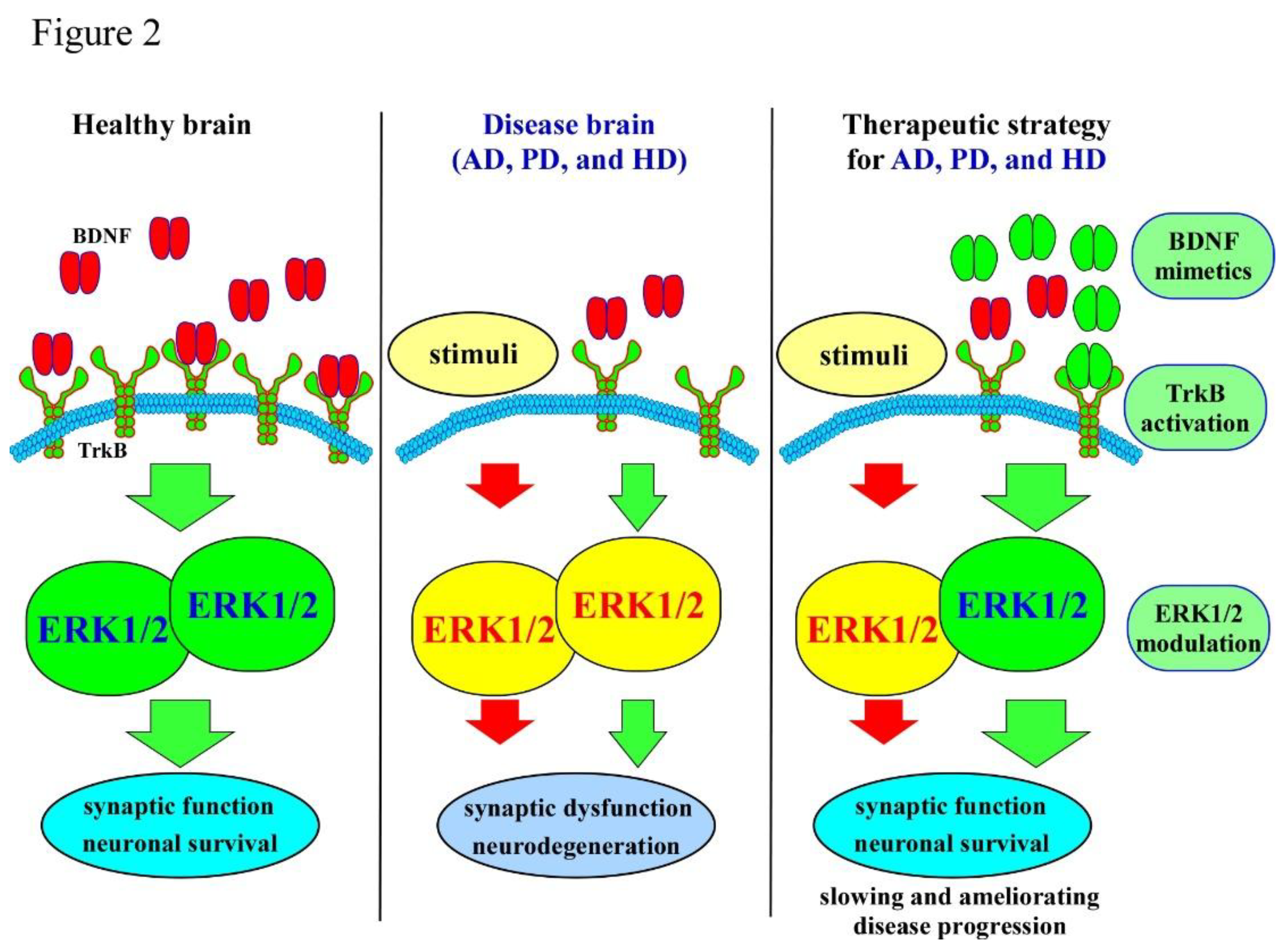
Figure 2.
Summary of BDNF–ERK signaling and its relevance to major neurodegenerative diseases. The schematic illustrates how BDNF activates ERK signaling through TrkB receptor engagement, supporting neuronal survival and synaptic plasticity (left panel). Dysregulation of this pathway contributes pathological outcomes across neurodegenerative disorders (middle panel). In AD, reduced BDNF–ERK signaling is associated with synaptic loss and cognitive decline. In PD, impaired pathway activity increases dopaminergic neuronal vulnerability within the nigrostriatal system. In HD, deficits in BDNF transport and ERK activation promote striatal neurodegeneration. The right panel summarizes key therapeutic strategies aimed at restoring pathway function, including enhancing BDNF expression, activating TrkB receptors, and modulating ERK activity.
Figure 2.
Summary of BDNF–ERK signaling and its relevance to major neurodegenerative diseases. The schematic illustrates how BDNF activates ERK signaling through TrkB receptor engagement, supporting neuronal survival and synaptic plasticity (left panel). Dysregulation of this pathway contributes pathological outcomes across neurodegenerative disorders (middle panel). In AD, reduced BDNF–ERK signaling is associated with synaptic loss and cognitive decline. In PD, impaired pathway activity increases dopaminergic neuronal vulnerability within the nigrostriatal system. In HD, deficits in BDNF transport and ERK activation promote striatal neurodegeneration. The right panel summarizes key therapeutic strategies aimed at restoring pathway function, including enhancing BDNF expression, activating TrkB receptors, and modulating ERK activity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



