
Submitted:
26 November 2025
Posted:
27 November 2025
You are already at the latest version
Abstract
Large language models (LLMs) hold considerable promise for automated extraction of structured biological information from scientific literature, yet their reliability in domain-specific tasks such as DNA probe parsing remains underexplored. We de-veloped a verification-focused, schema-guided extraction pipeline that transforms unstructured text from scientific articles into a normalized database of oligonucleotide probes, primers, and associated metadata. The system combines multi-turn JSON generation, strict schema validation, sequence-specific rule checks, and a post-processing recovery module that rescues systematically corrupted nucleotide outputs. Benchmarking across nine contemporary LLMs revealed distinct accuracy–hallucination trade-offs, with context-optimized Qwen3 model achieving the highest overall extraction efficiency while maintaining low hallucination rates. Iterative prompting substantially improved fidelity but introduced notable latency and vari-ance. Across all models, stable error profiles and the success of the recovery module indicate that most extraction failures stem from systematic and correctable formatting issues rather than semantic misunderstandings. These findings highlight both the potential and the current limitations of LLMs for structured scientific data extraction, and they provide a reproducible benchmark and extensible framework for future large-scale curation of molecular biology datasets.
Keywords:
large language model (LLM)
; data extraction
; nucleotide probe
1. Introduction
Scientific literature is a treasure trove of biological data. However, extracting the latter manually is a monumental task. Today Large Language Models (LLMs) predict remarkable proficiency in addressing this challenge. Emerging studies [1,2,3,4,5,6] underscore this potential: LLMs have successfully mined species interactions from tens of thousands of articles with high precision [7] and achieved near-expert-level accuracy in identifying biological entities [8]. Other work has demonstrated high accuracy in extracting complex data elements [9] and robust performance across diverse biological data types, from experimental parameters to database metadata [4,10,11].
Previous benchmarks show both efficiency and challenges with LLM data extraction. While these methods are much more efficient than SOTA fine-tuning approaches, they tend to be less precise in most cases [12]. LLMs exhibit several recurring error types in biological data extraction. These include missed data items (the most common error) [10], false positives/negatives in entity recognition [2,10], poor quantitative data extraction despite good performance on categorical data [6]. Furthermore, LLMs struggle with complex biological concepts, showing high error rates in causal inference and the semantic mapping of experimental conditions [1,3,13]. Beyond these specific errors, hallucination represents a critical and well-documented barrier to reliable use. Multiple studies identify the generation of fabricated information as a major obstacle in biomedical text mining [1,14], a problem exacerbated by the domain's complex terminology and the potential impact of errors [15].
Evidence demonstrates moderate to strong effectiveness for several verification approaches. A dual-stage verifier that identifies missing data before filtering incorrect extractions achieved up to a 20% F1-score improvement over standard methods [16]. Similarly, a collaborative approach using two LLMs for cross-critique yielded 94% accuracy on concordant responses and improved discordant response accuracy to 76% [17]. External validation, such as comparing extracted biological statements to web searches, has shown high precision (88%) though with limited recall (44%) [5]. While these methods are promising, with one large-scale application achieving 89.5% precision on biological interactions [18]. Therefore, studies achieving high accuracy still emphasize that human oversight remains essential for reliable results [13].
Despite these advances, the field lacks a specialized benchmark and tool for the automated extraction and, crucially, the verification of structured data for specific biological constructs like DNA probes. To address this gap we have developed a novel, verification-focused extraction pipeline and present a comprehensive benchmark of various LLMs on this specialized task. We hypothesize that for this domain, smaller, task-specific LLMs with constrained context windows will be efficient, and that structuring the extraction around JSON prompts combined with the stepped data extraction maximize accuracy. Our final approach, even if it sacrifices some recall and efficiency, achieves exceptionally high precision of the extracted data.
2. Materials and Methods
2.1. LLM Benchmarking
To evaluate the performance of our LLM-based data extraction pipeline, we established a comprehensive benchmarking framework. Model efficiency was assessed on two datasets. The first one, designed to test standard performance, consisted of 10 randomly selected articles (real data, “RD”) containing nucleotide probe information in their main or supplementary texts. The second one, an artificially constructed article (artificial data, “AD”) containing 100 nucleotide probes, was used to stress-test the pipeline, model error distributions, and evaluate scalability. The dataset details are available in Supplementary Methods 1.
A standardized JSON prompt (Supplementary Methods 2) was used to benchmark models, including both web-based interfaces and open-source implementations. Model outputs were compared against ground truth annotations to calculate a set of normalized key performance metrics (all ranging from 0 to 1): corrected extraction efficiency (cEEf), extraction errors (EEr), linking errors (LEr), experiment errors (ExEr), and hallucination rate (HR). We additionally defined a hallucination distance (HD) metric to quantify the severity of sequence hallucinations. The formulas for these metrics are provided and explained in the Supplementary Methods 3.
To handle the articles with extensive data, we employed an iterative prompting strategy (Supplementary Methods 2). We investigated the impact of several critical parameters on performance, including number of thinking tokens, input text preprocessing with the Marker tool, and the underlying LLM architecture.
Visualisations and statistical analysis were done using R 4.2 [19]. We used tidyverse [20], jsonlite [21] and stringdist [22] packages for the parsing. For visualisations we applied ggplot2-based graphics [23] with extensions ggpubr [24], aplot [25], ggviolinbox [26] and ggridges [27]. Mixed linear models were fitted with lme4 [28] and lmerTest [29].
2.2. Data Extraction Pipeline
Our automated pipeline for extracting the experimental data from scientific publications comprises three principal stages: document preparation, schema-guided LLM parsing, and data verification.
During document preparation, PDF articles are converted into a structured text format. Initial use of raw text layers from PDFs resulted in poor quality due to the loss of tabular structures and multi-column layouts. To mitigate this, we adopted the Marker tool [30], which converts PDFs into coherent Markdown while preserving structural elements. This step effectively reduces erratic line breaks, text duplication and other artifacts. We utilized the Force OCR mode to handle complex layouts, foregoing the built-in LLM correction due to its computational demands.
The second stage of the schema-guided LLM parsing involves LLM-based data extraction, which utilizes a combination of schema-guided JSON generation and multi-step chat completions using the Outlines library [31]. We defined a comprehensive JSON schema encapsulating all target data points, including article metadata, hybridization experiment parameters, and nucleotide sequences (probes and primers) in multiple formats. Direct extraction into this full schema proved unfeasible; therefore, we implemented a multi-step approach. Initially, the model extracts metadata, all nucleotide sequences, and a general experiment description using simplified schemas with fewer than five fields each. Subsequently, each identified sequence is processed in a dedicated chat session where the model, provided with the full article text, is queried to confirm the sequence's role, reformat it, and associate it with specific experimental parameters. All responses are constrained by small JSON schemas [32]. The results are aggregated into a final JSON object conforming to the complete schema and stored in a SQLite database [33]. More details on the iterative prompting approach are available in Supplementary Methods 2.
Given the long context required (often exceeding 32,000 tokens), we primarily used models from the Qwen family [34,35,36]. For comparative performance analysis, we also evaluated Gemma3 [37] and Phi-4 [38] models. All models were hosted locally using the Ollama inference server [39] on a dedicated machine featuring Ryzen 7900X CPU with 64 GB of RAM and RTX 3090Ti GPU. It provides an API for the request completion or chat completion.
Data verification stage helps to ensure the integrity of the automatically extracted data, and we implemented a rigorous three-stage validation process. First, syntactic validation checks for strict compliance with the predefined JSON schema, including correct syntax, presence of all required fields, and the absence of any extraneous fields; non-compliant records are rejected. Second, semantic validation applies custom regular expressions to nucleotide sequence fields to verify they contain only permitted characters and patterns. Finally, we perform a textual fidelity check, wherein each reported probe sequence must be at least 90% verifiable within the original article text to guard against hallucinations. Records failing any validation step were rejected from the final dataset.
3. Results
3.1. Benchmarking
Initial benchmarking on real data suggested that different LLMs might rely on distinct extraction vectors - that is, internally preferred patterns of probe recovery - despite achieving similar baseline efficiency. Without any task-specific prompting, most models retrieved up to 10 out of 20 probes per iteration (~50%). However, the specific probes extracted differed markedly between the GPT - Gemini group and the DeepSeek models, indicating that comparable accuracy can arise from divergent extraction pathways (Supplementary Figure S1). DeepSeek also generated the most structurally detailed outputs, returning the longest JSON objects, consistent with a richer internal representation of the input. Comparative analysis of the probe sets extracted by each model supported this interpretation. GPT-4o, Gemini, Sider, and chatPDF formed a tightly overlapping cluster, recovering largely similar subsets of probes, whereas DeepSeek and DeepSeek-R1 occupied a partially overlapping but predominantly distinct extraction space. SciSpace showed minimal overlap with any model, producing the lowest efficiency yet capturing several unique probes. These patterns indicated that the models differ not only in extraction success but also in the breadth and structural richness of the information they return.
Across all evaluated settings, the presence of Marker preprocessing had the strongest positive effect on extraction reliability. Although the Marker tool only restructures the PDF into a cleaner Markdown representation, this preprocessing substantially reduced noise in the model’s input and improved downstream consistency. Configurations using Marker consistently achieved higher extraction efficiency (EEf), corrected extraction efficiency (cEEf), and lower hallucination rate (HR) compared to runs without preprocessing. These improvements were evident both across iterative extraction steps (Supplementary Figure S2) and in summary metrics aggregated across sequence classes (Supplementary Figure S3).
Prompt length also proved critical. Shorter prompts produced better overall performance, even when longer versions included the full JSON-schema inline. While JSON-schema guidance was beneficial, embedding the entire schema directly into the prompt increased verbosity without improving accuracy. Instead, loading the JSON-schema into memory and passing only a minimal reference within the prompt provided a more compact, stable context for the model. This design preserved structural constraints while avoiding unnecessary linguistic overhead.
To select the model, we benchmark them on the standardised prompt (Figure 1). Its minimal version includes a JSON structure tailored for nucleotide-probe extraction (Supplementary Methods S3). Models with larger token capacities, such as DeepSeek and GPT-4o, immediately showed improved performance when this prompt was applied, extracting all probes from the real dataset. Building on this structured prompt, large-scale probe extraction became feasible, and we validated this capability using synthetic datasets.
To formally test the hypothesis of distinct extraction vectors, we evaluated whether multi-step (“stepped”) extraction would amplify or preserve these model-specific probe signatures. If models relied on reproducible extraction pathways, repeated sampling should reinforce their unique probe subsets. However, the UpSet analysis of correctly extracted probes across all models and iterations (Supplementary Figure S4) did not support this assumption. Intersection patterns were highly entangled: most probes were recovered by multiple models in various combinations, and only a small fraction of intersections were model-specific. Models that appeared distinct in single-step extraction did not maintain clearly separable extraction trajectories across repetitions. Instead, stepped extraction revealed substantial overlap between previously divergent model groups, suggesting that the hypothesized extraction vectors are not stable across iterations. Across models, the stepped-extraction analysis did not support the hypothesis that repeated prompting gradually amplifies errors or uncovers stable “extraction vectors.” Instead, performance across iterations was largely stable, and shifts in the extracted probe sets appeared stochastic rather than directional. Therefore, probably, only one LLM is sufficient to extract all probes.
Models differed systematically in how they balanced correct extraction and hallucinations. Glm, Gemini and Qwen showed the most favourable trade-off: high proportions of correct probes with relatively few hallucinations, and consistently low hallucination distance. DeepSeek and DeepSeek-R1 achieved stronger recall but at the cost of higher hallucination burden, producing many fabricated sequences per iteration - though these tended to remain close to the true sequence space. GPT-4o behaved conservatively, generating fewer hallucinations but also retrieving substantially fewer correct probes.
Error profiles showed two reproducible failure modes across all LLMs. First, models systematically struggled with lowercase sequences, frequently ignoring or transforming them, suggesting that orthography alone can trigger extraction failures. Second, all models showed predictable difficulty with repeat-encoded sequences (e.g., (NNN)x notation), often misinterpreting or expanding the repeat structure. These category-specific errors were more consistent than any iteration-dependent drift, further supporting the conclusion that extraction behaviour is shaped more by model-specific heuristics than by cumulative reasoning across steps.
Analysis of hallucination patterns revealed that most “hallucinations” were not de novo inventions but error-type distortions of true probes. Across all models, the majority of non-matching sequences fell into the low-HD range (0 < HD ≤ 0.5) - indicating that the models attempted to reproduce the correct sequence but introduced substitutions, dropped characters, or misinterpreted repeat blocks. Truly novel, high-HD fabrications (HD > 0.5) were comparatively rare and usually appeared in models with aggressive extraction behavior, such as early-iteration Qwen3-1k/3k or DeepSeek.
The distribution of Hallucination distances (Supplementary Figure S5A) illustrates this pattern clearly: nearly every model shows a dominant low-HD component, with DeepSeek and Qwen3-1k showing slightly broader tails. Error-type hallucinations also accounted for a large share of total extracted sequences, explaining why hallucination rates (HR; Figure 3B) are high for models with high recall - many errors represent “near-misses” rather than unrelated fabrications. Iterative reasoning (Supplementary Figure S5C) typically reduced the proportion of these errors over steps, especially in Qwen3-10k and Gemini, which showed the most consistent improvement in corrected extraction efficiency (cEEf).
Different “thinking tokens” amounts influence the extraction efficiency and hallucination rates of the model (Supplementary Figure S6A). To formally test this effect, we fitted linear mixed-effects models with iteration-dependent cumulative metrics as responses. The number of thinking tokens showed a significant negative effect on corrected extraction efficiency (cEEf) (Pr(>|t|) = 0.0016, df = 44.66, t = - 3.37), indicating that increasing the amount of chain-of-thought reasoning systematically reduced the proportion of correctly extracted items after adjusting for hallucinations. In contrast, hallucination rate (HR) was not affected by the number of thinking tokens (Pr(>|t|) = 0.943, df = 3.00, t = - 0.078), demonstrating that longer reasoning traces did not increase the likelihood of fabricating sequences.
We also hypothesized that increasing the number of iterations would lead to a higher probability of hallucinations (Supplementary Figure S6B). However, this expectation was not supported by statistical analysis. Across the full dataset, iteration count showed no significant effect on hallucination rate (mixed-effects model: Pr(>|t|) = 0.221, df = 48, t = 1.24). A separate analysis restricted to Qwen variants with different thinking-token configurations yielded the same conclusion (mixed-effects model: Pr(>|t|) = 0.476, df = 36, t = 0.72). These findings indicate that iterative querying of LLMs does not increase hallucination probability, thus enabling repeated extraction cycles until the model returns empty JSON responses.
However, different models showed comparable speeds on the full extraction task (Supplementary Figure S7). Iterative prompting introduces substantial overhead and dominates the temporal structure of the pipeline (Supplementary Figure S8). While a full end-to-end extraction requires on average 8.2 ± 4.4 s per sample (peaking at 19 s), most baseline extraction stages (A–F) remain close to zero after normalization. In contrast, the iterative steps - primarily SeqPrompt and SeqDesc - consistently emerge as the slowest segments across all models and articles.
Taken together, the results show that models differ not only in extraction capacity but also in how they trade recall for hallucination risk. Qwen3-1k, 10k and glm-4.6 provided the strongest overall balance, achieving high proportions of correct extraction with relatively controlled hallucination behavior. Context length emerges as a key driver of Qwen’s performance: insufficient context leads to aggressive but hallucination-prone extraction, while a well-scaled window supports both higher accuracy and greater reliability. So, we selected Qwen for further development.
3.2. Tool Architecture
We adopted an extraction architecture that pairs a schema-guided first pass with a controlled multi-turn refinement stage to balance recall and hallucination risk based on the Qwen. In the initial step, the model receives the full article and a minimal JSON schema that enforces basic nucleotide-sequence constraints, allowing it to enumerate all candidate probe sequences without prematurely filtering potentially valid items. Each candidate is then validated in an isolated chat session in which the full article is re-supplied as context and the model responds to a fixed sequence of narrowly scoped questions, each restricted by a compact JSON schema. This design forces the model to justify every probe individually, reducing hallucination rates while preserving the high recall observed in models with larger context windows. The outputs of these per-probe dialogues - structural attributes, modifications, targets, primer sets, and experimental conditions - are subsequently merged with article-level metadata to generate a complete, database-ready JSON entry. Although computationally slower due to the per-probe multi-turn structure, this architecture provides the most reliable balance between extraction efficiency and hallucination control observed in our benchmarks.
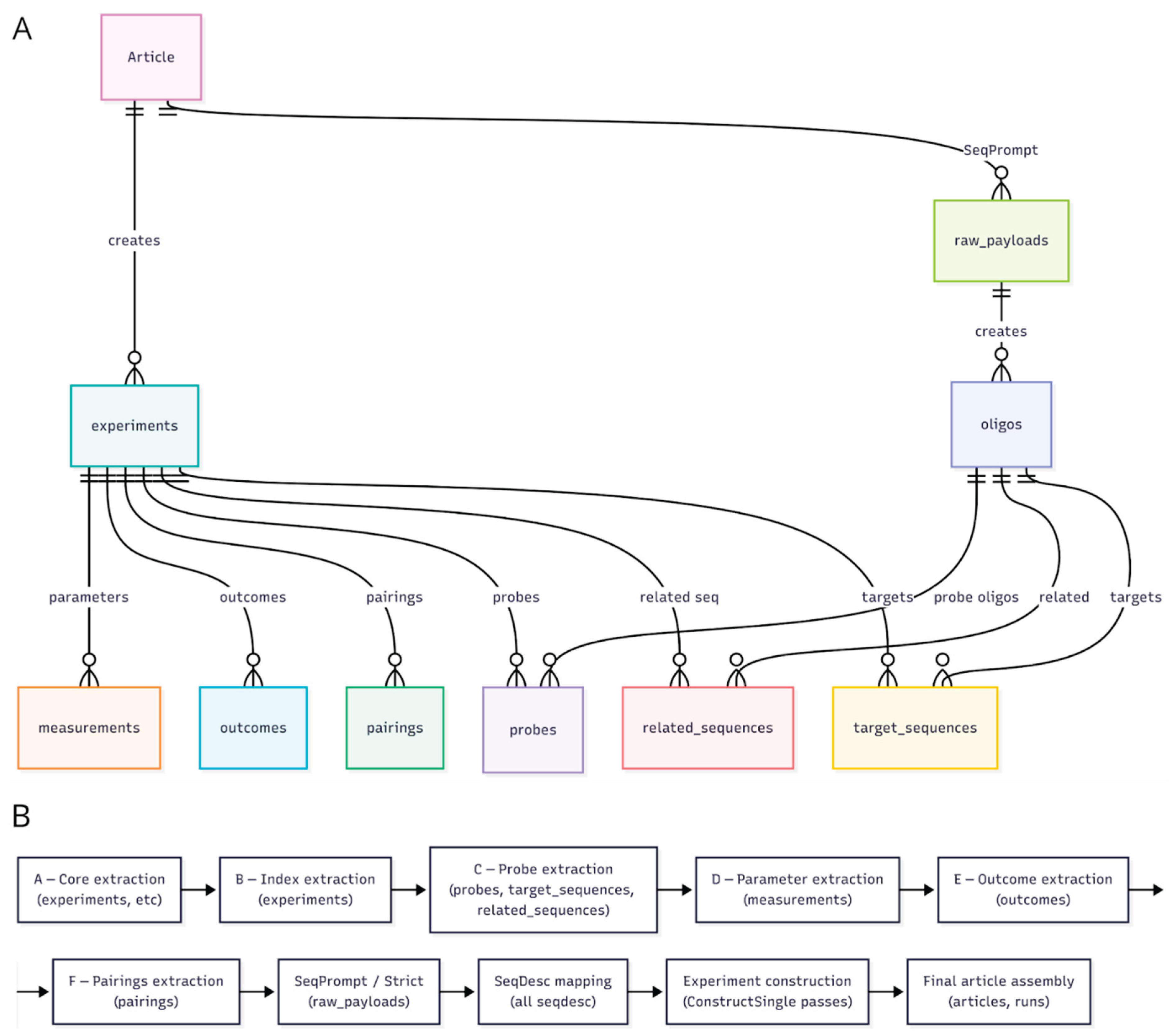
The resulting data model organizes the LLM-derived information into a coherent relational structure that links articles, extraction runs, experiments, and all sequence-level entities (Figure 2). Each article may generate multiple extraction runs, and each run yields a set of structured experiments. Within an experiment, all molecular entities are represented through a common table, which serves as the central sequence repository. Probes and primer pairs are associated with their corresponding experiments through direct references, while target sequences, outcomes, measurements, and pairwise relationships provide the detailed experimental context required for downstream analyses. Diagnostic extraction reports remain linked at the experiment level, allowing auditability and fine-grained error tracing. This schema enables robust integration of heterogeneous LLM outputs into a stable, queryable database suited for large-scale comparative studies.
Validation pipeline was implemented to assess the quality of the sequences extracted by the LLMs. The script connects directly to the SQL database generated in the previous step and evaluates each sequence following the procedure outlined in the Methods section. Out of approximately 1500 extracted sequences, only 210 passed the first structural and lexical validation filters, and 208 remained after the full validation workflow. This sharp reduction indicates that LLMs tend to produce either fully correct sequences or completely nonsensical strings, rather than generating hallucinations that closely resemble real probes. Importantly, these nonsensical outputs can often be matched to true sequences through reverse search, allowing recovery of the correct probe and substantially improving overall extraction performance. Incorporating this recovery step can possibly increase the final extraction efficiency up to approximately 82%.
4. Discussion
We developed and benchmarked a verification-centered pipeline for automated extraction of structured DNA probe data from scientific literature. The schema-guided, multi-turn parsing strategy - implemented with Qwen, Marker and Outlines - achieved an effective balance between recall and hallucination control. The final extraction efficiency of approximately 82%, reached only after a stringent three-stage validation and a post-processing recovery phase, demonstrates the necessity of verification for LLM-based data mining [17].
Benchmarking revealed distinct “extraction vectors” among different LLMs. This aligns with evidence that models with similar overall accuracy can rely on very different internal heuristics, attention patterns, and representational strategies. In our experiments, Qwen3’s performance improved significantly when the context window was scaled appropriately - an effect consistent with recent trends in LLM optimization that prioritize configuration and context over raw model size [3,31]. When context was insufficient, Qwen3 exhibited aggressive extraction but high hallucination rates; expanding the context window stabilized output quality and reliability. These observations suggest that for complex scientific tasks, task-specific tuning of context length, temperature, and reasoning budget can be more effective than default usage of the largest available models. Models such as DeepSeek, which prioritized high recall, often produced structured but hallucination-prone outputs - behavior consistent with well-characterized LLM failure modes where models "satisfice" uncertain prompts by producing plausible but incorrect outputs [15,32].
Single-pass outputs initially suggested the presence of model-specific extraction biases. However, repeated sampling revealed that these tendencies were not stable. Variability across iterations was mild, non-directional, and partially stochastic. Broader performance categories persisted across all runs: high-precision systems (Gemini, Qwen3-10k), high-recall but hallucination-prone systems (DeepSeek), and conservative, low-recall systems (GPT-4o). This suggests that iterative prompting does not substantially shift an LLM’s underlying extraction strategy.
Analysis of failure cases shows that hallucinations were typically due to fidelity breakdowns, not imaginative invention. Most incorrect outputs closely resembled valid probes but failed on specific formatting or lexical constraints (repeat encodings, case sensitivity). This observation is consistent with the broader literature: hallucination in structured-domain LLM applications often stems from boundary-condition failures rather than wholesale invention [12,14,15]. Slight modifications - such as enforcing stricter output formats, normalizing input, or adding explicit “if not found, output null” instructions - may substantially reduce error rates in future pipelines.
Runtime profiling showed that pure JSON-field extraction accounted for a negligible portion of wall-clock time. The majority of the latency stems from reasoning-heavy, multi-turn, or validation-regeneration stages, regardless of model architecture. Iterative prompting increased both runtime and variance, and was the major contributor to partial or empty JSON outputs; such failures tended to cluster in long articles and with models that processed chain-of-thought slowly. These findings identify iterative prompting as the primary scalability bottleneck of the system.
The three-stage validation pipeline - syntactic, semantic, and textual-fidelity checks - proved to be a critical strength. It mitigated the most problematic LLM failure mode: structured hallucination, where outputs appear confident and schema-valid but are factually incorrect [15,32]. By requiring strict JSON-schema conformance, nucleotide-pattern compliance, and at least 90% verifiability against the source text, we enforced high precision. The subsequent post-processing recovery step, which attempted to match nonsensical LLM output to true sequences via reverse search, was especially effective. This hybrid approach - combining statistical modeling (LLMs) with deterministic rule-based post-processing - leverages the strengths of both paradigms and improves robustness [31,32].
There remain important limitations. The computational overhead of the multi-turn per-probe refinement is substantial. While this design improves precision, it substantially slows throughput compared to single-pass extraction systems [40]. For applications demanding high-throughput or near real-time processing, this trade-off may be unacceptable. Future work could explore more efficient verification strategies. For example, a collaborative two-LLM “cross-critique” design - where two models independently extract data and then compare/harmonize their outputs - has shown promise in reducing errors in systematic review workflows [17]. External validation against public databases or web-based evidence could further improve reliability, though likely at the expense of recall [5]. Another promising direction is embedding extracted entities into biologically-informed semantic networks (e.g., ontology/knowledge graphs) to perform consistency and plausibility checks - not just at the sequence level but at the level of functional relationships [41]. Finally, the current benchmark focuses on nucleotide probes, which are highly structured and constrained. Extending evaluation to more abstract biological constructs - such as causal relationships, experimental conditions, or phenotype annotations - will be crucial to assess the generalizability of this verification-guided extraction paradigm [1,3,13].
5. Conclusions
Our research confirms that LLMs hold immense promise for automating the extraction of structured biological data. However, this promise is best realized not by treating LLMs as infallible oracles, but by embedding them within a robust, verification-centric architecture. The combination of schema-guided parsing, iterative refinement, multi-stage validation, and intelligent post-processing is a powerful way for achieving high-precision extraction. As LLM technology continues to evolve, with growing emphasis on efficiency, reasoning, and specialization, the principles and pipeline established here will provide a strong foundation for building ever more reliable and comprehensive scientific knowledge bases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, A.S. and S.M.; methodology, A.S. and V.D.; software, A.S.; validation, D.S.; formal analysis, V.D. and D.S.; investigation, A.S. and V.D.; resources, A.S.; data curation, A.T.; writing—original draft preparation, D.S.; writing—review and editing, A.T.; visualization, D.S.; supervision, S.M.; project administration, S.M.; funding acquisition, A.T.
Funding
This study was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement No 075-15-2024-563).
Data Availability Statement
Scripts used for the benchmarking and the final model can be found in the GitHub project https://github.com/CTLab-ITMO/PROBEst.
Acknowledgments
This research would not have been possible without the assistance of Prof. Anatoly Shalyto.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| AD | Artificial Data |
| API | Application Programming Interface |
| CPU | Central Processing Unit |
| cEEf | Corrected Extraction Efficiency |
| EEf | Extraction Efficiency |
| EEr | Extraction Errors |
| ExEr | Experiment Errors |
| ggplot2 | Grammar of Graphics plotting library for R |
| GPU | Graphics Processing Unit |
| HD | Hallucination Distance |
| HR | Hallucination Rate |
| JSON | JavaScript Object Notation |
| LEr | Linking Errors |
| LLM | Large Language Model |
| lme4 | Linear Mixed-Effects Models for R |
| OCR | Optical Character Recognition |
| Portable Document Format | |
| R | R programming language |
| RAM | Random Access Memory |
| RD | Real Data |
| SOTA | State of the Art |
| SQL | Structured Query Language |
| UML | Unified Modeling Language |
References
- Garcia, G.L.; Manesco, J.R.R.; Paiola, P.H.; Miranda, L.; de Salvo, M.P.; Papa, J.P. A Review on Scientific Knowledge Extraction Using Large Language Models in Biomedical Sciences 2024.
- Gartlehner, G.; Kugley, S.; Crotty, K.; Viswanathan, M.; Dobrescu, A.; Nussbaumer-Streit, B.; Booth, G.; Treadwell, J.R.; Han, J.M.; Wagner, J.; et al. AI-Assisted Data Extraction with a Large Language Model: A Study Within Reviews 2025.
- Schmidt, L.; Hair, K.; Graziosi, S.; Campbell, F.; Kapp, C.; Khanteymoori, A.; Craig, D.; Engelbert, M.; Thomas, J. Exploring the Use of a Large Language Model for Data Extraction in Systematic Reviews: A Rapid Feasibility Study. 2024. [CrossRef]
- Rettenberger, L.; Münker, M.F.; Schutera, M.; Niemeyer, C.M.; Rabe, K.S.; Reischl, M. Using Large Language Models for Extracting Structured Information From Scientific Texts. Curr. Dir. Biomed. Eng. 2024, 10, 526–529. [Google Scholar] [CrossRef]
- Adam, D.; Kliegr, T. Traceable LLM-Based Validation of Statements in Knowledge Graphs. Inf. Process. Manag. 2025, 62, 104128. [Google Scholar] [CrossRef]
- Gougherty, A.V.; Clipp, H.L. Testing the Reliability of an AI-Based Large Language Model to Extract Ecological Information from the Scientific Literature. Npj Biodivers. 2024, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Keck, F.; Broadbent, H.; Altermatt, F. Extracting Massive Ecological Data on State and Interactions of Species Using Large Language Models 2025, 2025. 01.24.63 4685.
- Jung, S.J.; Kim, H.; Jang, K.S. LLM Based Biological Named Entity Recognition from Scientific Literature. In Proceedings of the 2024 IEEE International Conference on Big Data and Smart Computing (BigComp); IEEE: Bangkok, Thailand, 18 February 2024; pp. 433–435. [Google Scholar]
- Konet, A.; Thomas, I.; Gartlehner, G.; Kahwati, L.; Hilscher, R.; Kugley, S.; Crotty, K.; Viswanathan, M.; Chew, R. Performance of Two Large Language Models for Data Extraction in Evidence Synthesis. Res. Synth. Methods 2024, 15, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Gartlehner, G.; Kahwati, L.; Hilscher, R.; Thomas, I.; Kugley, S.; Crotty, K.; Viswanathan, M.; Nussbaumer-Streit, B.; Booth, G.; Erskine, N.; et al. Data Extraction for Evidence Synthesis Using a Large Language Model: A Proof-of-concept Study. Res. Synth. Methods 2024, 15, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Zou, Z.; Bono, H.; Moriya, Y.; Kawashima, S.; Katayama, T.; Oki, S.; Ohta, T. Extraction of Biological Terms Using Large Language Models Enhances the Usability of Metadata in the BioSample Database. GigaScience 2025, 14, giaf070. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Hu, Y.; Peng, X.; Xie, Q.; Jin, Q.; Gilson, A.; Singer, M.B.; Ai, X.; Lai, P.-T.; Wang, Z.; et al. Benchmarking Large Language Models for Biomedical Natural Language Processing Applications and Recommendations. Nat. Commun. 2025, 16, 3280. [Google Scholar] [CrossRef] [PubMed]
- Konet, A.; Thomas, I.; Gartlehner, G.; Kahwati, L.; Hilscher, R.; Kugley, S.; Crotty, K.; Viswanathan, M.; Chew, R. Performance of Two Large Language Models for Data Extraction in Evidence Synthesis. Res. Synth. Methods 2024, 15, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Ivanisenko, T.V.; Demenkov, P.S.; Ivanisenko, V.A. An Accurate and Efficient Approach to Knowledge Extraction from Scientific Publications Using Structured Ontology Models, Graph Neural Networks, and Large Language Models. Int. J. Mol. Sci. 2024, 25, 11811. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Lee, N.; Frieske, R.; Yu, T.; Su, D.; Xu, Y.; Ishii, E.; Bang, Y.; Chen, D.; Dai, W.; et al. Survey of Hallucination in Natural Language Generation. ACM Comput. Surv. 2023, 55, 1–38. [Google Scholar] [CrossRef]
- Li, J.; Yuan, R.; Tian, Y.; Li, J. Towards Instruction-Tuned Verification for Improving Biomedical Information Extraction with Large Language Models. In Proceedings of the 2024 IEEE International Conference on Bioinformatics and Biomedicine (BIBM); IEEE: Lisbon, Portugal, December 3, 2024; pp. 6685–6692. [Google Scholar]
- Khan, M.A.; Ayub, U.; Naqvi, S.A.A.; Khakwani, K.Z.R.; Sipra, Z.B.R.; Raina, A.; Zhou, S.; He, H.; Saeidi, A.; Hasan, B.; et al. Collaborative Large Language Models for Automated Data Extraction in Living Systematic Reviews. J. Am. Med. Inform. Assoc. 2025, 32, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Keck, F.; Broadbent, H.; Altermatt, F. Extracting Massive Ecological Data on State and Interactions of Species Using Large Language Models 2025.
- R Core Team R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024.
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Ooms, J. The Jsonlite Package: A Practical and Consistent Mapping Between JSON Data and R Objects. ArXiv14032805 StatCO 2014. [Google Scholar]
- Loo, M.P.J. van der The Stringdist Package for Approximate String Matching. R J. 2014, 6, 111–122. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Kassambara, A. Ggpubr: “ggplot2” Based Publication Ready Plots; 2023.
- Yu, G. Aplot: Decorate a “ggplot” with Associated Information; 2025.
- Smutin, D. Ggviolinbox: Half-Violin and Half-Boxplot Geoms for Ggplot2 2025.
- Wilke, C.O. Ggridges: Ridgeline Plots in “Ggplot2”; 2025.
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using Lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef]
- Datalab-to/Marker 2025.
- Willard, B.T.; Louf, R. Efficient Guided Generation for Large Language Models 2023.
- Wright, A.; Andrews, H.; Hutton, B.; Dennis, G. JSON Schema: A Media Type for Describing JSON Documents; JSON Schema, 2020.
- Hipp, R.D. SQLite 2020.
- Yang, A.; Yu, B.; Li, C.; Liu, D.; Huang, F.; Huang, H.; Jiang, J.; Tu, J.; Zhang, J.; Zhou, J.; et al. Qwen2.5-1M Technical Report 2025.
- Yang, A.; Li, A.; Yang, B.; Zhang, B.; Hui, B.; Zheng, B.; Yu, B.; Gao, C.; Huang, C.; Lv, C.; et al. Qwen3 Technical Report 2025.
- Kwon, W.; Li, Z.; Zhuang, S.; Sheng, Y.; Zheng, L.; Yu, C.H.; Gonzalez, J.; Zhang, H.; Stoica, I. Efficient Memory Management for Large Language Model Serving with PagedAttention. In Proceedings of the Proceedings of the 29th Symposium on Operating Systems Principles; ACM: Koblenz Germany, October 23, 2023; pp. 611–626. [Google Scholar]
- Gemma Team; Kamath, A.; Ferret, J.; Pathak, S.; Vieillard, N.; Merhej, R.; Perrin, S.; Matejovicova, T.; Ramé, A.; Rivière, M.; et al. Gemma 3 Technical Report 2025.
- Abdin, M.; Aneja, J.; Behl, H.; Bubeck, S.; Eldan, R.; Gunasekar, S.; Harrison, M.; Hewett, R.J.; Javaheripi, M.; Kauffmann, P.; et al. Phi-4 Technical Report 2024.
- Ollama/Ollama 2025.
- Yin, Z. A Review of Methods for Alleviating Hallucination Issues in Large Language Models. Appl. Comput. Eng. 2024, 76, 258–266. [Google Scholar] [CrossRef]
- Abolhasani, M.; Pan, R. Leveraging LLM for Automated Ontology Extraction and Knowledge Graph Generation; 2024.
Figure 1.
Performance, hallucination behavior, and error structure of large language models (LLMs) in artificial probe-sequence extraction. (A) Bidirectional bar plot summarizing, for each LLM, the distribution of correctly extracted probes (right side) and hallucinated probes (left side). Transparent overlays denote variability across iterations; red-outlined segments represent de novo hallucinations with high perturbation (HP). (B) Heatmap of key extraction metrics: raw Extraction Efficiency (EEf), corrected Extraction Efficiency (cEEf), Hallucination Rate (HR), Hallucination Distance (HD), and high-perturbation proportion (HP). (C) Error-category heatmap showing the fraction of probes recovered within each synthetic degradation class (e.g., lowercase, repeats, short/long degeneration, modified). Values represent per-instrument recovery normalized by the number of true probes in each class.
Figure 1.
Performance, hallucination behavior, and error structure of large language models (LLMs) in artificial probe-sequence extraction. (A) Bidirectional bar plot summarizing, for each LLM, the distribution of correctly extracted probes (right side) and hallucinated probes (left side). Transparent overlays denote variability across iterations; red-outlined segments represent de novo hallucinations with high perturbation (HP). (B) Heatmap of key extraction metrics: raw Extraction Efficiency (EEf), corrected Extraction Efficiency (cEEf), Hallucination Rate (HR), Hallucination Distance (HD), and high-perturbation proportion (HP). (C) Error-category heatmap showing the fraction of probes recovered within each synthetic degradation class (e.g., lowercase, repeats, short/long degeneration, modified). Values represent per-instrument recovery normalized by the number of true probes in each class.
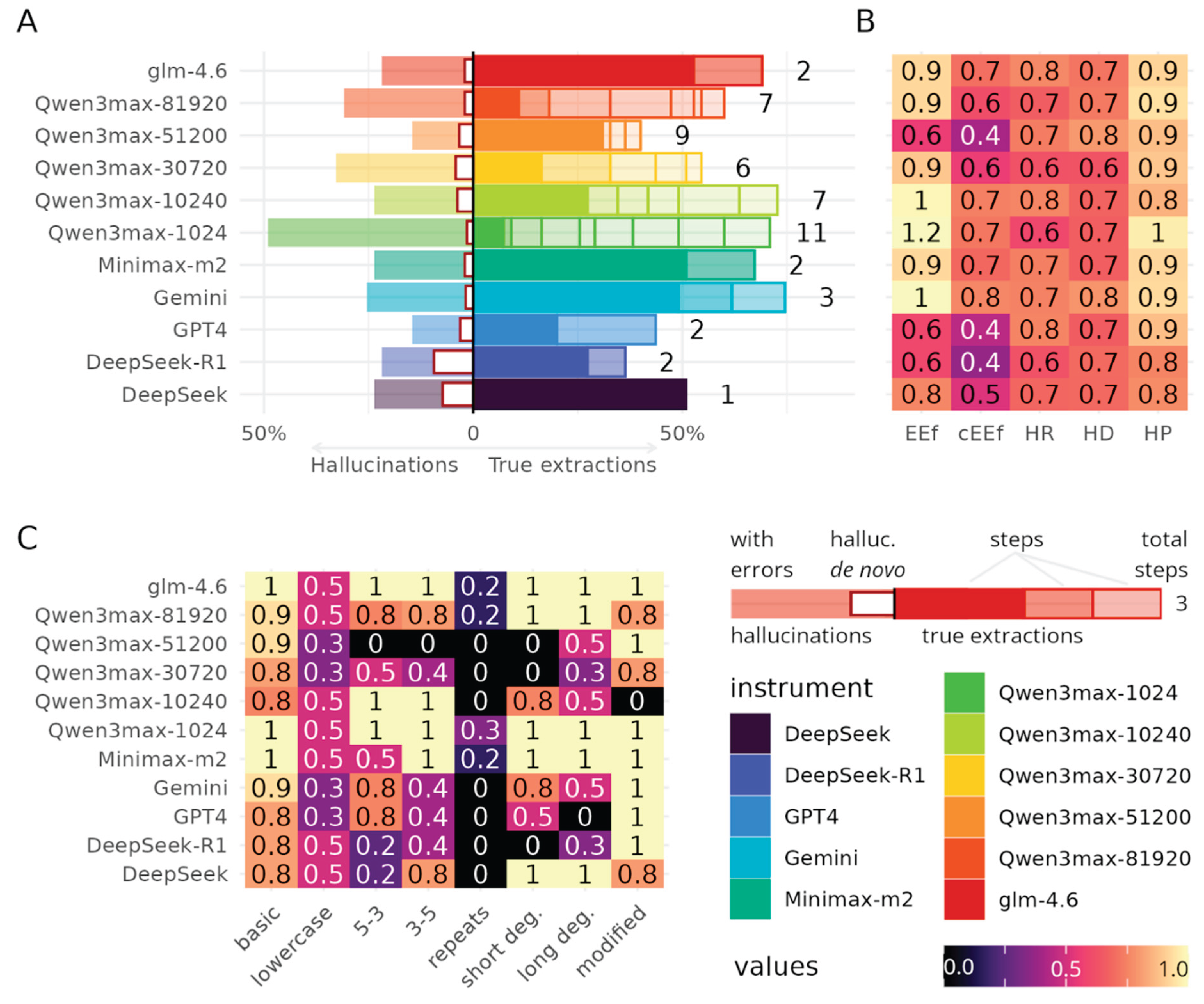
Figure 2.
Unified schema of the LLM-based extraction workflow and its execution pipeline. (A) Simplified UML-style entity–relationship diagram illustrating the structured JSON schema. Arrows show how higher-level objects create or reference lower-level components. (B) Overview of the sequential extraction pipeline, from core entity generation (A–F) through iterative prompting stages (SeqPrompt/Strict and SeqDesc), followed by experiment assembly and final article construction.
Figure 2.
Unified schema of the LLM-based extraction workflow and its execution pipeline. (A) Simplified UML-style entity–relationship diagram illustrating the structured JSON schema. Arrows show how higher-level objects create or reference lower-level components. (B) Overview of the sequential extraction pipeline, from core entity generation (A–F) through iterative prompting stages (SeqPrompt/Strict and SeqDesc), followed by experiment assembly and final article construction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



