Submitted:
24 November 2025
Posted:
26 November 2025
You are already at the latest version
Abstract
Autism Spectrum Disorder (ASD) is a genetically heterogeneous neurodevelopmental condition driven by rare de novo variants, copy number variations, and polygenic risk. SFARI-curated genes show high mutational constraint and enriched expression in cortical neurons and glia. This review highlights recent advances in CRISPR-based functional genomics using human pluripotent stem cells and induced pluripotent stem cells differentiated into neural progenitors, excitatory and inhibitory neurons, astrocytes, microglia, and brain organoids. CRISPR modalities including knockouts, CRISPRi and CRISPRa, base and prime editing, and Cas13 enable pooled and arrayed screens with high coverage at low multiplicity of infection. Integration of multimodal readouts such as Perturb-seq, single-cell and spatial transcriptomics, proximity labeling proteomics, and functional assays including microelectrode arrays and calcium imaging provides system-level insights into ASD gene function. Computational frameworks like MIMOSCA and SCEPTRE facilitate network reconstruction and pseudo-time inference. Case studies reveal Wnt and BAF complex dysregulation, microglial pruning deficits, and non-cell autonomous effects. Translational approaches target haplo-insufficient genes such as CHD8 and SCN2A using AAV or antisense oligonucleotides supported by isogenic iPSC models. Remaining challenges include model immaturity and scalability, while future directions focus on spatial perturb-omics, AI-driven causal inference, and standardized biobanks for precision ASD therapeutics.
Keywords:
Introduction
2. Autism Genetics Primer
2.1. Rare De Novo Variants, Inherited Risk, CNVs, and Common Polygenic Risk
2.2. High-Confidence vs Candidate ASD Genes
2.3. Functional Annotation and Population Resources
2.4. Challenges in ASD Genetics
3. Human Stem Cell Platforms for ASD Functional Genomics
3.1. Human Pluripotent Stem Cell Types: hESCs vs iPSCs
3.2. Differentiation Endpoints: Neural Lineages
3.3. Three-Dimensional Models
3.4. Co-Culture Systems and Microphysiological Platforms
3.5. Practical Considerations
4. The CRISPR Toolbox: Modalities and Deliverables
4.1. CRISPR-Cas Modalities and Their Application in ASD
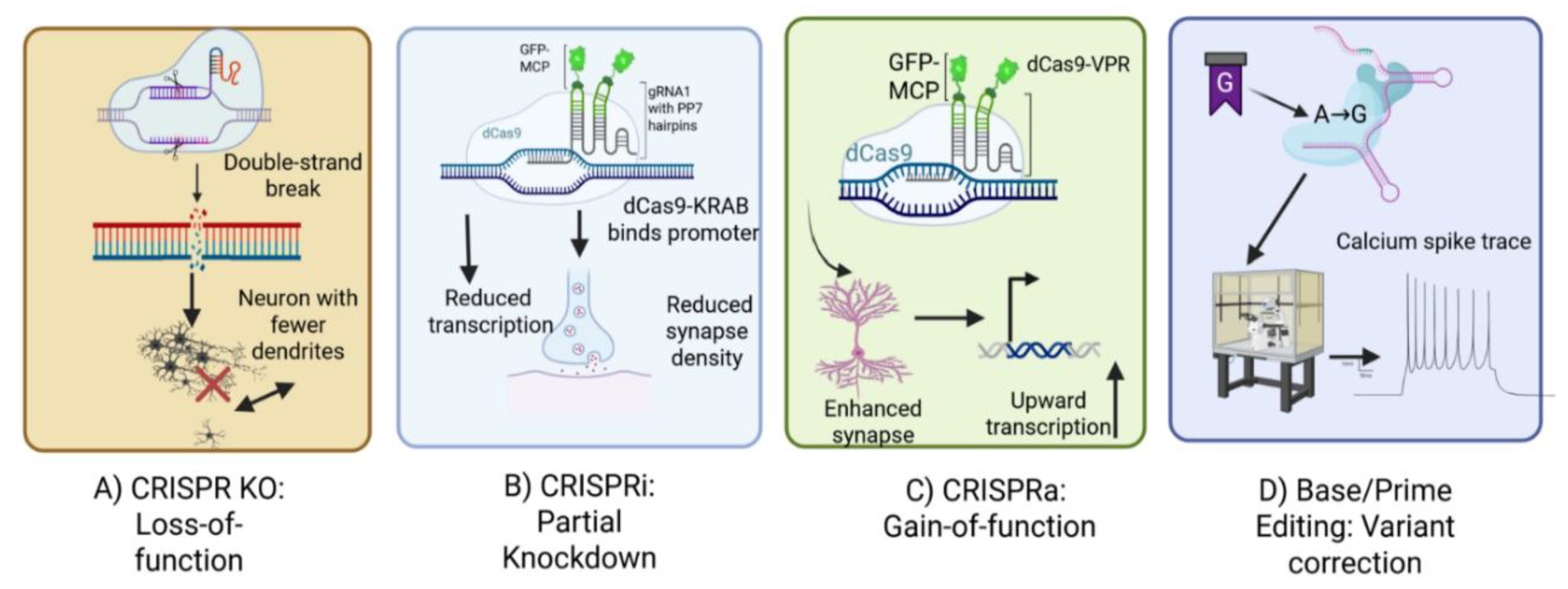
4.1.1. Nuclease-Based Editing (CRISPR-KO)
4.1.2. CRISPR Interference and Activation (CRISPRi/a)
4.1.3. Base and Prime Editors
4.1.4. Epigenetic and RNA Editors
4.2. Guide RNA Design
4.2.1. Tiling and Saturation Mutagenesis
4.3. CRISPR Library Design
4.3.1. Genome-Wide vs. Focused Libraries
4.3.2. Library Formats
4.4. Barcode Strategies and Readout Integration
4.5. Advantages for ASD Research
5. Building CRISPR-Engineered Stem Cell Libraries (Practical Guide)
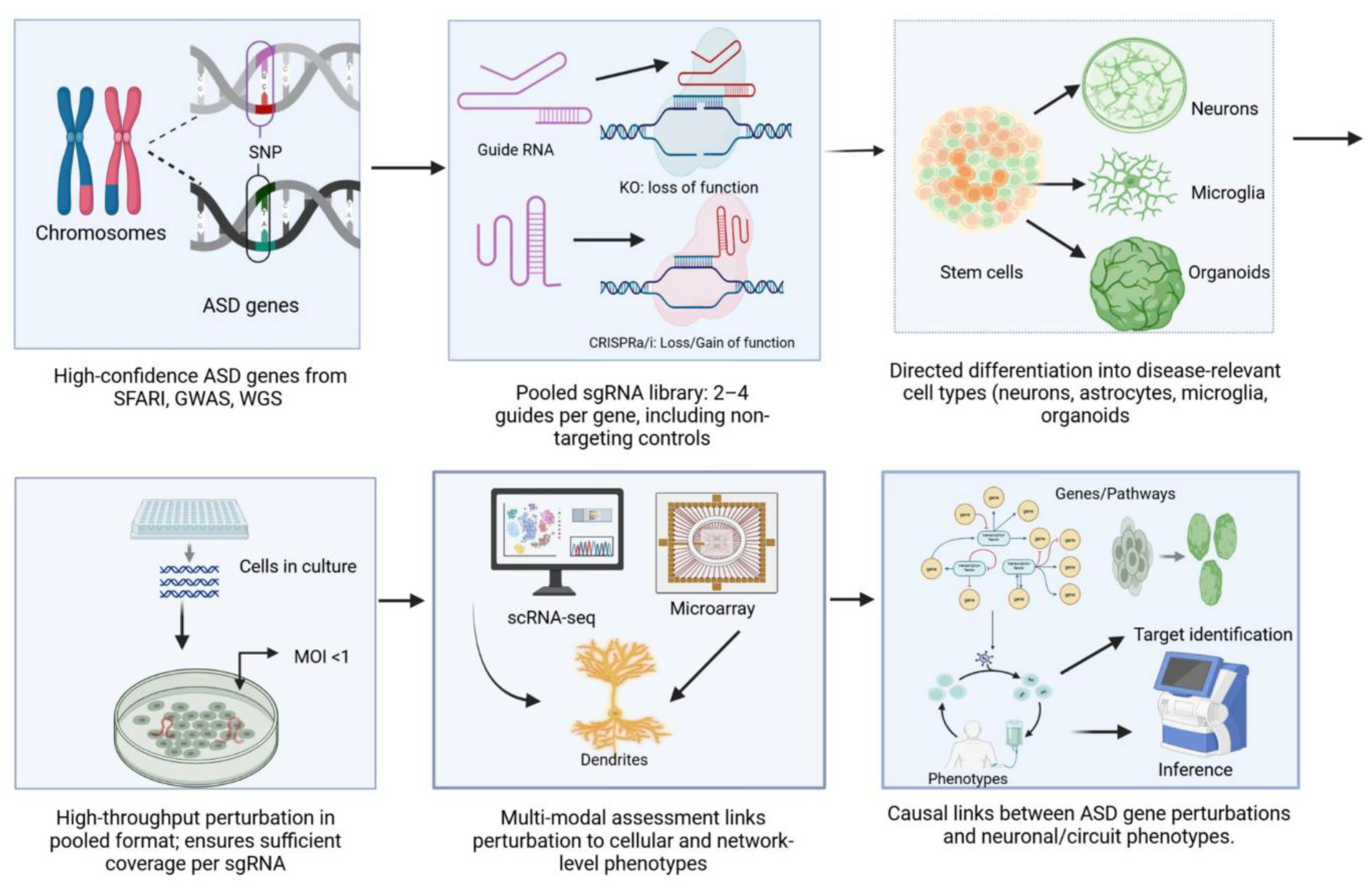
5.1. Gene/Variant Selection Strategy
5.2. Library Architecture
5.3. Cloning, Synthesis, and Quality Control
5.4. Delivery to hPSCs and Derivatives
5.5. Generation of Isogenic Clones for Validation
6. Screening Formats and Experimental Design
6.1. Pooled Negative/Positive Selection Screens
6.2. Phenotypic Pooled Screens with Bulk Readouts
6.3. High-Content Pooled Single-Cell Readouts (“Perturbomics”)
6.4. Spatially Resolved Pooled Screens and Integration with Spatial Transcriptomics
6.5. Arrayed Screening for Electrophysiology and Calcium Imaging Readouts
7. Readouts and Multimodal Profiling
7.1. Single-cell RNA-seq
7.2. Single-cell ATAC-seq and Multiome for Regulatory Effects
7.3. Spatial Transcriptomics
7.4. Proteomics and Proximity Labeling
7.5. Functional Phenotyping
7.6. Multimodal Integration and Inferences
8. Computational Pipelines and Statistical Analysis
8.1. Preprocessing and Quality Control
8.2. Differential Expression Analysis
8.3. Pseudotime and Trajectory Analyses
8.4. Network Reconstruction
8.5. Integration of Multi-Omic Datasets
8.6. Handling Batch Effects and Statistical Rigor
9. Validation Strategies and Orthogonal Assays
9.1. Single-Clone Validation
9.2. Orthogonal Perturbation to Confirm Directionality
9.3. Functional Assays: Electrophysiology, Synaptic, and Morphological Analysis
9.4. Cross-Model Validation (Patient iPSCs and Animal Models)
10. Case Studies: What's Been Done
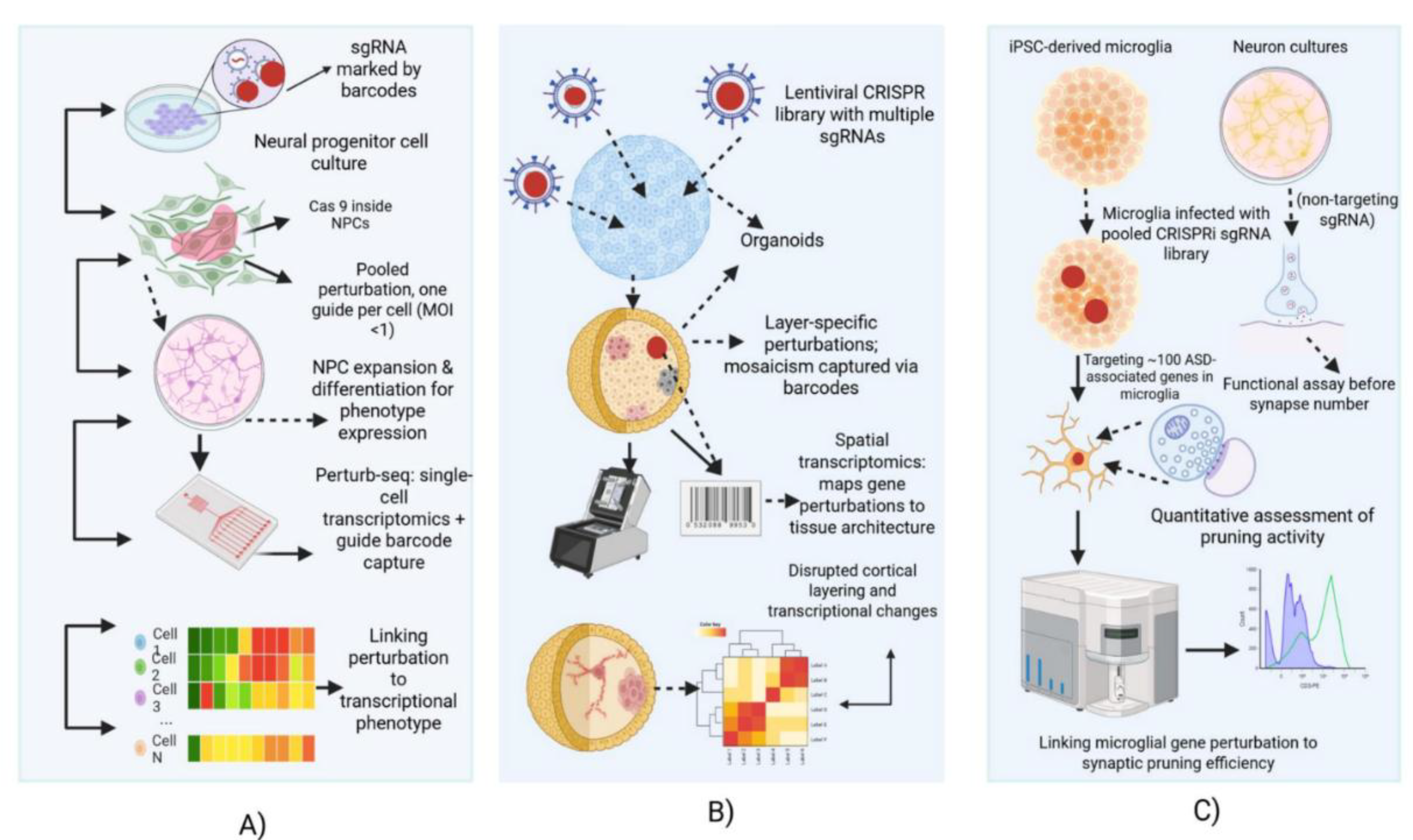
10.1. Pooled CRISPR Screens in Neural Cells: Historic Milestones
10.2. CRISPRi Screens in Microglia and Circuit-Relevant Phenotypes (Synaptic Pruning)
10.3. Pooled CRISPR Screens in Organoids — Successes, Bottlenecks, and Insights
10.4. Spatially Integrated CRISPR Screens: Mapping Non-Cell-Autonomous Effects
10.5. Inferences from Case Studies
11. Reproducibility, Standardization, and Best Practices
11.1. Experimental Reporting Standards
11.2. Minimum Metadata to Report (Cell Line Source, Passage, Differentiation Protocol Details, Batch IDs)
11.3. Data Deposition and Sharing (Raw FASTQ, Count Matrices, gRNA-Cell Maps, Metadata)
11.4. Recommended QC Checklists and Power/Coverage Calculators
11.5. Community Standards Initiatives and Consortia (Recommended Standardized Nomenclature for Perturbation IDs)
12. Ethical, Legal, and Social Implications (ELSI)
12.1. Human Stem Cell Research Governance and ISSCR Recommendations
12.2. Germline Editing vs. Somatic/Cell Models
12.3. Privacy and Data Sharing for Patient-Derived iPSCs
12.4. Societal Implications of Mechanistic Findings
12.5. Responsible Translational Pathways and Stakeholder Engagement
13. Translational Potential and Therapeutic Discovery
13.1. Target Discovery and Validation
13.2. Small-Molecule and Biologic Development Informed by Perturbation Phenotypes
13.3. Gene-Therapy Design Considerations
13.4. Personalized Medicine: Patient iPSC Panels and Isogenic Variant Correction
13.5. Regulatory Pathway for Therapeutic Translation
14. Challenges, Limitations, and Open Questions
14.1. Model Limitations
14.2. Technical Constraints for Scaling Pooled CRISPR in Organoids
14.3. Interpreting Pleiotropic Gene Effects and Cell Non-Autonomous Phenotypes
14.4. Statistical and Computational Challenges (False Positives/Negatives, Multiple Testing)
14.5. Prioritizing Hits for Translational Follow-Up
15. Future Directions and Roadmap
15.1. Integrating Spatially Resolved Perturbomics and Multi-Omics
15.2. Scaled Patient-Derived iPSC Biobanks and Standardized CRISPR Libraries for Population-Scale Functional Genomics
15.3. Use of Base and Prime Editors to Directly Model Patient Variants at Scale
15.4. AI/ML Integration for Prioritization and Causal Inference
15.5. Proposed Community Blueprint: Standard Reference Lines, Standard Readouts, and Common Data Formats
16. Conclusion
Funding
Authorship Contribution Statement
Declaration of Competing Interest
Acknowledgments
Ethical Statements
Declaration of generative AI and AI-assisted technologies in the writing process
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders [Internet]. Fifth Edition. American Psychiatric Association; 2013 [cited 2025 Sep 6]. Available online: https://psychiatryonline.org/doi/book/10.1176/appi.books.9780890425596. [CrossRef]
- Centers for Disease Control and Prevention. (2022). Prevalence and characteristics of autism spectrum disorder among children aged 8 years — Autism and Developmental Disabilities Monitoring Network, 16 sites, United States, 2022. Morbidity and Mortality Weekly Report Surveillance Summaries, 71(8), 1-23.
- MC, L.; MV, L.; Lancet, B.-C.S.A.T. 2014, 383(9920), 896–910.
- TA, L.; MC, W.; JP, N.; Munir K; KA, K., Prosser LA. Economic Burden of Childhood Autism Spectrum Disorders. Pediatrics. 2014, 133(3), e520–9.
- Lukmanji S; SA, M.; Kadhim S; KM, S.; EC, W.; CS, K., Jetté N. The co-occurrence of epilepsy and autism: A systematic review. Epilepsy Behav. 2019, 98, 238–48.
- Sandin S; Lichtenstein P; Kuja-Halkola R; Hultman C; Larsson H, Reichenberg A. The Heritability of Autism Spectrum Disorder. JAMA. 2017, 318(12), 1182.
- FK, S.; JA, K.; Wang J; MS, B.; Rubeis S, D.; JY, A.; Peng M; Collins R; Grove J; Klei L; Stevens C; Reichert J; MS, M.; Artomov M; Gerges S; Sheppard B; Xu X; Bhaduri A; Norman U; Brand H; Schwartz G; Nguyen R; EE, G.; Dias C; Betancur C; EH, C.; Gallagher L; Gill M; JS, S.; Thurm A; ME, Z.; AD, B.; MW, S.; AE, C.; ME, T.; DJ, C.; Devlin B; SJ, S.; Roeder K; MJ, D.; JD, B.; Aleksic B; Anney R; Barbosa M; Bishop S; Brusco A; Bybjerg-Grauholm J; Carracedo A; MCY, C.; AG, C.; BHY, C.; Coon H; ML, C.; Curró A; Bernardina B, D.; Doan R; Domenici E; Dong S; Fallerini C; Fernández-Prieto M; GB, F.; CM, F.; Fromer M; JJ, G.; Geschwind D; Giorgio E; González-Peñas J; Guter S; Halpern D; Hansen-Kiss E; He X; GE, H.; Hertz-Picciotto I; DM, H.; CM, H.; Ionita-Laza I; Jacob S; Jamison J; Jugessur A; Kaartinen M; GP, K.; Kolevzon A; Kushima I; SL, L.; Lehtimäki T; ET, L.; Lintas C; WI, L.; Lopergolo D; Lopes F; Ludena Y; Maciel P; Magnus P; Mahjani B; Maltman N; DS, M.; Meiri G; Menashe I; Miller J; Minshew N; EMS, M.; Moreira D; EM, M.; Mors O; PB, M.; Mosconi M; Muglia P; BM, N.; Nordentoft M; Ozaki N; Palotie A; Parellada M; MR, P.-B.; Pericak-Vance M; AM, P.; Pessah I; Puura K; Reichenberg A; Renieri A; Riberi E; EB, R.; KE, S.; Sandin S; SL, S.; Schellenberg G; SW, S.; Schlitt S; Schmidt R; Schmitt L; IMW, S.; Singh T; PM, S.; Smith M; Soares G; Stoltenberg C; Suren P; Susser E; Sweeney J; Szatmari P; Tang L; Tassone F; Teufel K; Trabetti E; MDP, T.; CA, W.; LA, W.; Werge T; DM, W.; EM, W.; Wilkinson E; AJ, W.; TW, Y.; MHC, Y.; Yuen R; Zachi E; Agerbo E; TD, A.; Appadurai V; Bækvad-Hansen M; Belliveau R; Buil A; CE, C.; Cerrato F; Chambert K; Churchhouse C; Dalsgaard S; Demontis D; Dumont A; Goldstein J; CS, H.; ME, H.; MV, H.; DP, H.; Huang H; Maller J; AR, M.; Martin J; Mattheisen M; Moran J; Pallesen J; DS, P.; CB, P.; MG, P.; Poterba T; JB, P.; Ripke S; AJ, S.; WK, T.; Turley P, Walters RK. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell. 2020, 180(3), 568-584.e23.
- DM, W.; Geschwind, D.H. Sex differences in autism spectrum disorders. Curr Opin Neurol. 2013, 26(2), 146–53.
- MJ, G.; Zhang P; Hadjimichael E; RL, W.; Chen C; Liu S; Won H; Bakel H, V.; Varghese M; Wang Y; AW, S.; Haney J; Parhami S; Belmont J; Kim M; Losada P, M.; Khan Z; Mleczko J; Xia Y; Dai R; Wang D; YT, Y.; Xu M; Fish K; PR, H.; Warrell J; Fitzgerald D; White K; AE, J.; Consortium, P.N.O.E.; MA, P.; Gerstein M; Liu C; LM, I.; Pinto D; DH, G.; AE, A.-K.; GE, C.; ME, G.; Song L; Safi A; GD, J.; GA, W.; TE, R.; FS, G.; Zandi P; Bryois J; AE, J.; AJ, P.; NA, I.; Collado-Torres L; TM, H.; EE, B.; JE, K.; Tao R; JH, S.; Akbarian S; Girdhar K; Jiang Y; Kundakovic M; Brown L; BS, K.; RB, P.; JR, W.; Zharovsky E; Jacobov R; Devillers O; Flatow E; GE, H.; BK, L.; DA, L.; Haroutunian V; CG, H.; AW, C.; Dracheva S; Kozlenkov A; Belmont J; DelValle D; Francoeur N; Hadjimichael E; Pinto D; Bakel H, V.; Roussos P; JF, F.; Bendl J; ME, H.; LM, M.; MA, P.; Chae Y; Peng J; Niu M; Wang X; MJ, W.; TG, B.; Chen C; Jiang Y; Dai R; AW, S.; Liu C; KS, G.; Xia Y; Vadukapuram R; Wang Y; Fitzgerald D; Cheng L; Brown M; Brown M; Brunetti T; Goodman T; Alsayed M; MJ, G.; DH, G.; Won H; Polioudakis D; Wamsley B; Yin J; Hadzic T, De La Torre Ubieta L, Swarup V, Sanders SJ, State MW, Werling DM, An JY, Sheppard B, Willsey AJ, White KP, Ray M, Giase G, Kefi A, Mattei E, Purcaro M, Weng Z, Moore J, Pratt H, Huey J, Borrman T, Sullivan PF, Giusti-Rodriguez P, Kim Y, Sullivan P, Szatkiewicz J, Rhie SK, Armoskus C, Camarena A, Farnham PJ, Spitsyna VN, Witt H, Schreiner S, Evgrafov OV, Knowles JA, Gerstein M, Liu S, Wang D, Navarro FCP, Warrell J, Clarke D, Emani PS, Gu M, Shi X, Xu M, Yang YT, Kitchen RR, Gürsoy G, Zhang J, Carlyle BC, Nairn AC, Li M, Pochareddy S, Sestan N, Skarica M, Li Z, Sousa AMM, Santpere G, Choi J, Zhu Y, Gao T, Miller DJ, Cherskov A, Yang M, Amiri A, Coppola G, Mariani J, Scuderi S, Szekely A, Vaccarino FM, Wu F, Weissman S, Roychowdhury T, Abyzov A. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018, 362(6420), eaat8127.
- Sanders SJ. Next-Generation Sequencing in Autism Spectrum Disorder. Cold Spring Harb Perspect Med. 2019, 9(8), a026872. [CrossRef]
- Patterson PH. Maternal infection and immune involvement in autism. Trends Mol Med. 2011, 17(7), 389–94. [CrossRef]
- KJ, K.; LC, F.; Tiao G; BB, C.; Alföldi J; Wang Q; RL, C.; KM, L.; Ganna A; DP, B.; LD, G.; Brand H; Solomonson M; NA, W.; Rhodes D; Singer-Berk M; EM, E.; EG, S.; JA, K.; RK, W.; Tashman K; Farjoun Y; Banks E; Poterba T; Wang A; Seed C; Whiffin N; JX, C.; KE, S.; Pierce-Hoffman E; Zappala Z; AH, O.-L.; EV, M.; Weisburd B; Lek M; JS, W.; Vittal C; IM, A.; Bergelson L; Cibulskis K; KM, C.; Covarrubias M; Donnelly S; Ferriera S; Gabriel S; Gentry J; Gupta N; Jeandet T; Kaplan D; Llanwarne C; Munshi R; Novod S; Petrillo N; Roazen D; Ruano-Rubio V; Saltzman A; Schleicher M; Soto J; Tibbetts K; Tolonen C; Wade G; ME, T., Genome Aggregation Database Consortium, Aguilar Salinas CA, Ahmad T, Albert CM, Ardissino D, Atzmon G, Barnard J, Beaugerie L, Benjamin EJ, Boehnke M, Bonnycastle LL, Bottinger EP, Bowden DW, Bown MJ, Chambers JC, Chan JC, Chasman D, Cho J, Chung MK, Cohen B, Correa A, Dabelea D, Daly MJ, Darbar D, Duggirala R, Dupuis J, Ellinor PT, Elosua R, Erdmann J, Esko T, Färkkilä M, Florez J, Franke A, Getz G, Glaser B, Glatt SJ, Goldstein D, Gonzalez C, Groop L, Haiman C, Hanis C, Harms M, Hiltunen M, Holi MM, Hultman CM, Kallela M, Kaprio J, Kathiresan S, Kim BJ, Kim YJ, Kirov G, Kooner J, Koskinen S, Krumholz HM, Kugathasan S, Kwak SH, Laakso M, Lehtimäki T, Loos RJF, Lubitz SA, Ma RCW, MacArthur DG, Marrugat J, Mattila KM, McCarroll S, McCarthy MI, McGovern D, McPherson R, Meigs JB, Melander O, Metspalu A, Neale BM, Nilsson PM, O’Donovan MC, Ongur D, Orozco L, Owen MJ, Palmer CNA, Palotie A, Park KS, Pato C, Pulver AE, Rahman N, Remes AM, Rioux JD, Ripatti S, Roden DM, Saleheen D, Salomaa V, Samani NJ, Scharf J, Schunkert H, Shoemaker MB, Sklar P, Soininen H, Sokol H, Spector T, Sullivan PF, Suvisaari J, Tai ES, Teo YY, Tiinamaija T, Tsuang M, Turner D, Tusie-Luna T, Vartiainen E, Vawter MP, Ware JS, Watkins H, Weersma RK, Wessman M, Wilson JG, Xavier RJ, Neale BM, Daly MJ, MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020, 581(7809), 434–43.
- ME, T.; EV, M., Gusella JF. Autism Spectrum Disorder Genetics: Diverse Genes with Diverse Clinical Outcomes. Harv Rev Psychiatry. 2014, 22(2), 65–75.
- BS, A.; DE, A.; DB, C.; HC, M.; EM, M.; LA, W.; Menashe I; Wadkins T; Banerjee-Basu S, Packer A. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism. 2013, 4(1), 36.
- Geschwind DH. Genetics of autism spectrum disorders. Trends Cogn Sci. 2011, 15(9), 409–16.
- TA, P.; Mediouni S; Leda A; RL, F.; ST, V.; HA, F.; DF, N., Ndhlovu LC. Next-Generation Human Cerebral Organoids as Powerful Tools To Advance NeuroHIV Research. Burdo TH, Prasad VR, editors. mBio. 2021, 12(4), e00680-21.
- Zeidan J; Fombonne E; Scorah J; Ibrahim A; MS, D.; Saxena S; Yusuf A; Shih A, Elsabbagh M. Global prevalence of autism: A systematic review update. Autism Res. 2022, 15(5), 778–90. [CrossRef]
- AVS, B.; Cidav Z; Knapp M, Mandell DS. Costs of Autism Spectrum Disorders in the United Kingdom and the United States. JAMA Pediatr. 2014, 168(8), 721. [CrossRef]
- Li C; JS, F.; Martins-Costa C; TR, B.; Themann J; Stuempflen M; AM, P.; Vertesy Á; JB, L.; Esk C; Elling U; Kasprian G; NS, C.; Treutlein B, Knoblich JA. Single-cell brain organoid screening identifies developmental defects in autism. Nature. 2023, 621(7978), 373–80. [CrossRef]
- Colizzi M; Lasalvia A, Ruggeri M. Prevention and early intervention in youth mental health: is it time for a multidisciplinary and trans-diagnostic model for care? Int J Ment Health Syst. 2020, 14(1), 23. [CrossRef]
- Nakanishi M; MP, A., Takumi T. Recent genetic and functional insights in autism spectrum disorder. Curr Opin Neurol. 2019, 32(4), 627–34. [CrossRef]
- Alonso-Gonzalez A; Rodriguez-Fontenla C, Carracedo A. De novo Mutations (DNMs) in Autism Spectrum Disorder (ASD): Pathway and Network Analysis. Front Genet. 2018, 9, 406. [CrossRef]
- Vysotskiy M; Zhong X; TW, M.-F.; Zhou D, Autism Working Group of the Psychiatric Genomics Consortium^, Bipolar Disorder Working Group of the Psychiatric Genomics Consortium^, Schizophrenia Working Group of the Psychiatric Genomics Consortium^, Cox NJ, Weiss LA. Integration of genetic, transcriptomic, and clinical data provides insight into 16p11.2 and 22q11.2 CNV genes. Genome Med. 2021, 13(1), 172.
- RP, M.; Warrier V; Brunel H; Buckingham C; Smith P; Allison C; Holt R; CR, B., Baron-Cohen S. Identifying rare genetic variants in 21 highly multiplex autism families: the role of diagnosis and autistic traits. Mol Psychiatry. 2023, 28(5), 2148–57. [CrossRef]
- Yousaf A; Waltes R; Haslinger D; SM, K.; Duketis E; Sachse M; Voran A; Biscaldi M; Schulte-Rüther M; Cichon S; Nöthen M; Ackermann J; Koch I; CM, F., Chiocchetti AG. Quantitative genome-wide association study of six phenotypic subdomains identifies novel genome-wide significant variants in autism spectrum disorder. Transl Psychiatry. 2020, 10(1), 215. [CrossRef]
- Group, P.S.C.-B.A., Psychiatric Genomics Consortium Autism Group, Weiner DJ, Wigdor EM, Ripke S, Walters RK, Kosmicki JA, Grove J, Samocha KE, Goldstein JI, Okbay A, Bybjerg-Grauholm J, Werge T, Hougaard DM, Taylor J, Skuse D, Devlin B, Anney R, Sanders SJ, Bishop S, Mortensen PB, Børglum AD, Smith GD, Daly MJ, Robinson EB. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet. 2017, 49(7), 978–85. [CrossRef]
- KA, S.; Williams S; ME, P.; Valencia-Prado M; MS, D.; EM, H.; CM, L.-A.; ET, P.; AV, B.; Bartholomew P; Nieves-Muñoz N; Sidwell K; Alford A; DA, B.; DiRienzo M; RT, F.; SM, F.; AE, H.; OM, P.; Shea L; SC, T.; Warren Z; Zahorodny W; Agosto-Rosa H; Anbar J; KY, C.; Esler A; Forkner A; Grzybowski A; AH, A.; Hallas L; Lopez M; Magaña S; RHN, N.; Parker J; Pierce K; Protho T; Torres H; SB, V.; Vehorn A; Zhang M; Andrews J; Greer F; Hall-Lande J; McArthur D; Mitamura M; AJ, M.; Pettygrove S; Shenouda J; Skowyra C; Washington A, Maenner MJ. Prevalence and Early Identification of Autism Spectrum Disorder Among Children Aged 4 and 8 Years — Autism and Developmental Disabilities Monitoring Network, 16 Sites, United States, 2022. MMWR Surveill Summ. 2025, 74(2), 1–22.
- Gudmundsson S; Singer-Berk M; NA, W.; Phu W; JK, G.; Solomonson M, Genome Aggregation Database Consortium, Rehm HL, MacArthur DG, O’Donnell-Luria A. Variant interpretation using population databases: Lessons from gnomAD. Hum Mutat. 2022, 43(8), 1012–30. [CrossRef]
- CA, A.-T.; Jobanputra V; DL, P.; Bick D; RJ, T.; Venner E; RA, G.; Young T; Barnett S; JW, B.; Boczek N; Chowdhury S; KA, E.; Guha S; Kulkarni S; Marcou C; Meng L; DR, M.; AU, R.; Spiteri E; Thomas-Wilson A; HM, K.; HL, R., Medical Genome Initiative*. Best practices for the interpretation and reporting of clinical whole genome sequencing. Npj Genomic Med. 2022, 7(1), 27.
- JA, M.; SL, D.; SM, S.; KA, S.; Ng L; Szafer A; Ebbert A; ZL, R.; JJ, R.; Aiona K; JM, A.; Bennet C; Bertagnolli D; Brouner K; Butler S; Caldejon S; Carey A; Cuhaciyan C; RA, D.; Dee N; TA, D.; BAC, F.; Feng D; TP, F.; Gee G; Goldy J; Gourley L; BW, G.; Gu G; RE, H.; JM, J.; CL, K.; Lau C; CK, L.; Lee F; TA, L.; Lesnar P; McMurray B; Mastan N; Mosqueda N; Naluai-Cecchini T; NK, N.; Nyhus J; Oldre A; Olson E; Parente J; PD, P.; SE, P.; Stevens A; Pletikos M; Reding M; Roll K; Sandman D; Sarreal M; Shapouri S; NV, S.; EH, S.; Sjoquist N; CR, S.; Smith M; AJ, S.; Williams D; Zöllei L; Fischl B; MB, G.; DH, G.; IA, G.; MJ, H.; RF, H.; Huang H; AR, J.; JA, K.; Levitt P; JW, P.; Šestan N; Wohnoutka P; Dang C; Bernard A; JG, H., Lein ES. Transcriptional landscape of the prenatal human brain. Nature. 2014, 508(7495), 199–206.
- AA, W.; RJ, U.; AC, N.; Genetic heterogeneity, M.J.H.; impacts, and methods through an associative lens. Genet Epidemiol. 2022, 46(8), 555–71.
- AM, F.; Schneider M; Zinkstok J; Baribeau D; SJRA, C., Vorstman JAS. Neurodevelopmental Trajectories and Psychiatric Morbidity: Lessons Learned From the 22q11.2 Deletion Syndrome. Curr Psychiatry Rep. 2021, 23(3), 13.
- KA, K.; TM, G.; CA, A., Williams AJ. SCN2A channelopathies in the autism spectrum of neuropsychiatric disorders: a role for pluripotent stem cells? Mol Autism. 2020, 11(1), 23.
- Tang D; Freudenberg J, Dahl A. Factorizing polygenic epistasis improves prediction and uncovers biological pathways in complex traits. Am J Hum Genet. 2023, 110(11), 1875–87. [CrossRef]
- Ardhanareeswaran K; Mariani J; Coppola G; Abyzov A, Vaccarino FM. Human induced pluripotent stem cells for modelling neurodevelopmental disorders. Nat Rev Neurol. 2017, 13(5), 265–78. [CrossRef]
- Nehme R, Barrett LE. Using human pluripotent stem cell models to study autism in the era of big data. Mol Autism. 2020, 11(1), 21. [CrossRef]
- Nomura T. Interneuron Dysfunction and Inhibitory Deficits in Autism and Fragile X Syndrome. Cells. 2021, 10(10), 2610. [CrossRef]
- Xu J, Wen Z. Brain Organoids: Studying Human Brain Development and Diseases in a Dish. Li X, editor. Stem Cells Int. 2021, 2021, 1–16. [CrossRef]
- Kshirsagar A; Mnatsakanyan H; Kulkarni S; Guo J; Cheng K; LD, O.; Bohra O; Sagar R; Mahairaki V; CE, B., Kathuria A. Multi-Region Brain Organoids Integrating Cerebral, Mid-Hindbrain, and Endothelial Systems. Adv Sci. 2025, 12(33), e03768.
- Zhao X; Xu Z; Xiao L; Shi T; Xiao H; Wang Y; Li Y; Xue F, Zeng W. Review on the Vascularization of Organoids and Organoids-on-a-Chip. Front Bioeng Biotechnol. 2021, 9, 637048. [CrossRef]
- JH, C.; Kim K, Cho IJ. Beyond Structure: Next-Generation Electrophysiological Platforms for Functional Brain Organoids. Int J Stem Cells. 2025, 18(3), 215–36.
- MD, D., Da Silva Chagas L, Traetta ME, Rodrigues RS, Acutain MF, Barykin E, Datusalia AK, German-Castelan L, Mattera VS, Mazengenya P, Skoug C, Umemori H. (Re)building the nervous system: A review of neuron–glia interactions from development to disease. J Neurochem. 2025, 169(1), e16258. [CrossRef]
- GG, D.S.; DP, S.; BG, D.C.; MA, P.; Gobbi A; RS, L., De La Torre LG. Microfluidic Systems to Mimic the Blood–Brain Barrier: from Market to Engineering Challenges and Perspectives. ACS Biomater Sci Eng. 2025, 11(7), 3789–815.
- AR, S.; RF, M.; MC, C.; HA, S., Sarmento B. Organ-on-chip platforms for nanoparticle toxicity and efficacy assessment: Advancing beyond traditional in vitro and in vivo models. Mater Today Bio. 2025 Aug;33:102053.
- Meng X; Yao D; Imaizumi K; Chen X; KW, K.; Reis N; MV, T.; McKinney A, A.; Kulkarni S; Panagiotakos G; MC, B., Pașca SP. Assembloid CRISPR screens reveal impact of disease genes in human neurodevelopment. Nature. 2023, 622(7982), 359–66.
- Volpato V, Webber C. Addressing variability in iPSC-derived models of human disease: guidelines to promote reproducibility. Dis Model Mech. 2020, 13(1), dmm042317. [CrossRef]
- Sanjana NE. Genome-scale CRISPR pooled screens. Anal Biochem. 2017, 532, 95–9. [CrossRef]
- Alda-Catalinas C; MA, E.-M., Reik W. Pooled CRISPR-activation screening coupled with single-cell RNA-seq in mouse embryonic stem cells. STAR Protoc. 2021, 2(2), 100426. [CrossRef]
- Dixit A; Parnas O; Li B; Chen J; CP, F.; Jerby-Arnon L; ND, M.; Dionne D; Burks T; Raychowdhury R; Adamson B; TM, N.; ES, L.; JS, W.; Friedman N, Regev A. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell. 2016, 167(7), 1853-1866.e17. [CrossRef]
- Haapaniemi E; Botla S; Persson J; Schmierer B, Taipale J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. 2018, 24(7), 927–30. [CrossRef]
- Binan L; Jiang A; SA, D.; Valakh V; Simonton B; Bezney J; RT, M.; KB, Y.; Nehme R; Cleary B, Farhi SL. Simultaneous CRISPR screening and spatial transcriptomics reveal intracellular, intercellular, and functional transcriptional circuits. Cell. 2025, 188(8), 2141-2158.e18. [CrossRef]
- Prilutsky D; NP, P.; Smedemark-Margulies N; TM, S.; DM, M., Kohane IS. iPSC-derived neurons as a higher-throughput readout for autism: promises and pitfalls. Trends Mol Med. 2014, 20(2), 91–104.
- Ferreira Da Silva J, Salic S, Wiedner M, Datlinger P, Essletzbichler P, Hanzl A, Superti-Furga G, Bock C, Winter G, Loizou JI. Genome-scale CRISPR screens are efficient in non-homologous end-joining deficient cells. Sci Rep. 2019, 9(1), 15751. [CrossRef]
- PI, T.; AM, D.; Song L; Safi A; NK, S.; AM, K.; TE, R.; GE, C., Gersbach CA. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods. 2015, 12(12), 1143–9.
- MG, K.; WS, L.; C lin, L.; BA, S.; Qiu X; NJ, S.; PN, C.V.; SP, P.; EJ, P.; MJ, V.; SC, L.; SP, B.; AA, A.; BR, W., Moriarity BS. CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat Commun. 2021, 12(1), 2437.
- Kantor A; McClements M, MacLaren R. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int J Mol Sci. 2020, 21(17), 6240. [CrossRef]
- Jeffries MA. Epigenetic editing: How cutting-edge targeted epigenetic modification might provide novel avenues for autoimmune disease therapy. Clin Immunol. 2018, 196, 49–58. [CrossRef]
- Zhu G; Zhou X; Wen M; Qiao J; Li G, Yao Y. CRISPR–Cas13: Pioneering RNA Editing for Nucleic Acid Therapeutics. BioDesign Res. 2024, 6, 0041. [CrossRef]
- DeBoever C; Li H; Jakubosky D; Benaglio P; Reyna J; KM, O.; Huang H; Biggs W; Sandoval E; D’Antonio M; Jepsen K; Matsui H; Arias A; Ren B; Nariai N; EN, S.; D’Antonio-Chronowska A; EK, F., Frazer KA. Large-Scale Profiling Reveals the Influence of Genetic Variation on Gene Expression in Human Induced Pluripotent Stem Cells. Cell Stem Cell. 2017, 20(4), 533-546.e7. [CrossRef]
- Wang X; Lalli M; Thopte U; A scalable, B.J.D., high-throughput neural development platform identifies shared impact of ASD genes on cell fate and differentiation [Internet]. 2024 [cited 2025 Sep 6]. Available online: http://biorxiv.org/lookup/doi/10.1101/2024.09.25.614184. [CrossRef]
- Navarro M, Simpson TI. SFARI Genes and where to find them; classification modelling to identify genes associated with Autism Spectrum Disorder from RNA-seq data [Internet]. 2021 [cited 2025 Sep 6]. Available online: http://biorxiv.org/lookup/doi/10.1101/2021.01.29.428754.
- Shin T; JHT, S.; Kosicki M; Kenny C; SG, B.; Kelley L; Qian X; Bonacina J; Papandile F; Antony I; Gonzalez D; Scotellaro J; EM, B.; RE, A.; Maury E; LA, P.; RN, D., Walsh CA. Rare variation in noncoding regions with evolutionary signatures contributes to autism spectrum disorder risk [Internet]. 2023 [cited 2025 Sep 6]. Available online: http://medrxiv.org/lookup/doi/10.1101/2023.09.19.23295780. [CrossRef]
- KD, H.; TM, B., Horn PS. Comparison and optimization of in silico algorithms for predicting the pathogenicity of sodium channel variants in epilepsy. Epilepsia. 2017, 58(7), 1190–8.
- SC, B.; Gandhi V; MC, T.; Marzette E; Teyssier N; JYY, C.; Hu X; Cramer A; Yadanar L; Shroff K; CG, J.; Eidenschenk C; JE, H.; Tian R, Kampmann M. A Massively Parallel CRISPR-Based Screening Platform for Modifiers of Neuronal Activity [Internet]. 2024 [cited 2025 Sep 6]. Available online: http://biorxiv.org/lookup/doi/10.1101/2024.02.28.582546. [CrossRef]
- Read A; Gao S; Batchelor E, Luo J. Flexible CRISPR library construction using parallel oligonucleotide retrieval. Nucleic Acids Res. 2017, 45(11), e101–e101. [CrossRef]
- RL, H.; Hayes M, Sugden B. Optimal LentiCRISPR-Based System for Sequential CRISPR/Cas9 Screens. ACS Synth Biol. 2022, 11(7), 2259–66. [CrossRef]
- OW, M.; Hebben M, Bovolenta C. Production of lentiviral vectors. Mol Ther - Methods Clin Dev. 2016, 3, 16017.
- CM, F., Schaffer DV. Adeno-associated virus-mediated delivery of CRISPR-Cas9 for genome editing in the central nervous system. Curr Opin Biomed Eng. 2018, 7, 33–41.
- Matsoukas IG. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front Genet. 2020, 11, 528. [CrossRef]
- Verstraelen P; Dyck M, V.; Verschuuren M; ND, K.; Nuydens R; JP, T., De Vos WH. Image-Based Profiling of Synaptic Connectivity in Primary Neuronal Cell Culture. Front Neurosci. 2018, 12, 389.
- Pogacar Z; Groot K; Jochems F, Dos Santos Dias M, Mulero-Sánchez A, Morris B, Roosen M, Wardak L, De Conti G, Velds A, Lieftink C, Thijssen B, Beijersbergen RL, Bernards R, Leite De Oliveira R. Genetic and compound screens uncover factors modulating cancer cell response to indisulam. Life Sci Alliance. 2022, 5(9), e202101348. [CrossRef]
- Feldman D; Funk L; Le A; RJ, C.; MD, L.; Tsai F; Soong B; Singh A, Blainey PC. Pooled genetic perturbation screens with image-based phenotypes. Nat Protoc. 2022, 17(2), 476–512. [CrossRef]
- RA, S.; WE, A.; Pan X; Sandhu J; Lu J; TK, L.; Smolyar K; ZA, S.; Dulac C; JS, W., Zhuang X. Perturb-Multimodal: A platform for pooled genetic screens with imaging and sequencing in intact mammalian tissue. Cell. 2025, 188(17), 4790-4809.e22.
- Cerneckis J; Cai H, Shi Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct Target Ther. 2024, 9(1), 112. [CrossRef]
- Dueck H; Khaladkar M; TK, K.; JM, S.; Francis C; Suresh S; SA, F.; Seale P; SG, B.; Bartfai T; Kuhn B; Eberwine J, Kim J. Deep sequencing reveals cell-type-specific patterns of single-cell transcriptome variation. Genome Biol. 2015, 16(1), 122. [CrossRef]
- Imaizumi K; Sone T; Ibata K; Fujimori K; Yuzaki M; Akamatsu W, Okano H. Controlling the Regional Identity of hPSC-Derived Neurons to Uncover Neuronal Subtype Specificity of Neurological Disease Phenotypes. Stem Cell Rep. 2015, 5(6), 1010–22. [CrossRef]
- Liu J; Tran V; VNP, V.; Byrne A; Borja M; YJ, K.; Agarwal S; Wang R; Awayan K; Murti A; Taychameekiatchai A; Wang B; Emanuel G; He J; Haliburton J; Pisco A, O., Neff NF. Concordance of MERFISH spatial transcriptomics with bulk and single-cell RNA sequencing. Life Sci Alliance. 2023, 6(1), e202201701. [CrossRef]
- Ishahak M; RH, H.; Annamalai D; Woodiwiss T; McCornack C; RT, C.; PA, D.; Qu X; Dahiya S; AH, K., Millman JR. Genetically Engineered Brain Organoids Recapitulate Spatial and Developmental States of Glioblastoma Progression. Adv Sci. 2025, 12(10), 2410110. [CrossRef]
- Yao D; Binan L; Bezney J; Simonton B; Freedman J; CJ, F.; Dey K; Geiger-Schuller K; Eraslan B; Gusev A; Regev A, Cleary B. Compressed Perturb-seq: highly efficient screens for regulatory circuits using random composite perturbations [Internet]. 2023 [cited 2025 Sep 6]. Available online: http://biorxiv.org/lookup/doi/10.1101/2023.01.23.525200. [CrossRef]
- GJ, B.; Liang R, Tian L. Monitoring activity in neural circuits with genetically encoded indicators. Front Mol Neurosci [Internet]. 2014 Dec 5 [cited 2025 Sep 6];7. Available online: http://journal.frontiersin.org/article/10.3389/fnmol.2014.00097/abstract. [CrossRef]
- Ahmed M; Muffat J, Li Y. Understanding neural development and diseases using CRISPR screens in human pluripotent stem cell-derived cultures. Front Cell Dev Biol. 2023, 11, 1158373. [CrossRef]
- Larsch J; Ventimiglia D; CI, B., Albrecht DR. High-throughput imaging of neuronal activity in Caenorhabditis elegans. Proc Natl Acad Sci [Internet]. 2013 Nov 5 [cited 2025 Sep 6];110(45). Available online: https://pnas.org/doi/full/10.1073/pnas.1318325110. [CrossRef]
- JA, M.; JS, S., Sanjana NE. Next-generation forward genetic screens: uniting high-throughput perturbations with single-cell analysis. Trends Genet. 2024, 40(2), 118–33.
- Kavaliauskaite G, Madsen JGS. Automatic quality control of single-cell and single-nucleus RNA-seq using valiDrops. NAR Genomics Bioinforma. 2023, 5(4), lqad101. [CrossRef]
- KM, M.; JK, S.; DG, M., Grueter BA. Patch-clamp and multi-electrode array electrophysiological analysis in acute mouse brain slices. STAR Protoc. 2021, 2(2), 100442.
- Lu C; Wei Y; Abbas M; Agula H; Wang E; Meng Z, Zhang R. Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects. Int J Mol Sci. 2024, 25(3), 1479. [CrossRef]
- Ogbeide S; Giannese F; Mincarelli L; Into the multiverse, M.I.C. 2022, 38(8), 831–43.
- D’haene E, Vergult S. Interpreting the impact of noncoding structural variation in neurodevelopmental disorders. Genet Med. 2021, 23(1), 34–46. [CrossRef]
- Baysoy A; Tian X; Zhang F; Renauer P; Bai Z; Shi H; Li H; Tao B; Yang M; Enninful A; Gao F; Wang G; Zhang W; Tran T; NH, P.; Bao S; Dong C; Xin S; Zhong M; Rankin S; Guy C; Wang Y; JP, C.; SM, P.-M.; Chi H; Chen S, Fan R. Spatially Resolved in vivo CRISPR Screen Sequencing via Perturb-DBiT [Internet]. 2024 [cited 2025 Sep 6]. Available online: http://biorxiv.org/lookup/doi/10.1101/2024.11.18.624106. [CrossRef]
- Song L; Chen W; Hou J; Guo M, Yang J. Spatially resolved mapping of cells associated with human complex traits. Nature. 2025, 641(8064), 932–41. [CrossRef]
- Qin W; KF, C.; PE, C., Ting AY. Deciphering molecular interactions by proximity labeling. Nat Methods. 2021, 18(2), 133–43.
- Ideker T; Thorsson V; JA, R.; Christmas R; Buhler J; JK, E.; Bumgarner R; DR, G.; Aebersold R, Hood L. Integrated Genomic and Proteomic Analyses of a Systematically Perturbed Metabolic Network. Science. 2001, 292(5518), 929–34. [CrossRef]
- CM, H.; Sivula M; JM, L.; DM, R.; Li B; AC, S.; Xia E; TA, R.; DJ, G., Cottrell JR. A System for Performing High Throughput Assays of Synaptic Function. Dryer SE, editor. PLoS ONE. 2011, 6(10), e25999.
- BF, V.; Gray E; Candalija A; KML, C.; Scaber J; Dafinca R; Katsikoudi A; Xu Y; Farrimond L; Wade-Martins R; WS, J.; MR, T.; SA, C., Talbot K. Human iPSC co-culture model to investigate the interaction between microglia and motor neurons. Sci Rep. 2022, 12(1), 12606.
- Wu X; Yang X; Dai Y; Zhao Z; Zhu J; Guo H, Yang R. Single-cell sequencing to multi-omics: technologies and applications. Biomark Res. 2024, 12(1), 110. [CrossRef]
- Cai L; Ma X, Ma J. Integrating scRNA-seq and scATAC-seq with inter-type attention heterogeneous graph neural networks. Brief Bioinform. 2024, 26(1), bbae711. [CrossRef]
- Schraivogel D; LM, S., Parts L. Pooled Genome-Scale CRISPR Screens in Single Cells. Annu Rev Genet. 2023, 57(1), 223–44. [CrossRef]
- Datlinger P; AF, R.; Schmidl C; Krausgruber T; Traxler P; Klughammer J; LC, S.; Kuchler A; Alpar D, Bock C. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods. 2017, 14(3), 297–301. [CrossRef]
- Rosati D; Palmieri M; Brunelli G; Morrione A; Iannelli F; Frullanti E, Giordano A. Differential gene expression analysis pipelines and bioinformatic tools for the identification of specific biomarkers: A review. Comput Struct Biotechnol J. 2024, 23, 1154–68. [CrossRef]
- PW, M.; CM, W., Califf RM. Clarifying Stem-Cell Therapy’s Benefits and Risks. N Engl J Med. 2017, 376(11), 1007–9.
- Gilis J; Perin L; Malfait M, Van Den Berge K, Assefa AT, Verbist B, Risso D, Clement L. Differential detection workflows for multi-sample single-cell RNA-seq data [Internet]. 2023 [cited 2025 Sep 6]. Available online: http://biorxiv.org/lookup/doi/10.1101/2023.12.17.572043. [CrossRef]
- ATL, L., Marioni JC. Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data. Biostatistics. 2017, 18(3), 451–64.
- Trapnell C; Cacchiarelli D; Grimsby J; Pokharel P; Li S; Morse M; NJ, L.; KJ, L.; TS, M., Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014, 32(4), 381–6. [CrossRef]
- Guo Y, Zhao X. CRISPR-based genetic screens in human pluripotent stem cells derived neurons and brain organoids. Cell Tissue Res. 2025, 399(1), 1–8. [CrossRef]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008, 9(1), 559.
- VY, K.; TS, A., Hemberg M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet. 2019, 20(5), 273–82.
- Hie B; Bryson B, Berger B. Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat Biotechnol. 2019, 37(6), 685–91. [CrossRef]
- HS, K.-O.; SJ, W.; CA, C.; ES, P.; TD, B.; JG, H.; Ahmad K, Henikoff S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 2019, 10(1), 1930.
- Regev A; SA, T.; ES, L.; Amit I; Benoist C; Birney E; Bodenmiller B; Campbell P; Carninci P; Clatworthy M; Clevers H; Deplancke B; Dunham I; Eberwine J; Eils R; Enard W; Farmer A; Fugger L; Göttgens B; Hacohen N; Haniffa M; Hemberg M; Kim S; Klenerman P; Kriegstein A; Lein E; Linnarsson S; Lundberg E; Lundeberg J; Majumder P; JC, M.; Merad M; Mhlanga M; Nawijn M; Netea M; Nolan G; Pe’er D; Phillipakis A; CP, P.; Quake S; Reik W; Rozenblatt-Rosen O; Sanes J; Satija R; TN, S.; Shalek A; Shapiro E; Sharma P; JW, S.; Stegle O; Stratton M; MJT, S.; FJ, T.; Uhlen M; Oudenaarden A, V.; Wagner A; Watt F; Weissman J; Wold B; Xavier R; Yosef N, Human Cell Atlas Meeting Participants. The Human Cell Atlas. eLife. 2017, 6, e27041.
- Jin Y, Marquardt S. Dual sgRNA-based Targeted Deletion of Large Genomic Regions and Isolation of Heritable Cas9-free Mutants in Arabidopsis. BIO-Protoc [Internet]. 2020 [cited 2025 Sep 6];10(20). Available online: https://bio-protocol.org/e3796. [CrossRef]
- YX, W.; Feige P; CE, B.; Hekmatnejad B; NA, D.; JM, R.; Faulkes S; DE, G., Rudnicki MA. EGFR-Aurka Signaling Rescues Polarity and Regeneration Defects in Dystrophin-Deficient Muscle Stem Cells by Increasing Asymmetric Divisions. Cell Stem Cell. 2019, 24(3), 419-432.e6.
- MA, L.; Renner M; CA, M.; Wenzel D; LS, B.; ME, H.; Homfray T; JM, P.; AP, J., Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013, 501(7467), 373–9.
- Lovell-Badge R; Anthony E; RA, B.; Bubela T; AH, B.; Carpenter M; RA, C.; Clark A; Clayton E; Cong Y; GQ, D.; Fu J; Fujita M; Greenfield A; SA, G.; Hill L; Hyun I; Isasi R; Kahn J; Kato K; JS, K.; Kimmelman J; JA, K.; Mathews D; Montserrat N; Mosher J; Munsie M; Nakauchi H; Naldini L; Naughton G; Niakan K; Ogbogu U; Pedersen R; Rivron N; Rooke H; Rossant J; Round J; Saitou M; Sipp D; Steffann J; Sugarman J; Surani A; Takahashi J; Tang F; Turner L; PJ, Z., Zhai X. ISSCR Guidelines for Stem Cell Research and Clinical Translation: The 2021 update. Stem Cell Rep. 2021, 16(6), 1398–408. [CrossRef]
- TE, L.; PW, A.; Barbaric I; Benvenisty N; Bhattacharyya A; JM, C.; LM, D.; JS, D.; LE, H.; Huch M; MS, I.; KB, J.; Kurtz A; MA, L.; Liberali P; MP, L.; CL, M.; MF, P.; Sato Y; Shimasaki N; AG, S.; Song J; Spits C; Stacey G; CA, W.; Zhao T, Mosher JT. ISSCR standards for the use of human stem cells in basic research. Stem Cell Rep. 2023, 18(9), 1744–52.
- Hu X; Jing M; Wang Y; Liu Y, Hua Y. Functional analysis of a novel de novo SCN2A variant in a patient with seizures refractory to oxcarbazepine. Front Mol Neurosci. 2023, 16, 1159649. [CrossRef]
- Jiangping H; Lihui L; Jiekai C, Center for Cell Lineage and Atlas (CCLA), Bioland Laboratory (Guangzhou Regenerative Medicine and Health Guangdong Laboratory), Guangzhou 510320, China, Key Laboratory of Regenerative Biology of the Chinese Academy of Sciences and Guangdong Provincial Key Laboratory of Stem Cell and Regenerative Medicine, Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, Guangzhou 510530, China. Practical bioinformatics pipelines for single-cell RNA-seq data analysis. Biophys Rep. 2022, 8(3), 158–69.
- IC, M., Voet T. Single Cell Genomics: Advances and Future Perspectives. Maizels N, editor. PLoS Genet. 2014, 10(1), e1004126.
- Vaidyanathan S; AA, S.; ZM, S.; DT, B.; SS, C.; Batish A; Le W; Baik R, De La O S, Kaushik MP, Galper N, Lee CM, Teran CA, Yoo JH, Bao G, Chang EH, Patel ZM, Hwang PH, Wine JJ, Milla CE, Desai TJ, Nayak JV, Kuo CJ, Porteus MH. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell. 2020, 26(2), 161-171.e4. [CrossRef]
- 119. Menyhart O; Weltz B, Győrffy B. MultipleTesting.com: A tool for life science researchers for multiple hypothesis testing correction. Delcea C, editor. PLOS ONE. 2021, 16(6), e0245824. [CrossRef]
- Hens K; Noens I; Peeters H, Steyaert J. The ethics of patenting autism genes. Nat Rev Genet. 2018, 19(5), 247–8.
- Qu H, Fang X. A Brief Review on the Human Encyclopedia of DNA Elements (ENCODE) Project. Genomics Proteomics Bioinformatics. 2013, 11(3), 135–41. [CrossRef]
- MNT, A., Simpson TI. SFARI genes and where to find them; modelling Autism Spectrum Disorder specific gene expression dysregulation with RNA-seq data. Sci Rep. 2022, 12(1), 10158.
- RJ, I.; KA, W.; MR, S.; Frias E; Ho D; Theriault K; Kommineni S; Chen J; Sondey M; Ye C; Randhawa R; Kulkarni T; Yang Z; McAllister G; Russ C; Reece-Hoyes J; Forrester W; GR, H.; Dolmetsch R, Kaykas A. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat Med. 2018, 24(7), 939–46.

| SFARI Category | Description | Example Genes |
|---|---|---|
| Syndromic (S) | Monogenic syndromes featuring ASD (often with ID, seizures, etc.) | FMR1, TSC2, MECP2, PTEN |
| 1 (High Confidence) | Strong evidence (≥3 de novo LoF mutations) | CHD8, SCN2A, SYNGAP1, ADNP |
| 2 (Strong Candidate) | Moderate evidence (e.g. ≥2 de novo LoF or replicated association) | GRIN2B, CTNND2, ANK2 |
| 3 (Suggestive) | Preliminary evidence (e.g. single de novo LoF or small studies) | CHRNA7, RAI1, DSCAM |
| Gene | SFARI Category | pLI Score (gnomAD v4.0) | Constraint Metric (o/e) | Developmental Brain Expression Pattern | Associated Phenotypes |
|---|---|---|---|---|---|
| FMR1 | Syndromic | 0.99 | 0.10 | Widely expressed in neurons; early development | Fragile X syndrome, intellectual disability, seizures |
| TSC2 | Syndromic | 0.99 | 0.08 | Neuronal progenitors; cortical regions | Tuberous sclerosis complex, epilepsy, ID |
| MECP2 | Syndromic | 0.98 | 0.09 | Methyl-CpG binding; glia and neurons | Rett syndrome, regression |
| PTEN | Syndromic | 0.98 | 0.07 | High in early cortical progenitors | Macrocephaly, ASD, tumor susceptibility |
| CHD8 | 1 (High Confidence) | 1.00 | 0.05 | High in prenatal cortex; excitatory neurons | Macrocephaly, intellectual disability |
| SHANK3 | 1 | 1.00 | 0.08 | Synaptic regions; postnatal peaks | Phelan-McDermid syndrome, social deficits |
| SCN2A | 1 | 0.99 | 0.12 | Neuronal ion channels; early development | Epilepsy, severe ASD |
| ARID1B | 1 | 1.00 | 0.04 | Chromatin remodeling; progenitors | Coffin-Siris syndrome, growth delays |
| SYNGAP1 | 1 | 1.00 | 0.06 | Synaptic plasticity; excitatory neurons | Epilepsy, intellectual disability |
| ADNP | 1 | 1.00 | 0.03 | Chromatin; neural progenitors | Helsmoortel-Van der Aa syndrome |
| GRIN2B | 2 (Strong Candidate) | 0.99 | 0.11 | Glutamate receptors; synapses | West syndrome, seizures |
| CTNND2 | 2 | 0.97 | 0.17 | Neuronal adhesion; cortical layers | Intellectual disability, ASD |
| ANK2 | 2 | 0.98 | 0.13 | Cytoskeleton; excitatory neurons | ASD, cardiac arrhythmias |
| FOXP1 | 2 | 0.98 | 0.15 | Transcription; cortical layers | Language impairments, motor delays |
| TCF4 | 2 | 0.97 | 0.18 | Neuronal differentiation; interneurons | Pitt-Hopkins syndrome, hypotonia |
| CHRNA7 | 3 (Suggestive) | 0.96 | 0.20 | Cholinergic neurons; hippocampus | Seizures, cognitive impairment |
| RAI1 | 3 | 0.95 | 0.22 | Chromatin regulator; multiple brain regions | Smith-Magenis syndrome, behavioral issues |
| DSCAM | 3 | 0.94 | 0.25 | Axon guidance; cortical layers | Down syndrome-related phenotypes |
| Aspect | hESCs | iPSCs | Notes |
|---|---|---|---|
| Source | Surplus embryos | Patient fibroblasts / somatic cells | Defines origin |
| Genetic Background | Limited diversity; allogenic | Patient-specific; captures variants | Impacts disease modeling |
| Reprogramming Artifacts | None | Epigenetic memory; somatic mutations | Only in iPSCs |
| Ethical Concerns | Embryo destruction | Minimal; informed consent | Critical for approval |
| Expansion Potential | High; stable karyotype | High; clonal instability possible | Affects scale-up |
| Differentiation Efficiency | Consistent across lines | Variable; donor-dependent | Important for reproducibility |
| Disease Modeling | Requires genome editing (e.g., CRISPR) | Direct from patients; idiopathic conditions (e.g., ASD) | Relevance to patient-specific studies |
| Immune Compatibility | Allogenic mismatch | Autologous potential | Consider for therapy |
| Cost / Accessibility | Restricted access | Widely available; biobanks | Affects practical use |
| Model / System | Key Features / Cell Types | Cellular Complexity | Structural Fidelity | Functional Readouts | Applications / Pros | Limitations / Cons |
|---|---|---|---|---|---|---|
| hESCs (Embryonic Stem Cells) | Naïve pluripotent stem cells from embryos | Single-cell pluripotent | Minimal | Differentiation potential; lineage tracing | Highly pluripotent, uniform; stable karyotype; high expansion potential | Limited lines; ethical concerns (embryo destruction); immune mismatch |
| iPSCs (Induced Pluripotent Stem Cells) | Reprogrammed adult somatic cells; patient genotype | Single-cell pluripotent | Minimal | Differentiation potential; disease modeling | Patient-specific; scalable; autologous potential; widely available from biobanks | Variability from reprogramming; epigenetic memory; donor-dependent differentiation efficiency |
| Neural Progenitor Cells (NPCs) | Expandable neural precursors (SOX2+, PAX6+) | Early neural lineage | Minimal | Proliferation, differentiation assays | Easily amplified; suitable for high-throughput screening | Lacks mature neuronal/glial functions |
| Cortical Excitatory Neurons | Glutamatergic neurons (TBR1+, VGLUT+) | Post-mitotic neurons | Minimal to 2D networks | Electrophysiology, synapse assays | Model cortical synapses; relevant to ASD | Require weeks to mature; fetal-like features |
| GABAergic Inhibitory Neurons | Interneurons (GAD67+, PVALB/SST) | Post-mitotic neurons | Minimal to 2D networks | Electrophysiology, migration assays | Study E/I balance and migration | Complex induction (SHH patterning); long maturation |
| Astrocytes | Glial cells (GFAP+, S100β+) | Glia | Minimal | Support synaptogenesis, calcium signaling | Support neurons and synapse formation; study cell-cell interactions | Require prolonged culture |
| Microglia | Brain macrophages (IBA1+, TMEM119+) | Myeloid-lineage glia | Minimal | Cytokine release, migration, synaptic pruning | Model neuroinflammation; study microglia-neuron interactions | Separate differentiation needed; no endogenous myeloid cells unless co-cultured |
| Oligodendrocytes | Myelinating glia (MBP+, OLIG2+) | Glia | Minimal | Myelination assays | Study myelination dynamics | Differentiation is very slow (months) |
| 2D Monolayer Culture | Neurons, astrocytes, glia | Low | Minimal | Single-cell imaging, electrophysiology, bulk/scRNA-seq | Cost-effective, scalable; suitable for high-throughput screens | Lacks tissue architecture; limited to cell-autonomous effects |
| 3D Brain Organoids | Self-organizing 3D tissues with neurons and glia | Multiple cell types | Recapitulates early development and cytoarchitecture | Gene expression, morphology, developmental trajectory | Models in vivo complexity; bridges 2D culture and in vivo brain | Diffusion limits; lack vascularization; limited long-term maturation |
| Assembloids / MRBO | Fused region-specific organoids (e.g., cortex + subpallium) | Multiple cell types, multiple regions | Models inter-regional connectivity | Electrophysiology, connectivity, gene expression | Model non-cell-autonomous effects and circuit pathology | High technical difficulty; costly; limited long-term stability |
| Vascularized Organoids | Organoids plus endothelial cells or implanted vasculature | Multiple cell types | Enhanced structural fidelity with vasculature | Long-term viability, nutrient diffusion | Improved nutrient supply; prolonged longevity | Difficult to reproduce uniform vasculature |
| Microfluidic Co-culture | 2–3 cell types (e.g., neurons + microglia) | Low to moderate | N/A | Microglial migration, inflammatory signaling, cytokine release | Enables controlled study of neuro-immune crosstalk | Does not model 3D architecture; limited to specific interactions |
| Modality | Mechanism | Perturbation Type | Library Design | ASD Use Cases | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
| CRISPR-KO (Nuclease-based editing) | Cas9 nuclease induces DSBs repaired by NHEJ, introducing indels | Complete loss-of-function | sgRNA libraries targeting coding exons | Modeling high-penetrance LGD variants (e.g., CHD8) | Robust LoF phenotypes, simple design, scalable | Potential lethality for essential genes; p53/DSB toxicity; mosaicism |
| CRISPRi (Interference) | dCas9-KRAB represses transcription | Tunable hypomorph (partial knockdown) | Promoter-proximal sgRNA libraries | Modeling dosage-sensitive or toxic LoF (e.g., SCN2A variants) | Non-lethal, reversible, dose-dependent effects | Variable repression efficiency; off-target chromatin effects |
| CRISPRa (Activation) | dCas9 fused to activators (VP64-p65-Rta) upregulates transcription | Gain-of-function / compensation | Promoter-proximal sgRNA libraries | Rescue of haploinsufficient genes; modeling GoF variants | Endogenous regulation, rescue strategies feasible | Limited to genes with accessible promoters; variable activation strength |
| Base Editors (CBE/ABE) | Cas9 nickase + deaminase mediates targeted base conversion (C→T or A→G) | Single-nucleotide substitution | sgRNA libraries with editable PAM-proximal sites | Missense or nonsense variants (e.g., SHANK3) | Precise, efficient point mutations; avoids DSBs | Window and PAM constraints; bystander edits |
| Prime Editors | Cas9 nickase + RT + pegRNA enables programmable substitutions, small indels | Any base substitution, small indel | pegRNA libraries with optimized design (nick-to-edit distance, PBS length) | Patient-specific VUS modeling; fine-tuned variant editing | Broad editing scope; reduced mosaicism | Lower efficiency; complex design; delivery challenges |
| Epigenetic Editors | dCas9 fused to epigenetic modifiers (e.g., DNMT3A, LSD1, p300) | Reversible transcriptional silencing/activation | sgRNA libraries targeting promoters, enhancers, non-coding elements | Modeling imprinting defects, non-coding variants | Reversible, non-genome-disruptive; context-specific modulation | Epigenetic changes may be unstable; off-target chromatin remodeling |
| RNA Editors (Cas13-based) | Cas13 targets RNA; with ADAR fusion mediates A→I editing | Transcript knockdown or RNA editing | sgRNA libraries targeting mRNA/transcripts | Transient knockdown or reversible editing in ASD genes | No DNA modification; reversible; RNA-level precision | Transient effects; off-target RNA cleavage; delivery limits |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).



