Submitted:
03 December 2025
Posted:
05 December 2025
You are already at the latest version
Abstract
Rett syndrome (RTT), an X-linked neurodevelopmental disorder predominantly arising from de novo MECP2 mutations, manifests with psychomotor regression, stereotypic hand movements, gait apraxia, and expressive aphasia, driven by dosage-sensitive epigenetic dysregulation via MeCP2's methyl-CpG-binding domain (MBD) and transcriptional repression domain (TRD). Isoform-specific expression (MeCP2-E1 neuronal predominance) and X-chromosome inactivation mosaicism underpin phenotypic variability, with missense (R133C, T158M) and nonsense (R168X, R255X) variants correlating to severity gradients. Multisystem pathophysiology encompasses brainstem-mediated respiratory dysrhythmias, QTc prolongation via ion channel perturbations, enteric hypomotility, osteopenic fractures, and mitochondrial bioenergetic deficits, exacerbated by glial-neuronal crosstalk and oxidative stress. Preclinical platforms, including Mecp2-null rodents, patient-derived iPSCs, and cerebral organoids, elucidate synaptic hyperexcitability, dendritic arborization deficits, and reversibility upon Mecp2 reactivation. Therapeutic modalities span supportive multidisciplinary interventions, FDA-approved trofinetide (IGF-1 analog modulating neurotrophic cascades), AAV-mediated gene replacement (NGN-401, TSHA-102 with miRARE autoregulation), ASOs for dosage normalization, and emerging PPAR-γ agonists targeting metabolic homeostasis. Prioritized research agendas emphasize validated biomarkers (BDNF/IGF-1 axes, miRNA signatures), combinatorial regimens, and equitable global access to mitigate caregiver burden and phenotypic heterogeneity.
Keywords:
MECP2 mutations
; epigenetic dysregulation
; neurodevelopmental disorder
; multisystem pathophysiology
; precision therapeutics
1. Introduction
Rett syndrome (RTT) is a rare, X-linked neurodevelopmental disorder that predominantly affects females, with a global prevalence of ~1 in 10,000 live female births, and is among the most common causes of complex disability in females [1,2,3]. Affected individuals exhibit apparently normal psychomotor development during the first 6–18 months, followed by regression in motor, cognitive, and social abilities, characterized by loss of purposeful hand use, emergence of stereotypic hand movements (wringing, clapping, or mouthing), gait abnormalities, and severe expressive language impairment. Atypical variants display milder or variable symptoms, sometimes retaining speech or ambulation [1,3]. Epidemiological data indicate that RTT occurs sporadically in most cases, with de novo MECP2 mutations accounting for over 95% of classic cases; males are rarely affected and usually present with severe neonatal encephalopathy due to hemizygosity, though survival is possible in some cases with somatic mosaicism [3,4]. The mutational spectrum of MECP2 is broad, encompassing missense, nonsense, frameshift mutations, and large deletions, and X-chromosome inactivation mosaicism contributes to phenotype variability [2,3,4]. Landmark studies in mice show that reactivation of a silent MECP2 allele can reverse symptoms, highlighting therapeutic potential [1,2,5]. Diagnosis is primarily clinical, based on revised consensus criteria requiring regression plus four core features—loss of purposeful hand use, loss of spoken language, hand stereotypies, and gait abnormalities—along with supportive features such as breathing disturbances, sleep disruption, scoliosis, growth retardation, and diminished pain sensitivity; genetic confirmation via MECP2 sequencing is recommended, though ~5–10% of clinically diagnosed cases lack detectable mutations, potentially involving CDKL5 or FOXG1 in atypical forms [3,5,6,7]. The natural history, defined by longitudinal studies including the RTT Natural History Study (RNHS), follows four stages: early developmental stagnation (6–18 months), rapid regression (1–4 years), pseudo-stationary phase (2–10 years), and late motor deterioration (beyond 10 years), with seizures affecting ~80% of individuals, often refractory, and median survival into the fifth decade, though sudden unexpected death accounts for ~20–30% of reported fatalities, primarily due to autonomic dysfunction [3,5,8,9]. RTT is associated with severe intellectual disability and profound deficits in language and motor skills, yet it is not classically degenerative, as many patients enter a prolonged “plateau” stage post-regression [1,3]. RTT exhibits functional compromise with preserved circuit plasticity; however, late motor loss reveals compounded network pathology, secondary orthopedic complications, and immobility—not intrinsic neurodegeneration. While traditionally considered a neuron-centric disorder, evidence indicates RTT is a multi-system disease, with MECP2 deficiency producing systemic morbidity by affecting autonomic, cardiac, gastrointestinal, musculoskeletal, endocrine, sensory, and sleep systems; complications include autonomic dysregulation, cardiac arrhythmias (QTc prolongation), gastrointestinal dysmotility affecting ~80% of patients, osteopenia, scoliosis in ~75%, growth failure, delayed puberty, and sensory disturbances, with ~70% of mortality attributed to non-neurological causes [1,2,5]. Molecular studies reveal altered lipid and mitochondrial metabolism, oxidative stress, and energy homeostasis deficits in peripheral tissues, and non-neuronal brain cells (astrocytes, microglia, oligodendrocytes) contribute to CNS and systemic pathology, indicating complex intercellular interactions [1,2,4,5,6,10,11]. Preclinical models, including mouse and patient-derived iPSC systems, together with multi-omic profiling, provide mechanistic insights and facilitate biomarker identification. Therapeutic strategies encompass supportive care, the first approved pharmacotherapy, trofinetide, gene therapy approaches including reactivation of silent MECP2 alleles, antisense oligonucleotides, and small molecules targeting downstream pathways; early intervention prior to plateau stages may maximize reversibility, as evidenced in adult mouse studies [1,2]. Comprehensive management addressing both neuronal and peripheral deficits is critical for optimizing outcomes, and ongoing research prioritizes disease modification, quality-of-life improvement, and equitable global access to care.
2. MECP2: Molecular Biology, Isoforms, and Genetics
2.1. Gene Structure, Isoforms, and Transcriptional Regulation
The MECP2 gene (methyl-CpG-binding protein 2) is located on the X chromosome at position Xq28 and spans approximately 76 kb across four exons. It encodes a highly abundant chromatin-associated protein that binds methylated CpG dinucleotides, acting as an “epigenetic reader” and a global regulator of gene expression. Alternative splicing of MECP2 generates two major mRNA transcripts, MECP2-E1 and MECP2-E2, which translate into the protein isoforms MeCP2-E1 and MeCP2-E2. MeCP2-E1 arises from exons 1, 3, and 4 and encodes a 492-amino acid protein, while MeCP2-E2 includes exons 2–4 and encodes a 486-residue protein. These isoforms differ only in their N-terminal sequences but are differentially regulated: MeCP2-E1 is the predominant isoform in neurons, whereas MeCP2-E2 is more abundant in glial and peripheral cells. Recent single-cell transcriptomics indicate that MeCP2-E1 accounts for over 90% of neuronal MECP2 expression, whereas MeCP2-E2 predominates in non-neuronal tissues [5,10,12,13]. Isoform-selective dosing, therefore, represents an important consideration for therapeutic design.
Both isoforms share the methyl-binding domain (MBD) and the transcriptional repression domain (TRD). The MBD enables binding to methylated DNA, while the TRD recruits co-repressor complexes, including Sin3A and histone deacetylases (HDACs), to mediate transcriptional repression. MeCP2 also participates in chromatin looping, alternative splicing, microRNA regulation, and RNA processing, and its activity is dynamically modulated by post-translational modifications such as phosphorylation at S421, which enables activity-dependent regulation [2,14,15].
2.2. Expression Patterns and Systemic Role
MECP2 expression is highest in the central nervous system (CNS), particularly in neurons, where its abundance rivals that of histones, reflecting the critical requirement for precise gene regulation in neuronal plasticity. Expression peaks postnatally during synaptogenesis but is also detectable at lower levels in peripheral tissues, including heart, lung, kidney, muscle, gut, and immune cells [14,16]. Proteomic studies indicate that MeCP2 stabilizes nucleosomes and facilitates long-range gene interactions, supporting a systemic role beyond neurons. This broad expression pattern is consistent with RTT being a multisystem disorder, with non-neuronal cell types (astrocytes, microglia, oligodendrocytes) and peripheral organs being directly affected by MeCP2 deficiency [15,17,18]. As shown in Figure 1, MeCP2 expression is predominantly enriched in the cerebral cortex and hippocampus, with moderate levels in the cerebellum and brainstem, while peripheral organs such as the lung and spleen show higher expression compared to the liver and intestine.
2.3. Functional Role and Dosage Sensitivity
MeCP2 functions as a dosage-sensitive transcriptional regulator, capable of both repressing and activating gene expression. Gene repression occurs via MBD-mediated DNA binding and recruitment of co-repressors, while transcriptional activation can involve binding to non-CG methylated DNA and recruitment of activators such as CREB. MeCP2 regulates neuronal gene networks, including BDNF, and affects metabolic homeostasis. Both deficiency and overexpression are pathogenic; expression control is therefore critical in all gene-targeting strategies. Loss-of-function mutations cause RTT, whereas duplications or triplications of the MECP2 locus lead to MECP2 duplication syndrome (MDS), characterized by hypotonia, intellectual disability, autism, and epilepsy, primarily in boys [10,17,19].
2.4. MECP2 Mutations and Genotype–Phenotype Correlations
MECP2 mutations are the primary cause of classic RTT, with over 95% of typical RTT cases harboring loss-of-function variants. Most mutations are de novo, though hotspots exist at CpG dinucleotides. Common recurrent mutations include missense changes (R106W, R133C, T158M), nonsense mutations (R168X), and truncating mutations (R255X, R270X, R294X), clustering in the MBD or TRD and impairing MeCP2 function. Mutational severity correlates with residual protein function: R133C and R306C are associated with milder RTT phenotypes, whereas nonsense or frameshift mutations generally produce more severe disease [4,11,20,21]. Table 1 summarizes the key MECP2 mutations identified in Rett syndrome, including their mutation types, common variants, approximate frequencies among classic MECP2 variants, associated phenotypic severity, additional clinical features, recent findings from 2024–2025, and relevant inferences.
X-chromosome inactivation (XCI) in females introduces cellular mosaicism, with approximately half the cells expressing the mutant allele and half the wild-type allele. Skewed XCI (>80% favoring the wild-type allele) is associated with milder clinical symptoms. Males, lacking a second X chromosome, typically exhibit severe neonatal encephalopathy and early mortality when carrying pathogenic MECP2 variants. Rare surviving males often have mosaic mutations or an extra X chromosome (Klinefelter syndrome), displaying a spectrum of phenotypes from severe cognitive impairment to attenuated RTT-like syndromes [13,19,22,23,24].
Recent analyses (2024–2025) indicate that MBD-disrupting mutations impair DNA binding more severely than TRD variants, linking residual function to clinical severity. Truncations in exons 3–4 are frequently associated with severe epilepsy and scoliosis, while C-terminal deletions often allow milder ambulation phenotypes. Genotype–phenotype correlations, however, remain imperfect due to XCI variability and other modulatory factors [15,18,24].
2.5. Preclinical Insights and Therapeutic Implications
A key insight from mouse genetics is that MeCP2 deficiency in mature neurons is not irreversibly degenerative. Landmark studies demonstrated that reactivation of Mecp2 in adult symptomatic knockout mice restored neuronal function, improved motor ability, normalized breathing, and extended lifespan. This highlights functional plasticity in affected neural circuits and implies that RTT is a disorder of functional impairment rather than permanent neuronal loss [7,25,26].
iPSC-derived neurons from RTT patients show reduced dendritic arborization and excitatory/inhibitory imbalance, which can be rescued by restoring MECP2 expression. These findings suggest that gene or protein replacement therapies, if isoform- and dose-calibrated to mimic endogenous patterns, have the potential to reverse, rather than merely halt, disease progression. Peripheral expression further implies non-cell-autonomous mechanisms, where MeCP2 deficiency in glial cells can exacerbate neuronal dysfunction via neuroinflammation [27,28,29].
MECP2 acts as a master regulator of gene expression, with critical roles in neuronal development, plasticity, and systemic function. Its dual activator/repressor functions, dosage sensitivity, and widespread expression contribute to the complex pathogenesis of RTT. Clinical severity is modulated by mutation type, XCI mosaicism, and residual protein function, while preclinical models provide strong evidence that re-establishing MeCP2 expression can restore neuronal health, opening avenues for disease-modifying therapies [4,15,16,30].
3. Central Nervous System Phenotype
3.1. Developmental Stages and Regression
RTT is a neurodevelopmental disorder characterized by a predictable, albeit variable, trajectory of developmental changes. The central nervous system (CNS) phenotype typically follows a stereotypical course: normal perinatal development is followed by stagnation between 6 and 18 months of age, regression between 1 and 4 years, and stabilization with persistent impairments in later stages [1,2,11,31].
3.2. Stage I: Early Onset / Developmental Arrest (6–18 months)
Stage I, also referred to as the early onset or developmental arrest stage, is marked by subtle, non-specific features such as deceleration of head growth (microcephaly), limited eye contact, and gross motor delays. Video analyses of home recordings suggest that hypotonia may be present even earlier than the overt clinical signs. Overall, development appears normal during the first year of life, but early stagnation in motor and social skills can be retrospectively identified [32,33].
3.3. Stage II: Rapid Progressive / Regression (1–4 years)
Stage II, the rapid regression stage, represents the hallmark of RTT and occurs typically between 1 and 4 years of age. During this period, children abruptly lose previously acquired skills, including purposeful hand use and spoken language. Stereotypic hand movements—such as wringing, squeezing, and mouthing—emerge to replace purposeful hand function. Gait abnormalities, including ataxia and dyspraxia, become evident, and autistic-like behaviors, irritability, and social withdrawal may manifest [27,28,34,35].
Seizures are common in this stage, affecting 60–80% of individuals, and include multiple types: focal, generalized tonic–clonic, atypical absence, and myoclonic. EEG findings may show slowing of background rhythms and paroxysmal discharges, though these are non-specific. Regression is often accompanied by the development of microcephaly, irritability, and deficits in eye contact [3,22,35,36].
The neurological regression in RTT has been linked to synaptic pruning failure due to loss of MeCP2 function, resulting in a disrupted excitatory/inhibitory balance in the CNS. Adult reversal studies suggest that neuronal plasticity may be retained despite early developmental deficits.
3.4. Stage III: Plateau / Pseudo-Stationary (2–10 years, extending into preadolescence)
Following regression, most individuals enter a plateau or pseudo-stationary stage. During this phase, further loss of previously acquired skills is typically absent, though some modest improvements in social engagement, eye contact, and nonverbal communication may occur. Motor difficulties persist: ataxic or dyspraxic gait is common, and rigid or spastic muscle tone may be present. Hand stereotypies remain prominent, sometimes intensifying.
Cognitive impairment remains severe and global, with most individuals demonstrating profound intellectual disability (IQ <50). Speech is profoundly affected; nearly all become non-verbal, though purposeful eye gaze and simple gestures are retained as primary modes of communication. Approximately half of girls achieve motor milestones such as independent walking in early childhood, but many subsequently lose ambulation and require wheelchairs. Scoliosis develops in approximately 80% of cases by adolescence [34,37,38].
3.5. Stage IV: Late Motor Deterioration (Post-10 years)
Stage IV, or late motor deterioration, is characterized by further mobility decline, bradykinesia, rigidity, dystonia, and progressive scoliosis [24,36,39]. Despite worsening motor function, cognitive abilities generally remain stable after the earlier regression stage, demonstrating a partial dissociation of motor and intellectual domains. Communication challenges remain, and the assessment of internal states such as pain or anxiety is complicated by severe motor apraxia [10,39].
3.6. Seizures and Electroencephalography
Seizures affect 60–80% of individuals with RTT, often presenting between 2 and 5 years of age, although onset can occur later. Seizure types include focal, generalized tonic–clonic, atypical absence, and myoclonic forms. While many respond to conventional anti-seizure medications, a subset develops drug-resistant epilepsy. EEG recordings reveal slowing of background rhythms and paroxysmal discharges; however, patterns are highly variable and non-diagnostic [6,16].
3.7. Neuroimaging and Brain Structure
Neuroimaging in RTT is generally unrevealing. MRI scans are typically normal or may show mild cerebral atrophy, reduction in white matter, or prefrontal/posterior frontal volume loss, without focal lesions. Magnetic resonance spectroscopy indicates mild alterations in neuronal metabolites, such as decreased N-acetylaspartate (NAA). There is no established imaging biomarker comparable to those in leukodystrophies or neurodegenerative disorders. EEG variability does not predict diagnosis, although some age-related changes in background rhythms have been reported [40,41].
3.8. Behavioral Features and Communication
Behavioral manifestations in RTT include anxiety, sleep disturbances, and autistic-like behaviors (e.g., avoidance of strangers). Sleep is frequently disrupted, with night waking and irregular architecture commonly reported. Despite profound disabilities, individuals often demonstrate social interest through eye gaze (“eye pointing”), which serves as a primary communication modality. Pain insensitivity has been anecdotally noted; quantitative studies suggest that normal pain perception is likely present, though motor apraxia masks typical pain expressions [1,9,19,25].
3.9. Natural History Data and Clinical Insights
The NIH Rett Natural History Study (RNHS) has provided comprehensive longitudinal data. In a cohort of over 600 girls (predominantly classic RTT, aged from infancy to ~64 years), approximately 60% had ever experienced seizures by parental report (48% on expert review). Universal features included developmental regression, stereotypic hand movements, and gait abnormalities. Scoliosis was present in over 60% by adolescence. Growth failure, manifesting as low height and weight z-scores, and breathing irregularities (episodic hyperventilation, apnea, or breath-holding while awake) were common. These high-quality data establish baselines for clinical understanding and inform therapeutic trials [22,42,43,44].
3.10. Summary of Clinical and CNS Phenotype
In summary, RTT presents with a stereotypical neurological trajectory: normal early development, stagnation by 6–18 months, regression between 1–4 years with loss of hand skills and language, emergence of hand stereotypies, and subsequent stabilization with persistent impairments. Intellectual disability is profound, with 90% of individuals non-verbal. Motor deficits include apraxia, dystonia, bradykinesia, and scoliosis. Communication is largely limited to eye gaze, and stereotypic hand movements affect all patients. Seizures are prevalent, and EEG abnormalities are common. Neuroimaging reveals mild reductions in brain volume (12–34%) but no diagnostic lesions. As shown in Figure 2, Rett syndrome progresses through distinct stages, beginning with early normal development, followed by stagnation, regression with loss of skills and emergence of stereotypies, stabilization with seizures and autonomic disturbances, and later complications such as scoliosis, gastrointestinal dysfunction, and increased caregiver burden in adulthood. The phenotype reflects underlying synaptic and network dysfunction due to MeCP2 deficiency, though retained plasticity offers avenues for potential therapeutic intervention [45,46,47].
4. Beyond the Brain — System-by-System Pathophysiology & Clinical Manifestations
Although RTT is classically defined by its neurological features, MeCP2 is ubiquitously expressed, and non-neuronal organs are measurably affected. The multi-systemic pathophysiology of RTT arises from two primary sources: direct effects of MeCP2 deficiency in non-neuronal tissues and cascading consequences of central autonomic dysfunction originating in the brainstem [27,31,48]. This interconnectedness transforms RTT from a simple neurological disorder into a complex systems-level disease requiring a holistic, multidisciplinary approach [13,18,48]. Below, we review the clinical manifestations, underlying mechanisms from animal and cell studies, and management issues for each system, with prevalence data drawn where available from the Rett Natural History Study (RNHS) and other registries. Table 2 provides an overview of key comorbidities and their approximate prevalence. Management of beyond-brain issues is multidisciplinary, with specialists in pulmonology, cardiology, gastroenterology, orthopedics, endocrinology, sleep medicine, dental care, and others working together to support these children.
4.1. Respiratory Control & Breathing Irregularities
Breathing irregularities affect 70–90% of individuals with RTT, predominantly during wakefulness [5,11,13,35]. Phenotypes include hyperventilation (60%), breath-holding (50–70%), apneas, and forceful expiratory events. These episodes can lead to cyanosis, oxygen desaturation, and seizures, contributing significantly to caregiver burden [49,50].
The pathophysiology involves MeCP2 deficiency within brainstem respiratory centers, including serotonergic and GABAergic circuits in the medulla and pons. iPSC studies indicate mitochondrial dysfunction exacerbates hypoxia, while Mecp2-null mouse models recapitulate spontaneous apneas and hyperventilation [51,52]. Reduced serotonin levels, impaired chemosensitivity, altered neuronal excitability, and glial dysfunction are implicated in the dysregulation. Stress and anxiety can further aggravate these episodes.
Management focuses on supportive measures: diaphragmatic breathing and relaxation techniques, oxygen supplementation, and non-invasive ventilation (BiPAP) for severe sleep apnea. Clinical trials are exploring pharmacological interventions, including 5-HT1A agonists, though results remain preliminary. Aspiration risk (~30%) and systemic oxidative stress are important considerations when managing respiratory dysfunction [53,54].
4.2. Cardiovascular System (Autonomic Dysfunction and Arrhythmia)
Autonomic instability is a hallmark of RTT, affecting 75–80% of patients, and includes erratic heart rate, blood pressure dysregulation, and temperature instability. Heart rate variability studies show reduced parasympathetic tone and relative sympathetic predominance. ECG abnormalities include QTc prolongation (20–30%) and T-wave alterations, correlating with a markedly increased risk of sudden, unexplained death (~25%)[55,56].
Mechanistically, MeCP2 loss disrupts ion channel expression (e.g., K^+ channels like KCNQ1), contributing to arrhythmogenesis. Animal studies demonstrate abnormal baroreflexes, sympathetic overdrive, and increased susceptibility to lethal arrhythmias [17,35,51,56]. Mitochondrial defects may also contribute to cardiac dysfunction.
Clinical management includes annual ECG monitoring, beta-blocker therapy, electrolyte correction, and, in severe cases, pacemaker or defibrillator consideration [57,58]. Retrospective analyses suggest certain sodium-channel-blocking antiepileptics may help normalize QTc intervals, complementing standard therapies.
4.3. Gastrointestinal System & Nutrition
Gastrointestinal (GI) complications are prevalent, with constipation affecting 80–90% of patients, GERD 40–50%, and dysphagia 60-70% [59,60,61]. Delayed gastric emptying and motility disorders lead to chronic malnutrition, growth failure (85%), and low body weight. Autonomic dysfunction and enteric MeCP2 deficiency are primary mechanisms, with iPSC gut organoid studies demonstrating reduced peristalsis.
Management includes dietary modifications (increased fiber and fluids), pharmacological interventions, swallow therapy, and timely enteral feeding (PEG tubes in ~50% of cases). Trofinetide (trade name DAYBUE), the first FDA-approved drug for RTT in 2023 for patients ≥2 years, frequently induces GI side effects (diarrhea in ~80%, vomiting/nausea ~25%), requiring careful titration and anti-diarrheal support. Systemic oxidative stress and microbiome alterations may further exacerbate GI dysfunction [62,63,64,65].
4.4. Skeletal & Musculoskeletal System
Skeletal abnormalities are common and progressive in RTT. Scoliosis affects up to ≥60–85% by 25 years, often requiring surgical intervention in ~30%. Contractures and abnormal gait patterns are frequent due to spasticity and hypotonia transitioning to hypertonia.
Bone health is compromised, with 50–60% exhibiting BMD z-scores ≈45–60% experiencing fragility fractures. Low BMD arises from immobility, poor nutrition (vitamin D/ D/calcium deficiency), anticonvulsant use, and potential direct effects of MeCP2 on osteoblast function. Bisphosphonates, physiotherapy, and nutritional supplementation are key management strategies [66,67,68].
4.5. Metabolic & Mitochondrial Dysfunction
RTT exhibits systemic metabolic abnormalities and mitochondrial dysfunction in ~50% of patients [2,69,70]. Structural mitochondrial defects (enlarged/swollen organelles, irregular cristae) lead to ATP deficiency, oxidative stress, and hypoxia vulnerability. Lactate elevation and dyslipidemia have been reported in subsets, while fibroblast studies show altered bioenergetics and mitochondrial gene expression.
Restoring MeCP2 in preclinical models reduces oxidative stress, indicating partial reversibility [10,44]. Nutritional strategies, coenzyme Q10, L-carnitine, B-vitamin supplementation, and trials of IGF-1 or trofinetide target both neuronal and systemic metabolic deficits [52,69,70]. Ketogenic diets and other metabolic interventions are under investigation.
4.6. Immune System & Glial/Peripheral Immune Interactions
MeCP2 deficiency in glial and peripheral immune cells (astrocytes, microglia, macrophages) contributes to systemic inflammation and non-cell-autonomous neuronal dysfunction [67,70,71,72]. Hyper-responsive microglia release excess TNF-α, IL-6, and glutamate, exacerbating CNS pathology. Peripheral cytokine profiles are also skewed (IL-6, IL-8), and subtle immune dysregulation may manifest as fatigue, eosinophilia, or hyper-IgE.
While gross immunodeficiency is uncommon, neuroimmune crosstalk is a key pathogenic component. Current clinical management is supportive, correcting specific nutritional deficiencies and monitoring for infections [61].
4.7. Endocrine & Growth / Reproductive Health
Growth failure affects ~80% of RTT patients, with microcephaly nearly universal. Poor nutrition, high energy expenditure, and feeding difficulties contribute, alongside endocrine disruption. IGF-1 deficiency is notable, and trofinetide (IGF-1 analog) shows promise in improving growth and neurological outcomes [73,74].
Puberty may be delayed, with early adrenarche and late or absent menarche. Menstrual irregularities, including secondary amenorrhea, are common. Endocrine management focuses on optimizing nutrition, monitoring growth and puberty, and maintaining bone health, potentially with hormone replacement if indicated.
4.8. Sleep, Sensory Systems & Pain
Sleep disturbances affect 70–80% of patients, including insomnia, nocturnal awakenings, sleep-disordered breathing, and bruxism. These issues impair daytime function and caregiver burden. Sensory processing can be altered: some patients show hypoalgesia or hyperalgesia, complicating pain assessment, particularly in non-verbal individuals. Management includes sleep hygiene, melatonin, or low-dose sedatives (trazodone, clonidine)[75,76,77].
4.9. Oral/Dental & Dental Health
Bruxism affects 80% of patients, leading to tooth wear, jaw pain, and oral health challenges [78,79]. High-arched palate, tongue thrust, and poor oral hygiene contribute to dental caries. Management requires early and regular dental visits, preventive care, and customized interventions like mouth guards.
4.10. Other Organ Systems (Renal, Dermatologic, Ophthalmologic)
Renal complications are rare (~10%) but may include UTIs, kidney stones, or urine retention. Dermatologic findings include cold extremities, vasomotor instability, eczema, and Raynaud-like symptoms [80,81]. Ophthalmologic manifestations include strabismus (30%), refractive errors, and occasional cortical visual deficits [82,83]. Retinal changes have been reported in research imaging studies, but are not yet clinically actionable.
While RTT is primarily neurodevelopmental, MeCP2 deficiency impacts virtually every organ system. Effective management requires a multidisciplinary approach encompassing pulmonology, cardiology, gastroenterology, orthopedics, endocrinology, immunology, sleep medicine, dental care, and supportive therapies. Clinical surveillance, proactive interventions, and ongoing research into targeted therapies (IGF-1 analogs, 5-HT1A agonists, metabolic and mitochondrial support) remain essential to improving outcomes and quality of life for patients [44,51,61,84].
5. Biomarkers, Outcome Measures, and Trial Endpoints
A major challenge in conducting clinical trials for RTT is the lack of validated, objective, and sensitive outcome measures and biomarkers [85]. Historically, trial endpoints have relied heavily on subjective scales completed by caregivers or clinicians. The RTT Behavior Questionnaire (RSBQ), a 45-item scale assessing common symptoms, and the Clinical Global Impression-Improvement (CGI-I) scale, which measures overall clinical status, have been widely used in trials [86,87]. While these scales demonstrated statistical significance in the first approved RTT drug trial, they are subjective and may not capture the full scope of a multi-systemic disorder [88,89]. Clinical severity scales such as the Motor Behavioral Assessment (MBA) and the Clinical Severity Score (CSS) quantify global RTT severity by rating key domains, including motor skills, hand use, communication, and autonomic signs, whereas the RSBQ focuses on behaviors such as mood, hyperventilation, and self-injury. These scales are widely used in natural history studies and trials, but are relatively coarse. Objective physiological endpoints have also been explored; breathing irregularities can be quantified using apnea counts or respiratory inductance plethysmography during wakefulness, polysomnography measures sleep quality and nocturnal respiratory events, and cardiac monitoring through ECG or Holter recordings provides arrhythmia and heart rate variability data. Periodic EEGs have been used to assess background rhythm changes, although no specific EEG marker is established as a definitive endpoint, with spike density sometimes serving as a surrogate for neuronal health. Near-term candidate or under-validation clinical endpoints include awake breathing event rates (apnea counts, respiratory inductance plethysmography), heart rate variability (HRV) and QTc interval on ECG/Holter recordings, sleep quality and nocturnal respiratory metrics via polysomnography, and caregiver-reported frequency of purposeful hand use. There is a critical need for reliable molecular biomarkers to provide objective measures of disease progression and therapeutic response. Candidate molecular biomarkers under investigation include serum or cerebrospinal fluid (CSF) levels of MeCP2-regulated proteins such as BDNF and IGF-1, neurotransmitter metabolites, certain miRNAs or lipids, blood markers of oxidative stress or inflammation, and CSF MeCP2 levels. Exploratory biomarkers currently under validation include the BDNF/IGF-1 axis (serum/CSF levels), circulating cytokines and inflammatory mediators, neurotransmitter metabolites, miRNA and lipidomic signatures, metabolomics-based readouts of systemic dysfunction, and blood-based oxidative stress markers. Neuroimaging biomarkers, such as brain volume on MRI, diffusion tensor imaging metrics, and MR spectroscopy metabolites, are experimental but promising, with high-resolution MRI and PET offering further insights despite requiring sedation in young children. Digital wearables are emerging for real-time monitoring of breathing and seizures, complementing these physiological and molecular measures. Caregiver-reported outcomes, including daily logs and quality-of-life questionnaires such as the Pediatric Quality of Life Inventory (PedsQL) and Caregiver Strain Index, capture patients’ overall function and the multidimensional impact. Recently, an international workshop of clinicians, researchers, and families highlighted the need for “fit-for-purpose” endpoints, emphasizing that no single measure is sufficient and that trials should include a composite of scales, biomarkers, and caregiver assessments focused on the most meaningful outcomes such as hand function, communication, seizure control, and safety [90,91,92]. Candidate endpoints derived from the Rett Natural History Study (RNHS) include post-regression head growth velocity, frequency of hand use, and breathing event rates. Trial-readiness initiatives now also include digital wearables for breathing and seizure monitoring. Ultimately, validating new biomarkers—imaging- or blood-based—that correlate with clinical state remains a priority, and multi-modal endpoints integrating clinical, physiological, molecular, and caregiver-reported measures are essential for sensitive, objective, and reliable evaluation of therapeutic efficacy in RTT.
6. Models and Mechanistic Tools (Preclinical & Translational Platforms)
6.1. Rodent Models of RTT
Preclinical research into RTT has heavily relied on rodent models, particularly mice, which have been instrumental in elucidating disease mechanisms and testing therapeutic strategies. Mecp2-null, heterozygous, and transgenic mouse models have successfully recapitulated many core neurological and behavioral features of RTT, including regression, stereotypies, seizures, tremors, gait ataxia, and breathing abnormalities[31]. The hemizygous Mecp2^–/Y mice appear normal at birth but develop progressive neurological deficits by 4–6 weeks, culminating in death around 10–12 weeks. Heterozygous females (Mecp2+/–) model the mosaicism of human RTT females, displaying a milder, later-onset phenotype with motor and respiratory deficits over months. Conditional knockouts, in which Mecp2 is selectively deleted in specific cell types, have revealed cell-specific roles: neuronal deletions largely reproduce RTT-like behavior, whereas astrocyte deletions produce subtler effects, and microglial deletions affect synaptic pruning. These findings highlight neurons as the critical site of pathology [93,94].
Transgenic overexpression models have also been generated to study MECP2 duplication syndrome, which causes hypotonia, motor deficits, and epilepsy. These models underscore that both insufficient and excessive MeCP2 are deleterious and have been used to test antisense oligonucleotide (ASO) therapies aimed at reducing MeCP2 expression. Rat models, developed more recently, provide advantages such as larger brains for intracerebroventricular (ICV) drug delivery and neurophysiologic monitoring. While non-human primate (NHP) or other large-animal models are limited, 2025 efforts aim to explore biodistribution and human-like physiology if feasible [95,96].
Mouse and rat models have been pivotal in demonstrating that adult plasticity allows reversal of RTT phenotypes following Mecp2 reactivation, highlighting that therapeutic interventions need not be limited to neonatal stages. Many candidate therapies—including IGF-1, BDNF mimetics, and gene therapy—have been tested in these models, yielding critical insights into neurotransmitter alterations, synaptic function, and histological changes.
6.2. Human Cell-Based Models
To overcome species differences inherent in rodent models and bridge the gap to human pathology, patient-derived induced pluripotent stem cells (iPSCs) have emerged as powerful tools [97]. The iPSCs can be differentiated into neurons, astrocytes, and microglia, allowing in vitro investigation of human-specific disease mechanisms such as dendritic branching deficits, synaptic impairments, and altered gene expression profiles. Isogenic control lines, genetically identical except for the MECP2 mutation, provide a high-fidelity system to study mutation-specific effects [97,98,99].
Three-dimensional cerebral organoids derived from RTT iPSCs further recapitulate aspects of tissue architecture and cellular complexity in the developing human brain. These “mini-brains” enable studies of early neurodevelopment, neuronal network activity, and abnormal connectivity, providing physiologically relevant platforms for drug screening and biomarker discovery. Although organoids lack full circuit maturation, they consistently reflect RTT-associated phenotypes, such as impaired neuron growth and synaptic connectivity [100,101,102].
6.3. Molecular and Multi-Omic Insights
Advances in high-resolution molecular profiling have greatly enhanced the understanding of RTT pathophysiology. Single-cell transcriptomics, proteomics, and metabolomics reveal cell-type-specific alterations in gene expression, protein abundance, and metabolic pathways that contribute to disease [103,104,105]. These studies identify isoform-specific roles of MeCP2, elucidate downstream molecular cascades, and facilitate the discovery of objective biomarkers of disease state or severity. Integrating multi-omic data enables stratification of patients for personalized therapeutic approaches, moving beyond the causal gene to a comprehensive understanding of molecular dysfunction.
6.4. Comparative Strengths and Limitations of Models
Each model system offers unique advantages and limitations. Mouse models provide controlled genetics, reproducibility, and in vivo testing, but cannot capture certain human features, such as speech. iPSC-derived neurons and glia offer human-specific mechanistic insights but lack whole-body context. “As summarized in Table 3, various preclinical and translational model systems for MECP2-related disorders are compared in terms of their genetic alterations, key features, strengths, limitations, and recent inferences (2024–2025). This comparison highlights the utility and constraints of each model in studying disease mechanisms and evaluating potential therapeutic strategies. Cerebral organoids emulate aspects of early brain development and network formation but display variability and incomplete maturation. Despite these limitations, convergence of findings across models—such as synaptic dysfunction, impaired plasticity, and metabolic deficits—strengthens confidence in mechanistic inferences and therapeutic targets. Notably, the profound phenotypic rescue achieved by Mecp2 reactivation in adult mice underlines the translational relevance of these preclinical models [106,107].
7. Therapeutic Landscape: Symptom Control to Disease Modification
7.1. Standard Supportive Care
Management of Rett syndrome (RTT) centers on comprehensive symptomatic and supportive care. Because RTT affects virtually every organ system, a multidisciplinary approach is essential, involving pediatric neurologists, pulmonologists, gastroenterologists, orthopedists, endocrinologists, dietitians, and therapists (physical, occupational, and speech) [67,90]. As shown in Figure 3, the timeline highlights key milestones in Rett syndrome research, from the discovery of the MECP2 gene to the recent development of gene therapy approaches. Respiratory support includes monitoring children for breath-holding episodes and sleep apnea, with severe episodes potentially requiring supplemental oxygen or manual ventilation support. Families are trained to recognize and manage apnea and clear secretions, and vaccination against respiratory pathogens, including influenza and pneumococcus, is recommended. Cough assist devices are used to prevent pneumonia. Seizure management involves prescribing antiepileptic drugs (AEDs) based on seizure type, with valproate, levetiracetam, or lamotrigine commonly used, although response is often incomplete. Benzodiazepines may be employed for refractory status epilepticus, and vagus nerve stimulation has been trialed in a few cases with limited data. Certain drugs, such as sodium channel blockers, may exacerbate gait or breathing issues, necessitating individualized therapy. EEG monitoring guides treatment, and the ketogenic diet may be considered for drug-resistant epilepsy, although data in RTT remain anecdotal [92,107,108]. Gastrointestinal care and nutrition involve aggressive management of constipation using laxatives such as polyethylene glycol, senna, and stool softeners, while liquid polyethylene glycol can simultaneously address constipation and nutritional needs. Gastroesophageal reflux disease (GERD) is treated with acid suppression via proton-pump inhibitors and feeding modifications, including thickened feeds and positioning. Swallowing therapy from speech-language pathologists prevents aspiration, and texture-modified diets are used as necessary. When weight falters, gastrostomy tube feeding ensures stable nutrition, reduces aspiration risk, and improves growth parameters. Dietitians calculate caloric requirements (often 110–120 kcal/kg/day due to hypermetabolism) and monitor micronutrients, supplementing vitamin D, calcium, and multivitamins as needed, while weight-bearing activities such as standing frames support muscle tone and bone health [59,109]. Orthopedic and neuromotor care includes early initiation of physical and occupational therapy to maintain mobility and prevent contractures, stretching exercises for ankles and knees, use of orthoses such as ankle-foot orthoses, and hip surveillance imaging. Scoliosis may require instrumented spinal fusion, which often improves pulmonary function and sitting balance, and caregivers may employ transfer devices such as Hoyer lifts for safe handling in later childhood [107]. Communication and developmental support rely on augmentative and alternative communication (AAC) strategies, including eye-tracking technology, picture boards, and gestures, while early intervention programs optimize cognitive and social development. Behavioral and psychological support for anxiety or mood swings may involve low-dose SSRIs or alpha-agonists, though evidence is limited [92,109]. Dental care and other considerations include proactive dental cleanings, fluoride use, and management of bruxism. Finally, caregiver support encompasses comprehensive care plans, respite services, and caregiver education, with palliative care principles applied to focus on quality of life and symptom relief; severe agitation or pain may occasionally be treated with low-dose opioids or neuromodulators [90,91].
7.2. Approved Pharmacotherapy: Trofinetide (DAYBUE™)
Trofinetide, a synthetic analog of the N-terminal tripeptide of IGF-1, became the first FDA-approved therapy for Rett syndrome (RTT) in 2023 for patients aged ≥2 years. Its design was inspired by IGF-1 trials and observations that Mecp2-deficient neurons exhibit reduced IGF-1 signaling. Trofinetide appears to promote neuronal growth and synaptic function, although its precise mechanism of action is not fully elucidated. In pivotal clinical trials involving patients aged 5–20 years, trofinetide demonstrated statistically significant improvements on coprimary endpoints, including caregiver-rated behavior scales (RSBQ) and the clinical global impression of improvement (CGI-I). Secondary outcomes, such as communication and motor behavior, exhibited positive trends but frequently did not reach statistical significance [110,111]. Real-world studies conducted between 2024 and 2025 reported behavioral gains of 20–50% as noted by caregivers, contributing to improvements in overall quality of life. Gastrointestinal adverse events were the most commonly observed, with diarrhea occurring in over 80% of patients compared to less than 20% on placebo, nausea in approximately 25%, and occasional weight loss or decreased appetite. These side effects can be mitigated through dose titration and the use of anti-diarrheal or anti-nausea agents, with nutritional monitoring recommended throughout treatment. Access to trofinetide remains constrained by its high cost, amounting to hundreds of thousands of USD per year, and limited global availability, with insurance coverage and patient assistance programs influencing patient access. Despite its modest efficacy, trofinetide represents the first disease-directed therapy for RTT.
7.3. Gene Replacement / Gene Therapy
Gene therapy for Rett syndrome aims to deliver a functional copy of MECP2 to neurons using adeno-associated virus (AAV) vectors, such as NGN-401 and TSHA-102, representing a potential disease-modifying approach [112,113,114]. Preclinical studies in Mecp2-deficient mice demonstrated that intravenous or intracerebroventricular delivery of AAV9-Mecp2 improved survival, motor function, and respiratory parameters. However, MECP2 is dosage-sensitive, and overexpression can lead to toxicity, as observed in MECP2 duplication syndrome. Consequently, dose titration is critical, and regulatory elements, including EXACT™ and miRARE, are employed to control expression. Early-stage AAV-MECP2 programs have shown a narrow therapeutic window, highlighting the importance of precise dose-finding and expression control to ensure safety. Clinical trials indicate that low-dose NGN-401 is safe, with interim efficacy demonstrating RSBQ improvements ranging from 28–52%. Safety monitoring in these trials includes immune surveillance and hepatic enzyme assessment [51,115]. Translating preclinical success to humans involves significant challenges, including achieving sufficient CNS penetration, maintaining long-term expression control, mitigating cardiopulmonary risks, and manufacturing high-titer AAV vectors. Consequently, early-phase trials remain cautious, often prioritizing patients with severe phenotypes or early diagnosis. Alternative strategies, including non-viral gene delivery approaches using nanoparticles or exosomes, as well as gene editing tools such as CRISPR and base editors, are under investigation but remain experimental.
7.4. Antisense Oligonucleotides and Dosage Normalization
ASOs are being investigated to normalize MECP2 levels in duplication syndromes and may inform RTT therapies. Preclinical studies in MDS mice demonstrated phenotypic improvement with MECP2 knockdown. Early 2024 data support precise titration strategies to avoid inducing RTT-like symptoms. Lessons from ASO development—including intrathecal delivery, off-target monitoring, and durability assessment—guide all genetic approaches in RTT [116,117].
7.5. Small Molecules, Neurotrophic Strategies, and Repurposing
Pharmacologic agents for Rett syndrome (RTT) primarily target disease pathophysiology or provide symptomatic relief. Agents acting on the IGF-1 pathway, such as trofinetide and full-length IGF-1 (mecasermin), have been shown in preclinical studies to cross the blood-brain barrier, improving gait and lifespan in mice [110], however, placebo-controlled IGF-1 trials have indicated potential breathing issues, necessitating further evaluation. The BDNF pathway is regulated by MeCP2, and enhancing BDNF signaling with small molecules, including LM22A-4 and selective serotonin reuptake inhibitors (SSRIs), has improved outcomes in murine models, although translation to humans remains limited [73]. Serotonergic agents, including SSRIs or 5-HT_1A agonists such as sarizotan, aim to modulate mood and respiratory function, but large clinical trials have failed to demonstrate efficacy; nonetheless, some 5-HT modulators are used off-label to manage gastrointestinal or behavioral symptoms. Synaptic modulators, including NMDA receptor antagonists (memantine, ketamine) and stimulants such as modafinil, have been investigated for behavioral and alertness improvements, yet results remain inconclusive. Metabolic modulators, including supplements such as CoQ10, L-carnitine, and triheptanoin, exhibit minimal clinical effects, while newer agents, including leriglitazone (2025), are currently under investigation. Symptomatic management is achieved through low-dose benzodiazepines or SSRIs for anxiety and mood disturbances, glycopyrrolate for drooling, and baclofen or diazepam for spasticity. To date, trofinetide represents the only pharmacotherapy demonstrating disease-modifying potential, whereas other agents, including sigma-1 receptor agonists (blarcamesine) and neuromuscular modulators (ezogabine), are in early Phase 2/3 clinical trials [109,117,118].
7.6. Cellular and Other Novel Strategies
Emerging interventions for neurological disorders include stem cell therapy, exosome-based approaches, and neuromodulation. Neural precursor or mesenchymal stem cell (MSC) transplantation aims to provide trophic support, with early studies in mouse models demonstrating transient benefits; however, translation to humans remains speculative and is associated with safety concerns. Exosomes, derived from MSCs or other cell types, have the potential to deliver RNAs and proteins to modulate neuronal growth or inflammation, but these approaches are still in the early discovery phase. Neuromodulation strategies, including trigeminal nerve stimulation and vagus nerve stimulation, have shown anecdotal improvements in alertness or breathing, although controlled clinical trials are required to validate these observations.
7.7. Clinical Trial Design and Operational Considerations
Rett syndrome (RTT) trials face several unique challenges. Phenotypic heterogeneity, including variations in age, mutation type, and X-chromosome inactivation, necessitates careful stratification or randomization of participants. Small patient populations, due to the ultra-rare nature of the disease, limit sample sizes, with international trials often enrolling only tens of patients; innovative designs such as Bayesian adaptive trials and N-of-1 trials have been employed to address this limitation. Outcome sensitivity is a critical consideration, as standard measures may lack the precision needed to detect subtle changes; consequently, natural history studies (RNHS) and wearable monitoring devices are increasingly utilized to track nuanced clinical outcomes. Safety monitoring is essential, particularly for gene and novel therapies, given MECP2’s narrow therapeutic window. Ethical considerations are paramount, as pediatric patients and cognitively impaired individuals require guardian consent, and the progressive nature of the disease raises concerns regarding placebo use. Finally, global access remains limited, with most trials conducted in North America and Europe; international collaboration and registry sharing, such as through the RTT Global Registry, are critical to ensure diverse patient inclusion.
This structure maintains all scientific details, data points, references, clinical observations, and trial results, while presenting a clear flow from supportive care to cutting-edge therapies and trial design [88]. An overview of current and emerging therapeutic approaches in RTT, along with their mechanisms and efficacy, is summarized in Table 4.
Therapy design principles for RTT emphasize precise dose control to regulate MECP2 and pathway-targeting therapies and avoid toxicity, brain-wide coverage to ensure adequate distribution across affected neuronal populations, and consideration of timing and age of intervention, with earlier initiation correlating with better outcomes. Isoform targeting, accounting for MECP2 isoforms and cell-type specificity, and vigilant monitoring of peripheral safety to detect systemic and off-target effects are also critical components of effective therapeutic strategies.
8. Safety & Regulatory Considerations
The development of novel therapies for RTT, particularly AAV-based gene therapies, introduces significant safety and regulatory considerations, including immunogenicity, dose-dependent toxicity, biodistribution concerns, and unintended off-target effects. AAV vectors can trigger immune responses, especially in patients with pre-existing neutralizing antibodies, requiring pre-enrollment screening for antibody titers. Acute reactions such as cytokine release and organ toxicity, notably liver injury, have been observed in high-dose AAV trials for other diseases, highlighting the need for careful stepwise dosing and monitoring of liver function tests and inflammatory markers. Long-term follow-up is essential to detect delayed adverse events, including rare risks such as insertional mutagenesis and unforeseen effects of MECP2 expression in peripheral cells, with current guidelines recommending up to 15 years of surveillance due to the permanent nature of AAV therapy. Small-molecule therapies such as trofinetide, which has received Orphan Drug and Rare Pediatric Disease designations, emphasize careful patient selection (ages 5–20) and monitoring for gastrointestinal adverse events, with post-marketing surveillance tracking efficacy and safety in the wider population. Other emerging agents, like Blarcamesine, similarly benefit from Orphan and Fast-Track designations, but require attention to off-target CNS effects, liver enzyme elevations, and potential drug interactions. Regulatory agencies, including the FDA and EMA, actively engage with RTT therapies, especially MECP2 gene therapy, due to the risk of overexpression, recommending low initial doses and built-in stopping rules for first-in-human trials. Patient advocacy groups (IRSF, RSRT) have played a crucial role in defining trial endpoints and guiding therapy development, exemplified by the breakthrough designation of trofinetide. Overall, the regulatory framework promotes patient-centered endpoints while emphasizing meticulous safety assessments, long-term monitoring, and responsible risk–benefit evaluation, with ultimate responsibility resting on clinical researchers and therapy developers [119,120].
9. Quality of Life, Caregiver Burden, and Health-Services Issues
RTT has a profound and pervasive impact on patients and their caregivers, significantly affecting quality of life [90,91]. The care required ranges from basic daily living activities to complex medical needs, is continuous, and often exceeds the capacity of standard health services[88]. Caregivers, often mothers, experience high levels of physical, emotional, and social burden, with surveys in 2024 reporting high Zarit scores (>30) and substantial mental health strain [115,121,122]. The inability of individuals with RTT to communicate pain or discomfort adds further anxiety to routine care.20 Daily tasks, including feeding, toileting, and mobility assistance, often require 24/7 attention, and sleep disturbances in children contribute to parental fatigue, anxiety, and depression. RTT health services vary by region: in countries with specialized clinics (US, Europe, Australia), multidisciplinary teams and supportive therapies are accessible, whereas in other areas, care may be fragmented, with children sometimes misdiagnosed with cerebral palsy or autism and missing the benefits of targeted interventions such as scoliosis monitoring or gastrostomy planning. Early genetic diagnosis increasingly enables timely access to resources. Transition from pediatric to adult care poses additional challenges, as adult specialists are often less familiar with RTT, leading to gaps in long-term management of orthopedic, seizure, and cardiac issues, though multidisciplinary clinics can reduce caregiver burden by ~20%. RNHS surveys highlight these adult-care gaps, emphasizing the need for dedicated adult RTT clinics and guidelines for lifelong management. Palliative care, extending beyond end-of-life care, involves holistic symptom management and family support, including feeding tube discussions, advanced care planning for respiratory illnesses, and pain management, with ethical considerations around aggressive interventions versus comfort care. Professional guidelines advocate informed, family-centered decision-making that respects patient and caregiver values, integrating pediatric palliative principles into adult care. Effective RTT management requires a collaborative, team-based approach involving multiple specialists, advocacy organizations, and social support networks, which together enhance patient quality of life and mitigate caregiver burden [123].
10. Global Health, Equity & Access to Specialized Care
RTT occurs worldwide, yet its diagnosis and management are highly uneven across regions. In high-income countries, genetic testing is standard when RTT is suspected, and specialized multidisciplinary centers provide comprehensive care, whereas in low- and middle-income countries (LMICs), limited awareness, lack of testing, and insufficient specialized care result in many girls being unrecognized or misdiagnosed, with delays often exceeding five years. Estimates suggest hundreds of thousands of individuals with RTT exist globally, but many have never been formally diagnosed, hindering epidemiological studies and equitable enrollment in clinical trials. Access to approved therapies, such as trofinetide, or emerging interventions like gene therapy, remains restricted in LMICs due to cost and infrastructure limitations. Even in high-income countries, obtaining treatments can involve lengthy insurance approvals, and families outside major metropolitan areas may need to travel long distances to reach RTT centers; telemedicine has partially mitigated these barriers, especially during the COVID-19 pandemic, by enabling remote consultation with RTT experts. Addressing these inequities is a moral and ethical imperative, requiring policy interventions and international collaboration. Recommended strategies include universal newborn screening when cost-effective, training healthcare workers to recognize RTT, educational campaigns to reduce late diagnoses, and advocacy for subsidized or compassionate access to new treatments. International registries, such as the RTT Global Registry, consolidate knowledge, enable data sharing, and facilitate global access to research and clinical trials, while rare disease initiatives can ensure funding for research, clinical support, and infrastructure development. Telemedicine expansion, registry participation, and policy-driven universal screening have been emphasized in the 2025 recommendations to reduce disparities. Achieving equity in RTT care, therefore, demands a multi-faceted approach, integrating improved diagnosis, broader access to supportive services, and innovative solutions to make new therapies affordable and available worldwide, ensuring that all individuals with RTT, regardless of geographic or socioeconomic status, benefit from advancements in the field [124,125].
11. Gaps, Controversies & Prioritized Research Agenda
Despite significant progress, many questions in Rett syndrome (RTT) remain unanswered. A critical gap exists in the identification of validated peripheral biomarkers, including blood metabolites, cytokines, or imaging signals, which accurately reflect central nervous system (CNS) MeCP2 function, disease progression, and therapeutic response. Although multi-omics studies have identified promising candidates, further validation is required.45 Safe and controllable systemic delivery of MECP2 remains essential, as recent trial failures emphasize the need to achieve functional expression without toxicity. Approaches under investigation include non-viral vectors, precise gene-editing techniques, and inducible or self-regulating expression systems [112]. Given the multi-systemic nature of RTT, clinical trials should integrate endpoints beyond cognitive and motor scales, incorporating measures such as respiratory event rates, ECG changes, gastrointestinal function, and other peripheral assessments to comprehensively evaluate therapeutic efficacy [69,92,126]. Phenotypic heterogeneity driven by MECP2 mutation type and X-chromosome inactivation (XCI) patterns further complicates trial design, underscoring the importance of stratification by genotype and XCI for precision medicine approaches and predicting treatment outcomes [118,122]. Considering RTT’s lifelong trajectory, long-term follow-up and the establishment of global registries are necessary to track disease progression, aging-related complications—including Parkinsonian features and osteoporosis—and the real-world safety and effectiveness of both approved and emerging therapies [34]. Research has predominantly focused on pediatric females, highlighting the need for dedicated studies in adult females and mosaic males, who exhibit distinct clinical trajectories, therapeutic responses, and safety considerations [55,69]. The molecular mechanisms underlying non-neuronal pathology, including cardiomyopathy, immune dysregulation, and gastrointestinal dysfunction, remain poorly understood and necessitate further basic mechanistic research. Current mouse models have limitations in recapitulating human RTT, and the development of engineered large animal or primate models is critical for translational studies and systemic evaluation of therapies. Exploration of combination therapies integrating symptomatic and disease-modifying treatments, as well as gene reactivation approaches to unsilence the inactive X chromosome, offers potential for improved outcomes but remains largely untested. Equally important are considerations of global equity in diagnostics, therapies, and trials, along with the incorporation of patient and family input to define meaningful outcomes. Finally, ongoing controversies include the extent of symptom reversibility and isoform-specific targeting, while emerging priorities involve omics-based RTT subtype characterization and artificial intelligence-driven multi-system endpoint analysis for precise therapeutic evaluation.
12. Conclusions
RTT caused by mutations in the MECP2 gene is a complex monogenic disorder that extends far beyond the central nervous system, affecting respiratory, cardiac, gastrointestinal, skeletal, and metabolic systems, thereby necessitating a holistic, “whole-person” perspective. MeCP2 functions as a global epigenetic regulator, and its absence results in widespread gene expression changes; preclinical studies have shown that restoring MeCP2, even in adulthood, can reverse disease phenotypes, highlighting the remarkable reversibility of RTT. The recent FDA approval of trofinetide provides symptomatic relief and represents a historic milestone, while gene therapies hold promise for disease modification, though clinical translation is challenged by MeCP2’s narrow therapeutic window, necessitating precise dosage control, safe delivery mechanisms, and integration of objective biomarkers. Effective RTT management will require combinatorial strategies addressing both central and peripheral dysfunctions, supported by robust natural history studies, well-characterized outcome measures, and global collaboration. By prioritizing multi-systemic clinical trials, equitable access, and biomarker-guided interventions, the field aims not only to extend life but also to improve communication, health, and overall quality of life, offering a tangible path toward meaningful disease modification and potentially curative therapies.
13. Declaration
13.1. Funding
The author(s) acknowledge the funds received by Uddalak Das from the Department of Biotechnology (Grant No: DBTHRDPMU/JRF/BET-24/I/2024-25/376), the Council of Scientific and Industrial Research (Grant No: 24J/01/00130), and the Indian Council of Medical Research (Grant No: 3/1/3/BRET-2024/HRD (L1)).
13.1. Authorship Contribution Statement
Madhumita Saha: Conceptualization, Validation, Writing Original Draft, Writing Review & Editing, Visualization, Supervision, Project Administration; Sahil Bhardwaj: Writing Original Draft, Writing Review & Editing, Visualization; Kesavaperumal Gopalakrishnan: Writing Review & Editing; Tanveen Kaur Soni: Writing Original Draft, Writing Review & Editing; Samer Shamshad: Writing - Review & Editing; Uddalak Das: Conceptualization, Project Administration, Supervision, Funding Acquisition, Review & Editing. All authors critically revised the paper and approved the final version
13.1. Declaration of Competing Interest
The author(s) report no conflict of interest.
13.1. Acknowledgement
None
13.1. Ethical Statements
None
13.1. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
The writing of this review paper involved the use of generative AI and AI-assisted technologies only to enhance the clarity, coherence, and overall quality of the manuscript. The authors acknowledge the contributions of AI in the writing process while ensuring that the final content reflects the author's own insights and interpretations of the literature. All interpretations and conclusions drawn in this manuscript are the sole responsibility of the author.
References
- Bricker K, Vaughn BV. Rett syndrome: a review of clinical manifestations and therapeutic approaches. Front Sleep [Internet]. 2024 May 21 [cited 2025 Sep 3];3. Available from: https://www.frontiersin.org/journals/sleep/articles/10.3389/frsle.2024.1373489/full.
- Kyle SM, Vashi N, Justice MJ. Rett syndrome: a neurological disorder with metabolic components. Open Biol. 2018 Feb;8(2):170216. [CrossRef]
- May D, Kponee-Shovein K, Neul JL, Percy AK, Mahendran M, Downes N, Chen G, Watson T, Pichard DC, Kennedy M, Lefebvre P. Characterizing the journey of Rett syndrome among females in the United States: a real-world evidence study using the Rett syndrome natural history study database. J Neurodev Disord. 2024 Jul 26;16(1):42. [CrossRef]
- Pejhan S, Rastegar M. Role of DNA Methyl-CpG-Binding Protein MeCP2 in Rett Syndrome Pathobiology and Mechanism of Disease. Biomolecules. 2021 Jan 8;11(1):75. [CrossRef]
- Gold WA, Percy AK, Neul JL, Cobb SR, Pozzo-Miller L, Issar JK, Ben-Zeev B, Vignoli A, Kaufmann WE. Rett syndrome. Nat Rev Dis Primer. 2024 Nov 7;10(1):84.
- Carvalho MR de, Cavalcante TT, Oliveira PS, Naves PVF, Cunha PEL. Rett syndrome due to mutation in the MECP2 gene and electroencephalographic findings. Arq Neuropsiquiatr. 2024 Aug;82(8):1–2.
- Ribeiro MC, MacDonald JL. Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 2020 Feb 15;1729:146644. [CrossRef]
- Buchanan CB, Stallworth JL, Scott AE, Glaze DG, Lane JB, Skinner SA, Tierney AE, Percy AK, Neul JL, Kaufmann WE. Behavioral profiles in Rett syndrome: Data from the natural history study. Brain Dev. 2019 Feb 1;41(2):123–34. [CrossRef]
- Percy AK, Benke TA, Marsh ED, Neul JL. Rett syndrome: The Natural History Study journey. Ann Child Neurol Soc. 2024;2(3):189–205.
- Chin EWM, Goh ELK. MeCP2 Dysfunction in Rett Syndrome and Neuropsychiatric Disorders. Methods Mol Biol Clifton NJ. 2019;2011:573–91.
- Vidal S, Xiol C, Pascual-Alonso A, O’Callaghan M, Pineda M, Armstrong J. Genetic Landscape of Rett Syndrome Spectrum: Improvements and Challenges. Int J Mol Sci. 2019 Aug 12;20(16):3925. [CrossRef]
- Fang X, Baggett LM, Caylor RC, Percy AK, Neul JL, Lane JB, Glaze DG, Benke TA, Marsh ED, Motil KJ, Barrish JO, Annese FE, Skinner SA. Parental age effects and Rett syndrome. Am J Med Genet A. 2024 Feb;194(2):160–73. [CrossRef]
- Kim JA, Kwon WK, Kim JW, Jang JH. Variation spectrum of MECP2 in Korean patients with Rett and Rett-like syndrome: a literature review and reevaluation of variants based on the ClinGen guideline. J Hum Genet. 2022 Oct;67(10):601–6.
- Ip JPK, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nat Rev Neurosci. 2018 Jun;19(6):368–82. [CrossRef]
- Liu Y, Flamier A, Bell GW, Diao AJ, Whitfield TW, Wang HC, Wu Y, Schulte F, Friesen M, Guo R, Mitalipova M, Liu XS, Vos SM, Young RA, Jaenisch R. MECP2 directly interacts with RNA polymerase II to modulate transcription in human neurons. Neuron. 2024 Jun 19;112(12):1943-1958.e10. [CrossRef]
- Ali NE, Tariq N, Naz G, Abalkhail A, Kausar T, Mazhar I, Zia S, Aqib AI, Khan NU. Rett syndrome: advances in Understanding MeCP2 function, potential gene therapies, and public health implications. Mol Biol Rep. 2025 Jul 8;52(1):687.
- Golubiani G, van Agen L, Tsverava L, Solomonia R, Müller M. Mitochondrial Proteome Changes in Rett Syndrome. Biology. 2023 Jul 3;12(7):956.
- Pascual-Alonso A, Xiol C, Smirnov D, Kopajtich R, Prokisch H, Armstrong J. Identification of molecular signatures and pathways involved in Rett syndrome using a multi-omics approach. Hum Genomics. 2023 Sep 15;17(1):85. [CrossRef]
- Qian J, Guan X, Xie B, Xu C, Niu J, Tang X, Li CH, Colecraft HM, Jaenisch R, Liu XS. Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Sci Transl Med. 2023 Jan 18;15(679):eadd4666.
- Collins BE, Neul JL. Rett Syndrome and MECP2 Duplication Syndrome: Disorders of MeCP2 Dosage. Neuropsychiatr Dis Treat. 2022;18:2813–35.
- Zahorakova D, Lelkova P, Gregor V, Magner M, Zeman J, Martasek P. MECP2 mutations in Czech patients with Rett syndrome and Rett-like phenotypes: novel mutations, genotype-phenotype correlations and validation of high-resolution melting analysis for mutation scanning. J Hum Genet. 2016 Jul;61(7):617–25. [CrossRef]
- Abo Zeid M, Elrosasy A, Mohamed RG, Ghazou A, Goufa E, Hassan N, Abuzaid Y. A meta-analysis of the efficacy and safety of trofinetide in patients with rett syndrome. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. 2024 Oct;45(10):4767–78.
- Lou S, DJiake Tihagam R, Wasko UN, Equbal Z, Venkatesan S, Braczyk K, Przanowski P, Il Koo B, Saltani I, Singh AT, Likhite S, Powers S, Souza GMPR, Maxwell RA, Yu J, Zhu LJ, Beenhakker M, Abbott SBG, Lu Z, Green MR, Meyer KC, Tushir-Singh J, Bhatnagar S. Targeting microRNA-dependent control of X chromosome inactivation improves the Rett Syndrome phenotype. Nat Commun. 2025 Jul 4;16(1):6169. [CrossRef]
- Takahashi S, Takeguchi R, Kuroda M, Tanaka R. Atypical Rett syndrome in a girl with mosaic triple X and MECP2 variant. Mol Genet Genomic Med. 2020 Mar;8(3):e1122. [CrossRef]
- Collins BE, Merritt JK, Erickson KR, Neul JL. Safety and efficacy of genetic MECP2 supplementation in the R294X mouse model of Rett syndrome. Genes Brain Behav. 2022 Jan;21(1):e12739.
- Powers S, Likhite S, Gadalla KK, Miranda CJ, Huffenberger AJ, Dennys C, Foust KD, Morales P, Pierson CR, Rinaldi F, Perry S, Bolon B, Wein N, Cobb S, Kaspar BK, Meyer KC. Novel MECP2 gene therapy is effective in a multicenter study using two mouse models of Rett syndrome and is safe in non-human primates. Mol Ther J Am Soc Gene Ther. 2023 Sep 6;31(9):2767–82. [CrossRef]
- Akaba Y, Takahashi S. MECP2 duplication syndrome: Recent advances in pathophysiology and therapeutic perspectives. Brain Dev. 2025 Aug;47(4):104371.
- Chen X, Han X, Blanchi B, Guan W, Ge W, Yu YC, Sun YE. Graded and pan-neural disease phenotypes of Rett Syndrome linked with dosage of functional MeCP2. Protein Cell. 2021 Aug;12(8):639–52.
- Haase F, Gloss BS, Tam PPL, Gold WA. WGCNA Identifies Translational and Proteasome-Ubiquitin Dysfunction in Rett Syndrome. Int J Mol Sci. 2021 Sep 15;22(18):9954. [CrossRef]
- Anitha A, Poovathinal SA, Viswambharan V, Thanseem I, Iype M, Anoop U, Sumitha PS, Parakkal R, Vasu MM. MECP2 Mutations in the Rett Syndrome Patients from South India. Neurol India. 2022;70(1):249–53. [CrossRef]
- Chin EWM, Goh ELK. Behavioral Characterization of MeCP2 Dysfunction-Associated Rett Syndrome and Neuropsychiatric Disorders. Methods Mol Biol Clifton NJ. 2019;2011:593–605.
- Li Y, Anderson AG, Qi G, Wu SR, Revelli JP, Liu Z, Zoghbi HY. Early transcriptional signatures of MeCP2 positive and negative cells in Rett syndrome. BioRxiv Prepr Serv Biol. 2025 Jun 26;2025.06.26.661761.
- Müller M. Disturbed redox homeostasis and oxidative stress: Potential players in the developmental regression in Rett syndrome. Neurosci Biobehav Rev. 2019 Mar;98:154–63. [CrossRef]
- Chin Wong L, Hung PL, Jan TY, Lee WT, Taiwan Rett Syndrome Association. Variations of stereotypies in individuals with Rett syndrome: A nationwide cross-sectional study in Taiwan. Autism Res Off J Int Soc Autism Res. 2017 Jul;10(7):1204–14.
- Lazek R, Karoum A, Fathalla W. Genotype-Phenotype Correlation and Therapeutic Amenability in a Cohort of Rett Syndrome Patients: A Single-Center Study. Cureus. 2025 Jun;17(6):e86953.
- Zhou J, Cattoglio C, Shao Y, Tirumala HP, Vetralla C, Bajikar SS, Li Y, Chen H, Wang Q, Wu Z, Tang B, Zahabiyon M, Bajic A, Meng X, Ferrie JJ, LaGrone A, Zhang P, Kim JJ, Tang J, Liu Z, Darzacq X, Heintz N, Tjian R, Zoghbi HY. A novel pathogenic mutation of MeCP2 impairs chromatin association independent of protein levels. Genes Dev. 2023 Oct 1;37(19–20):883–900. [CrossRef]
- Bahram Sangani N, Koetsier J, Gomes AR, Diogo MM, Fernandes TG, Bouwman FG, Mariman ECM, Ghazvini M, Gribnau J, Curfs LMG, Reutelingsperger CP, Eijssen LMT. Involvement of extracellular vesicle microRNA clusters in developing healthy and Rett syndrome brain organoids. Cell Mol Life Sci CMLS. 2024 Sep 21;81(1):410.
- Lopes AG, Loganathan SK, Caliaperumal J. Rett Syndrome and the Role of MECP2: Signaling to Clinical Trials. Brain Sci. 2024 Jan 24;14(2):120. [CrossRef]
- Pascual-Alonso A, Martínez-Monseny AF, Xiol C, Armstrong J. MECP2-Related Disorders in Males. Int J Mol Sci. 2021 Sep 4;22(17):9610. [CrossRef]
- Shiohama T, Tsujimura K. Quantitative Structural Brain Magnetic Resonance Imaging Analyses: Methodological Overview and Application to Rett Syndrome. Front Neurosci. 2022;16:835964. [CrossRef]
- Kong Y, Li QB, Yuan ZH, Jiang XF, Zhang GQ, Cheng N, Dang N. Multimodal Neuroimaging in Rett Syndrome With MECP2 Mutation. Front Neurol. 2022;13:838206.
- Buchanan CB, Stallworth JL, Joy AE, Dixon RE, Scott AE, Beisang AA, Benke TA, Glaze DG, Haas RH, Heydemann PT, Jones MD, Lane JB, Lieberman DN, Marsh ED, Neul JL, Peters SU, Ryther RC, Skinner SA, Standridge SM, Kaufmann WE, Percy AK. Anxiety-like behavior and anxiolytic treatment in the Rett syndrome natural history study. J Neurodev Disord. 2022 May 14;14(1):31. [CrossRef]
- Stallworth JL, Dy ME, Buchanan CB, Chen CF, Scott AE, Glaze DG, Lane JB, Lieberman DN, Oberman LM, Skinner SA, Tierney AE, Cutter GR, Percy AK, Neul JL, Kaufmann WE. Hand stereotypies: Lessons from the Rett Syndrome Natural History Study. Neurology. 2019 May 28;92(22):e2594–603.
- Vilvarajan S, McDonald M, Douglas L, Newham J, Kirkland R, Tzannes G, Tay D, Christodoulou J, Thompson S, Ellaway C. Multidisciplinary Management of Rett Syndrome: Twenty Years’ Experience. Genes. 2023 Aug 11;14(8):1607.
- Andoh-Noda T, Inouye MO, Miyake K, Kubota T, Okano H, Akamatsu W. Modeling Rett Syndrome Using Human Induced Pluripotent Stem Cells. CNS Neurol Disord Drug Targets. 2016;15(5):544–50. [CrossRef]
- Parent H, Ferranti A, Niswender C. Trofinetide: a pioneering treatment for Rett syndrome. Trends Pharmacol Sci. 2023 Oct;44(10):740–1.
- Sweatt JD, Tamminga CA. An epigenomics approach to individual differences and its translation to neuropsychiatric conditions. Dialogues Clin Neurosci. 2016 Sep;18(3):289–98. [CrossRef]
- Vogel Ciernia A, Yasui DH, Pride MC, Durbin-Johnson B, Noronha AB, Chang A, Knotts TA, Rutkowsky JR, Ramsey JJ, Crawley JN, LaSalle JM. MeCP2 isoform e1 mutant mice recapitulate motor and metabolic phenotypes of Rett syndrome. Hum Mol Genet. 2018 Dec 1;27(23):4077–93.
- Marano D, Fioriniello S, D’Esposito M, Della Ragione F. Transcriptomic and Epigenomic Landscape in Rett Syndrome. Biomolecules. 2021 Jun 30;11(7):967. [CrossRef]
- Sandweiss AJ, Brandt VL, Zoghbi HY. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: prospects for future therapies. Lancet Neurol. 2020 Aug;19(8):689–98.
- Haase FD, Coorey B, Riley L, Cantrill LC, Tam PPL, Gold WA. Pre-clinical Investigation of Rett Syndrome Using Human Stem Cell-Based Disease Models. Front Neurosci. 2021;15:698812.
- Pramanik S, Bala A, Pradhan A. Zebrafish in understanding molecular pathophysiology, disease modeling, and developing effective treatments for Rett syndrome. J Gene Med. 2024 Feb;26(2):e3677.
- MacKay J, Leonard H, Wong K, Wilson A, Downs J. Respiratory morbidity in Rett syndrome: an observational study. Dev Med Child Neurol. 2018 Sep;60(9):951–7. [CrossRef]
- Rashid N, Darer JD, Ruetsch C, Yang X. Aspiration, respiratory complications, and associated healthcare resource utilization among individuals with Rett syndrome. Orphanet J Rare Dis. 2025 May 15;20(1):232.
- Crosson J, Srivastava S, Bibat GM, Gupta S, Kantipuly A, Smith-Hicks C, Myers SM, Sanyal A, Yenokyan G, Brenner J, Naidu SR. Evaluation of QTc in Rett syndrome: Correlation with age, severity, and genotype. Am J Med Genet A. 2017 Jun;173(6):1495–501.
- Rodrigues GD, Cordani R, Veneruso M, Chiarella L, Prato G, Ferri R, Carandina A, Tobaldini E, Nobili L, Montano N. Predominant cardiac sympathetic modulation during wake and sleep in patients with Rett syndrome. Sleep Med. 2024 Jul;119:188–91. [CrossRef]
- Meyyazhagan A, Balasubramanian B, Kathannan S, Alagamuthu KK, Easwaran M, Shanmugam S, Pappusamy M, Bhotla HK, Mustaqahamed S, Arumugam VA, Kaul T, Keshavarao S. Scrutinizing the molecular, biochemical, and cytogenetic attributes in subjects with Rett syndrome (RTT) and their mothers. Epilepsy Behav EB. 2020 Oct;111:107277.
- Singh J, Lanzarini E, Santosh P. Autonomic dysfunction and sudden death in patients with Rett syndrome: a systematic review. J Psychiatry Neurosci JPN. 2020 May 1;45(3):150–81. [CrossRef]
- Berger TD, Fogel Berger C, Gara S, Ben-Zeev B, Weiss B. Nutritional and gastrointestinal manifestations in Rett syndrome: long-term follow-up. Eur J Pediatr. 2024 Sep;183(9):4085–91.
- Caputi V, Hill L, Figueiredo M, Popov J, Hartung E, Margolis KG, Baskaran K, Joharapurkar P, Moshkovich M, Pai N. Functional contribution of the intestinal microbiome in autism spectrum disorder, attention deficit hyperactivity disorder, and Rett syndrome: a systematic review of pediatric and adult studies. Front Neurosci. 2024;18:1341656. [CrossRef]
- Wang Q, Yang Q, Liu X. The microbiota-gut-brain axis and neurodevelopmental disorders. Protein Cell. 2023 Oct 25;14(10):762–75.
- Borghi E, Vignoli A. Rett Syndrome and Other Neurodevelopmental Disorders Share Common Changes in Gut Microbial Community: A Descriptive Review. Int J Mol Sci. 2019 Aug 26;20(17):4160. [CrossRef]
- Downs J, Wong K, Leonard H. Associations between genotype, phenotype and behaviours measured by the Rett syndrome behaviour questionnaire in Rett syndrome. J Neurodev Disord. 2024 Oct 25;16(1):59.
- Moore R, Poulsen J, Reardon L, Samples-Morris C, Simmons H, Ramsey KM, Whatley ML, Lane JB. Managing Gastrointestinal Symptoms Resulting from Treatment with Trofinetide for Rett Syndrome: Caregiver and Nurse Perspectives. Adv Ther. 2024 Apr;41(4):1305–17.
- Motil KJ, Beisang A, Smith-Hicks C, Lembo A, Standridge SM, Liu E. Recommendations for the management of gastrointestinal comorbidities with or without trofinetide use in Rett syndrome. Expert Rev Gastroenterol Hepatol. 2024 Jun;18(6):227–37. [CrossRef]
- Borloz E, Villard L, Roux JC. Rett syndrome: think outside the (skull) box. Fac Rev. 2021;10:59.
- Singh J, Fiori F, Law ML, Ahmed R, Ameenpur S, Basheer S, Chishti S, Lawrence R, Mastroianni M, Mosaddegh A, Santosh P. Development and Psychometric Properties of the Multi-System Profile of Symptoms Scale in Patients with Rett Syndrome. J Clin Med. 2022 Aug 30;11(17):5094. [CrossRef]
- Y L, R G, R J. Rett Syndrome: Thinking Beyond Brain Borders. Adv Exp Med Biol [Internet]. 2025 [cited 2025 Sep 3];1477. Available from: https://pubmed.ncbi.nlm.nih.gov/40442389/.
- Wang W, Li H, Xiao M, Mu M, Xu H, Wang B. Clinical Research on Rett Syndrome: Central Hypoxemia and Hypokalemic Metabolic Alkalosis. Altern Ther Health Med. 2024 Jan;30(1):167–71.
- Zlatic SA, Duong D, Gadalla KKE, Murage B, Ping L, Shah R, Fink JJ, Khwaja O, Swanson LC, Sahin M, Rayaprolu S, Kumar P, Rangaraju S, Bird A, Tarquinio D, Carpenter R, Cobb S, Faundez V. Convergent cerebrospinal fluid proteomes and metabolic ontologies in humans and animal models of Rett syndrome. iScience. 2022 Sep 16;25(9):104966. [CrossRef]
- A V, V C, A P, G V. Ox-inflammasome involvement in neuroinflammation. Free Radic Biol Med [Internet]. 2023 Oct [cited 2025 Sep 3];207. Available from: https://pubmed.ncbi.nlm.nih.gov/37442280/.
- Gonçalez JL, Shen J, Li W. Molecular Mechanisms of Rett Syndrome: Emphasizing the Roles of Monoamine, Immunity, and Mitochondrial Dysfunction. Cells. 2024 Dec 17;13(24):2077.
- Das DK, Raha S, Sanghavi D, Maitra A, Udani V. Spectrum of MECP2 gene mutations in a cohort of Indian patients with Rett syndrome: report of two novel mutations. Gene. 2013 Feb 15;515(1):78–83. [CrossRef]
- Zepeda-Mendoza CJ, Bardon A, Kammin T, Harris DJ, Cox H, Redin C, Ordulu Z, Talkowski ME, Morton CC. Phenotypic interpretation of complex chromosomal rearrangements informed by nucleotide-level resolution and structural organization of chromatin. Eur J Hum Genet EJHG. 2018 Mar;26(3):374–81.
- Huang CH, Wong LC, Chu YJ, Hsu CJ, Wang HP, Tsai WC, Lee WT. The sleep problems in individuals with Rett syndrome and their caregivers. Autism Int J Res Pract. 2024 Dec;28(12):3118–30. [CrossRef]
- Kay C, Leonard H, Smith J, Wong K, Downs J. Genotype and sleep independently predict mental health in Rett syndrome: an observational study. J Med Genet. 2023 Oct;60(10):951–9. [CrossRef]
- Tascini G, Dell’Isola GB, Mencaroni E, Di Cara G, Striano P, Verrotti A. Sleep Disorders in Rett Syndrome and Rett-Related Disorders: A Narrative Review. Front Neurol. 2022;13:817195.
- Lai YYL, Downs J, Zafar S, Wong K, Walsh L, Leonard H. Oral health care and service utilisation in individuals with Rett syndrome: an international cross-sectional study. J Intellect Disabil Res JIDR. 2021 Jun;65(6):561–76. [CrossRef]
- Neul JL, Percy AK, Benke TA, Berry-Kravis EM, Glaze DG, Marsh ED, Lin T, Stankovic S, Bishop KM, Youakim JM. Trofinetide for the treatment of Rett syndrome: a randomized phase 3 study. Nat Med. 2023 Jun;29(6):1468–75.
- Hirano D, Goto Y, Shoji H, Taniguchi T. Relationship between hand stereotypies and purposeful hand use and factors causing skin injuries and joint contractures in individuals with Rett syndrome. Early Hum Dev. 2023 Aug;183:105821. [CrossRef]
- Zhang E, Zhao T, Sikora T, Ellaway C, Gold WA, Van Bergen NJ, Stroud DA, Christodoulou J, Kaur S. CHD8 Variant and Rett Syndrome: Overlapping Phenotypes, Molecular Convergence, and Expanding the Genetic Spectrum. Hum Mutat. 2025;2025:5485987.
- Migovich M, Ullal A, Fu C, Peters SU, Sarkar N. Feasibility of wearable devices and machine learning for sleep classification in children with Rett syndrome: A pilot study. Digit Health. 2023;9:20552076231191622. [CrossRef]
- Wandin H, Lindberg P, Sonnander K. Aided language modelling, responsive communication and eye-gaze technology as communication intervention for adults with Rett syndrome: three experimental single case studies. Disabil Rehabil Assist Technol. 2023 Oct;18(7):1011–25.
- Monteiro-Fernandes D, Charles I, Guerreiro S, Cunha-Garcia D, Pereira-Sousa J, Oliveira S, Teixeira-Castro A, Varney MA, Kleven MS, Newman-Tancredi A, P Sheikh Abdala A, Duarte-Silva S, Maciel P. Rescue of respiratory and cognitive impairments in Rett Syndrome mice using NLX-101, a selective 5-HT1A receptor biased agonist. Biomed Pharmacother Biomedecine Pharmacother. 2025 May;186:117989.
- Cordone V. Biochemical and molecular determinants of the subclinical inflammatory mechanisms in Rett syndrome. Arch Biochem Biophys. 2024 Jul;757:110046. [CrossRef]
- Kaufmann WE, Oberman LM, Downs J, Leonard H, Barnes KV. Rett Syndrome Behaviour Questionnaire: Variability of Scores and Related Factors. J Child Adolesc Psychopharmacol. 2025 Feb 5; [CrossRef]
- Percy AK, Neul JL, Benke TA, Marsh ED, Glaze DG. A review of the Rett Syndrome Behaviour Questionnaire and its utilization in the assessment of symptoms associated with Rett syndrome. Front Pediatr. 2023;11:1229553.
- Neul JL, Percy AK, Benke TA, Berry-Kravis EM, Glaze DG, Peters SU, Jones NE, Youakim JM. Design and outcome measures of LAVENDER, a phase 3 study of trofinetide for Rett syndrome. Contemp Clin Trials. 2022 Mar;114:106704. [CrossRef]
- Oberman LM, Leonard H, Downs J, Cianfaglione R, Stahlhut M, Larsen JL, Madden KV, Kaufmann WE. Rett Syndrome Behaviour Questionnaire in Children and Adults With Rett Syndrome: Psychometric Characterization and Revised Factor Structure. Am J Intellect Dev Disabil. 2023 May 1;128(3):237–53. [CrossRef]
- Cohen SR, Helbig I, Kaufman MC, Schust Myers L, Conway L, Helbig KL. Caregiver assessment of quality of life in individuals with genetic developmental and epileptic encephalopathies. Dev Med Child Neurol. 2022 Aug;64(8):957–64.
- Killian JT, Lane JB, Lee HS, Pelham JH, Skinner SA, Kaufmann WE, Glaze DG, Neul JL, Percy AK. Caretaker Quality of Life in Rett Syndrome: Disorder Features and Psychological Predictors. Pediatr Neurol. 2016 May;58:67–74.
- McGraw SA, Smith-Hicks C, Nutter J, Henne JC, Abler V. Meaningful Improvements in Rett Syndrome: A Qualitative Study of Caregivers. J Child Neurol. 2023 Apr;38(5):270–82.
- Bajikar SS, Zhou J, O’Hara R, Tirumala HP, Durham MA, Trostle AJ, Dias M, Shao Y, Chen H, Wang W, Yalamanchili HK, Wan YW, Banaszynski LA, Liu Z, Zoghbi HY. Acute MeCP2 loss in adult mice reveals transcriptional and chromatin changes that precede neurological dysfunction and inform pathogenesis. Neuron. 2025 Feb 5;113(3):380-395.e8. [CrossRef]
- Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron. 2015 Nov 18;88(4):651–8.
- Sadhu C, Lyons C, Oh J, Jagadeeswaran I, Gray SJ, Sinnett SE. The Efficacy of a Human-Ready miniMECP2 Gene Therapy in a Pre-Clinical Model of Rett Syndrome. Genes. 2023 Dec 24;15(1):31.
- Yang K, Li T, Geng Y, Zhang R, Xu Z, Wu J, Yuan Y, Zhang Y, Qiu Z, Li F. Protocol for the neonatal intracerebroventricular delivery of adeno-associated viral vectors for brain restoration of MECP2 for Rett syndrome. STAR Protoc. 2024 Dec 20;5(4):103344. [CrossRef]
- Gomathi M, Balachandar V. Novel therapeutic approaches: Rett syndrome and human induced pluripotent stem cell technology. Stem Cell Investig. 2017;4:20.
- Huber A, Sarne V, Beribisky AV, Ackerbauer D, Derdak S, Madritsch S, Etzler J, Huck S, Scholze P, Gorgulu I, Christodoulou J, Studenik CR, Neuhaus W, Connor B, Laccone F, Steinkellner H. Generation and Characterization of a Human Neuronal In Vitro Model for Rett Syndrome Using a Direct Reprogramming Method. Stem Cells Dev. 2024 Mar;33(5–6):128–42. [CrossRef]
- Samarasinghe RA, Miranda OA, Buth JE, Mitchell S, Ferando I, Watanabe M, Allison TF, Kurdian A, Fotion NN, Gandal MJ, Golshani P, Plath K, Lowry WE, Parent JM, Mody I, Novitch BG. Identification of neural oscillations and epileptiform changes in human brain organoids. Nat Neurosci. 2021 Oct;24(10):1488–500.
- Gomes AR, Fernandes TG, Vaz SH, Silva TP, Bekman EP, Xapelli S, Duarte S, Ghazvini M, Gribnau J, Muotri AR, Trujillo CA, Sebastião AM, Cabral JMS, Diogo MM. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front Cell Dev Biol. 2020;8:610427.
- Mok RSF, Zhang W, Sheikh TI, Pradeepan K, Fernandes IR, DeJong LC, Benigno G, Hildebrandt MR, Mufteev M, Rodrigues DC, Wei W, Piekna A, Liu J, Muotri AR, Vincent JB, Muller L, Martinez-Trujillo J, Salter MW, Ellis J. Wide spectrum of neuronal and network phenotypes in human stem cell-derived excitatory neurons with Rett syndrome-associated MECP2 mutations. Transl Psychiatry. 2022 Oct 18;12(1):450.
- Pradeepan KS, McCready FP, Wei W, Khaki M, Zhang W, Salter MW, Ellis J, Martinez-Trujillo J. Calcium-Dependent Hyperexcitability in Human Stem Cell-Derived Rett Syndrome Neuronal Networks. Biol Psychiatry Glob Open Sci. 2024 Mar;4(2):100290. [CrossRef]
- Aldosary M, Al-Bakheet A, Al-Dhalaan H, Almass R, Alsagob M, Al-Younes B, AlQuait L, Mustafa OM, Bulbul M, Rahbeeni Z, Alfadhel M, Chedrawi A, Al-Hassnan Z, AlDosari M, Al-Zaidan H, Al-Muhaizea MA, AlSayed MD, Salih MA, AlShammari M, Faiyaz-Ul-Haque M, Chishti MA, Al-Harazi O, Al-Odaib A, Kaya N, Colak D. Rett Syndrome, a Neurodevelopmental Disorder, Whole-Transcriptome, and Mitochondrial Genome Multiomics Analyses Identify Novel Variations and Disease Pathways. Omics J Integr Biol. 2020 Mar;24(3):160–71.
- Karaosmanoglu B, Imren G, Ozisin MS, Reçber T, Simsek Kiper PO, Haliloglu G, Alikaşifoğlu M, Nemutlu E, Taskiran EZ, Utine GE. Ex vivo disease modelling of Rett syndrome: the transcriptomic and metabolomic implications of direct neuronal conversion. Mol Biol Rep. 2024 Sep 13;51(1):979. [CrossRef]
- Pascual-Alonso A, Xiol C, Smirnov D, Kopajtich R, Prokisch H, Armstrong J. Multi-omics in MECP2 duplication syndrome patients and carriers. Eur J Neurosci. 2024 Jul;60(2):4004–18.
- Baroncelli L, Auel S, Rinne L, Schuster AK, Brand V, Kempkes B, Dietrich K, Müller M. Oral Feeding of an Antioxidant Cocktail as a Therapeutic Strategy in a Mouse Model of Rett Syndrome: Merits and Limitations of Long-Term Treatment. Antioxid Basel Switz. 2022 Jul 20;11(7):1406.
- Panayotis N, Ehinger Y, Felix MS, Roux JC. State-of-the-art therapies for Rett syndrome. Dev Med Child Neurol. 2023 Feb;65(2):162–70.
- Persico AM, Ricciardello A, Cucinotta F. The psychopharmacology of autism spectrum disorder and Rett syndrome. Handb Clin Neurol. 2019;165:391–414.
- Kaufmann WE, Stallworth JL, Everman DB, Skinner SA. Neurobiologically-based treatments in Rett syndrome: opportunities and challenges. Expert Opin Orphan Drugs. 2016 Oct 2;4(10):1043–55.
- Percy AK, Neul JL, Benke TA, Berry-Kravis EM, Glaze DG, Marsh ED, An D, Bishop KM, Youakim JM. Trofinetide for the treatment of Rett syndrome: Results from the open-label extension LILAC study. Med N Y N. 2024 Sep 13;5(9):1178-1189.e3. [CrossRef]
- Singh A, Balasundaram MK, Gupta D. Trofinetide in Rett syndrome: A brief review of safety and efficacy. Intractable Rare Dis Res. 2023 Nov;12(4):262–6.
- Coorey B, Haase F, Ellaway C, Clarke A, Lisowski L, Gold WA. Gene Editing and Rett Syndrome: Does It Make the Cut? CRISPR J. 2022 Aug;5(4):490–9. [CrossRef]
- Fonzo M, Sirico F, Corrado B. Evidence-Based Physical Therapy for Individuals with Rett Syndrome: A Systematic Review. Brain Sci. 2020 Jun 30;10(7):410.
- Saby JN, Peters SU, Roberts TPL, Nelson CA, Marsh ED. Evoked Potentials and EEG Analysis in Rett Syndrome and Related Developmental Encephalopathies: Towards a Biomarker for Translational Research. Front Integr Neurosci. 2020;14:30.
- Buckley N, Stahlhut M, Elefant C, Leonard H, Lotan M, Downs J. Parent and therapist perspectives on “uptime” activities and participation in Rett syndrome. Disabil Rehabil. 2022 Dec;44(24):7420–7. [CrossRef]
- Bajikar SS, Sztainberg Y, Trostle AJ, Tirumala HP, Wan YW, Harrop CL, Bengtsson JD, Carvalho CMB, Pehlivan D, Suter B, Neul JL, Liu Z, Jafar-Nejad P, Rigo F, Zoghbi HY. Modeling antisense oligonucleotide therapy in MECP2 duplication syndrome human iPSC-derived neurons reveals gene expression programs responsive to MeCP2 levels. Hum Mol Genet. 2024 Nov 8;33(22):1986–2001.
- Gogliotti RG, Senter RK, Rook JM, Ghoshal A, Zamorano R, Malosh C, Stauffer SR, Bridges TM, Bartolome JM, Daniels JS, Jones CK, Lindsley CW, Conn PJ, Niswender CM. mGlu5 positive allosteric modulation normalizes synaptic plasticity defects and motor phenotypes in a mouse model of Rett syndrome. Hum Mol Genet. 2016 May 15;25(10):1990–2004.
- Merritt JK, Fang X, Caylor RC, Skinner SA, Friez MJ, Percy AK, Neul JL. Normalized Clinical Severity Scores Reveal a Correlation between X Chromosome Inactivation and Disease Severity in Rett Syndrome. Genes. 2024 May 8;15(5):594. [CrossRef]
- Abbas A, Fayoud AM, El Din Moawad MH, Hamad AA, Hamouda H, Fouad EA. Safety and efficacy of trofinetide in Rett syndrome: a systematic review and meta-analysis of randomized controlled trials. BMC Pediatr. 2024 Mar 23;24(1):206.
- Cherchi C, Chiappini E, Amaddeo A, Chiarini Testa MB, Banfi P, Veneselli E, Cutrera R, panel for the Problems in Patients with Rett Syndrome. Management of respiratory issues in patients with Rett syndrome: Italian experts’ consensus using a Delphi approach. Pediatr Pulmonol. 2024 Jul;59(7):1970–8.
- Furley K, Mehra C, Goin-Kochel RP, Fahey MC, Hunter MF, Williams K, Absoud M. Developmental regression in children: Current and future directions. Cortex J Devoted Study Nerv Syst Behav. 2023 Dec;169:5–17.
- Xiol C, Vidal S, Pascual-Alonso A, Blasco L, Brandi N, Pacheco P, Gerotina E, O’Callaghan M, Pineda M, Armstrong J, Rett Working Group. X chromosome inactivation does not necessarily determine the severity of the phenotype in Rett syndrome patients. Sci Rep. 2019 Aug 19;9(1):11983.
- Lim J, Greenspoon D, Hunt A, McAdam L. Rehabilitation interventions in Rett syndrome: a scoping review. Dev Med Child Neurol. 2020 Aug;62(8):906–16.
- Downs J, Pichard DC, Kaufmann WE, Horrigan JP, Raspa M, Townend G, Marsh ED, Leonard H, Motil K, Dietz AC, Garg N, Ananth A, Byiers B, Peters S, Beatty C, Symons F, Jacobs A, Youakim J, Suter B, Santosh P, Neul JL, Benke TA. International workshop: what is needed to ensure outcome measures for Rett syndrome are fit-for-purpose for clinical trials? June 7, 2023, Nashville, USA. Trials. 2024 Dec 21;25(1):845. [CrossRef]
- Howell KB, White SM, McTague A, D’Gama AM, Costain G, Poduri A, Scheffer IE, Chau V, Smith LD, Stephenson SEM, Wojcik M, Davidson A, Sebire N, Sliz P, Beggs AH, Chitty LS, Cohn RD, Marshall CR, Andrews NC, North KN, Cross JH, Christodoulou J, Scherer SW. International Precision Child Health Partnership (IPCHiP): an initiative to accelerate discovery and improve outcomes in rare pediatric disease. NPJ Genomic Med. 2025 Feb 27;10(1):13. [CrossRef]
- Smith M, Arthur B, Cikowski J, Holt C, Gonzalez S, Fisher NM, Vermudez SAD, Lindsley CW, Niswender CM, Gogliotti RG. Clinical and Preclinical Evidence for M1 Muscarinic Acetylcholine Receptor Potentiation as a Therapeutic Approach for Rett Syndrome. Neurother J Am Soc Exp Neurother. 2022 Jul;19(4):1340–52. [CrossRef]
Figure 1.
Distribution of MeCP2 gene expression across CNS regions and peripheral organs. The gradient bar denotes relative intensity, with darker shades indicating higher expression. MeCP2 expression is enriched in the cerebral cortex and hippocampus, with moderate levels in the cerebellum and brainstem. In peripheral tissues, higher expression is observed in the lung and spleen, whereas comparatively lower levels are detected in the liver and intestine.
Figure 1.
Distribution of MeCP2 gene expression across CNS regions and peripheral organs. The gradient bar denotes relative intensity, with darker shades indicating higher expression. MeCP2 expression is enriched in the cerebral cortex and hippocampus, with moderate levels in the cerebellum and brainstem. In peripheral tissues, higher expression is observed in the lung and spleen, whereas comparatively lower levels are detected in the liver and intestine.
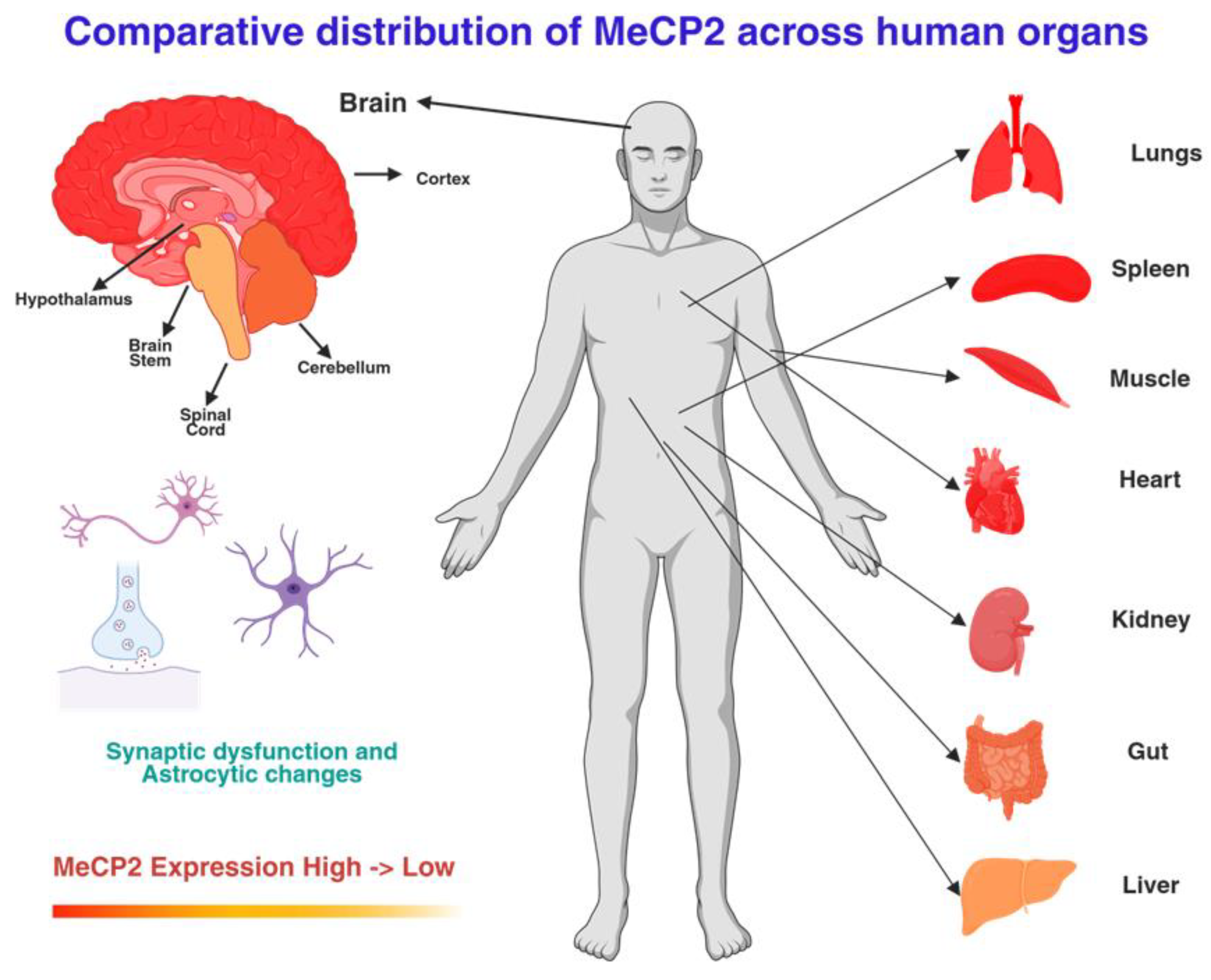
Figure 2.
Progression of Rett syndrome across the lifespan. The schematic outlines stages from early normal development to stagnation, regression with skill loss and stereotypies, stabilization with seizures and autonomic disturbances, and later complications, including scoliosis, gastrointestinal dysfunction, and increased caregiver burden into adulthood.
Figure 2.
Progression of Rett syndrome across the lifespan. The schematic outlines stages from early normal development to stagnation, regression with skill loss and stereotypies, stabilization with seizures and autonomic disturbances, and later complications, including scoliosis, gastrointestinal dysfunction, and increased caregiver burden into adulthood.
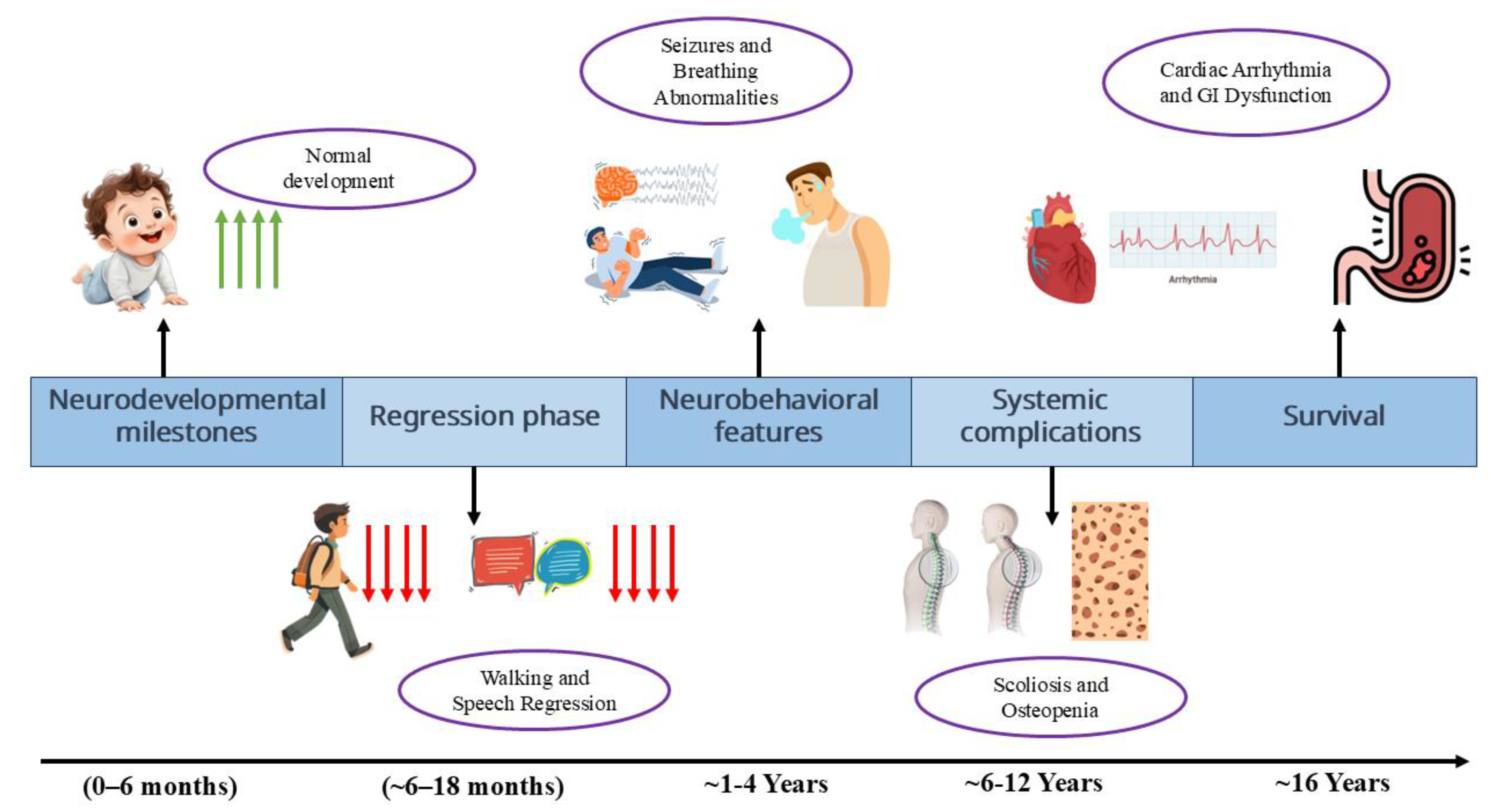
Figure 3.
Timeline of major milestones in Rett syndrome research and therapy development. The schematic illustrates the discovery of the MECP2 gene in 1999, followed by advancements in symptomatic and supportive care, the initiation of clinical trials for potential drugs, and the approval of trofinetide as the first targeted treatment. More recently, gene therapy approaches have entered early-phase clinical trials, offering the potential for disease-modifying interventions.
Figure 3.
Timeline of major milestones in Rett syndrome research and therapy development. The schematic illustrates the discovery of the MECP2 gene in 1999, followed by advancements in symptomatic and supportive care, the initiation of clinical trials for potential drugs, and the approval of trofinetide as the first targeted treatment. More recently, gene therapy approaches have entered early-phase clinical trials, offering the potential for disease-modifying interventions.
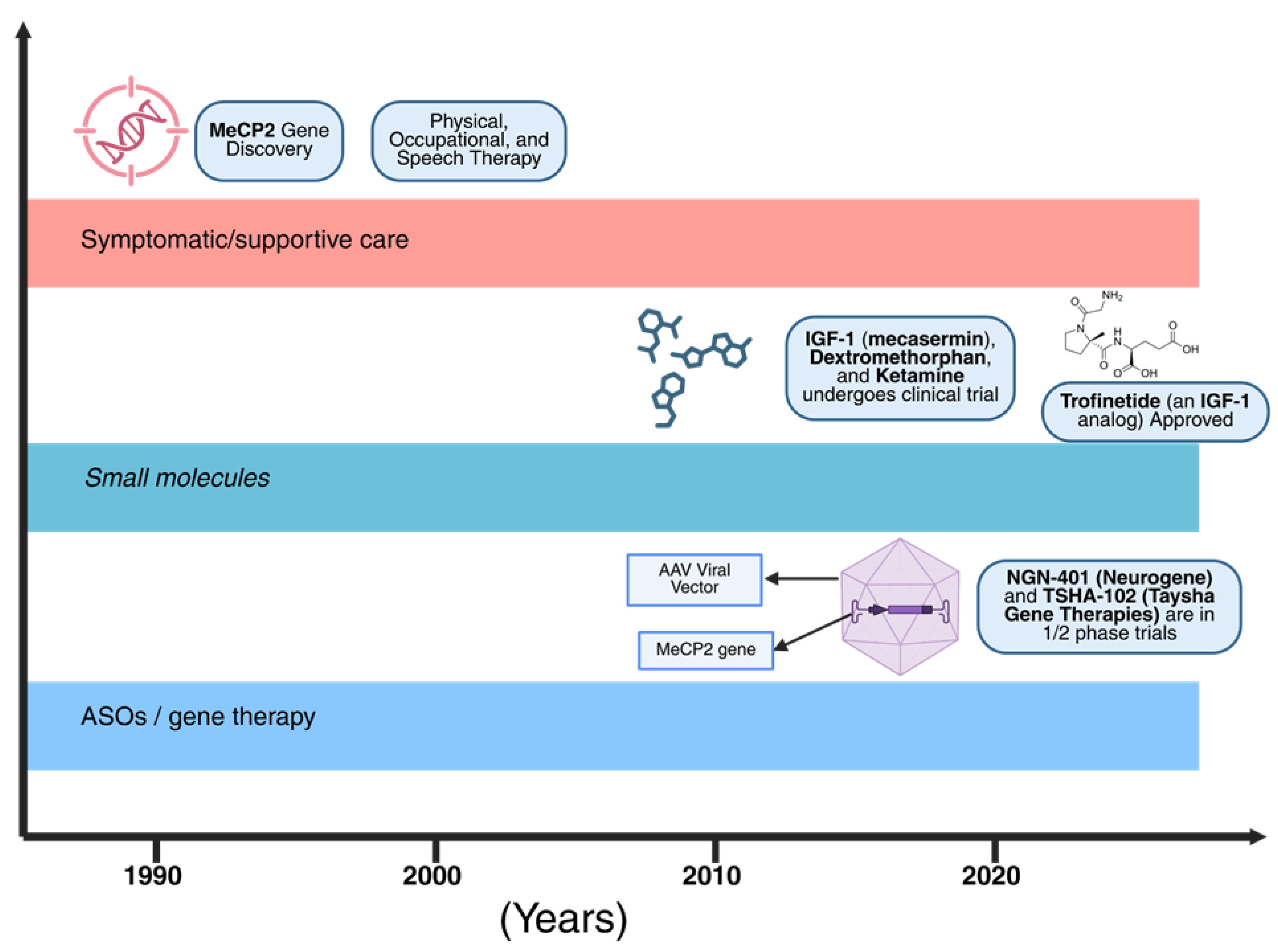

Table 1.
Key MECP2 Mutations and Genotype-Phenotype Correlations.
| Mutation Type | Common Variants | Approximate frequency among classic MECP2 variants in RTT (%)—cohort-dependent | Phenotypic Severity | Associated Features | Recent Data | Inference |
|---|---|---|---|---|---|---|
| Missense | R106W, R133C, T158M | 40–50 | Moderate to Severe | Early regression, epilepsy (60–80%), scoliosis | Disrupt methyl-binding domain; partial function retained. | R133C shows a milder phenotype due to residual binding. RNHS: Better ambulation in R133C (30% vs. 10% in R106W). |
| Nonsense | R168X, R255X, R270X | 30–40 | Severe | Profound intellectual disability, non-ambulatory, respiratory issues | Early truncation leading to a null function. | High sudden death risk (25%). 2025 data: Correlation with mitochondrial dysfunction suggests metabolic crisis. |
| Frameshift/Deletions | C-terminal deletions | 10–15 | Mild to Moderate | Preserved speech variant, later onset | Retain partial domains; 2024 iPSC data show reduced synaptic loss. | Therapeutic window exists for partial restoration. |
| Duplications | MECP2 duplication syndrome | <5 (males predominant) | Severe (males lethal) | Hypotonia, infections, autism-like features | Overexpression toxicity; mouse models demonstrate anxiety phenotypes. | Dosage sensitivity inferred. |
Table 2.
Multisystem Clinical Features, Prevalence, Mechanistic Insights, and Management Approaches in RTT.
Table 2.
Multisystem Clinical Features, Prevalence, Mechanistic Insights, and Management Approaches in RTT.
| System | Key Features | Prevalence | Mechanisms (Animal/iPSC Evidence) | Clinical Management | Inference |
|---|---|---|---|---|---|
| Respiratory / Breathing Dysrhythmia | Awake hyperventilation, apnea, breath-holds; brainstem circuit disruption; mitochondrial hypoxia | ~60–80% (RNHS 2024–2025: 70–80%) | Brainstem circuit disruption; mitochondrial hypoxia | NIV, oxygen; training | Central–peripheral interplay; oxidative stress target for therapies. |
| Cardiovascular / Autonomic | QTc prolongation, arrhythmia, HRV ↓ vagal tone; sympathetic overdrive | ~20% QTc↑ (RNHS: 75% instability; 20–30% QTc) | Ion channel dysregulation; sympathetic overdrive; mitochondrial contribution | Annual ECG; beta-blockers | Mitochondrial role inferred; sudden death prevention priority. |
| Gastrointestinal (Constipation, GERD, Dysphagia)* | Severe chronic constipation; reflux disease; swallowing incoordination | Constipation ~80–90% (RNHS: 80%); GERD/Dysphagia ~60–70% (RNHS: 70%) | Enteric neuron hypofunction; microbiome shifts | Laxatives, gastrostomy | Inflammation link; trofinetide GI AEs highlight need for adjuncts. |
| Skeletal (Scoliosis, Low BMD/Fractures)* | Scoliosis often progressive (>40°); osteopenia; frequent fractures | Scoliosis ≥60–85% (RNHS: 85%); Low BMD ≈45–60%; fractures ~30% (RNHS: 20%) | Osteoblast defects; endocrine (low Vitamin D) | Bisphosphonates; surgery | Immobility exacerbates; early PT inferred to mitigate. |
| CNS – Seizures | Onset 2–5 yrs; mix of focal and generalized; treatable | ~60–90% lifetime | Cortical hyperexcitability; neuronal circuit dysfunction | Antiepileptic drugs (AEDs); multidisciplinary seizure management | Seizure control impacts quality of life and neurodevelopment. |
| Growth / Nutrition | Short stature, poor weight gain, microcephaly | ~80–100% | Nutritional/metabolic dysfunction; growth hormone/endocrine contribution | Nutritional support; multidisciplinary monitoring | Growth failure central to prognosis; metabolic basis |
| Sleep | Insomnia, night awakenings, non-restorative sleep | ~60–80% | Circadian rhythm disruption; neurotransmitter imbalance | Behavioral interventions; melatonin | Sleep disturbance linked to cognition and seizures. |
| Anxiety / Mood | Excessive fear, anxious behaviors, mood swings | ~50–70% | Amygdala circuit dysfunction; altered GABA/glutamate signaling | Behavioral therapy; SSRIs (case-based) | Mental health central to QoL; neurochemical imbalance inferred. |
| Dental – Bruxism | Daytime teeth grinding | ~80–100% | Abnormal brainstem reflex pathways; neuromuscular dysregulation | Mouthguards; dental monitoring | Contributes to dental wear; symptomatic care only. |
| Peripheral / Autonomic – Cold Extremities | Vasomotor instability (cyanosis of hands/feet) | ~50% | Autonomic dysregulation; vascular tone impairment | Supportive management; warming measures | Peripheral autonomic dysfunction reinforces systemic involvement |
Note: Prevalence values represent harmonized estimates from multiple registries, with variability depending on cohort size, age distribution, and diagnostic methodology (“registry variability”).
Table 3.
Comparative Overview of Preclinical and Translational Model Systems for MECP2-Related Disorders.
Table 3.
Comparative Overview of Preclinical and Translational Model Systems for MECP2-Related Disorders.
| Model System | Genetic Alteration / Type | Key Features / Uses | Strengths | Limitations | Inferences |
|---|---|---|---|---|---|
| Mouse Mecp2-null (Bird) males | Mecp2 null (hemizygote) | Rapid-onset RTT-like phenotype; seizures; early death (~10 wks) | High phenotypic fidelity; reproducible phenotype | Small brain size; not representative of female mosaicism | Adult re-expression reverses ~80% of phenotypes; omics show metabolic shifts. |
| Mouse Mecp2-null (heterozygous females) | Mecp2+/– (female heterozygote) | Mosaic neuronal populations; slower progression | Models clinical female RTT; long survival | Variable expressivity due to XCI | Omics reveal compensatory networks in mosaic populations. |
| Mouse Mecp2-duplication | Multiple Mecp2 transgene copies | Models MECP2 duplication syndrome; seizures | Relevant for antisense therapy | Overexpression artifacts; limited survival | Overexpression risks validated in vivo. |
| Conditional knockout mice | Mecp2 deleted in neuron/astrocyte/microglia | Cell-type specific phenotypes | Dissects MECP2 roles | Restricted scope | Highlights glial contributions to disease. |
| Rat Mecp2 knockout | Mecp2 deletion | RTT-like features; larger size | Larger brain; better dosing/autonomic profiling | Fewer strains; less genetic versatility | Enhanced autonomic profiling; better for gene therapy dosing. |
| Human iPSC-derived neurons | Patient fibroblasts reprogrammed | Reduced dendritic arborization, synaptic deficits | Human genotype/phenotype context | No systemic environment | Synaptic rescue demonstrated; 2025: mitochondrial targets identified. |
| Brain organoids (human iPSC) | 3D cortical cultures | Reveal early cortical networks, activity patterns | Human-specific; network-level | Limited maturation | Organoid models validate early neurodevelopmental signatures. |
| Large Animal / NHP Mecp2 models | Transgenic/gene-edited | Translational biodistribution, pharmacology | Closer to human brain | Ethical and costly | AAV safety validated; overexpression risks confirmed. |
| Zebrafish Mecp2-null | MecP2 mutant fish | Transparent larvae; rapid assays | Fast, scalable | Limited behavioral fidelity | Useful for rapid drug screening and in vivo imaging approaches. |
Table 4.
Overview of Therapeutic Approaches in RTT.
| Therapeutic Class / Strategy | Example(s) / Compound(s) | Mechanism of Action | Status | Key Efficacy / Safety Notes | Inference |
|---|---|---|---|---|---|
| Pharmacotherapy | Trofinetide | IGF-1 analog; neurotrophic/anti-inflammatory | FDA-Approved (2023) | Efficacy on RSBQ, CGI-I, CSBS. GI side effects (diarrhea, vomiting). | Symptom modifier; real-world reports suggest sustained benefits with careful GI management. |
| Neurotrophic / Growth Factor Strategies | IGF-1 (mecasermin) | Neurotrophic support | Phase 2 | Mixed efficacy; not advanced. | Highlights limitations of systemic IGF-1 therapy. |
| Gene Replacement Therapy | NGN-401 (Neurogene) | AAV-based MECP2 delivery | Phase I/II | High-dose halted (fatal inflammatory syndrome). Low-dose: 28–52% RSBQ improvement. | Potential disease modifier; tight dose regulation essential. |
| Dosage Normalization | ASOs (e.g., ISIS-LEGRO) | Silence MECP2 mRNA | Preclinical/early trials | Dosage titration possible; risk of over-suppression. | Conceptually validates dosage control; requires precision for safety. |
| Metabolic / Mitochondrial Modulators | Leriglitazone | PPAR-γ agonist; mitochondrial targeting | Phase 2a | Ongoing; systemic benefit expected. | Mitochondrial/metabolic pathways are relevant therapeutic targets. |
| Cell-based Approaches | Stem cell transplantation, exosome therapy | Replacement/trophic support | Investigational | Safety and targeting limitations. | Currently experimental; requires advanced targeting. |
| Neuromodulation | TNS, VNS | Electrical network modulation | Experimental | Small studies: improvements in arousal/respiration. | Adjunct potential but preliminary. |
| Symptomatic / Supportive Care | PT, OT, speech, nutritional support | Multidisciplinary | Standard of care | Improves quality of life. | Remains essential alongside all experimental therapies. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



