
Submitted:
23 November 2025
Posted:
26 November 2025
You are already at the latest version
Abstract
Artificial intelligence (AI) technologies have recently undergone a transformative growth in capabilities. In human genetics, AI is rapidly advancing our ability to reveal the effects of genetic variation. This review explores recent progress and remaining challenges across the diverse applications of AI in genotype-to-phenotype mapping, from predicting the functional and clinical consequences of mutations, to identifying causal genes, to estimating disease risk. Particular emphasis is placed on the growing utility of general-purpose foundation models trained on massive genomic data, including DNA and protein language models, alongside areas where narrower machine-learning approaches still dominate. The review concludes with key considerations for future progress and impact.
Keywords:
machine learning
; deep learning
; genomic foundation models
; variant effect prediction
; protein language model
; DNA language model
1. Why AI? Why genetics?
In November 2020, DeepMind unveiled AlphaFold, an artificial intelligence (AI) model capable of accurately predicting protein structures [1]. The Nobel Prize committee later described this breakthrough as “solving a 50-year-old problem” [2], a milestone many had believed to be decades away [3]. Similar advances in AI-driven protein engineering shared the same Nobel Prize for enabling “the almost impossible feat of building entirely new kinds of proteins” [2]. With the current rate of progress, it is widely believed that AI is bound to fundamentally reshape biomedicine. But to seriously discuss the implications of these advances, we must ask: what specific problems can AI solve, and how exactly will it help us fight disease? The most popular narrative highlights the potential of AI in protein and drug design, but here I wish to explore another, underappreciated angle: human genetics.
Genetics is a science of changes and their consequences, which presents profound challenges. Investigating the effects of mutations, especially ones never observed before, requires more than finding statistical correlations; it requires reasoning. As we will see, many AI applications struggle to deal with genetic changes. AlphaFold, for example, relies too heavily on naturally-occurring structures and struggles to predict the structures of mutated protein sequences [4]. But other AI applications rise to the challenge.
AI offers an imperfect but practical approach to studying genetic effects, thanks to its ability to piece together and transfer knowledge learned directly from genomic data. This review explores how such AI is reshaping our decades-long quest to decipher the link between genotype and phenotype. The primary audience is geneticists interested in learning how AI can assist their investigations. The review is also intended for AI researchers looking for a deeper understanding of this unique domain and its challenges. To make it accessible to readers from diverse backgrounds, a glossary of AI and genetics terms (Table 1) and a summary of genomic AI models (Table 2) mentioned throughout the review are provided below.
2. Terminology & AI Trends
The term artificial intelligence (AI) is somewhat fluid. Here, it will be used interchangeably with machine learning (ML), referring to any computer program that learns from data rather than relying on explicitly programmed rules. Classical ML aims to learn patterns from a given dataset that would generalize to similar data in order to perform a well-defined task (Figure 1A). Modern AI, on the other hand, aims for broader generality. A key development is the rise of foundation models, a somewhat loosely defined term describing models trained on massive datasets to support a wide range of possible applications (Figure 1B). While the label is overused and many so-called foundation models are of little use in practice, the best ones have demonstrated remarkable capabilities. These models currently hold the most promise, so they receive special attention in this review. Yet in some areas, as we will see, narrower models continue to deliver the best results.
Covered topics
Throughout this review, we will explore how AI is used to uncover the molecular and clinical consequences of genetic variation, spanning both coding and non-coding genetic effects and both monogenic and polygenic diseases (Figure 2). While much of this discussion applies across species, the primary focus is human. We will start with one of the earliest applications of ML in genetics: predicting whether a given coding mutation is pathogenic. From there, we will turn to more specific structural and molecular phenotypes, and expand the discussion to non-coding variants affecting gene regulation. The final sections will shift to downstream applications in statistical genetics, including genetic association studies and disease risk prediction. We will conclude with a brief overview of the main challenges facing the field.
3. Identifying Disease-Causing Protein Mutations
To begin our exploration of AI applications in human genetics, let us start with a longstanding problem that forms the basis for many others: determining whether mutations are pathogenic.
The clinical gap: Rare genetic disorders typically follow Mendelian inheritance, where inheriting one or two copies of a pathogenic mutation will cause the condition [34,35]. Despite rapid advances in sequencing technologies and diagnostic protocols, only ~50% of patients currently end up with a clear molecular diagnosis, due to uncertainty over which mutations are pathogenic [36,37]. To help close this diagnostic gap, pathogenicity prediction models are trained to predict whether a given mutation is pathogenic or benign [23,38].
Training strategies: Most variant pathogenicity prediction models, including popular methods such as PolyPhen [20] and REVEL [22], rely on supervised ML: a model is trained on variants explicitly labeled as pathogenic or benign. An alternative strategy, employed by methods like SIFT [23] and phyloP [19], predicts variant pathogenicity based on evolutionary conservation. These methods infer that variants at highly conserved sites, or variants rarely observed across species, are more likely to be deleterious. More recent methods build on this principle by training deep neural networks on families of homologous protein sequences across different species. EVE, for example, models the variation of sequences within each protein family and infers how compatible variants are with these models [14]. This approach allows models to learn not only which specific sites are conserved, but also what general types of sequence variations are likely to be negatively selected within a given protein family.
Protein language modeling: Protein language models take this approach further, training a single universal model on the entire space of protein sequences known across all species and protein families (Figure 3A). This simple training task, when applied over hundreds of millions of protein sequences, gives rise to a foundation model that has acquired general knowledge about the complex interplay between protein sequence, structure and function [5,6,39,40,41,42] (Figure 3B). Unlike structure prediction models like AlphaFold, protein language models can predict function directly from sequence without explicitly dealing with protein structure.
Once trained, protein language models can predict variant effects “zero shot” (i.e., without additional training) by assuming that sequence likelihood, which they are trained to predict, is a good proxy for fitness [43]. Protein language models such as ESM1b have shown high accuracy and generalizability at variant effect prediction, including in the context of complex (non-missense) variants and alternative protein isoforms [13]. However, this approach may not scale indefinitely. While sequence likelihood correlates with fitness, it is ultimately a different prediction target. For example, a protein language model may predict amino acids by simply memorizing portions of the phylogenetic tree [44]. Indeed, recent works have shown that beyond a certain point, further scaling of protein language models only degrades their variant effect predictions [45]. Designing better training objectives that reliably track sequence fitness is an important open problem [46].
Hybrid methods: Recent variant pathogenicity prediction models such as AlphaMissense [9] and PrimateAI-3D [21] aim to get the best of both worlds by combining protein language modeling with supervised learning and explicit structure inference. AlphaMissense in particular is highly accurate at predicting pathogenic mutations [47]. However, the higher accuracy of this approach comes at the cost of reduced generality. For example, both AlphaMissense and PrimateAI-3D are restricted to single-letter substitutions, and cannot generalize to types of mutations they have not explicitly trained on such as insertions and deletions.
Clinical impact: Since the publication of clinical guidelines for variant classification about a decade ago [34], computational variant pathogenicity predictions have improved substantially. Presented with a random pathogenic and a random benign mutation from a clinical benchmark such as ClinVar [35], a modern AI model will correctly rank the pathogenic mutation as more damaging with over 90% probability across many types of variants [47]. These advances have led to much stronger clinical evidence being assigned to computational predictions [48,49] and improved methodology for deriving clinical evidence from predictions [50,51]. However, despite this significant progress, clinical guidelines still center on other sources of evidence and treat computational predictions as secondary. In light of the new state of the art, it may be time to come up with entirely new variant classification guidelines where computational predictions play a more central role.
Limitations: Despite the enormous progress we have seen, variant pathogenicity prediction models still have substantial limitations. First, the data used for training and evaluating these models gives a somewhat biased view of all the pathogenic mutations that truly exist. While ClinVar and other clinical datasets are quite comprehensive with respect to coding variants, they do not uniformly cover the entire coding genome and all types of genetic effects [47]. Additionally, pathogenicity is a rather abstract phenotype, and models that predict it directly from sequence are very likely to end up as “black boxes”. Users are left with little guidance on when to trust these predictions. As a starting point, more research is needed to characterize how prediction accuracy varies across genomic and disease contexts. Another limitation is that most models are optimized to detect mutations that inhibit molecular functions rather than enhance or change them. As a result, existing models tend to underperform on mutations with gain-of-function and dominant-negative effects [52]. Addressing these limitations calls for more efforts to predict concrete molecular and cellular phenotypes, thereby providing more context to the useful but abstract label of “pathogenic” or “damaging”.
3.1. The pursuit of mechanism: predicting genetic effects at the molecular level
Protein structure: To understand the consequences of a coding mutation at the molecular level, a good place to start is asking how it affects the structure of the protein. The 2024 Nobel Prize Committee described AlphaFold as having solved the problem of protein structure prediction [2,3]; but while a large chunk of the problem can indeed be considered solved, state-of-the-art models such as AlphaFold and RoseTTAFold still face major limitations [53]. In particular, it has been observed that AlphaFold performs poorly when dealing with mutated sequences [4], probably due to its training on predominantly wildtype proteins that fold correctly. In other words, protein structure prediction in its most general form – predicting the structure that an arbitrary amino-acid sequence will fold into – remains far from solved. This is especially true from the perspective of human genetics, where the object of interest is sequences that deviate from wildtype. For example, we do not yet have the ability to accurately predict how patient-specific mutations affect protein structure.
An alternative structure prediction approach was introduced by ESMFold based on protein language modeling [5]. Unlike AlphaFold, protein language models are not dependent on homology, giving ESMFold a significant advantage when dealing with mutated and de-novo sequences. However, ESMFold is overall less accurate than AlphaFold and RoseTTAFold at wildtype protein structure prediction, and a systematic evaluation of structure prediction models over mutated proteins is yet to be carried out.
An important dimension of structure prediction is the availability of structural data. The Protein Data Bank (PDB) [54], the main repository of protein structures, is dominated by proteins crystallized under artificial conditions. As a result, it does not fully reflect the actual protein conformations adopted in the aqueous environment of living cells, which include aspects such as conformational flexibility (e.g., fold switching) and intrinsically disordered regions. It also underrepresents many transient or complex protein assemblies that are too challenging to resolve with current experimental techniques [55,56]. Structure databases are further dominated by functional proteins, with few examples of destabilizing mutations that could reveal how sequence variation impacts protein structure [53]. All of these biases likely affect the predictions made by AlphaFold and other models.
Molecular phenotypes: Variant effect prediction can also be directed at specific molecular and cellular phenotypes. Data on these phenotypes often comes from multiplexed assays of variant effect (MAVE), a family of experimental methods for measuring the functional impact of tens of thousands of variants relative to a reference sequence [57,58]. In the context of coding mutations, deep mutational scans (DMS) can measure phenotypes such as protein expression, enzymatic activity, ligand binding, or cell proliferation. The molecular phenotypes measured by these methods often serve as proxies for variant pathogenicity [13,14,43]. Functional assays can also help uncover the mechanism underlying genetic effects or guide genetic engineering.
Computational prediction of molecular phenotypes can expedite these efforts by allowing experimentalists to assay a subset of mutations and computationally impute the rest. Several models have been proposed for this task, but accurately predicting molecular phenotypes in a robust and general way remains an open challenge. A recent study even found that many of these complex models are outperformed by a simple linear model [59].
The approach taken by existing phenotype prediction methods is to treat each assay as an independent prediction task. However, if the goal is to train models that capture the complex mechanisms of genetic effects, it might be necessary to move past one-dimensional scores toward predictions of richer phenotypes. For example, to distinguish between mutations that cause protein misfolding (resulting in low protein activity and expression) and mutations that disrupt protein activity through other mechanisms (resulting in low activity but normal expression), it may be necessary to consider readouts from both expression and activity assays. Modern experimental designs now allow high-throughput measurements of multiple phenotypes over the same set of mutations [60], opening the door to modeling variant effects as an inherently high-dimensional phenomenon. Unfortunately such rich datasets are still rare. There is a pressing need for more molecular data that goes beyond one-dimensional loss-of-function effects.
3.2. Predicting regulatory effects
Our main focus so far has been coding variants that affect phenotypes through changes to protein sequences. But the vast majority of variants in the human genome are in non-coding regions. Collectively, non-coding genetic variation accounts for most of the heritability for common diseases. Non-coding variants primarily affect gene regulation, namely how genes are expressed across different cells and biological contexts. Interpreting these complex effects is more challenging than analyzing protein sequence changes [61], and computational tools are generally less reliable in this context [62]. Nonetheless, recent progress gives reason for optimism.
Splicing: One major way in which variants influence gene regulation is by altering splicing. Several methods have been developed to predict splicing activity directly from DNA sequences. SpliceAI, for example, predicts the probabilities of each nucleotide in the input sequence acting as a splice acceptor, splice donor or neither [30]. By comparing these predictions between wildtype and mutated sequences, the model can identify splice mutations. Predicted probabilities from SpliceAI are also used as features by other methods (e.g., CADD) to determine whether intronic variants are pathogenic [10]. Recently, DeepMind’s AlphaGenome model was reported to provide improved predictions of splice variant effects compared to SpliceAI and other models [24].
Gene-expression phenotypes: Most non-coding variants are thought to affect phenotypes by modulating gene expression. Gene expression changes can in turn be mediated by chromatin modifications such as DNA methylation or histone modifications. Accordingly, many AI models, including DeepSEA [26], ExPecto [28] and Enformer [27], have been developed to predict chromatin marks from DNA sequences. Commonly predicted chromatin labels include histone modifications, transcription factor binding, and chromatin accessibility across cell types (catalogued by the ENCODE project) [63].
A common strategy, employed by ExPecto and Enformer, is to pretrain a model to predict chromatin labels and then fine-tune it to predict gene expression (e.g., over GTEx [64]). However, because these models are trained on wildtype sequences from the reference genome, they are primarily useful for predicting baseline (population-average) gene expression, and generally fail to capture individual-level differences driven by genetic variation [25,63].
A few recent models have taken different approaches. Rather than predicting normalized transcript counts, the Borzoi model directly predicts RNA sequencing coverage across the genome [65]. This allowed unified modeling of multiple layers of gene regulation, including splicing, and marginally improved variant effect prediction. Building and expending on this approach, AlphaGenome was trained as a large-scale foundation model to simultaneously predict 5,930 human and 1,128 mouse genomic annotations across 11 modalities, including gene expression, splicing, chromatin states, and chromatin contact maps [24]. It was reported to moderately improve over Borzoi in predicting the effect sizes and inferred causality of genetic associations with gene expression changes (eQTLs). Unfortunately, AlphaGenome is not yet available as an open-source model and there are no plans to release its training code. Other models, such as PrediXcan [29], are directly trained to predict individual-level gene expression from genotyped variants, but are limited to variants present in the training data (e.g., GTEx).
Importantly, the majority of benchmarks for genetic effects on gene expression are based on association studies. To make progress on models that predict causal genetic effects, we need data that includes not only natural genetic variation but also synthetic perturbations (e.g., from massively parallel reporter assays and Perturb-Seq) [63]. Such experimental data remains limited, and the data we do have is insufficiently utilized by existing benchmarks. Another open challenge is modeling long-distance genomic interactions. Enformer, for example, has a context size of 100K nt, which in principle should allow it to capture many distal enhancers, but it has been shown to mostly focus on nearby promoters while largely ignoring more distant dependencies [66]. Another dimension that is not fully accounted for is cell-type differences [67]. Existing models are trained over a predefined set of cell types, but this approach cannot account for the full repertoire of cells in the body. On top of that, gene expression and chromatin states, like most phenotypes, are influenced by non-genetic factors that should be accounted for.
Non-coding genes: A handful of computational tools have been designed for predicting the consequences of genetic variation affecting non-coding genes (defined as genes that produce functional RNA but are not coding for proteins). These include models for variants affecting microRNAs [68] and their 3’ untranslated region targets [69], mitochondrial tRNA genes [70], and RNA base-pairing (secondary structure) disruptions [71]. Despite these efforts, genetic changes in non-coding genes remains an underexplored area of genomic AI. Likewise, there are very few annotated variant effects (e.g., in ClinVar [35]) in non-coding genes.
Pathogenicity of non-coding mutations: Several pathogenicity prediction models are applicable to non-coding variants. CADD, first released in 2019 [11] and last updated in 2024 [12], predicts variant pathogenicity based on dozens of coding and non-coding genomic annotations, including outputs from underlying AI models (e.g., SpliceAI). Other methods, including DANN [72], FATHMM-MKL [16] and FATHMM-XF [15], implement similar strategies. While these tools can in principle score any variant, their performance is strongest in well-characterized parts of the genome that have extensive features (e.g., coding and intronic regions) and likely much worse in poorly understood areas (e.g., lncRNA genes). A more unbiased approach is taken by PhyloP which, similar to SIFT, uses a phylogenetic model to compare observed mutation frequencies to a baseline model of neutral evolution [19]. However, PhyloP reflects only whether a given genomic site is conserved, without accounting for the specific base changes introduced by a particular variant.
Evaluation of variant pathogenicity prediction relies on clinical databases such as ClinVar [35]. Some types of variants, such as those in splice sites or untranslated regions of messenger RNAs, are sufficiently common to allow reliable estimates of model accuracy. Others, including variants in promoters or distant regulatory elements, are exceedingly rare in clinical databases, making it difficult to draw conclusions about model performance in these genomic regions.
4. Universal Genomic Models
There is an overall trend in AI towards increasing generality, where more general models often come to outperform specialized ones over time. We should expect a similar trend in genetics. A single general model could potentially handle all types of genetic effects, avoiding the need to design a separate algorithm for each case. Moreover, because our understanding of the genome is incomplete, we often do not know which genomic features are relevant or what should even be predicted. By taking a more general approach, we can train models that discover the rules of genomics on their own, sidestepping these uncertainties.
DNA sequences, like proteins, are natural targets for language modeling. Whereas protein language models are trained to predict protein sequences (Figure 3A) and thereby implicitly learn the evolutionary forces shaping protein structure and function (Figure 3B), DNA language models do the same at the DNA level. By predicting the repertoire of DNA sequences found across species, they implicitly learn about the forces that shape genomes, including natural selection and the diverse functions encoded within them. Training directly on DNA sequences allows the development of general-purpose foundation models that learn about the genome without being tied to any particular task [7]. DNA language models are, in principle, applicable to all genomic regions and can take into account all potential effects of a variant without resorting to hard-coded rules. This can be important, for example, for variants with unexpected effects (e.g., a coding variant that also affects splicing or gene expression).
While the concept of DNA language modeling appears promising, early implementations showed mixed results. One preprint even reported that many DNA language models perform just as well when initialized with random weights before fine-tuning on specific tasks, suggesting that DNA language modeling itself does not contribute much to the performance of these specific models [73].
A more recent DNA language model called Evo2 appears to be a significant leap forward, demonstrating clear and useful applications [7]. In particular, Evo2 shows close to state-of-the-art accuracy at variant effect prediction with respect to deep mutational scans and variant pathogenicity. For coding variants, Evo2 is approaching the performance of protein language models, and it appears to also perform well on non-coding variants [47].
Another recent model called GPN-MSA shows even better accuracy and robustness at variant effect prediction across both coding and non-coding regions [17,47]. While the basic idea resembles DNA language modeling, GPN-MSA is dependent on multiple sequence alignment (MSA) inputs, where a target human sequence is interpreted alongside homologous sequences from other species. This approach is beneficial for overall prediction accuracy, but limits the model to naturally occurring sequences with good MSA coverage. A similar model called PhyloGPN used MSA only during training, thereby allowing the model to make predictions from a single input sequence [18], but at the cost of reduced accuracy [47].
Overall, our ability to predict the damaging effect of every possible variant in the human genome has greatly improved, but this has mostly been demonstrated in the context of Mendelian monogenic diseases. More work is needed to expand the applicability of genomic AI to other contexts and types of genetic effects. It might also be beneficial to allow models to learn from human genetic variation (e.g., allele frequencies) in addition to wildtype sequences across different species [74].
An important unresolved question for genome modeling is when genomes can be treated as self-contained: what can be inferred from DNA sequence alone, and when do we need other types of data? Some problems can only be posed with respect to multiple modalities. For example, predicting DNA-protein binding requires training on both DNA and protein sequences. Very recent studies have begun to explore multimodal genomic foundation models, such as language models trained jointly on nucleotide and amino-acid sequences [75], but their practical value remains to be seen. A key challenge is how to design training objectives that require foundation models to learn general relationships between modalities that remain useful across tasks. For example, training a foundation model that takes DNA-protein pairs requires specifying which DNA and protein sequences to pair, which is not straightforward because proteins and DNA can interact in many different ways.
5. Informing Genetic Association Studies with Genomic Predictions
Much of our focus so far has been on variant pathogenicity as a generic phenotype, putting aside the question of which specific disease we are talking about. Of course, a big part of human genetics is identifying which genetic elements affect which specific traits. AI is useful for these efforts as well.
Genome-wide association studies (GWAS) provide a data-driven approach to identify variants associated with a given trait. However, complex correlations – both among genetic variants and between genetic and environmental factors – make it extremely challenging to distinguish causal from non-causal genetic associations [76]. Purely statistical methods also lack the power to implicate variants that are not sufficiently common in the population. Genomic AI can address these challenges by providing complementary evidence about the likelihood of variants and genes being causal based on their genomic context.
Fine-mapping GWAS variants: Given that most GWAS associations are not causal, substantial efforts have been dedicated to developing methods for identifying the causal variants underlying genetic associations, a process known as fine-mapping. Most fine-mapping approaches remain purely statistical, while sometimes incorporating genomic annotations [77]. A few studies have attempted to use variant effect predictions as priors to obtain smaller credible sets of variants with high likelihood of containing the causal variants [78]. The rationale is that predictions based on the genomic sequence indicate whether a variant affects DNA patterns linked to gene regulation. Such predictions place the purely statistical GWAS associations in a genomic context and help identify variants that are more likely to be causal. Many of the AI models for predicting the chromatin and gene-expression effects of non-coding variants, including DeepSEA, ExPecto, Enformer and PrediXcan, were originally developed for the purpose of determining which GWAS variants are more likely to be causal. Unfortunately, these models have a limited capacity to capture gene-expression differences between individuals [25,63], limiting their utility for fine-mapping.
Other models, including GWAVA [32] and cV2F [31], were explicitly trained to predict whether GWAS variants are causal. However, independent evaluations and comparisons of fine-mapping algorithms have largely focused on the purely statistical approaches while neglecting the ML-based methods. These efforts also suffer from a data limitation problem, as ground-truth causal variants are not easy to come by.
Gene-level approaches: Some methods have shifted focus from variants to genes. Since each causal gene likely involves multiple causal variants, it is statistically easier to implicate them. For example, a model named FLAMES was trained to predict the most likely causal gene at a GWAS-significant region by integrating gene features (e.g., gene expression and gene ontology) and variant-to-gene features (e.g., eQTLs and chromatin interactions) [33]. However, because very few ground-truth causal genes are known, gene prioritization models are often trained on proxies for causality. FLAMES, for example, used genes with a significant burden of loss-of-function mutations in disease carriers as proxy causal genes.
Some burden tests leverage variant effect predictions to discover gene-trait associations directly. By using genomic predictions as priors, these methods benefit from increased sensitivity to genetic effects that can be detected by AI models (Figure 4). However, this comes at the cost of bias toward the type of effects that the genomic AI is capable of detecting. Transcriptome-wide association study (TWAS) [79] and PrediXcan [29] train models to impute gene expression from genotypes, and then apply the trained models to identify genes whose predicted gene expression correlates with a phenotype. Similarly, proteome-wide association study (PWAS) uses a variant pathogenicity prediction model to identify proteins whose predicted functional damage due to coding mutations is correlated with a phenotype [80,81].
Using the right genomic priors: Many of the mentioned methods use variant effect predictions as priors for how likely variants are to be causal. While this seems like a natural strategy, it is important to recognize that existing variant effect predictions are typically optimized for variants linked to Mendelian diseases. Some studies suggest that these predictions do not generalize well to complex (i.e., non-Mendelian) traits [82,83]. More general models that are not specifically trained on labels from Mendelian diseases (e.g., DNA language models) could prove more appropriate in that context, but further research is needed.
6. Disease Risk Estimation
Looking at an individual’s genome and accurately estimating their chances of encountering different medical conditions has long been a holy grail in human genetics. Predicting an outcome (disease) as a function of observed variables (genetics) is a typical machine-learning task. Some direct-to-consumer genetic testing services like 23andMe offer such disease risk reports. When considering common diseases such as cancer, cardiovascular, neurodegenerative and psychiatric conditions, these estimates are typically based on polygenic scores which combine the estimated effects of all variants present in an individual's genome into an overall risk score [84].
Contrary to the trend in other domains of ML, polygenic scores are still dominated by simple linear models [76,84]. Attempts to account for nonlinearities between variants have so far failed to provide significant improvements in phenotype predictions. A mixture of empirical [85] and theoretical [86,87] justifications have been articulated to argue that, statistically speaking, the variance of most traits can be explained by an additive model of variant effects. But others do not see it as a settled case and advocate for more exploration of nonlinear genetic models [76,88,89,90,91].
Another notable departure from modern AI trends is that most polygenic scores are purely statistical, treating variants as random variables we have no prior knowledge about. That is likely suboptimal. Some methods have tried supplementing variants with predefined genomic annotations, but the models are still left to learn from scratch how these annotations relate to the target phenotype [92]. Experience from other domains of AI suggests that training on additional tasks beyond just the target phenotype would be beneficial. For example, fine-tuning genomic foundation models to predict disease risk would take advantage of strong genomic priors while also allowing nonlinear modeling. One study used PrimateAI-3D [21] to quantify the contributions of rare variants to disease risk. They found that while common variants explain more phenotypic variance overall, rare-variant polygenic scores had more power at the ends of the distribution to identify individuals at the greatest risk for disease. This suggests that rare-variant polygenic scores may be more relevant for population genetic screening and risk management [93]. Another underutilized source of information that could be integrated into polygenic scores is clinical markers and other non-genetic factors, as most common diseases are strongly influenced by an individual’s lifestyle and history of environmental exposures [94,95,96].
After several years of developing and studying polygenic scores, we are finally starting to see strong evidence for their clinical utility, particularly for identifying high-risk individuals who may benefit from additional medical screenings [96,97,98]. But the medical community is still reluctant to fully implement these genetic scores. For example, the American College of Medical Genetics and Genomics (ACMG) does not yet recommend widespread use of polygenic scores [99]. One of the main barriers to adoption is the limited robustness of polygenic scores when applied over genetic cohorts different from the ones they were originally trained on [76]. In particular, individuals of non-European ancestry tend to receive much poorer predictions due to their underrepresentation in genetic studies [99,100].
7. Challenges for Real-World Impact
Let us conclude with a brief overview of challenges that will be critical in the coming years to unlock the full potential of AI in human genetics.
Benchmarks make the AI world go around: It is difficult to exaggerate the role that well-designed datasets play in driving AI progress. As the AlphaFold team noted: “We’re indebted to [...] the whole community, not least the experimentalists whose structures enable this kind of rigorous assessment” [1]. AI researchers tend to be opportunistic and go after problems where high-quality data and benchmarks are available. This largely explains why databases such as ClinVar [35] and the Protein Data Bank (PDB) [54] have produced some of the most capable genomic AI. In other areas we are still lacking sufficiently challenging benchmarks that reliably track important open problems. Without such benchmarks, AI developers may be misled into believing that they are making progress [73].
Foundation models in particular, precisely because they can be applied to so many different tasks, need to be critically evaluated on important problems and compared to strong baselines. The importance of strong baselines was recently exemplified by the finding that many sophisticated models for predicting the functional effects of mutations were outperformed by a simple linear model [59].
Beware of homology: The exceptional predictive power of homologous sequences sharing an evolutionary origin with the target sequence is one of the most reliable guiding principles in computational genomics [14,17,39]. Simple homology-based methods such as SIFT [23] and phyloP [19] remain competitive even more than 15-25 years after their development [7]. It is safe to say that including homology information will make almost any model perform better on benchmarks. However, relying on homology too much can prevent models from generalizing. Dependence on homology is especially dangerous in the context of synthetic or mutated sequences that deviate from naturally-occurring or wildtype sequences. Since different variations of the same gene share the same homologues, a homology-based approach may not distinguish between them. AlphaFold, for example, performs poorly on mutated sequences, in part due to its dependence on multiple sequence alignment inputs [4]. Dependence on homology can also preclude genomic targets without a sufficient number of close homologues [13,14].
Interpretability matters: One of the greatest barriers to widespread use of AI in genetics is the “black box” nature of many models, especially frontier deep-learning models with billions of inscrutable parameters. In the context of clinical genetics, most clinicians would be reluctant to diagnose a patient based on predictions from an inscrutable deep-learning model. Genetic discovery too would progress much faster if we had more clarity about what patterns the models pick up on.
Thankfully, the mechanistic interpretability field of AI has progressed a lot over the past few years. For example, a few recent works have extracted human-interpretable features and explained parts of the computation of frontier large language models using sparse autoencoders, which decompose trained models into sparse features [101,102]. This approach is entirely unsupervised, relying on the assumption that sparse features, where only a few features are active for each data point, are more likely to be interpretable. Some of these techniques have been applied to genomic models. For example, sparse features extracted from Evo2 were found to match recognizable genomic elements such as exons and introns [7]. Another line of work explores inherently interpretable model designs, for example to discover RNA motifs that affect splicing [103].
That said, interpreting AI is fundamentally challenging. Models often find creative ways to process information, which can be quite different from what their designers intended (e.g., taking advantage of unintended correlations in the data [104]). For example, most language models contain self-attention layers designed to model the extent to which each element in the sequence depends on each other element. It used to be fashionable to try to interpret these numbers, but growing evidence suggests that they do not provide meaningful explanations for predictions [105]. Despite the enormous challenge, understanding the complex logic trained into deep-learning models is one of the most important problems to work on.
We need more clinical implementation work: Once a model has been developed and benchmarked, it is tempting to consider the work complete. But if the goal is to benefit real patients, then the hardest part has not even begun. For instance, it is one thing to show that a polygenic score has some predictive power for disease risk, and another thing entirely to establish how it can meaningfully improve clinical decision making. Variant pathogenicity scores, too, need to be calibrated according to guidelines before they can be applied for variant classification in clinical settings [48,49,51]. These efforts require knowledge of both machine learning and clinical guidelines, which is not a common intersection of skills.
Academic funding is ill-suited for serious AI work: Judging by where AI investments are going, one might conclude that our society values cat videos more than life-saving biomedical breakthroughs. Frontier genomic models remain orders-of-magnitude smaller than their text and image counterparts [106], and academia is struggling to keep up with for-profit AI companies [7,42]. Meanwhile, models that do come out of academic labs rarely meet basic infrastructure standards, largely due to limited engineering talent. Unfortunately, it is exceedingly difficult to convince funding agencies that such talent is required and needs to be paid competitive salaries. This funding gap could in principle be met if government and philanthropic funders better appreciated the importance of state-of-the-art hardware and skilled engineers for AI progress.
Biosecurity needs to be taken seriously: As genomic AI becomes more powerful, it also becomes more dangerous in the hands of bad actors. As a recent policy article put it, “the same biological model able to design a benign viral vector to deliver gene therapy could be used to design a more pathogenic virus capable of evading vaccine-induced immunity” [107]. While it is not realistic to expect model developers to be fully responsible for every possible abuse of their work, researchers should exercise caution and foresight to help mitigate such risks. For example, it might be a good idea to exclude eukaryote-infecting viruses from the training of public foundation models [7], although this alone is likely insufficient [107].
Acknowledgements
I would like to thank Po-Yu Lin (NYU), Anushka Sinha (NYU), Juan Irizarry Cole (NYU), Omer Weissbrod (Eleven Therapeutics), Oded Regev (NYU) and Yaniv Shmueli (Cassidy Bio) for valuable feedback on earlier drafts of this review.
References
- DeepMind. AlphaFold: A Solution to a 50-Year-Old Grand Challenge in Biology [Internet]. 2020. Available from: https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology.
- Outreach, NP. The Nobel Prize in Chemistry 2024 [Internet]. 2024. Available from: https://www.nobelprize.org/prizes/chemistry/2024/press-release/.
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; et al. Highly accurate protein structure prediction with AlphaFold. nature. 2021, 596, 583–9. [Google Scholar] [CrossRef] [PubMed]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; et al. Using AlphaFold to predict the impact of single mutations on protein stability and function. Plos One. 2023, 18, e0282689. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. 2023, 379, 1123–30. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci. 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Brixi G, Durrant MG, Ku J, Poli M, Brockman G, Chang D, et al. Genome modeling and design across all domains of life with Evo 2. bioRxiv. 2025;2025–02.
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021, 373, 871–6. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science. 2023, 381, eadg7492. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Schubach, M.; Shendure, J.; Kircher, M. CADD-Splice—improving genome-wide variant effect prediction using deep learning-derived splice scores. Genome Med. 2021, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–94. [Google Scholar] [CrossRef] [PubMed]
- Schubach, M.; Maass, T.; Nazaretyan, L.; Röner, S.; Kircher, M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024, 52, D1143–54. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Goldman, G.; Wang, C.H.; Ye, C.J.; Ntranos, V. Genome-wide prediction of disease variant effects with a deep protein language model. Nat Genet. 2023, 55, 1512–22. [Google Scholar] [CrossRef] [PubMed]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; et al. Disease variant prediction with deep generative models of evolutionary data. Nature. 2021, 599, 91–5. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.F.; Shihab, H.A.; Mort, M.; Cooper, D.N.; Gaunt, T.R.; Campbell, C. FATHMM-XF: accurate prediction of pathogenic point mutations via extended features. Bioinformatics. 2018, 34, 511–3. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; et al. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics. 2015, 31, 1536–43. [Google Scholar] [CrossRef] [PubMed]
- Benegas G, Albors C, Aw AJ, Ye C, Song YS. A DNA language model based on multispecies alignment predicts the effects of genome-wide variants. Nat Biotechnol. 2025;1–6.
- Albors C, Li JC, Benegas G, Ye C, Song YS. A Phylogenetic Approach to Genomic Language Modeling. ArXiv Prepr ArXiv250303773. 2025.
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–21. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Gao H, Hamp T, Ede J, Schraiber JG, McRae J, Singer-Berk M, et al. The landscape of tolerated genetic variation in humans and primates. Science. 2023 ;380(6648):eabn8153. 2 June.
- Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99(4):877–85.
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001 May;11(5):863–74.
- Avsec Ž, Latysheva N, Cheng J, Novati G, Taylor KR, Ward T, et al. AlphaGenome: advancing regulatory variant effect prediction with a unified DNA sequence model. bioRxiv. 2025;2025–06.
- Sasse A, Ng B, Spiro AE, Tasaki S, Bennett DA, Gaiteri C, et al. Benchmarking of deep neural networks for predicting personal gene expression from DNA sequence highlights shortcomings. Nat Genet. 2023 Dec 1;55(12):2060–4.
- Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning–based sequence model. Nat Methods. 2015 Oct 1;12(10):931–4.
- Avsec Ž, Agarwal V, Visentin D, Ledsam JR, Grabska-Barwinska A, Taylor KR, et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat Methods. 2021 Oct 1;18(10):1196–203.
- Zhou J, Theesfeld CL, Yao K, Chen KM, Wong AK, Troyanskaya OG. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat Genet. 2018 Aug 1;50(8):1171–9.
- Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015 Sept 1;47(9):1091–8.
- Jaganathan K, Panagiotopoulou SK, McRae JF, Darbandi SF, Knowles D, Li YI, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535–48.
- Fabiha T, Evergreen I, Kundu S, Pampari A, Abramov S, Boytsov A, et al. A consensus variant-to-function score to functionally prioritize variants for disease. bioRxiv. 2024;2024–11.
- Ritchie GRS, Dunham I, Zeggini E, Flicek P. Functional annotation of noncoding sequence variants. Nat Methods. 2014 Mar 1;11(3):294–6.
- Schipper M, de Leeuw CA, Maciel BAPC, Wightman DP, Hubers N, Boomsma DI, et al. Prioritizing effector genes at trait-associated loci using multimodal evidence. Nat Genet. 2025 Feb 1;57(2):323–33.
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015 ;17(5):405–23. 1 May.
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8.
- Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet. 2015;97(2):199–215.
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med. 2016;18(7):696–704.
- Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Med. 2020 Dec 2;12(1):103.
- Ofer D, Brandes N, Linial M. The language of proteins: NLP, machine learning & protein sequences. Comput Struct Biotechnol J. 2021;19:1750–8.
- Brandes N, Ofer D, Peleg Y, Rappoport N, Linial M. ProteinBERT: a universal deep-learning model of protein sequence and function. Bioinformatics. 2022;38(8):2102–10.
- Elnaggar A, Heinzinger M, Dallago C, Rehawi G, Wang Y, Jones L, et al. Prottrans: Toward understanding the language of life through self-supervised learning. IEEE Trans Pattern Anal Mach Intell. 2021;44(10):7112–27.
- Hayes T, Rao R, Akin H, Sofroniew NJ, Oktay D, Lin Z, et al. Simulating 500 million years of evolution with a language model. Science. 2025;eads0018.
- Meier J, Rao R, Verkuil R, Liu J, Sercu T, Rives A. Language models enable zero-shot prediction of the effects of mutations on protein function. Adv Neural Inf Process Syst. 2021;34:29287–303.
- Weinstein E, Amin A, Frazer J, Marks D. Non-identifiability and the blessings of misspecification in models of molecular fitness. Adv Neural Inf Process Syst. 2022;35:5484–97.
- Hou C, Liu D, Zafar A, Shen Y. Understanding Protein Language Model Scaling on Mutation Effect Prediction. bioRxiv. 2025;2025–04.
- Pugh CW, Nuñez-Valencia PG, Dias M, Frazer J. From Likelihood to Fitness: Improving Variant Effect Prediction in Protein and Genome Language Models. bioRxiv. 2025;2025–05.
- Lu B, Liu X, Lin PY, Brandes N. Genomic heterogeneity inflates the performance of variant pathogenicity predictions. bioRxiv. 2025;2025–09.
- Wilcox EH, Sarmady M, Wulf B, Wright MW, Rehm HL, Biesecker LG, et al. Evaluating the impact of in silico predictors on clinical variant classification. Genet Med. 2022;24(4):924–30.
- Bergquist T, Stenton SL, Nadeau EA, Byrne AB, Greenblatt MS, Harrison SM, et al. Calibration of additional computational tools expands ClinGen recommendation options for variant classification with PP3/BP4 criteria. bioRxiv. 2024.
- Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med. 2018;20(9):1054–60.
- Lin PY, Brandes N. P-KNN: Maximizing variant classification evidence through joint calibration of multiple pathogenicity prediction tools. bioRxiv. 2025.
- Gerasimavicius L, Livesey BJ, Marsh JA. Loss-of-function, gain-of-function and dominant-negative mutations have profoundly different effects on protein structure. Nat Commun. 2022 ;13(1):3895. 6 July.
- Fadini A, Li M, McCoy AJ, Terwilliger TC, Read RJ, Hekstra D, et al. AlphaFold as a Prior: Experimental Structure Determination Conditioned on a Pretrained Neural Network. bioRxiv. 2025;2025–02.
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–42.
- Terwilliger TC, Liebschner D, Croll TI, Williams CJ, McCoy AJ, Poon BK, et al. AlphaFold predictions are valuable hypotheses and accelerate but do not replace experimental structure determination. Nat Methods. 2024;21(1):110–6.
- Chakravarty D, Lee M, Porter LL. Proteins with alternative folds reveal blind spots in AlphaFold-based protein structure prediction. Curr Opin Struct Biol. 2025;90:102973.
- Esposito D, Weile J, Shendure J, Starita LM, Papenfuss AT, Roth FP, et al. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol. 2019;20:1–11.
- Notin P, Kollasch A, Ritter D, Van Niekerk L, Paul S, Spinner H, et al. Proteingym: Large-scale benchmarks for protein fitness prediction and design. Adv Neural Inf Process Syst. 2023;36:64331–79.
- Hsu C, Nisonoff H, Fannjiang C, Listgarten J. Learning protein fitness models from evolutionary and assay-labeled data. Nat Biotechnol. 2022 ;40(7):1114–22. 1 July.
- LaFlam TN, Billesbølle CB, Dinh T, Wolfreys FD, Lu E, Matteson T, et al. Phenotypic pleiotropy of missense variants in human B cell-confinement receptor P2RY8. bioRxiv. 2025;2025–02.
- Ellingford JM, Ahn JW, Bagnall RD, Baralle D, Barton S, Campbell C, et al. Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Med. 2022;14(1):73.
- Wang Z, Zhao G, Li B, Fang Z, Chen Q, Wang X, et al. Performance comparison of computational methods for the prediction of the function and pathogenicity of non-coding variants. Genomics Proteomics Bioinformatics. 2023;21(3):649–61.
- Sasse A, Chikina M, Mostafavi S. Unlocking gene regulation with sequence-to-function models. Nat Methods. 2024;21(8):1374–7.
- Consortium Gte. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318–30.
- Linder J, Srivastava D, Yuan H, Agarwal V, Kelley DR. Predicting RNA-seq coverage from DNA sequence as a unifying model of gene regulation. Nat Genet [Internet]. 2025 Jan 8. [CrossRef]
- Karollus A, Mauermeier T, Gagneur J. Current sequence-based models capture gene expression determinants in promoters but mostly ignore distal enhancers. Genome Biol. 2023 Mar 27;24(1):56.
- Regev A, Teichmann SA, Lander ES, Amit I, Benoist C, Birney E, et al. The human cell atlas. elife. 2017;6:e27041.
- Cammaerts S, Strazisar M, Dierckx J, Del Favero J, De Rijk P. miRVaS: a tool to predict the impact of genetic variants on miRNAs. Nucleic Acids Res. 2016;44(3):e23–e23.
- Barenboim M, Zoltick BJ, Guo Y, Weinberger DR. MicroSNiPer: a web tool for prediction of SNP effects on putative microRNA targets. Hum Mutat. 2010;31(11):1223–32.
- Sonney S, Leipzig J, Lott MT, Zhang S, Procaccio V, Wallace DC, et al. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput Biol. 2017;13(12):e1005867.
- Sabarinathan R, Tafer H, Seemann SE, Hofacker IL, Stadler PF, Gorodkin J. RNA snp: efficient detection of local RNA secondary structure changes induced by SNP s. Hum Mutat. 2013;34(4):546–56.
- Quang D, Chen Y, Xie X. DANN: a deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics. 2014;31(5):761–3.
- Vishniakov K, Viswanathan K, Medvedev A, Kanithi PK, Pimentel MA, Rajan R, et al. Genomic Foundationless Models: Pretraining Does Not Promise Performance. bioRxiv. 2024;2024–12.
- He AY, Palamuttam NP, Danko CG. Training deep learning models on personalized genomic sequences improves variant effect prediction. United States; 2025.
- He Y, Fang P, Shan Y, Pan Y, Wei Y, Chen Y, et al. Generalized biological foundation model with unified nucleic acid and protein language. Nat Mach Intell. 2025;1–12.
- Brandes N, Weissbrod O, Linial M. Open problems in human trait genetics. Genome Biol. 2022;23(1):131.
- Schaid DJ, Chen W, Larson NB. From genome-wide associations to candidate causal variants by statistical fine-mapping. Nat Rev Genet. 2018 Aug 1;19(8):491–504.
- Kathail P, Bajwa A, Ioannidis NM. Leveraging genomic deep learning models for non-coding variant effect prediction. ArXiv Prepr ArXiv241111158. 2024.
- Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016 Mar 1;48(3):245–52.
- Brandes N, Linial N, Linial M. PWAS: proteome-wide association study—linking genes and phenotypes by functional variation in proteins. Genome Biol. 2020;21(1):173.
- Kelman G, Zucker R, Brandes N, Linial M. PWAS Hub for exploring gene-based associations of common complex diseases. Genome Res. 2024;34(10):1674–86.
- Kim SS, Dey KK, Weissbrod O, Márquez-Luna C, Gazal S, Price AL. Improving the informativeness of Mendelian disease-derived pathogenicity scores for common disease. Nat Commun. 2020 Dec 7;11(1):6258.
- Benegas G, Eraslan G, Song YS. Benchmarking DNA Sequence Models for Causal Variant Prediction in Human Genetics.
- Choi SW, Mak TSH, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15(9):2759–72.
- Polderman TJC, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015 ;47(7):702–9. 1 July.
- Mäki-Tanila A, Hill WG. Influence of Gene Interaction on Complex Trait Variation with Multilocus Models. Genetics. 2014 July;198(1):355–67.
- Moore JH, Williams SM. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. Bioessays. 2005;27(6):637–46.
- Génin, E. Missing heritability of complex diseases: case solved? Hum Genet. 2020 Jan 1;139(1):103–13.
- Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci. 2012;109(4):1193–8.
- Li J, Li X, Zhang S, Snyder M. Gene-environment interaction in the era of precision medicine. Cell. 2019;177(1):38–44.
- Brandes N, Linial N, Linial M. Genetic association studies of alterations in protein function expose recessive effects on cancer predisposition. Sci Rep. 2021;11(1):14901.
- Zheng Z, Liu S, Sidorenko J, Wang Y, Lin T, Yengo L, et al. Leveraging functional genomic annotations and genome coverage to improve polygenic prediction of complex traits within and between ancestries. Nat Genet. 2024 ;56(5):767–77. 1 May.
- Fiziev PP, McRae J, Ulirsch JC, Dron JS, Hamp T, Yang Y, et al. Rare penetrant mutations confer severe risk of common diseases. Science. 2023;380(6648):eabo1131.
- Moldovan A, Waldman YY, Brandes N, Linial M. Body mass index and birth weight improve polygenic risk score for type 2 diabetes. J Pers Med. 2021;11(6):582.
- Lin J, Mars N, Fu Y, Ripatti P, Kiiskinen T, Tukiainen T, et al. Integration of Biomarker Polygenic Risk Score Improves Prediction of Coronary Heart Disease. JACC Basic Transl Sci. 2023 Dec;8(12):1489–99.
- Samani NJ, Beeston E, Greengrass C, Riveros-McKay F, Debiec R, Lawday D, et al. Polygenic risk score adds to a clinical risk score in the prediction of cardiovascular disease in a clinical setting. Eur Heart J. 2024 Sept 7;45(34):3152–60.
- McHugh JK, Bancroft EK, Saunders E, Brook MN, McGrowder E, Wakerell S, et al. Assessment of a polygenic risk score in screening for prostate cancer. N Engl J Med. 2025;392(14):1406–17.
- Padrik P, Tõnisson N, Hovda T, Sahlberg KK, Hovig E, Costa L, et al. Guidance for the Clinical Use of the Breast Cancer Polygenic Risk Scores. Cancers [Internet]. 2025;17(7). Available from: https://www.mdpi.com/2072-6694/17/7/1056.
- Abu-El-Haija A, Reddi HV, Wand H, Rose NC, Mori M, Qian E, et al. The clinical application of polygenic risk scores: A points to consider statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2023;25(5):100803.
- Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019 Apr 1;51(4):584–91.
- Templeton A, Conerly T, Marcus J, Lindsey J, Bricken T, Chen B, et al. Scaling Monosemanticity: Extracting Interpretable Features from Claude 3 Sonnet. Transform Circuits Thread [Internet]. 2024; Available from: https://transformer-circuits.pub/2024/scaling-monosemanticity/index.html.
- Ameisen E, Lindsey J, Pearce A, Gurnee W, Turner NL, Chen B, et al. Circuit Tracing: Revealing Computational Graphs in Language Models. Transform Circuits Thread [Internet]. 2025; Available from: https://transformer-circuits.pub/2025/attribution-graphs/methods.html.
- Liao SE, Sudarshan M, Regev O. Deciphering RNA splicing logic with interpretable machine learning. Proc Natl Acad Sci. 2023;120(41):e2221165120.
- Xiao K, Engstrom L, Ilyas A, Madry A. Noise or signal: The role of image backgrounds in object recognition. ArXiv Prepr ArXiv200609994. 2020.
- Jain S, Wallace BC. Attention is not explanation. ArXiv Prepr ArXiv190210186. 2019.
- Ma Z, He J, Qiu J, Cao H, Wang Y, Sun Z, et al. BaGuaLu: targeting brain scale pretrained models with over 37 million cores. In: Proceedings of the 27th ACM SIGPLAN Symposium on Principles and Practice of Parallel Programming. 2022. p. 192–204.
- Bloomfield D, Pannu J, Zhu AW, Ng MY, Lewis A, Bendavid E, et al. AI and biosecurity: The need for governance. Science. 2024;385(6711):831–3.
Figure 1.
From task-specific machine learning to general-purpose AI. (A) Classical machine learning models are trained for a specific task (e.g., predicting the effects of mutations) using carefully curated datasets tailored to that task. (B) The modern AI approach, in contrast, centers around general-purpose foundation models trained on massive amounts of data. The pretrained model can then be applied to a wide range of applications either directly (zero-shot) or via additional training (fine-tuning). New capabilities of a model are sometimes discovered years after its release.
Figure 1.
From task-specific machine learning to general-purpose AI. (A) Classical machine learning models are trained for a specific task (e.g., predicting the effects of mutations) using carefully curated datasets tailored to that task. (B) The modern AI approach, in contrast, centers around general-purpose foundation models trained on massive amounts of data. The pretrained model can then be applied to a wide range of applications either directly (zero-shot) or via additional training (fine-tuning). New capabilities of a model are sometimes discovered years after its release.
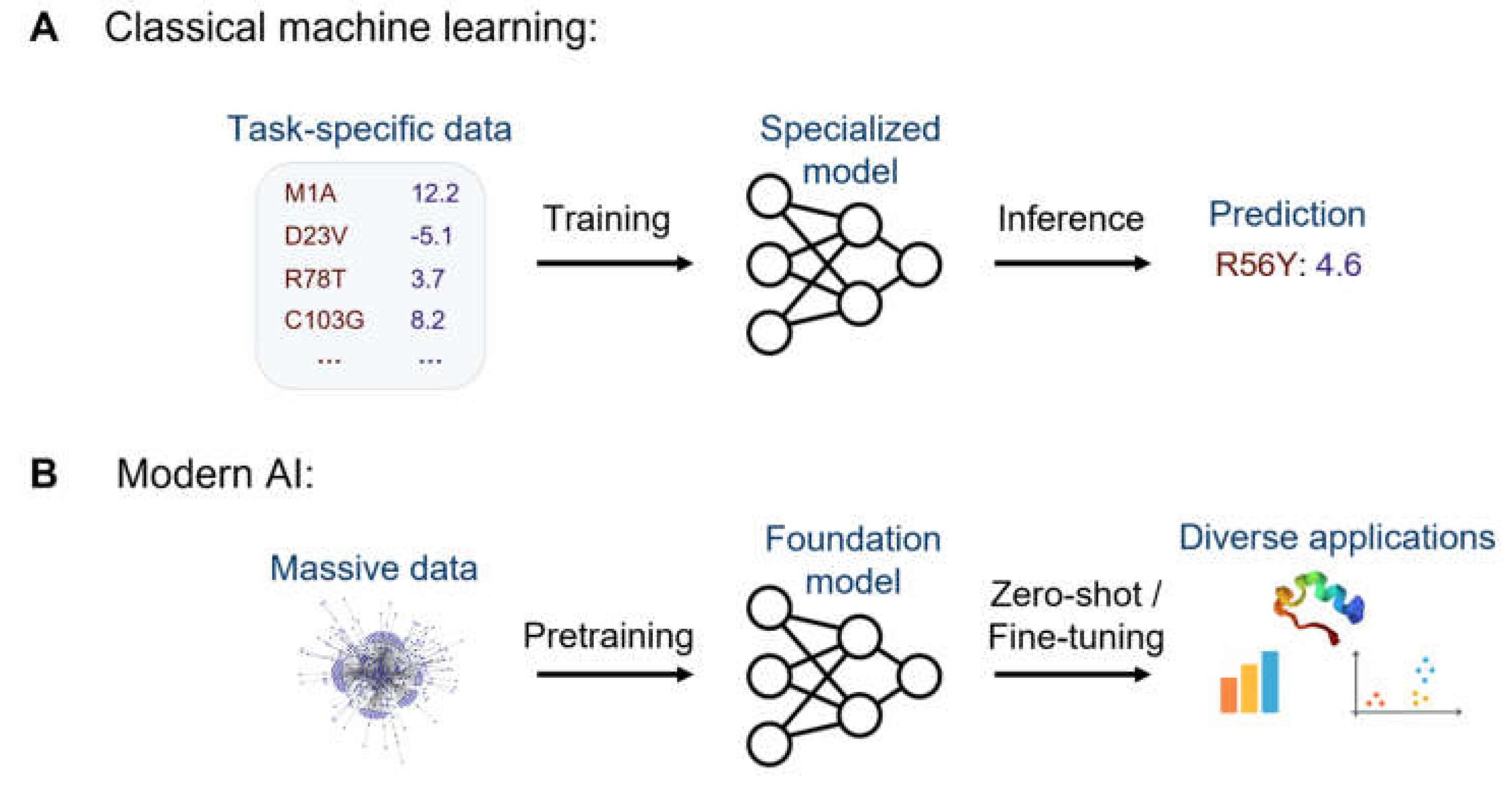
Figure 2.
Applications of AI in human genetics. AI can inform a wide range of genotype-to-phenotype mapping tasks pertaining to both molecular and clinical phenotypes.
Figure 2.
Applications of AI in human genetics. AI can inform a wide range of genotype-to-phenotype mapping tasks pertaining to both molecular and clinical phenotypes.
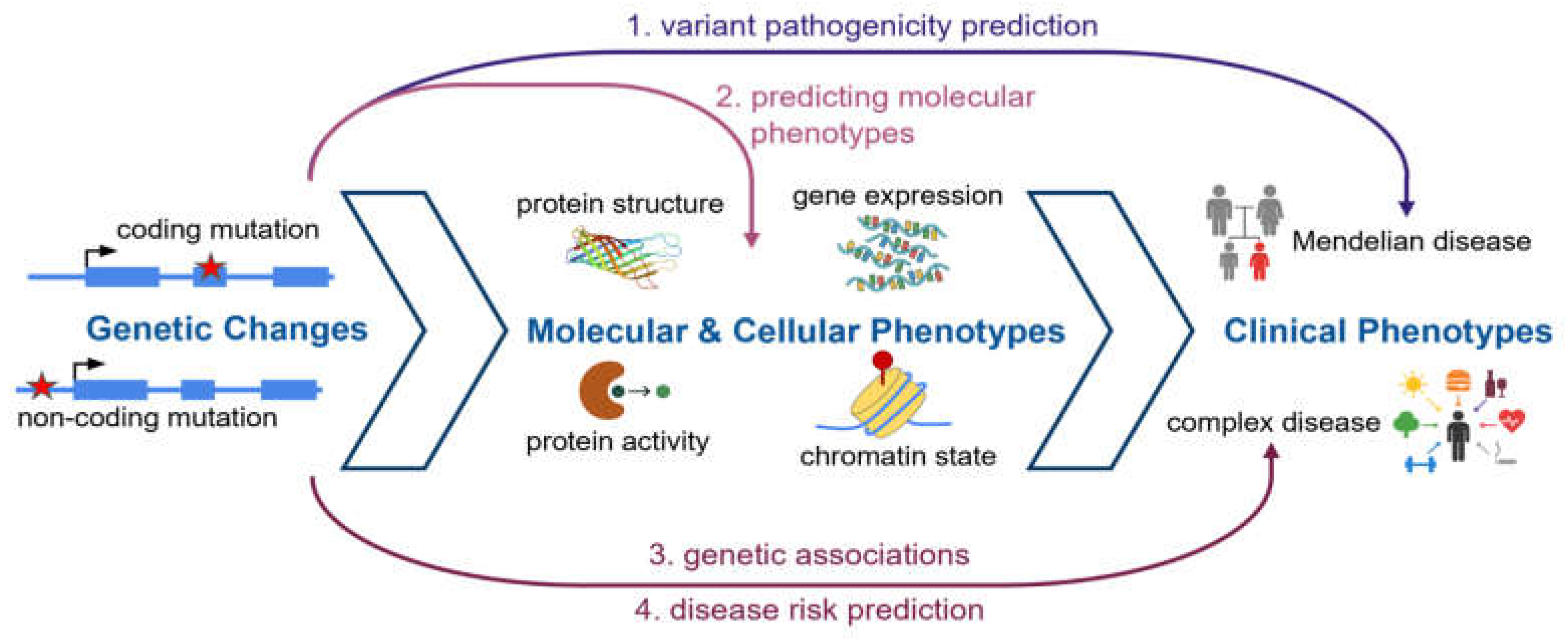
Figure 3.
Protein language models. (A) Analogous to large language models (LLMs) of text, protein language models are trained to predict the correct amino acid at each protein position over hundreds of millions of protein sequences. (B) Protein language models implicitly learn about the underlying processes that have generated the sequences they are trained on, including protein structure, function and fitness.
Figure 3.
Protein language models. (A) Analogous to large language models (LLMs) of text, protein language models are trained to predict the correct amino acid at each protein position over hundreds of millions of protein sequences. (B) Protein language models implicitly learn about the underlying processes that have generated the sequences they are trained on, including protein structure, function and fitness.
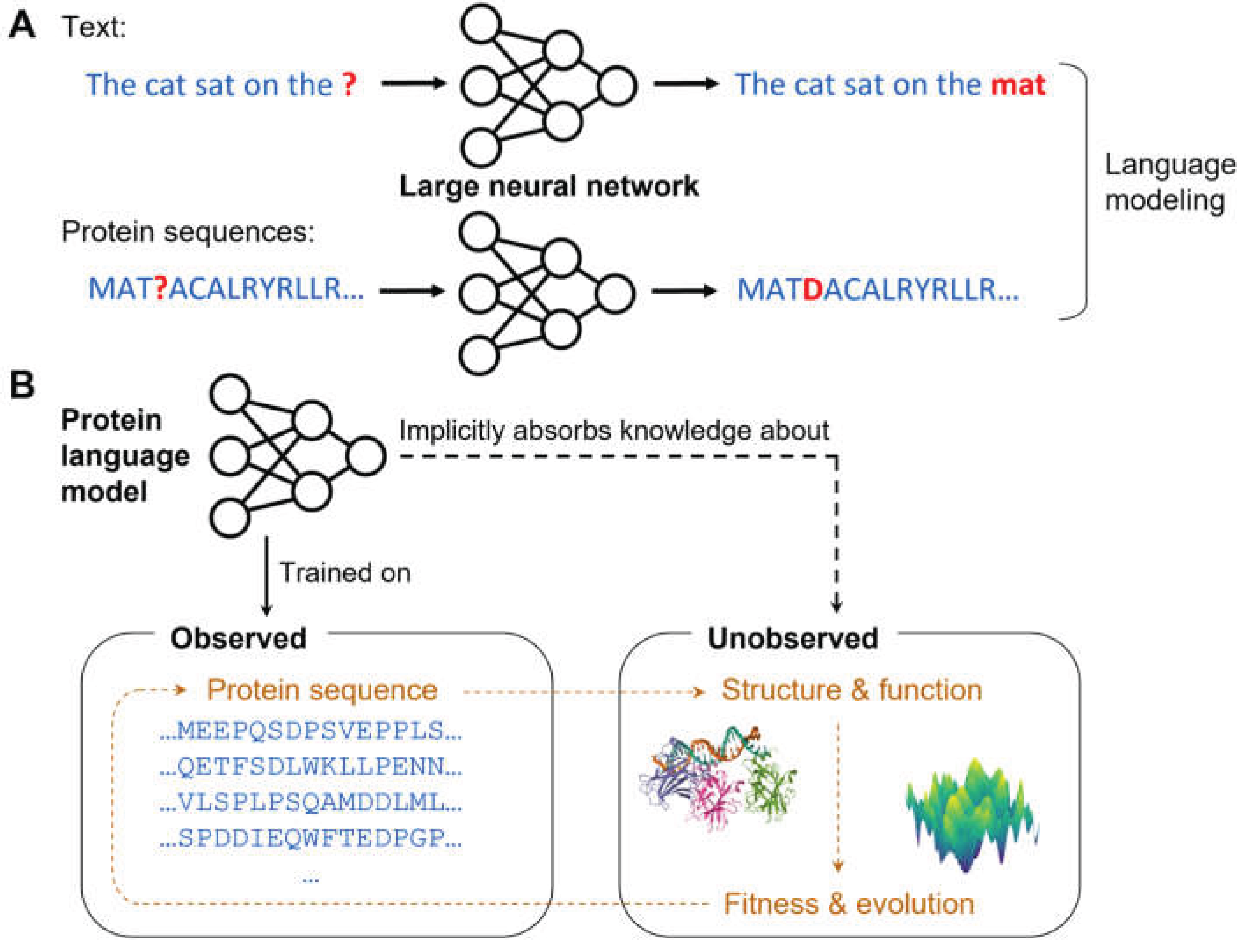
Figure 4.
Informing genetic associations with genomic predictions. Whereas standard burden tests only consider the frequencies of variants in cases and controls (and predefined genomic annotations), methods informed by machine-learning predictions can test whether variants in cases and controls show different predicted effects (e.g., on gene expression or protein function).
Figure 4.
Informing genetic associations with genomic predictions. Whereas standard burden tests only consider the frequencies of variants in cases and controls (and predefined genomic annotations), methods informed by machine-learning predictions can test whether variants in cases and controls show different predicted effects (e.g., on gene expression or protein function).

Table 1.
Glossary of AI and genetics terms.
| Term | Definition |
| Amino acid | The molecular building block of proteins. There are 20 standard amino acids (and a few non-standard ones) which can be chained together in numerous arrangements to form protein sequences. |
| Burden test | A statistical method used to detect genes or other genomic regions associated with a trait. Instead of testing each variant separately, burden tests combine multiple variants into a single score that represents the overall burden of variants in a region per individual. The burden score is then tested for correlation with the trait across individuals. |
| Coding & non-coding genomic region | Coding regions in the genome contain instructions for creating proteins, whereas non-coding genomic regions have other functions, usually related to gene regulation. Mutations are also described as coding or non-coding, depending on whether they affect the sequence of a protein product. |
| Chromatin mark | A chemical modification of the DNA complex, known as chromatin. Common examples include DNA methylation and histone modifications. The state of the chromatin has a critical role in gene expression regulation. |
| Deep learning | A machine-learning approach based on artificial neural networks. Each artificial neuron is represented by a number and can affect the numeric values of downstream neurons in the network, depending on the strength of connections (weights) between pairs of neurons. The more neuron layers there are between the input and output, the deeper the network is. |
| Exon & intron | Genes are made up of exons and introns. Exons are the parts of a gene that remain in the final RNA, while introns are removed during RNA processing in a process called splicing. |
| Feature | In machine learning, a feature is an individual measurable property given as input to a model. Features can be anything from the age of a patient to the GC content of a DNA sequence, depending on the problem. The choice and design of features heavily influence model performance, especially in classical machine learning. |
| Fitness | A quantitative measure of an organism’s reproductive success. Mutations that improve survival or reproduction increase fitness and are favored by natural selection. |
| Foundation model | A large, general-purpose AI model trained on massive data and designed to be highly adaptable. Instead of being trained for one specific task, foundation models learn broad knowledge and representations that can be repurposed for diverse applications. Large language models are a notable example. |
| Gene | A segment of the genome that contains the instructions for making an RNA product, which in turn is often translated to a protein product. |
| Homology | Similarity between genomic sequences due to shared evolutionary origin. Homologous sequences often have related functions, meaning that knowledge about one can be used to make inferences about the other. |
| Genetic effect | The influence that a genetic factor, such as a variant or a gene, has on a trait. Some genetic effects can single-handedly lead to severe disease, while others are more subtle. |
| Genome | The complete set of DNA in an organism, including all the genes and non-coding regions. |
| Genome-wide association study (GWAS) | A study design used to identify genetic variants associated with a trait by scanning the entire genome in a large population. Each variant is tested for statistical correlation with the trait, for example whether it is more common in cases (people with a given disease) than controls (people without it). |
| Genotype & phenotype | Genotype refers to an individual’s genetic makeup, including the specific variants they carry. Phenotype refers to an observable trait, such as height, disease status, or gene expression levels. Much of human genetics is studying how specific genotypes affect specific phenotypes. |
| Large language model (LLM) | A deep-learning model trained to predict the next piece of text in a given source (usually taken from the internet) given all the text that came before it. This simple training task, if applied over huge amounts of text and compute, has given rise to some of the most capable models (such OpenAI’s GPT models which power ChatGPT). |
| Monogenic / Mendelian & polygenic / complex disease | Monogenic (or Mendelian) diseases are caused by mutations in a single gene and follow clear inheritance patterns. Polygenic (or complex) diseases, in contrast, are influenced by many genetic variants, most of which have small effects, along with environmental factors (and their complex interactions). |
| Multiple sequence alignment | A method for lining up multiple genomic sequences so that similar positions across sequences are aligned. This helps identify and characterize conserved regions of shared origin and infer evolutionary constraints. |
| Mutation / variant | A change in a DNA sequence, considered the basic unit of genetic difference between individuals. The terms “mutation” and “variant” sometimes carry different nuances, but are used largely interchangeably in this review. |
| Nucleotide (nt) | The building block of DNA and RNA molecules. DNA and RNA sequences can be represented as strings in a four-letter alphabet consisting of the four nucleotides: A, C, G, and T (for DNA) or U (for RNA). Sequence lengths are measured in nucleotides (e.g., 120 nt). |
| Promoter & enhancer | Regulatory DNA sequences that control when and how much genes are expressed (i.e., transcribed into RNA). A promoter is located near the start of a gene and is required to initiate transcription. Enhancers can be farther away and boost gene activity. |
| Protein | The molecules that carry out most biological functions that sustain life, from catalyzing chemical reactions to connecting tissues. Proteins are made of long chains of amino acids folded into specific 3D structures. A protein’s sequence determines its structure and function. Mutations that change that sequence can disrupt the protein and cause disease. |
| Phylogeny | The evolutionary relationships among species or genes, often represented as a tree. Phylogenies are reconstructed by comparing genomic sequences to infer how species or sequences have diverged and evolved over time. |
| Splicing | A biological process that removes specific parts of an RNA (called introns) and joins the remaining pieces (called exons) to create a mature RNA molecule. Splicing is guided by specific sequences, primarily the splice donor at the start of an intron and the splice acceptor at its end. Errors in splicing can change the resulting RNA molecule and cause disease. |
| Supervised learning | Training machine-learning models on labeled data, where each sample has a label indicating the desired prediction value. |
| Transcript / RNA | A biological molecule similar to DNA, produced by transcribing the sequence of a gene. Some RNA molecules code for proteins; others help regulate genes or support other biological functions. |
| Wildtype | An unmutated sequence, used as a reference point when studying genetic variation. A wildtype gene, protein or organism is one without the specific changes being investigated. |
| Zero-shot prediction | Application of an AI model to a task it hasn’t been explicitly trained to perform without fine-tuning or changing the model. For example, it turns out that protein language models, which have been trained to predict wild-type protein sequences across different species, can predict whether coding mutations in the human genome are pathogenic or benign. |
Table 2.
Genomic AI models used in human genetics.
| Task | Model | Repository / Server | Ref |
| Protein / DNA language modeling | ESM | github.com/facebookresearch/esm | [5,6] |
| Evo2 | github.com/ArcInstitute/evo2 | [7] | |
| Protein structure prediction | AlphaFold |
alphafoldserver.com alphafold.ebi.ac.uk |
[3] |
| ESMFold |
github.com/facebookresearch/esm esmatlas.com/resources |
[5] | |
| RoseTTAFold | github.com/RosettaCommons/RoseTTAFold | [8] | |
| Variant pathogenicity prediction | AlphaMissense | zenodo.org/records/8360242 | [9] |
| CADD | cadd.gs.washington.edu | [10,11,12] | |
| ESM1b |
github.com/ntranoslab/esm-variants huggingface.co/spaces/ntranoslab/esm_variants |
[13] | |
| EVE | evemodel.org | [14] | |
| Evo2 | github.com/ArcInstitute/evo2 | [7] | |
| FATHMM | fathmm.biocompute.org.uk | [15,16] | |
| GPN-MSA | huggingface.co/collections/songlab/gpn-msa-65319280c93c85e11c803887 | [17] | |
| PhyloGPN | huggingface.co/songlab/PhyloGPN | [18] | |
| phyloP | compgen.cshl.edu/phast | [19] | |
| PolyPhen-2 | genetics.bwh.harvard.edu/pph2 | [20] | |
| PrimateAI-3D | primateai3d.basespace.illumina.com | [21] | |
| REVEL | sites.google.com/site/revelgenomics | [22] | |
| SIFT | sift.bii.a-star.edu.sg | [23] | |
| Predicting variant effects on chromatin & gene regulation | AlphaGenome | deepmind.google.com/science/alphagenome | [24] |
| Borzoi | github.com/calico/borzoi | [25] | |
| DeepSEA | deepsea.princeton.edu | [26] | |
| Enformer | github.com/google-deepmind/deepmind-research/tree/master/enformer | [27] | |
| ExPecto | hb.flatironinstitute.org/expecto | [28] | |
| PrediXcan | github.com/hakyimlab/PrediXcan | [29] | |
| SpliceAI | github.com/Illumina/SpliceAI | [30] | |
| Fine-mapping GWAS results | cV2F | github.com/Deylab999MSKCC/cv2f | [31] |
| GWAVA | www.sanger.ac.uk/tool/gwava | [32] | |
| FLAMES | github.com/Marijn-Schipper/FLAMES | [33] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



