Submitted:
17 November 2025
Posted:
18 November 2025
You are already at the latest version
Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is the most prevalent chronic liver disorder in both children and adults. Pediatric MASLD, however, is not simply an early form of adult disease, as it exhibits distinct developmental, histological, and metabolic features. These characteristics reflect the complex, multi-hit, developmental continuum that begins in utero. Maternal obesity, gestational diabetes, and poor diet quality during pregnancy are linked to hepatic steatosis in offspring, implicating intra-uterine exposure to dyslipidemia, hyperglycemia, and excess free fatty acid flux as ini-tiators of fetal hepatic lipid deposition. After birth, feeding behaviors such as a prolonged duration of breastfeeding appears protective, while formula feeding, especially high added-sugar formulations, may accelerate rapid weight gain and predispose to later steatosis. Early childhood diets high in added sugars, saturated fats, and ultra-processed foods may further promote hepatic lipogenesis and inflammation and reveal underlying genetic susceptibility to metabolic dysfunction. This narrative review summarizes recent human and translational studies examining the relationship between prenatal, postnatal, and early childhood nutrition and offspring hepatic lipid accumulation, emphasizing early-life windows for intervention to reduce the burden of pediatric MASLD.
Keywords:
pediatric MASLD
; developmental programming
; maternal obesity
; gestational diabetes
; breastfeeding
; infant formula
; added sugars
; early-life nutrition
; hepatic steatosis
; metabolic imprinting
1. Introduction
Over the past two decades, the landscape of pediatric chronic disease has been profoundly shaped by metabolic dysfunction-associated steatotic liver disease (MASLD), now recognized as the most common chronic liver condition among children worldwide [1,2]. MASLD encompasses a spectrum of progressive liver disorders defined by excessive hepatic fat accumulation in the context of metabolic dysfunction. In children, MASLD is diagnosed when hepatic steatosis is detected by imaging or histology in the presence of at least one cardiometabolic risk factor, such as obesity, dyslipidemia, dysglycemia, or hypertension [3]. While many children exhibit only simple steatosis, a clinically meaningful subset progress to metabolic dysfunction-associated steatohepatitis (MASH). This more severe condition is characterized by steatosis, hepatocellular injury (e.g., ballooning degeneration), and lobular inflammation, often accompanied by fibrosis.
The development of inflammation and fibrosis in adult MASLD patients usually takes decades [4]. In contrast, MASH can develop in children and adolescents within only a few years of the onset of hepatic steatosis or injury [5,6,7]. This accelerated and more aggressive trajectory suggests that it is not merely an earlier manifestation of adult disease, but rather, a distinct entity. Features of pediatric MASLD, such as portal-predominant inflammation and periportal fibrosis [8], as opposed to the lobular inflammation typical of adults, are consistent with age-specific mechanisms of injury and repair, and this diverse pattern of pathophysiology may partially explain why MASLD confers a higher mortality risk in young adulthood [9,10,11,12]. These histological differences underscore the fundamental divergence in pediatric and adult disease pathogenesis.
In specific, pediatric disease often presents with portal-based (zone 1) inflammation and fibrosis, whereas adult disease typically shows pericentral (zone 3) injury and perisinusoidal fibrosis [8,13,14]. Schwimmer et al. [14] delineated two histologic sub-types of pediatric steatohepatitis: one characterized by ballooning degeneration with zone 3 fibrosis and another defined by zone 1 portal inflammation and fibrosis, the latter associated with younger age and more rapid progression of fibrosis. Additionally, zone 1 involvement has been found to correlate with advanced fibrosis and earlier disease onset, while zone 3 predominance has been linked to steatohepatitis [13]. These zonal patterns may reflect developmental differences in hepatic metabolism, oxygen and nutrient gradients, and immune-parenchymal signaling [8].
The public health implications of pediatric MASLD are considerable. In the United States, MASLD affects 5-20% of children and adolescents [15,16,17,18]. Among U.S. adolescents aged 12-17, approximately 20% meet the criteria for obesity, and of these, 70% have MASLD [19]. Internationally, the prevalence approaches 13% in the general pediatric population and up to 47% among youth with obesity [2]. While obesity is a strong risk factor for pediatric MASLD, a significant proportion of young patients have a healthy weight [20,21], mirroring patterns observed in adult disease [22]. However, these figures likely underestimate the true burden of pediatric MASLD, as most children are asymptomatic and diagnosis is often incidental, revealed by elevated alanine aminotransferase (ALT) levels or imaging abnormalities in the course of obesity screening [23].
Pediatric MASLD is a multifactorial disease resulting from the dynamic interplay among genetic predisposition, early developmental programming, and environmental exposures. A strong heritable component for pediatric MASLD is supported by familial clustering [24,25], and the association of variants in PNPLA3, TM6SF2, GCKR–known to confer susceptibility in adults–with hepatic steatosis and fibrosis progression in children [26,27,28,29]. Furthermore, ethnic and racial background significantly influences disease risk, with higher prevalence in Hispanic and Asian youth [19,25]. Early-life factors such as maternal obesity, insulin resistance, gestational diabetes, and abnormal birth weight further modulate susceptibility [8]. These differences persist even after accounting for obesity and likely reflect a combination of genetic, and social, and metabolic determinants, including dietary patterns and adipose tissue distribution.
As in adult disease, environmental and lifestyle factors are key drivers and important targets for preventing and managing pediatric MASLD. For example, poor diet quality [30,31,32,33,34,35,36,37] and low physical activity levels, common in children with MASLD, contribute to hepatic fat accumulation and metabolic dysfunction [35,38,39]. These modifiable factors provide essential targets for early, non-pharmacologic intervention. Given the critical role of modifiable factors in early life, the purpose of this brief narrative review is to synthesize the evidence linking prenatal, postnatal, and early childhood nutrition to the developmental programming of hepatic lipid accumulation.
To narrow the scope, this review focuses on nutritional and metabolic determinants of pediatric MASLD and does not explore mechanistic domains such as epigenetic regulation, paternal contributions, or microbiome-mediated effects, for which readers are referred to recent comprehensive reviews [8,40,41]. For consistency with current nomenclature, the term MASLD is used throughout this review. However, most of the cited literature predates the 2023 consensus reclassification [3], defining hepatic outcomes as nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH). Thus, while the terminology has evolved, the underlying phenotype of excess hepatic fat accumulation in the context of metabolic dysfunction remains largely comparable.
2. Methods
This narrative review was conducted through comprehensive searches of the PubMed and Scopus databases, encompassing literature published up to October 2025. The search strategy was designed to systematically identify human and relevant animal studies linking prenatal, postnatal, and early childhood nutrition with later-life hepatic outcomes.
Search terms included combinations of “maternal obesity”, “maternal diet”, “gestational diabetes”, “fetal programming”, “breastfeeding”, “infant formula”, “added sugars”, “postnatal diet”, “pediatric nutrition”, “hepatic steatosis”, “liver fat”, “liver fibrosis”, “NAFLD”, “NASH”, “MASLD”, “MASH”, and “MAFLD”. Longitudinal cohort studies, randomized controlled trials, and recent systematic reviews were prioritized. Mechanistic animal studies were selectively included to provide biological context.
3. A Brief Overview of Pediatric MASLD Pathophysiology
MASLD is ultimately a systemic disorder of energy metabolism [42]. Insulin resistance in adipose tissue and muscle elevates circulating fatty acids and alters adipokine profiles, while gut dysbiosis increases intestinal permeability and exposure to gut-derived endotoxins [43]. These extrahepatic disturbances create a feed-forward loop of lipotoxicity, inflammation, and fibrogenesis [44].
Early-stage MASLD is characterized by hepatic steatosis, which arises from an imbalance between lipid accretion and disposal. Excess delivery of free fatty acids from adipose tissue, enhanced de novo lipogenesis driven by both hyperinsulinemia and high dietary fructose intake, and impaired β-oxidation collectively promote triglyceride accumulation in hepatocytes [45]. When lipid storage capacity in hepatocytes is exceeded, metabolic pathways become dysregulated, leading to the generation of toxic lipid intermediates such as ceramides, diacylglycerols, and free cholesterol, and inducing mitochondrial dysfunction, oxidative stress, and endoplasmic reticulum stress.
These processes activate the unfolded protein response, generate reactive oxygen species, and trigger hepatocyte injury accompanied by the release of damage-associated molecular patterns (DAMPs) [46,47,48,49]. DAMPs are then recognized by pattern recognition receptors on immune cells, which triggers inflammatory cascades [50]. This process stimulates hepatocytes to release cytokines and chemokines that subsequently recruit Kupffer cells and infiltrating macrophages. Concurrently, mitochondrial DNA and lipid peroxidation products activate innate immune pathways such as TLR4 and NLRP3 inflammasome signaling [51]. Chronic inflammation further disrupts insulin signaling, perpetuating a cycle of metabolic stress and cellular injury. Persistent hepatocyte stress also activates hepatic stellate cells, promoting extracellular matrix deposition and fibrogenesis [49].
4. Maternal Nutrition and Offspring Hepatic Steatosis
Observational studies consistently support associations between maternal obesity, gestational diabetes mellitus (GDM), and increased hepatic fat in offspring [8,41] (Table 1). Using magnetic resonance spectroscopy (MRS), Modi et al. [52] observed an 8.6% increase in neonatal intrahepatic lipid per unit increase in maternal BMI, independent of neonatal body weight. Similarly, Brumbaugh et al. [53] reported 68% higher hepatic fat in neonates born to obese mothers with GDM compared to those of normal-weight women, with intrahepatic lipid content correlating with maternal BMI rather than neonatal adiposity. These findings were among the first to suggest that fetal hepatic steatosis can occur independently of generalized fat accretion and imply that lipid partitioning to the fetal liver is affected by adipose storage-independent pathways.
In the longitudinal Western Australia Pregnancy (Raine) Cohort study, maternal pre-pregnancy obesity predicted MASLD at 17 years of age, independent of a Western-style diet consumption [54]. Likewise, in the Shanghai Prenatal Cohort (SPCS), maternal obesity and GDM were independently associated with hepatic steatosis in 8-year-old offspring [58]. The prevalence of hepatic steatosis rose across maternal BMI quartiles, and children of women with both obesity and GDM exhibited an eightfold-greater odds of steatotic liver compared to those of normal weight, normoglycemic mothers.
Comparable associations have been documented in large longitudinal cohorts with extended follow-up, including the Avon Longitudinal Study of Parents and Children (ALSPAC) and the ESPRESSO (Epidemiology Strengthened by Histopathology Reports in Sweden) studies. The ALSPAC cohort, with follow-up into young adulthood, demonstrated a persistent association between maternal adiposity and offspring hepatic fat [55,56]. Maternal pre-pregnancy overweight and obesity conferred higher risk of hepatic steatosis, whether assessed by ultrasound in adolescence (17-18 years) [55] or transient elastography in young adulthood (24 years) [56]. At both time points, this association was largely mediated by the offspring’s concurrent adiposity, but not by birthweight or breastfeeding, suggesting that the principal mechanism involves interplay between intrauterine metabolic programming and postnatal lifestyle trajectories. In contrast, the influence of maternal diabetes or glycosuria appeared to diminish over time: it was a strong, independent predictor of adolescent hepatic steatosis [aOR 6.74 (95% CI 2.47, 18.40)] [55], but was substantially attenuated and nonsignificant by early adulthood [OR 1.39 (95% CI 0.87, 2.21)] [56]. Excess gestational weight also increased steatosis risk, though this effect was primarily mediated by offspring BMI. In the ESPRESSO study, a population-based case-control design leveraging national registry data, maternal obesity was associated with biopsy-proven MASLD and greater disease severity in adult offspring (≤25 years of age), independent of GDM, smoking, and socioeconomic factors [57].
4.1. Connecting Human Observational Findings and Mechanisms
Human studies indicate that both maternal metabolic status and diet quality during pregnancy independently modulate offspring risk of hepatic steatosis. However, heterogeneity exists in the timing and modality of hepatic outcome assessment, ranging from infancy to adulthood and from ultrasound to biopsy. Mediation analyses suggest that while postnatal adiposity accounts for part of the association, the persistence of prenatal effects even after adjustment supports the idea that intrauterine exposure may establish a “metabolic memory” within the developing liver. The consistency of findings across ethnically diverse cohorts supports an independent effect of maternal metabolic dysregulation on offspring hepatic lipid handling. Rare histologic evidence from autopsy studies of stillborn infants reinforces this link by eliminating postnatal confounding. In a retrospective autopsy study, Patel et al. [60] found macrovesicular steatosis in 79% of stillborn infants (n=33) of diabetic mothers, compared to 17% of infants (n=48) born to normoglycemic mothers. The severity of steatosis correlated with gestational age and fetal weight. Although this study was limited by the grouping of all maternal diabetes types (type 1, type 2, and gestational) into a single category and potential postmortem lipid redistribution, its findings suggest that maternal hyperglycemia and dyslipidemia alone can induce substantial fetal hepatic lipid deposition, independently of shared postnatal factors.
During normal pregnancy, maternal triglycerides and free fatty acids rise to support fetal growth. However, mothers with obesity and gestational diabetes experience a sharper increase, with triglyceride levels ~40-50% higher than those of normal-weight women throughout early to late gestation [61]. This maternal hypertriglyceridemia, influenced by both maternal body composition and dietary fat consumption, modulates placental lipid processing and contributes significantly to offspring metabolic health [62]. Barbour and Hernandez [61] termed the resulting process “making fat from fat” whereby elevated maternal triglycerides, hydrolyzed by placental lipases, are transferred as free fatty acids to the developing fetus (Figure 1). Because the fetal liver has limited oxidative capacity and inefficient VLDL transport, it is poorly equipped to manage this significant lipid flux. Ultimately, this excess lipid accretion drives increased fetal adiposity, a key predictor of childhood obesity and lifelong metabolic dysfunction [63,64].
4.2. Insights from Animal Models
Understanding the mechanisms underlying fetal metabolic programming is an ongoing area of investigation, necessitating the use of animal models to define placental transport and fetal organ responses in utero [62]. However, species differences in placental architecture and placental lipid handling complicate extrapolation from animal models. For example, while rodent models have been vital for understanding the long-term metabolic programming in offspring, they frequently differ from the human phenotype, sometimes resulting in a fetal growth restricted phenotype rather than overgrowth [65,66,67,68,69].
Despite these caveats, non-human primate studies have yielded informative insights into maternal programming of offspring hepatic steatosis. Studies in macaques and baboons exposed to a high calorie, high fat, or high fructose diet observed a substantial increase in fetal hepatic triglycerides, oxidative stress, and upregulation of lipogenic and gluconeogenic genes during late gestation that often persisted after birth [70,71,72]. A Western-style (i.e., high-sugar, high-fat, high-calorie, low fiber) maternal diet also induced bile acid dysregulation, mitochondrial impairment, and altered immune programming in macaque offspring [73] and resulted in periportal collagen deposition and stellate cell activation in fetal macaque liver [74,75]. This pattern persisted in juvenile offspring even after weaning to a control diet [74]. However, modification of the obesogenic diet back to a control diet prior to a second pregnancy normalized fetal oxygenation and reduced markers of hepatic lipotoxicity and oxidative stress [75], indicating that maternal nutrition exerts lasting, diet-responsive effects on offspring hepatic physiology.
Recent studies in other animal models provide additional mechanistic context for maternal dietary influences on offspring hepatic metabolism. In rats, high-fat feeding during gestation and lactation induced hepatic steatosis in offspring, accompanied by increased body weight, impaired glucose tolerance, elevated serum cholesterol, and altered expression of lipogenic and beta-oxidation genes at weaning [76,77]. Similar effects were observed following maternal consumption of junk food or high-fat/high-sucrose diets, which promoted hepatic triglyceride accumulation, oxidative stress, insulin resistance, and downregulation of genes involved in lipid oxidation and VLDL transport [78,79]. When these diets were consumed during lactation, offspring exhibited greater total body fat and impaired liver function [80].
Excess maternal sugar intake and obesity exert additional diet-independent programming effects. Maternal fructose feeding, ranging from 10% to 63% of energy, induced maternal hypertriglyceridemia, fetal hyperinsulinemia, and altered leptin, SREBP-1c, and ACC2 signaling in rat offspring [81,82]. Perinatal obesity in mice primed a persistent hepatic metabolic stress response, characterized by impaired oxidative phosphorylation, dysregulated hepatokine expression, and long-lasting alterations in lipid metabolism, even in the absence of histological steatosis [83]. Postnatal exposure to a high-fat diet amplified these effects, exacerbating hepatic lipid accumulation and promoting ceramide deposition in offspring [84].
4.3. Macronutrient Composition and Hepatic Steatosis Risk
Overall, findings from human studies complement animal evidence. In the Healthy Start Study, poor maternal diet quality, characterized by high intakes of sugar and “empty calories”, and low consumption of green vegetables and legumes, was associated with greater hepatic fat in children aged 4-8 years [59]. Conversely, higher maternal fiber intake and adherence to a Mediterranean-style dietary pattern during pregnancy were linked to lower hepatic fat, independent of pre-pregnancy BMI [59]. Findings from the ALSPAC cohort showed a link between high free-sugar intake during pregnancy and hepatic steatosis in young adulthood [56]. Together, these studies suggest that maternal nutritional quality, in additional to maternal metabolic status, influences offspring hepatic lipid handling.
Emerging evidence further implicates maternal overnutrition in altering fetal immune and microbial programming. Fecal microbiota from infants of obese mothers induced hepatic inflammation, gut barrier dysfunction, and periportal injury in germ-free mice, recapitulating features of pediatric MASLD [85]. Related studies suggest that maternal insulin resistance and hypertriglyceridemia activate de novo lipogenic and inflammatory pathways in the fetal liver that persist despite post-weaning dietary normalization [86,87,88]. These findings support the hypothesis that the “first hit” in pediatric MASLD originates in utero through enduring metabolic and immunologic imprinting.
4.4. Modifiability
Interventional animal studies provide crucial proof-of-principle that the programmed metabolic trajectory may be modifiable. For example, supplementation with the antioxidant pyrrolquinoline quinone during gestation and lactation in mice restored mitochondrial fatty acid oxidation, increased protective n-3 polyunsaturated fatty acids, and reduced triglyceride accumulation in offspring [89]. Furthermore, studies in non-human primates show the efficacy of dietary change: while maternal high-fat feeding decreased fetal n-3 fatty acids, increased the n-6:n-3 ratio, and promoted hepatic apoptosis, switching dams to a control diet prior to subsequent pregnancy normalized these adverse fetal outcomes [90]. Similarly, pre-conception diet reversal in obese macaques partially normalized fetal hepatic triglycerides, oxidative stress, lipogenic gene expression, and key metabolites, although effects like elevated ceramides persisted [75,91]. Targeted interventions, such as treating obese macaques on a Western-style diet with resveratrol, have also been found to reduce markers of hepatic injury including fetal hepatic collagen deposition, portal triad fibrosis, and oxidative stress, and fetal hypoxemia [91]. Collectively, these findings demonstrate that the adverse fetal hepatic programming induced by maternal overnutrition is not permanent and can be significantly mitigated through pre-conception or gestational nutritional and therapeutic interventions.
4.5. The DOHaD Framework
The Developmental Origins of Health and Disease (DOHaD) theory posits that early nutritional and metabolic exposures establish long-term physiological set points, influencing later disease susceptibility [92,93]. While the hypothesis was initially framed around cardiovascular disease and type 2 diabetes, MASLD has now become accepted as an organ-specific extension of this concept. It is through the combination of intrauterine nutrient oversupply and subsequent postnatal dietary excess that the long-term hepatic lipid metabolism, insulin sensitivity, and inflammatory tone are programmed across the life course in offspring. Specifically, clinical, imaging, and experimental data confirm that maternal metabolic dysregulation can initiate hepatic lipid deposition and fibrogenic signaling in utero, thereby predisposing offspring to metabolic dysfunction later in life.
This developmental continuum, linking maternal status and early feeding to MASLD risk, is supported by a recent systematic review of maternal and perinatal factors influencing pediatric liver health [94]. Across studies, several risk and protective factors align with the DOHaD model (Table 2). While findings regarding gestational diabetes, preterm birth, and small-for-gestational-ages remain inconclusive or inconsistent, the collective evidence strengthens the developmental linkage between maternal metabolic status and early feeding patterns and subsequent pediatric MASLD risk.
5. Early Childhood Nutrition and Liver Fat Accretion
5.1. Postnatal and Infant Dietary Factors
The transition from exclusive milk feeding to complementary foods represents a sensitive window for shaping hepatic metabolism. Evidence from the Raine cohort indicates that breastfeeding without supplemental milk for ≥ 6 months reduces the odds of adolescent MASLD by 36%, independent of Western dietary pattern at 17 years of age [54]. Early introduction of formula or supplemental milk (< 6 months) was associated with a higher prevalence and severity of steatosis and a more adverse metabolic profile. These findings are consistent with those of Nobili et al. [95], which showed a protective role for infant nutrition, with a longer duration of breastfeeding independently associated with a lower risk of biopsy-proven MASH and MASH fibrosis in later adolescence, even when accounting for age, waist circumference, gestational age, and birth weight. However, an unrelated study showed that associations between infant feeding and general, visceral, and hepatic fat at age 10 were largely attenuated after adjustment for sociodemographic and maternal factors [96]. Examination at earlier postnatal timepoints (i.e., two months of age) detected no differences in adipose tissue or intrahepatocellular lipid accumulation between breastfed and formula-fed infants [97]. This observation may suggest that the substantial intrahepatocellular lipid increase seen early in life, independent of method of feeding, may be a normal physiological process or that the adverse effects of formula on liver fat are delayed beyond this early neonatal window.
Infant formula composition can further influence hepatic lipid accumulation. A recent analysis of commercial products showed that infant formulas contain significantly more medium-chain fatty acids compared with human milk [98]. Feeding high-energy, medium-chain fatty acid-rich formula to neonatal pigs induced hepatic steatosis and altered hepatocyte intermediary metabolism compared with isocaloric long-chain fatty acid formula [99]. Although extrapolation to human infants is limited, these findings indicate that the quality and chain length of dietary fats can influence hepatic lipid storage during early development.
Early sugar exposure may also be detrimental. In a population-based cohort of nearly 2,000 infants, those consuming >2 servings/day of sugar-containing beverages had a ~3-fold higher odds of MASLD at 10 years compared to infants consuming <1 serving/day, with strongest effects among children of mothers with lower education levels and those who developed excess adiposity [100]. Although added sugar is not recommended for children < 2 years of age [101], most infant formulas sold in the United States primarily contain added sugars rather than naturally occurring lactose [102]. In an analysis of 73 commercial formulas, the median proportion of added sugars was ~60% in standard formula and 85-90% in gentle and lactose-free formulations [102]. Formula feeding is a predictor of rapid weight gain [101,103], a strong driver of later obesity and insulin resistance that promotes ectopic fat deposition and hepatic de novo lipogenesis [104]. Indeed, rapid weight gain in the first three months of life was associated with later fatty liver index in young adulthood [105]. Mechanistically, hepatic metabolism of rapidly absorbed simple sugars, particularly fructose, stimulates de novo lipogenesis and impairs insulin signaling, accelerating hepatic fat accumulation [106]. Experimental sugar-reduction studies in children lower de novo lipogenesis and liver fat, supporting a causal role for dietary sugars in hepatic steatosis [107,108,109]. Studies such as these raise concerns that the high added sugar content of many commercial formulas could contribute to rapid weight gain, early adiposity, increased cardiometabolic risk, and downstream liver fat accumulation.
5.2. Childhood and Early Adolescent Dietary Influences
As children transition into middle childhood and early adolescence, cumulative dietary patterns, environmental exposures, and genetic susceptibility begin to interact. The EPOCH (Exploring Perinatal Outcomes Among Children) study found that increased consumption of fiber, vegetable protein, and polyunsaturated fats from childhood (~10 years) to adolescence (~16 years) was associated with lower hepatic fat, whereas greater intake of animal protein predicted higher hepatic fat [110]. Examination of gene-diet interaction revealed that the PNPLA3 rs738409 risk variant strengthened the dietary associations with hepatic fat [110]. Specifically, the inverse associations of fiber and vegetable protein and a positive association of saturated fat were markedly stronger in carriers of the risk allele.
The evidence linking nutrient intakes to hepatic fat primarily derives from cross-sectional studies, which cannot assess causality [32,111,112,113,114,115]. Longitudinal studies are scarce. No associations were observed between energy-adjusted macronutrient intakes at different time points in childhood and hepatic fat at 17 years [116]. Likewise, no association between dietary pattern in childhood with later hepatic fat was found in the EPOCH cohort, although adherence to a healthier diet was associated with lower hepatic fat [117].
Complementary evidence links dietary inflammatory potential with disease severity. In a cross-sectional cohort of 125 children and adolescents with MASLD, higher Dietary Inflammatory Index (DII) scores were associated with 4-fold greater odds of severe steatosis and higher FIB-4 scores [118]. Each unit increase in DII corresponded to a 2.6-fold increase in odds of more severe steatosis, suggesting that pro-inflammatory dietary patterns exacerbate hepatic injury even after MASLD onset.
Although randomized trials in older children demonstrate that both Mediterranean and low-fat diets reduce hepatic steatosis and improve insulin sensitivity [119,120], such studies primarily target secondary prevention. The current review aims to focus on early-life determinants –prenatal through childhood–when hepatic metabolic pathways are still being established. Dietary exposures occurring after approximately three years of age increasingly reflect environmental and behavioral influences more than developmental programming. Readers interested in dietary interventions in older children and adolescents are referred to recent comprehensive reviews [36,37,108].
6. Conclusions
6.1. Conclusions: A Life Course Developmental Continuum
The collective evidence reviewed here supports the classification of pediatric MASLD as a developmental disease evolving from the prenatal and early postnatal nutritional environments. Maternal metabolic dysfunction, diet quality, and gestational weight gain represent determinants of hepatic lipid metabolism in offspring, initiating a cascade of lipogenic and inflammatory adaptations that can persist long after birth.
Human and non-human primate studies demonstrate that maternal obesity and consumption of a Western-style diet not only drive fetal hepatic lipid accumulation, and initiate fibrogenic changes in utero, revealing how susceptible the fetal liver is to excess maternal lipids. The subsequent postnatal period represents a second critical window in this continuum (Figure 2). Longitudinal studies suggest that breastfeeding provides sustained protection against hepatic fat accumulation, while sucrose-containing formula feeding can promote rapid weight gain and prime the liver for increased de novo lipogenesis and insulin resistance. As children grow, cumulative dietary exposures further influence hepatic lipotoxicity and inflammation, while nutrient-dense dietary patterns and genetic variants modulate the extent of these effects.
From a mechanistic perspective, the overlap between maternal and postnatal exposures implies that pediatric MASLD results, in part, from maladaptive hepatic and systemic responses to energy excess during sensitive developmental stages. Altered placental lipid transport initiates early hepatic fat deposition, which is later compounded by overnutrition and accelerated growth. The predominance of portal-based injury in pediatric MASLD supports the idea that early nutritional exposures may sensitize periportal hepatocytes to metabolic stress, creating a biological imprint that predisposes to fibrosis later in life.
6.2. Methodological Limitations and Future Directions
Despite evidence for a developmental continuum, the current body of literature faces methodological limitations. Variability in study design, dietary assessment, outcome measures, and follow-up duration complicates comparisons and limits causal inference [41]. Moreover, because most of the studies summarized here used imaging-based measures of hepatic steatosis or older NAFLD terminology, extrapolation to MASLD as currently defined needs to be made with caution. Human studies are further limited by their reliance on self-reported intake and lack of biochemical validation, while controlled feeding studies that could establish causality remain difficult to perform for ethical and logistic reasons.
6.3. Opportunities for Prevention and Intervention
The reversibility of developmental programming provides a potential path for intervention. Animal models show that improving pre-conception diet or supplementing with compounds such as resveratrol attenuates fetal hepatic injury. Translating these insights into clinical practice requires a life-course prevention approach that emphasizes optimal maternal nutrition before and during pregnancy, supports breastfeeding for at least six months, limits added sugars in infant formulas and complementary foods, and fosters nutritious dietary habits from infancy onward [121,122]. Thus, prioritizing nutrition across the lifespan, beginning prior to conception, represents the single most effective strategy for reducing the global burden of pediatric MASLD.
Funding
This research was funded by National Institutes of Health, grant number R01DK127015.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACC2 | acetyl-CoA carboxylase 2 |
| ALSPAC | Avon Longitudinal Study of Parents and Children |
| ALT | alanine aminotransferase |
| aOR | adjusted odds ratio |
| BMI | body mass index |
| CD36 | cluster of differentiation 36 |
| CI | confidence interval |
| DAMPs | damage-associated molecular patterns |
| DII | Dietary Inflammatory Index |
| EPOCH | Exploring Perinatal Outcomes Among Children |
| ESPRESSO | Epidemiology Strengthened by Histopathology Reports in Sweden |
| FABP | fatty acid binding protein |
| FATP | fatty acid transport protein |
| FATs | fatty acid transporters |
| FFAs | free fatty acids |
| FIB-4 | fibrosis-4 index |
| GCKR | glucokinase regulatory protein |
| GDM | gestational diabetes mellitus |
| IHCL | intra-hepatocellular lipid |
| MAFLD | metabolic associated fatty liver disease |
| MASH | metabolic dysfunction-associated steatohepatitis |
| MASLD | metabolic dysfunction-associated steatotic liver disease |
| MRS | magnetic resonance spectroscopy |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| NLRP3 | NLR family pyrin domain containing 3 |
| OR | odds ratio |
| PNPLA3 | patatin-like phospholipase domain-containing 3 |
| SPCS | Shanghai Prenatal Cohort Study |
| SREBP-1c | sterol regulatory element binding protein 1c |
| TGs | triglycerides |
| TM6SF2 | transmembrane 6 superfamily 2 |
| TLF4 | toll-like receptor 4 |
| VLDL | very low-density lipoprotein |
References
- Jia, S.; Ye, X.; Wu, T.; Wang, Z.; Wu, J. Global prevalence of metabolic dysfunction-associated fatty liver disease in children and adolescents with overweight and obesity: a systematic review and meta-analysis. BMC Gastroenterology 2025, 25, 691. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Kalligeros, M.; Henry, L. Epidemiology of metabolic dysfunction-associated steatotic liver disease. Clin Mol Hepatol 2025, 31, S32–S50. [Google Scholar] [CrossRef]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P. , et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 2015, 13. 643-654 e641-649; quiz e639-640. [Google Scholar] [CrossRef]
- Draijer, L.; Voorhoeve, M.; Troelstra, M.; Holleboom, A.; Beuers, U.; Kusters, M.; Nederveen, A.; Benninga, M.; Koot, B. A natural history study of paediatric non-alcoholic fatty liver disease over 10 years. JHEP Rep 2023, 5, 100685. [Google Scholar] [CrossRef]
- Alves, V.P.V.; Mouzaki, M.; Xanthakos, S.A.; Zhang, B.; Tkach, J.A.; Ouyang, J.; Dillman, J.R.; Trout, A.T. Longitudinal evaluation of pediatric and young adult metabolic dysfunction-associated steatotic liver disease defined by MR elastography. Eur Radiol 2025, 35, 2474–2486. [Google Scholar] [CrossRef]
- Xanthakos, S.A.; Lavine, J.E.; Yates, K.P.; Schwimmer, J.B.; Molleston, J.P.; Rosenthal, P.; Murray, K.F.; Vos, M.B.; Jain, A.K.; Scheimann, A.O. , et al. Progression of Fatty Liver Disease in Children Receiving Standard of Care Lifestyle Advice. Gastroenterology 2020, 159, 1731–1751.e1710. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Biddinger, S.B.; Ibrahim, S.H. MASLD in children: integrating epidemiological trends with mechanistic and translational advances. J Clin Invest 2025, 135. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Charatcharoenwitthaya, P.; Treeprasertsuk, S.; Benson, J.T.; Enders, F.B.; Angulo, P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut 2009, 58, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Thai, N.Q.N.; Noon, S.L.; Ugalde-Nicalo, P.; Anderson, S.R.; Chun, L.F.; David, R.S.; Goyal, N.P.; Newton, K.P.; Hansen, E.G. , et al. Long-term mortality and extrahepatic outcomes in 1096 children with MASLD: A retrospective cohort study. Hepatology 2025. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Roelstraete, B.; Hartjes, K.; Shah, U.; Khalili, H.; Arnell, H.; Ludvigsson, J.F. Non-alcoholic fatty liver disease in children and young adults is associated with increased long-term mortality. J Hepatol 2021, 75, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Mouzaki, M.; Hassan, S.; Kehar, M.; Mysore, K.R.; Mauney, E.; Nonga, D.; Karjoo, S.; Sood, S.; Tou, A. , et al. Call to action-Pediatric MASLD requires immediate attention to curb health crisis. Hepatology 2025. [Google Scholar] [CrossRef]
- Africa, J.A.; Behling, C.A.; Brunt, E.M.; Zhang, N.; Luo, Y.; Wells, A.; Hou, J.; Belt, P.H.; Kohil, R.; Lavine, J.E. , et al. In Children With Nonalcoholic Fatty Liver Disease, Zone 1 Steatosis Is Associated With Advanced Fibrosis. Clin Gastroenterol Hepatol 2018, 16, 438–446.e431. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef]
- Sahota, A.K.; Shapiro, W.L.; Newton, K.P.; Kim, S.T.; Chung, J.; Schwimmer, J.B. Incidence of Nonalcoholic Fatty Liver Disease in Children: 2009-2018. Pediatrics 2020, 146. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.L.; Schwimmer, J.B. Epidemiology of Pediatric Nonalcoholic Fatty Liver Disease. Clin Liver Dis (Hoboken) 2021, 17, 196–199. [Google Scholar] [CrossRef]
- Perumpail, B.J.; Manikat, R.; Wijarnpreecha, K.; Cholankeril, G.; Ahmed, A.; Kim, D. The prevalence and predictors of metabolic dysfunction-associated steatotic liver disease and fibrosis/cirrhosis among adolescents/young adults. J Pediatr Gastroenterol Nutr 2024, 79, 110–118. [Google Scholar] [CrossRef]
- Shi, G.X.; Qian, Y.S.; Jiang, C.M.; Liu, Z.Z.; Yang, X.; Xu, Y.W.; Jin, S.S.; Chu, J.G.; Qian, G.Q.; Yang, N.B. Prevalence of steatotic liver disease (MASLD, MetALD, ALD) and clinically significant fibrosis in US adolescents : Authors’ name. Sci Rep 2024, 14, 25724. [Google Scholar] [CrossRef]
- Arshad, T.; Paik, J.M.; Biswas, R.; Alqahtani, S.A.; Henry, L.; Younossi, Z.M. Nonalcoholic Fatty Liver Disease Prevalence Trends Among Adolescents and Young Adults in the United States, 2007-2016. Hepatol Commun 2021, 5, 1676–1688. [Google Scholar] [CrossRef]
- Yu, E.L.; Golshan, S.; Harlow, K.E.; Angeles, J.E.; Durelle, J.; Goyal, N.P.; Newton, K.P.; Sawh, M.C.; Hooker, J.; Sy, E.Z. , et al. Prevalence of Nonalcoholic Fatty Liver Disease in Children with Obesity. J Pediatr 2019, 207, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Mischel, A.K.; Liao, Z.; Cao, F.; Dunn, W.; Lo, J.C.; Newton, K.P.; Goyal, N.P.; Yu, E.L.; Schwimmer, J.B. Prevalence of Elevated ALT in Adolescents in the US 2011-2018. J Pediatr Gastroenterol Nutr 2023, 77, 103–109. [Google Scholar] [CrossRef]
- DiStefano, J.K.; Gerhard, G.S. NAFLD in normal weight individuals. Diabetol Metab Syndr 2022, 14, 45. [Google Scholar] [CrossRef]
- Li, J.; Cheung, R. Nonalcoholic Fatty Liver Disease in Children: Where Are We? Clin Gastroenterol Hepatol 2022, 20, 2210–2215. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Hagstrom, H.; Sun, J.; Bergman, D.; Shang, Y.; Yang, W.; Roelstraete, B.; Ludvigsson, J.F. Familial coaggregation of MASLD with hepatocellular carcinoma and adverse liver outcomes: Nationwide multigenerational cohort study. J Hepatol 2023, 79, 1374–1384. [Google Scholar] [CrossRef]
- Zhang, X.; Chang, K.M.; Yu, J.; Loomba, R. Unraveling Mechanisms of Genetic Risks in Metabolic Dysfunction-Associated Steatotic Liver Diseases: A Pathway to Precision Medicine. Annu Rev Pathol 2025, 20, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, A.; Pacifico, L.; Chiesa, C.; Perla, F.M.; Ceci, F.; Angeloni, A.; D’Erasmo, L.; Di Martino, M.; Arca, M. Genetic and metabolic predictors of hepatic fat content in a cohort of Italian children with obesity. Pediatr Res 2019, 85, 671–677. [Google Scholar] [CrossRef]
- Zusi, C.; Mantovani, A.; Olivieri, F.; Morandi, A.; Corradi, M.; Miraglia Del Giudice, E.; Dauriz, M.; Valenti, L.; Byrne, C.D.; Targher, G. , et al. Contribution of a genetic risk score to clinical prediction of hepatic steatosis in obese children and adolescents. Dig Liver Dis 2019, 51, 1586–1592. [Google Scholar] [CrossRef]
- Hudert, C.A.; Selinski, S.; Rudolph, B.; Blaker, H.; Loddenkemper, C.; Thielhorn, R.; Berndt, N.; Golka, K.; Cadenas, C.; Reinders, J. , et al. Genetic determinants of steatosis and fibrosis progression in paediatric non-alcoholic fatty liver disease. Liver Int 2019, 39, 540–556. [Google Scholar] [CrossRef] [PubMed]
- Riccio, S.; Melone, R.; Vitulano, C.; Guida, P.; Maddaluno, I.; Guarino, S.; Marzuillo, P.; Miraglia Del Giudice, E.; Di Sessa, A. Advances in pediatric non-alcoholic fatty liver disease: From genetics to lipidomics. World J Clin Pediatr 2022, 11, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Faienza, M.F.; Baima, J.; Cecere, V.; Monteduro, M.; Farella, I.; Vitale, R.; Antoniotti, V.; Urbano, F.; Tini, S.; Lenzi, F.R. , et al. Fructose Intake and Unhealthy Eating Habits Are Associated with MASLD in Pediatric Obesity: A Cross-Sectional Pilot Study. Nutrients 2025, 17. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Y.; Lim, J.H.; Joung, H.; Yoon, D. Association Between Ultraprocessed Food Consumption and Metabolic Disorders in Children and Adolescents with Obesity. Nutrients 2024, 16. [Google Scholar] [CrossRef]
- Nier, A.; Brandt, A.; Conzelmann, I.B.; Ozel, Y.; Bergheim, I. Non-Alcoholic Fatty Liver Disease in Overweight Children: Role of Fructose Intake and Dietary Pattern. Nutrients 2018, 10. [Google Scholar] [CrossRef]
- Cohen, C.C.; Li, K.W.; Alazraki, A.L.; Beysen, C.; Carrier, C.A.; Cleeton, R.L.; Dandan, M.; Figueroa, J.; Knight-Scott, J.; Knott, C.J. , et al. Dietary sugar restriction reduces hepatic de novo lipogenesis in adolescent boys with fatty liver disease. J Clin Invest 2021, 131. [Google Scholar] [CrossRef]
- Zeng, X.F.; Varady, K.A.; Wang, X.D.; Targher, G.; Byrne, C.D.; Tayyem, R.; Latella, G.; Bergheim, I.; Valenzuela, R.; George, J. , et al. The role of dietary modification in the prevention and management of metabolic dysfunction-associated fatty liver disease: An international multidisciplinary expert consensus. Metabolism 2024, 161, 156028. [Google Scholar] [CrossRef] [PubMed]
- Mager, D.R.; Patterson, C.; So, S.; Rogenstein, C.D.; Wykes, L.J.; Roberts, E.A. Dietary and physical activity patterns in children with fatty liver. Eur J Clin Nutr 2010, 64, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Sandel, P.; Ma, L.; Wang, H.; Pasman, E.A. You Are What You Eat: A Review on Dietary Interventions for Treating Pediatric Nonalcoholic Fatty Liver Disease. Nutrients 2023, 15. [Google Scholar] [CrossRef]
- Jamil, A.; Chivese, T.; Elshaikh, U.; Sendall, M. Efficacy of the Mediterranean diet in treating metabolic dysfunction-associated steatotic liver disease (MASLD) in children and adolescents: a systematic review and meta-analysis. BMC Public Health 2024, 24, 2701. [Google Scholar] [CrossRef]
- Medrano, M.; Arenaza, L.; Migueles, J.H.; Rodriguez-Vigil, B.; Ruiz, J.R.; Labayen, I. Associations of physical activity and fitness with hepatic steatosis, liver enzymes, and insulin resistance in children with overweight/obesity. Pediatr Diabetes 2020, 21, 565–574. [Google Scholar] [CrossRef]
- Li, S.; Jin, S.; Fang, P.; Pan, C.; Huang, S. Association between excessive screen time and steatotic liver disease in adolescents: Findings from the 2017-2018 National Health and Nutrition Examination Survey. Pediatr Obes 2025, 20, e70010. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Li, L.; Cheng, L.; Gu, Z.; Hong, Y. Maternal obesity and offspring metabolism: revisiting dietary interventions. Food Funct 2025, 16, 3751–3773. [Google Scholar] [CrossRef]
- Mouzaki, M.; Woo, J.G.; Divanovic, S. Gestational and Developmental Contributors of Pediatric MASLD. Semin Liver Dis 2024, 44, 43–53. [Google Scholar] [CrossRef]
- Stefan, N.; Yki-Jarvinen, H.; Neuschwander-Tetri, B.A. Metabolic dysfunction-associated steatotic liver disease: heterogeneous pathomechanisms and effectiveness of metabolism-based treatment. Lancet Diabetes Endocrinol 2025, 13, 134–148. [Google Scholar] [CrossRef]
- Schnabl, B.; Damman, C.J.; Carr, R.M. Metabolic dysfunction-associated steatotic liver disease and the gut microbiome: pathogenic insights and therapeutic innovations. J Clin Invest 2025, 135. [Google Scholar] [CrossRef]
- Iturbe-Rey, S.; Maccali, C.; Arrese, M.; Aspichueta, P.; Oliveira, C.P.; Castro, R.E.; Lapitz, A.; Izquierdo-Sanchez, L.; Bujanda, L.; Perugorria, M.J. , et al. Lipotoxicity-driven metabolic dysfunction-associated steatotic liver disease (MASLD). Atherosclerosis 2025, 400, 119053. [Google Scholar] [CrossRef]
- Koo, S.H. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial Adaptation in Nonalcoholic Fatty Liver Disease: Novel Mechanisms and Treatment Strategies. Trends Endocrinol Metab 2017, 28, 250–260. [Google Scholar] [CrossRef]
- Brankovic, M.; Jovanovic, I.; Dukic, M.; Radonjic, T.; Opric, S.; Klasnja, S.; Zdravkovic, M. Lipotoxicity as the Leading Cause of Non-Alcoholic Steatohepatitis. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Krahmer, N.; Walther, T.C.; Farese, R.V., Jr. The pathogenesis of hepatic steatosis in MASLD: a lipid droplet perspective. J Clin Invest 2025, 135. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Jiang, W.; Zhou, R. DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity 2024, 57, 752–771. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Modi, N.; Murgasova, D.; Ruager-Martin, R.; Thomas, E.L.; Hyde, M.J.; Gale, C.; Santhakumaran, S.; Dore, C.J.; Alavi, A.; Bell, J.D. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res 2011, 70, 287–291. [Google Scholar] [CrossRef]
- Brumbaugh, D.E.; Tearse, P.; Cree-Green, M.; Fenton, L.Z.; Brown, M.; Scherzinger, A.; Reynolds, R.; Alston, M.; Hoffman, C.; Pan, Z. , et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J Pediatr 2013, 162, 930–936.e931. [Google Scholar] [CrossRef] [PubMed]
- Ayonrinde, O.T.; Oddy, W.H.; Adams, L.A.; Mori, T.A.; Beilin, L.J.; de Klerk, N.; Olynyk, J.K. Infant nutrition and maternal obesity influence the risk of non-alcoholic fatty liver disease in adolescents. J Hepatol 2017, 67, 568–576. [Google Scholar] [CrossRef]
- Patel, S.; Lawlor, D.A.; Callaway, M.; Macdonald-Wallis, C.; Sattar, N.; Fraser, A. Association of maternal diabetes/glycosuria and pre-pregnancy body mass index with offspring indicators of non-alcoholic fatty liver disease. BMC Pediatr 2016, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Sekkarie, A.; Welsh, J.A.; Northstone, K.; Stein, A.D.; Ramakrishnan, U.; Vos, M.B. Associations of maternal diet and nutritional status with offspring hepatic steatosis in the Avon longitudinal study of parents and children. BMC Nutr 2021, 7, 28. [Google Scholar] [CrossRef]
- Hagstrom, H.; Simon, T.G.; Roelstraete, B.; Stephansson, O.; Soderling, J.; Ludvigsson, J.F. Maternal obesity increases the risk and severity of NAFLD in offspring. J Hepatol 2021, 75, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Shen, F.; Zou, Z.Y.; Yang, R.X.; Jin, Q.; Yang, J.; Chen, G.Y.; Fan, J.G. Association of maternal obesity and gestational diabetes mellitus with overweight/obesity and fatty liver risk in offspring. World J Gastroenterol 2022, 28, 1681–1691. [Google Scholar] [CrossRef]
- Cohen, C.C.; Perng, W.; Sauder, K.A.; Shapiro, A.L.B.; Starling, A.P.; Friedman, C.; Felix, J.F.; Kupers, L.K.; Moore, B.F.; Hebert, J.R. , et al. Maternal Diet Quality During Pregnancy and Offspring Hepatic Fat in Early Childhood: The Healthy Start Study. J Nutr 2023, 153, 1122–1132. [Google Scholar] [CrossRef]
- Patel, K.R.; White, F.V.; Deutsch, G.H. Hepatic steatosis is prevalent in stillborns delivered to women with diabetes mellitus. J Pediatr Gastroenterol Nutr 2015, 60, 152–158. [Google Scholar] [CrossRef]
- Barbour, L.A.; Hernandez, T.L. Maternal Lipids and Fetal Overgrowth: Making Fat from Fat. Clin Ther 2018, 40, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Easton, Z.J.W.; Regnault, T.R.H. The Impact of Maternal Body Composition and Dietary Fat Consumption upon Placental Lipid Processing and Offspring Metabolic Health. Nutrients 2020, 12. [Google Scholar] [CrossRef]
- Moore, B.F.; Harrall, K.K.; Sauder, K.A.; Glueck, D.H.; Dabelea, D. Neonatal Adiposity and Childhood Obesity. Pediatrics 2020, 146. [Google Scholar] [CrossRef]
- Amati, F.; McCann, L.; Castaneda-Gutierrez, E.; Prior, E.; van Loo-Bouwman, C.A.; Abrahamse-Berkeveld, M.; Oliveros, E.; Ozanne, S.; Symonds, M.E.; Chang, C.Y. , et al. Infant fat mass and later child and adolescent health outcomes: a systematic review. Arch Dis Child 2024, 109, 125–129. [Google Scholar] [CrossRef]
- Aguilera, N.; Salas-Perez, F.; Ortiz, M.; Alvarez, D.; Echiburu, B.; Maliqueo, M. Rodent models in placental research. Implications for fetal origins of adult disease. Anim Reprod 2022, 19, e20210134. [Google Scholar] [CrossRef]
- Li, M.; Sloboda, D.M.; Vickers, M.H. Maternal obesity and developmental programming of metabolic disorders in offspring: evidence from animal models. Exp Diabetes Res 2011, 2011, 592408. [Google Scholar] [CrossRef]
- Farley, D.; Tejero, M.E.; Comuzzie, A.G.; Higgins, P.B.; Cox, L.; Werner, S.L.; Jenkins, S.L.; Li, C.; Choi, J.; Dick, E.J., Jr. , et al. Feto-placental adaptations to maternal obesity in the baboon. Placenta 2009, 30, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Animal models of human pregnancy and placentation: alternatives to the mouse. Reproduction 2020, 160, R129–R143. [Google Scholar] [CrossRef] [PubMed]
- Marzetta, C.A.; Rudel, L.L. A species comparison of low density lipoprotein heterogeneity in nonhuman primates fed atherogenic diets. J Lipid Res 1986, 27, 753–762. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J Clin Invest 2009, 119, 323–335. [Google Scholar] [CrossRef]
- Puppala, S.; Li, C.; Glenn, J.P.; Saxena, R.; Gawrieh, S.; Quinn, A.; Palarczyk, J.; Dick, E.J., Jr.; Nathanielsz, P.W.; Cox, L.A. Primate fetal hepatic responses to maternal obesity: epigenetic signalling pathways and lipid accumulation. J Physiol 2018, 596, 5823–5837. [Google Scholar] [CrossRef]
- Thorn, S.R.; Baquero, K.C.; Newsom, S.A.; El Kasmi, K.C.; Bergman, B.C.; Shulman, G.I.; Grove, K.L.; Friedman, J.E. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes 2014, 63, 2702–2713. [Google Scholar] [CrossRef]
- Nash, M.J.; Dobrinskikh, E.; Al-Juboori, S.I.; Janssen, R.C.; Fernandes, J.; Argabright, A.; D’Alessandro, A.; Kirigiti, M.A.; Kievit, P.; Aagaard, K.M. , et al. Maternal Western Diet Programmes Bile Acid Dysregulation and Hepatic Fibrosis in Fetal and Juvenile Macaques. Liver Int 2025, 45, e16236. [Google Scholar] [CrossRef] [PubMed]
- Nash, M.J.; Dobrinskikh, E.; Janssen, R.C.; Lovell, M.A.; Schady, D.A.; Levek, C.; Jones, K.L.; D’Alessandro, A.; Kievit, P.; Aagaard, K.M. , et al. Maternal Western diet is associated with distinct preclinical pediatric NAFLD phenotypes in juvenile nonhuman primate offspring. Hepatol Commun 2023, 7, e0014. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, S.R.; Mulligan, C.M.; Janssen, R.C.; Baker, P.R., 2nd; Bergman, B.C.; D’Alessandro, A.; Nemkov, T.; Maclean, K.N.; Jiang, H.; Dean, T.A. , et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Mol Metab 2018, 18, 25–41. [Google Scholar] [CrossRef]
- Huang, Y.; Ye, T.; Liu, C.; Fang, F.; Chen, Y.; Dong, Y. Maternal high-fat diet during pregnancy and lactation affects hepatic lipid metabolism in early life of offspring rat. J Biosci 2017, 42, 311–319. [Google Scholar] [CrossRef]
- Kereliuk, S.M.; Brawerman, G.M.; Dolinsky, V.W. Maternal Macronutrient Consumption and the Developmental Origins of Metabolic Disease in the Offspring. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef]
- Bayol, S.A.; Simbi, B.H.; Fowkes, R.C.; Stickland, N.C. A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic Fatty liver disease in rat offspring. Endocrinology 2010, 151, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, M.; Nilsson, C.; Rosendal, A.; Nielsen, M.O.; Raun, K. Maternal chocolate and sucrose soft drink intake induces hepatic steatosis in rat offspring associated with altered lipid gene expression profile. Acta Physiol (Oxf) 2014, 210, 142–153. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Zeng, J.J.; Kjaergaard, M.; Guan, N.; Raun, K.; Nilsson, C.; Wang, M.W. Effects of a maternal diet supplemented with chocolate and fructose beverage during gestation and lactation on rat dams and their offspring. Clin Exp Pharmacol Physiol 2011, 38, 613–622. [Google Scholar] [CrossRef]
- Sloboda, D.M.; Li, M.; Patel, R.; Clayton, Z.E.; Yap, C.; Vickers, M.H. Early life exposure to fructose and offspring phenotype: implications for long term metabolic homeostasis. J Obes 2014, 2014, 203474. [Google Scholar] [CrossRef]
- Vickers, M.H.; Clayton, Z.E.; Yap, C.; Sloboda, D.M. Maternal fructose intake during pregnancy and lactation alters placental growth and leads to sex-specific changes in fetal and neonatal endocrine function. Endocrinology 2011, 152, 1378–1387. [Google Scholar] [CrossRef]
- Stegmann, S.K.; Vohlen, C.; Im, N.G.; Niehues, J.; Selle, J.; Janoschek, R.; Kuiper-Makris, C.; Lang, S.; Demir, M.; Steffen, H.M. , et al. Perinatal obesity primes the hepatic metabolic stress response in the offspring across life span. Sci Rep 2025, 15, 6416. [Google Scholar] [CrossRef]
- Hellgren, L.I.; Jensen, R.I.; Waterstradt, M.S.; Quistorff, B.; Lauritzen, L. Acute and perinatal programming effects of a fat-rich diet on rat muscle mitochondrial function and hepatic lipid accumulation. Acta Obstet Gynecol Scand 2014, 93, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K. , et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat Commun 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.S.; Heerwagen, M.J.; Friedman, J.E. Developmental programming of pediatric nonalcoholic fatty liver disease: redefining the”first hit”. Clin Obstet Gynecol 2013, 56, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.M.; Reynolds, R.M.; Prescott, S.L.; Nyirenda, M.; Jaddoe, V.W.; Eriksson, J.G.; Broekman, B.F. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol 2017, 5, 53–64. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clement, K. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol 2020, 17, 279–297. [Google Scholar] [CrossRef]
- Mandala, A.; Dobrinskikh, E.; Janssen, R.C.; Fiehn, O.; D’Alessandro, A.; Friedman, J.E.; Jonscher, K.R. Maternal Pyrroloquinoline Quinone Supplementation Improves Offspring Liver Bioactive Lipid Profiles throughout the Lifespan and Protects against the Development of Adult NAFLD. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Grant, W.F.; Gillingham, M.B.; Batra, A.K.; Fewkes, N.M.; Comstock, S.M.; Takahashi, D.; Braun, T.P.; Grove, K.L.; Friedman, J.E.; Marks, D.L. Maternal high fat diet is associated with decreased plasma n-3 fatty acids and fetal hepatic apoptosis in nonhuman primates. PLoS One 2011, 6, e17261. [Google Scholar] [CrossRef]
- Nash, M.J.; Dobrinskikh, E.; Newsom, S.A.; Messaoudi, I.; Janssen, R.C.; Aagaard, K.M.; McCurdy, C.E.; Gannon, M.; Kievit, P.; Friedman, J.E. , et al. Maternal Western diet exposure increases periportal fibrosis beginning in utero in nonhuman primate offspring. JCI Insight 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. Maternal nutrition, fetal nutrition, and disease in later life. Nutrition 1997, 13, 807–813. [Google Scholar] [CrossRef]
- Barker, D.J. The origins of the developmental origins theory. J Intern Med 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Querter, I.; Pauwels, N.S.; De Bruyne, R.; Dupont, E.; Verhelst, X.; Devisscher, L.; Van Vlierberghe, H.; Geerts, A.; Lefere, S. Maternal and Perinatal Risk Factors for Pediatric Nonalcoholic Fatty Liver Disease: A Systematic Review. Clin Gastroenterol Hepatol 2022, 20, 740–755. [Google Scholar] [CrossRef]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Alterio, A.; Tiribelli, C.; Agostoni, C. A protective effect of breastfeeding on the progression of non-alcoholic fatty liver disease. Arch Dis Child 2009, 94, 801–805. [Google Scholar] [CrossRef]
- Vogelezang, S.; Santos, S.; van der Beek, E.M.; Abrahamse-Berkeveld, M.; Duijts, L.; van der Lugt, A.; Felix, J.F.; Jaddoe, V.W.V. Infant breastfeeding and childhood general, visceral, liver, and pericardial fat measures assessed by magnetic resonance imaging. Am J Clin Nutr 2018, 108, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Gale, C.; Thomas, E.L.; Jeffries, S.; Durighel, G.; Logan, K.M.; Parkinson, J.R.; Uthaya, S.; Santhakumaran, S.; Bell, J.D.; Modi, N. Adiposity and hepatic lipid in healthy full-term, breastfed, and formula-fed human infants: a prospective short-term longitudinal cohort study. Am J Clin Nutr 2014, 99, 1034–1040. [Google Scholar] [CrossRef] [PubMed]
- Purkiewicz, A.; Pietrzak-Fiecko, R. Determination of the Fatty Acid Profile and Lipid Quality Indices in Selected Infant Formulas. Molecules 2024, 29. [Google Scholar] [CrossRef]
- Gerrard, S.D.; Yonke, J.A.; Seymour, K.A.; Sunny, N.E.; El-Kadi, S.W. Feeding medium-chain fatty acid-rich formula causes liver steatosis and alters hepatic metabolism in neonatal pigs. Am J Physiol Gastrointest Liver Physiol 2023, 325, G135–G146. [Google Scholar] [CrossRef]
- Geurtsen, M.L.; Santos, S.; Gaillard, R.; Felix, J.F.; Jaddoe, V.W.V. Associations Between Intake of Sugar-Containing Beverages in Infancy With Liver Fat Accumulation at School Age. Hepatology 2021, 73, 560–570. [Google Scholar] [CrossRef]
- Fidler Mis, N.; Braegger, C.; Bronsky, J.; Campoy, C.; Domellof, M.; Embleton, N.D.; Hojsak, I.; Hulst, J.; Indrio, F.; Lapillonne, A. , et al. Sugar in Infants, Children and Adolescents: A Position Paper of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition Committee on Nutrition. J Pediatr Gastroenterol Nutr 2017, 65, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Rips-Goodwin AR, J.D. , Griebel-Thompson A, Kang KL, Fazzino TL. US infant formulas contain primarily added sugars: An analysis of infant formulas on the US market. Journal of Food Composition and Analysis 2025, 141, 107369. [Google Scholar] [CrossRef]
- Appleton, J.; Russell, C.G.; Laws, R.; Fowler, C.; Campbell, K.; Denney-Wilson, E. Infant formula feeding practices associated with rapid weight gain: A systematic review. Matern Child Nutr 2018, 14, e12602. [Google Scholar] [CrossRef]
- Arisaka, O.; Ichikawa, G.; Koyama, S.; Sairenchi, T. Childhood obesity: rapid weight gain in early childhood and subsequent cardiometabolic risk. Clin Pediatr Endocrinol 2020, 29, 135–142. [Google Scholar] [CrossRef]
- Breij, L.M.; Kerkhof, G.F.; Hokken-Koelega, A.C. Accelerated infant weight gain and risk for nonalcoholic fatty liver disease in early adulthood. J Clin Endocrinol Metab 2014, 99, 1189–1195. [Google Scholar] [CrossRef]
- DiStefano, J.K.; Shaibi, G.Q. The relationship between excessive dietary fructose consumption and paediatric fatty liver disease. Pediatr Obes 2020. [Google Scholar] [CrossRef]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O. , et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Distefano, J.K.; Gerhard, G.S. Effects of dietary sugar restriction on hepatic fat in youth with obesity. Minerva Pediatr (Torino) 2024, 76, 439–448. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K. , et al. Effects of Dietary Fructose Restriction on Liver Fat, De Novo Lipogenesis, and Insulin Kinetics in Children With Obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef]
- Cohen, C.C.; Perng, W.; Sauder, K.A.; Ringham, B.M.; Bellatorre, A.; Scherzinger, A.; Stanislawski, M.A.; Lange, L.A.; Shankar, K.; Dabelea, D. Associations of Nutrient Intake Changes During Childhood with Adolescent Hepatic Fat: The Exploring Perinatal Outcomes Among CHildren Study. J Pediatr 2021, 237, 50–58.e53. [Google Scholar] [CrossRef]
- Papandreou, D.; Rousso, I.; Malindretos, P.; Makedou, A.; Moudiou, T.; Pidonia, I.; Pantoleon, A.; Economou, I.; Mavromichalis, I. Are saturated fatty acids and insulin resistance associated with fatty liver in obese children? Clin Nutr 2008, 27, 233–240. [Google Scholar] [CrossRef]
- Papandreou, D.; Karabouta, Z.; Pantoleon, A.; Rousso, I. Investigation of anthropometric, biochemical and dietary parameters of obese children with and without non-alcoholic fatty liver disease. Appetite 2012, 59, 939–944. [Google Scholar] [CrossRef]
- Arenaza, L.; Medrano, M.; Oses, M.; Huybrechts, I.; Diez, I.; Henriksson, H.; Labayen, I. Dietary determinants of hepatic fat content and insulin resistance in overweight/obese children: a cross-sectional analysis of the Prevention of Diabetes in Kids (PREDIKID) study. Br J Nutr 2019, 121, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.N.; Le, K.A.; Walker, R.W.; Vikman, S.; Spruijt-Metz, D.; Weigensberg, M.J.; Allayee, H.; Goran, M.I. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr 2010, 92, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.S.; Lang, S.; Gilbert, M.; Kamat, D.; Bansal, S.; Ford-Adams, M.E.; Desai, A.P.; Dhawan, A.; Fitzpatrick, E.; Moore, J.B. , et al. Assessment of Diet and Physical Activity in Paediatric Non-Alcoholic Fatty Liver Disease Patients: A United Kingdom Case Control Study. Nutrients 2015, 7, 9721–9733. [Google Scholar] [CrossRef]
- Anderson, E.L.; Howe, L.D.; Fraser, A.; Macdonald-Wallis, C.; Callaway, M.P.; Sattar, N.; Day, C.; Tilling, K.; Lawlor, D.A. Childhood energy intake is associated with nonalcoholic fatty liver disease in adolescents. J Nutr 2015, 145, 983–989. [Google Scholar] [CrossRef]
- Perng, W.; Harte, R.; Ringham, B.M.; Baylin, A.; Bellatorre, A.; Scherzinger, A.; Goran, M.I.; Dabelea, D. A Prudent dietary pattern is inversely associated with liver fat content among multi-ethnic youth. Pediatr Obes 2021, 16, e12758. [Google Scholar] [CrossRef]
- Amiri, F.; Moludi, J.; Jouybari, T.A.; Ghasemi, M.; Sharifi, M.; Mahaki, B.; Soleimani, D. Relationship between dietary inflammatory index and metabolic dysfunction associated steatotic liver disease in children. Sci Rep 2025, 15, 5081. [Google Scholar] [CrossRef]
- Akbulut, U.E.; Isik, I.A.; Atalay, A.; Eraslan, A.; Durmus, E.; Turkmen, S.; Yurttas, A.S. The effect of a Mediterranean diet vs. a low-fat diet on non-alcoholic fatty liver disease in children: a randomized trial. Int J Food Sci Nutr 2022, 73, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Yurtdas, G.; Akbulut, G.; Baran, M.; Yilmaz, C. The effects of Mediterranean diet on hepatic steatosis, oxidative stress, and inflammation in adolescents with non-alcoholic fatty liver disease: A randomized controlled trial. Pediatr Obes 2022, 17, e12872. [Google Scholar] [CrossRef]
- Tanvig, M.; Vinter, C.A.; Jorgensen, J.S.; Wehberg, S.; Ovesen, P.G.; Beck-Nielsen, H.; Christesen, H.T.; Jensen, D.M. Effects of lifestyle intervention in pregnancy and anthropometrics at birth on offspring metabolic profile at 2.8 years: results from the Lifestyle in Pregnancy and Offspring (LiPO) study. J Clin Endocrinol Metab 2015, 100, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Tanvig, M.; Vinter, C.A.; Jorgensen, J.S.; Wehberg, S.; Ovesen, P.G.; Lamont, R.F.; Beck-Nielsen, H.; Christesen, H.T.; Jensen, D.M. Anthropometrics and body composition by dual energy X-ray in children of obese women: a follow-up of a randomized controlled trial (the Lifestyle in Pregnancy and Offspring [LiPO] study). PLoS One 2014, 9, e89590. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mechanisms of fetal lipid overload in maternal obesity. Elevated maternal triglycerides (TGs) and free fatty acids (FFAs) characteristic of excess adiposity and gestational diabetes, are delivered to the placenta. Placental lipases hydrolyze TGs, generating FFAs that are transported across the syncytiotrophoblast via fatty acid transporters (FATs), such as FATP, FABP, and CD36. The resulting increase in lipid flux to the fetus overwhelms hepatic oxidative and VLDL export capacities, leading to intrahepatic lipid accumulation and fetal hepatic steatosis.
Figure 1.
Mechanisms of fetal lipid overload in maternal obesity. Elevated maternal triglycerides (TGs) and free fatty acids (FFAs) characteristic of excess adiposity and gestational diabetes, are delivered to the placenta. Placental lipases hydrolyze TGs, generating FFAs that are transported across the syncytiotrophoblast via fatty acid transporters (FATs), such as FATP, FABP, and CD36. The resulting increase in lipid flux to the fetus overwhelms hepatic oxidative and VLDL export capacities, leading to intrahepatic lipid accumulation and fetal hepatic steatosis.
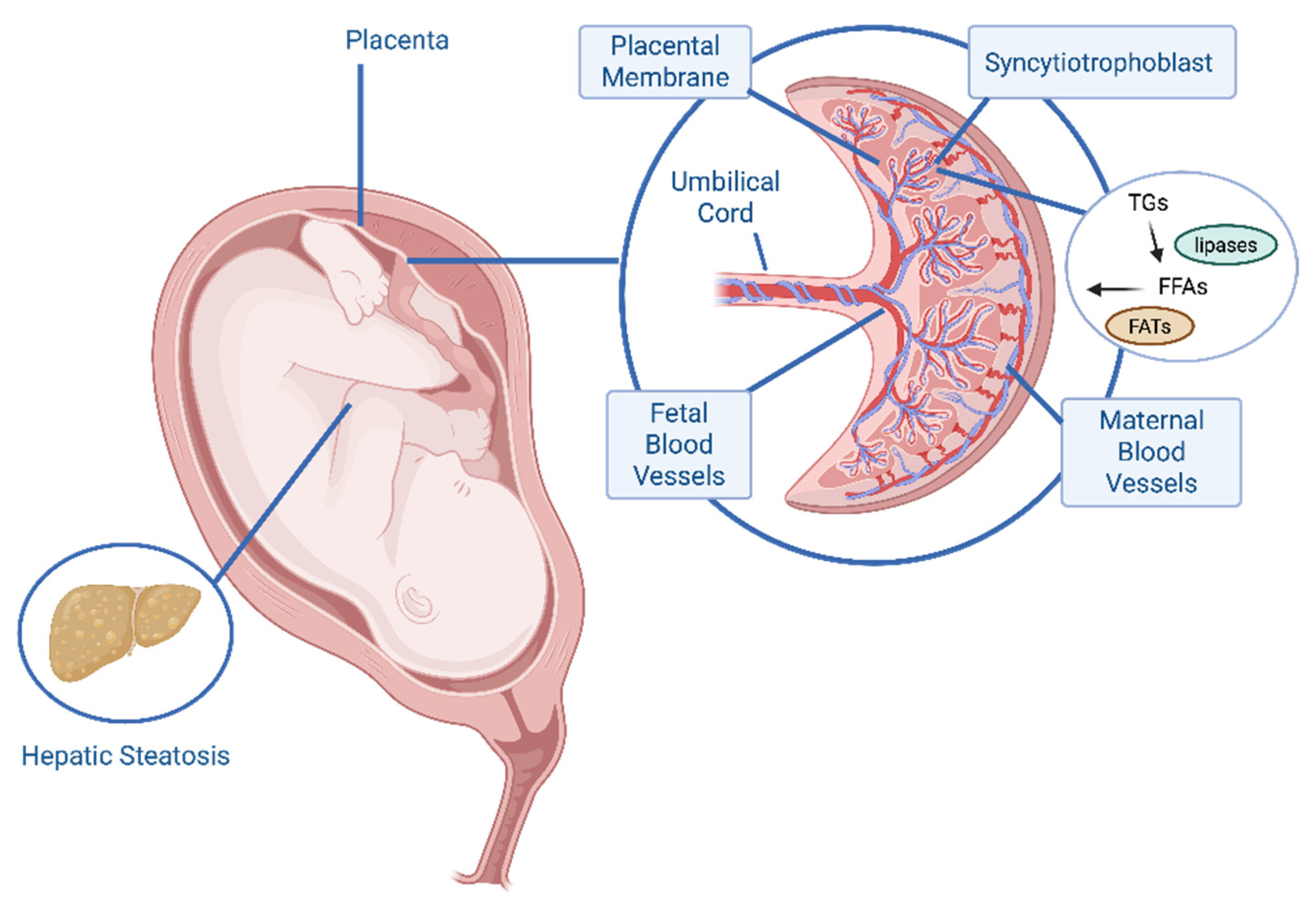
Figure 2.
Cumulative trajectory from early metabolic programming. This figure illustrates the cumulative and chronological exposure to risk factors across developmental stages that converge to increase lifetime risk of pediatric MASLD. Prenatal: Fetal exposures, including maternal obesity, gestational diabetes mellitus (GDM), maternal diet, and exposures to smoking, stress, environmental toxins, and medications, result in fetal hepatic overload; the initial metabolic injury that programs subsequent disease risk. Postnatal: Early infancy practices, such as feeding methods and rapid weight gain, contribute to established metabolic dysfunction. Childhood: Continuing factors, such as poor diet quality, genetic susceptibility, lifestyle (e.g., sedentary behavior), and adverse environmental exposures (e.g., pollutants, endocrine disrupting compounds), compounded by increasing adiposity, finalize the trajectory towards MASLD. The curved arrow represents the cumulative pathway of metabolic programming. It signifies that the effects of exposures from all preceding stages (prenatal, postnatal, childhood) build upon each other, resulting in the development of pediatric MASLD.
Figure 2.
Cumulative trajectory from early metabolic programming. This figure illustrates the cumulative and chronological exposure to risk factors across developmental stages that converge to increase lifetime risk of pediatric MASLD. Prenatal: Fetal exposures, including maternal obesity, gestational diabetes mellitus (GDM), maternal diet, and exposures to smoking, stress, environmental toxins, and medications, result in fetal hepatic overload; the initial metabolic injury that programs subsequent disease risk. Postnatal: Early infancy practices, such as feeding methods and rapid weight gain, contribute to established metabolic dysfunction. Childhood: Continuing factors, such as poor diet quality, genetic susceptibility, lifestyle (e.g., sedentary behavior), and adverse environmental exposures (e.g., pollutants, endocrine disrupting compounds), compounded by increasing adiposity, finalize the trajectory towards MASLD. The curved arrow represents the cumulative pathway of metabolic programming. It signifies that the effects of exposures from all preceding stages (prenatal, postnatal, childhood) build upon each other, resulting in the development of pediatric MASLD.
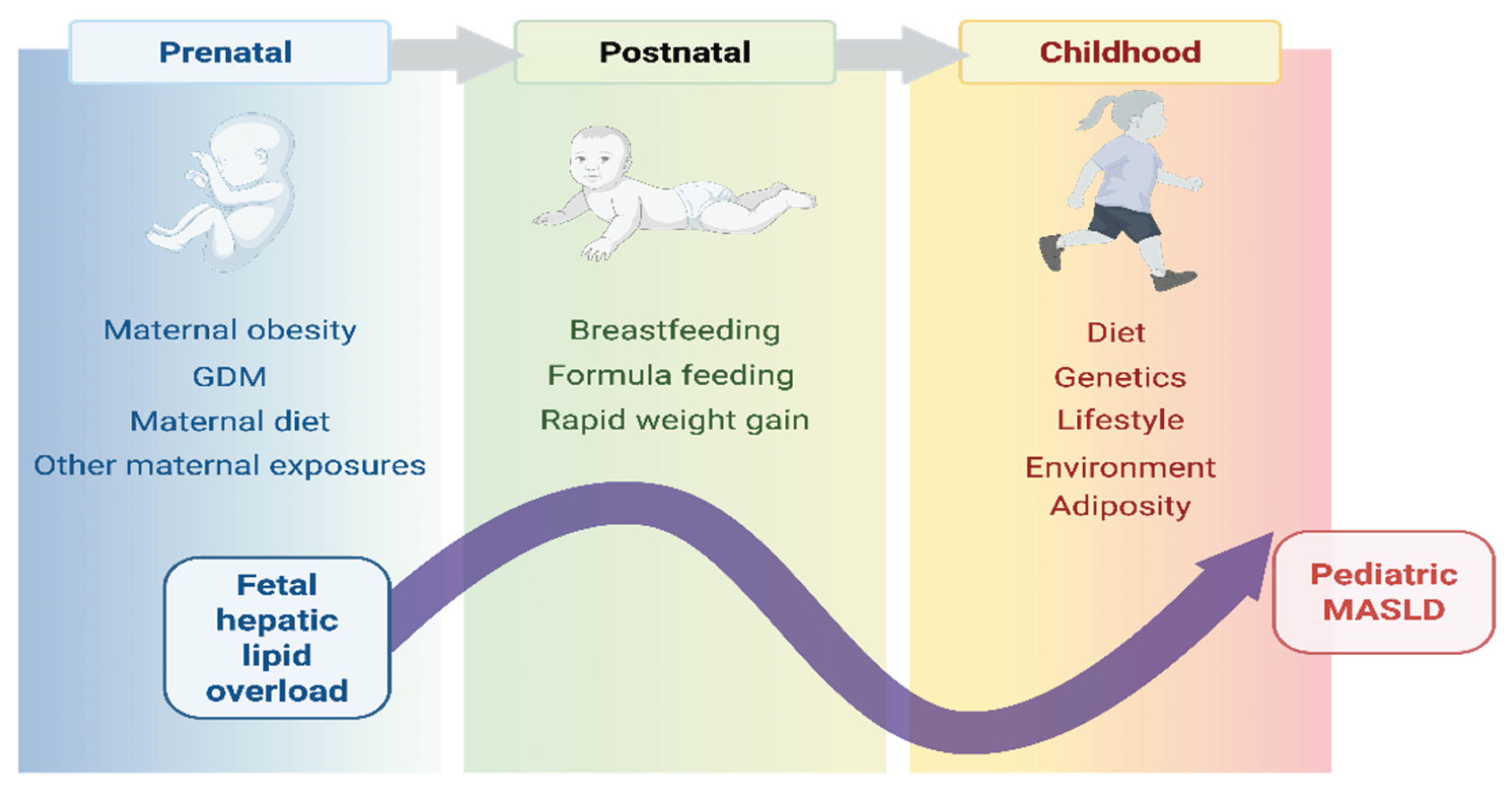

Table 1.
Observational studies of maternal BMI or diet quality and offspring hepatic fat.
| Cohort | Loc | n | Age | Modality | Main Findings | Ref |
| Children’s Hospital CO | USA | 25 | 1-3 wks | MRS | Greater IHCL content in neonates born to obese diabetic mothers | [53] |
| Chelsea & Westminster Hospital | UK | 105 | 11.7 d | MRS | 8.6 % IHCL increase per BMI unit | [52] |
| Raine | Aus | 1170 | 17 y | USS | Maternal obesity increases adolescent MASLD risk, breastfeeding > 6 months confers protection | [54] |
| ALSPAC | UK | 1,215 | 17-18 y | USS | Offspring adiposity mediates maternal obesity/diabetes steatosis risk | [55] |
| ALSPAC | UK | 3,353 | 24 y | TE | [56] | |
| ESPRESSO | Sweden | 165 | <25 y | Biopsy | Maternal obesity increases MASLD/severe MASLD in young adults | [57] |
| SPCS | China | 430 | 8 y | TE | Offspring steatosis aOR 8.26 for maternal obesity and GDM | [58] |
| Healthy Start | USA | 278 | 4-8 y | MRI | Poor maternal diet increases offspring steatosis susceptibility | [59] |
Table 2.
Key Risk and Protective Factors Aligned with DOHaD Model.
| Factor | Association with pediatric MASLD risk | Potential role in DOHaD programming |
| Maternal pre-pregnancy overweight/obesity | Consistently identified as a modifiable risk factor | Programs fetal liver for high lipid storage due to nutrient oversupply |
| Breastfeeding (≥6 months) | Frequently associated with duration-dependent protective effect | Promotes a slower, healthier growth trajectory and provides bioactive factors that modulate metabolism |
| Rapid post-natal catch-up growth | May contribute to later hepatic steatosis | Exacerbates metabolic stress on an in utero-programmed liver, accelerating fat accumulation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



