
Submitted:
13 November 2025
Posted:
14 November 2025
You are already at the latest version
Abstract
Platinum-based drugs play a pivotal role in contemporary cancer treatment, but their therapeutic utility is often limited by acquired resistance. The diiodido analogue, cis-PtI2(NH3)2 is a promising derivative that has demonstrated the ability to overcome cis-platin resistance in vitro. To establish the molecular basis for this superior activity, we integrated experimental 14N Nuclear Magnetic Resonance (NMR) spectroscopy with computational density functional theory (DFT) methods to precisely and comparatively understand the drug activation mechanisms. Comparative 14N NMR experiments elucidated the initial ligand substitution step, confirming halide displacement and a markedly higher tendency for ammonia release from cis-PtI2(NH3)2, particularly when reacting with sul-fur-containing amino acids. Complementary DFT calculations determined the substitution energy values, revealing that the superior leaving-group ability of iodide results in a thermodynamically more favorable activation. Conceptual DFT parameters (softness, hardness, and Fukui indices) further demonstrated that initial substitution induces a strong trans effect, leading to the electronic sensitization of the remaining iodide ligand. This strong agreement between computational predictions and experimental data establishes a coherent molecular activation mechanism for cis-PtI2(NH3)2 demonstrating that iodide substitution promotes both thermodynamic and electronic activation of the plati-num center, which is the key to its distinct pharmacological profile and ability to circumvent resistance.
Keywords:
platinum complexes
; cancer
; cisplatin analogue
; NMR
; DFT
1. Introduction
Platinum-based drugs represent one of the cornerstones of contemporary anticancer chemotherapy, with cisplatin being the paradigm and still one of the most widely used agents in clinical practice [1]. Despite its remarkable efficacy, the therapeutic utility of cisplatin is limited by intrinsic and acquired resistance, as well as by severe dose-dependent side effects [2]. For this reason, the development and mechanistic investigation of cisplatin analogues remain a highly active area of research, aiming both at improving pharmacological performance and at disclosing the molecular principles underlying platinum–biomolecule interactions [3,4].
In this context, the diiodido analogue of cisplatin, cis-[PtI2(NH3)2] (hereafter cisPtI2, Figre 1), has long been neglected, mainly due to early and largely unsubstantiated claims of poor pharmacological activity [5]. Our recent systematic studies, however, have contributed to reappraising the chemical and biological profile of cisPtI2. In particular, we have shown that this compound displays cytotoxicity comparable to cisplatin against several tumor cell lines and, importantly, is able to overcome platinum resistance in vitro [6]. Furthermore, while the DNA platination pattern of cisPtI2 closely mirrors that of cisplatin, subtle yet significant differences in its solution behavior and reactivity appear to be responsible for its distinct biological effects, notably its ability to overcome platinum resistance [6,7].
Figure 1.
Chemical structure of cisplatin (left) and its iodide analogue (right).
Previous studies highlighted how cisPtI2 differs from cisplatin in its interactions with proteins and model amino acid sidechains. Comparative experiments with lysozyme and cytochrome c revealed a divergent reactivity pattern, which was further rationalized by density functional theory (DFT) calculations on sulfur- and nitrogen-containing ligands. These studies demonstrated that while both cisplatin and cisPtI2 readily undergo halide substitution, the subsequent steps, including possible ammonia release, are markedly influenced by the nature of the incoming ligand [8]. Such findings pointed to the protein environment as a crucial determinant of the peculiar reactivity of cisPtI2 and of the nature of its adducts.
Building on these foundations, the present study aims to provide a deeper mechanistic understanding of the activation processes of cisplatin and cisPtI2 by combining complementary experimental and computational approaches. In particular, we employed 14N nuclear magnetic resonance (NMR) spectroscopy to monitor the earliest substitution steps, focusing on halide displacement and the potential release of coordinated ammonia. Complementary to this experimental work, DFT was used to calculate the Gibbs free energy of substitution – which assesses the thermodynamic stability of the Pt-biomolecule conjugates – along with conceptual DFT descriptors such as softness, hardness, and Fukui indices. These calculations, performed using small-molecule models, correlate theoretical reactivity parameters with the experimentally observed ligand exchange behaviour.
The integration of NMR spectroscopy with computational chemistry offers a coherent and complementary perspective on the ligand substitution processes of these platinum complexes. By elucidating the molecular basis of halide and ammonia release, this work contributes to a more precise definition of the activation pathways of cisplatin and cisPtI2. Ultimately, such knowledge is essential for rationalizing the distinct pharmacological properties of these complexes and for guiding the design of new platinum-based drugs with improved efficacy and resistance profiles.
2. Results and discussion
2.1. Cisplatin vs. cisPtI2 : NMR Investigation of Ligand Exchange with Histidine, Cysteine, and Methionine
The ligand substitution reactions of cisplatin and cisPtI2 with three representative amino acids (histidine, cysteine, and methionine) were investigated using NMR spectroscopy. Due to technical constraints and the risk of salt precipitation, the study was limited to the first substitution step, which is nevertheless the most relevant for understanding the distinct reactivity of these Pt complexes with small proteins, as previously reported [9,10]. All experiments were conducted in a 50% DMF/water mixture to ensure the solubility of all reagents throughout the reaction. The coordination behavior of the ammonia ligand, including its potential release from the platinum center, was monitored for both complexes by 14N NMR spectroscopy. 14N is a magnetically active nucleus (spin = 1) with a quadrupolar moment. Although this feature is often considered disadvantageous, we selected 14N because its faster magnetization recovery (T1 of 14N in ammonia is ~25 times shorter than that of 15N) enables the acquisition of thousands of scans within a relatively short time, yielding spectra with an excellent signal-to-noise ratio. However, the linewidth of 14N signals increases proportionally with lattice anisotropy around the observed nucleus, restricting the applicability of this technique mainly to highly symmetrical molecules. In the case of the two Pt complexes, the 14N NMR spectra typically display a broad resonance attributable to platinum-coordinated ammonia. Upon release of ammonia from the metal center, a sharp signal corresponding to the NH4+ cation emerges at –23 ppm, allowing a qualitative assessment of ammonia release by NMR spectroscopy (Table 1).
As shown in Figs. S1-S4, it is evident that the two complexes upon dissolution in the reference medium are stable over time and that no ammonia release occurs. The stability of the 14NNMR signal also suggests that no halide release occurs under the applied experimental conditions. This observation contrasts with our previous findings on halide release from the two Pt compounds under physiological-like conditions [6]. However, in our case, the different behavior is attributable to the relatively high percentage of DMF that disfavors halide displacement and to the relatively large concentration of the two complexes.
Upon addition of 1 equivalent of each amino acid to either Cisplatin and cis-[PtI2(NH3)2] the 14NNMR spectra depicted in Figure 2, Figure 3, Figure 4 are obtained.
14N NMR experiments confirmed the occurrence of ammonia release in almost all cases, with the only exception being the incubation of cisplatin with histidine. Quantitatively, the preferential ammonia release from cisPtI2 with respect to cisplatin is clearly demonstrated by the significantly higher NH4+ peak heights.
2.2. Computational Assessment
The present study employs a purely thermodynamic approach, calculating the Gibbs free energy of substitution (GFE) for the reaction in which reactants and products are infinitely separated. This methodology differs significantly from the pseudounimolecular approach used in our previous work on cisplatin and cisPtI2 [8], which focused exclusively on the reaction profile including reactant-adduct, transition-state, and product-adduct species. While the prior work provided insight into kinetic barriers [8], the current free energy values reflect the inherent thermodynamic driving force for the overall substitution process, providing important insight into the relative stability of the resulting Pt-biomolecule conjugates at equilibrium.
A systematic comparison of the substitution reaction GFE energies (Table 2 and Table 3) immediately highlights a substantial thermodynamic advantage for iodoplatin over cisplatin. For all three biologically relevant nucleophile models – thiol (CH3SH), dimethyl sulfur (CH3SCH3), and imidazole (imi) – substitution is thermodynamically more favorable for the iodo-complexes. This thermodynamic advantage stems directly from the intrinsically superior leaving group ability of the iodide (I−) ligand compared to chloride (Cl−). This fundamental difference shifts the substitution reaction to become significantly less endothermic or more exothermic in the iodoplatin series. For instance, substitution by the thiol model is highly endothermic for cisplatin (GFE = +4.9 kcal/mol), yet for iodoplatin, the reaction is only mildly endothermic (GFE = +2.2 kcal/mol), representing an approximate 2.7 kcal/mol thermodynamic driving force differential. Crucially, the overall order of substitution favorability remains consistent for both parent compounds: imidazole ≫ dimethyl sulfur > thiol. The most thermodynamically favorable substitution observed is the replacement of iodide by imidazole (GFE = −7.4 kcal/mol), reflecting the strong binding ability of the imidazole nitrogen to Pt(II). This robust thermodynamic preference for N-donor sites supports and quantitatively extends observations learned from the kinetic analysis in the iodoplatin study [8].
Iodoplatin exhibits a significantly lower chemical hardness ( = 0.148) and a commensurately higher global softness ( = 3.386) compared to cisplatin ( = 0.169, = 2.953). This electronic profile indicates that iodoplatin is an intrinsically softer molecule, which aligns with the principle of hard-soft acid-base (HSAB) theory and suggests a strong preference for interaction with soft, polarizable nucleophiles, such as the sulfur donors (CH3SH, CH3SCH3). The increased softness quantitatively supports the kinetic lability implied by the GFE data, confirming that iodoplatin is electronically primed for substitution. Furthermore, the chemical potential () shifts to more negative values upon substitution by sulfur ligands for both cisplatin and iodoplatin, suggesting the resulting cationic products are stronger electron acceptors. The electrophilicity index (), a measure of the complex's capacity to acquire electrons, generally increases upon substitution, with the substituted iodoplatin complexes, [Pt(NH3)2I(L)]+, showing the highest values (up to =0.087 for the CH3SCH3 adduct). This heightened electrophilicity indicates that the positively charged substitution products are not inert, but rather highly reactive species with an increased propensity for subsequent binding events, such as coordination to a second nucleophile or cross-linking DNA.
To understand the specific electronic reorganization driving the substitution, we analyzed the local reactivity using Fukui indices ( and ), which quantify the change in electron density at specific atomic sites upon gaining or losing an electron (Table 4 and Table 5). The index describes local electrophilicity (susceptibility to nucleophilic attack at the Pt center), and describes local nucleophilicity (electron-donating ability, relevant for the halide leaving groups). The calculated Fukui indices confirm the electronic basis for the thermodynamic preference of ligand substitution in cisplatin and iodoplatin.
Analysis of the leaving group character, represented by , reveals that for the parent compounds Pt(NH3)2 × 2, the negative value of is significantly greater on the iodide atoms (X = I, = -0.278) than on the chloride atoms (X=Cl, = -0.172). This 62% larger magnitude of on the iodide atom signifies a correspondingly greater capacity to donate electron density (be lost as I−), confirming its superior leaving group ability in the initial substitution step.
In terms of the central Pt atom's reactivity, the Pt center in cisplatin exhibits a more negative value (-0.414) than that in iodoplatin (-0.302), suggesting the Pt atom in cisplatin is, unexpectedly, a slightly stronger electron acceptor in its initial state. However, the overall reaction outcome is decisively dominated by the facile release of the iodide ligand, as confirmed by the favorable GFE data.
Upon monosubstitution to form [Pt(NH3)2X(L)]+, a notable electronic and mechanistic consequence is observed: the value on the remaining halide (X) is significantly enhanced in magnitude, particularly for the remaining iodide atom (X=I). The on I drops to values as low as -0.633 when the nucleophile is CH3SH or CH3SCH3. This almost 130% increase in the magnitude of the index on the remaining iodide signifies a powerful electronic activation, essentially demonstrating an augmented trans-effect from the newly bound sulfur ligand. This substantial increase in the leaving group character of the remaining iodide atom explains the heightened lability and enhanced reactivity observed in iodoplatin-based complexes following initial substitution. This potent local electronic effect provides a robust molecular explanation for the distinct reactivity pattern of iodoplatin with proteins observed in initial experimental studies (as discussed in [8]), where the substitution leads to a product that is not just thermodynamically stable but is also electronically primed for subsequent, rapid binding events, a characteristic essential for achieving potent biological activity.
4. Materials and Methods
Synthesis of compounds and 14N NMR experiments. Cisplatin was purchased from Merck and used without further purification, cisPtI2 was synthesized as previously reported by some of us [6]. NMR spectra were recorded on a Bruker Avance III 400 spectrometer equipped with a Bruker Ultrashield 400 Plus superconducting magnet (resonating frequencies: 400.13 and 28.89 MHz for 1H and 14N respectively) and a 5 mm PABBO BB-1H/D Z-GRD Z108618/0049 probe. All experiments were run at room temperature (25 ± 2 °C) with a standard 1D sequence with inverse gated decoupling (zgig). All spectra were recorded as a mean of 2000 scans, with recycle delay of 2 seconds. All samples used in 14NNMR experiments were prepared in a solvent mixture of 250 µl of DMF-d7 and 250 µl of Buffer Acetate 100 mM pH 6.9. Quantitative determination of ammonia release at 24h was performed comparing absolute integrals of NH4+ signals with respect to a 100mM NH4Br standard solution. The comparison was performed through nmrq function of Topspin 4.0.1 software.
Computational details. Our computations were based upon density functional theory (DFT), a proven methodology for accurately modeling the geometry and reaction characteristics of transition metal compounds [11,12,13], including those containing platinum [14,15]. All calculations were carried out employing the Gaussian 16 quantum chemistry package [16] We utilized the range-corrected density functional ωB97X-D [17] throughout, selecting it for its known precision in determining both molecular structures and electronic energies [18]. Geometries of all molecules were optimized, and their initial electronic and solvation energies calculated, using the ωB97X-D functional paired with the def2SVP basis set [19,20] in water. To account for the surrounding solvent environment, the polarizable continuum model (PCM) in its integral equation formalism variant (IEFPCM) [21] was applied. This specific model was chosen because it has recently demonstrated significantly reduced errors for the aqueous solvation free energies of both neutral and ionic species compared to alternative continuum models [22]. Following geometry optimization, frequency calculations were performed to confirm the stability of the optimized structures and to obtain the necessary Zero-Point Energy (ZPE) and thermal corrections for calculating thermodynamic properties. The final, high-level single-point electronic and solvation energies were then computed on the optimized geometries using the ωB97X-D functional in conjunction with the larger def2TZVP basis set [19,20].
To comprehensively characterize the electronic reactivity of the compounds, we employed conceptual DFT (cDFT) [23,24]. The global reactivity parameters—specifically the chemical potential (), hardness (), softness (), and electrophilicity ()—were derived from the energies of the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO), as determined at the ωB97X-D/def2SVP level of theory in water. These parameters are reported in hartrees (Ha), with the exception of softness (), which is in Ha–1. The parameters were calculated using the following established formulas: the chemical potential μ was approximated as ; the hardness η was approximated as ; the softness S was defined as ; and the electrophilicity ω was calculated as .
The local reactivity at each atomic center was assessed by calculating the dimensionless Fukui indices () [25]. These indices were determined from the Mulliken charges () for a given atom , which were calculated at the same ωB97X-D/def2SVP level of theory in water. The formulas used for the Fukui indices for nucleophilic () and electrophilic () attack were: and , respectively, where , , and denote the number of electrons in the neutral, anionic, and cationic species.
5. Conclusions
The combined experimental and computational investigation presented here provides new mechanistic insights into the activation pathways of cisplatin and its iodide analogue, cisPtI2. 14N NMR results revealed a markedly higher tendency of the iodide derivative to undergo ammonia release, particularly in the presence of sulfur-containing amino acids. Complementary DFT calculations demonstrated that the enhanced reactivity of cisPtI2 arises from its lower chemical hardness and the superior leaving-group ability of iodide, resulting in significantly more favorable substitution thermodynamics. Conceptual DFT descriptors and Fukui function analysis confirmed this, highlighting the intrinsic electronic softness of the iodinated species and the strong trans effect induced by the newly bound nucleophile on the remaining iodide ligand. Together, these findings outline a consistent picture in which iodide substitution promotes both thermodynamic and electronic activation of the platinum center, ultimately explaining the distinct reactivity and potent biological activity, including its ability to overcome cisplatin resistance, compared to cisplatin. The present results contribute to a deeper understanding of halide-dependent activation mechanisms and may guide the rational design of next-generation platinum-based anticancer agents with improved performance and resistance profiles.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1-S4.
Author Contributions
Conceptualization, T.M. and D.C.; methodology, D.C., T.M., A.P., D.L.M.; investigation,R.D.L., E.B, M.P., D.C., L.C., L.F.,D.P., A.M.; data curation, T.M., A.M., I.T., D.C.; writing—original draft preparation, T.M., D.C., I.T.; writing—review and editing, T.M., I.T, A.Z., D.L.M, L.C., L.F., A.M; A.Z., T.M., A.N., P.F.; All authors have read and agreed to the published version of the manuscript.
Funding
T.M. thanks the financial support from Ministero Italiano dell’Universitàe della Ricerca (MUR) under the program PRIN 2022-Progetti di Rilevante Interesse Nazionale, project code: 2022ALJRPL “Biocompatible nanostructures for the chemotherapy treatment of prostate cancer” and from University of Pisa, SPARK-Proof of Concept 2025.
Data Availability Statement
The data underlying this article will be shared on reasonable re- quest to the corresponding authors.
Acknowledgments
We acknowledge the CINECA award under the ISCRA initiative, for the availability of high performance computing resources and support. I.T. gratefully acknowledges the usage of HPC resources from Direction du Numérique – Centre de Calcul de l’Université de Bourgogne (DNUM CCUB).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Linares, J.; Sallent-Aragay, A.; Badia-Ramentol, J.; Recort-Bascuas, A.; Méndez, A.; Manero-Rupérez, N.; Re, D.L.; Rivas, E.I.; Guiu, M.; Zwick, M.; et al. Long-Term Platinum-Based Drug Accumulation in Cancer-Associated Fibroblasts Promotes Colorectal Cancer Progression and Resistance to Therapy. Nat Commun 2023, 14, 746. [Google Scholar] [CrossRef]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef]
- Cirri, D.; Chiaverini, L.; Pratesi, A.; Marzo, T. Is the Next Cisplatin Already in Our Laboratory? Comments on Inorganic Chemistry 2023. [Google Scholar] [CrossRef]
- Chiaverini, L.; Leo, R.D.; Famlonga, L.; Pacini, M.; Baglini, E.; Barresi, E.; Peana, M.F.; Tolbatov, I.; Marrone, A.; La Mendola, D.; et al. The Metal(Loid)s’ Dilemma. What’s the next Step for a New Era of Inorganic Molecules in Medicine? Metallomics. 2025, 17, mfaf013. [Google Scholar] [CrossRef]
- Musumeci, D.; Platella, C.; Riccardi, C.; Merlino, A.; Marzo, T.; Massai, L.; Messori, L.; Montesarchio, D. A First-in-Class and a Fished out Anticancer Platinum Compound: Cis-[PtCl2(NH3)2] and Cis-[PtI2(NH3)2] Compared for Their Reactivity towards DNA Model Systems. Dalton Trans 2016, 45, 8587–8600. [Google Scholar] [CrossRef]
- Marzo, T.; Pillozzi, S.; Hrabina, O.; Kasparkova, J.; Brabec, V.; Arcangeli, A.; Bartoli, G.; Severi, M.; Lunghi, A.; Totti, F.; et al. Cis-Pt I2(NH3)2: A Reappraisal. Dalton Trans. 2015, 44, 14896–14905. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, A.G.; Cama, M.; Pajuelo-Lozano, N.; Álvarez-Valdés, A.; Perez, I.S. New Findings in the Signaling Pathways of Cis and Trans Platinum Iodido Complexes’ Interaction with DNA of Cancer Cells. ACS Omega 2019, 4, 21855–21861. [Google Scholar] [CrossRef]
- Tolbatov, I.; Marzo, T.; Cirri, D.; Gabbiani, C.; Coletti, C.; Marrone, A.; Paciotti, R.; Messori, L.; Re, N. Reactions of Cisplatin and Cis-[PtI2(NH3)2] with Molecular Models of Relevant Protein Sidechains: A Comparative Analysis. J Inorg Biochem 2020, 209, 111096. [Google Scholar] [CrossRef]
- Wang, J.; Tao, J.; Jia, S.; Wang, M.; Jiang, H.; Du, Z. The Protein-Binding Behavior of Platinum Anticancer Drugs in Blood Revealed by Mass Spectrometry. Pharmaceuticals (Basel) 2021, 14, 104. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Chval, Z.; Burda, J.V. Cisplatin Interaction with Cysteine and Methionine in Aqueous Solution: Computational DFT/PCM Study. J. Phys. Chem. B 2009, 113, 3139–3150. [Google Scholar] [CrossRef] [PubMed]
- Tolbatov, I.; Marzo, T.; Umari, P.; Mendola, D.L.; Marrone, A. Detailed Mechanism of a DNA/RNA Nucleobase Substituting Bridging Ligand in Diruthenium(II,III) and Dirhodium(II,II) Tetraacetato Paddlewheel Complexes: Protonation of the Leaving Acetate Is Crucial. Dalton Trans. 2025, 54, 662–673. [Google Scholar] [CrossRef]
- Tolbatov, I.; Umari, P.; Marrone, A. The Binding of Diruthenium (II,III) and Dirhodium (II,II) Paddlewheel Complexes at DNA/RNA Nucleobases: Computational Evidences of an Appreciable Selectivity toward the AU Base Pairs. J Mol Graph Model 2024, 131, 108806. [Google Scholar] [CrossRef]
- Tolbatov, I.; Marrone, A. Reactivity of N-Heterocyclic Carbene Half-Sandwich Ru-, Os-, Rh-, and Ir-Based Complexes with Cysteine and Selenocysteine: A Computational Study. Inorg. Chem. 2022, 61, 746–754. [Google Scholar] [CrossRef]
- Scoditti, S.; Dabbish, E.; Russo, N.; Mazzone, G.; Sicilia, E. Anticancer Activity, DNA Binding, and Photodynamic Properties of a N∧C∧N-Coordinated Pt(II) Complex. Inorg. Chem. 2021, 60, 10350–10360. [Google Scholar] [CrossRef]
- Tolbatov, I.; Cirri, D.; Tarchi, M.; Marzo, T.; Coletti, C.; Marrone, A.; Messori, L.; Re, N.; Massai, L. Reactions of Arsenoplatin-1 with Protein Targets: A Combined Experimental and Theoretical Study. Inorg. Chem. 2022, 61, 3240–3248. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets Tor Molecular Calculations. Springer Vol.3.
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Remya, K.; Suresh, C.H. Which Density Functional Is Close to CCSD Accuracy to Describe Geometry and Interaction Energy of Small Noncovalent Dimers? A Benchmark Study Using Gaussian09. Journal of Computational Chemistry 2013, 34, 1341–1353. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theoret. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M.; Tomasi, J. A New Definition of Cavities for the Computation of Solvation Free Energies by the Polarizable Continuum Model. J. Chem. Phys. 1997, 107, 3210–3221. [Google Scholar] [CrossRef]
- Klamt, A.; Moya, C.; Palomar, J. A Comprehensive Comparison of the IEFPCM and SS(V)PE Continuum Solvation Methods with the COSMO Approach. J. Chem. Theory Comput. 2015, 11, 4220–4225. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Chattaraj, P.K. Conceptual Density Functional Theory Based Electronic Structure Principles. Chem. Sci. 2021, 12, 6264–6279. [Google Scholar] [CrossRef] [PubMed]
- Yang, Weitao. ; Mortier, W.J. The Use of Global and Local Molecular Parameters for the Analysis of the Gas-Phase Basicity of Amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef]
Figure 2.
14N NMR spectra of cis-PtCl2(NH3)2 17 mM (A) and cisPtI2 34 mM (B) incubated up to 24 h with 1 equivalent of histidine.
Figure 2.
14N NMR spectra of cis-PtCl2(NH3)2 17 mM (A) and cisPtI2 34 mM (B) incubated up to 24 h with 1 equivalent of histidine.

Figure 3.
14N NMR: 14N NMR spectra of cis-PtCl2(NH3)2 17 mM (A) and cisPtI2 34 mM (B) incubated up to 24 h with 1 equivalent of methionine.
Figure 3.
14N NMR: 14N NMR spectra of cis-PtCl2(NH3)2 17 mM (A) and cisPtI2 34 mM (B) incubated up to 24 h with 1 equivalent of methionine.
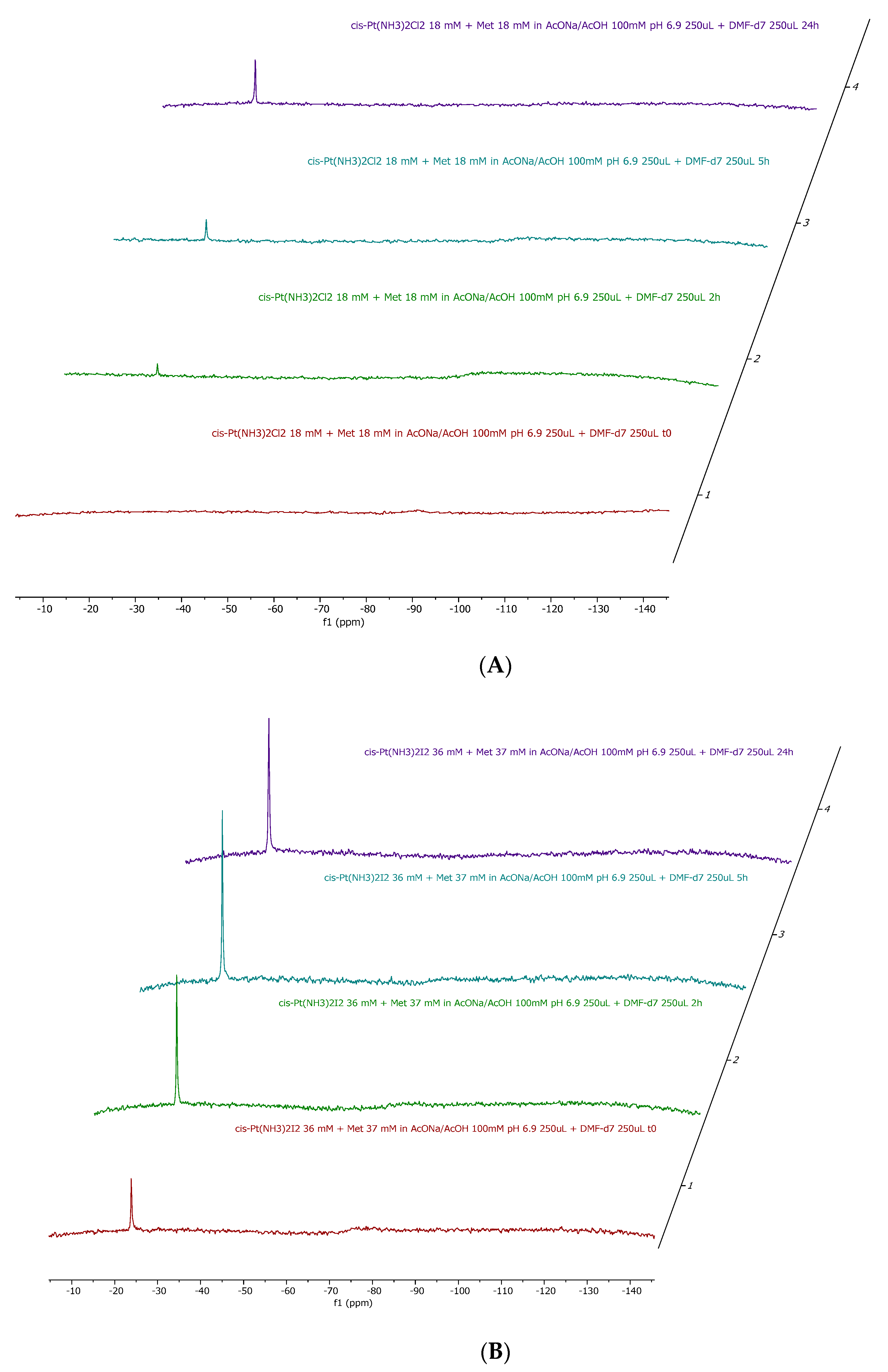
Figure 4.
14N NMR spectra of cis-PtCl2(NH3)2 17 mM (A) and cisPtI2 34 mM (B) incubated up to 24 h with 1 equivalent of cysteine.
Figure 4.
14N NMR spectra of cis-PtCl2(NH3)2 17 mM (A) and cisPtI2 34 mM (B) incubated up to 24 h with 1 equivalent of cysteine.
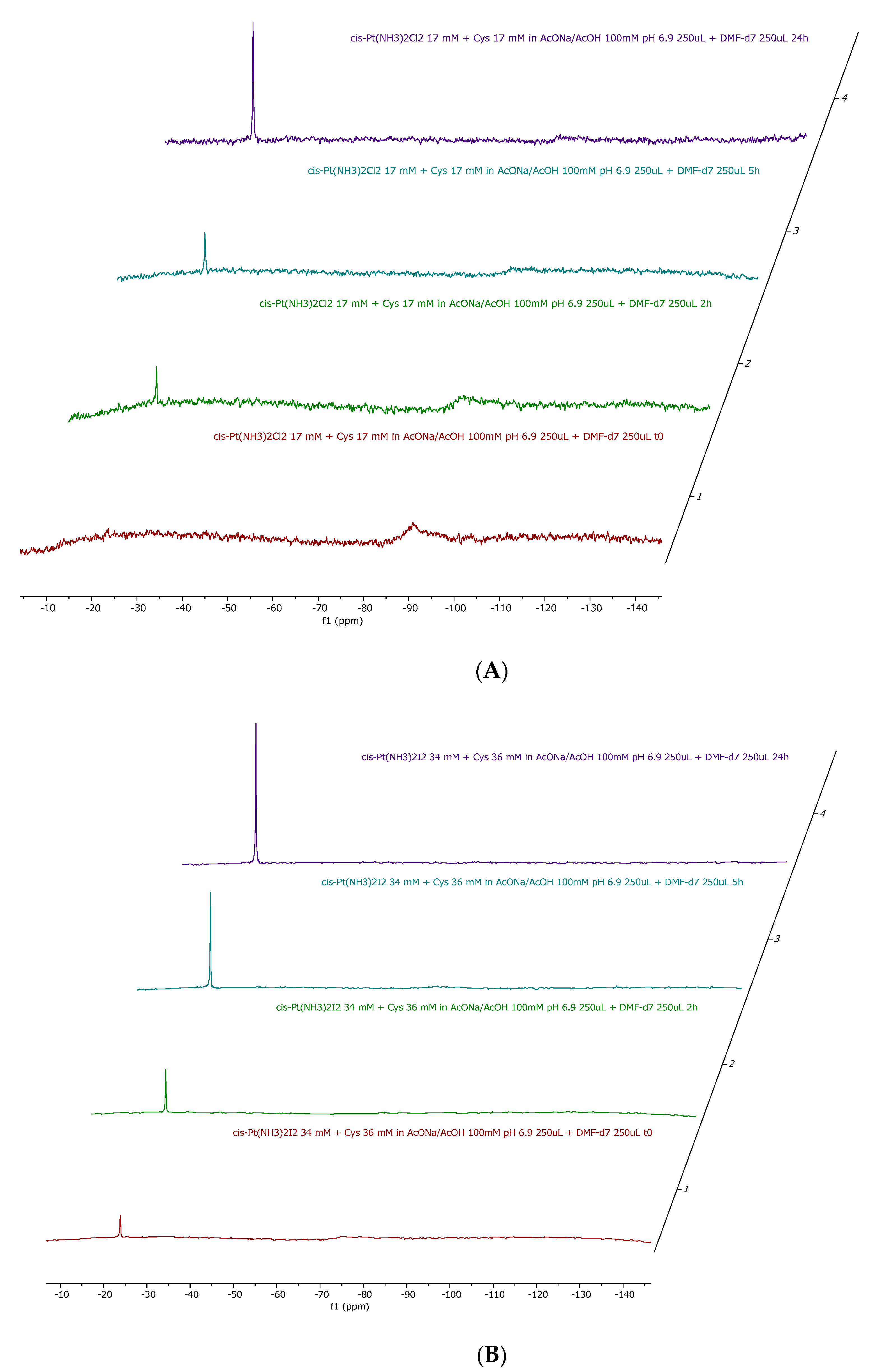

Table 1.
Quantitative determination (%) of released ammonia at 24h of incubation.
| Cisplatin + His | Cisplatin + Met | Cisplatin + Cys | cis-[PtI2(NH3)2] + His | cis-[PtI2(NH3)2] + Met | cis-[PtI2(NH3)2] + Cys |
|---|---|---|---|---|---|
| No release | 11% | 14% | 54% | 41% | 100% |
Table 2.
Gibbs free energies of substitution (GFE in kcal/mol) and conceptual DFT global descriptors (,,,) for cisplatin and monosubstituted analogues.
Table 2.
Gibbs free energies of substitution (GFE in kcal/mol) and conceptual DFT global descriptors (,,,) for cisplatin and monosubstituted analogues.
| Complex | GFE | ||||
|---|---|---|---|---|---|
| Pt(NH3)2Cl2 | 0 | -0.140 | 0.169 | 2.953 | 0.058 |
| Pt(NH3)2Cl(CH3SH) | 4.9 | -0.161 | 0.169 | 2.966 | 0.077 |
| Pt(NH3)2Cl(CH3SCH3) | 0.2 | -0.160 | 0.170 | 2.942 | 0.075 |
| Pt(NH3)2Cl(imi) | -4.2 | -0.144 | 0.177 | 2.819 | 0.059 |
| Pt(NH3)Cl2(CH3SH) | 5.1 | -0.154 | 0.163 | 3.071 | 0.072 |
| Pt(NH3)Cl2(CH3SCH3) | 1.0 | -0.153 | 0.164 | 3.053 | 0.071 |
| Pt(NH3)Cl2(imi) | -1.6 | -0.138 | 0.169 | 2.954 | 0.056 |
Table 3.
Gibbs free energies of substitution (GFE in kcal/mol) and conceptual DFT global descriptors (μ, , , ) for cisPtI2 and monosubstituted analogues.
Table 3.
Gibbs free energies of substitution (GFE in kcal/mol) and conceptual DFT global descriptors (μ, , , ) for cisPtI2 and monosubstituted analogues.
| Complex | GFE | μ | η | S | ω |
|---|---|---|---|---|---|
| Pt(NH3)2I2 | 0 | -0.149 | 0.148 | 3.386 | 0.075 |
| Pt(NH3)2I(CH3SH) | 2.2 | -0.161 | 0.152 | 3.279 | 0.085 |
| Pt(NH3)2I(CH3SCH3) | -2.3 | -0.159 | 0.153 | 3.259 | 0.082 |
| Pt(NH3)2I(imi) | -7.4 | -0.146 | 0.160 | 3.117 | 0.067 |
| Pt(NH3)I2(CH3SH) | 4.0 | -0.159 | 0.142 | 3.528 | 0.089 |
| Pt(NH3)I2(CH3SCH3) | -0.1 | -0.157 | 0.142 | 3.511 | 0.087 |
| Pt(NH3)I2(imi) | -2.9 | -0.146 | 0.149 | 3.352 | 0.071 |
Table 4.
Calculated Fukui Indices ( and ) for atomic centers in cisplatin and its monosubstituted analogues.
Table 4.
Calculated Fukui Indices ( and ) for atomic centers in cisplatin and its monosubstituted analogues.
| complex | Fukui index | Pt | N | N | Cl | Cl | N(imi) | S |
|---|---|---|---|---|---|---|---|---|
| Pt(NH3)2Cl2 |
-0.414 | -0.024 | -0.024 | -0.169 | -0.169 | n/a | n/a | |
| -0.517 | -0.001 | -0.001 | -0.172 | -0.172 | n/a | n/a | ||
| Pt(NH3)2Cl(CH3SH) | -0.361 | -0.024 | -0.031 | -0.172 | n/a | n/a | -0.136 | |
| -0.462 | -0.001 | -0.009 | -0.286 | n/a | n/a | -0.054 | ||
| Pt(NH3)2Cl(CH3SCH3) | -0.362 | -0.022 | -0.030 | -0.166 | n/a | n/a | -0.121 | |
| -0.459 | 0.000 | -0.009 | -0.274 | n/a | n/a | -0.053 | ||
| Pt(NH3)2Cl(imi) | -0.424 | -0.011 | -0.041 | -0.189 | n/a | 0.002 | n/a | |
| -0.485 | 0.004 | -0.024 | -0.292 | n/a | 0.016 | n/a | ||
| Pt(NH3)Cl2(CH3SH) | -0.340 | -0.024 | n/a | -0.161 | -0.158 | n/a | -0.134 | |
| -0.442 | 0.002 | n/a | -0.275 | -0.100 | n/a | -0.044 | ||
| Pt(NH3)Cl2(CH3SCH3) | -0.344 | -0.023 | n/a | -0.154 | -0.155 | n/a | -0.116 | |
| -0.443 | -0.003 | n/a | -0.258 | -0.104 | n/a | -0.043 | ||
| Pt(NH3)Cl2(imi) | -0.404 | -0.020 | n/a | -0.173 | -0.168 | 0.006 | n/a | |
| -0.470 | 0.000 | n/a | -0.112 | -0.247 | 0.026 | n/a |
Table 5.
Calculated Fukui indices ( and ) for atomic centers in iodoplatin and its monosubstituted analogues.
Table 5.
Calculated Fukui indices ( and ) for atomic centers in iodoplatin and its monosubstituted analogues.
| complex | Fukui index | Pt | N | N | I | I | N(imi) | S |
|---|---|---|---|---|---|---|---|---|
| Pt(NH3)2I2 |
-0.302 | -0.022 | -0.022 | -0.245 | -0.245 | n/a | n/a | |
| -0.359 | 0.004 | 0.004 | -0.278 | -0.278 | n/a | n/a | ||
| Pt(NH3)2I(CH3SH) | -0.308 | -0.025 | -0.028 | -0.278 | n/a | n/a | -0.116 | |
| -0.226 | -0.011 | 0.004 | -0.633 | n/a | n/a | -0.025 | ||
| Pt(NH3)2I(CH3SCH3) | -0.312 | -0.025 | -0.026 | -0.273 | n/a | n/a | -0.100 | |
| -0.226 | -0.010 | 0.004 | -0.630 | n/a | n/a | -0.023 | ||
| Pt(NH3)2I(imi) | -0.345 | -0.017 | -0.035 | -0.304 | n/a | 0.004 | n/a | |
| -0.249 | -0.009 | -0.002 | -0.635 | n/a | 0.029 | n/a | ||
| Pt(NH3)I2(CH3SH) | -0.254 | -0.021 | n/a | -0.232 | -0.232 | n/a | -0.107 | |
| -0.212 | -0.005 | n/a | -0.571 | -0.114 | n/a | -0.019 | ||
| Pt(NH3)I2(CH3SCH3) | -0.261 | -0.019 | n/a | -0.228 | -0.230 | n/a | -0.089 | |
| -0.216 | -0.004 | n/a | -0.562 | -0.118 | n/a | -0.018 | ||
| Pt(NH3)I2(imi) | -0.289 | -0.021 | n/a | -0.252 | -0.236 | 0.009 | n/a | |
| -0.097 | 0.000 | n/a | -0.354 | -0.482 | 0.014 | n/a |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



