
Submitted:
06 November 2025
Posted:
10 November 2025
You are already at the latest version
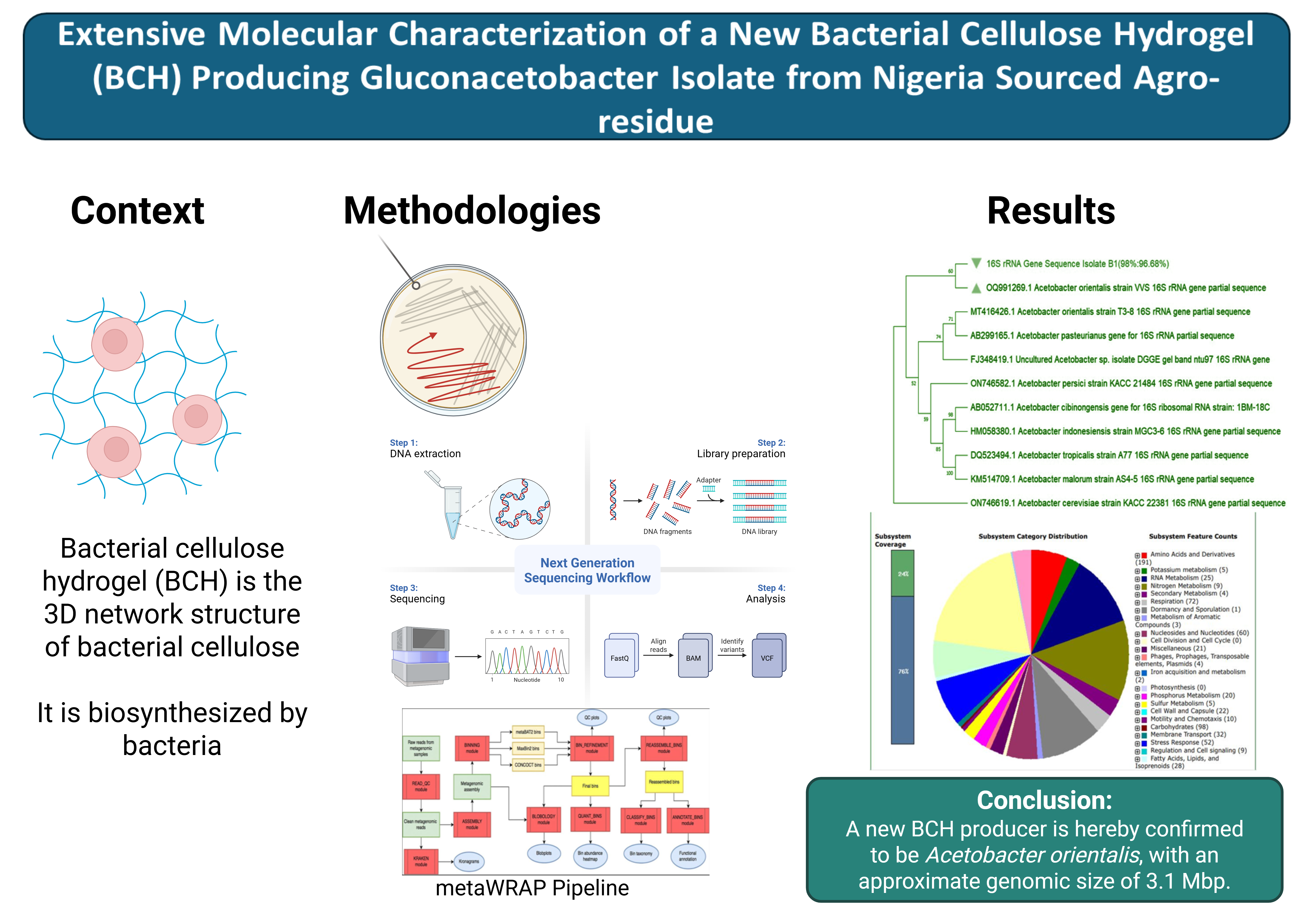
Abstract
Bacterial cellulose hydrogels (BCH) are characterized as exopolysaccharides of glucose polymer consisting of β-1-4– glycosidic linkage with various degrees of polymerizations, which are biosynthesized by bacteria. Information on the molecular characterization of a Nigeria BCH producer isolate is completely unavailable. The study therefore aimed to characterize a new Acetobacter species that had previously been confirmed to produce BCH. The BCH-producing isolate was characterized by PCR amplification of its 16S rRNA gene, followed by sequencing and construction of phylogenetic tree. The whole-genome sequence of the isolate was also determined using the NGS Illumina sequencing, with downstream analysis of metagenomic reads through the metaWRAP pipeline. The BCH producer isolate was identified to be Acetobacter orientalis strain Zaria-B1, based on sequence identity with the reference Acetobacter orientalis strain VVS. Based on its annotated genome, the isolate was found to have an approximate genomic size of 3.1 Mbp, 45 total number of RNAs, GC content of 52.5%, 3,046 total number of protein-encoding genes, N50 of 253,774 bp, L50 of 4, as well as 30 number of contigs. Nucleotide BLAST of the cellulose synthase gene sequence confirmed the bin to be Acetobacter orientalis. The whole-genome characterization alongside the 16S rRNA genotyping confirmed the BCH-producing isolate to be Acetobacter orientalis.
Keywords:
bacterial cellulose hydrogel (BCH)
; Acetobacter orientalis
; molecular characterization
; Nigeria
; metaWRAP
; 16S rRNA gene
1. Introduction
Cellulose is the most common renewable biopolymer (homopolysaccharide) produced in the biosphere (C6H10O5) and is essentially made up of glucose monomers joined together by β (1-4) glycosidic linkages. It is produced by microorganisms, animals, and plants [1,2]. Bacterial cellulose hydrogel (BCH) is the three-dimensional (3-D) network structure of naturally occurring bacterial cellulose that can absorb and retain significant volumes of water. The purification processes for plant-based cellulose such as logging, debarking, chipping, mechanical pulping, screening, chemical pulping, and bleaching consume a lot of energy and are not environmentally friendly [3]. By contrast, just a few contaminants, such as cells and/or medium components, are present in bacterial cellulose (BC) that is formed during fermentation. There is a growing interest in developing fully bio-based cellulosic polymer with excellent properties, such as tensile strength and Young's modulus. BCH has good mechanical properties, positioning it as a choice bioresource for reinforcing agents in composite materials [4]. BCH is devoid of contaminating substances including lignin, hemicellulose, pectin, wax, and other challenging-to-remove plant components [5].
BCH is suited for the creation of artificial skin and wound dressings due to the biocompatibility of its nanofibers and their high water-holding capacity [3]. The biomaterial has been commercialized as high-end products for health, food, high-strength papers, audio speakers, filtration membranes, wound dressing materials, artificial skin, artificial blood vessels, and other biomedical devices due to desirable properties like its three-dimensional nanomeric structures, unique physical, mechanical, and thermal properties, and its higher purity [6].
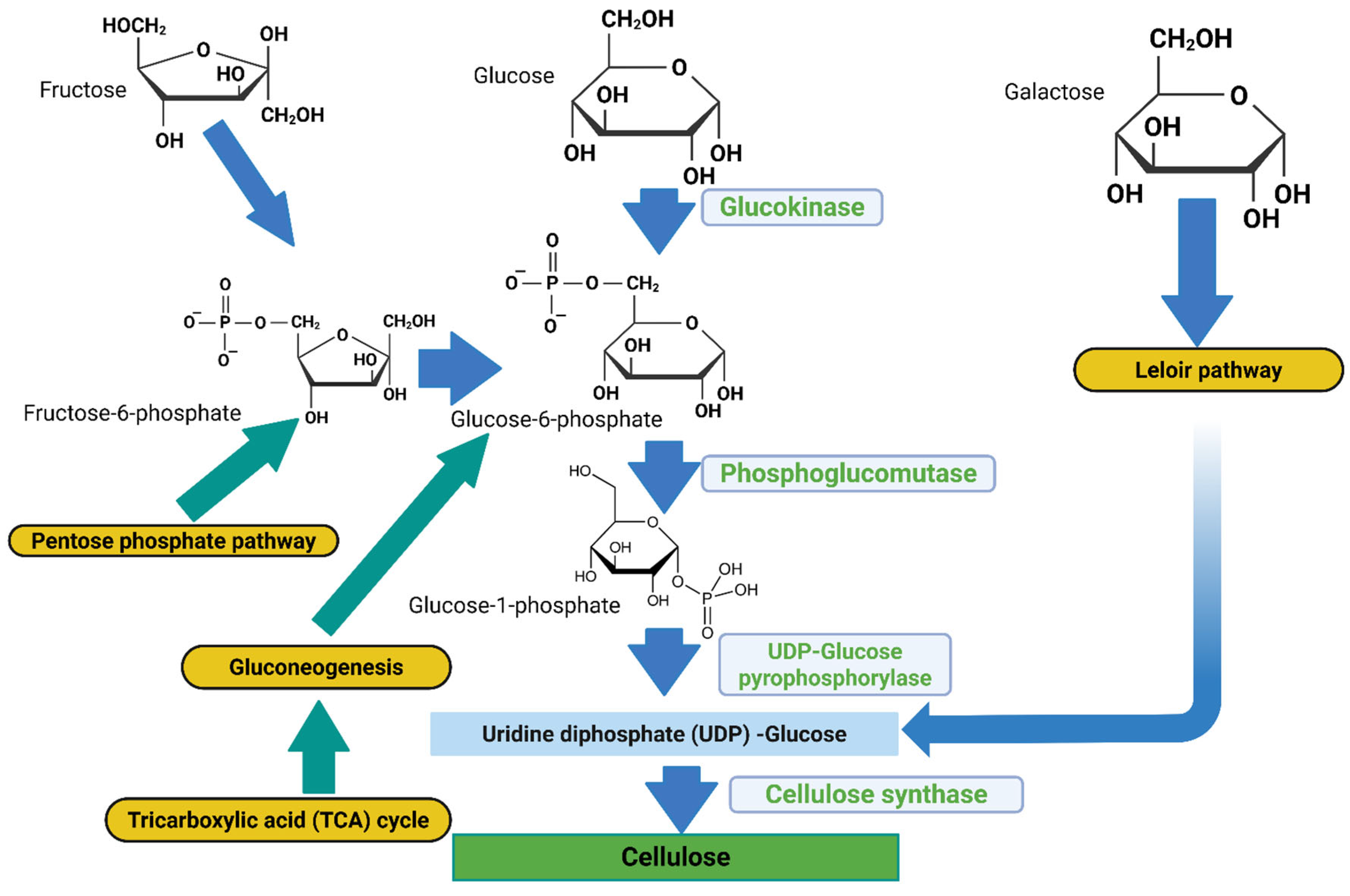
Gluconacetobacter species use a multi-step metabolic route involving different enzymes, catalytic complexes, and regulatory proteins at each stage for cellulose production [7]. The biosynthetic pathway, assuming glucose is the carbon source, consists of four major enzymatic steps: (i) glucose phosphorylation by glucokinase; (ii) isomerization of glucose-6-phosphate (Glc-6-P) to glucose-1-phosphate (Glc-1-P) by phosphoglucomutase; (iii) synthesis of UDP-glucose (UDPGlc) by UDPG pyrophosphorylase (UGPase); and (iv) cellulose synthase reaction. In all Gluconacetobacter species and other BCH-producing bacterial species, the bacterial cellulose synthesis (bcs) operon encodes the biosynthesis of cellulose [8]. This operon has four subunits (bcsA, bcsB, bcsC, and bcsD) which are necessary for full BCH biosynthesis. The catalytic subunit of cellulose synthase is encoded by the first gene of the bcsABCD operon, bcsA. The second messenger that initiates the cellulose synthesis process, cyclic diguanylate monophosphate (c-di-GMP), is bound by the regulatory subunit of cellulose synthase, which is encoded by the second gene, bcsB. [7,9].
Figure 1.
Bacterial cellulose hydrogel biosynthesis pathway (metabolite denoted by blue and grey arrows, metabolic pathways as yellow boxes, and enzymes involved in the respective reactions denoted in green). Figure generated using Biorender.com.
Figure 1.
Bacterial cellulose hydrogel biosynthesis pathway (metabolite denoted by blue and grey arrows, metabolic pathways as yellow boxes, and enzymes involved in the respective reactions denoted in green). Figure generated using Biorender.com.
Acetic acid bacteria (AAB) are a group of microorganisms that belong to the family Acetobacteraceae of the class Alphaproteobacteria and made up of 14 genera. Due to its capacity to produce comparatively high amounts of BCH in liquid culture from a variety of carbon and nitrogen sources, Gluconacetobacter xylinus (previously known as Acetobacter xylinum) is one of the most researched species [11]. Despite extensive studies on bacterial BCH producers, none has been isolated or identified here in Nigeria. Moreover, the Acetobacter orientalis species has not been reported or documented in literature to produce BCH. Therefore, the search for other species of Acetobacter (Gluconacetobacter) that is able to produce BCH has become imperative. Additionally, the complete genome sequencing analysis of local strains of Gluconacetobacter [11], as well as characterization of subunit genes in the cellulose-synthesizing operon (acs operon) will serve as genomic data that provide a viable platform that can be used to understand and modify the phenotype of the bacterial cellulose synthase (bcs) genes to further improve BCH production.
Molecular identification techniques have revolutionized bacterial identification studies, which enable the rapid and accurate identification of certain species. Numerous uses exist for DNA sequencing methods, including the identification of bacterial species and the tracking of the transmission of genes encoding antibiotic resistance. Examples of these uses include Sanger sequencing, NGS, and third-generation sequencing [12]. While there are many methods available in modern bacterial taxonomy to ascertain both genotypic and phenotypic traits, whole genome DNA-DNA hybridization and 16S rRNA gene sequencing are essential [13]. One method used to ascertain an organism's entire DNA sequence is called Whole Genome Sequencing (WGS). The method uses high-throughput sequencing to generate a lot of data in a short amount of time. DNA extraction, library preparation, sequencing, and bioinformatics analysis are the steps involved in WGS [14]. The identification and characterization of bacteria has been transformed by WGS. This technique has several significant applications, one of which is that it makes it easier to identify the genes that code for certain essential metabolic proteins, including bacterial cellulose synthase [12].
To identify the most similar biological sequences, the BLAST (Basic Local Alignment Search Tool) takes a query sequence - either a DNA or protein sequence - provided by the researcher and compares it to a library of biological sequences on the National Center for Biotechnology Information (NCBI) database [15]. BLAST is an algorithm or piece of software used for pairwise sequence (nucleotide or protein) alignment [16]. The extraction and interpretation of high-quality metagenomic bins are greatly enhanced by MetaWRAP, an intuitive modular pipeline that automates the core tasks in metagenomics analysis. Modern software use metaWRAP to manage metagenomic data processing, which begins with raw sequence reads and ends with metagenomic bins and their analysis [17,18,19].
2. Materials and Methods
2.1. Retrieval and culturing of the BCH-producing isolate on Hestrin-Schrann (HS) media
The Acetobacter orientalis Zaria-B1 isolate was retrieved from a preliminary study which involved isolation from banana peel agro-residue using the method described by [20]. This was then followed by subsequent screening for BCH production using the HS broth and agar media, as well as further characterization of the BCH-producing isolate through microscopic examination, morphological, and biochemical characterizations. The HS media had the following components: 2% (w/v) Glucose, 0.5% (w/v) Yeast extract, 0.5% (w/v) Peptone, 0.27% (w/v) Na2HPO4 (Disodium hydrogen phosphate), 0.15% (w/v) Citric acid, and 1.8% (w/v) agar. 10 mL of the screened isolate was inoculated into 90 mL of the broth media. The mixture was then incubated on a shaker incubator at 30 °C for 48 hrs at 150 rpm. The resulting culture was used for extraction of genomic DNA. For the agar plates, 100 µL preculture of the isolate was aliquoted into each of the prepared sterile agar plates and spread out using a sterile inoculating loop spreader. The plates were then incubated at 30 °C for 48 hours and were observed for colony growth. Colonies from each plate were randomly picked and used for genomic DNA extraction
2.2. Genomic DNA extraction from broth and agar cultures
Prior to DNA extraction, 200 µL of the broth culture from each Erlenmeyer flask was dispensed into 1.5 mL microcentrifuge tubes. For samples used for bacterial DNA extraction from agar plates, scoops of bacterial smear (colony) were picked randomly from the agar plates into 1.5 mL Eppendorf tubes containing 200 µL of DNA Elution Buffer using P100 pipette tips. The mixture was vortexed, and the genomic DNA was extracted using the Zymo Research Quick-DNATM Miniprep Plus Kit (USA) following the manufacturer’s instructions. The extracted genomic DNAs were quantified on a Nanodrop spectrophotometer, and their concentrations and purities (A260/A280) were determined.
2.3. Polymerase Chain Reaction (PCR) amplification of the 16S rRNA gene
The bacterial species was identified by PCR amplification of the 16S rRNA gene from the genomic DNA using generic forward 48A (5'-AGAGTTTGATCCTGGCTCAG-3') and reverse 48B (5'-TACGGCTACCTTGTTACGACTT-3') primers. The PCR mix was prepared with a total reaction volume of 25 µL, comprising 9.5 µL of nuclease-free water (NFW), 10 µM of each primer, 12.5 µL of Quick-load 2× Mastermix, and 1 µL of template DNA. A negative control reaction contained NFW in place of template DNA. The PCR cycling conditions are: initial denaturation at 94 °C for 5 minutes, and 35 cycles of denaturation at 94 °C for 30 seconds, annealing at 48 °C for 1 minute, extension at 68 °C for 1 minute, and final extension step at 68 °C for 10 minutes.
The PCR amplicons from the experiments were analyzed on 1% agarose gel stained with ethidium bromide visualization dye to ascertain positive PCR amplicons. Gel from the electrophoresis was visualized in a GelDoc system under the UV (Ultraviolet) light.
2.4. Sequencing of positive PCR amplicon
The amplified PCR amplicon with the most distinct clear band, was sequenced by the Dye-terminator method with an AB1 XL3500 Genetic Analyzer according to the service provider’s instruction (Inqaba Biotec West Africa, IBWA, Ibadan). This summarily includes preparation of the BigDye mix, preparation of the sequencing reaction, thermal cycling, precipitation reaction (DNA sequencing cleanup), and capillary electrophoresis (actual sequencing).
2.5. Bioinformatics analysis of sequencing Result
The AB1 data of the sequenced amplicons were generated. This data contained the chromatograms of the individual sequences alongside some ambiguous nucleotide codes. The ambiguous codes were edited and replaced with standard nucleotide codes using BioEdit Sequence Alignment Editor v7.2.5. Edited reverse sequence was reverse complemented using the reverse complement function. Thereafter, edited forward and reverse sequences were then aligned together using the program Pairwise Alignment function. Finally, a consensus nucleotide sequence representative of the two aligned sequences was generated with the program’s Create Consensus Sequence algorithm.
For molecular identification of the isolate, the generated consensus sequence was subjected to the NCBI nucleotide BLAST, and ten (10) sequences from the BLAST hits (output), were selected based on the highest query cover and percentage identity indexes, as well as the lowest E-values, and downloaded in FASTA format. The 10 sequences with the query consensus sequence were subjected to ClustalW multiple sequence alignment (MSA) using MEGA11 software. A phylogenetic tree was constructed to determine the most recent and closely related ancestry of the isolate. The evolutionary history was inferred by using the Maximum Likelihood method and Tamura-Nei model. A bootstrap value of 1000 was set for the phylogeny. The phylogenetic tree was further edited using the Fig Tree software v1.4.4. The consensus sequence of the 16S rRNA gene of the characterized isolate was deposited in the NCBI GenBank Database and was assigned accession number.
2.6. Whole-Genome sequencing of the isolate
The whole-genome sequencing of the BCH producing isolate was carried out by Inqaba Biotec. Library preparation for the whole-genome sequencing was achieved as follows: Genomic DNA was fragmented using an enzymatic approach (NEB Ultra II FS kit). Resulting DNA fragments were size selected (200 – 700bp), using AMPure XP beads, the fragments were end repaired and Illumina-specific adapter sequences were ligated to each fragment. Each sample was individually indexed, and a second size selection step was performed. Samples were then quantified using a fluorometric method. The samples were diluted to a standard concentration (4 nM) and then sequenced on Illumina NextSeq500 platform, using a NextSeq mid out kit (300 cycle), following a standard protocol as described by the manufacturer. 2x150 bp paired end read data was produced for each sample. The library preparation method used can be found at: https://international.neb.com/protocols/2017/10/25/protocol-for-fs-dna-library-prep-kit-e7805-e6177-with-inputs-less-than-or-equal-to-100-ng
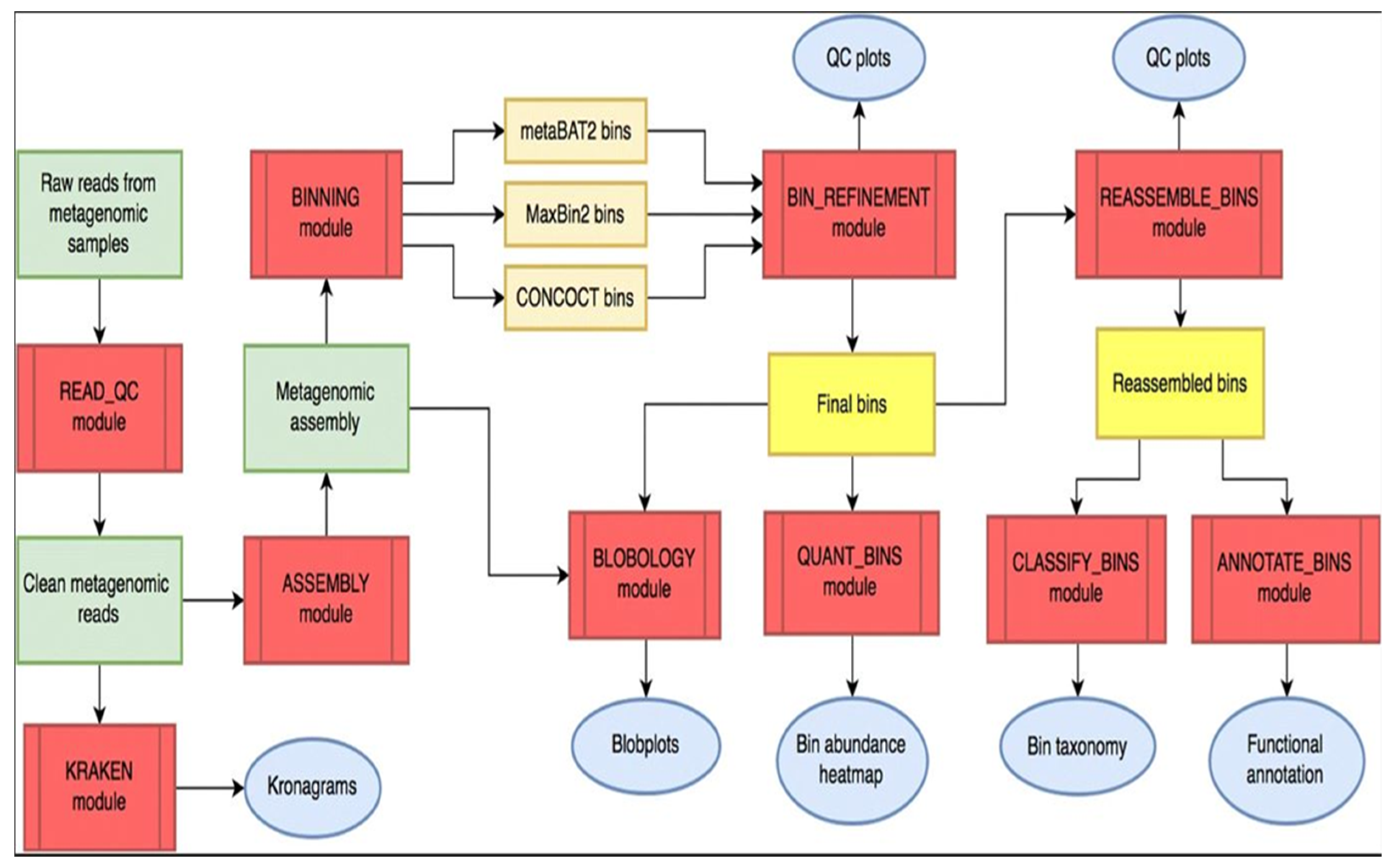
2.7. Data analysis of the Whole-Genome Sequence (WGS) Reads using the metaWRAP pipeline modules
MetaWRAP is an easy-to-use modular pipeline that automates the core tasks in metagenomics analysis and is an open-source program available at https://github.com/bxlab/metaWRAP [18]. After downloading the metagenomics reads of the NGS, the metaWRAP-Read_qc module was run to trim the reads and remove human contamination, using the “bmtagger hg38”. The metagenomes (reads) were then assembled with the metaWRAP-Assembly module, using the “metaSPADES or MegaHIT” program (software). This was executed following concatenation of the sample reads. To know the taxonomic composition of communities (reads), the “Kraken” module was run on both the reads as well as the assembly. After assembly, the co-assembly was binned with three different algorithms (CONCOCT, MaxBin and MetaBAT) with the metaWRAP-Binning module [17].
Next, the concoct, maxbin, and the metabat bin sets were consolidated into a single, stronger bin set, with the metaWRAP Bin-Refinement module. The CheckM database was employed for this module. By default, the minimum completion of 70%, and maximum contamination of 5%, were deployed by the program. The community (consolidated bin set) and the extracted bins were visualized with the Blobology module. This module was used to project the entire assembly onto a GC vs Abundance plane and annotate them with taxonomy and bin information. Thereafter, the abundances of the draft genomes (bins) across the sample were determined with the Quant module. Next, the consolidated bin set was re-assembled with the Reassemble-bins module.
The Reassemble-bins module collects reads belonging to each bin and then reassemble them separately with a "permissive" and a "strict" algorithm. Only the bins that improved through reassembly were altered in the final set. Thereafter, the taxonomy of each bin was inferred with the Classify-bins module using the Taxator-tk program [21]. For this module, the NCBI_nt and NCBI_tax databases were required. The Classify_bins module used Taxator-tk to accurately assign taxonomy to each contig and then consolidated the results to estimate the taxonomy of the whole bin.
No doubt the success and accuracy of the predictions would rely heavily on the query cover and percentage sequence identity indexes of the existing database. Finally, the generated bin (bins) was functionally annotated with the Annotate-bins module using the RAST (Rapid Annotation using Subsystem Technology) online tool [17,18,21]. The annotated bin was then visualized on the SEED viewer online tool
Figure 2.
Overall Workflow of metaWRAP Pipeline. Modules (red); metagenomics data (green); intermediate (orange) and final bin sets (yellow), and data reports and figures (blue) (Adapted from https://github.com/bxlab/metaWRAP).
Figure 2.
Overall Workflow of metaWRAP Pipeline. Modules (red); metagenomics data (green); intermediate (orange) and final bin sets (yellow), and data reports and figures (blue) (Adapted from https://github.com/bxlab/metaWRAP).
3. Results
3.1. Molecular characterization of the BCH-producing isolate using the 16S rRNA gene
3.1.1. Genomic DNA extraction and quantification
The concentrations of the extracted DNAs are within the standard expected range of 5-100 ng/µL for genomic DNA, while the purity level also fell within the expected perfect A260/A280 ratio of 1.80 as shown in Table 1 below. Notable among the extracted DNA samples is sample PLT2, in which PCR amplicon was eventually sequenced and used for downstream analysis. Moreover, the same genomic material was used for the whole-genome sequencing.
Parameters in Table 1 were generated from the Nanodrop spectrophotometer readings
3.1.2. Gel electrophoresis of the 16S rRNA PCR amplicons
Four (4) of the extracted genomic DNA were randomly selected based on the Nanodrop quality check readings and subjected to PCR amplification of the 16S rRNA gene, using a combination of both the agar and broth cultures. Out of these 4, 3 were successfully amplified as shown in Figure 3 below. From the gel, well PLT2 had the clearest distinct band, and therefore, was sequenced. Nonetheless, wells PLT1 and BRT1 also had some distinct bands, and were all the same amplicon size (1,400 bp).
Keys: M = 100 basepair Molecular marker; bp =basepair, kb = kilobase; PLT1 & PLT2 = Amplicons of extracted genomic DNA from agar plates; BRT1 & BRT2 = Amplicons of extracted template DNA from broth cultures; NTC = Non-template Control
3.1.3. Nucleotide BLAST output with parameters of the sequenced 16S rRNA gene
The sequences of the PCR amplicons were analyzed and aligned using the BioEdit Sequence Alignment Editor v7.2.5. A consensus sequence of the 16S rRNA gene was generated and subjected to NCBI nucleotide BLAST. From the BLAST result, different species of the genus Acetobacter were returned, including orientalis, tropicalis, persici, pasteurianus, cibinongensis, indonesiensis, malorum, and cerevisiae. Nevertheless, the species “orientalis”, constituted majority of the hits. Based on the BLAST output, the first hit, Acetobacter orientalis strain VVS isolated in India, had the highest percentage identity of 96.68%, E-value of 0.0, and query cover of 98%, being shared parameters together with the second reference Uncultured Acetobacter sp. isolate DGGE.
3.1.4. Evolutionary relatedness (Phylogeny) of the BCH-producing isolate Zaria-B1
The evolutionary relatedness and ancestry of the isolate were predicted from the BLAST output of its 16S rRNA gene sequence as well as its relative positioning on the phylogenetic tree. This finding made possible the accurate prediction and confirmation of the genus as well as species of the study organism. Furthermore, the constructed phylogenetic tree for the isolate and other reference Acetobacter species placed the query sequence in the same clade with the reference Acetobacter orientalis strain VVS, as indicated with the yellow triangle in Figure 4. This is an obvious indication that the isolate had the most recent and closely related ancestry with the reference species. Based on the highest percentage identity shared with this reference species, and the evolutionary relatedness predicted from the positioning in the phylogeny, the BCH producer isolate was identified as Acetobacter orientalis.
3.1. Whole-genome characterization of the Acetobacter orientalis Zaria-B1
3.2.1. Completion and contamination indexes of reassembled bin
For a comprehensive molecular characterization of the isolate, and to investigate the gene for cellulose synthase catalyzing the biosynthesis of BCH in the isolate, whole-genome sequencing analysis of the isolate was conducted. Reads (short fragments of DNA) generated from the Next Generation Sequencing (NGS) of the whole genome were subjected to the metaWRAP pipeline modules for analysis. For accurate assessment of the Re-assembly module, three important indices (N50, Completion and Contamination) were used. N50 statistics defines assembly quality in terms of contiguity. The N50 is defined as the sequence length of the shortest contig at 50% of the total assembly length. Based on the chart in Figure 5a, the reassembled Bin had an N50 of 253,774 bp. The expected minimum Bin completion (-C) was set at 70%, and there was a Bin completion of approximately 100% as indicated by the red curve in chart b. While the expected maximum contamination rate (-X) ranged from 5 - 10%, it returned a contamination of about 0.7%, as each bar on chart c stands for a contamination rate of 1%. These three indices are a clear indication that the reassembly of the bin was successful.
3.2.2. Hierarchical taxonomic classification of Bin from the metaWRAP classify-Bin module
Another important module of the metaWRAP pipeline is the “CLASSIFY_Bin Module”, which is used to assign taxonomic hierarchies to the Bin under analysis. First, the contigs (a set of DNA segments or sequences that overlap in a way that provides a contiguous representation of a genomic region) in all bins are combined into one file, and blastn v2.7.1 was used to align the contigs to the NCBI_nt database. The alignment results were then used by taxator-kt v1.3.3e to estimate the taxonomy of each contig. The most likely taxonomy of each bin is then estimated from individual contig predictions. Once no further taxonomic ranking can be estimated, the final taxonomy of the bin is reported (Adapted from https://github.com/bxlab/metaWRAP) [18,22].
The reported final taxonomy is presented on Clustered heatmaps in Figure 6, Figure 7 and Figure 8. Figure 6 shows that majority of the scattered dots are colored blue, which was assigned the super-kingdom (Domain) taxonomy of bacteria, and the other group in this ranking is eukaryote (red); while Figure 7 also reveals that most of the dots are colored blue, which corresponds to a phylum taxonomy of proteobacteria, the other taxonomic groups in this ranking are arthropoda (red) and firmicutes (yellow). Then Figure 8 gave similar results, in which more than 95% of the dots (corresponding to the different contigs) are colored blue, in which case, the blue color stands for the order taxonomy of rhodospirillales, and the other taxonomic group in this rank is the sphingomonadales (red). Additionally, in each of the taxonomic assignments, the blue dots also had the highest contig abundance as shown on the vertical axes.
3.2.3. Genome annotation using the metaWRAP annotate_bin module
Annotation of the assembled genome was achieved by the metaWRAP “Annotate_BINS module”, using the RAST and the SEED Viewer online tools (Figure 9 and Figure 10). Based on the annotation, the complete genome size of the isolate is 3,084,067 bp (3.1 Mbp); GC content of 52.5%; N50 = 253,774 bp; L50 = 4; number of contigs (with PEGs) =30; number of coding sequences = 3,046; and number of RNAs = 45. Figure 10 shows the metabolic orientation of the Acetobacter orientalis Zaria-B1 isolate from the annotated genome. The figure shows genes connected to the subsystem and their category distribution. The categories are expandable down to the specific genes (Secondary Metabolism). These are some of the predicted metabolisms of the isolate, alongside their respective number of protein-encoding genes (PEGs) shown in brackets, that translate into gene products that participate in each metabolic pathway. The Acetobacter orientalis Zaria-B1 was predicted to exhibit both primary and secondary metabolisms, based on its annotated genome.
3.2.4. NCBI nucleotide BLAST of the cellulose synthase gene
The hierarchical taxonomies of the bin from the isolate Zaria-B1 all corresponded to the ones assigned by the NCBI GenBank database after deposition of its 16S rRNA gene sequence. However, the system could not go beyond this level of classification, and based on the developer’s instruction, it was recommended to use one of the most conserved (marker) gene sequence of the target organism from the annotated genome for nucleotide BLAST to identify the species. Therefore, the cellulose synthase catalytic subunit gene was adopted for the BLAST. Figure 11 shows the BLAST result of the cellulose synthase gene, which returned only one hit, and that comes from Acetobacter orientalis, strain FAN1 with percentage identity of 98.44% and query cover of 100%. This is an apparent confirmation that the assembled genome comes from Acetobacter orientalis, in addition to the previous 16S rRNA characterization and identification. Based on the foregoing, the isolate Zaria-B1 was confirmed to be Acetobacter orientalis. The gene sequence for the cellulose synthase is the coding sequence, having a start codon ATG and a stop codon TAG at the flanking ends of the sequence (Accession: PP393681). The bcs gene sequence had a length of 4,608 bp, and an amino acid sequence length of 1,536 aa. Following the whole-genome characterization, the cellulose synthase catalytic subunit gene and the beta-1,4-endoglucanase gene sequences of the isolate were deposited in the NCBI GenBank database and were assigned accession numbers PP393681 and PP393682, respectively. The products of the two genes are essential for the functional biosynthesis of the bacterial cellulose hydrogel (BCH).
NCBI nucleotide BLAST output of the cellulose synthase gene retrieved from the annotated genome. The BLAST returned only one hit, the Acetobacter orientalis cellulose synthase catalytic gene, with a query cover of 100%, E-value of 0.0 and percentage identity of 98.44%.
4. Discussion
The ultrapure and nanofibrillar structure of BCH differentiates it from plant cellulose. BCH is well known for its strength, flexibility and high water holding capacity reaching up to ~90% of its weight. Therefore, it comes as no surprise that BCH attracts significant attention, and numerous approaches have been pursued for research and development of the biomaterial. Synthesis of BCH is a multistep process, involving many enzymes and proteins [23]. In BCH synthesis, glucose monomers linked by β-1,4-glucan chains are simultaneously polymerized by cellulose synthase, a membrane bound enzyme, which utilizes UDP-glucose as substrate. According to [24], genes for bacterial cellulose hydrogel (BCH) biosynthesis are localized in an operon (the bcs operon) in the Gluconacetobacter species. Bacterial cellulose is chemically the same in its primary composition as cellulose produced by other organisms (higher plants, algae), but the degree of polymerization and crystallinity as well as other physical properties are unique to each organism [12].
In modern bacterial taxonomy, a range of techniques to determine both phenotypic and genotypic characteristics are available, but whole genome DNA-DNA hybridization and 16S rRNA gene sequencing play a central role. The overall 16S rRNA gene sequence similarity between the type strains and the type species of the 10 genera of acetic acid bacteria (AAB) ranges from 92.1 to 99.0 %, the latter found between Asaia and Swaminathania [12].
Molecular characterization of microorganisms has become necessary to complement other preliminary characterizations (such as microscopic, morphological, and biochemical) which are not sensitive enough to determine the species and strains of target organisms. In this study, molecular techniques were used to characterize a new BCH producing isolate, identified as Acetobacter orientalis, via PCR amplification of bacterial 16S rRNA gene and sequencing. The findings from this study are similar to several other studies that have reported the characterization of different species of the Acetobacteraceae family, using similar techniques. [24] isolated and characterized a bacterial nanocellulose (BNC) producer from rotten fruits in Malaysia as Gluconacetobacter xylinus; the species Acetobacter orientalis was isolated from Indonesian flowers, fruits and fermented foods and characterized as orientalis species [25]. Similarly, [26] and [27] also isolated Acetobacter tropicalis from fermented juice of mango, capable of producing vinegar in Burkina Faso, and a cellulolytic and ligninolytic bacterium Acetobacter orientalis XJC-C from a marine soft coral in China, respectively, and characterized same. All these studies employed PCR amplification of the bacterial 16S rRNA gene, sequencing and construction of phylogenetic trees to ascertain the genus and species of the organisms. In these studies, the amplified 16S rRNA gene amplicon had nucleotide sizes ranging from 1.3 -1.5 kbp.
The N50 is a statistical index that defines assembly quality in terms of contiguity. It can be thought of as the point of half of the mass of the distribution; the number of bases from all contigs longer than the N50 will be close to the number of bases from all contigs shorter than the N50. While N50 corresponds to the sequence length in base pairs, L50 represents the number of sequences. Since contigs are ordered according to their length when calculating N50, we can say that L50 is simply the rank of the contig that gives us the N50 length [21]. Two additional parameters that are used to assess the qualities of a metagenomics bin are completion and contamination. From Figure 5, the estimated completion of the bin is approximately 100%, while the contamination was found to be 0.7%. Completion refers to the level of coverage of the population genome during assembly and reassembly, while contamination is the amount of sequence that does not belong to this population from another genome [21]. In the metaWRAP reassembly module, the expected minimum completion is 50-70%, while the maximum contamination is 5-10%. These two metrics are usually estimated by counting universal single-copy genes within each bin. The percentage of expected single-copy genes that are found within a bin is interpreted as its completion, while the contamination is calculated from the percentage of single-copy genes that are found in duplicate [21]. Based on the 4 parameters discussed, the metaWRAP analysis of the metagenome was accurately and successfully carried out.
Findings from the annotated genome of the isolate Zaria-B1 correspond to the works of [23], who characterized the whole-genome of Acetobacter orientalis strain FAN1 and obtained a total genomic size of 3,041,114 bp; 3,563 protein-encoding genes (PEGs); GC content of 52.34%; total of 70 RNAs (15 rRNAs and 55 tRNAs). However, their strain was reported to be involved in the fermentation of yoghurt and production of lactobionic acid, rather than BCH. This confirms that this species has not been reported to be involved in the production of BCH. The genomic size obtained in this study is slightly different from the results obtained by [11], who reported from the annotated genome of Gluconacetobacter xylinus CGMCC 2955 a total genomic size of 3,563,314 bp, 3,193 protein-encoding genes (PEGs), GC content of 63.29%, 117 non-coding RNAs (ncRNAs), 45 ribosomal RNAs (rRNAs). However, the Gluconacetobacter xylinus was identified as a BCH producer, which is one of the species of Gluconacetobacter, that has been reported to produce BCH. Similarly, the work of [14] which characterized the whole-genome of Komagataeibacter sp. strain CGMCC 17276 and found a somewhat different genomic size of 3,983,026 bp; 4,107 protein-encoding genes (PEGs); a GC content of 62.21%, total number of RNAs being 72 (57 tRNAs and 15 rRNAs). Furthermore, [8] isolated the acetic acid bacterium Acetobacter pasteurianus 386B from cocoa bean heap in Ghana, which was reported to be involved in the fermentation of cocoa bean and carried out 454 Pyrosequencing of the genome of the isolate, and the sequenced genome was annotated with the GenDB software v2.2. Based on the annotation, the acetic acid bacterium was found to have a genomic size of 2,818,679 bp; a total of 7 plasmids; 118 number of contigs; a GC content of 52.19%; 2,595 number of protein-encoding sequences (PEGs), 5 rRNAs, and 57 tRNAs.
Figure 11 shows the NCBI nucleotide BLAST output for the cellulose synthase catalytic gene retrieved from the annotated genome of the isolate. This BLAST was necessary as the metaWRAP pipeline program could only classify the reassembled bin to the Order level of taxonomy. As a result, and based on the developer’s recommendation, manually looking at marker genes of the target organism of interest (such as the cellulose synthase gene, being the most conserved gene for all BCH producers), can result in much more specific taxonomy assignment. The BLAST result returned only one hit, which corresponded to the Acetobacter orientalis strain FAN1 cellulose synthase gene, with a query cover of 100% and percentage identity of 98.44%. Based on this result, and the taxonomy assigned to the isolate by the NCBI GenBank database, the BCH producer isolate was confirmed to be Acetobacter orientalis.
5. Conclusions
This study successfully identified and characterized a new BCH-producing Gluconacetobacter orientalis strain, Zaria-B1 based on the bacterial 16S rRNA genotyping, sequencing and phylogeny. The whole genome sequence of the isolate was successfully analyzed and annotated with both functional and hypothetical proteins. This is the first report of Acetobacter orientalis to produce BCH. Additionally, this is the first research to successfully identify and characterize a BCH producer here in Nigeria and, the whole genome sequence of the isolate Zaria-B1 is one of the few available data globally, and obviously, the only one here in the country. Gene sequences of the isolate have been deposited in the NCBI GenBank database with accession numbers: OR835989, PP393681, and PP393682. Interestingly, research output from this study had been successfully patented through a Germany based IP filling company- Ideas2ipr. It is hereby recommended that predicted hypothetical proteins from the annotated genome of the isolate should be explored to predict their structure and functions, and its cellulose synthase genes expressed in another fast-growing bacterial host such as Escherichia coli for enhanced recombinant production of the biomaterial.
6. Patents
Research output from this study had been successfully patented through a Germany based IP filling company- Ideas2ipr, with patent model no 20 2024 102 959. The patent certificate can be found in supplementary material Figure S1.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Certificate of Patent.
Author Contributions
Conceptualization, E.O.B. and Y.K.E.I.; methodology, S.C.O.; software, S.C.O.; validation, S.C.O., J.L., B.Y.L., and I.Z.W.; formal analysis, S.C.O. and R.B.M.; investigation, E.O.B.; resources, E.O.B. and Y.K.E.I.; data curation, S.C.O.; writing—original draft preparation, S.C.O.; writing—review and editing, E.O.B. and Y.K.E.I.; visualization, S.C.O.; supervision, E.O.B., Y.K.E.I., A.A.S., M.N.S., A.B.S., M.T.T. and M.H.S.; project administration, S.C.O.; funding acquisition, E.O.B. and Y.K.E.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Research Funds (NRF) under the Tertiary Education Trust Funds (TETFUND), Nigeria. The grant was released under grant number TETF/DR&D-CE/NRF/2020/SETI/572/VOL.1.
Data Availability Statement
Data are contained within the article. Deposited sequence data in the GenBank repository can be accessed on the National Center for Biotechnology Information (NCBI) database with accession numbers; OR835989, PP393681, and PP393682.
Acknowledgments
The authors acknowledge the financial support of the NRF TETFUND Grant, which has made possible a successful execution of the research. Utmost acknowledgement also goes to the Africa Center of Excellence for Neglected Tropical Diseases and Forensic Biotechnology (ACENTDFB), for providing the required facility for the wet lab, as well as funding. We also acknowledge and thank Dr. Youssouf Mouliom Mfopit of Institute of Agricultural Research for Development (IRAD) Yaounde, Cameroon, for establishing the connection for analysis of the metagenomics reads generated from the NGS Illumina sequencing.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| BCH | Bacterial cellulose hydrogel |
| PCR | Polymerase chain reaction |
| rRNA | Ribosomal RNA |
| WGS | Whole-genome sequencing |
References
- Castro, C., Zuluaga, R., Álvarez, C., Putaux, J. L., Caro, G., Rojas, O. J., Mondragon, I., & Gañán, P. (2012). Bacterial Cellulose Produced by a New Acid-resistant Strain of Gluconacetobacter Genus: Carbohydrate Polymers. 89(4), 1033-1037. [CrossRef] [PubMed]
- Qiu, X., & Hu, S. (2013). “Smart” Materials Based on Cellulose: A Review of the Preparations, Properties, and Applications: Materials, 6(3), 738-781. [CrossRef] [PubMed]
- Gizem, B., & Athanasios, M. (2021). Systematic Understanding of Recent Developments in Bacterial Cellulose Biosynthesis at Genetic, Bioprocess and Product Levels. International Journal of Molecular Sciences. 22(7192). 1-18. Retrieved from . [CrossRef]
- Singh, O., Panesar, P.S., & Chopra, H.K. (2017). Isolation and Characterization of Cellulose Producing Bacterial Isolate from Rotten Grapes. Biosciences Biotechnology Research Asia, 14(1). 373-380. Retrieved from . [CrossRef]
- Jelenko, K., Cepec, E., Nascimento, F. X., & Trček, J. (2023). Comparative genomics and phenotypic characterization of Gluconacetobacter entanii, a highly acetic acid-tolerant bacterium from vinegars. Foods, 12(1), 214. [CrossRef]
- McManus, J. B., Deng, Y., Nagachar, N., Kao, T., & Tien, M. (2016). Acsa–acsb: The core of the cellulose synthase complex from Gluconacetobacter hansenii ATCC23769. Enzyme and Microbial Technology, 82, 58-65. [CrossRef]
- Liu, D., Cao, Y., Qu, R., Gao, G., Chen, S., Zhang, Y., Wu, M., Ma, T., & Li, G. (2019). Production of bacterial cellulose hydrogels with tailored crystallinity from Enterobacter Sp. FY-07 by the controlled expression of colanic acid synthetic genes. Carbohydrate Polymers, 207, 563-570. [CrossRef]
- Illeghems, K., De Vuyst, L., & Weckx, S. (2013). Complete genome sequence and comparative analysis of Acetobacter pasteurianus 386B, a strain well-adapted to the cocoa bean fermentation ecosystem. BMC Genomics, 14(1). [CrossRef]
- Singhania, R. R., Patel, A. K., Tsai, M., Chen, C., & Di Dong, C. (2021). Genetic modification for enhancing bacterial cellulose production and its applications. Bioengineered, 12(1), 6793-6807. [CrossRef]
- Abhay, P., & Rakesh, K. (2021). A Review on Production, Characterization and Application of Bacterial Cellulose and its Biocomposites: Journals of Polymers and the Environment. (January). [CrossRef]
- Liu, M., Lingpu, L., Shiru, J., Siqi, L., Yang, Z., & Cheng, Z. (2018). Complete Genome Analysis of Gluconacetobacter xylinus CGMCC 2955 for Elucidating Bacterial Cellulose Biosynthesis and Metabolic Regulation: Scientific Reports. (2018) 8:6266. [CrossRef]
- Abdulateef, A.S., Owaif, H.A., and Hadi, N.A. (2023). Molecular Identification Techniques of Bacteria: A Review. International Journal of Research Publications and Reviews; 4(5), pp. 388-393.
- Cleenwerck, I. (2008). Improved classification and identification of acetic acid bacteria based on molecular techniques [Unpublished doctoral dissertation]. UNIVERSITEIT, GENT.
- Lu, T., Gao, H., Liao, B., Wu, J., Zhang, W., Huang, J., Liu, M., Huang, J., Chang, Z., Jin, M., Yi, Z., & Jiang, D. (2020). Characterization and optimization of production of bacterial cellulose from strain CGMCC 17276 based on whole-genome analysis. Carbohydrate Polymers, 232, 115788. [CrossRef]
- Samal, K. C., Sahoo, J. P., Behera, L., & Dash, T. (2021). Understanding the BLAST (Basic local alignment search tool) program and a step-by-step guide for its use in life science research. Bhartiya Krishi Anusandhan Patrika, (Of). [CrossRef]
- Staton, J.L. (2015). Understanding Phylogenies: Constructing and Interpreting Phylogenetic Trees. Journal of the South Carolina Academy of Science; 13(1), 24-29.
- Aziz, R.K., Bartels, D., Best, A.A., DeJongh, M., Disz, T., Edwards, R.A., Formsman, K., Gerdes, S… and Zagnitko, O. (2008). The RAST Server: Rapid Annotations using Subsystem Technology. BMC Genomics, 9(75); 1-15. [CrossRef]
- Overbeek, R., Olson, R., Pusch, G.D., Olsen, G.J., Davis, J.J., Disz, T., Edwards, R.A., Gerdes, S… and Stevens, R. (2013). The SEED and the Rapid Annotations of Microbial Genomes using Subsystem Technology (RAST). Nucleic Acids Research, 42(1); D206-D214. [CrossRef]
- Swingler, S., Gupta, A., Gibson, H., Kowalczuk, M., Heaselgrave, W., & Radecka, I. (2021). Recent Advances and Applications of Bacterial Cellulose in Biomedicine. Polymers (Basel), 13(3). [CrossRef]
- Revin, V., Liyaskina, E., Nazarkina, M., Bogatyreva, A. and Shchankin, M. (2018). Cost-effective production of bacterial cellulose using acidic food industry by-products. Brazilian Journal of Microbiology, 49S (2018): 151–159. 1517-8382. [CrossRef]
- Uritskiy, G.V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP- A Flexible Pipeline for Genome-Resolved Metagenomic Data Analysis. Microbiome, 6(158); 1-13. Retrieved from . [CrossRef]
- Gizem, B., Alexander, B., & Athanasios, M. (2017). Recombinant Biosynthesis of Bacterial Cellulose in Genetically Modified Escherichia coli. Bioprocess and Biosystems Engineering. 41:265-279. [CrossRef]
- Nakashima, N., & Tamura, T. (2018). Whole-genome sequence of Acetobacter orientalis strain FAN1, isolated from Caucasian yogurt. Genome Announcements, 6(13). [CrossRef]
- Abba, M., Abdullahi, M., Md Nor, M. H., Chong, C. S., & Ibrahim, Z. (2017). Isolation and characterisation of locally isolated Gluconacetobacter xylinus BCZM Sp. with nanocellulose producing potentials. IET Nanobiotechnology, 12(1), 52-56. [CrossRef]
- Lisdiyanti, P., Kawasaki, H., Seki, T., Yamada, Y., Uchimura, T., & Komagata, K. (2001). Identification of Acetobacter strains isolated from Indonesian sources, and proposals of Acetobacter syzygii Sp. Nov., Acetobacter cibinongensis Sp. Nov., and Acetobacter orientalis Sp. Nov. The Journal of General and Applied Microbiology, 47(3), 119-131. [CrossRef]
- Assiètta, O., K., M. S., A., T. C., Bassirou, N., Alfred, T., & S., A. O. (2019). Molecular identification of acetic acid bacteria isolated from fermented mango juices of Burkina Faso: 16S rRNA gene sequencing. African Journal of Biotechnology, 18(29), 766-773. [CrossRef]
- Chen, Y., Wang, W., Zhou, D., Cai, B., Zhang, M., Qi, D., Jing, T., Zang, X., Zhang, L., & Xie, J. (2021). Acetobacter orientalis XJC-C with a high lignocellulosic biomass-degrading ability improves significantly composting efficiency of banana residues by increasing metabolic activity and functional diversity of bacterial community. Bioresource Technology, 324, 124661. [CrossRef]
- Al-Deresawi, T.S., Mohammed, M.K., & Khudhair, S.H. (2022). Isolation, Screening, and Identification of Local Bacterial Isolates Producing Bio-Cellulose. Journal of Medicinal and Chemical Sciences, 6(2023). 622-633. Retrieved from http://www.jmchemsci.com/.
- Al-Janabi, S. S., Shawky, H., El-Waseif, A. A., Farrag, A. A., Abdelghany, T. M., & El-Ghwas, D. E. (2022). Stable, efficient, and cost-effective system for the biosynthesis of recombinant bacterial cellulose in Escherichia coli DH5α platform. Journal of Genetic Engineering and Biotechnology, 20(1). [CrossRef]
- Gupta, N. (2019). DNA extraction and polymerase chain reaction. Journal of Cytology, 36(2), 116. [CrossRef]
- Guzman, J. D., & Vilcinskas, A. (2021). Genome analysis suggests the bacterial family Acetobacteraceae is a source of undiscovered specialized metabolites. [CrossRef]
- Kiryu, T., Kiso, T., Nakano, H., Ooe, K., Kimura, T., & Murakami, H. (2009). undefined. Journal of Dairy Science, 92(1), 25-34. [CrossRef]
- Matsutani, M., Hirakawa, H., Yakushi, T., & Matsushita, K. (2010). Genome-wide phylogenetic analysis of Gluconobacter, Acetobacter, and Gluconacetobacter. FEMS Microbiology Letters, 315(2), 122-128. [CrossRef]
- Qiu, X., & Hu, S. (2013). “Smart” Materials Based on Cellulose: A Review of the Preparations, Properties, and Applications: Materials, 6(3), 738-781. [CrossRef] [PubMed]
- Saxena, I.M., Krystyna, K., Kazuo, O., & Malcom, R.B. JR. (1994). Characterization of Genes in the Cellulose-synthesizing Operon (acs Operon) of Acetobacter xylinum: Implications for Cellulose Crystallization: Journal of Bacteriology. Vol. 176, No. 18. p 5735-5752.
Figure 3.
Gel electrophoregram of 16S rRNA gene PCR Amplicons.
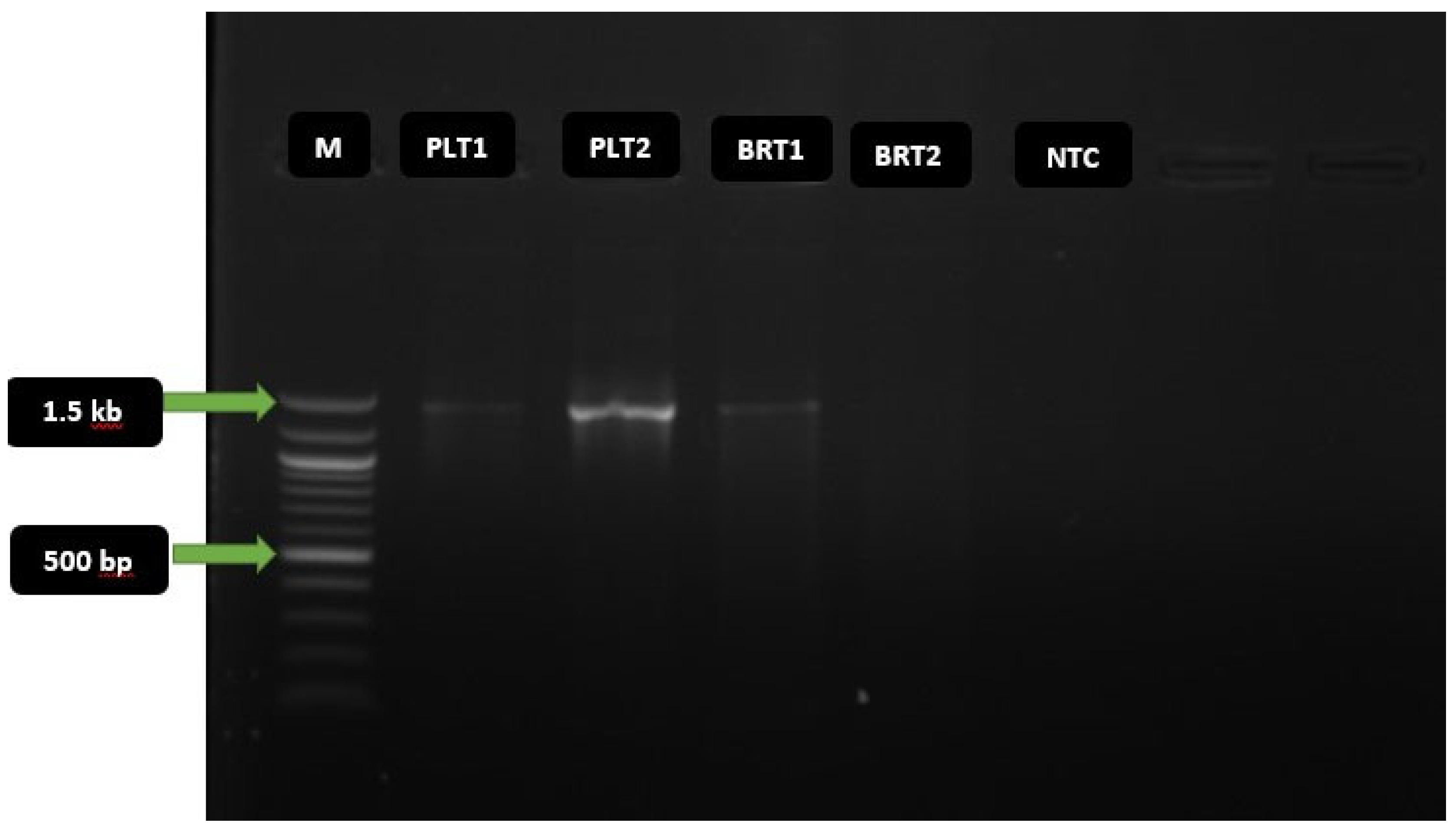
Figure 4.
Phylogenetic tree of the 16S rRNA gene sequence of the best BCH-producing isolate Zaria-B1.
Figure 4.
Phylogenetic tree of the 16S rRNA gene sequence of the best BCH-producing isolate Zaria-B1.

Figure 5.
(A) Reassembled N50 plot; (B) Completion plot, and (C) Contamination plot, of reassembled bin (The readings on the vertical axis of chart c in Figure 5 were not displayed due to software errors. Hence, the values of each bar were predicted from the standard contamination chart of the metaWRAP pipeline).
Figure 5.
(A) Reassembled N50 plot; (B) Completion plot, and (C) Contamination plot, of reassembled bin (The readings on the vertical axis of chart c in Figure 5 were not displayed due to software errors. Hence, the values of each bar were predicted from the standard contamination chart of the metaWRAP pipeline).
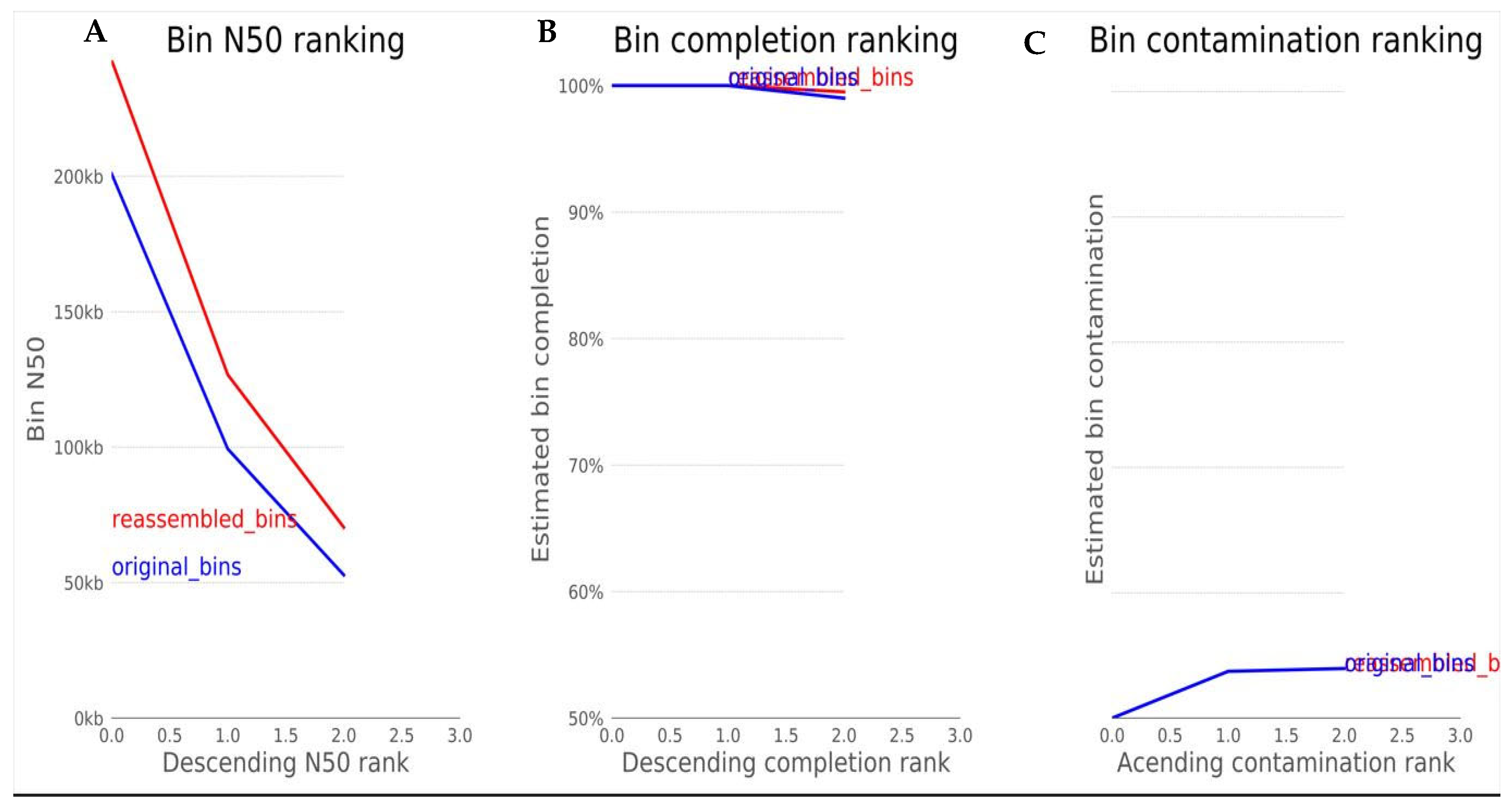
Figure 6.
Blobology clustered heatmap for Super-Kingdom (Domain) hierarchical taxonomy.

Figure 7.
Blobology clustered heatmap for phylum hierarchical taxonomy of bin.
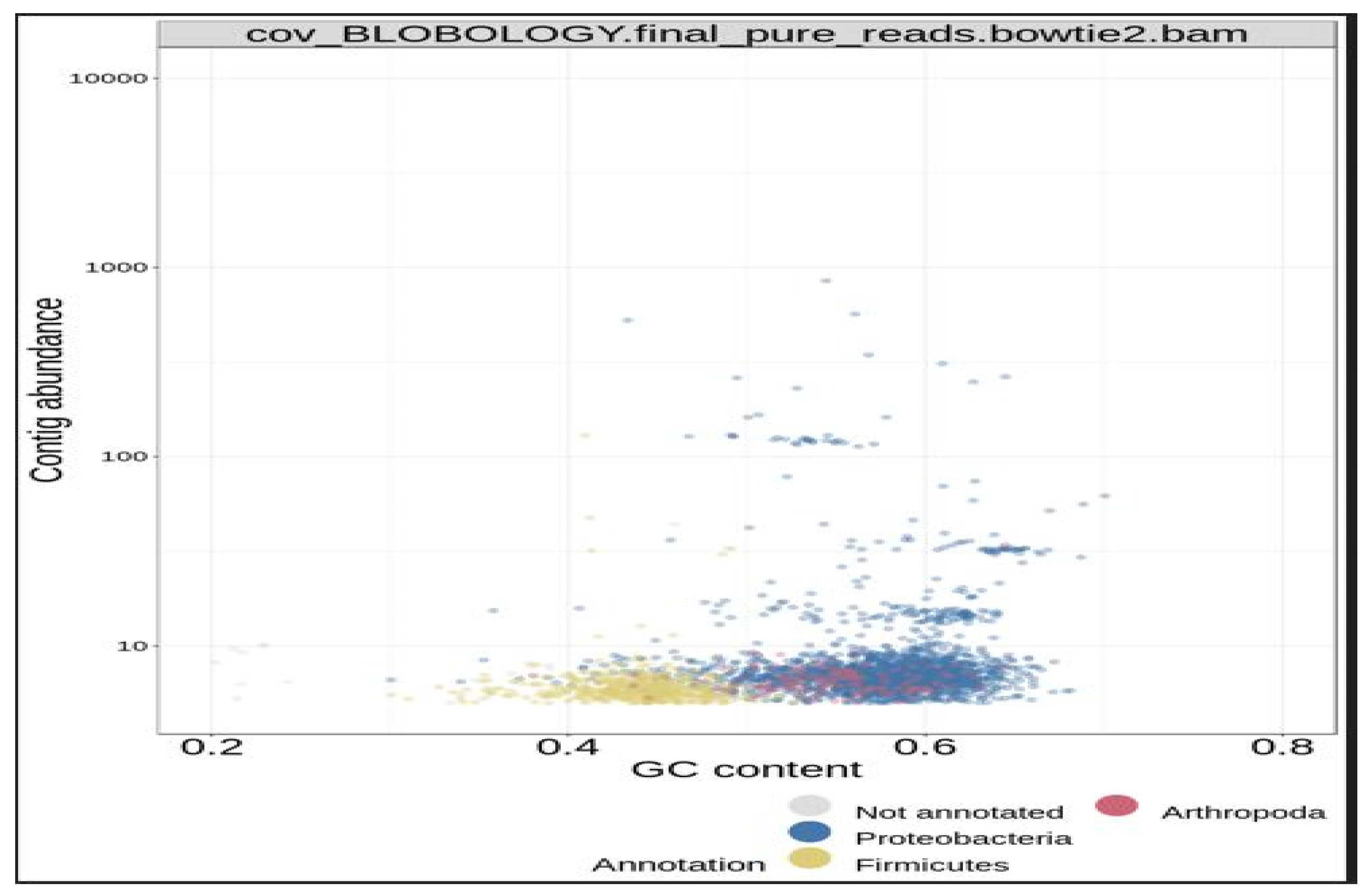
Figure 8.
Blobology clustered heatmap for Order hierarchical taxonomy of bin
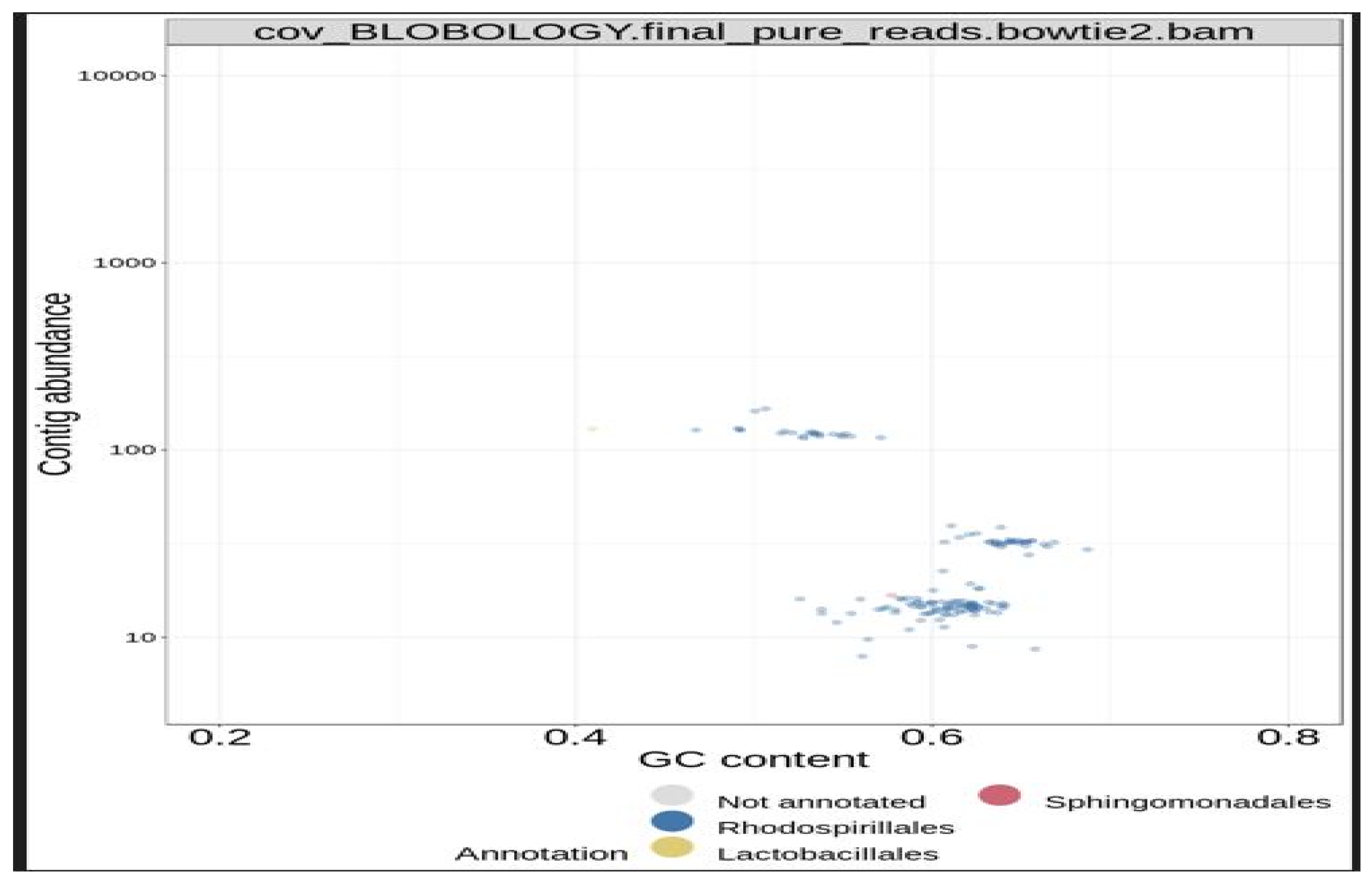
Figure 9.
The Seed viewer homepage showing the annotated genome with various parameters generated (Domain, Taxonomy, Size, GC Content, N50, L50, Number of contigs, Number of subsystems, Number of coding sequences, and Number of RNAs).
Figure 9.
The Seed viewer homepage showing the annotated genome with various parameters generated (Domain, Taxonomy, Size, GC Content, N50, L50, Number of contigs, Number of subsystems, Number of coding sequences, and Number of RNAs).
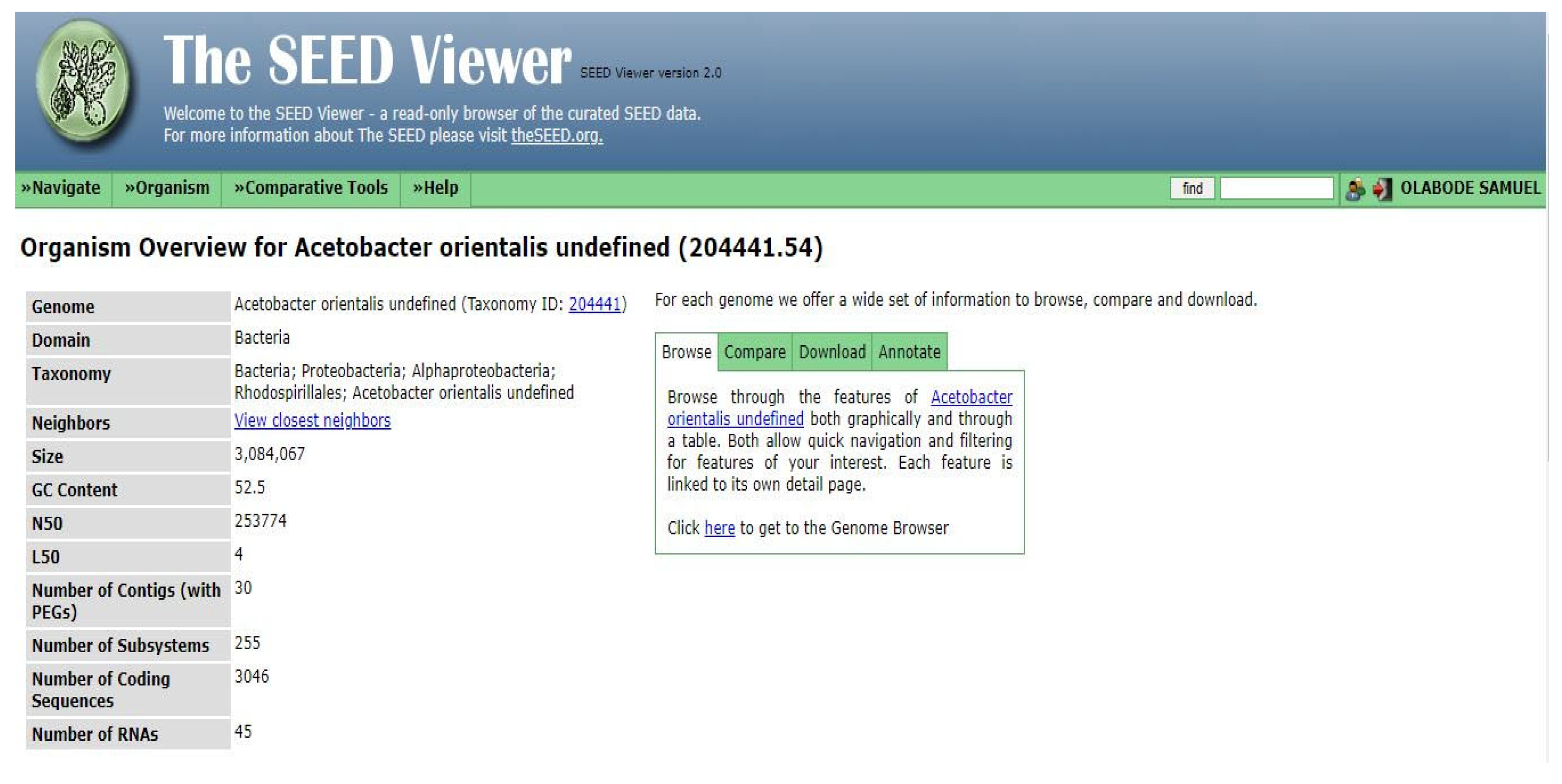
Figure 10.
Genome overview and metabolisms of the isolate Zaria-B1.
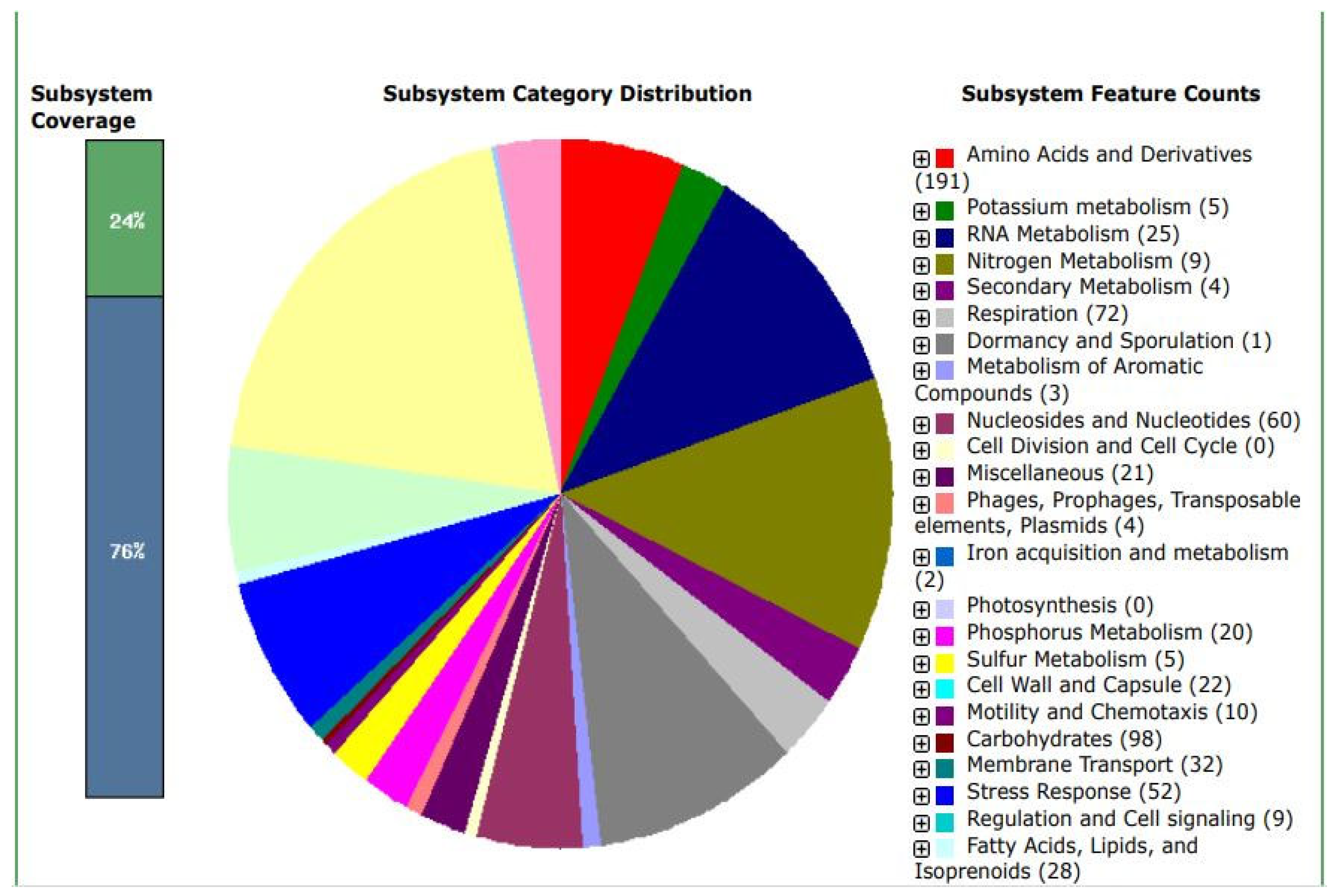
Figure 11.
NCBI Nucleotide BLAST Output of the Cellulose Synthase Catalytic Subunit Gene Sequence.
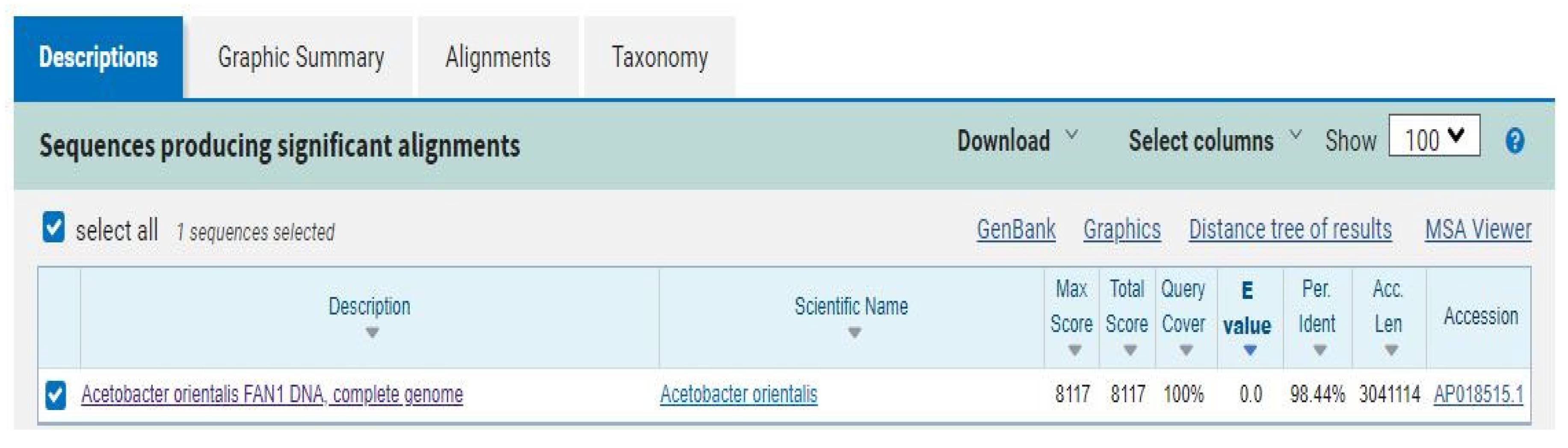

Table 1.
Extracted genomic DNA with their concentrations and purity level (A260/A280).
| Sample ID | ng/µL | A260/A280 | A260/A230 |
| PLT1 | 32.3 | 1.73 | 0.26 |
| PLT2 | 15.3 | 1.79 | 0.22 |
| PLT3 | 17.3 | 1.71 | 0.12 |
| PLT4 | 27.7 | 1.60 | 0.08 |
| BRT1 | 66.7 | 1.70 | 0.07 |
| BRT2 | 15.2 | 1.81 | 0.08 |
| BRT3 | 68.0 | 1.93 | 0.25 |
| BRT4 | 17.1 | 2.01 | 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



