Submitted:
16 October 2025
Posted:
03 November 2025
You are already at the latest version
Abstract
Background. Colorectal cancer (CRC) remains a major global public health problem, particularly in Western countries. The imple-mentation of next-generation sequencing (NGS) has enabled multigene panel analysis to identify cancer-predisposing variants and characterize molecular alterations in tumors.
To investigate molecular differences in CRC between patients younger than 50 years and those older than 50 years.
Methods. Inclusion criteria were the absence of prior radio- or chemotherapy and availability of DNA suitable for NGS. Patients were divided into two age groups: ≤50 years and >50 years. DNA was extracted from FFPE tissue samples. Targeted sequencing was performed using an Illumina hotspot cancer panel comprising 50 genes (700 amplicons).
Results. Mutation frequencies in younger vs. older patients were: TP53 (76% vs. 64%), APC (57% vs. 45%), KRAS (43% vs. 73%), NRAS (29% vs. 0%), SMAD4 (9% vs. 15%), PIK3CA (14% vs. 33%), FBXW7 (5% vs. 15%). Co-occurrence of APC/KRAS/TP53 mutations was observed in 20% of cases. KRAS mutations were significantly more frequent in older patients (p=0.001), while NRAS mutations occurred exclusively in younger patients (29% vs. 0%, p=0.021). Overall, 46% of patients exhibited multiple gene alter-ations (≥3 mutations). Notably, IDH1 and CTBX1 mutations were found only in patients with better prognosis, whereas TP53 mu-tations were nearly twice as common in patients with worse outcomes.
Conclusions. Multigene panel sequencing revealed distinct age-related molecular patterns in CRC. Younger patients were more likely to harbor NRAS mutations, whereas KRAS alterations predominated in older individuals. These findings underscore the relevance of NGS-based multigene profiling for risk stratification and personalized therapy in colorectal cancer.
Keywords:
genomic era
; next-generation sequencing (NGS)
; personalized medicine
; colorectal cancer
1. Introduction
Colorectal cancer (CRC) is the third most diagnosed malignancy and the second leading cause of cancer-related mortality worldwide, accounting for nearly 2 million new cases and 1 million deaths annually [1]. Early-onset colorectal cancer (EOCRC) is defined as colorectal cancer diagnosed in individuals younger than 50 years of age. This definition is widely used in medical literature and aligns with the age threshold at which most population-based colorectal cancer screening programs begin for average-risk individuals [2,3,4,5,6].
The rising incidence of colorectal cancer (CRC) among young adults was first identified in 2003 in the United States. This finding was derived from data collected by the nine Surveillance, Epidemiology, and End Results (SEER) registries covering the years 1973–1999. During this period, colon cancer rates among older adults remained stable, and rectal cancer rates declined; however, incidence rates among younger adults increased by 17% for colon cancer and 75% for rectal cancer. Although the total number of CRC cases in younger individuals was relatively low, the upward trend is noteworthy. Moreover, younger patients tended to present with more advanced disease stages, both in the colon (25.8% vs. 35.3%, P < 0.001) and the rectum (38.4% vs. 41.7%, P = 0.005). [7]
It is well-established that early-onset CRC incidence has increased over the past three decades in the United States. CRC incidence rates have increased by nearly 45% in adults ages 20–49 years, from 8.6 per 100,000 in 1992 to 13.1 per 100,000 in 2016 in the United States. Equally concerning is that CRC mortality rates among adults younger than 50 years increased by 1.3% per year from 2008–2017, whereas CRC mortality rates declined by 3% per year in individuals aged 65 years and older; decreases in mortality have slowed to 0.6% per year in individuals ages 50 to 64 years. Although incidence patterns for early-onset CRC are similar in men and women over the past few decades, incidence varies by site (predominantly rectal and distal colon), stage (more late-stage disease), race, ethnicity, and geographic residence [8]
The incidence of early-onset colorectal cancer has been increasing globally over the past several decades, particularly in high-income countries, and now accounts for approximately 10–14% of all new colorectal cancer Most cases are sporadic, but a higher proportion of patients with early-onset disease have hereditary cancer syndromes—most commonly Lynch syndrome—compared to later-onset cases [2,3,4]. Early-onset colorectal cancer is more likely to be present at an advanced stage and with tumors located in the distal colon or rectum [9,10,11].
Risk factors include both non-modifiable factors (family history, hereditary syndromes, inflammatory bowel disease) and modifiable factors (obesity, poor diet, sedentary lifestyle smoking, alcohol) [3]. The American Cancer Society recommends starting colorectal cancer screening at age 45 for average-risk individuals to address the rising incidence of early-onset disease [2]. Case–control studies have identified several potential risk factors for early-onset colorectal cancer (CRC), including male sex, race (particularly Black and Asian ethnicities), a family history of CRC, alcohol consumption, weight loss of ≥5 kg within five years prior to colonoscopy, processed meat intake, and inflammatory bowel disease (IBD). In contrast, regular aspirin use and higher consumption of vegetables, citrus fruits, fish, β-carotene, vitamin C, vitamin E, and folate have been linked to a reduced risk of early-onset CRC [10,11].
Early-onset CRC most frequently arises in the rectum (35–37%), followed by the distal colon (25–26%) and the proximal colon (22–23%). In comparison, among individuals aged ≥50 years (later-onset CRC), approximately 29% of cases occur in the rectum, 27% in the distal colon, and 29% in the proximal colon [12].
Patients with early-onset CRC are more likely to present with advanced disease, poor tumor differentiation, and aggressive clinical features, indicating potential biological differences compared with late-onset CRC [13,14].The mutational patterns observed in EOCRC differ from those in later-onset disease in several key respects. EOCRC demonstrates a higher prevalence of pathogenic germline variants, particularly in DNA mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2), which are characteristic of Lynch syndrome and result in microsatellite instability-high (MSI-H) tumors. Approximately 16–25% of EOCRC cases harbor germline variants, with about half attributable to Lynch syndrome [11,12,13,14,15,16].
In sporadic, microsatellite-stable (MSS) EOCRC, somatic mutational profiles show increased frequency of TP53 and CTNNB1 mutations, and decreased frequency of APC, KRAS, and BRAF mutations compared to later-onset CRC. Notably, BRAF V600E mutations and CpG island methylator phenotype (CIMP-high) are less common in EOCRC, especially in rectal and left-sided tumors. KRAS codon 12 mutations and enrichment of NOTCH1, FBXW7, PIK3CA, and FGFR3 mutations are more frequent in EOCRC than in adult-onset cases [17,18,19,20].
Hypermutated EOCRC tumors exhibit higher tumor mutational burden (TMB) and increased mutation rates in APC, KRAS, CTNNB1, and TCF7L2, while BRAF and RNF43 mutations are less frequent compared to late onset hypermutated CRC [3]. Co-mutation networks in EOCRC MSS tumors include FBXW7 with NOTCH3, RB1, and PIK3R1.
In summary, EOCRC is characterized by a higher rate of germline MMR mutations, increased TP53 and CTNNB1 mutations, lower APC, KRAS, and BRAF mutation rates, and unique co-mutational patterns, supporting the need for age-specific molecular profiling.
While these mechanisms are well established, accumulating evidence suggests that the distribution and frequency of driver mutations may vary by age. Understanding these age-related genomic differences is critical for risk stratification, early detection, and individualized therapeutic strategies [20,21,22,23,24].
Next-generation sequencing (NGS) has revolutionized cancer genomics by enabling high-throughput multigene profiling with improved sensitivity and cost-effectiveness. Compared with conventional sequencing, NGS allows for comprehensive detection of single nucleotide variants, insertions and deletions, copy number variations, and gene fusions across large cancer-associated panels. The recent NGS approaches and their revolutionary role in the identification of novel genomic CRC characteristics lead to advancement of understanding the CRC carcinogenesis and the screening of clinically actionable targets for personalized medicine. In CRC, NGS has been widely applied to identify clinically actionable alterations in genes such as KRAS, NRAS, BRAF, PIK3CA, TP53, and mismatch repair genes [25]. However, systematic age-stratified multigene analyses of CRC using NGS remain limited.
This study aims to characterize age-related genomic alterations in colorectal cancer through multigene analysis using next-generation sequencing. By comparing mutational landscapes between younger and older patients, we seek to elucidate potential biological differences that may underlie variations in clinical presentation and outcomes. Such insights may contribute to the refinement of age-specific screening strategies, prognostic models, and precision therapeutic approaches.
2. Materials and Methods
The study was conducted at the Department of Clinical and Experimental Pathology, Jan Kochanowski University, Kielce, Poland.
2.1. Inclusion and Exclusion Criteria
Patients with histopathologically confirmed colorectal adenocarcinoma, NOS were included. Other histological subtypes were excluded due to their distinct molecular pathways. Moreover, all immunohistochemistry confirmed microsatelility instable tumors were rejected. Additional exclusion criteria included prior radiotherapy or chemotherapy. Only patients with DNA of sufficient quality for next-generation sequencing (NGS) were eligible.
Patients were divided into two age-based groups:
≤50 years (n = 21)
50 years (n = 33)
In total, the study cohort comprised 54 patients (29 men and 25 women).
DNA Extraction
Genomic DNA was extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissue. Extraction was performed using the MagCore Automated Extraction Kit No. 405 (MagCore, RBC Bioscience, New Taipei, Taiwan), following the manufacturer’s protocol. DNA concentration and purity were assessed using the Quantus® Fluorometer (Promega, Madison, WI, USA) and the QuantiFluo ONE dsDNA Kit.
2.2. Library Preparation and Sequencing
Amplicon-based analysis was performed to target hotspot regions in 50 oncogenes and tumor suppressor genes. Libraries were prepared with the AmpliSeq Library PLUS for Illumina® kit (San Diego, CA, USA) according to the AmpliSeq for Illumina Cancer HotSpot Panel v2 Reference Guide. This panel generates 207 gene-specific amplicons and covers approximately 2800 clinically relevant mutations.
Amplification: Multiplex PCR (HotSpot Panel v2) was performed using DNA diluted to a final concentration of ~30 ng/reaction.
Adapter ligation: Adapters from the AmpliSeq CD Indexes Set A were ligated to the amplified DNA.
Purification: Amplicons were purified using NucleoMag® NGS Clean-up and Size Select beads (Macherey-Nagel GmbH & Co., Düren, Germany).
Quantification and pooling: Libraries were quantified with the QuantiFluo® ONE dsDNA System (Promega, USA), normalized to 4 nM, and equimolarly pooled.
Size analysis: Fragment distribution was assessed with the 4150 TapeStation System (Agilent Technologies, Santa Clara, CA, USA).
Sequencing was carried out on the Illumina MiSeq Dx platform using the MiSeq Reagent Micro Kit v2 (300 cycles). Libraries were denatured with NaOH and diluted to a final concentration of 20 pM. A 9 pM working library was prepared with a 5% spike-in of PhiX Control v2 (Illumina, San Diego, CA, USA) to monitor sequencing quality.
2.3. Statistical Analysis
Clinicopathological and molecular data were analyzed using SPSS Statistics v22 (IBM Corp., Armonk, NY, USA). Continuous variables are presented as mean ± SD and range; categorical variables are expressed as percentages. Group comparisons were performed using:
χ² test or Fisher’s exact test for categorical variables
Mann–Whitney U test or Student’s t-test for continuous variables
A two-sided p-value ≤ 0.05 was considered statistically significant.
3. Results
3.1. Clinical Features
Patients were divided into two age groups: ≤50 years (n=21) and >50 years (n=33). Clinicopathological features are summarized in Table 1.
The median age was 43 years in the younger group and 63 years in the older group. The female-to-male ratio was 1.2:1. No significant differences were observed in mean age or sex distribution between groups. However, younger patients are more frequently presented with advanced-stage disease, whereas earlier stages predominated among older patients (p < 0.05).
3.2. Somatic Mutations in the Whole Cohort
The most frequently mutated genes were TP53 (64%), KRAS (60%), and APC (51%), followed by PIK3CA (25%) and SMAD4 (13%).
3.3. Mutations by Age Group
Mutation frequencies stratified by age are shown in Table 3.
In patients ≤50, the most common mutations were TP53 (76%), APC (57%), and KRAS (43%). In patients >50, the most common mutations were KRAS (73%), TP53 (64%), and APC (45%). KRAS mutations were significantly more frequent in older patients (p = 0.001). NRAS mutations were detected exclusively in younger patients (29% vs. 0%, p = 0.021).
Co-occurrence of Mutations As shown in Figure 2, nearly half of patients (47%) presented with multiple mutations: Three concurrent mutations: 28% Four mutations: 13% Five or more mutations: 6% No significant differences in co-occurrence patterns were observed between age groups.
3.4. Mutations and Prognosis
Mutations stratified by prognosis are summarized in Table 4.
TP53 mutations were nearly twice as common in patients with worse prognosis (23 vs. 13). IDH1, CTBX1, HNF1A, and ALK mutations occurred only in patients with favorable prognosis. ERBB4, GNAS, and ATM mutations occurred only in patients with poor prognosis.
4. Discussion
Clinical outcomes associated with early-onset colorectal cancer are characterized by a higher likelihood of advanced stage (III/IV) at diagnosis, more aggressive histopathologic features (including signet-ring cell and mucinous adenocarcinoma, and poor differentiation), and a greater frequency of recurrence, particularly in stage I disease. Despite these adverse features, crude overall survival and cancer-specific survival are generally similar to, or marginally better than, those seen in later-onset CRC after adjustment for stage and other prognostic factors [26,27,28,29,30].
Younger patients with EOCRC are more likely to receive intensive multimodality therapy, including higher rates of adjuvant and multiagent chemotherapy, but this does not translate into a substantial improvement in adjusted survival outcomes. In metastatic disease, EOCRC patients have similar overall survival compared to older patients, despite more aggressive treatment regimens. Long-term excess mortality remains elevated for both EOCRC and late-onset CRC, even beyond five years post-diagnosis, underscoring the need for extended survivorship care [30].
In a large study of 648 patients, Wang et al. reported mutation frequencies of TP53 (52.8%), KRAS (46.7%), APC (24.1%), PIK3CA (18.9%), SMAD4 (9.5%), BRAF (6.1%), FBXW7 (5.3%), and NRAS (4.1%) (11). Our results are in close agreement, with TP53 (64%), KRAS (60%), APC (51%), PIK3CA (25%), SMAD4 (13%), FBXW7 (11%), NRAS (11%), and BRAF (7%), confirming cross-population consistency in CRC mutational patterns. Although the overall incidence of CRC is decreasing, the burden among young adults has grown significantly in recent decades [31]. Risk factors such as obesity, alcohol, smoking, sedentary lifestyle, and diets high in red meat may contribute disproportionately to this rise in younger populations [32,33].
These results support findings by Chang et al. in Taiwan (n=1,475), who also concluded that molecular differences between young and old CRC patients are limited, except for KRAS and NRAS, which showed significant age-dependent differences. Specifically, KRAS mutations were significantly more frequent in older patients (p=0.001), whereas NRAS mutations were restricted to younger patients (p=0.021).
TP53 and KRAS mutations in early-onset colorectal cancer (EOCRC) are significant because they reflect distinct molecular features and have prognostic implications. In EOCRC, TP53 mutations are more frequent than in later-onset cases, particularly in microsatellite-stable tumors, suggesting a greater role for TP53-driven tumorigenesis in younger patients. KRAS mutations, while common in colorectal cancer overall, are somewhat less frequent in EOCRC compared to late-onset disease but still represent a substantial proportion of cases and may be more prevalent in certain EOCRC cohorts [34,35,36].
Functionally, TP53 mutations are associated with loss of tumor suppressor activity, contributing to genomic instability and aggressive tumor behavior. KRAS mutations, particularly in codons 12 and 13, activate downstream signaling pathways (MAPK, PI3K), promoting cell proliferation and resistance to anti-EGFR therapies [36]. The co-occurrence of TP53 and KRAS mutations is associated with worse prognosis, including higher risk of recurrence and decreased survival, especially in metastatic settings. Specific combinations, such as KRAS codon 13 and TP53 L3 domain mutations, further stratify risk for poor outcomes [37].
In summary, TP53 mutations are more common in EOCRC and are linked to aggressive tumor biology, while KRAS mutations drive oncogenic signaling and predict resistance to targeted therapies; their co-occurrence portends a worse clinical outcome. The prevalence of multiple gene alterations (co-alterations) is higher in younger patients, with early-onset CRC showing more frequent co-occurrence of mutations such as KRAS with ATM, ARID1A, CREBBP, FAT1, KMT2B, and KMT2D, whereas these gene pairs are more often exclusive in late-onset CRC [37]. Younger patients also show enrichment of NOTCH1, FBXW7, PIK3CA, and FGFR3 mutations.
Regarding prognosis, TP53 mutations are associated with more aggressive tumor biology and worse clinical outcomes. IDH1, CTBX1, HNF1A, and ALK mutations are also linked to poor prognosis in CRC, with patients harboring these mutations experiencing shorter progression-free and overall survival. The presence of multiple co-mutations, such as TP53/IDH1 or TP53/ALK, further correlates with adverse outcomes other studies echo these observations. Chatsirisupachai et al. demonstrated that TP53 and CTNNB1 mutations were more common in younger CRC patients, while APC, KRAS, and BRAF V600E mutations predominated in older ones. Our study partially confirms these findings: TP53 dominated in ≤50 patients, while KRAS dominated in >50 patients [38].
Most CRC cases are diagnosed in patients >50 years, but early-onset cases are increasing, now representing a substantial public health concern. Early detection and personalized treatment approaches are critical for this population [34,35,36]. Large retrospective studies, including one analyzing ~36,000 patients, have shown that early-onset CRC is more often associated with microsatellite instability (MSI), CTNNB1 and ATM mutations, and CIMP hypermethylation [39].
Our results also align with reports by Kim et al., identifying recurrent alterations in known cancer-related genes (APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, NRAS). Prognostic implications are notable: TP53 mutations were almost twice as frequent in patients with worse prognosis, while IDH1, CTBX1, HNF1A, and ALK mutations occurred only in patients with better outcomes [40,41,42]. These findings agree with Lipsyc, Kalady, and Shen, who linked KRAS, NRAS, BRAF, and PIK3CA mutations with poor survival [43,44,45]. NRAS has been implicated in inflammation-driven tumorigenesis, which may explain why we observe NRAS mutations only in younger patients.
Interestingly, IDH1 mutations have been associated with younger age, favorable prognosis, and better therapeutic response [46,47]. Consistently, in our cohort, IDH1 mutations appeared exclusively in patients with better prognosis.
Taking together, our results emphasize the clinical value of targeted NGS panels in CRC. Technology not only provides a comprehensive overview of recurrent mutations but also identifies rare genetic alterations that may carry prognostic or therapeutic implications. Future challenges lie in integrating NGS data into clinical workflows to refine patient stratification, improve early detection, and enable more precise treatment selection.
4.1. The Question Is—Does This Matter for Patient’s Management?
Molecular profiling has played a crucial role in identifying biomarkers that may enhance clinical outcomes for colorectal cancer (CRC) patients and improve survival rates among those with metastatic disease. Numerous molecular alterations have been characterized as potential biomarkers and are being evaluated in both completed and ongoing clinical trials alongside targeted and immunotherapeutic approaches.
4.2. Targeting the CIN Pathway
Extensive research is focused on developing synthetic modulators of Wnt signaling, including small molecules, peptides, and inhibitory antibodies that suppress this pathway. Lithium chloride, approved by the U.S. Food and Drug Administration (FDA), is already used clinically and is known to activate CTNNB1 by inhibiting GSK3. Additionally, nonsteroidal anti-inflammatory drugs (NSAIDs) and the selective COX-2 inhibitor celecoxib have been shown to block CTNNB1-dependent transcription in CRC [48,49,50,51].
4.3. Targeting the MSI Pathway
Microsatellite instability (MSI) has proven to be a reliable biomarker for predicting immunotherapy response in metastatic CRC. Immune checkpoint inhibitors have demonstrated promising results across various solid tumors. Pembrolizumab, a humanized IgG4 monoclonal antibody, became the first FDA-approved PD-1 inhibitor for metastatic CRC treatment [52,53]. A phase I clinical trial further confirmed pembrolizumab’s antitumor activity in patients with MSI-H CRC.
4.4. Targeting the CIMP Pathway
Therapeutic strategies targeting the EGFR pathway typically utilize anti-EGFR monoclonal antibodies and tyrosine kinase inhibitors (TKIs) directed against intracellular kinases. Cetuximab was the first monoclonal antibody developed to target EGFR; upon binding to its extracellular domain, it induces receptor internalization and degradation. Multiple studies have demonstrated the positive impact of cetuximab on clinical outcomes in CRC patients [54,55].
5. Conclusions
Molecular and clinicopathological differences between younger and older CRC patients were limited. However, KRAS mutations were significantly more frequent in older patients, and NRAS mutations were exclusive to younger patients.
In younger patients, the most common mutation was TP53 (76%), while in older patients, KRAS (73%) predominated.
Almost half of patients (46%) harbored multiple gene alterations, most often three to five mutations.
TP53 mutations were almost twice as frequent in patients with worse prognosis, while IDH1, CTBX1, HNF1A, and ALK mutations were confined to patients with better prognosis.
The exclusive presence of NRAS mutations in young patients (29%) suggests an age-specific molecular pattern.
This targeted NGS assay proved suitable for clinical application, with potential relevance for diagnosis and prognosis in Polish CRC patients.
Author Contributions
Conceptualization: Monika Kozłowska-Geller; data curation: Łukasz Nawacki, formal analysis: Monika Kozłowska-Geller; funding acquisition: Łukasz Nawacki, investigation: Monika Wawszczak-Kasza; methodology: Monika Kozłowska-Geller, Piotr Lewitowicz; project administration: Monika Kozłowska-Geller, Piotr Lewitowicz; resources: Monika Wawszczak-Kasza, Wojciech Lewitowicz, Jacek Bicki; supervision: Piotr Lewitowicz; writing—original draft: Monika Kozłowska-Geller; writing—review & editing: Piotr Lewitowicz.
Funding
This study was supported by a grant from Jan Kochanowski University (Grant No. SUPB.RN.22.067).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Collegium Medicum Jan Kochanowski University in Kielce 11.04.2012 (project No 03.2012).
Informed Consent Statement
Written informed consent has been obtained from the patient(s) to perform the molecular analysis and to publish it.
Data Availability Statement
Research data supporting this publication are available upon request from Monika Kozłowska-Geller (monika.kozlowska.chir@onet.pl.
Conflicts of Interest
The authors declare no conflict of interest.
Ethical Compliance
All procedures involving human participants were conducted in accordance with institutional/national ethical standards, the 1975 Helsinki Declaration, and its later amendments.
References
- Baidoun, F.; Elshiwy, K.; Elkeraie, Y.; Merjaneh, Z.; Khoudari, G.; Sarmini, M.T.; Gad, M.; Al-Husseini, M.; Saad, A. Colorectal Cancer Epidemiology: Recent Trends and Impact on Outcomes. Curr. Drug Targets 2021, 22, 998–1009. [Google Scholar] [CrossRef]
- Jayakrishnan, T.; Ng, K. Early-Onset Gastrointestinal Cancers. JAMA 2025, 334, 1373. [Google Scholar] [CrossRef]
- REACCT Collaborative; Zaborowski, A.M.; Abdile, A.; Adamina, M.; Aigner, F.; D’allens, L.; Allmer, C.; Álvarez, A.; Anula, R.; Andric, M.; et al. Characteristics of Early-Onset vs Late-Onset Colorectal Cancer. JAMA Surg. 2021, 156, 865–874. [CrossRef]
- Spaander, M.C.W.; Zauber, A.G.; Syngal, S.; Blaser, M.J.; Sung, J.J.; You, Y.N.; Kuipers, E.J. Young-onset colorectal cancer. Nat. Rev. Dis. Prim. 2023, 9, 1–21. [Google Scholar] [CrossRef]
- Medici, B.; Riccò, B.; Caffari, E.; Zaniboni, S.; Salati, M.; Spallanzani, A.; Garajovà, I.; Benatti, S.; Chiavelli, C.; Dominici, M.; et al. Early Onset Metastatic Colorectal Cancer: Current Insights and Clinical Management of a Rising Condition. Cancers 2023, 15, 3509. [Google Scholar] [CrossRef]
- Gausman, V.; Dornblaser, D.; Anand, S.; Hayes, R.B.; O'COnnell, K.; Du, M.; Liang, P.S. Risk Factors Associated With Early-Onset Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 2752–2759.e2. [Google Scholar] [CrossRef] [PubMed]
- O'COnnell, J.B.; Maggard, M.A.; Liu, J.H.; Etzioni, D.A.; Livingston, E.H.; Ko, C.Y. Rates of Colon and Rectal Cancers are Increasing in Young Adults. Am. Surg. 2003, 69, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Siegel RL, Miller KD, Goding Sauer A, et al. Colorectal cancer statistics, 2020. CA Cancer J Clin 2020.
- Mauri, G.; Sartore-Bianchi, A.; Russo, A.; Marsoni, S.; Bardelli, A.; Siena, S. Early-onset colorectal cancer in young individuals. Mol. Oncol. 2018, 13, 109–131. [Google Scholar] [CrossRef]
- Spaander, M.C.W.; Zauber, A.G.; Syngal, S.; Blaser, M.J.; Sung, J.J.; You, Y.N.; Kuipers, E.J. Young-onset colorectal cancer. Nat. Rev. Dis. Prim. 2023, 9, 1–21. [Google Scholar] [CrossRef]
- Rosato, V.; Bosetti, C.; Levi, F.; Polesel, J.; Zucchetto, A.; Negri, E.; La Vecchia, C. Risk factors for young-onset colorectal cancer. Cancer Causes Control. 2012, 24, 335–341. [Google Scholar] [CrossRef]
- Gausman, V.; Dornblaser, D.; Anand, S.; Hayes, R.B.; O'COnnell, K.; Du, M.; Liang, P.S. Risk Factors Associated With Early-Onset Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2020, 18, 2752–2759.e2. [Google Scholar] [CrossRef]
- Archambault, A.N.; Su, Y.-R.; Jeon, J.; Thomas, M.; Lin, Y.; Conti, D.V.; Win, A.K.; Sakoda, L.C.; Lansdorp-Vogelaar, I.; Peterse, E.F.; et al. Cumulative Burden of Colorectal Cancer–Associated Genetic Variants Is More Strongly Associated With Early-Onset vs Late-Onset Cancer. Gastroenterology 2020, 158, 1274–1286.e12. [Google Scholar] [CrossRef]
- Spaander, M.C.W.; Zauber, A.G.; Syngal, S.; Blaser, M.J.; Sung, J.J.; You, Y.N.; Kuipers, E.J. Young-onset colorectal cancer. Nat. Rev. Dis. Prim. 2023, 9, 1–21. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Escobar, D.; Jones, R.; Gao, J.; Sun, L.; Liao, J.; Yang, G.-Y. Unique clinicopathologic and genetic alteration features in early onset colorectal carcinoma compared with age-related colorectal carcinoma: a large cohort next generation sequence analysis. Hum. Pathol. 2020, 105, 37–46. [Google Scholar] [CrossRef]
- Willauer, A.N.; Liu, Y.; Pereira, A.A.L.; Lam, M.; Morris, J.S.; Raghav, K.P.S.; Morris, V.K.; Menter, D.; Broaddus, R.; Meric-Bernstam, F.; et al. Clinical and molecular characterization of early-onset colorectal cancer. Cancer 2019, 125, 2002–2010. [Google Scholar] [CrossRef]
- Ugai, T.; Haruki, K.; Harrison, T.A.; Cao, Y.; Qu, C.; Chan, A.T.; Campbell, P.T.; Akimoto, N.; Berndt, S.; Brenner, H.; et al. Molecular Characteristics of Early-Onset Colorectal Cancer According to Detailed Anatomical Locations: Comparison With Later-Onset Cases. Am. J. Gastroenterol. 2022, 118, 712–726. [Google Scholar] [CrossRef]
- Ponvilawan, B.; Sakornsakolpat, P.; Pongpaibul, A.; Roothumnong, E.; Akewanlop, C.; Pithukpakorn, M.; Korphaisarn, K. Comprehensive genomic analysis in sporadic early-onset colorectal adenocarcinoma patients. BMC Cancer 2025, 25, 1–7. [Google Scholar] [CrossRef]
- Matsuda, T.; Fujimoto, A.; Igarashi, Y. Colorectal Cancer: Epidemiology, Risk Factors, and Public Health Strategies. Digestion 2025, 106, 91–99. [Google Scholar] [CrossRef]
- Saraiva, M.R.; Rosa, I.; Claro, I. Early-onset colorectal cancer: A review of current knowledge. World J. Gastroenterol. 2023, 29, 1289–1303. [Google Scholar] [CrossRef]
- Venugopal, A.; Carethers, J.M. EPIDEMIOLOGY AND BIOLOGY OF EARLY ONSET COLORECTAL CANCER. 2022, 21, 162–182. [CrossRef]
- Siegel, R.L.; Jakubowski, C.D.; Fedewa, S.A.; Davis, A.; Azad, N.S. Colorectal Cancer in the Young: Epidemiology, Prevention, Management. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, e75–e88. [Google Scholar] [CrossRef]
- Nfonsam, V.; Wusterbarth, E.; Gong, A.; Vij, P. Early-Onset Colorectal Cancer. Surg. Oncol. Clin. North Am. 2022, 31, 143–155. [Google Scholar] [CrossRef]
- Abbes, S.; Baldi, S.; Sellami, H.; Amedei, A.; Keskes, L. Molecular methods for colorectal cancer screening: Progress with next-generation sequencing evolution. World J. Gastrointest. Oncol. 2023, 15, 425–442. [Google Scholar] [CrossRef]
- Liao, C.-K.; Hsu, Y.-J.; Chern, Y.-J.; Yu, Y.-L.; Lin, Y.-C.; Hsieh, P.-S.; Chiang, J.-M.; You, J.-F. Differences in characteristics and outcomes between early-onset colorectal cancer and late-onset colorectal cancers. Eur. J. Surg. Oncol. (EJSO) 2024, 50, 108687. [Google Scholar] [CrossRef]
- Jayakrishnan, T.; Ng, K. Early-Onset Gastrointestinal Cancers. JAMA 2025, 334, 1373. [Google Scholar] [CrossRef]
- Foppa, C.; Maroli, A.; Lauricella, S.; Luberto, A.; La Raja, C.; Bunino, F.; Carvello, M.; Sacchi, M.; De Lucia, F.; Clerico, G.; et al. Different Oncologic Outcomes in Early-Onset and Late-Onset Sporadic Colorectal Cancer: A Regression Analysis on 2073 Patients. Cancers 2022, 14, 6239. [Google Scholar] [CrossRef]
- Carbone, F.; Spinelli, A.; Ciardiello, D.; Luc, M.R.; de Pascale, S.; Bertani, E.; Fazio, N.; Romario, U.F. Prognosis of early-onset versus late-onset sporadic colorectal cancer: Systematic review and meta-analysis. Eur. J. Cancer 2024, 215, 115172. [Google Scholar] [CrossRef]
- Barot, S.; Liljegren, A.; Nordenvall, C.; Blom, J.; Radkiewicz, C. Incidence trends and long-term survival in early-onset colorectal cancer: a nationwide Swedish study. Ann. Oncol. 2025, 36, 1400–1408. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, H.; Hou, Y.; Zhou, X.; Liang, L.; Zhang, Z.; Shi, H.; Xu, S.; Hu, P.; Zheng, Z.; et al. Performance validation of an amplicon-based targeted next-generation sequencing assay and mutation profiling of 648 Chinese colorectal cancer patients. Virchows Arch. 2018, 472, 959–968. [Google Scholar] [CrossRef]
- Kim BJ, Hanna MH. Colorectal cancer in young adults. J Surg Oncol. 2023;127(8):1231–1309.
- Alshenaifi, J.Y.; Vetere, G.; Maddalena, G.; Yousef, M.; White, M.G.; Shen, J.P.; Vilar, E.; Parseghian, C.; Dasari, A.; Morris, V.K.; et al. Mutational and co-mutational landscape of early onset colorectal cancer. Biomarkers 2025, 30, 64–76. [Google Scholar] [CrossRef]
- Escobar, D.; Jones, R.; Gao, J.; Sun, L.; Liao, J.; Yang, G.-Y. Unique clinicopathologic and genetic alteration features in early onset colorectal carcinoma compared with age-related colorectal carcinoma: a large cohort next generation sequence analysis. Hum. Pathol. 2020, 105, 37–46. [Google Scholar] [CrossRef]
- Willauer, A.N.; Liu, Y.; Pereira, A.A.L.; Lam, M.; Morris, J.S.; Raghav, K.P.S.; Morris, V.K.; Menter, D.; Broaddus, R.; Meric-Bernstam, F.; et al. Clinical and molecular characterization of early-onset colorectal cancer. Cancer 2019, 125, 2002–2010. [Google Scholar] [CrossRef]
- Ugai, T.; Haruki, K.; Harrison, T.A.; Cao, Y.; Qu, C.; Chan, A.T.; Campbell, P.T.; Akimoto, N.; Berndt, S.; Brenner, H.; et al. Molecular Characteristics of Early-Onset Colorectal Cancer According to Detailed Anatomical Locations: Comparison With Later-Onset Cases. Am. J. Gastroenterol. 2022, 118, 712–726. [Google Scholar] [CrossRef]
- Ponvilawan, B.; Sakornsakolpat, P.; Pongpaibul, A.; Roothumnong, E.; Akewanlop, C.; Pithukpakorn, M.; Korphaisarn, K. Comprehensive genomic analysis in sporadic early-onset colorectal adenocarcinoma patients. BMC Cancer 2025, 25, 1–7. [Google Scholar] [CrossRef]
- Chatsirisupachai K, Lagger C, de Magalhães JP. Age-associated differences in the cancer molecular landscape. Trends Cancer. 2022;8(11):962–971.Spaander MCW, Zauber AG, Syngal S. Young-onset colorectal cancer. Nat Rev Dis Primers. 2023;9(1):21.
- Chang, C.-C.; Lin, P.-C.; Lin, C.-C.; Lan, Y.-T.; Lin, H.-H.; Lin, C.-H.; Yang, S.-H.; Liang, W.-Y.; Chen, W.-S.; Jiang, J.-K.; et al. Molecular and Clinicopathological Differences by Age at the Diagnosis of Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 1441. [Google Scholar] [CrossRef]
- Cheong, C.; Oh, S.Y.; Kim, Y.B.; Suh, K.W. Differences in biological behaviors between young and elderly patients with colorectal cancer. PLOS ONE 2019, 14, e0218604. [Google Scholar] [CrossRef]
- Khan, S.A.; Morris, M.; Idrees, K.; Gimbel, M.I.; Rosenberg, S.; Zeng, Z.; Li, F.; Gan, G.; Shia, J.; LaQuaglia, M.P.; et al. Colorectal cancer in the very young: a comparative study of tumor markers, pathology and survival in early onset and adult onset patients. J. Pediatr. Surg. 2016, 51, 1812–1817. [Google Scholar] [CrossRef]
- Youssef, A.S.E.-D.; Abdel-Fattah, M.A.; Lotfy, M.M.; Nassar, A.; Abouelhoda, M.; Touny, A.O.; Hassan, Z.K.; Eldin, M.M.; Bahnassy, A.A.; Khaled, H.; et al. Multigene Panel Sequencing Reveals Cancer-Specific and Common Somatic Mutations in Colorectal Cancer Patients: An Egyptian Experience. Curr. Issues Mol. Biol. 2022, 44, 1332–1352. [Google Scholar] [CrossRef]
- Shen, Y.; Han, X.; Wang, J.; Wang, S.; Yang, H.; Lu, S.-H.; Shi, Y. Prognostic impact of mutation profiling in patients with stage II and III colon cancer. Sci. Rep. 2016, 6, 24310–24310. [Google Scholar] [CrossRef]
- Lipsyc, M.; Yaeger, R. Impact of somatic mutations on patterns of metastasis in colorectal cancer. 2015, 6, 645–649–649. [CrossRef]
- Kalady, M.F.; DeJulius, K.L.B.; Sanchez, J.A.; Jarrar, A.; Liu, X.; Manilich, E.; Skacel, M.; Church, J.M.M. BRAF Mutations in Colorectal Cancer Are Associated With Distinct Clinical Characteristics and Worse Prognosis. Dis. Colon Rectum 2012, 55, 128–133. [Google Scholar] [CrossRef]
- de Botton, S.; Mondesir, J.; Willekens, C.; Touat, M. IDH1 and IDH2 mutations as novel therapeutic targets: current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.-L.; Xiong, Y. IDH1andIDH2Mutations in Tumorigenesis: Mechanistic Insights and Clinical Perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef]
- Ghosh, N.; Hossain, U.; Mandal, A.; Sil, P.C. The Wnt signaling pathway: a potential therapeutic target against cancer. Ann. New York Acad. Sci. 2019, 1443, 54–74. [Google Scholar] [CrossRef]
- Dihlmann, S.; Siermann, A.; Doeberitz, M.v.K. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate β-catenin/TCF-4 signaling. Oncogene 2001, 20, 645–653. [Google Scholar] [CrossRef]
- Lewitowicz, P.; Koziel, D.; Gluszek, S.Z.; Matykiewicz, J.; Wincewicz, A.; Horecka-Lewitowicz, A.; Nasierowska-Guttmejer, A. Is there a place for practical chemoprevention of colorectal cancer in light of COX-2 heterogeneity? Pol. J. Pathol. 2014, 4, 276–282. [Google Scholar] [CrossRef]
- Tuynman, J.B.; Vermeulen, L.; Boon, E.M.; Kemper, K.; Zwinderman, A.H.; Peppelenbosch, M.P.; Richel, D.J. Cyclooxygenase-2 Inhibition Inhibits c-Met Kinase Activity and Wnt Activity in Colon Cancer. Cancer Res. 2008, 68, 1213–1220. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- O'Neil, B.H.; Wallmark, J.M.; Lorente, D.; Elez, E.; Raimbourg, J.; Gomez-Roca, C.; Ejadi, S.; A Piha-Paul, S.; Stein, M.N.; Razak, A.R.A.; et al. Safety and antitumor activity of the anti–PD-1 antibody pembrolizumab in patients with advanced colorectal carcinoma. PLOS ONE 2017, 12, e0189848. [Google Scholar] [CrossRef]
- Mendelsohn, J.; Prewett, M.; Rockwell, P.; Goldstein, N.I. CCR 20th Anniversary Commentary: A Chimeric Antibody, C225, Inhibits EGFR Activation and Tumor Growth. Clin. Cancer Res. 2015, 21, 227–229. [Google Scholar] [CrossRef]
- Jonker, D.J.; O'Callaghan, C.J.; Karapetis, C.S.; Zalcberg, J.R.; Tu, D.; Au, H.-J.; Berry, S.R.; Krahn, M.; Price, T.; Simes, R.J.; et al. Cetuximab for the Treatment of Colorectal Cancer. New Engl. J. Med. 2007, 357, 2040–2048. [Google Scholar] [CrossRef]
Figure 1.
Oncoplot displays the somatic mutations distribution of the top highly mutated genes.
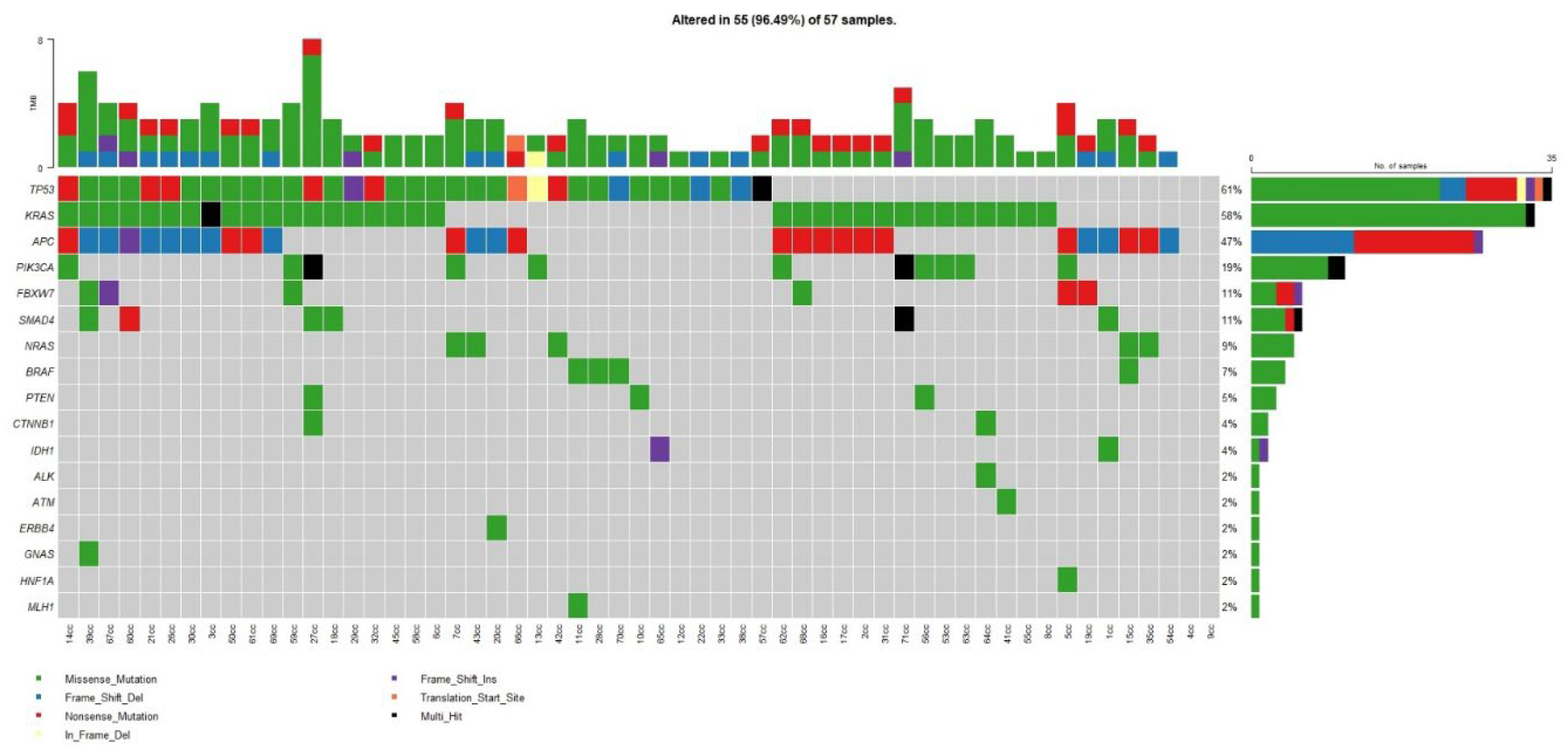
Figure 2.
Co-occurence of mutations in the studied group of patients.
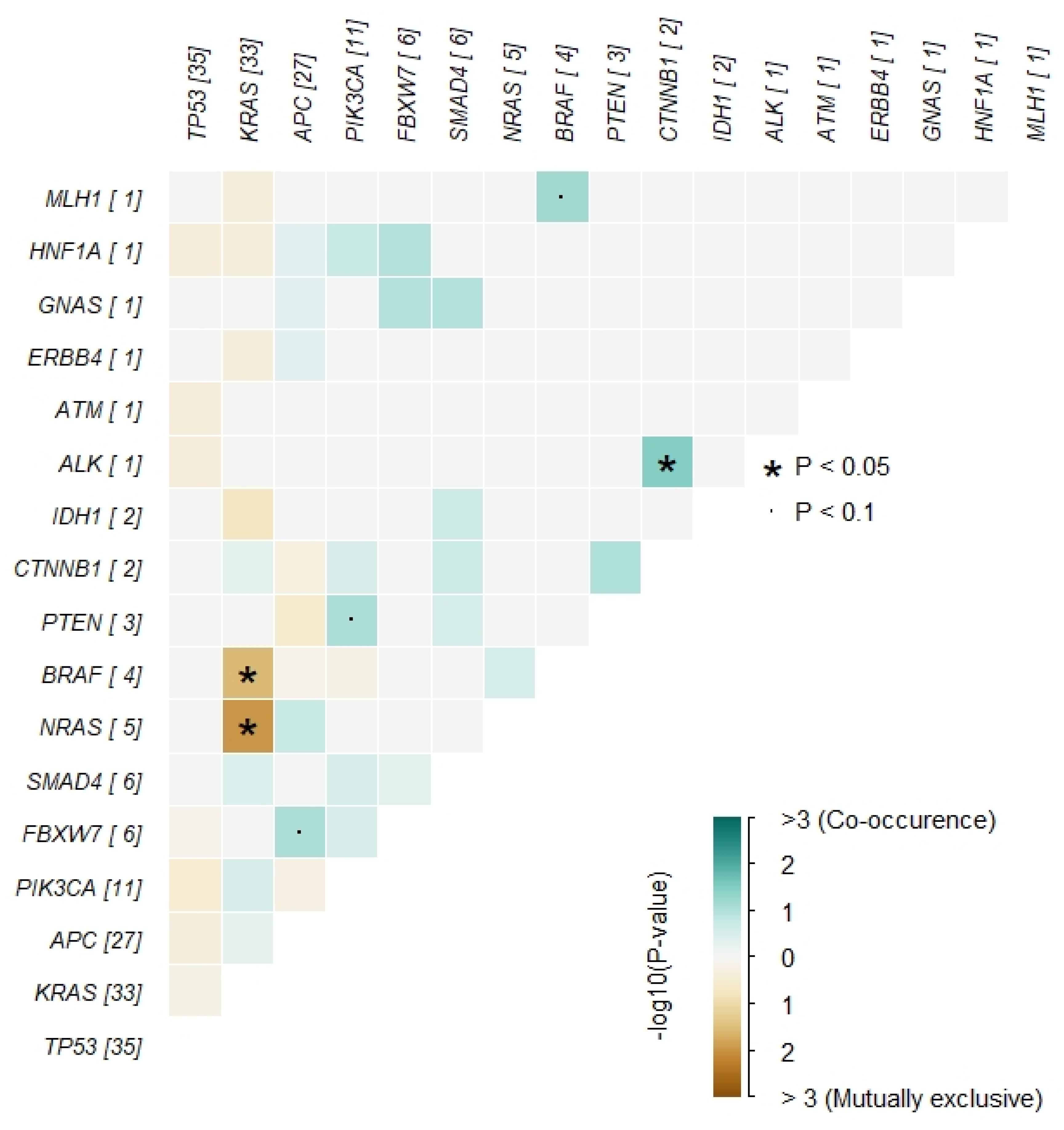

Table 1.
Clinicopathological features of the study cohort.
| Variable | All patients (n=54) | ≤50 years (n=21) | >50 years (n=33) |
|---|---|---|---|
| Median age (range) | 55 (31–92) | 43 (31–50) | 63 (51–92) |
| Sex | |||
| Male | 29 (54%) | 10 | 19 |
| Female | 25 (46%) | 11 | 14 |
| Stage | I: 9; II: 14; III: 17; IVA: 6; IVB: 8 | Higher proportion of advanced stages | Lower proportion of advanced stages |
Table 2.
Distribution of mutations in all patients (n=54).
| Gene | % | n |
|---|---|---|
| TP53 | 64% | 35 |
| KRAS | 60% | 33 |
| APC | 51% | 28 |
| PIK3CA | 25% | 14 |
| SMAD4 | 13% | 7 |
| NRAS | 11% | 6 |
| FBXW7 | 11% | 6 |
| BRAF | 7% | 4 |
Table 3.
Mutation distribution in ≤50 and >50 groups.
| Gene | ≤50 years (n=21) |
|---|---|
| TP53 | 16 (76%) |
| APC | 12 (57%) |
| KRAS | 9 (43%) |
| NRAS | 6 (29%) |
| PIK3CA | 3 (14%) |
| SMAD4 | 2 (9%) |
| FBXW7 | 1 (5%) |
| Other (HNF1A, ERBB4, PTEN, BRAF, CTNNB1, ALK, IDH1, GNAS, ATM) | sporadic |
Table 4.
Mutation distribution in patients with better vs. worse prognosis.
| Gene | Better prognosis (Stage I–II) | Worse prognosis (Stage III–IVB) |
|---|---|---|
| TP53 | 13 | 23 |
| KRAS | 17 | 15 |
| APC | 16 | 13 |
| PIK3CA | 9 | 6 |
| NRAS | 2 | 4 |
| IDH1 | 2 | 0 |
| CTBX1 | 2 | 0 |
| HNF1A | 1 | 0 |
| ALK | 1 | 0 |
| ERBB4 | 0 | 1 |
| GNAS | 0 | 1 |
| ATM | 0 | 1 |
| Others (FBXW7, SMAD4, BRAF, PTEN) | similar frequencies across groups |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



