
Submitted:
01 November 2025
Posted:
03 November 2025
You are already at the latest version
Abstract
Predictive multiscale cellular modeling redefines precision medicine by integrating hypothesis grammars, digital twins, and integrative genomics to forecast tumor–immune dynamics, therapeutic resistance, and cellular plasticity. Hypothesis grammars translate mechanistic theories into executable agent-based models (ABMs) and hybrid ODE–PDE systems, enabling rapid in silico hypothesis testing. Patient-specific digital twins, driven by multi-omics data, employ stochastic ensemble methods to simulate clonal evolution and microenvironmental interactions. Integrative genomics, leveraging algorithms like SCODE and SimiC, infers causal gene regulatory networks (GRNs) using Bayesian variational autoencoders, embedding dynamic intracellular logic into tissue-scale simulations. Applications include in silico oncology trials optimizing checkpoint blockade and combination therapies. Large language models enhance rule induction, while FAIR-compliant digital cell repositories ensure reproducibility. Verification, validation, and uncertainty quantification (VVUQ) via Sobol sensitivity and Kennedy–O’Hagan calibration address non-identifiability. Federated learning mitigates privacy and bias concerns. This framework enables virtual clinical trials and adaptive theranostics, transforming biological understanding into actionable, equitable precision medicine.
Keywords:
multiscale modeling
; hypothesis grammars
; digital twins
; integrative genomics
; in silico oncology
1. Introduction
The landscape of cellular and cancer biology is undergoing a profound transformation, driven by advances in high-throughput sequencing, spatial omics, artificial intelligence, and computational modeling. Traditional reductionist approaches that examine isolated molecular alterations are insufficient to explain emergent behaviors, such as tumor–immune interactions, therapeutic resistance, or cellular plasticity, which are inherently multiscale and context-dependent [1]. Although genomics and single-cell transcriptomics have provided unprecedented molecular snapshots, these static datasets cannot predict the dynamic trajectories that shape disease progression or therapeutic outcomes [2]. A new paradigm of predictive modeling has thus emerged, integrating hypothesis-driven reasoning with patient-specific multi-omics data to generate simulations capable of forecasting cellular futures with fidelity comparable to meteorological models [3].
At the center of this paradigm are three converging pillars. The first is the hypothesis grammar, a plain-language, rule-based framework that translates biological hypotheses into executable mathematical or agent-based models [4]. By lowering the technical barrier of coding, these grammars democratize modeling and enable biologists to formalize mechanistic theories that can be directly tested in silico. Applications include modeling tumor–immune ecosystems, where spatial genomics and agent-based rules generate forecasts of clonal evolution and immune evasion under therapeutic pressure [5]. The second pillar is the digital twin, a virtual patient-specific replica of cellular systems that integrates genomic, imaging, and clinical datato simulate disease progression, therapeutic response, and emergent behaviors in silico [6]. In oncology, digital twins have been used to predict tumor evolution under targeted and immunotherapies, optimize treatment combinations, and design adaptive clinical strategies [7]. The third pillar, integrative genomics, anchors these models in biological reality by combining transcriptomic, proteomic, epigenomic, and spatial data layers to calibrate and validate simulations [8]. When coupled with machine learning, integrative frameworks enhance predictive fidelity and scalability, thereby reducing computational costs while maintaining mechanistic interpretability [9]. Emerging approaches are beginning to integrate large language models with hypothesis grammars, enabling automated hypothesis generation, refinement of simulation rules, and community repositories of reusable “digital cell lines” [10].
Together, these innovations represent more than incremental advances in computational biology; they constitute a conceptual reimagining of how biological knowledge is generated, tested, and translated. Predictive multiscale cellular modeling promises to accelerate discovery, democratize model creation, and enable in silico clinical trials that complement traditional bench and bedside research. Yet, realizing this vision requires addressing outstanding challenges, including the standardization of data formats, model reproducibility, the interpretability of AI-driven predictions, and the ethical implications of deploying patient-specific simulations in clinical decision-making [6,11]. By synthesizing recent progress across hypothesis grammars, digital twins, multiscale modeling, and integrative genomics, this review aims to provide a state-of-the-art perspective on the opportunities, challenges, and future trajectories of predictive cellular modeling in precision medicine.
2. Foundations: Hypothesis Grammars for Cell Behavior Modeling
2.1. Concept and Rationale
Hypothesis grammars represent a novel paradigm for translating qualitative biological knowledge into quantitative computational frameworks. Traditional systems biology relies heavily on bespoke coding or differential equations manually constructed by computational specialists, creating a bottleneck between experimental discovery and model implementation. Hypothesis grammars address this gap by introducing plain-language rule-based representations of cellular interactions, where statements such as “activated T cells secrete IFN- γ, inducing macrophage polarization” or “tumor cells upregulate PD-L1 in response to cytokine exposure” are compiled into underlying mathematical or algorithmic processes [4]. This democratizes access by enabling biologists to directly encode hypotheses into simulations without requiring extensive programming expertise [12].
The rationale for this framework lies in its ability to accelerate the hypothesis–test–refine cycle. Instead of months of programming, researchers can now iterate through hypotheses in minutes, running in silico experiments that generate “virtual data” and refining rules accordingly [13]. Critically, these grammars incorporate stochastic elements, capturing biological noise and phenotypic heterogeneity in processes such as gene expression or clonal expansion [14]. This makes them particularly suitable for modeling emergent phenomena, including immune escape, epithelial–mesenchymal transition, and microbial community interactions. By bridging human-readable logic and machine-executable models, hypothesis grammars provide a foundational epistemological basis for predictive multiscale modeling.
2.2. Implementation Frameworks and Platforms
The operationalization of hypothesis grammars has been exemplified by open-source simulation environments, such as PhysiCell, a physics-based three-dimensional agent-based modeling (ABM) platform [15]. PhysiCell allows rules to be specified in spreadsheets or XML-based syntax, which are automatically translated into biochemical and biomechanical interactions at runtime. For example, a grammar rule such as "if oxygen < 5 mmHg, then increase VEGF secretion rate" is converted into diffusion equations and signaling rules within the simulation engine. This enables the large-scale modeling of tumors, immune infiltration, and therapy responses with millions of cells, without requiring direct coding [15,16].
Recent versions of PhysiCell have extended grammar-based modeling to include predefined templates for apoptosis, proliferation, chemotaxis, and phagocytosis [17]. Hybrid approaches now integrate machine learning into grammar systems, where experimental data guide rule optimization through adaptive fitting [9]. Cloud-based implementations further scale these frameworks, allowing real-time simulations of organoids or patient-derived xenograft models. Together, these platforms constitute a “code-free” modeling ecosystem that empowers both computational and experimental scientists to collaboratively develop multiscale simulations as observed in Table 1.
2.3. Applications and Case Studies
The versatility of hypothesis grammars has been demonstrated across oncology, immunology, and developmental biology. In cancer research, grammar-based ABMs have simulated tumor–immune interactions by encoding rules for antigen recognition, immune evasion, and cytokine-mediated feedback loops [23]. Models have successfully replicated clinical observations, such as resistance to checkpoint blockade, tumor-associated macrophage polarization, and the heterogeneous efficacy of T-cell infiltration [24]. In silico trials based on patient-derived multi-omics data have been used to predict therapeutic outcomes, highlighting how microenvironmental heterogeneity shapes immune dynamics [25].
Beyond oncology, grammars have modeled immune responses in sepsis, recapitulating the plasticity of macrophages and neutrophils under inflammatory signals [18]. Neurodevelopmental studies have applied grammars to simulate cortical layering, where rules governing progenitor division orientation and migration generated accurate reproductions of brain architecture [26]. These case studies illustrate how grammar-driven models not only reproduce known biology but also generate testable predictions about emergent behaviors, thereby functioning as virtual laboratories.
2.4. Toward Digital Cell Repositories
An important conceptual advance emerging from hypothesis grammar frameworks is the notion of the "digital cell," a modular, reusable computational unit that encodes cellular rules and behaviors [27]. Much like physical cell banks, digital cell lines can be validated, shared, and adapted across laboratories, creating standardized repositories that enhance reproducibility and accelerate model development. Initiatives are already underway to establish community-driven libraries of grammar-encoded models, analogous to open-source software repositories [28].
These repositories would enable researchers to access pre-validated digital macrophages, fibroblasts, or tumor cells and assemble them into larger simulations without having to recreate the basic rules from scratch. Standardization is expected to significantly reduce duplication of effort, enhance transparency, and promote cross-institutional collaboration. In the long term, digital repositories integrated with FAIR (Findable, Accessible, Interoperable, Reusable) data principles could form the backbone of predictive digital twins, linking reusable grammar modules with patient-specific data for clinical applications [29].
3. Digital Twins: Predicting Cellular Futures with Patient Genomics
3.1. Weather-like Cellular Forecasts
The concept of the digital twin has introduced a paradigm shift in biomedical modeling, reframing disease prediction as a forecasting problem analogous to meteorology. Just as weather prediction relies on initial atmospheric states and underlying physical laws, digital twins harness patient-specific molecular and cellular data as initial conditions, coupled with mechanistic or agent-based rules, to forecast disease trajectories [7]. In oncology, this analogy has been particularly powerful: high-dimensional tumor snapshots derived from bulk or single-cell genomics, transcriptomics, proteomics, and imaging can serve as the "atmospheric inputs," while hypothesis grammars or systems biology models provide the governing rules of tumor–immune dynamics [30]. Simulations initialized with such inputs can then project tumor growth, immune evasion, or therapeutic responses over time, producing forecasts that evolve dynamically as new patient data become available [3].
The weather analogy also highlights the probabilistic, ensemble-based nature of predictive modeling. Just as meteorologists run ensembles of climate models to capture uncertainties, digital twins often simulate multiple parallel trajectories using stochastic variants of patient data and interaction rules. This yields probabilistic forecasts of disease progression, identifying likely outcomes as well as low-probability but clinically significant events, such as rapid relapse or catastrophic treatment failure [31]. In practice, iterative updating is central to this methodology: as patients undergo biopsies, imaging scans, or liquid biopsy profiling, these new data refine the initial conditions of the twin, recalibrating forecasts in real time [32]. Such continuous calibration enables digital twins to function as “living” predictive systems, evolving alongside the patient and offering clinicians dynamic decision support.
The predictive power of weather-like cellular forecasts has been demonstrated in recent computational oncology studies. For example, agent-based models initialized with single-cell transcriptomics from glioblastoma samples successfully predicted clonal expansions and resistance trajectories under radiotherapy, thereby anticipating patient outcomes more accurately than bulk genomic predictors [33]. Similarly, simulations of metastatic melanoma incorporating spatial transcriptomics predicted the emergence of immune-excluded niches weeks before they became clinically detectable [34]. These examples underscore the transformative clinical potential of treating patient data not as static descriptors but as evolving states that feed into time-forward models. The ultimate goal is to embed these forecasts into precision medicine pipelines, allowing oncologists to anticipate cellular futures with the same granularity with which meteorologists anticipate weather events.
3.2. Digital Tumor Twins
Among the most compelling applications of digital twin technology is the construction of digital tumor twins—virtual representations of patient tumors that simulate their dynamics under therapeutic perturbations. Building a digital tumor twin involves three interdependent steps. First, comprehensive patient-specific data are collected, including genomic alterations, transcriptomic states, proteomic signatures, radiological imaging, histopathological assessments, and even wearable sensor data [35]. Second, multiscale modeling frameworks—often agent-based or hybrid ODE–PDE–ABM systems—translate these data into simulations of cellular behavior, tissue-level architecture, and microenvironmental constraints [16,31]. Finally, these twins are subjected to virtual perturbations, where candidate therapies, drug combinations, or radiation schedules are simulated in silico to identify optimal strategies for individual patients [36]. Table 2 mentions digital twin frameworks and their applications in developing digital tumor twins.
Unlike static biomarkers or risk scores, digital tumor twins are dynamic and adaptive. As treatment progresses, longitudinal data streams—including circulating tumor DNA (ctDNA), single-cell liquid biopsies, or follow-up imaging—are integrated into the twin, updating its state and modifying its predictions [21]. This bidirectional loop establishes a continuous partnership between the physical patient and their virtual counterpart, allowing oncologists to “query” the twin before making therapeutic adjustments. In pancreatic cancer, for instance, patient-derived transcriptomes have been used to initialize tumor twins, which were then updated with longitudinal ctDNA data to successfully predict chemotherapy response and relapse intervals [40]. In breast cancer, ModCell™, a mechanistic digital twin engine developed by ‘Alacris Theranostics’, has demonstrated clinical feasibility by simulating therapy responses across large patient cohorts, suggesting drug combinations that were later validated in ex vivo organoid assays [41]. A representative workflow of predictive cellular modeling using digital twins is illustrated in Figure 2, which outlines the sequential process from patient data intake through hypothesis-driven modeling, simulation, and iterative recalibration.”
The adaptability of digital tumor twins is not limited to therapy prediction but extends to prognostication and clinical trial design. In silico trials using tumor twins can test hundreds of therapeutic regimens across thousands of virtual patients, identifying likely responders and non-responders before real-world implementation [22]. This dramatically reduces costs, ethical concerns, and time burdens associated with early-phase clinical trials. Moreover, tumor twins can be shared across institutions as standardized computational assets, promoting reproducibility and cross-validation. The integration of FAIR (Findable, Accessible, Interoperable, Reusable) data principles ensures interoperability between platforms, further catalyzing community adoption [29]. Ultimately, digital tumor twins represent a cornerstone of predictive oncology, moving beyond population averages toward individualized, dynamically updated models that anticipate disease progression and therapeutic outcomes.
Figure 1.
Workflow of predictive cellular modeling using digital twins. Panel A shows patient-to-genomics intake, where multi-omics and imaging data are collected and processed into model-ready features. Panel B illustrates hypothesis grammar compilation into mechanistic/multiscale models. Panel C depicts digital twin simulations of tumor–immune interactions and forecasts under candidate regimens. Panel D highlights the update loop with virtual cohorts, clinical decision support, and recalibration with new biopsy or imaging data.
Figure 1.
Workflow of predictive cellular modeling using digital twins. Panel A shows patient-to-genomics intake, where multi-omics and imaging data are collected and processed into model-ready features. Panel B illustrates hypothesis grammar compilation into mechanistic/multiscale models. Panel C depicts digital twin simulations of tumor–immune interactions and forecasts under candidate regimens. Panel D highlights the update loop with virtual cohorts, clinical decision support, and recalibration with new biopsy or imaging data.
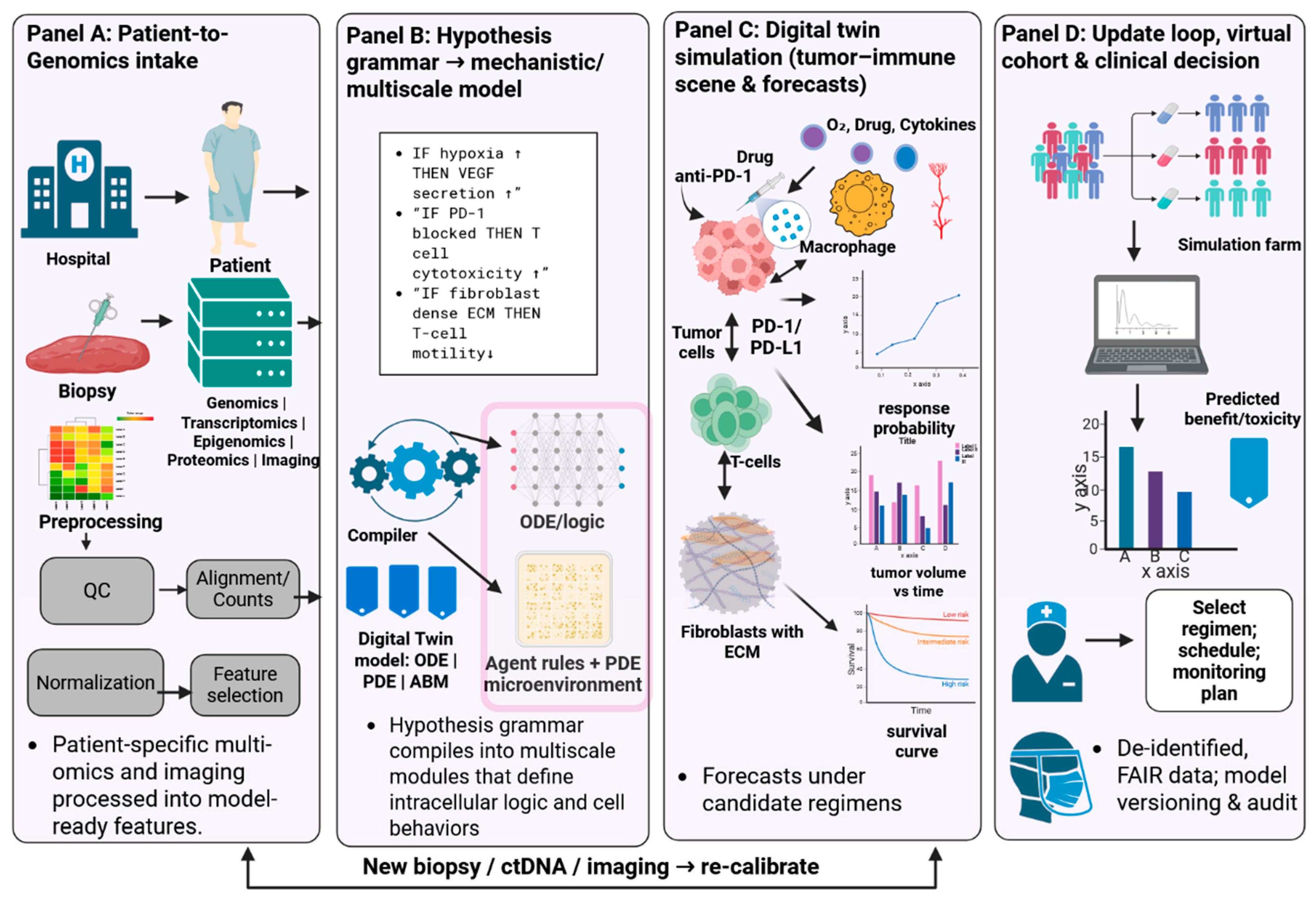
3.3. Integrative Modeling of Tumor–Immune Evolution
A key strength of digital twins lies in their ability to capture the co-evolutionary dynamics of tumors and their surrounding immune microenvironments, often referred to as the tumor–immune ecosystem or Tumor Immune Micro-Environment (TiME). Cancer progression is shaped not only by intrinsic tumor genetics but also by interactions with stromal cells, fibroblasts, vascular networks, and diverse immune populations [42]. Digital twins encode these interactions as rule-based grammars or mechanistic equations, informed by patient-derived omics and spatial data. For example, rules such as "hypoxic tumor cells secrete VEGF, inducing angiogenesis" or "MDSCs suppress cytotoxic T-cell activity via arginase production" are formalized into simulations that recapitulate known immunological phenomena [43].
Integrative twins have been used to explore immune escape mechanisms in melanoma, lung, and pancreatic cancers. Simulations incorporating spatial transcriptomics revealed how fibroblast-rich stroma physically excluded T cells, predicting non-responsiveness to checkpoint blockade therapies [44]. Similarly, digital twins of triple-negative breast cancer demonstrated that even minor variations in antigen presentation pathways could lead to qualitatively distinct immune trajectories, from full clearance to complete immune escape [45]. These models have also been extended to simulate therapeutic interventions by encoding checkpoint inhibition as a rule ("anti-PD-1 antibody blocks PD-L1 engagement, restoring T-cell cytotoxicity"). Twins forecasted heterogeneous responses across virtual patients, mirroring clinical trial outcomes [37].
Importantly, tumor–immune twin models are increasingly multi-omics, integrating genomic, epigenomic, transcriptomic, and metabolomic features to calibrate immune rules. Blood-derived cytokine profiles have been used to tune immune cell secretion rates, while TCR-seq data informed T-cell clonotype diversity within twins [46]. These integrative frameworks offer mechanistic insights into why some patients mount durable responses, while others relapse rapidly, informing the rational design of combination therapies. By functioning as virtual laboratories, digital twins enable researchers to test hypotheses about immune modulation at a scale and granularity that is impossible in vivo, guiding the development of next-generation immunotherapies.
4. Mechanistic and Multiscale Modeling in Systems Biology
4.1. Systems-Scale Modeling Foundations
Mechanistic systems biology seeks to explain how perturbations at molecular scales propagate upward to cellular phenotypes, tissue remodeling, and clinical outcomes by encoding causal structure (biochemistry, biophysics, and control) into quantitative models. Rather than treating tumors or immune responses as collections of independent parts, multiscale models assert that cross-talk between intracellular networks, cell mechanics, and microenvironmental transport generates emergent behaviors—e.g., phenotypic plasticity, therapy resistance, and spatial heterogeneity—that cannot be inferred from single-layer correlations [13]. A canonical workflow links data to mechanism through (i) model structure selection (e.g., signaling logic or kinetic graphs), (ii) parameterization constrained by multi-omics and imaging, (iii) uncertainty quantification and sensitivity analysis to identify governing processes, and (iv) prospective validation against independent perturbations [47]. Multiscale modeling approaches comprise several representative tools highlighting cellular growth and their spatial scales as mentioned in Table 3.
At intracellular scales, ordinary differential equation (ODE) and rule-based formalisms capture combinatorial signaling and post-translational control; at the cell–tissue interface, agent rules encode proliferation, migration, death, and adhesion; at tissue scales, reaction–diffusion partial differential equations (PDEs) describe nutrients, cytokines, and drugs; and at organ scales continuum mechanics captures growth-induced stress and perfusion limits [48]. “Bridging” operators propagate information across scales—for example, kinase activity computed from ODE signaling modules modulates an agent’s fate decision; secreted cytokines from many agents source diffusion fields; and evolving stress fields feedback on integrin signaling to alter motility. Critically, model identifiability and predictive value depend on data that span scales: phospho-proteomics and live-cell reporters for kinetics, single-cell omics for state distributions, multiplex/spatial imaging for neighborhood context, and longitudinal radiology or pathology for macroscopic constraints [49]. This integration, together with the explicit propagation of parametric and structural uncertainty, distinguishes mechanistic multiscale models from descriptive statistical predictors and underpins their use in hypothesis testing, the design of combination therapies, and the calibration of “digital twin” in the clinic [22]. Representative modeling paradigms are illustrated in Figure 3.
4.2. Rule-Based, Agent-Based, and Hybrid Multiscale Formalisms
Complex signaling exhibits a combinatorial explosion (multi-site phosphorylation, multi-valent complexes). Rule-based languages (BioNetGen, Kappa, PySB) address this by specifying interaction rules at the site/motif level rather than enumerating reactions, generating executable reaction networks on demand, and enabling rigorous simulation/analysis of high-dimensional pathway logic with far fewer equations [56,57,58]. Rule-based modules are frequently embedded within larger multiscale constructs to transmit intracellular decisions (e.g., commitment to apoptosis, entry into or exit from EMT) to cell-level behaviors. Boolean and stochastic logical models provide complementary coarse-grained alternatives when kinetic constants are uncertain, with tools such as ‘MaBoSS’ enabling probabilistic state-transition dynamics suitable for single-cell variability and rare-event exploration [15,59].
At the cellular and tissue scales, agent-based models (ABMs) explicitly represent individual cells with states, shapes, and local rules governing contact, chemotaxis, haptotaxis, secretion, and fate transitions. Open platforms—including PhysiCell, CompuCell3D, and Morpheus—simulate hundreds of thousands to millions of agents interacting via shared microenvironmental PDEs for oxygen, drugs, and cytokines [51,60,61]. ABMs excel when spatial context, clonal competition, and stochasticity dominate (e.g., immune infiltration/avoidance, niche construction, invasive fingering). For example, coupling tumor agents to macrophage and T-cell agents with checkpoint and metabolic rules reproduces immune-excluded versus inflamed phenotypes and clarifies how stromal architecture or acidification blocks effector penetration [62].
Hybrid models couple discrete agents to continuum PDEs and, increasingly, embed intracellular logic within each agent—yielding truly multiscale architectures. PhysiBoSS (PhysiCell + MaBoSS) assigns a cell-specific logical network that drives agent actions, so that extracellular cues (diffusing ligands, hypoxia) feed intracellular decisions (cell cycle arrest, apoptosis, cytokine secretion) which then modify the extracellular milieu [19]. Chaste and related frameworks support the tight integration of electrophysiology, mechanics, and growth laws with cell-based dynamics to model, for instance, ductal morphogenesis or solid-stress-limited perfusion in tumors [63]. Such hybrids enable mechanistic "what-if" experiments—e.g., the knock-in of a phosphatase site in a signaling rule, or stiffening of the extracellular matrix—and the observation of their amplified consequences for invasion fronts or drug penetration. With careful calibration and verification make these models testbeds for therapy design (dose, schedule, and spatial targeting) under realistic constraints of diffusion, binding, and clearance [15,19,51,59,60,62,63].
4.3. Machine Learning with Mechanistic Multiscale Models
Data-driven learning augments mechanistic modeling along three axes: (i) parameter inference and model selection, (ii) fast emulation/surrogates for expensive simulations, and (iii) physics-informed predictors that embed biological laws in the learning objective. First, hierarchical Bayesian calibration and global sensitivity analysis identify influential parameters and quantify posterior uncertainty; approximate Bayesian computation and likelihood-free inference can be paired with ABMs when analytic likelihoods are intractable [64]. Second, neural surrogates and Gaussian-process emulators learn mappings from parameters/initial conditions to summary outcomes (e.g., invasion speed, immune-cell density at the front), enabling rapid exploration, multi-objective optimization, and robust control under uncertainty; active-learning loops adaptively sample parameter space where the surrogate is uncertain to minimize simulation budgets [65]. Third, physics-informed neural networks (PINNs) and universal differential equations impose ODE/PDE constraints during training, ensuring that learned predictors respect conservation, transport, and reaction kinetics, thereby improving data efficiency and extrapolation beyond the training manifold [9,66].
These synergies directly serve multiscale oncology and immunology. For example, ABM-PDE tumor models calibrated with spatial transcriptomics and multiplex imaging via differentiable or emulator-based inference recover patient-specific chemotaxis, kill rates, and secretion fluxes, then propagate uncertainty to predict distributions over treatment outcomes (response vs. non-response) [34]. Reinforcement learning layered on mechanistic simulators identifies adaptive therapy schedules (dose holidays, drug switching) that exploit eco-evolutionary trade-offs, while constraint terms penalize clinically infeasible regimens (toxicity, cumulative dose) and maintain biological plausibility [67,68]. Hybrid “gray-box” architectures—in which a neural component approximates a poorly known sub-process (e.g., stromal remodeling) while the remainder is governed by equations—balance interpretability with flexibility, preserving causal levers for interventional design [9,34,66,67,69]. Finally, mechanistic simulators can generate large, labeled synthetic cohorts to pre-train ML models or to stress-test prognostic signatures under controlled distribution shifts (e.g., varying hypoxia or mutation burden), thereby improving robustness prior to deployment on scarce clinical datasets [34,65].
Together, these developments delineate a principled path from molecular mechanism to clinical prediction: rule-based or logical signaling determines cell-fate propensities; agents enact these propensities within mechanically and chemically coupled tissues; PDEs transport resources and therapies; and ML closes the loop by calibrating, accelerating, and constraining models with patient data. The result is a cohesive, testable, and extensible framework for studying disease mechanisms, prioritizing drug combinations, and powering patient-specific digital twins with quantified uncertainty and mechanistic interpretability [9,13,15,22,47,48,49,51,56,57,58,60,61,62,63,64,65,66,67,69].
4.4. Inference of Biological Networks & Regulatory Programs
The inference of biological networks and regulatory programs has become one of the most powerful approaches for understanding cellular decision-making and predicting phenotypic outcomes. Central to this effort is the reconstruction of gene regulatory networks (GRNs), which represent the interconnected transcription factors, target genes, and regulatory elements that define a cell's state and behavior. Advances in single-cell technologies, particularly single-cell RNA sequencing (scRNA-seq), have provided unprecedented resolution of cellular heterogeneity, revealing transcriptional variability that is masked in bulk assays [70]. However, the sparsity of scRNA-seq data—manifested as dropout events where expressed transcripts are not detected—and high levels of biological and technical noise make GRN inference a non-trivial computational challenge [71]. Addressing these issues has required the development of novel algorithmic strategies that incorporate temporal dynamics, probabilistic inference, and integration of prior biological knowledge.
Early approaches to GRN inference relied on co-expression patterns, correlation-based networks, and mutual information to connect genes with shared activity. While informative, these methods produced static, undirected graphs that lacked causal directionality and were prone to false positives in noisy datasets [72]. The advent of single-cell time-course data and pseudo-temporal ordering has provided a transformative advance, allowing researchers to infer causal relationships based on the dynamic progression. Algorithms such as SCODE, which uses matrix decomposition to infer transcription factor activity trajectories, and SINCERITIES, which incorporates pseudo-time and regression models, have begun to reveal the dynamic rewiring of regulatory architecture during processes like differentiation [73]. More recently, advanced frameworks such as ‘SimiC’ employ sparse regression with similarity constraints to jointly infer networks across multiple contiguous cell states, thereby identifying conserved and rewired regulatory edges that drive transitions between states [74]. Bayesian approaches, including dynamic Bayesian networks and variational autoencoders combined with Granger causality, have also demonstrated effectiveness in capturing directionality while explicitly modeling uncertainty [75].
The significance of network inference extends far beyond reconstructing topology. A static map of regulatory connections provides only limited biological insight; the true value lies in transforming networks into predictive engines of cellular dynamics. A useful analogy is that a static GRN is like a roadmap, whereas a dynamic GRN resembles a GPS navigation system with real-time traffic information. Dynamic networks not only identify regulatory hubs but also describe how the strength and direction of connections evolve over time, especially under perturbations [76]. For example, inference of dynamic GRNs from scRNA-seq and RNA velocity data has revealed bistable transcriptional switches in hematopoietic stem cells, highlighting molecular checkpoints that determine lineage commitment [77]. Similarly, network inference in T cells exposed to checkpoint blockade has identified rewired transcription factor modules underlying immune exhaustion, pinpointing novel intervention targets [78]. Such predictions are invaluable for therapeutic discovery, as they reveal vulnerabilities in regulatory architecture that may be exploited pharmacologically. Crucially, inferred GRNs can be embedded within larger mechanistic or multiscale models, functioning as the intracellular “engines” that dictate cell fate decisions in tissue simulations. For example, in agent-based tumor twins, each simulated cell may carry an individualized GRN that governs its proliferation, apoptosis, or cytokine secretion rules, thereby linking intracellular regulation to emergent tumor–immune dynamics [19]. Similarly, Boolean or Petri-net models of inferred GRNs can be used to simulate attractor landscapes of cell states, identifying stable phenotypes (e.g., effector versus exhausted T cells) and predicting how perturbations shift the distribution of states [79]. This capacity to couple inferred regulatory programs with higher-order tissue and organ-level models is a cornerstone of predictive cellular modeling, allowing the integration of molecular information into clinically actionable frameworks.
Beyond gene expression, modern inference methods are expanding to incorporate epigenomic, proteomic, and spatial transcriptomic data. Chromatin accessibility profiles from single-cell ATAC-seq provide direct information on regulatory potential, guiding the identification of active transcription factors and enhancer–promoter interactions [80]. Proteomic data, particularly from mass cytometry and imaging mass spectrometry, introduce post-transcriptional and signaling constraints into GRNs, while spatial transcriptomics provides the necessary context to infer regulatory differences between cells in distinct microenvironments [81]. These multi-modal approaches enhance the robustness and biological plausibility of inferred networks, offering a more complete view of regulatory control.
The field is now advancing toward community standards and benchmarking initiatives. Projects such as the DREAM challenges have established gold-standard datasets for network inference, enabling the systematic evaluation of algorithms across diverse biological contexts [82]. FAIR-compliant repositories are emerging for inferred networks, ensuring transparency, reproducibility, and reuse [29]. These developments mirror earlier progress in other areas of computational biology and are essential for translating GRN inference into reliable clinical tools.
The inference of biological networks and regulatory programs has matured from correlation-based maps into dynamic, predictive frameworks that capture causal regulation at single-cell resolution. The integration of temporal information, probabilistic modeling, and multi-omics data is driving unprecedented insights into how cells transition between states and respond to perturbations. Embedding these networks into mechanistic and multiscale simulations further amplifies their utility, enabling the translation of molecular regulation into predictions at the tissue and patient levels. As methods become standardized and repositories of validated regulatory networks expand, GRN inference will increasingly serve as a foundation for precision medicine, identifying critical regulatory nodes that guide therapy and illuminating the dynamic rules that govern cellular fates.
5. Predictive Genomics & Clinical Translation
5.1. Genomic Risk and Predictive Models
Predictive genomics aims to harness the information encoded within an individual’s genome to forecast disease susceptibility, treatment response, and long-term health trajectories. Central to this effort has been the development of polygenic risk scores (PRS), which aggregate the contributions of thousands to millions of single nucleotide polymorphisms (SNPs) to estimate genetic predisposition to complex diseases [29]. PRS have demonstrated predictive value for conditions such as coronary artery disease, breast cancer, and type 2 diabetes, but their discriminatory capacity remains modest, with area under the receiver operating characteristic curve (AUC) values typically in the 0.6–0.7 range [83]. While adding clinical covariates, such as age, sex, family history, and environmental exposures, can improve performance, the ceiling for predictive accuracy is constrained by the underlying heritability of the trait [84]. Furthermore, population bias remains a persistent challenge: the vast majority of PRS are derived from European cohorts, leading to reduced transferability and poorer accuracy in underrepresented populations, which exacerbates health disparities when implemented clinically [85].
These limitations underscore the gap between statistical correlation and actionable prediction. Traditional GWAS-based risk models can indicate that an individual is at elevated genetic risk, but they often cannot explain the mechanisms by which risk alleles exert their effects, nor do they directly suggest therapeutic strategies. To overcome these challenges, predictive genomics is shifting from population-level statistical risk estimation to individual-level mechanistic prediction. Dynamic genomic predictors now integrate multi-omics data—including transcriptomic, epigenomic, proteomic, and metabolomic profiles—alongside germline and somatic variation to provide a more context-dependent and causal representation of disease risk [86]. Such integrative approaches allow not only risk stratification but also forecasting of likely disease trajectories and intervention points. For example, models that incorporate somatic mutation burden and clonal heterogeneity can anticipate resistance mechanisms to targeted therapy, while those integrating epigenomic context can reveal regulatory vulnerabilities that are invisible to purely sequence-based predictors [54]. By transitioning from a "what is the probability of disease?" framework to a "how can we intervene to change the outcome?" paradigm, predictive genomics is aligning more closely with the goals of precision medicine.
5.2. Mechanistic Predictive Platforms and Clinical Translation
The emergence of mechanistic predictive platforms represents a major advance in translating genomic insights into actionable clinical decisions. Unlike PRS, which remains fundamentally correlational, mechanistic simulators embed patient-specific omics data into dynamic, executable models of cellular signaling and regulatory networks. A leading example is ModCell™, a clinically deployed platform that integrates exome sequencing and transcriptomics into a curated pan-cancer signaling network to generate “virtual tumors” [87]. Within this framework, genomic alterations are mapped to their mechanistic consequences—such as altered kinase activity or disrupted feedback loops—and then simulated under various therapeutic perturbations. Drugs are modeled as interventions on network nodes or edges, enabling predictions of how targeted therapies, immunotherapies, or combinations will alter cellular behavior [87]. Clinical reports from ModCell™ have been used to inform tumor board decisions, demonstrating that mechanistic models can provide interpretable, patient-specific guidance that complements empirical treatment selection [41].
Beyond oncology, Table 4 represents the predictive genomics platforms that are being deployed in cardiovascular diseases, neurology, and the treatment of rare genetic disorders. For example, integrative risk models for atrial fibrillation combine polygenic risk with electrophysiological simulations, enabling the prediction of arrhythmic events and the stratification of patients for preventive interventions [89]. In neurodegenerative diseases, patient-derived iPSC models have been linked to digital twins that simulate protein aggregation and neuronal network disruption, guiding early intervention strategies [90]. Hybrid platforms, such as ISPOT-K, which couple organoid-based drug testing with digital twin modeling, illustrate the convergence of experimental and computational approaches for personalized therapy design [91].
A critical consideration for clinical translation is the interpretability of results. Black-box deep learning models, although powerful, often lack transparency, creating barriers to physician adoption and regulatory approval. Rule-based or glass-box machine learning approaches—including decision trees, generalized rule lists, and causal graph-based predictors—provide interpretable outputs, identifying which mutations, expression patterns, or pathway alterations drive a prediction [92]. Such interpretability is vital in a clinical context, as observed in Table 5, where treatment decisions must be justified to patients and multidisciplinary teams. Moreover, interpretable models facilitate discovery: for example, rule-based classifiers applied to microbial genomics have revealed novel resistance determinants, a principle readily transferable to cancer or rare disease genomics [93]. An overview of predictive genomics, mechanistic platforms, and clinical translation is illustrated in Figure 3.
The path to routine implementation of predictive genomics requires overcoming technical, clinical, and ethical challenges. Data harmonization across sequencing platforms, integration of diverse omics modalities, and rigorous benchmarking of predictive models remain priorities. Clinically, prospective validation in large, diverse cohorts is essential to ensure reproducibility and generalizability. Ethically, issues of consent, privacy, and equitable access must be addressed to prevent the exacerbation of disparities in genomic medicine [94]. Despite these hurdles, predictive genomics is rapidly approaching a translational inflection point. By embedding genomic data into mechanistic frameworks and ensuring interpretability, predictive models are evolving from risk estimators into actionable clinical decision-support systems. This transition promises to accelerate the development of personalized therapies, optimize trial design through in silico cohorts, and ultimately improve patient outcomes across diverse diseases.
6. Emerging Technologies & Future Directions
6.1. Integrating LLMs with Hypothesis Grammars
A near-term frontier is the tight coupling of large language models (LLMs) with hypothesis grammars to accelerate knowledge extraction, model construction, and iterative refinement. Decades of work in machine reading for biology already demonstrate that textual claims can be lifted into structured, executable statements (e.g., INDRA assembling causal “subject– relation–object” mechanisms from papers and converting them into dynamical models) [55,95]. Modern LLMs extend this pipeline: they can parse heterogeneous literature, protocols, and database annotations, propose candidate rules (“IF hypoxia THEN ↑VEGF secretion; IF PD-1 blockade THEN ↑T-cell cytotoxic probability”), and draft grammars that mechanistic simulators can immediately execute. Crucially, LLM output should serve as a hypothesis generator rather than a ground truth; the grammar plus simulator acts as a "mechanistic truth filter," stress-testing LLM-proposed rules against known biophysics, unit constraints, and empirical priors. Weak or non-identifiable rules are pruned by sensitivity analysis and falsification loops. Retrieval-augmented generation (RAG) can further ground LLM proposals in citable sources, reducing hallucinations by forcing model induction from curated corpora (databases, guidelines, pathway resources) [96,97], while tool-use–enabled LLMs can call ontology resolvers, unit checkers, and SBML/CellML validators during rule synthesis [98]. In practice, the workflow is a closed loop: (i) LLM extracts or proposes rules with citations; (ii) the hypothesis grammar compiles and executes them; (iii) simulation diagnostics (fit, stability, constraint violations) feed back to the LLM to refine or discard rules; and (iv) curated rules are archived with full provenance. Because grammars operate in natural language and map to formal operators, they are uniquely positioned for LLM co-pilot integration—bridging unstructured biological text and rigorously testable models at scale [38,55]. As this ecosystem matures, expect automated assembly of intracellular modules (SBML), cell-behavior grammars for ABMs, and protocol-aware treatment nodes (dose, schedule, route), all logged with machine-readable provenance to support auditability and regulatory review [53,96]. hese emerging directions, including AI-assisted hypothesis grammars, standardized repositories, and digital clinical trials, are summarized in Figure 4.
6.2. Community Repositories, Standards, and Reproducibility
Sustained progress depends on shared, versioned repositories of “digital cell lines,” tissue microenvironments, and therapy modules that are findable, accessible, interoperable, and reusable (FAIR). Existing standards provide the scaffolding: SBML and CellML for biochemical and electrophysiological networks [53,96]; COMBINE archives for packaging models and data with simulation specifications; and public deposition in BioModels for curation and reuse [20]. At the multicellular level, MultiCellDS defines structured representations for phenotypes, microenvironmental context, and cell behaviors to support exchange across ABM platforms [99]. Embedding hypothesis grammars within these ecosystems enables a uniform representation of rules ("IF…THEN…WITH rate k") alongside parameter priors, units, and evidence links, and allows direct composition—e.g., swapping a macrophage digital line or drug-PK module without re-coding. To ensure credibility, repositories should mandate: (i) executable examples and unit tests; (ii) machine-actionable provenance (what text/evidence generated each rule); (iii) parameter priors with uncertainty;(iv) simulation manifests (solver, tolerances); and (v) persistent identifiers. Community benchmarking—à la DREAM challenges for network inference—should be extended to multiscale grammars and digital twins, with shared tasks (e.g., reproducing spatial T-cell exclusion patterns, predicting response to checkpoint blockade in held-out cohorts) and pre-registered scoring metrics [82]. Finally, FAIR data principles must be enforced across modalities (single-cell/spatial omics, imaging, longitudinal labs) to enable robust, cross-site calibration of digital twins and to avoid silent dataset shift that would erode predictive validity [100].
6.3. Benchmarking, Verification/Validation, and Regulatory Pathways
For clinical impact, models must be not only accurate but also credible under recognized verification, validation, and uncertainty quantification (VVUQ) frameworks. Global sensitivity analysis (e.g., Sobol/variance-based indices) should rank influential rules and parameters, guiding experimental design toward measurements that maximize the reduction of predictive uncertainty [101]. Bayesian calibration—ideally following Kennedy–O’Hagan’s treatment of model discrepancy—provides posterior parameter distributions and explicit residual structure, which can be propagated to generate prediction intervals for patient-level outcomes [102]. Credibility assessment should follow risk-based standards, such as ASME V&V 40, which aligns the rigor of verification/validation activities with the clinical decision risk class of the model's intended use (screening vs. dosing vs. life-critical control) [103]. On the regulatory front, model-informed drug development (MIDD) initiatives recognize the role of mechanistic models across preclinical and clinical phases, and agencies are increasingly providing pathways for leveraging in silico evidence in trial design, dose selection, and device/software evaluation [104]. In oncology, “in silico clinical trials” populated by digital twins can de-risk protocol choices and stratification strategies before expensive enrollment, provided that external validation on both retrospective and prospective cohorts is documented and that uncertainty bounds accompany any decision support [105]. Looking forward, converging technologies—LLM-assisted rule induction, standards-compliant repositories, and rigorous VVUQ—will enable continuously learning digital twins that update with new patient data, carry transparent mechanistic rationales, and produce auditable, uncertainty-aware recommendations. This synthesis will transform hypothesis grammars into living clinical assets: models that are buildable by many, reusable across contexts, testable against reality, and acceptable to regulators because their assumptions, evidence, and risks are explicit and systematically managed [100,101,102,103,104,105].
7. Challenges, Limitations, Ethical Considerations & Conclusions
7.1. Technical and Clinical Challenges
Despite the rapid progress in predictive cellular modeling, several technical bottlenecks constrain scalability and translation. A fundamental limitation is data granularity: single-cell and spatial omics technologies provide unprecedented resolution, yet they remain snapshots that fail to fully capture dynamic rates of signaling, transcription, or epigenetic modifications [105]. Integrating heterogeneous data modalities—genomics, transcriptomics, proteomics, metabolomics, imaging—into coherent models is non-trivial due to batch effects, variable coverage, and incompatible measurement scales [106]. Model calibration also faces the issue of non-identifiability, where multiple parameters set produce equally plausible fits, thereby undermining mechanistic interpretability and predictive robustness [54]. These technical, ethical, and clinical challenges raise several predictive cellular modeling concerns. However, there are proposed directions for resolving these challenges as mentioned in Table 6.
Computational demands remain substantial. Simulating millions of agents in 3D over clinically relevant time horizons requires high-performance computing, advanced solvers, and memory-efficient data structures [107]. Emerging hybrid architectures that integrate GPUs, distributed computing, and cloud-based infrastructures alleviate some constraints, but resource availability remains a limiting factor for widespread adoption. Additionally, hybrid models that combine mechanistic equations with machine learning often loses transparency as they grow in scope, creating “gray boxes” that are harder to interpret for clinicians [15,59].
From a clinical standpoint, the most pressing challenge is validation. Predictive models must demonstrate accuracy and reproducibility in prospective cohorts before influencing patient care [69]. Yet longitudinal datasets linking multi-omics, treatment regimens, and outcomes remain scarce, fragmented, and often locked behind institutional or commercial barriers. Without such data, model generalizability and reliability cannot be rigorously assessed. Moreover, regulatory frameworks for in silico diagnostics are still evolving, creating uncertainty in pathways for approval and adoption [6,114,115].
7.2. Ethical and Societal Considerations
Predictive genomics and digital twins raise pressing ethical questions that extend beyond traditional biomedical research. Algorithmic bias is a central concern: genomic datasets are heavily skewed toward individuals of European ancestry, resulting in risk scores and models that underperform in underrepresented populations [105]. Without corrective action, predictive modeling could exacerbate healthcare disparities by offering unequal accuracy across demographic groups. Ensuring representativeness in training datasets and applying fairness-aware machine learning approaches are urgent priorities [86].
Data privacy poses another challenge. Genomic and multi-omics data are inherently identifiable, making true anonymization impossible [112]. Sharing data for collaborative modeling thus risks exposing sensitive information not only about individuals but also about their relatives, who did not explicitly consent to such sharing. Advanced cryptographic methods (federated learning, homomorphic encryption, secure multiparty computation) are being explored to reconcile data sharing with privacy protection [111].
Consent frameworks must evolve as well. Patients contributing to digital twin models may not fully anticipate downstream uses of their data, such as model sharing, cross-institutional reuse, or application to secondary research questions. Transparent consent processes, ongoing communication, and patient engagement in governance structures are critical for maintaining trust [109]. Finally, accountability must be clarified: when a digital twin informs a treatment recommendation, responsibility remains with the clinician; however, transparency in how the model reached its output is vital to support decision-making and mitigate liability concerns [116].
8. Conclusions and Future Outlook
The convergence of hypothesis grammars, digital twins, multiscale mechanistic models, and predictive genomics heralds a transformative shift in biomedical research and clinical practice. These technologies collectively enable dynamic, causal, and interpretable simulations of cellular systems that can be continuously updated with patient-specific data. Already, platforms such as PhysiCell and ModCell™ demonstrate the feasibility of embedding omics-driven rules into clinically actionable simulations [15,107]. The long-term vision is to extend these frameworks into digital clinical trials, where thousands of virtual patients are simulated to optimize therapeutic strategies before enrollment in clinical trials, thereby reducing costs, risks, and time to approval [6,115].
Realizing this vision, however, requires addressing key challenges: ensuring computational scalability, standardizing data and model repositories, establishing regulatory credibility, mitigating bias, and safeguarding privacy. It also demands a culture of collaboration across computational scientists, clinicians, ethicists, and policymakers. By embedding transparency, fairness, and patient-centered values into model development, predictive cellular modeling can avoid the pitfalls of inequity and mistrust that have plagued earlier digital health innovations.
Predictive cellular modeling is poised to evolve from a research frontier into a cornerstone of precision medicine. Its trajectory will depend on balancing technical rigor with ethical responsibility, and innovation with inclusivity. If these challenges are met, the integration of hypothesis grammars, digital twins, and predictive genomics could redefine not only how we understand disease but also how we design therapies and deliver care—transforming in silico simulations from experimental tools into trusted clinical allies for generations to come [86,105,109,111,112,113,116].
9. Declaration
9.1. Funding
The author(s) acknowledge the funds received by Uddalak Das from the Department of Biotechnology (Grant No: DBTHRDPMU/JRF/BET-24/I/2024-25/376), the Council of Scientific and Industrial Research (Grant No: 24J/01/00130), and the Indian Council of Medical Research (Grant No: 3/1/3/BRET-2024/HRD (L1)).
9.2. Authorship Contribution Statement
S.T. Gopukumar: Writing - Review & Editing; Dyumn Dwivedi: Writing - Original Draft, Writing - Review & Editing; Tanveen Kaur Soni: Visualization; Praveen Ganesh Natarajan: Writing - Review & Editing, S. Ramkanth: Writing - Review & Editing; Arpita Mitra: Writing - Review & Editing; Uddalak Das: Conceptualization, Project Administration, Supervision, Funding Acquisition, Review & Editing. All authors critically revised the paper and approved the final version
9.3. Declaration of Competing Interest
The author(s) report no conflict of interest.
9.4. Acknowledgement
None
9.5. Ethical Statements
None
9.6. Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
The writing of this review paper involved the use of generative AI and AI-assisted technologies only to enhance the clarity, coherence, and overall quality of the manuscript. The authors acknowledge the contributions of AI in the writing process while ensuring that the final content reflects the author's own insights and interpretations of the literature. All interpretations and conclusions drawn in this manuscript are the sole responsibility of the author.
References
- Altschuler, S.J.; Wu, L.F. Cellular Heterogeneity: Do Differences Make a Difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef]
- Macklin, P.; Edgerton, M.E.; Thompson, A.M.; Cristini, V. Patient-calibrated agent-based modelling of ductal carcinoma in situ (DCIS): From microscopic measurements to macroscopic predictions of clinical progression. J. Theor. Biol. 2012, 301, 122–140. [Google Scholar] [CrossRef]
- Johnson, J.A.I.; Bergman, D.R.; Rocha, H.L.; Zhou, D.L.; Cramer, E.; Mclean, I.C.; Dance, Y.W.; Booth, M.; Nicholas, Z.; Lopez-Vidal, T.; et al. Human interpretable grammar encodes multicellular systems biology models to democratize virtual cell laboratories. Cell 2025, 188, 4711–4733. [Google Scholar] [CrossRef]
- Rahbar, S.; Shafiekhani, S.; Allahverdi, A.; Jamali, A.; Kheshtchin, N.; Ajami, M.; Mirsanei, Z.; Habibi, S.; Makkiabadi, B.; Hadjati, J.; et al. Agent-based Modeling of Tumor and Immune System Interactions in Combinational Therapy with Low-dose 5-fluorouracil and Dendritic Cell Vaccine in Melanoma B16F10. Iran. J. Allergy Asthma Immunol. 2022, 21, 151–166. [Google Scholar] [CrossRef]
- Stamatakos, G.; Kolokotroni, E.; Panagiotidou, F.; Tsampa, S.; Kyroudis, C.; Spohn, S.; Grosu, A.L.; Baltas, D.; Zamboglou, C.; Sachpazidis, I. In silico oncology: a mechanistic multiscale model of clinical prostate cancer response to external radiation therapy as the core of a digital (virtual) twin. Sensitivity analysis and a clinical adaptation approach. Front. Physiol. 2025, 16. [Google Scholar] [CrossRef]
- Corral-Acero, J.; Margara, F.; Marciniak, M.; Rodero, C.; Loncaric, F.; Feng, Y.; Gilbert, A.; Fernandes, J.F.; Bukhari, H.A.; Wajdan, A.; et al. The ‘Digital Twin’ to enable the vision of precision cardiology. Eur. Heart J. 2020, 41, 4556–4564. [Google Scholar] [CrossRef]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. eLife 2017, 6, e27041. [Google Scholar] [CrossRef]
- Rackauckas, C.; Ma, Y.; Martensen, J.; Warner, C.; Zubov, K.; Supekar, R.; Skinner, D.; Ramadhan, A.; Edelman, A. Universal Differential Equations for Scientific Machine Learning. arXiv 2020, arXiv:2001.04385. [Google Scholar]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Topol, E.J. High-performance medicine: the convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Glen, C.M.; Kemp, M.L.; Voit, E.O. Agent-based modeling of morphogenetic systems: Advantages and challenges. PLOS Comput. Biol. 2019, 15, e1006577. [Google Scholar] [CrossRef]
- Wolkenhauer, O.; Auffray, C.; Jaster, R.; Steinhoff, G.; Dammann, O. The road from systems biology to systems medicine. Pediatr. Res. 2013, 73, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Wolkenhauer, O. Stochastic approaches in systems biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 385–397. [Google Scholar] [CrossRef]
- Ghaffarizadeh, A.; Heiland, R.; Friedman, S.H.; Mumenthaler, S.M.; Macklin, P. PhysiCell: An open source physics-based cell simulator for 3-D multicellular systems. PLoS Comput. Biol. 2018, 14, e1005991. [Google Scholar] [CrossRef]
- Alamoudi, E.; Schälte, Y.; Müller, R.; Starruß, J.; Bundgaard, N.; Graw, F.; Brusch, L.; Hasenauer, J. FitMultiCell: simulating and parameterizing computational models of multi-scale and multi-cellular processes. Bioinformatics 2023, 39, btad674. [Google Scholar] [CrossRef] [PubMed]
- Macklin, P.; Frieboes, H.B.; Sparks, J.L.; Ghaffarizadeh, A.; Friedman, S.H.; Juarez, E.F.; Jonckheere, E.; Mumenthaler, S.M. Progress Towards Computational 3-D Multicellular Systems Biology. Adv. Exp. Med. Biol. 2016, 936, 225–246. [Google Scholar]
- Vodovotz, Y.; An, G. Systems Biology and Inflammation. In: Yan Q, editor. Systems Biology in Drug Discovery and Development [Internet]. Totowa, NJ: Humana Press; 2010 [cited 2025 Sept 4]. p. 181–201. Available from: http://link.springer.com/10.1007/978-1-60761-800-3_9.
- Letort, G.; Montagud, A.; Stoll, G.; Heiland, R.; Barillot, E.; Macklin, P.; Zinovyev, A.; Calzone, L. PhysiBoSS: a multi-scale agent-based modelling framework integrating physical dimension and cell signalling. Bioinformatics 2019, 35, 1188–1196. [Google Scholar] [CrossRef]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; et al. BioModels—15 years of sharing computational models in life science. Nucleic Acids Res. 2019, 48, D407–D415. [Google Scholar] [CrossRef]
- Cristini, V.; Lowengrub, J. Multiscale Modeling of Cancer: An Integrated Experimental and Mathematical Modeling Approach; Cambridge University Press: Cambridge, UK, 2010; p. 278. [Google Scholar]
- Viceconti, M.; Hunter, P. The Virtual Physiological Human: Ten Years After. Annu. Rev. Biomed. Eng. 2016, 18, 103–123. [Google Scholar] [CrossRef]
- Van Liedekerke, P.; Palm, M.M.; Jagiella, N.; Drasdo, D. Simulating tissue mechanics with agent-based models: concepts, perspectives and some novel results. Comput. Part. Mech. 2015, 2, 401–444. [Google Scholar] [CrossRef]
- Cess, C.G.; Finley, S.D. Multi-scale modeling of macrophage—T cell interactions within the tumor microenvironment. PLoS Comput. Biol. 2020, 16, e1008519. [Google Scholar] [CrossRef]
- An, G.; Mi, Q.; Dutta-Moscato, J.; Vodovotz, Y. Agent-based models in translational systems biology. WIREs Mech. Dis. 2009, 1, 159–171. [Google Scholar] [CrossRef]
- Stein-O’Brien, G.L.; Clark, B.S.; Sherman, T.; Zibetti, C.; Hu, Q.; Sealfon, R.; Liu, S.; Qian, J.; Colantuoni, C.; Blackshaw, S.; et al. Decomposing Cell Identity for Transfer Learning across Cellular Measurements, Platforms, Tissues, and Species. Cell Syst. 2019, 8, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Karlsson, J.; Ferro, M.; Mañas, A.; Kanzaki, R.; Fredlund, E.; Murphy, A.J.; Morton, C.L.; Andersson, N.; Woolard, M.A.; et al. Evolutionary unpredictability in cancer model systems. Sci. Rep. 2025, 15, 20334. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, K.; Kananathan, S.; Roberts, M.G.; Meyer, J.P.; Sharif Shohan, M.U.; Xavier, A.; Maire, M.; Zyoud, A.; Men, J.; Ng, S.; et al. Reproducibility in systems biology modelling. Mol. Syst. Biol. 2021, 17, e9982. [Google Scholar] [CrossRef] [PubMed]
- Sansone, S.A.; McQuilton, P.; Rocca-Serra, P.; Gonzalez-Beltran, A.; Izzo, M.; Lister, A.L.; Thurston, M. FAIRsharing as a community approach to standards, repositories and policies. Nat. Biotechnol. 2019, 37, 358–367. [Google Scholar] [CrossRef]
- Stamatakos, G. The Oncosimulator - Combining Clinically Driven and Clinically Oriented Multiscale Cancer Modeling with Information Technology in the In Silico Oncology Context. Research Journal of Clinical Pediatrics [Internet]. 2021 Oct 1 [cited 2025 Sept 14];2021. Available from: https://www.scitechnol.com/abstract/the-oncosimulator-combining-clinically-driven-and-clinically-oriented-multiscale-cancer-modeling-with-information-technology-in-th-17191.html.
- Mollica, L.; Leli, C.; Sottotetti, F.; Quaglini, S.; Locati, L.D.; Marceglia, S. Digital twins: a new paradigm in oncology in the era of big data. ESMO Real World Data Digit. Oncol. 2024, 5, 100056. [Google Scholar] [CrossRef]
- Björnsson, B.; Borrebaeck, C.; Elander, N.; Gasslander, T.; Gawel, D.R.; Gustafsson, M.; Jörnsten, R.; Lee, E.J.; Li, X.; Lilja, S.; et al. Digital twins to personalize medicine. Genome Med. 2020, 12, 4. [Google Scholar] [CrossRef]
- Jenner, A.L.; Smalley, M.; Goldman, D.; Goins, W.F.; Cobbs, C.S.; Puchalski, R.B.; Chiocca, E.A.; Lawler, S.; Macklin, P.; Goldman, A.; et al. Agent-based computational modeling of glioblastoma predicts that stromal density is central to oncolytic virus efficacy. iScience 2022, 25, 104395. [Google Scholar] [CrossRef]
- Love, N.R.; Williams, C.; Killingbeck, E.E.; Merleev, A.; Saffari Doost, M.; Yu, L.; McPherson, J.D.; Mori, H.; Borowsky, A.D.; Maverakis, E.; et al. Melanoma progression and prognostic models drawn from single-cell, spatial maps of benign and malignant tumors. Sci. Adv. 2024, 10, eadm8206. [Google Scholar] [CrossRef]
- Stamatakos, G.S.; Perez, M.A.; Radhakrishnan, R. Editorial: Multiscale cancer modeling, in silico oncology and digital (virtual) twins in the cancer domain. Front. Physiol. 2025, 16, 1614235. [Google Scholar] [CrossRef]
- Alarcón, T.; Byrne, H.M.; Maini, P.K. A mathematical model of the effects of hypoxia on the cell-cycle of normal and cancer cells. J. Theor. Biol. 2004, 229, 395–411. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models are Few-Shot Learners. arXiv 2020, arXiv:2005.14165. [Google Scholar] [CrossRef]
- Pleyer, J.; Fleck, C. Agent-based models in cellular systems. Front. Phys. 2023, 10, 968409. [Google Scholar] [CrossRef]
- Altrock, P.M.; Liu, L.L.; Michor, F. The mathematics of cancer: integrating quantitative models. Nat. Rev. Cancer 2015, 15, 730–745. [Google Scholar] [CrossRef]
- ModCell [Internet]. ALACRIS Theranostics. 2024 [cited 2025 Sept 14]. Available from: https://www.alacris.de/modcell/.
- Fridman, W.H.; Zitvogel, L.; Sautès–Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Bagaev, A.; Kotlov, N.; Nomie, K.; Svekolkin, V.; Gafurov, A.; Isaeva, O.; Osokin, N.; Kozlov, I.; Frenkel, F.; Gancharova, O.; et al. Conserved pan-cancer microenvironment subtypes predict response to immunotherapy. Cancer Cell 2021, 39, 845–865. [Google Scholar] [CrossRef]
- Li, B.; Li, T.; Pignon, J.C.; Wang, B.; Wang, J.; Shukla, S.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 2016, 48, 725–732. [Google Scholar] [CrossRef]
- Raue, A.; Schilling, M.; Bachmann, J.; Matteson, A.; Schelke, M.; Kaschek, D.; Hug, S.; Kreutz, C.; Harms, B.D.; Theis, F.J.; et al. Lessons Learned from Quantitative Dynamical Modeling in Systems Biology. PLoS ONE 2013, 8, e74335. [Google Scholar] [CrossRef]
- Deisboeck, T.S.; Wang, Z.; Macklin, P.; Cristini, V. Multiscale cancer modeling. Annu. Rev. Biomed. Eng. 2011, 13, 127–155. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Norton, K.A.; Gong, C.; Jamalian, S.; Popel, A.S. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes 2019, 7, 37. [Google Scholar] [CrossRef]
- Swat, M.H.; Thomas, G.L.; Belmonte, J.M.; Shirinifard, A.; Hmeljak, D.; Glazier, J.A. Multi-scale modeling of tissues using CompuCell3D. Methods Cell Biol. 2012, 110, 325–366. [Google Scholar]
- Stoll, G.; Caron, B.; Viara, E.; Dugourd, A.; Zinovyev, A.; Naldi, A.; Kroemer, G.; Barillot, E.; Calzone, L. MaBoSS 2.0: an environment for stochastic Boolean modeling. Bioinformatics 2017, 33, 2226–2228. [Google Scholar] [CrossRef]
- Hucka, M.; Finney, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Gyori, B.M.; Bachman, J.A.; Subramanian, K.; Muhlich, J.L.; Galescu, L.; Sorger, P.K. From word models to executable models of signaling networks using automated assembly. Mol. Syst. Biol. 2017, 13, 954. [Google Scholar] [CrossRef]
- Rule-Based Modelling of Cellular Signalling. In: Caires L, Vasconcelos VT, editors. CONCUR 2007 – Concurrency Theory [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 2007 [cited 2025 Sept 4]. p. 17–41. Available from: http://link.springer.com/10.1007/978-3-540-74407-8_3.
- Harris, L.A.; Hogg, J.S.; Tapia, J.J.; Sekar, J.A.P.; Gupta, S.; Korsunsky, I.; Arora, A.; Barua, D.; Sheehan, R.P.; Faeder, J.R. BioNetGen 2.2: advances in rule-based modeling. Bioinformatics 2016, 32, 3366–3368. [Google Scholar] [CrossRef]
- Lopez, C.F.; Muhlich, J.L.; Bachman, J.A.; Sorger, P.K. Programming biological models in Python using PySB. Mol. Syst. Biol. 2013, 9, 646. [Google Scholar] [CrossRef]
- Heiland, R.; Bergman, D.; Lyons, B.; Waldow, G.; Cass, J.; Rocha, H.L.; Ruscone, M.; Noël, V.; Macklin, P. PhysiCell Studio: a graphical tool to make agent-based modeling more accessible. Gigabyte 2024, 2024, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Starruß, J.; de Back, W.; Brusch, L.; Deutsch, A. Morpheus: a user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Viara, E.; Barillot, E.; Calzone, L. Continuous time boolean modeling for biological signaling: application of Gillespie algorithm. BMC Syst. Biol. 2012, 6, 116. [Google Scholar] [CrossRef] [PubMed]
- Norton, K.A.; Gong, C.; Jamalian, S.; Popel, A.S. Multiscale Agent-Based and Hybrid Modeling of the Tumor Immune Microenvironment. Processes 2019, 7, 37. [Google Scholar] [CrossRef]
- Mirams, G.R.; Arthurs, C.J.; Bernabeu, M.O.; Bordas, R.; Cooper, J.; Corrias, A.; Davit, Y.; Dunn, S.J.; Fletcher, A.G.; Harvey, D.G.; et al. Chaste: An Open Source C++ Library for Computational Physiology and Biology. PLoS Comput. Biol. 2013, 9, e1002970. [Google Scholar] [CrossRef]
- Toni, T.; Welch, D.; Strelkowa, N.; Ipsen, A.; Stumpf, M.P.H. Approximate Bayesian computation scheme for parameter inference and model selection in dynamical systems. J. R. Soc. Interface 2009, 6, 187–202. [Google Scholar] [CrossRef]
- Razavi, S.; Tolson, B.A.; Burn, D.H. Review of surrogate modeling in water resources. Water Resour. Res. 2012, 48, W07401. [Google Scholar] [CrossRef]
- Raissi, M.; Perdikaris, P.; Karniadakis, G.E. Physics-informed neural networks: A deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J. Comput. Phys. 2019, 378, 686–707. [Google Scholar] [CrossRef]
- Ebrahimi, S.; Lim, G.J. A reinforcement learning approach for finding optimal policy of adaptive radiation therapy considering uncertain tumor biological response. Artif. Intell. Med. 2021, 121, 102193. [Google Scholar] [CrossRef]
- Yang, C.Y.; Shiranthika, C.; Wang, C.Y.; Chen, K.W.; Sumathipala, S. Reinforcement learning strategies in cancer chemotherapy treatments: A review. Comput. Methods Programs Biomed. 2023, 229, 107280. [Google Scholar] [CrossRef]
- Willard, J.; Jia, X.; Xu, S.; Steinbach, M.; Kumar, V. Integrating Physics-Based Modeling with Machine Learning: A Survey. arXiv 2020, arXiv:2003.04919. [Google Scholar]
- Chen, S.; Mar, J.C. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformatics 2018, 19, 232. [Google Scholar] [CrossRef]
- Andrews, T.S.; Hemberg, M. Identifying cell populations with scRNASeq. Mol. Aspects Med. 2018, 59, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context. BMC Bioinformatics 2006, 7, S7. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Kiryu, H.; Furusawa, C.; Ko, M.S.H.; Ko, S.B.H.; Gouda, N.; Hayashi, T.; Nikaido, I. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33, 2314–2321. [Google Scholar] [CrossRef]
- Peng, J.; Serrano, G.; Traniello, I.M.; Calleja-Cervantes, M.E.; Chembazhi, U.V.; Bangru, S.; Ezponda, T.; Rodriguez-Madoz, J.R.; Kalsotra, A.; Prosper, F.; et al. SimiC enables the inference of complex gene regulatory dynamics across cell phenotypes. Commun. Biol. 2022, 5, 351. [Google Scholar] [CrossRef]
- Lopez, R.; Regier, J.; Cole, M.B.; Jordan, M.I.; Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 2018, 15, 1053–1058. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring Regulatory Networks from Expression Data Using Tree-Based Methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Weinreb, C.; Wolock, S.; Klein, A.M. SPRING: a kinetic interface for visualizing high dimensional single-cell expression data. Bioinformatics 2018, 34, 1246–1248. [Google Scholar] [CrossRef]
- Singer, M.; Wang, C.; Cong, L.; Marjanovic, N.D.; Kowalczyk, M.S.; Zhang, H.; Nyman, J.; Sakuishi, K.; Kurtulus, S.; Gennert, D.; et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 2016, 166, 1500–1511. [Google Scholar] [CrossRef]
- Saez-Rodriguez, J.; Simeoni, L.; Lindquist, J.A.; Hemenway, R.; Bommhardt, U.; Arndt, B.; Haus, U.U.; Weismantel, R.; Gilles, E.D.; Klamt, S.; et al. A Logical Model Provides Insights into T Cell Receptor Signaling. PLoS Comput. Biol. 2007, 3, e163. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Hill, A.J.; Aghamirzaie, D.; Daza, R.M.; Pliner, H.A.; Berletch, J.B.; Filippova, G.N.; Huang, X.; Christiansen, L.; DeWitt, W.S.; et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 2018, 174, 1309–1324. [Google Scholar] [CrossRef]
- Eng, C.H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef]
- Marbach, D.; Costello, J.C.; Küffner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Kellis, M.; Collins, J.J.; Stolovitzky, G. Wisdom of crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.M.; Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020, 12, 44. [Google Scholar] [CrossRef] [PubMed]
- Inouye, M.; Abraham, G.; Nelson, C.P.; Wood, A.M.; Sweeting, M.J.; Dudbridge, F.; Lai, F.Y.; Kaptoge, S.; Brozynska, M.; Wang, T.; et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults. J. Am. Coll. Cardiol. 2018, 72, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Shi, J.; García-Closas, M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat. Rev. Genet. 2016, 17, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, J.; Schönhuth, A. Genetic features for drug responses in cancer — Investigating an ensemble-feature-selection approach. Comput. Biol. Med. 2025, 196, 110572. [Google Scholar] [CrossRef]
- Willard, J.; Jia, X.; Xu, S.; Steinbach, M.; Kumar, V. Integrating Scientific Knowledge with Machine Learning for Engineering and Environmental Systems. arXiv 2020, arXiv:2003.04919. [Google Scholar] [CrossRef]
- Lorenzo, G.; Ahmed, S.R.; Hormuth, D.A.; Vaughn, B.; Kalpathy-Cramer, J.; Solorio, L.; Yankeelov, T.E.; Gomez, H. Patient-Specific, Mechanistic Models of Tumor Growth Incorporating Artificial Intelligence and Big Data. Annu. Rev. Biomed. Eng. 2024, 26, 529–560. [Google Scholar] [CrossRef]
- Pellman, J.; Sheikh, F. Atrial Fibrillation: Mechanisms, Therapeutics, and Future Directions. Compr. Physiol. 2015, 5, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Bassett, A.R.; Wood, M.J.A. Precision Modulation of Neurodegenerative Disease-Related Gene Expression in Human iPSC-Derived Neurons. Sci. Rep. 2016, 6, 28420. [Google Scholar] [CrossRef] [PubMed]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell. 2019, 1, 206–215. [Google Scholar] [CrossRef]
- Kim, J.I.; Maguire, F.; Tsang, K.K.; Gouliouris, T.; Peacock, S.J.; McAllister, T.A.; McArthur, A.G.; Beiko, R.G. Machine Learning for Antimicrobial Resistance Prediction: Current Practice, Limitations, and Clinical Perspective. Clin. Microbiol. Rev. 2022, 35, e00179–21. [Google Scholar] [CrossRef]
- Thorogood, A.; Dalpé, G.; Knoppers, B.M. Return of individual genomic research results: are laws and policies keeping step? Eur. J. Hum. Genet. 2019, 27, 535–546. [Google Scholar] [CrossRef]
- Cuellar, A.A.; Lloyd, C.M.; Nielsen, P.F.; Bullivant, D.P.; Nickerson, D.P.; Hunter, P.J. An Overview of CellML 1.1, a Biological Model Description Language. SIMULATION 2003, 79, 740–747. [Google Scholar] [CrossRef]
- Lewis, P.; Perez, E.; Piktus, A.; Petroni, F.; Karpukhin, V.; Goyal, N.; Küttler, H.; Lewis, M.; Yih, W.; Rocktäschel, T.; et al. Retrieval-Augmented Generation for Knowledge-Intensive NLP Tasks. arXiv 2020, arXiv:2005.11401. [Google Scholar]
- Schick, T.; Dwivedi-Yu, J.; Dessì, R.; Raileanu, R.; Lomeli, M.; Zettlemoyer, L.; Cancedda, N.; Scialom, T. Toolformer: Language Models Can Teach Themselves to Use Tools. arXiv 2023, arXiv:2302.04761. [Google Scholar] [CrossRef]
- Friedman, S.H.; Anderson, A.R.A.; Bortz, D.M.; Fletcher, A.G.; Frieboes, H.B.; Ghaffarizadeh, A.; Grimes, D.R.; Hawkins-Daarud, A.; Hoehme, S.; Juarez, E.F.; et al. MultiCellDS: a standard and a community for sharing multicellular data. bioRxiv 2016, 090696. [Google Scholar]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.j.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; Da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 160018. [Google Scholar] [CrossRef] [PubMed]
- Saltelli, A.; Ratto, M.; Andres, T.; Campolongo, F.; Cariboni, J.; Gatelli, D.; Saisana, M.; Tarantola, S. Global Sensitivity Analysis. The Primer [Internet]. 1st ed. Wiley; 2007 [cited 2025 Sept 4]. Available from: https://onlinelibrary.wiley.com/doi/book/10.1002/9780470725184.
- Kennedy, M.C.; O’Hagan, A. Bayesian calibration of computer models. J. R. Stat. Soc. Ser. B Stat. Methodol. 2001, 63, 425–464. [Google Scholar] [CrossRef]
- American Society of Mechanical Engineers. Assessing Credibility of Computational Modeling Through Verification and Validation: Application to Medical Devices: ASME V&V 40 - 2018; ASME: New York, NY, USA, 2018; p. 47. [Google Scholar]
- Galluppi, G.R.; Ahamadi, M.; Bhattacharya, S.; Budha, N.; Gheyas, F.; Li, C.C.; Chen, Y.; Dosne, A.G.; Kristensen, N.R.; Magee, M.; et al. Considerations for Industry—Preparing for the FDA Model-Informed Drug Development (MIDD) Paired Meeting Program. Clin. Pharmacol. Ther. 2024, 116, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Viceconti, M.; Pappalardo, F.; Rodriguez, B.; Horner, M.; Bischoff, J.; Musuamba Tshinanu, F. In silico trials: Verification, validation and uncertainty quantification of predictive models used in the regulatory evaluation of biomedical products. Methods 2021, 185, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Raue, A.; Kreutz, C.; Maiwald, T.; Klingmüller, U.; Timmer, J. Addressing parameter identifiability by model-based experimentation. IET Syst. Biol. 2011, 5, 120–130. [Google Scholar] [CrossRef]
- Gutenkunst, R.N.; Waterfall, J.J.; Casey, F.P.; Brown, K.S.; Myers, C.R.; Sethna, J.P. Universally Sloppy Parameter Sensitivities in Systems Biology Models. PLoS Comput. Biol. 2007, 3, e189. [Google Scholar] [CrossRef]
- Kammoun, A.; Slama, R.; Tabia, H.; Ouni, T.; Abid, M. Generative Adversarial Networks for face generation: A survey. ACM Comput. Surv. 2022, 55, 1–37. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, Y.; Chen, T.; Tong, Y. Federated Machine Learning: Concept and Applications. ACM Trans. Intell. Syst. Technol. 2019, 10, 1–19. [Google Scholar] [CrossRef]
- Stamatakos, G.S.; Dionysiou, D.D.; Graf, N.M.; Sofra, N.A.; Desmedt, C.; Hoppe, A.; Uzunoglu, N.K.; Tsiknakis, M. The “Oncosimulator”: a multilevel, clinically oriented simulation system of tumor growth and organism response to therapeutic schemes. In Towards the clinical evaluation of in silico oncology. In Proceedings of the 2007 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Lyon, France, 22–26 August 2007; pp. 6629–6632. [Google Scholar]
- Gymrek, M.; McGuire, A.L.; Golan, D.; Halperin, E.; Erlich, Y. Identifying Personal Genomes by Surname Inference. Science 2013, 339, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef]
- Morley, J.; Machado, C.C.V.; Burr, C.; Cowls, J.; Joshi, I.; Taddeo, M.; Floridi, L. The ethics of AI in health care: A mapping review. Soc. Sci. Med. 2020, 260, 113172. [Google Scholar] [CrossRef]
- Bekisz, S.; Geris, L. Cancer modeling: From mechanistic to data-driven approaches, and from fundamental insights to clinical applications. J. Comput. Sci. 2020, 46, 101198. [Google Scholar] [CrossRef]
- Brown, L.V.; Gaffney, E.A.; Wagg, J.; Coles, M.C. Applications of mechanistic modelling to clinical and experimental immunology: an emerging technology to accelerate immunotherapeutic discovery and development. Clin. Exp. Immunol. 2018, 193, 284–292. [Google Scholar] [CrossRef]
- Hunter, A.; Lewis, C.; Hill, M.; Chitty, L.S.; Leeson-Beevers, K.; McInnes-Dean, H.; Harvey, K.; Pichini, A.; Ormondroyd, E.; Thomson, K. Public and patient involvement in research to support genome services development in the UK. J. Transl. Genet. Genom. 2023, 7, 17–26. [Google Scholar] [CrossRef]
Figure 2.
Mechanistic and multiscale modeling approaches in systems biology. Intracellular mechanistic models use ordinary differential equations (ODEs), rule-based, or stochastic kinetics to represent molecular interactions. Cellular/agent-based models capture heterogeneity, stochasticity, and spatial interactions via agent rules. Hybrid/multiscale frameworks integrate ODE/PDE kinetics with agent-based models (ABMs), linking intracellular mechanisms to tissue-level and organ-scale outcomes.
Figure 2.
Mechanistic and multiscale modeling approaches in systems biology. Intracellular mechanistic models use ordinary differential equations (ODEs), rule-based, or stochastic kinetics to represent molecular interactions. Cellular/agent-based models capture heterogeneity, stochasticity, and spatial interactions via agent rules. Hybrid/multiscale frameworks integrate ODE/PDE kinetics with agent-based models (ABMs), linking intracellular mechanisms to tissue-level and organ-scale outcomes.
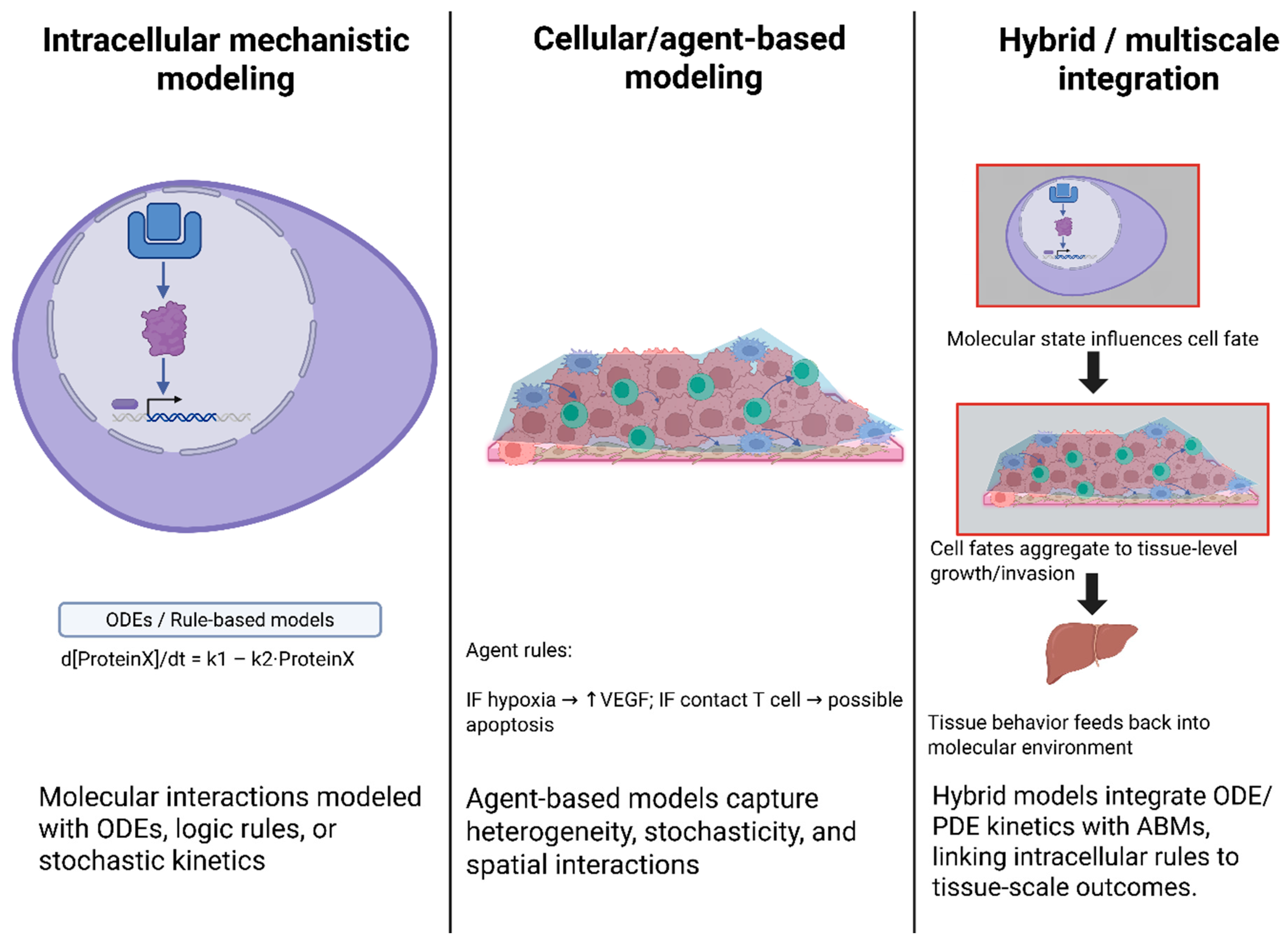
Figure 3.
Predictive genomics and clinical translation workflow. Genomic risk models (e.g., polygenic risk scores) estimate disease susceptibility but have limited accuracy and ancestry bias. Mechanistic predictive platforms (e.g., ModCell™) embed patient omics into signaling networks to simulate treatment responses in silico. Clinical translation integrates predictive genomics and digital twins into therapy selection, adaptive management, and virtual clinical trials.
Figure 3.
Predictive genomics and clinical translation workflow. Genomic risk models (e.g., polygenic risk scores) estimate disease susceptibility but have limited accuracy and ancestry bias. Mechanistic predictive platforms (e.g., ModCell™) embed patient omics into signaling networks to simulate treatment responses in silico. Clinical translation integrates predictive genomics and digital twins into therapy selection, adaptive management, and virtual clinical trials.
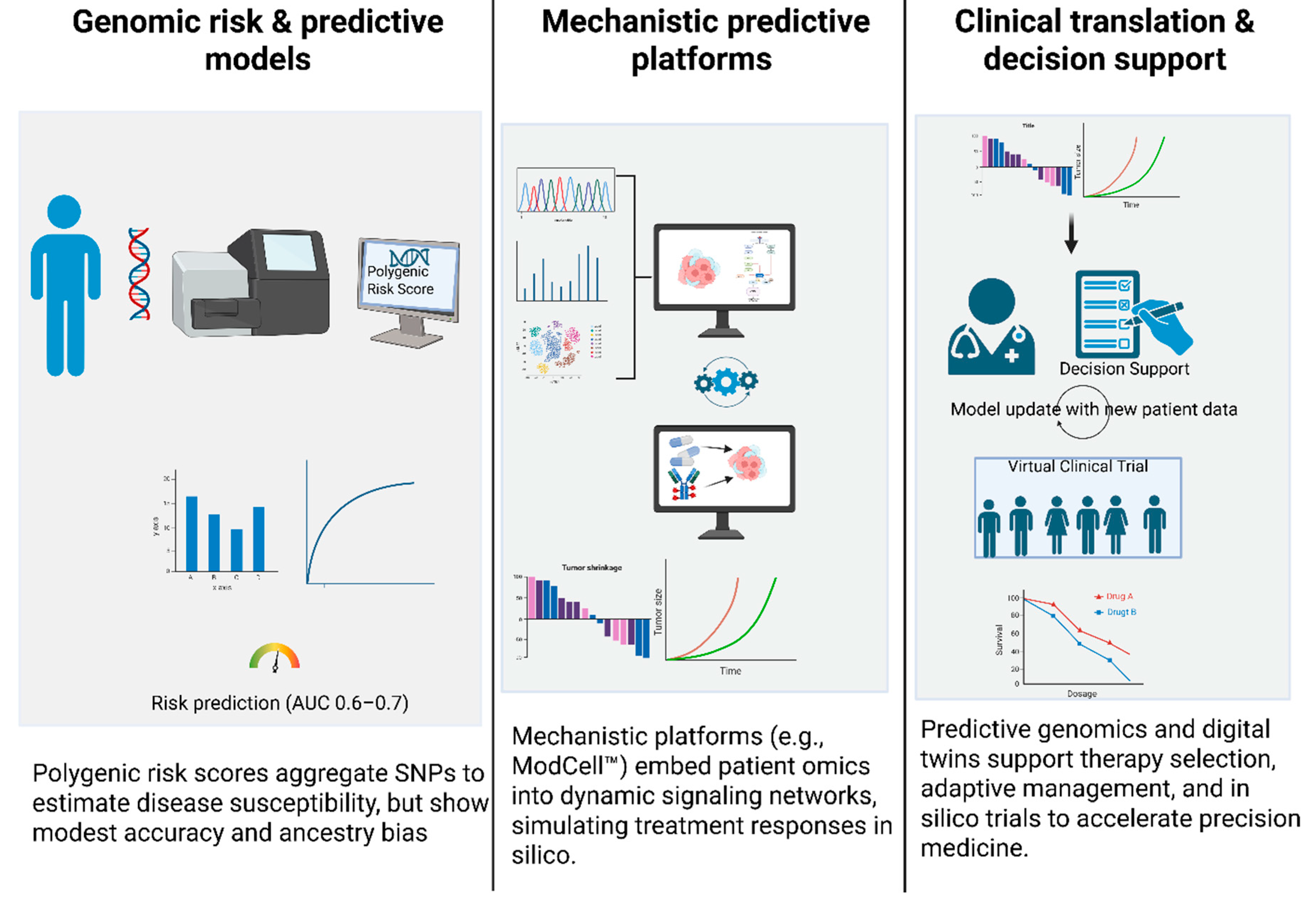
Figure 4.
Emerging technologies and future directions in predictive cellular modeling. Large language models (LLMs) extract biological hypotheses and compile them into mechanistic grammars for simulation. Standardized repositories of digital cell lines enable reuse, benchmarking, and cross-lab validation. Digital clinical trials integrate virtual patient cohorts and digital twins to support adaptive therapy optimization and precision care.
Figure 4.
Emerging technologies and future directions in predictive cellular modeling. Large language models (LLMs) extract biological hypotheses and compile them into mechanistic grammars for simulation. Standardized repositories of digital cell lines enable reuse, benchmarking, and cross-lab validation. Digital clinical trials integrate virtual patient cohorts and digital twins to support adaptive therapy optimization and precision care.
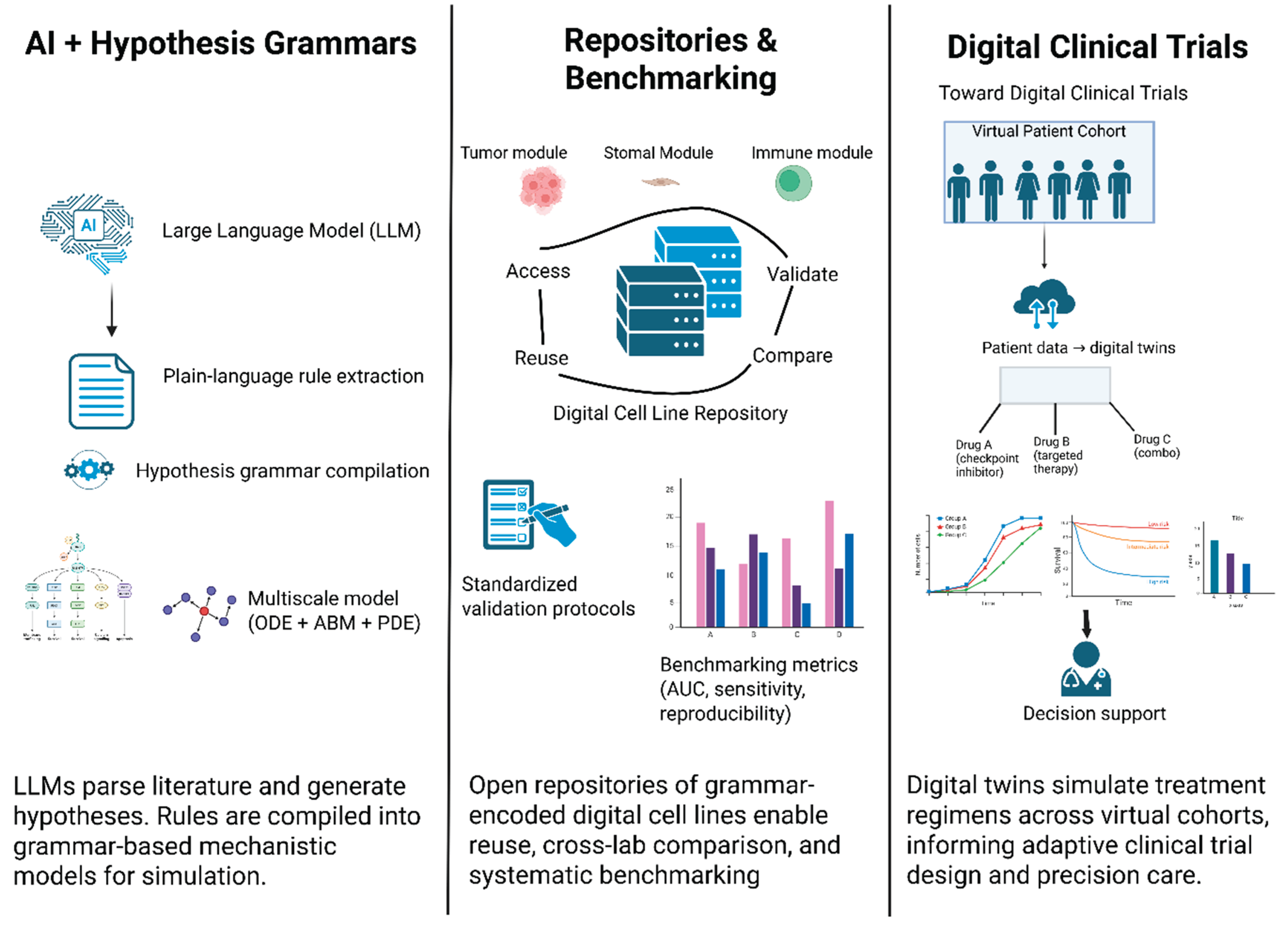

Table 1.
Examples of Hypothesis-Grammar Modeling Platforms and Applications.
| Framework/Tool | Description | Application Examples | Key Features |
|---|---|---|---|
| PhysiCell (CBHG) | Agent-based modeling platform with built-in cell-behavior hypothesis grammar [15]. | Cancer-immune simulations; liver metastasis; brain cortical development. | Human-readable rule language; handles diffusion, mechanics, and cell rules [15,18]. |
| PhysiBoSS | ABM platform coupling PhysiCell with MaBoSS (Boolean network simulator) [19]. | Multi-scale cancer models combining cell agents with intracellular signaling. | Integrates Boolean GRNs into each agent; enables cell decisions based on signaling [19,20]. |
| Custom Hypothesis Engines | Standalone scripts or libraries that parse plain-language rules into ODEs/ABMs [21,22]. | Prototype tools in research labs for specialized tissue models (no community name). | Often tailored to a project, focus on readability over performance [17]. |
Table 2.
Digital Twin Frameworks and Applications in Oncology.
| Framework/Platform | Modeling Approach | Data Inputs | Representative Application |
|---|---|---|---|
| TumorTwin | Modular Python toolkit for patient-specific tumor models. Combines PDE solvers (diffusion, reaction) with imaging-derived geometries [7,37]. | Patient imaging (MRI/CT) + molecular profiles (for parameter fitting). | In silico high-grade glioma growth (radiation therapy planning). |
| ModCell™ (Alacris) | Large-scale ODE signaling network mechanistic model. High-dimensional parametric model of pathways with omics-driven parameterization [20,38]. | Tumor omics (genome, transcriptome, proteome) + known drug targets/kinetics. | Predicted to kill the patient's tumor. |
| PhysiCell + Grammar | Agent-based modeling with hypothesis-grammar rules. Spatial tumor microenvironment with immune/CAF agents (cells) and diffusing molecules [25,39]. | Patient biopsy data (cell counts, expression) to set rule parameters. | Breast and pancreatic tumor models combining patient transcriptomics and spatial data to predict invasion and therapy response |
| Digital Twin Cloud Platforms | (Emerging) Hybrid models integrating ML forecasts with mechanistic simulators [7,8,37]. | Real-time patient monitoring data (future). | Promised for predictive monitoring (no specific example yet in published literature). |
Table 3.
Multiscale Modelling Approaches in Cancer Systems Biology.
| Modeling Approach | Spatial Scale | Key Components | Representative Tools |
|---|---|---|---|
| Agent-Based (Discrete) | Cellular (µm) | Individual cells serve as agents, governed by rules that regulate proliferation, death, motility, and signaling. Nutrient/cytokine fields via PDEs [21]. | PhysiCell, CompuCell3D, Morpheus; used in GBM invasion, TME studies. |
| Reaction–Diffusion PDE | Tissue (mm) | Continuum fields for growth factors, drugs, and oxygen. Averaged cell densities or ignored individual cells [21,50,51]. | Custom PDE solvers, often coupled with ABM for nutrients. |
| ODE/Boolean (Intracellular) | Subcellular | Gene/protein network dynamics within each cell (mass-action ODEs or Boolean rules) [38,51,52]. | COPASI, MaBoSS, BioNetGen (Rule-based), CellNOpt. |
| Hybrid (ABM + Signaling) | Multi-scale | Coupled systems: e.g., ABM for cell positions + Boolean/ODE networks per agent [40,51,53,54]. | PhysiCell+MaBoSS, CHASTE (multi-scale configs), Elecans. |
| Machine Learning (Data-driven) | Varies | Statistical/ML models trained on data (lacking explicit physics). May incorporate mechanistic features [32,51,54,55]. | Random forests, Neural nets, and Physics-Informed neural networks. |
Table 4.
Comparative Analysis of Multiscale Cellular Modeling Paradigms.
| Modeling Paradigm | Core Mechanism | Scale of Focus | Key Strengths | Key Weaknesses |
|---|---|---|---|---|
| Agent-Based Models (ABMs) | Individual, rule-based agents (cells) interacting locally. | Cellular, Tissue | Captures emergent properties, exhibits high biological interpretability, is modular, and is flexible [25,39]. | High computational cost, difficult to calibrate parameters, not feasible for large-scale, homogeneous systems. |
| Continuum Models | PDEs/ODEs representing tissue as a continuous medium. | Tissue, Organ | Computationally efficient, well-established mathematical theory, effective for bulk phenomena [21,22]. | Obscures individual cell heterogeneity and is limited in capturing stochasticity and emergent behaviors. |
| Hybrid Models | Combines discrete agents with a continuous field. | Cellular to Tissue | Balances computational efficiency and mechanistic detail, allowing for the modeling of heterogeneous and critical regions [38,45,88]. | Increased model complexity, difficult to integrate different mathematical formalisms seamlessly. |
Table 5.
Interpretable Rule-Based Models in Genomics.
| Method | Type | Interpretability Strength | Example Usage |
|---|---|---|---|
| Decision Trees | Tree-based (white-box) | Moderate (paths give logic) | Classify tumor subtypes by gene expression thresholds [76]. |
| Rule Lists | Sequence of if-then rules | High (short, readable rules) | Identified antibiotic-resistance markers via k-mer rules [56,58]. |
| Rule Ensembles (e.g., RuleFit) | Weighted rules | Moderate (composite rules) | Aggregate multiple simple rules for robust classification [56,57,58]. |
| Sparse Linear Models | e.g., LASSO (intrinsic) | High (coefficients as effects) | PRS models: linear combo of variant effects [83]. |
| Global Surrogate | Interpretable model fitted to black-box outputs | Moderate | Fit a decision tree on predictions of a deep net to approximate its logic [4,93]. |
Table 6.
Technical and Ethical Challenges in Predictive Cellular Modeling.
| Challenge Category | Specific Barrier | Description of Issue | Proposed Direction for Resolution |
|---|---|---|---|
| Technical | Parameter Identifiability | High-dimensional models often have multiple parameter sets that fit the data, leading to non-unique solutions [7,108,109]. | Development of new computational methods (e.g., Bayesian approaches, ML integration) that can handle large parameter spaces and incorporate prior knowledge to constrain solutions. |
| Data Granularity | Current omics data are often static "snapshots" that fail to capture the full temporal dynamics of cellular processes[54,110,111,112]. | Increased focus on time-series and longitudinal data collection, as well as new methods to infer dynamic trajectories from static data (e.g., pseudo-time analysis). | |
| Computational Complexity | Large-scale, 3D simulations are computationally prohibitive, limiting the scope of models and the speed of simulations [22,25,44]. | Investment in high-performance computing (HPC) infrastructure and the use of ML to create efficient "surrogate models" that can run simulations at a fraction of the cost. | |
| Ethical & Clinical | Algorithmic Bias | Models trained on unrepresentative or biased data can perpetuate or amplify existing health inequities [113]. | A multidisciplinary approach to model development that includes diverse teams, representative datasets, and a focus on transparency and fairness metrics. |
| Patient Privacy & Consent | Genomic data is intrinsically identifiable, making it difficult to balance data sharing for research with a patient's right to privacy [109]. | Adoption of ethical frameworks like GDPR, robust de-identification and encryption techniques, and new models for data ownership and federated learning that keep data local. | |
| Regulatory Validation | Lack of a clear, standardized framework for validating and approving predictive models for clinical use[8,105]. | Collaborative efforts between academia, industry, and regulatory bodies (e.g., FDA) to establish clear guidelines, standardized benchmarking protocols, and community-wide acceptance criteria. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



