Submitted:
01 November 2025
Posted:
03 November 2025
Read the latest preprint version here
Abstract
Acute myeloid leukemia (AML) and myelodysplastic neoplasms (MDS) are clonal hematopoietic malignancies in which next-generation sequencing (NGS) has become integral for diagnosis, classification, risk stratification, and measurable residual dis-ease (MRD) monitoring. Traditional cytogenetic and PCR-based assays remain useful, but targeted NGS panels now represent the standard of care, providing rapid and sen-sitive detection of recurrent gene mutations, structural variants, and gene fusions. Whole-genome, whole-exome, RNA sequencing, and long-read platforms expand the spectrum of detectable alterations, though targeted panels remain most practical for routine diagnostics. Bioinformatic pipelines and quality metrics—including read length, sequencing depth, and coverage—are critical for accurate variant calling, with validation often required for variants of uncertain significance or those near detection thresholds.
NGS is now embedded in diagnostic frameworks, including the WHO 2022 and ICC classifications, which incorporate recurrently mutated genes such as TP53, ASXL1, RUNX1, and FLT3. These data inform prognostic models, with ELN-2022 defining ad-verse-risk AML subgroups for patients treated with intensive chemotherapy, ELN-2024 AML patients treated with less-intensive therapies, and the IPSS-M refining MDS risk categories by integrating mutational data. NGS also enables MRD monitor-ing, with gene panels and PCR-NGS hybrid approaches (e.g., for FLT3-ITD) showing increasing clinical utility, though standardization is still lacking. Furthermore, diag-nostic NGS frequently uncovers germline predisposition syndromes (e.g., DDX41, GATA2), with significant implications for treatment decisions and donor selection in transplantation.
We discuss advantages, limitations, and future perspectives of NGS in clinical hema-tology, with the aim of providing a practical guide for clinicians navigating the grow-ing complexity of molecular data in daily practice.
Keywords:
acute myeloid leukemia (AML)
; next-generation sequencing (NGS)
; NGS-report
; myelodysplastic neoplasms (MDS)
; measurable residual disease (MRD)
; germline predisposition to myeloid neoplasms
1. Introduction
Acute myeloid leukemia (AML) and myelodysplastic neoplasms (abbreviated MDS) are clonal hematopoietic neoplasms, characterized by at least one somatic mutation in 96% of all cases [1]. Although the WHO fifth edition still allows diagnosis of AML by differentiation for cases that lack defining genetic abnormalities, and dysplasia in MDS also corresponds to morphological criteria [2], for both diseases the diagnosis relies increasingly on extensive laboratory workups to identify genetic characteristics. In this contest, the classical PCR-based and cytogenetic techniques have been rapidly corroborated by Next-generation sequencing (NGS). It is now standard of care to obtain mutational analysis using NGS on every patient with AML, with direct implications for treatment selection and prognostication [3,4]. Indeed, NGS enables the rapid sequencing of millions of DNA fragments simultaneously, providing comprehensive insights into the genome structure, genetic variations, gene expression profiles, and epigenetic modifications of neoplastic cells [5]. Unfortunately, the easy availability at reduced costs of NGS in AML/MDS patients has increased the uncertainty for both hematologists and patients about the widespread availability of extensive information provided by reading the NGS report. Therefore, we conducted this review to facilitate clinicians, similar to what was published for oncologists and ESMO recommendations for the use of NGS in advanced cancers [6,7].
2. NGS Techniques
NGS is based on the principle of massively parallel sequencing, where millions of DNA fragments are sequenced simultaneously [8]. Different NGS approaches can be applied depending on clinical and research needs, allowing the sequencing of the whole genome, exome, or transcriptome [9]. Whole genome sequencing (WGS) provides a complete picture of genetic variations; whole exome sequencing (WES) focuses on protein-coding regions; RNA-sequencing can be useful for identification of fusion genes, whereas targeted sequencing panels, the most widely used in clinical practice, concentrate on sets of genes recurrently mutated in MDS and AML [10].
2.1. NGS Workflow
The typical NGS workflow involves extraction of DNA or RNA from the sample, library preparation through fragmentation and adaptor ligation (hybridization capture-based assay) or through target enrichment in amplification-based assays, followed by sequencing [9].
In hybridization capture, the protocol involves random shearing of DNA followed by the ligation of sequencing adapters to the ends of the resulting fragments. Regions of interest within the library are then selectively captured using biotinylated oligonucleotide probes.
In contrast, amplicon sequencing uses primers specifically designed for the regions of interest to enable targeted PCR amplification. Adapters are then added to the amplified products to prepare them for sequencing.
The method chosen for performing targeted sequencing can be determined by considering several factors such as number of targets, number of steps and cost per sample.
In addition to the adapters required for sequencing on the instrument, patient-specific index sequences are incorporated. These indexes enable the pooling of multiple libraries into a single sequencing run, allowing for the simultaneous analysis of multiple samples, even from different patients [11].
Different platforms use different sequencing techniques. The most common ones include sequencing by synthesis, sequencing by hybridization, sequencing by ligation, sequence detection through electric impedance [12]. The most widely adopted approach in clinical practice is sequencing by synthesis (SBS), used by Illumina instruments, where fluorescently labelled nucleotides are incorporated and detected in real time; a semiconductor-based variation of SBS through detection of H⁺ rather than fluorescence released after nucleotide incorporation, is implemented in Ion Torrent systems [13]. In both cases, amplification of the DNA fragments is necessary before sequencing. In the first case, the fragments are loaded onto a flow cell where they fold over to form bridges, generating the so-called bridge amplification; in the second case, the fragments are amplified once each is bound to a magnetic bead [14].
More recently, long-read technologies have emerged: single-molecule real-time sequencing (SMRT) by Pacific Biosciences, which enables highly accurate “HiFi” long reads, and nanopore sequencing (Oxford Nanopore Technologies), which directly measures changes in ionic current as nucleic acids pass through protein pores, providing ultra-long reads and real-time analysis [13]. While short-read SBS remains the gold standard for routine AML and MDS diagnostics, long-read platforms are increasingly explored for detecting structural variants, fusion genes, and transcript isoforms [9,15]. In these cases, no amplification of the fragments is required prior to sequencing [11].
Bioinformatic pipelines then align reads to a reference genome, call and annotate variants, and generate interpretable data. The key biological metrics used in NGS after alignment to a reference genome, omitting the run quality-check, include the read length, the sequencing depth and the coverage, the duplication rate, the level of detection (LoD), the CG-bias, as the CG-rich fragments may show a lower coverage, and some other assay-specific parameters [9,11,13]. Below we will shortly describe these parameters. In the clinical setting, results often require validation, and variant interpretation must integrate allele frequency, functional annotation, and clinical relevance [6,9].
In next-generation sequencing (NGS), a read represents the nucleotide sequence obtained from a single DNA or RNA fragment during the sequencing process. Read length depends on the platform used, but in clinical oncology and hematology, the read length in most targeted DNA and RNA panels varies generally between 50-300 bp (commonly 100-250 bp) which are sufficient to detect point mutations, indels, gene fusions but may create challenges for precise detection of long insertions like long FLT3 ITD variants and long deletions like CALR type 1 mutations [12,16,17,18]. The whole-genome sequencing and long-read technologies may produce much longer reads, typically in the range of 10–100 kilobases and in some cases extending to several megabases, which facilitate the detection of structural variants, complex rearrangements, and full-length transcript isoforms [19,20,21]. Moreover, from technical point of view, reads can fail to be mapped uniquely, to remain unmapped or to be mapped to uninformative transcripts, in case of RNA-seq [22].
The accuracy of each read in sequencing is evaluated using the Phred Quality Score (Q score), a probabilistic indicator, expressed on a logarithmic scale, of the likelihood that a base has been incorrectly assigned. A Q score of 30, for example, indicates an error probability of 1 in 1000, corresponding to a specificity of 99.9% [23].
Low Q score values increase the risk of false positives in variant identification, compromising data reliability and requiring additional confirmatory analyses.
The sequencing coverage refers to the percentage of a known reference base (genome/exome/transcriptome/targeted gene panel) that has been sequenced at least once. Insufficient coverage can introduce sequence errors when the absence of a gene or a gene fragment may require disambiguation between an assembly error or a relevant finding. Moreover, insufficient coverage can produce additional complications since it can be further misunderstood during the bioinformatical analysis [15]. Some fragments like CG-rich regions or polyploid genes or investigating very rare events may require increased sequencing depth or coverage [22].
The sequencing depth, defined theoretically as LN/G, where L is the read length, N is the number of reads and G is the haploid genome length, means the average number of times each nucleotide in a DNA or RNA sample is sequenced during a NGS run, typically expressed as a multiple of 'x' (e.g., 100x) [22]. For RNA-seq and targeted RNA-sequencing approaches, it is essential to report the total number of sequencing reads per sample, as this strongly impacts sensitivity for gene expression profiling, detection of fusion transcripts, and low-abundance isoform identification [24]. For standard whole-transcriptome (mRNA) RNA-seq in human samples, a depth of 30–50 million reads per sample is recommended, with a minimum of 20–30 million reads considered sufficient for general expression analysis; scenarios requiring detection of low-expression transcripts or detailed splicing may require significantly deeper sequencing, while targeted RNA panels may need as few as 1-3 million reads per sample due to focused transcript coverage [24,25,26,27].
Different NGS types have different sequencing depth ranges. Generally, WGS provides the sequencing depth that ranges at 30–60× and the exome-only coverage with WES permits the depth range at 100-200× that are insufficient for low frequency variant detection. In the meanwhile, the narrow genome coverage using the panel sequencing allows reaching a higher depth of sequencing (typically at 500–1000×) [28].
The sequencing depth required for the reliable detection of low-frequency variants remains a critical issue in NGS diagnostics. According to the Association for Molecular Pathology and College of American Pathologists (AMP/CAP) guidelines, a minimum coverage of >250× per tested amplicon is recommended for somatic variant detection in targeted sequencing panels used in routine oncology diagnostics (variant allele frequencies (VAFs) 5%). VAF is defined as the percentage of reads reporting a specific genetic variant relative to the total number of reads covering that particular gene locus. This threshold is considered a baseline standard and is not intended to ensure sensitivity for very low VAFs or for applications such as minimal residual disease monitoring [9]. More detailed evaluations demonstrate that substantially higher coverage is needed to confidently detect variants at low allele frequencies: for example, Petrackova et al. showed that detection of a 3% VAF with >99% confidence requires approximately 1,650× coverage and at least 30 supporting reads [29]. Similarly, systematic analyses of sequencing error rates suggest that to achieve robust sensitivity for a 2% VAF, sequencing depths on the order of 1,000× or higher are generally necessary [28]. The necessity of providing excessively high sequencing depth to detect low-frequency variants can be overcome through the selective amplification of target regions that allows enrichment of variant-containing loci within the sequencing library. This approach is widely used in targeted sequencing panels [30].
In addition to sequencing depth and VAF, most NGS protocols require a minimum number of supporting reads—that is, individual sequencing reads that contain the variant allele. This requirement is essential to distinguish true variants from sequencing artifacts, particularly at low VAF. For example, thresholds of minimum 10 and better ≥ 20–30 supporting reads are commonly applied in clinical and research settings to ensure reliable variant calling and reduce the number of false positives [31,32]. At least 3 supporting reads are necessary for detection of a fusion variant [31].
2.2. NGS Types in Clinical Practice
Whole genome sequencing (WGS) delivers a comprehensive view of genetic variation, capturing both coding and non-coding regions along with structural variants and copy number alterations, thus providing unparalleled breadth in genomic analysis. Whole exome sequencing (WES), in contrast, targets only the protein-coding exons—about 1–2.5% of the genome—offering a more cost-effective approach while still maintaining a high yield of clinically relevant variant detection [12,33,34].
RNA-sequencing (RNA-seq or transcriptome-wide sequencing) is particularly valuable for identifying fusion genes and for capturing gene expression profiles and transcriptomic alterations that DNA-based assays miss [33]. Moreover, RNA-seq may be required for definition of one of multiple possible fusion partner genes of a biologically dominant gene like in KM2TA rearrangements (e.g., KMT2A::MLLT3, KMT2A::MLLT1, KMT2A::ELL) that have a prognostic importance and are impossible to be identified with cytogenetic assays like FISH alone [2].
Finally, targeted sequencing panels—the most used in clinical diagnostics—focus on a curated set of genes recurrently mutated in a distinct pathology (usually 20-50 genes for MDS and AML), providing higher coverage and analytic sensitivity at reduced cost and turnaround time [10]. In targeted sequencing panels, the amplification of specific genomic regions of interest against the whole genomic background increases the relative representation of these loci within the sequencing library thereby reducing the need for extremely high overall sequencing depth and allowing detection of variants with relatively low VAF [30,34]. The information generated by targeted gene panels can influence diagnostic decisions, guide therapeutic choices, and/or provide prognostic insights for a particular malignancy. Target panels can be designed to detect relevant single nucleotide variants (SNV), small insertions and deletions (indels), copy number alterations, fusion genes, and structural variants [9]. Notably, targeted sequence panel may have a limited sensitivity for detection of large insertions or deletions including FLT3 ITD, MLL PTD, and 52bp CALR deletions and may require more sophisticated bioinformatical approaches or use of other molecular biology diagnostic assays due to the discrepancy between the large and/or variable size of these alterations and relatively small size of NGS reads (50-300 bp) [17,35,36,37]. This pitfall, however, seemed to be resolved even allowing patient-tailored measurement of FLT3-ITD MRD by PCR-NGS in the latest clinical trials [38]. On the other hand, genomic regions of high GC content like CEBPA variants can pose a challenge for amplification during a library preparation and require higher depth of sequencing [17,39]. However, these problems could also be overcome using long-read NGS targeted sequence panels designed for AML diagnosis [19]. Indeed, numerous recent studies have proved the high diagnostic sensitivity and concordance with traditional analytic methods of NGS panels that allow generation of target-enriched libraries with increased amplicon specificity appropriate for FLT3 ITD and CEBPA diagnostic testing [9,18,37,40].
Most panels have been modified with addition of new relevant genes according to the most recent WHO/ICC 2022 classifications [41]. The core genes most frequently recommended for inclusion in targeted NGS panels for myeloid malignancies typically include: NPM1, CEBPA, ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2, TP53, JAK2, CALR, MPL, CSF3R, KIT, FLT3, IDH1/2, quite frequently they include also ETV6, KRAS, NRAS, and WT1 [2,18] (Table 1).
However, the numbers of genes (core and/or additional genes) and gene fragments (hotspot regions of a single gene relevant for a distinct gene and pathological context, flanking regions, coding/non coding sequences, whole gene sequence) included in panels can differ substantially between laboratories, especially in the absence of a standardized NGS technique for AML/MDS diagnosis [27,42,43].
2.3. Sample Type and Quantity
In case of most haematological samples, including MDS or AML, tumor cells are obtained from bone marrow or from peripheral blood (Figure 2). In myeloproliferative diseases, there is a high degree of concordance between the results obtained on peripheral blood samples and on bone marrow samples making a peripheral blood NGS a reliable and less invasive diagnostic tool, yet bone marrow samples appear to be superior to peripheral blood for MRD measurement [44,45]. White blood cell count, or flow cytometry data can provide an approximate cell number in the sample. In case if bone marrow aspirate in unavailable NGS can be performed using formalin-fixed paraffin-embedded (FFPE) BM trephine biopsies, however, this type of sample may produce underperforming reads as compared with fresh samples since the chemical preparation with formaldehyde induces cross-linking between cellular macromolecules and amino groups of DNA bases thus reducing DNA sample quality [42]. The necessary sample amount varies between different assays and studies, Illumina recommends 80 ng of total DNA or RNA per sample, however robust results may be obtained with smaller nucleic acid quantities like 40-60 ng or higher like 100-120 ng per sample [31,32,46]. One study that compared several diagnostic panels appropriate for FLT3-ITD and CEBPA diagnostic testing reported that 200 ng of DNA per sample were essential for optimal library preparation [18]. Long-read and ultra-long-read NGS assays can require higher DNA quantity varying from several hundred nanograms to micrograms [19].
2.4. Results Validation
Due to the complexity of NGS workflow and the possibility of false positives due to technical artifacts, bioinformatic errors, or low-level variants close to the detection threshold, NGS results may require a confirmatory testing with other molecular biology techniques like Sanger sequencing, droplet digital PCR, quantitative PCR or capillary electrophoresis fragment analysis, to ensure the accuracy and reliability of molecular findings. Current guidelines, including those from the Association for Molecular Pathology (AMP) and the College of American Pathologists (CAP), recommend orthogonal confirmation in selected cases, particularly for ambiguous results, variants near the assay’s limit of detection, variants of uncertain significance (VUS), or when results have immediate therapeutic implications [9,47]. In well-validated, high-performing targeted NGS panels, confirmatory testing may not be necessary for recurrent hotspot mutations especially considering the high concordance between NGS and other molecular diagnostic methods [18,40]. Overall, validation strategies should be tailored to the assay design, variant type, and clinical context, balancing analytical rigor with turnaround time and cost-effectiveness [28].
2.5. Standardization
The integration of next-generation sequencing (NGS) into the diagnostic workflow for hematologic malignancies has highlighted the critical need for standardization across laboratories and platforms. Variability in sequencing chemistry, library preparation, bioinformatics pipelines, and reporting can lead to significant differences in variant detection and interpretation [18,41]. International guidelines, such as those issued by the Association for Molecular Pathology (AMP) and the College of American Pathologists (CAP), recommend harmonization of procedures from pre-analytical steps (sample collection, nucleic acid extraction, library preparation) to analytical performance metrics (read length, depth of coverage, uniformity, error rates, variant allele frequency thresholds) and post-analytical processes (variant annotation, classification, and reporting) [1, 39]. Proficiency testing programs and inter-laboratory comparisons have also been attempted to ensure reproducibility and clinical reliability [9,39]. Despite the fact that ELN 2022 recommendations for AML, ESMO Clinical Practice Guidelines for MDS and WHO 2022 and ICC 2022 diagnostic classifications highlight the integration of NGS into diagnostic and prognostic workflows of AML and MDS and establish the minimal set of relevant genes, the insufficient standardization of diagnostic NGS limits its implementation in clinical practice [2,3,29,43,48]. Indeed, challenges remain in establishing consensus on minimum gene panels, defining limits of detection for clinically actionable variants, and standardizing the use of NGS for measurable residual disease (MRD) monitoring [49]. Moreover, the existing guidelines published several years ago should be updated according with the most recent advantages in NGS technique that have been implemented into clinical diagnostic practice since then. Notably, Italy has recently issued specific guidelines through SIES addressing the role of advanced molecular diagnostics, including NGS, in adult AML, and similar recommendations have been published in several other countries [50,51].
2.6. Type of Alterations
NGS enables detection of single nucleotide variants (SNVs), small insertions and deletions (indels), and, depending on the platform, copy number variations (CNVs) and structural rearrangements. RNA-based sequencing can further identify fusion transcripts, which are highly relevant in AML.
It is important to use nomenclature that is as uniform as possible, allowing for unambiguous designation and effective sharing. The Human Genome Variation Society (HGVS) provides standard nomenclature: its use is recommended as a guideline for variant nomenclature. Clinical reports should include references to the sequence, the nomenclature for the coding sequence, and the protein to facilitate functional interpretations. For the description of coding variants, the longest known reference transcript for each gene should be used and indicated in the report.
Some commonly used resources include RefSeq (https://www.ncbi.nlm.nih.gov/refseq), Ensembl (http://www.ensembl.org/index.html), and Locus Reference Genomic (LRG; https://www.lrg-sequence.org) [47].
2.7. Result Interpretation
Bioinformatic analysis can be divided into three levels:
- Primary Analysis:
Digital images generated by detecting fluorescence from labeled nucleotides are converted into a text file (FASTQ) representing the sequence of individual bases.
- Secondary Analysis
Sequences obtained from samples pooled in a single sequencing run are filtered and separated based on their indices. This process of sorting reads is called demultiplexing.
Variant identification, also known as variant calling, involves detecting differences between the reference genome and the aligned reads. This process can identify thousands of genetic variants, which are compiled into a single file for each patient called a VCF (“Variant Call Format”).
- Tertiary Analysis
The final stage focuses on interpreting the genetic results in the context of the clinical phenotype, aiming to establish potential links and causal relationships between the identified genetic variants and the diseases of interest.
Variants can be classified according to their actionability or pathogenicity.
Currently, two major schemes are proposed: the AMP/ ASCO/CAP tier system and the ClinGen/CGC/VICC for the classification of oncogenicity of somatic variants.
In the first one, somatic variants are grouped into four tiers according to their clinical relevance for cancer diagnosis, prognosis, and treatment: Tier I includes variants with strong clinical significance; Tier II comprises variants with potential clinical significance; Tier III covers variants of uncertain clinical significance; Tier IV consists of variants that are benign or likely benign [47].
In the second one, the classification relies on an integrated evaluation of multiple criteria, which include population data, computational predictions or data derived from predictive algorithms and information regarding the variant’s location within relevant or non-relevant functional domains of the protein. Importantly, no single criterion alone is sufficient to determine the pathogenicity of a variant; instead, pathogenicity must be supported by multiple lines of evidence. This comprehensive integration of criteria enables the classification of variants into five categories: 1-Benign; 2-Likely benign; 3- Variant of uncertain significance; 4-Likely pathogenic; 5-Pathogenic.
Based on the strength and extent of available scientific evidence, pathogenicity criteria are further ranked as oncogenic very strong (OVS1), strong (OS1-3), moderate (OM1-4), or supporting (OP1-4). Conversely, benignity criteria are classified as somatic benign very strong (SBVS1), strong (SBS1-2), or supporting (BP1-2) [52].
2.8. Useful Database
Following a growing number of large-scale genome sequencing projects for a variety of cancer types, including hematological cancers, a large amount of data has been produced and incorporated into various genomic databases.
The relevance of an identified variant is closely related to how often it has already been found in the context of cancer. It is important to assess the incidence and prevalence of tumor variants identified in different tumor entities, using databases and published literature. For the annotation of somatic variants, there are numerous cancer databases, such as the Cancer Gene Census (http://cancer.sanger.ac.uk/cancergenome/projects/census/), the Catalogue of Somatic Mutations in Cancer (COSMIC; http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), and The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/). These databases provide information on sequence variants in different cancer types and subtypes, cross-references to other genomic databases, as well as references to published literature, cellular pathways, targeted therapies, clinical trials, and outcome data [52].
Probable germline variants that may reside in genes associated with cancer predisposition syndromes can emerge during the sequencing of the tumor sample. Nevertheless, it is important to correlate this data with tumor cellularity [47].
In this regard, it may be useful to consult databases, such as Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/omim) and Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php) that provide information helpful for evaluating the potential nature of these variants. Among these, a commonly used database is ClinVar (http://www.ncbi.nlm.nih.gov/clinvar). It lists germline variants found in various tissues, from pathogenic to benign, providing clinical evidence and associated clinical studies, as well as indicating any classification by expert panels. Sometimes, it also reports somatic occurrences for certain variants [53].
When the germline nature of a variant is suspected, it is appropriate, through a properly validated germline test, after consulting with the physician and obtaining a properly completed and signed informed consent from the patient, to test it on a sample of tissue not affected by the disease for which the somatic analysis was initially requested.
Therefore, it is recommended to indicate in the issued report that the test is not able to distinguish somatic variants from benign ones, in cases where there may be suspicion of germline origin [47].
To evaluate the frequency of variants in large populations, population databases are used, which provide information on a given allele at a given locus in a large cohort of individuals belonging to distinct subpopulations. When using these databases, it is necessary to determine whether groups of healthy or sick individuals were used, whether more than one individual from the same family was included, and the age range of the subjects. They are frequently used to characterize benign variants, based on an arbitrary cut-off (Minor Allele Frequency, MAF). There is no standardized MAF threshold: it is recommended to use 1%, possibly based on the patient's specific ethnicity. Among the most used are: 1000 Genomes Project (http://browser.1000genomes.org), Exome Variant Server (http://evs.gs.washington.edu/EVS), dbSNP (http://www.ncbi.nlm.nih.gov/snp), dbVar (http://www.ncbi.nlm.nih.gov/dbvar), ExAC (http://exac.broadinstitute.org/ [47].
Finally, when evaluating a variant in a gene, in silico prediction algorithms are used to assess whether it could cause alterations in splicing or in the structure and function of the encoded protein. The degree of evolutionary conservation of the amino acid or nucleotide residue, the biochemical impact of amino acid substitution considering specific physicochemical properties, and the position of the variant in the translated protein are some of the main criteria used by various algorithms to predict functional impact.
Some of the most used in silico tools are PolyPhen2 (http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), MutationTaster (http://www.mutationtaster.org), and CADD (http://cadd.gs.washington.edu).
However, these are only predictions, and their use in interpreting sequence variants should be done with caution. It is not recommended that these predictions be used as the sole source of evidence for making a clinical decision [47].
2.9. Report Structure
At the end of the NGS analysis process, it is essential to prepare a report summarizing the results obtained. It is important that this report should be complete, providing the technical details necessary for an evaluation of what has been done, understandable to non-specialist professionals and patients, and concise enough, to avoid an overload of information that makes it difficult to read and understand. The report must include information about the laboratory that performed the analysis, the patient's personal details, the sample's traceability, the clinical question, the variants identified with their interpretation, and the method used.
It is important that the report clearly displays at the top the name of the laboratory with its certification (e.g., UNI EN ISO 9001) and accreditation (e.g., UNI EN ISO 15189, UNI CEI EN ISO/IEC 17025). Additionally, it’s preferable to include the name of the director, the laboratory's contact information, as well as the name of the referring clinician and the department of origin. [23].
It is important that the patient's personal details are included: surname, first name, date of birth, gender, and ethnicity.
The report must also include information about the biological sample: a unique identifier, date of collection, type (e.g., peripheral blood, bone marrow) and, if it’s possible, an estimate of the percentage of blasts, the diagnosis, and/or the clinical indication for the test.
The report must contain information on the NGS assay used, including sequencing technology or platform, the name of panel, and its version. In addition, a list of the analyzed genes and the covered regions must be provided.
A brief description of the methodology and its limitations must be included. This includes a list of all software tools used to obtain the results (including version numbers) as well as the reference genome employed (GRCh37-hg19 or GRCh38) [54].
It is important to indicate the limitations of the method: indicate what type of approach is unable to identify, which alterations will not be reported (also in relation to their oncogenicity/benignity and actionability). It is possible to report the overall coverage obtained specifically for each sample, measured as the average coverage over the entire sequenced genomic region. According to AMP/CAP guidelines, an average coverage of at least 200X is recommended to reliably detect somatic variants. In this context, VAF threshold of 5% is commonly used for variant calling and should be reported in the analysis [23].
The results section is the central part of the report.
For clarity, it may be preferable to present the variants in order of clinical relevance, with the most pathogenic or actionable variants listed first.
All reported variants must be described using HGVS.c and HGVS.p notation, together with their VAF and classification. The identified variants are summarized in a dedicated final section, structured to ensure immediate understanding, and supplemented with references to relevant literature where appropriate [47]. The report concludes with the date of reporting, the full name of the biologist who performed the analyses, and the signature of the laboratory manager. Each page of the report must be appropriately numbered [6] (Figure 1).
2.10. NGS vs. Quantitative-PCR vs. Digital-PCR
Today, laboratories have access to various technologies that can be used in combination to achieve their experimental objectives. These include long-established methods such as quantitative PCR (q-PCR) and more recent technologies such as digital PCR (dPCR) and NGS.
The qPCR allows real-time measurement of DNA amplification through fluorescence monitoring, providing relative or absolute quantification.
In dPCR, the sample is divided into thousands of independent partitions: the ratio between the positive partitions, which show a fluorescent signal, and the total number of partitions is used to calculate the concentration of the target in the sample. Through absolute quantification, it allows the detection of a low number of copies of the target molecule without interference from PCR inhibitors and without the need for a standard curve, making it a sensitive and accurate technique [55,56]. Although QPCR has a more limited resolution, it remains a faster, less expensive technique, allowing simultaneous analysis of numerous samples and multiple targets.In both cases, knowledge of the mutation to be investigated is required, but this then allows monitoring over time.
NGS allows for large-scale studies, both quantitative and qualitative, enabling analysis with a large and more complex amount of data to be managed and processed, thereby increasing costs and reporting times. It is useful for detecting multiple mutations simultaneously from a larger number of samples, albeit with lower sensitivity than dPCR [57,58].
3. NGS and Impact on Diagnosis and Classification
3.1. NGS Role in MDS: From Diagnosis to Classification
The transition from the last WHO2016 to modern ICC 2022 and WHO 5th classifications was marked by the introduction of novel genetically defined MDS categories [61,2,48,62]. In this context, the availability of NGS testing is crucial not only to correctly identify patient category but also to address patients to genetically-driven therapies. Innovative changes in the WHO 5th include the definition of MDS with low blast count and SF3B1 mutations, which has been added to the former category of MDS LB with ring sideroblasts (RS) [2]. This important new category has been implemented also in the ICC classification, thereby substituting the previous MDS with RS [48]. WHO-2022 has implemented another new genetically-driven category defined as MDS with bi-allelic TP53 mutations, irrespective of blast count [48]. Similarly, ICC introduced the diagnostic categories of MDS and MDS/AML with mutated TP53., which differ on blast counts. [2]. The new “MDS/AML” is an overlap category that includes MDs with 10 to 19% blasts which replaces the former WHO category of MDS with excess-blast 2 to better reflect a continuum between myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) [61]. NGS is indeed necessary to define MDS/AML category, as a number of mutations need to be either detected or excluded to correctly classify patients. In particular, MDS/AML cases must not harbor a genetic abnormality that by itself defines AML (NPM1, CEBPA); additionally, ICC identifies subcategories of MDS/AML including i) MDS/AML with mutated TP53, ii) MDS/AML with myelodysplasia-related gene mutations or cytogenetic abnormalities and iii) MDS/AML, NOS (not otherwise specified) where information on mutational status of a number of genes is required in order to address proper diagnosis ( Tp53, ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2 and RUNX1) [48]. Finally, WHO 2022 and the ICC classification both introduced relevant updates related to clonal hematopoiesis, including the formal recognition of Clonal Hematopoiesis of Indeterminate Potential (CHIP) and clonal cytopenia of undetermined significance (CCUS) entities as part of the spectrum of myeloid neoplasm [2,48].
3.2. CHIP and CCUS, the Role of NGS
CHIP is currently defined by the presence of somatic mutations that are recurrently mutated in hematologic malignancies (e.g., DNMT3A, TET2, ASXL1) in individuals without evidence of cytopenia or dysplasia. This condition is usually detected through NGS in otherwise healthy individuals and is more common in older adults [63,64]. Several studies have demonstrated an increased risk of developing hematologic malignancies (especially MDS/AML) in individuals with CHIP and an increased risk of atherosclerotic cardiovascular disease [65,66,67]. Variant allele frequency ≥2% is considered significant in CHIP mutations. While in WHO 2022 CCUS is not a diagnostic category, CCUS is considered a distinct diagnostic entity in ICC, and its recognition requires close follow-up, especially in case high-risk mutations are detected (e.g., TP53) [2,48] [68]. CCUS as per ICC is defined ad persistent unexplained cytopenia in at least one lineage in the absence of morphologic dysplasia or increase in blasts [48], and NGS is crucial in this definition by detecting at least one somatic mutation in genes commonly mutated in myeloid neoplasms (e.g., DNMT3A, TET2, ASXL1, SF3B1, SRSF2, etc.). The type of mutations and VAF deserve special consideration in these conditions. CHIP is asymptomatic and carries a 1% per year risk of progression to hematologic malignancy. However, multiple studies have identified high-risk features in CHIP, including specific mutations (TP53, U2AF1, SRSF2, IDH1/2), the presence of concomitant multiple mutations, the detection of larger clones (e.g. 10-15% VAF) or the expansion of these mutant clones over time [68,69]. Regarding CCUS, mutations in spliceosome and epigenetic regulators are considered at higher risk of progression when compared to typical CHIP mutations and the presence of ≥2 pathogenic mutations confers a ~50% risk of progression at 5 years [70].
3.3. Novel Genetically-Based Categories in MDS
3.3.1. SF3B1 in MDS
SF3B1 (splicing factor 3b subunit 1) is the largest subunit of the SF3B complex and serves as a core component of the U2 snRNP, whose function results in critical branch site recognition in the early stages of spliceosome assembly. Mutations in SF3B1 were originally identified in 2011 and present across all spectrum of myeloid malignancies, including MDS, MDS/AML and AML [71]. However, mutations in this gene were enriched in MDS and particularly enriched in MDS with ringed sideroblasts which showed up to 65% of patients mutated for SF3B1 [71,72]. It has been confirmed by several studies that the detection of SF3B1 mutation includes over 90% of MDS with ≥5% ring sideroblasts. The majority of SF3B1 gene mutations are heterozygous substitutions clustered in exons 12–15 of the gene, with K700E accounting for one-third of the variants. SF3B1 mutations alter the function of the protein, which, however retains structural integrity [72]. According to ICC classification, MDS with SF3B1 mutation is now a distinct entity and does not even require any more the presence of ringed sideroblasts [48]. MDS with SF3B1 mutations must present with <5% in the bone marrow and <2% in the peripheral blood and specific cytogenetics (isolated del(5q), −7/del(7q), abn3q26.2, or complex) and molecular abnormalities (TP53 multi-hit and RUNX1) should be excluded [48]. WHO 2022 is more conservative and still retains the definition of MDS with low blasts and ring sideroblasts for those cases with wild-type SF3B1 and ≥15% ring sideroblasts [2]. This specific MDS category is not only homogeneous in terms of genetics but is also associated with a low rate of progression and better OS when detected in the context of low blast counts and without additional genetic abnormalities [72].
3.3.2. TP53 in MDS
TP53 is a tumor suppressor gene located on chromosome 17p13.1, frequently mutated in human cancers and associated with poor outcomes in the majority of neoplastic diseases. Pathogenic TP53 alterations are detected in 7-11% of MDS. TP53 alterations not only include mutations but also other loss of genetic material, including copy-neutral loss of heterozygosity and segmental deletions [73,74]. The majority of TP53 mutations (-80%) are missense substitutions scattered from exons 5 through 8, causing amino-acid changes, while the remaining are truncating alterations. Active p53 controls transcription in more than 150 genes implicated in DNA damage repair, apoptosis, senescence, and cell cycle arrest. Mutations produce nonfunctional p53, disrupt self-renewal and induce proliferation of hematopoietic stem cell. The WHO2022 classification defined a specific diagnostic MDS category defined as “MDS with biallelic TP53 inactivation (MDS-biTP53)” where patients present with multiple TP53 alterations [2]. Biallelic TP53 (biTP53) alterations may consist of multiple mutations or mutation with concurrent deletion of the other allele (TP53 copy number loss or copy neutral loss of heterozygosity). The detection of biallelic inactivation of TP53 is complex and requires more than one technique. NGS analysis is essential to detect single mutation or multiple mutations in the gene, covering at least exons 4 to 11 [75]. The first choice to identify loss of genetic material at the TP53 site is fluorescence in situ hybridization with specific probe sets for the TP53 locus on 17p13.1. However, NGS results may help in the identification of multi-hit TP53 variants. First, the presence of more than one TP53 mutation usually implies the involvement of both alleles of the gene; second, the detection of TP53 mutations with VAF ≥ 50% can be used as a surrogate (although not certain) of a loss of genetic material on the other TP53 allele [2]. The ICC classification has introduced two distinct categories of MDs with TP53 mutations: MDS with mutated TP53 mutation and MDS/AML with mutated TP53. The first includes MDS cases with blasts up to 9% with either multi-hit TP53 mutation or TP53 mutation (VAF > 10%) and complex karyotype often with loss of 17 [48]. The multi-hit TP53 mutation is defined as the presence of either 2 distinct TP53 mutations (each VAF > 10%), a single TP53 mutation with 17p deletion or copy-neutral LOH at the TP53 locus or a TP53 mutant VAF of >50% [48]. MDS/AML with TP53 is more simply defined as those cases with 10 to 19% blasts and any TP53 mutation >10% [48]. Overall, these MDS categories, particular multi-hit TP53 are often characterized by complex karyotype and dismal prognosis [76].
3.4. Impact of NGS in the Definition of AML, Novel Genetic Classifications
Nowadays, AML can be classified according to two systems: the WHO fifth edition [2] or the International Consensus Classification (ICC) of Myeloid Neoplasms and Acute Leukemias [48]. Genetic entities have overcome morphologic and cytometric features, thereby definitely introducing NGS analysis as a cornerstone in AML diagnosis. The ICC classification has fostered the role of genetics by lowering the blasts limit to 10% for cases presenting with recurrent genetic abnormalities (BCR::ABL1 excluded). In addition, ICC has introduced MDS/AML category in cases with 10 to 19% of blasts. The ICC approach might be figured as hierarchical. In the first step, recurrent genetic abnormalities should be identified, and if not present, the second step requires research of TP53 mutations (VAF > 10%) and myelodysplasia-related gene mutations or cytogenetics abnormalities. Therefore, the second step of the ICC relies almost entirely on NGS for both identification and quantification of TP53 mutations and also for detection of ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2 and RUNX1 mutations, present as myelodysplasia-related genes [55].
3.4.1. TP53 Mutations in AML
[56] In de novo AML, mutated TP53 is detected in 5-10% of cases and is enriched in elderly patients and those with therapy-related AML. Similarly to MDS, TP53 mutations present as monoallelic or biallelic mutations; in the latter case, the second TP53 allele is inactivated by either point mutations, copy neutral LOH (cnLOH) or loss of heterozygosity (LOH). TP53 mutations define aggressive AML subset either if presenting as de novo, arising from previous MDS or as therapy-related and frequently associated with a complex karyotype. Different from MDS, a single TP53 mutation with VAF>10% is sufficient to define AML with TP53 mutations (> 20% blasts) or MDS/AML with TP53 mutation (10-19%) according to ICC [2,48].
3.4.2. MDS-Related Mutations
ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, and ZRSR2 genes. ICC classification but also ELN2022 guidelines have implemented a 9-gene signature to define the new category of AML (>20% blasts) or MDS/AML (10-19%) with myelodysplasia-related mutations [2,48]. These mutations were introduced in ELN 2022 in the adverse risk given their strong association with secondary AML that transformed from myelodysplastic syndrome (MDS) or chronic myelomonocytic leukemia (CMML) [77]. This category is introduced in the ICC together with the MDS-related cytogenetic abnormalities, thus expanding the previous WHO 2016 category of AML with myelodysplasia related changes based on the sole morphologic analysis [48,61]. WHO 5th edition also identifies the category of AML, myelodysplasia-related (WHO-AML-MR) defined by specific molecular and cytogenetic abnormalities but still including AML transformation of MDS and MDS/MPN [2]. More in details, the WHO 5th edition includes only 8 of the 9 genes detailed in ICC classification, excluding RUNX1. These 8 genes have been demonstrated to retain >95% specificity for the diagnosis of secondary AML evolving from previous MDS, thereby defining the ontogeny of s-AML vs de novo AML [1]. More in details SRSF2, SF3B1, U2AF1, and ZRSR2 are mutated in nearly half of s-AML and encode for proteins involved in the spliceosome machinery essential in RNA processing, in particular those participating in the pre-mRNA splicing. The majority of SRSF2 results in alterations at the P95 residue, while SF3B1 mutations cluster in K700 residue, with a minority of cases involving K666 residue. U2AF1 gene also presents mutational hotspot in S34F and Q157 residues, while ZRZS2 mutations are scattered throughout the gene and preferentially occurred in the Pre-ZF1 (49%), UHM (27%), and Post-ZF2 (13%) domains [78,79]. ASXL1, EZH2 and BCOR belong to the class of chromatin modifiers. ASXL1 regulates epigenetics and transcription through interaction with polycomb complex proteins and various transcription activators and repressors. This gene is ubiquitously mutated in myeloid neoplasms and mutations are mostly located in the final exon of the gene (exon 13), with one of the most common mutation being G646Xfs*12 [78,80]. 10.3324/haematol.2014.114157). EZH2 is a lysine methyltransferase found as the central core protein of the polycomb repressive complex 2 (PRC2). Most of the mutations are located in the SET ([Su(var)3-9, Enhancer-of-zeste and Trithorax]) or CXC (cysteine-rich region) domain at the C-terminus of the protein and are responsible for the catalytic activity of the methyltransferase. Half of mutations cause a stop-gain or frameshift, resulting in a truncated protein [81]. BCOR is a transcription factor involved in the control of embryogenesis, mesenchymal stem cell function, hematopoiesis, and lymphoid development. It acts as a tumor suppressor genes and mutations are localized in the entire gene and might be either nonsense, frameshift, missense or splice mutations [82]. STAG2 is s a core component of the cohesin complex participating in chromatin organization, transcriptional regulation, and DNA repair. Mutations are scattered throughout the gene but the majority are located in the N-terminus and in the STAG domain and are either nonsense, frameshift or splice-site [83]. The identification of MDS-related mutations by NGS, is clinically relevant because it can reveal a sub-group of high-risk AML patients that could benefit from a different frontline treatment (e.g., CPX351 [84]) and consolidation with HSCT.
3.4.3. RUNX1
Mutations are enriched in s-AML ontogeny and ICC has included this gene in the category of AML MDS-related mutations. RUNX1 is a transcription factor, key regulator of hematopoiesis and involved in hematopoietic stem cell emergence and regulation. The mutational spectrum includes missense mutations in the N-terminal domain, mostly located in the RUNT domain, and truncating mutations C-terminal within the transactivation domain (TAD). RUNX1 is associated with poor outcomes across the spectrum of all myeloid malignancies and in AML [85].
3.4.4. AML-Defining Mutations
CEBPA. CEBPA gene is located on chromosome 19q13.11 and is an important transcription factor involved in myeloid differentiation. 5 to 15% of AML cases harbor CEBPA mutations and are more frequent in younger adults [86]. CEBPA mutations are located both in the N-terminal TAD and C-terminal bZIP domains and can be mono or bi-allelic [86]. Both ICC and WHO 2022 classification recognize CEBPA-mutant AML as a distinct entity but differ as ICC includes only AML cases with in-frame b-zip domain mutations (but with lower blast count to 10%) [2,48]. WHO defines the broader category of AML with CEBPA mutations (including both biallelic and b-zip monoallelic mutations). CEBPA mutations can be identified by PCR, NGS is a useful tool which enables to identify both the number of mutations (mono vs biallelic) and the location of the mutations sequencing the N-terminal and the C-terminal regions of the genes [86]. The latter information is relevant as the majority of familiar mutations in CEBPA gene are located in the N-terminal region and are often accompanied by a “second-hit” in the C-terminal domain of the gene [87]. The detection of such a variant (accompanied by VAF≥30% or 40%) should always encourage germ-line testing. Some studies have highlighted the difficulty of NGS in detecting CEBPA mutations when biallelic [57].
NPM1. The nucleophosmin (NPM1) gene encodes for the most abundant nucleolar protein. Thanks to its property to act as a histone chaperone and to shuttle between the nucleus and cytoplasm, the NPM1 protein is involved in multiple cellular functions, including the formation of the nucleolus through liquid-liquid phase separation, regulation of ribosome biogenesis and transport, control of DNA repair and centrosome duplication, and response to nucleolar stress. NPM1 is the most frequently mutated gene in about 30-35% of AML [88,89]. Due to its unique biological and clinical features, NPM1-mutated AML is regarded as a distinct leukemia entity in the WHO 5th edition and ICC classifications of myeloid malignancies [2,48]. The NPM1 mutant undergoes changes at the C-terminus of the protein that leads to its delocalization in the cytoplasm of the leukemic cells [88]. A new class of drugs, named ‘menin inhibitors’, like revumenib, showed effect in r/r AML [90,91,92]. Several studies compared detection of NPM1 by PCR and NGS [93,94,95] highlighting some differences; but in practice, PCR is available faster than the usual turnaround times of NGS [59,96].
3.5. AML Mutations Driving Target Therapies (IDH1/2 and FLT3)
At diagnosis of AML, the presence of mutations in FLT3 and IDH1 or IDH2, represents actionable targets for patients eligible for intensive chemotherapy and ineligible for intensive therapy, respectively [97]. Therefore, it is of utmost importance detecting the presence of these mutations; PCR is still considered the gold standard (e.g., Abbott RealTime IDH1 [98]) mainly for reduced times of response and limited costs. Indeed, we can add to the standard 7+3 backbone midostaurin in presence of FLT3-ITD or FLT3-TKD or add quizartinib only for FLT3-ITD [99,100]. Similarly, in patients ineligible for IC, IDH1 positive cases can benefit from the addition of ivosidenib to a backbone of azacitidine [101]. NGS methods correlate with PCR quite well, but sensitivity is slightly decreased for FLT3-ITD [95].
4. NGS and Impact on Risk Stratification
When hematologists approach to a new patient with AML or MDS, one of the first questions is “What is the prognosis of this neoplasia?”. Several prognostic tools have been developed to answer this key question: obviously age, comorbidity, geriatric score, fitness of the immune system, genetic characteristics AML or MDS, and effectiveness of available therapies play a critical role [102,103,104]. The past scoring systems also employed cytogenetical data (e.g., ELN2017) [105], but the more recent stratification is based on multiple genetic alterations, that require NGS for their detection. Therefore, NGS analysis is becoming mandatory for patients at their diagnosis of AML or MDS. Moreover, initial stratification strategies are important also for therapeutical management (Which patients should undergo allo-SCT?). In this paragraph, we will consider the impact of NGS results on the risk-stratification.
4.1. Myelodysplastic Syndromes: Prognostic Impact of Molecular Data
Myelodysplastic syndromes represent both a diagnostic and classification challenge, mainly due to the genetic complexity of the disease. Genetic mutations are quickly acquiring importance in modern WHO and ICC classifications, and this is paralleled by an effort to improve traditional risk stratification tools in MDS by including mutational asset of the most frequently mutated genes in MDS [106]. Myelodysplastic syndromes (MDS) are indeed a heterogeneous cancer type and accurate survival prediction models are important for therapeutic allocation. Traditionally, the IPSS-R has been the most used risk score to predict MDS patient outcome. IPSS-R stratifies patients by scoring cytopenias and blast % in the bone marrow, but also includes cytogenetic alterations [107]. IPSS-R has been widely validated [108], however the evolution of MDS can be greatly variable, depending on both disease- and patient-related factors. In recent years, the knowledge of the genetic landscape in MDS has increased and multiple studies have investigated the role of somatic mutation in MDS patient outcome. Based on data from 3711 patients with MDS who were profiled for mutations in 152 genes, the IPSS-M is a recently developed score for risk stratification in MDS, which incorporates molecular in addition to more traditional cytopenias, blast counts, and cytogenetics. MDS patients are classified into six categories based on the mutational status of a list of 31 genes that are included in routine diagnostic NGS myeloid panels, divided in 16 main effect genes and 15 residual genes. Multi-hit TP53, FLT3-ITD/TKD, and KMT2A-PTD played major roles in predicting adverse outcomes [109]. Interestingly, 46% of MDS patients were re-stratified with IPSS-M from the original IPSS-R classifications, with the majority upgraded to higher risk categories. IPSS-M has been validated in large patient cohorts for its prognostic value; however, there is still limited data on the therapeutic implication of the current reclassification.
4.2. Acute Myeloid Leukemia
ELN recommendations for AML risk stratification have evolved since the 2010 edition, progressively including the growing evidence and impact of gene mutations in prognostication. Compared to the previous ELN 2010 and 2017, the most recent 2022 update requires mutational data on 13 genes to correctly allocate AML patients in favorable, intermediate or adverse risk categories. In particular, the adverse-risk category has been enriched with 7 gene mutations: BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, ZRSR2 added to ASXL1 and RUNX1 (present in previous ELN 2017) forming the group “myelodysplasia-related” (MR) gene group [6,7]. Therefore, the use of NGS has become mandatory to correctly stratify these patients.
The presence of one of these mutations allocates patients to the adverse-risk category when not accompanied with favorable-risk alterations. The importance of TP53 mutations has been in ELN2022. ELN Several studies have validated ELN2022 guidelines further exploring the role of MR mutations. Most studies highlight the heterogeneity of outcome among different MR mutations allocated to the adverse-risk category, with some of these (i.e., RUNX1, ASXL1, U2AF1) showing worse outcomes while others (i.e., EZH2, STAG2, or ZRSR2) showed an intermediate-risk outcome. The evolving treatment landscape of AML has recently prompted the introduction of low intensity regimens for older or unfit AML patients [110]. Hypomethylating agents (HMA), azacitidine and decitabine, and the HMA-based combination therapies with venetoclax or ivosidenib in IDH1-mutated AML have improved the outcome in these patient categories [101]. However, previous risk stratification, including ELN 2022, was based on clinical trials including exclusively intensively treated patients and the attempt to employ this classification for lower-intensity regimens has been unsatisfactory.
4.2.1. Stratification of AML for Patients Treated with less-Intensive Therapy
In 2024, ELN proposed a genetic-based risk classification (2024 ELN Less-Intensive) for patients with newly diagnosed AML receiving less-intensive regimens based on HMAs [111]. Data were derived from large trial cohorts, including ASTRAL-1 randomizing guadecitabine or other therapies (guadecitabine vs azacitidine, decitabine, LDAC) [112], VIALE-A trial (NCT02993523) randomizing AZA-VEN vs. AZA placebo and phase 3 AGILE study (NCT03173248) adding IDH1-inhibitor IVOSIDENIB to azacytidine (compared to placebo-AZA). Karyotype status together with a small number of genes including NPM1, IDH1 and 2, FLT3-ITD, DDX41, NRAS, KRAS and TP53, was sufficient to create a score to dissect patients into favorable, intermediate and adverse risk categories. Intermediate-risk category in ELN 2024 remains still quite broad, as many cytogenetic and molecular abnormalities do not have solid prognostic data in the context of AML receiving less-intensive therapies. In addition, following the path of IDH1 mutations which are now considered favorable risk after the introduction of specific inhibitors, other gene mutations could be reclassified as new target therapies become available.
The Role of NGS in MRD Measurement and Monitoring
Measurable residual disease (MRD) has become very important in AML patients to guide therapy (e.g., selection of the best consolidation strategies, maintenance, follow-up, DLI, etc.), monitoring for relapses, and as a prognostic biomarker [113,114].
The 2021 Update on MRD in AML, from the European LeukemiaNet MRD working party, considered NGS-MRD monitoring as useful to refine prognosis in addition to MFC, but to date, there are insufficient data to recommend NGS-MRD as a stand-alone technique [115]. The main pitfall is the relatively low sensitivity of NGS methods to detect little clones of MRD. Indeed, in diagnostic test the significativity is considered >5%; while NGS-MRD test positivity (measured on genomic DNA) is provisionally defined as ≥0.1% VAF. Although NGS-MRD test negativity is defined as <0.1% VAF, results <0.1% may still be associated with adverse outcomes [115].
The advantage of NGS-MRD is the potential to detect clonal heterogeneity and gain/loss of mutations that anticipate relapse of AML. For example, NPM1mut, which is considered a stable marker for qRT-PCR detection [116], can negativize at relapse in about 10% of patients [117,118]. Therefore, studying a panel of genes can better monitor the clonal shift of AML and detect new genes predicting imminent morphologic relapse. Nevertheless, NGS-MRD has several disadvantages: the low sensitivity due to error rates unless error-correction method included [119,120], some mutated genes found in healthy people without hematologic abnormalities, mutations in DNMT3A, TET2, and ASXL1 (DTA) can be found in age-related clonal hematopoiesis, Germline mutations (VAF of about 50%) are noninformative for MRD [115]; genetic clonal heterogeneity and evolution over time [121,122,123]; emergence or selection of sub-clone(s) at relapse [124]. In a post hoc analysis of the data from the MORPHO trial, the level of MRD detected with this approach correlated remarkably with RFS and the risk of relapse, and that MRD positivity at any level negatively affected RFS [125].
Despite the potentials of MRD-NGS, it remains an investigational technique, not yet standardized (also the correct timepoint is controversial [126]) and widespread in all laboratories; we will report two exceptions (FLT3 MRD and a panel of 5 genes) that are near to entering into clinical practice.
The gene encoding fms-like tyrosine kinase-3 (FLT3) is the most frequently (25%) mutated gene in AML, with internal tandem duplications (ITDs) varying in length from three base pairs (bp) to more than 400 bp, expected to result in a functional protein due to their in-frame nature [127]. Therefore, each patient with FLT3-mut has a peculiar ITD, that is difficult to detect and monitoring by PCR-MRD, during the disease timepoints. An interesting laboratory technique produced by Invivoscribe® -and approved by FDA- combines an initial PCR to amplify FLT3-ITD mutations. Barcoded primers target the area around the juxtamembrane (JM) region of the FLT3 gene to amplify the sequences interested by duplication in DNA. The PCR products are then purified and prepared for sequencing in NGS [128]. This assay detects ITDs from 9 base pairs up to a length of 252 base pairs and insertions from 3 base pairs to less than the maximum ITD size with an analytical sensitivity of 5 x 10-5 mutant alleles per total alleles. A powerful demonstration of the NGS/PCR method for the detection of FLT3 MRD was established with the Morhpo trial. In this study, patients received gilteritinib for 2 years as post allo-HSCT; half of the participants had MRD detectable pre- or post-HSCT, and, in a prespecified subgroup analysis, gilteritinib was beneficial in this population (HR, 0.515 [95% CI, 0.316 to 0.838]; P = .0065), but not in those patients with undetectable MRD [129]. It is forecasted that this NGS/PCR technique for FLT3 may be recommended in subsequent MRD testing guidelines [113].
The last example of NGS-MRD is the pre-MEASURE study, testing in patients with AML the value of a panel of 5 genes: DNA sequencing was performed on pretransplant blood from patients before allogeneic hematopoietic cell transplant in variants in FLT3, NPM1, IDH1, IDH2, and KIT using targeted error–corrected DNA sequencing. The persistence of FLT3 internal tandem duplication or NPM1 variants in the blood at an allele fraction of 0.01% or higher was associated with increased relapse and worse survival compared with those without these variants [130]. Moreover, in the ALFA0702 study (NCT00932412) an error-corrected-NGS-MRD showed that the persistence of non-DTA mutations (HR = 2.23 for RFS and 2.26 for OS), and DTA mutations (HR = 2.16 for OS) were associated with poorer prognosis in multivariate analysis [131].
6. The Role of NGS in the Detection of Germline Predisposition [132]
When we approach a new patient with AML, we have an unprecedented opportunity to detect germline predisposition simply by requesting a diagnostic panel of NGS. In a Korean group of patients with AML, the prevalence of germline mutations was 7.2% [133]. The presence of a mutation in gene in germline and not as somatic acquisition can be suspected when VAF is higher ≥30% or 40%. [134,135] Therefore, NGS is a revolution to our past clinical concept of “familial MDS/AML” where at least two or more first- and/ or second-degree relatives in the same family develop acute leukemia (AL), myeloid malignancy or characteristic cytopenias, with one case being MDS or AML [136]. Conversely, when we perform diagnostic NGS, we should be aware, and possibly always correctly inform patients, that an NGS performed at diagnosis of AML/MDS could disclose germline mutations that could reveal familial MDS/AML with clinical, psychological, and social implications for the patients and his/her relatives [136,137]. Moreover, the presence of familial mutations with germline predisposition can have an impact on the choice of donor source when hematopoietic cell transplant is considered from a relative of the patient, albeit donor derived leukemia is a rare event (about 1%) and case reports of MDS derived from donor have been reported [16,138,139,140]. The ELN-2022 guidelines list the clinical features that should prompt testing of germline predisposition: (at least 2 cancers, age of diagnosis of AML/MDS earlier than average, and personal history of hematologic cancer and another relative within 2 generations with a hematologic cancer or a solid tumor <50 years, or with other hematopoietic abnormalities) [3]. Conversely, the Nordic MDS study group (NMDSG) suggested in their guidelines a slightly different group of patients, that should be studied for myeloid neoplasms with germline predisposition, and recommended confirming the genes identified by NGS ad diagnosis on a different tissue like fibroblast or peripheral blood in remission [135,141].
The 5th edition of WHO classification of hematolymphoid tumors has organized myeloid neoplasms associated with germline predisposition into three different subgroups: Myeloid neoplasms with germline predisposition without a preexisting platelet disorder or organ dysfunction (DDX41, TP53, CEBPA); Myeloid neoplasms with germline predisposition and pre-existing platelet disorder (RUNX1, ANKRD26, ETV6); and Myeloid neoplasms with germline predisposition and potential organ dysfunction (GATA2, Shwachman-Diamond syndrome (SDS) [142], Fanconi anemia (FA) [143], Down syndrome [144], etc.) [2,145].
6.1. DDX41
The most frequent genetic alteration in the general population is the DDX41, which accounts for about 80% of the germinal myeloid neoplasms [146]. DEAD-box helicase 41 (DDX41), localized on chromosome 5q35, in particular has an important role in innate immune sensing and hematopoietic homeostasis. DDX41 recognizes foreign or self-nucleic acids generated during microbial infection, to initiate an anti-pathogen response [147]. In one of the largest studies conducted on international cohorts of 346 patients with DDX41 from 9082 myeloid neoplasms (MN) patients, DDX41 explained 80% of the germline predisposition to MN. In particular, in the Japanese male population, patients had a 10-folds higher incidence of DDX41 than the general population. Moreover, until 40 years of age, people showed low risk of AML/MDS, but it increased to about 50% by 90 years of age [148]. Moreover, DDX41 seems associated with favorable prognosis when AML is treated with HMA + venetoclax and this category is categorized as favorable risk by ELN2024 [111]; however, there are very limited numbers of these patients [149,150] that make this findings preliminary. Similarly, in patients treated with intensive chemotherapy was reported the favorable prognostic role of mDDX41 in MDS/AML with longer OS when compared to DDX41 wild-type AML and favorable risk AML [151,152,153]. The favorable effect of DDX41 mitigates other negative markers like TP53 [148]. Recommendations from the Nordic working group on germline predisposition for myeloid neoplasms have been published, and include information on symptomatic carriers of germline DDX41 variants to include in surveillance programs [146].
6.2. TP53
The TP53 gene encodes a protein considered one of the cell’s most crucial transcription factors, often referred to as the “guardian of the genome.” Due to its key functions, TP53 is the most commonly mutated tumor suppressor gene in the development of cancers. Li-Fraumeni syndrome is caused by a pathogenic germline mutation in TP53 and is linked to a markedly increased risk of various solid tumors—such as those of the breast, pancreas, central nervous system, and sarcomas—as well as blood cancers like MDS, AML, and ALL. By the age of 70, the likelihood of developing cancer in affected individuals approaches nearly 100%. In these patients, myeloid hematologic malignancies often arise secondary to prior radiation or chemotherapy treatments administered for other cancers. Mutations in TP53 greatly increase the risk associated with these blood cancers and are consistently correlated with a poor prognosis [154].
6.3. GATA2
The GATA-binding protein 2 (GATA2) has a central role in blood stem cell generation and maintenance; GATA2 plays a direct role in modulating p53-induced apoptosis via the regulation of the MDM2 modulator RASSF4, explaining chemo-resistance [155]. The GATA2 syndrome was first described in 2011 in patients with absence of monocytes, B cells, B-cell precursors, NK cells and/ or plasmacytoid dendritic cells in blood; late onset infections (non-tuberculous mycobacterial infections, fungal infections, and human papillomavirus (HPV) infections); pulmonary alveolar proteinosis; risk to progress to MDS or AML, and a pattern of inheritance autosomal dominant with mutations in one allele of GATA2 (haplo-insufficient) [156]. Germline GATA2 mutations accounted for 15% of advanced and 7% of all primary MDS cases in a population of pediatric patients, so GATA2 is the most common pediatric germline mutation [157]. Albeit data on the efficacy of allo-transplant derives from small groups of patients with somatic GATA2-mutated AML, its role seems beneficial, also to reduce the risk of opportunistic infections [158]. Given that there are almost 200 unique (likely) pathogenic variants described that can be classified into four groups: 1) truncating mutations (splice site, nonsense, frameshift, and whole-gene deletions) proximal to or within the ZF2 domain; 2) missense mutations within the ZF2 domain; 3) mutations resulting in aberrant mRNA splicing (e.g., synonymous changes); 4 other regulatory variants [159]; it is mandatory to perform an NGS study of the whole gene to detect all these variants.
6.4. RUNX1
The gene encoding RUNX1 (a transcription factor) was identified in AML carrying translocation t(8,21), resulting in its previous name AML1, but later, for homology to the Drosophila transcription factor runt, the name of this transcription factor family was changed to runt-related proteins, or “Runx” [160]. In 1999, a familial platelet disorder with associated myeloid malignancy, a rare autosomal dominant disease associated with low platelets, was identified. In a study of 27 families with RUNX1 mutations, half of the patients had a CHIP and sequential sequencing data from 19 patients demonstrated dynamic changes of somatic mutations over time [161]. AML patients with RUNX1 mutations have a dismal prognosis [162]. Since multiple alterations in RUNX1 have been found, including multiexon or whole gene deletions, frameshifts, missense and nonsense mutations, [163] NGS is required. Moreover, the differential diagnosis of other familial disorders associated with thrombocytopenia can be achieved by sequencing ETV6 [164,165] and ANKRD26 [166,167,168].
6.5. ETV6
The ETV6 gene produces a transcriptional repressor belonging to the ETS family, playing a key role in the maturation of red blood cells and megakaryocytes [165]. Germline mutations in ETV6 are inherited in an autosomal dominant fashion and can lead to thrombocytopenia type 5, with risk—approximately 30%—of developing blood cancers. These malignancies can be either lymphoid (such as acute lymphoblastic leukemia, ALL) or myeloid (including acute myeloid leukemia, AML, and myelodysplastic syndromes, MDS). Similar to other hereditary blood disorders, the onset of cancer usually requires at least one additional acquired mutation in genes commonly linked to leukemia development.
7. The Impact of NGS in the Detection of Druggable Mutations at Relapse
In patients with AML relapsed after several lines of therapy or after allogeneic transplantation, the prognosis is dismal, with necessity to activate early palliative care or enroll patients in clinical trials [169,170,171]. However, in the near future, NGS might become a compass to guide targeting actionable genes detected in r/r AML patients. Indeed, an Italian case report of two patients revealed clonal and sub-clonal evolution that could favor uncovering new therapeutic targets [172]. In a more robust dataset of 1470 AML patients, NGS was set to explore 17 potentially actionable genes (ALK, CSF1R, FGFR1/2/3, FLT3, IDH1/2, JAK2, KDR, KRAS/NRAS, NPM1, PDGFRA, PTPN11, RET and TP53), because they are directly or indirectly targeted with standard (6% received off-label agents) or investigational agents (53% of these enrolled in clinical trials) [173]. The main pitfall of this approach is that many mutations identified by NGS are either linked to drugs not yet approved, or cannot be targeted by currently available treatments (e.g., TP53), but NGS promises to improve survival and quality of life of older adults [174].
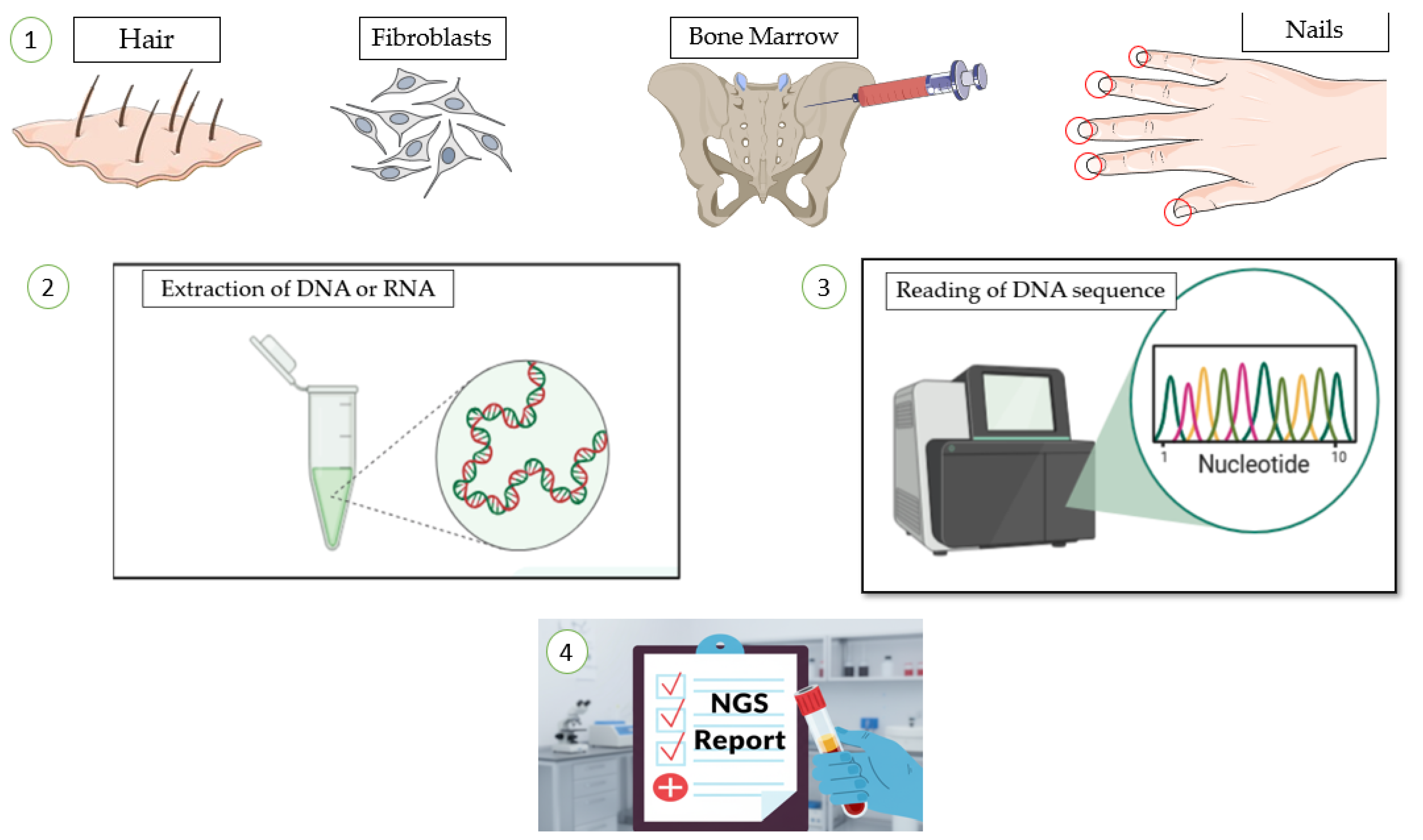
Figure 2.
The workflow of genetic analysis NGS, from sample collection to result reporting. 1) Biological specimens, such as hair, fibroblasts, bone marrow, or nails, are first collected from the patient. 2) nucleic acids (DNA or RNA) are isolated from these samples in the laboratory. 3) The extracted genetic material is then processed and sequenced to determine the precise order of nucleotides. This process generates data that are analyzed by bio-informatics to interpret the genetic sequence. 4) The findings are compiled into a formal laboratory report for clinical assessment.
Figure 2.
The workflow of genetic analysis NGS, from sample collection to result reporting. 1) Biological specimens, such as hair, fibroblasts, bone marrow, or nails, are first collected from the patient. 2) nucleic acids (DNA or RNA) are isolated from these samples in the laboratory. 3) The extracted genetic material is then processed and sequenced to determine the precise order of nucleotides. This process generates data that are analyzed by bio-informatics to interpret the genetic sequence. 4) The findings are compiled into a formal laboratory report for clinical assessment.
8. Discussion
The advent of faster and cheapest NGS techniques promises to revolutionize the field of MN. The availability of several gene Panels (e.g., the Illumina TruSight Myeloid panel, the Archer VariantPlex Core Myeloid panel, the Human Myeloid Neoplasms QIASeq DNA Panel and the AmpliSeq for Illumina Myeloid panel). The presence of several translocations can be revealed using a single diagnostic test with RNA-based NGS (RNA sequencing), saving money and lab resources from extensive cytogenetic studies, and the few specific real-time quantitative PCR for selected transcripts [175]. With RNA sequencing of the known gene partner, finding other rare partner has become easier (e.g., KMT2A) [176]. Moreover, the possibility to sequence long genes, that present technical difficulty to sequence (e.g., CEBPA is a GC-rich gene which is particularly difficult to amplify by PCR or TP53), can help clinicians to assign a patient to a different risk-group. Nevertheless, we still have a limited number of drugs in the first line setting, to offer each patient a personalized approach and precision medicine. In a retrospective study, patients were reevaluated in light of new findings after NGS/cytogenetic results, but they showed no significant statistical difference of survival between the two groups [96].
For AML/MDS patients, probably TP53 mutations represent one of the most negative prognostic markers, given the low sensitivity to chemotherapy (including venetoclax-based treatments) and the substantial limited activity of HSCT [177,178,179,180]. Therefore, sequencing TP53 is of utmost importance, albeit technical challenges are present (e.g., the presence of VUS not covered in actual databases) [181]. Unfortunately, no p53-based therapeutics have been approved, yet, probably because, as a nuclear transcription factor, p53 does not possess typical drug target features and has therefore long been considered undruggable [182]. Another hot topic is the role of NGS as MRD marker, that is important to refine the selection of AML patients to transplant in 1st CR, and to maintain with pharmacological intervention (e.g., gilteritinib in MRD+ patients after transplant) [129]. Moreover, patients who relapse show clonal evolution and biological shifts that harden treatment, but NGS could represent an opportunity to study specific mechanisms of resistance to targeted drugs such as FLT3-inhibitors [183,184]. In the future, personalized medicine could apply to patients rapidly identified by NGS; for example, Menin inhibitors for KMT2A-rearranged AML [90].
9. Conclusions
Reading the final report of an NGS exam in a patient with AML or MDS is a challenging task. Clinicians should be aware of the potential and pitfall of the technique employed by their laboratory, including the platform used, the depth of readings, and the interpretation of variants identified. VAF is another critical concept that should be considered especially in patient who present with the typical mutation of clonal hemopoiesis (e.g., DNMT3A, SRSF2, TET2), to take in mind the dimension of the clone present at the diagnosis and in the follow-up timepoints. Another important concept is that not all the mutation in the same gene are the same, and some cases still have unknown clinical significance (VUS), that require access to databases, but limited info could be available [185]. We recognize that currently clinicians are navigating the growing complexity of molecular data in daily practice, therefore we encourage a direct interaction with biotechnologists and bioinformatics involved in the process of NGS and writing of reports. Moreover, for patients with suspected germline predisposition to MN a genetic counseling and testing for a multi-gene panel is advised [186]. In conclusion, the cross-talk between hematologists and biotechnologists about the NGS report is not intended as the endpoint, but the beginning of a multi-disciplinary discussion, in particular in most challenging cases.
10. Future Directions
The field of NGS in AML and MDS is changing fast, with a broader availability of cheaper, simpler and faster sequencing platforms. Clinicians are growing their ability and experience to interpret NGS reports from their patients. We believe this will change profoundly our clinical practice, because each patient (including elderly patients) will undergo NGS. The complexity of the field, might appear overwhelming at first glance, including consultation with website-based databases of pathologic variants. Nevertheless, the integration of algorithms of Artificial Intelligence, could simplify the task of hematologist and avoid the stuck into analysis paralysis [187,188].
Author Contributions
Conceptualization, S.R., S.P. and L.C.; writing—original draft preparation, N.Z, S.P., L.C.; writing—review and editing, all.; visualization, S.P.; supervision, C.T.; project. All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AML | Acute myeloid leukemia |
| CCUS | Clonal cytopenia of undetermined significance |
| CHIP | Clonal Hematopoiesis of Indeterminate Potential |
| HSCT | Hematopoietic stem cell transplantation |
| ICC | International Consensus Conference |
| MDS | Myelodysplastic neoplasms |
| MN | Myeloid neoplasms |
| MRD | Measurable residual disease |
| NGS | Next generation sequencing |
| SBS | Sequencing by synthesis |
| VAF | Variant allele frequencies |
| VUS | Variants of uncertain significance |
| WES | Whole exome sequencing |
| WGS | Whole genome sequencing |
| WHO | World Health Organization |
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. New England Journal of Medicine 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Gooptu, M.; Murdock, H.M.; Soiffer, R.J. How I Treat AML Relapse after Allogeneic HSCT. Blood 2025, 145, 2128–2137. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Schmid, S.; Jochum, W.; Padberg, B.; Demmer, I.; Mertz, K.D.; Joerger, M.; Britschgi, C.; Matter, M.S.; Rothschild, S.I.; Omlin, A. How to Read a Next-Generation Sequencing Report—What Oncologists Need to Know. ESMO Open 2022, 7, 100570. [Google Scholar] [CrossRef]
- Mosele, M.F.; Westphalen, C.B.; Stenzinger, A.; Barlesi, F.; Bayle, A.; Bièche, I.; Bonastre, J.; Castro, E.; Dienstmann, R.; Krämer, A.; et al. Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Advanced Cancer in 2024: A Report from the ESMO Precision Medicine Working Group. Annals of Oncology 2024, 35, 588–606. [Google Scholar] [CrossRef]
- Kim, Y.J.; Takahashi, K. Emerging Technologies of Single-Cell Multi-Omics. Haematologica 2025, 110, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed]
- Duncavage, E.J.; Bagg, A.; Hasserjian, R.P.; DiNardo, C.D.; Godley, L.A.; Iacobucci, I.; Jaiswal, S.; Malcovati, L.; Vannucchi, A.M.; Patel, K.P.; et al. Genomic Profiling for Clinical Decision Making in Myeloid Neoplasms and Acute Leukemia. Blood 2022, 140, 2228–2247. [Google Scholar] [CrossRef] [PubMed]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library Construction for Next-Generation Sequencing: Overviews and Challenges. Biotechniques 2014, 56, 61–64. [Google Scholar] [CrossRef]
- Satam, H.; Joshi, K.; Mangrolia, U.; Waghoo, S.; Zaidi, G.; Rawool, S.; Thakare, R.P.; Banday, S.; Mishra, A.K.; Das, G.; et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology 2023, 12, 997. [Google Scholar] [CrossRef]
- Lee, J.Y. The Principles and Applications of High-Throughput Sequencing Technologies. Dev Reprod 2023, 27, 9–24. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat Rev Genet 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Peroni, E.; Randi, M.L.; Rosato, A.; Cagnin, S. Acute Myeloid Leukemia: From NGS, through scRNA-Seq, to CAR-T. Dissect Cancer Heterogeneity and Tailor the Treatment. Journal of Experimental & Clinical Cancer Research 2023, 42, 259. [Google Scholar] [CrossRef]
- O’Connor, T.E.; Shaw, R.; Madero-Marroquin, R.; Roloff, G.W. Clinical Considerations at the Intersection of Hematopoietic Cell Transplantation and Hereditary Hematopoietic Malignancy. Front. Oncol. 2023, 13, 1180439. [Google Scholar] [CrossRef]
- Thomas, M.; Sukhai, M.A.; Zhang, T.; Dolatshahi, R.; Harbi, D.; Garg, S.; Misyura, M.; Pugh, T.; Stockley, T.L.; Kamel-Reid, S. Integration of Technical, Bioinformatic, and Variant Assessment Approaches in the Validation of a Targeted Next-Generation Sequencing Panel for Myeloid Malignancies. Arch Pathol Lab Med 2017, 141, 759–775. [Google Scholar] [CrossRef] [PubMed]
- Akabari, R.; Qin, D.; Hussaini, M. Technological Advances: CEBPA and FLT3 Internal Tandem Duplication Mutations Can Be Reliably Detected by Next Generation Sequencing. Genes (Basel) 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Cumbo, C.; Minervini, C.F.; Orsini, P.; Anelli, L.; Zagaria, A.; Minervini, A.; Coccaro, N.; Impera, L.; Tota, G.; Parciante, E.; et al. Nanopore Targeted Sequencing for Rapid Gene Mutations Detection in Acute Myeloid Leukemia. Genes (Basel) 2019, 10. [Google Scholar] [CrossRef]
- Warburton, P.E.; Sebra, R.P. Long-Read DNA Sequencing: Recent Advances and Remaining Challenges. Annu Rev Genomics Hum Genet 2023, 24, 109–132. [Google Scholar] [CrossRef]
- Moustakli, E.; Christopoulos, P.; Potiris, A.; Zikopoulos, A.; Mavrogianni, D.; Karampas, G.; Kathopoulis, N.; Anagnostaki, I.; Domali, E.; Tzallas, A.T.; et al. Long-Read Sequencing and Structural Variant Detection: Unlocking the Hidden Genome in Rare Genetic Disorders. Diagnostics (Basel) 2025, 15. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing Depth and Coverage: Key Considerations in Genomic Analyses. Nat Rev Genet 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Anu, R.I.; Patel, A.; Pathak, N.; Mehta, P.; Chougule, A.; Sheth, H.; Veldore, V.; Suryavanshi, M.; Gupta, V.G.; Disel, U.; et al. Uniform Reporting of Next Generation Sequencing: Indian Society of Medical and Pediatric Oncology. Indian J Med Paediatr Oncol 2025, 46, 351–362. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol 2016, 17, 13. [Google Scholar] [CrossRef]
- Dillon, L.W.; Hayati, S.; Roloff, G.W.; Tunc, I.; Pirooznia, M.; Mitrofanova, A.; Hourigan, C.S. Targeted RNA-Sequencing for the Quantification of Measurable Residual Disease in Acute Myeloid Leukemia. Haematologica 2019, 104, 297–304. [Google Scholar] [CrossRef]
- Sa, A.C.C.; Sadee, W.; Johnson, J.A. Whole Transcriptome Profiling: An RNA-Seq Primer and Implications for Pharmacogenomics Research. Clin Transl Sci 2018, 11, 153–161. [Google Scholar] [CrossRef]
- Baccarella, A.; Williams, C.R.; Parrish, J.Z.; Kim, C.C. Empirical Assessment of the Impact of Sample Number and Read Depth on RNA-Seq Analysis Workflow Performance. BMC Bioinformatics 2018, 19, 423. [Google Scholar] [CrossRef]
- Shin, H.T.; Choi, Y.L.; Yun, J.W.; Kim, N.K.D.; Kim, S.Y.; Jeon, H.J.; Nam, J.Y.; Lee, C.; Ryu, D.; Kim, S.C.; et al. Prevalence and Detection of Low-Allele-Fraction Variants in Clinical Cancer Samples. Nat Commun 2017, 8, 1377. [Google Scholar] [CrossRef] [PubMed]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front Oncol 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R. Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics (Basel) 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Al-Kateb, H.; Knight, S.M.; Sivasankaran, G.; Voss, J.S.; Pitel, B.A.; Blommel, J.H.; Jerde, C.R.; Rumilla, K.M.; Lee, J.L.; Mattson, N.R.; et al. Clinical Validation of the TruSight Oncology 500 Assay for the Detection and Reporting of Pan-Cancer Biomarkers. J Mol Diagn 2025, 27, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Ananth, S.; Njau, A.; Ahluwalia, P.; Jones, K.; Ahluwalia, M.; Okechukwu, N.; Savage, N.M.; Kota, V.; et al. Clinical Performance and Utility of a Comprehensive Next-Generation Sequencing DNA Panel for the Simultaneous Analysis of Variants, TMB and MSI for Myeloid Neoplasms. PLoS One 2020, 15, e0240976. [Google Scholar] [CrossRef]
- Serrati, S.; De Summa, S.; Pilato, B.; Petriella, D.; Lacalamita, R.; Tommasi, S.; Pinto, R. Next-Generation Sequencing: Advances and Applications in Cancer Diagnosis. Onco Targets Ther 2016, 9, 7355–7365. [Google Scholar] [CrossRef]
- Koboldt, D.C. Best Practices for Variant Calling in Clinical Sequencing. Genome Med 2020, 12, 91. [Google Scholar] [CrossRef]
- Sun, Q.Y.; Ding, L.W.; Tan, K.T.; Chien, W.; Mayakonda, A.; Lin, D.C.; Loh, X.Y.; Xiao, J.F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of Mutations in Acute Myeloid Leukemia with Partial Tandem Duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Abel, H.J.; Lockwood, C.M.; Payton, J.E.; Szankasi, P.; Kelley, T.W.; Kulkarni, S.; Pfeifer, J.D.; Duncavage, E.J. Detection of FLT3 Internal Tandem Duplication in Targeted, Short-Read-Length, next-Generation Sequencing Data. J Mol Diagn 2013, 15, 81–93. [Google Scholar] [CrossRef]
- Aguilera-Diaz, A.; Vazquez, I.; Ariceta, B.; Manu, A.; Blasco-Iturri, Z.; Palomino-Echeverria, S.; Larrayoz, M.J.; Garcia-Sanz, R.; Prieto-Conde, M.I.; Del Carmen Chillon, M.; et al. Assessment of the Clinical Utility of Four NGS Panels in Myeloid Malignancies. Suggestions for NGS Panel Choice or Design. PLoS One 2020, 15, e0227986. [Google Scholar] [CrossRef]
- Levis, M.; Shi, W.; Chang, K.; Laing, C.; Pollner, R.; Gocke, C.; Adams, E.; Berisha, F.; Lameh, J.; Lesegretain, A. FLT3 Inhibitors Added to Induction Therapy Induce Deeper Remissions. Blood 2020, 135, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Keegan, A.; Bridge, J.A.; Lindeman, N.I.; Long, T.A.; Merker, J.D.; Moncur, J.T.; Montgomery, N.D.; Nagarajan, R.; Rothberg, P.G.; Routbort, M.J.; et al. Proficiency Testing of Standardized Samples Shows High Interlaboratory Agreement for Clinical Next Generation Sequencing-Based Hematologic Malignancy Assays With Survey Material-Specific Differences in Variant Frequencies. Arch Pathol Lab Med 2020. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, K.S.; Lee, T.G.; Lee, S.; Shin, S.; Lee, S.T. A Comparative Study of Next-Generation Sequencing and Fragment Analysis for the Detection and Allelic Ratio Determination of FLT3 Internal Tandem Duplication. Diagn Pathol 2022, 17, 14. [Google Scholar] [CrossRef]
- Chaudhary, S.; Chaudhary, P.; Ahmad, F.; Arora, N. Acute Myeloid Leukemia and Next-Generation Sequencing Panels for Diagnosis: A Comprehensive Review. J Pediatr Hematol Oncol 2024, 46, 125–137. [Google Scholar] [CrossRef]
- Hobeck, A.D.; Wendt, S.; Krohn, S.; Knuebel, G.; Bartels, S.; Schipper, E.; Junghanss, C.; Murua Escobar, H. Comparative Analyses of Targeted Myeloid Cancer Next-Generation Sequencing Panel in Fresh Blood, Bone Marrow and FFPE Material. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Fenaux, P.; Haase, D.; Santini, V.; Sanz, G.F.; Platzbecker, U.; Mey, U.; Esmo Guidelines Committee. Electronic address: clinicalguidelines@esmo. org Myelodysplastic Syndromes: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-up(Dagger☆). Ann Oncol 2021, 32, 142–156. [Google Scholar] [CrossRef]
- Jumniensuk, C.; Nobori, A.; Lee, T.; Senaratne, T.N.; Rao, D.; Pullarkat, S. Concordance of Peripheral Blood and Bone Marrow Next-Generation Sequencing in Hematologic Neoplasms. Adv Hematol 2022, 2022, 8091746. [Google Scholar] [CrossRef] [PubMed]
- Lucas, F.; Michaels, P.D.; Wang, D.; Kim, A.S. Mutational Analysis of Hematologic Neoplasms in 164 Paired Peripheral Blood and Bone Marrow Samples by Next-Generation Sequencing. Blood Adv 2020, 4, 4362–4365. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Zheng, Z.; Wu, Y.; Zhang, Z.; Xu, Z.; Liu, Y.; Jiang, L.; Wu, J. NGS Panel Enhance Precise Diagnosis of Myeloid Neoplasms under WHO-HAEM5 and International Consensus Classification: An Observational Study. Medicine (Baltimore) 2024, 103, e38556. [Google Scholar] [CrossRef] [PubMed]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn 2017, 19, 4–23. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Kwon, R.; Yeung, C.C.S. Advances in Next-Generation Sequencing and Emerging Technologies for Hematologic Malignancies. Haematologica 2024, 109, 379–387. [Google Scholar] [CrossRef]
- Horgan, D.; Curigliano, G.; Riess, O.; Hofman, P.; Buttner, R.; Conte, P.; Cufer, T.; Gallagher, W.M.; Georges, N.; Kerr, K.; et al. Identifying the Steps Required to Effectively Implement Next-Generation Sequencing in Oncology at a National Level in Europe. J Pers Med 2022, 12. [Google Scholar] [CrossRef]
- Darlington, M.; Sujobert, P.; Kosmider, O.; Luque Paz, D.; Kaltenbach, S.; Figeac, M.; Hayette, S.; Mezaour, N.; Coquerelle, S.; Alary, A.S.; et al. Targeted High-Throughput Sequencing for Hematological Malignancies: A GBMHM Survey of Practice and Cost Evaluation in France. Hemasphere 2023, 7, e943. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the Classification of Pathogenicity of Somatic Variants in Cancer (Oncogenicity): Joint Recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genetics in Medicine 2022, 24, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Christinat, Y.; Hamelin, B.; Alborelli, I.; Angelino, P.; Barbié, V.; Bisig, B.; Dawson, H.; Frattini, M.; Grob, T.; Jochum, W.; et al. Reporting of Somatic Variants in Clinical Cancer Care: Recommendations of the Swiss Society of Molecular Pathology. Virchows Arch 2024, 485, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Balducci, S.; Guerrini, F.; Del Re, M.; Cacciola, R. Digital Droplet PCR in Hematologic Malignancies: A New Useful Molecular Tool. Diagnostics 2022, 12, 1305. [Google Scholar] [CrossRef]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute Quantification by Droplet Digital PCR versus Analog Real-Time PCR. Nat Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the Introduction of Next-Generation Sequencing (NGS) for Diagnostics of Myeloid Malignancies into Clinical Routine Use. Blood Cancer J 2018, 8, 113. [Google Scholar] [CrossRef]
- Panuzzo, C.; Jovanovski, A.; Ali, M.S.; Cilloni, D.; Pergolizzi, B. Revealing the Mysteries of Acute Myeloid Leukemia: From Quantitative PCR through Next-Generation Sequencing and Systemic Metabolomic Profiling. JCM 2022, 11, 483. [Google Scholar] [CrossRef]
- Wurm, S.; Waltersdorfer, M.; Loindl, S.; Moritz, J.M.; Herzog, S.A.; Bachmaier, G.; Berghold, A.; Kashofer, K.; Beham-Schmid, C.; Hoefler, G.; et al. Acute Myeloid Leukemia in the Next-Generation Sequencing Era: Real-World Data from an Austrian Tertiary Cancer Care Center. Wien Klin Wochenschr 2024. [Google Scholar] [CrossRef]
- da Rosa, S.E.A.; de Lima, L.B.; Silveira, C.N.; Cortes, L.G.F.; de Oliveira Filho, J.B.; de Souza Reis, R.; Cervato, M.C.; Rodrigues, P.H.S.; de Oliveira Pelegrino, K.; Petroni, R.C.; et al. Real-World Genomic Profiling of Acute Myeloid Leukemia and the Impact of European LeukemiaNet Risk Stratification 2022 Update. Clin Transl Oncol 2023, 25, 3431–3436. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, J.; Qin, T.; Xu, Z.; Qu, S.; Pan, L.; Li, B.; Wang, H.; Zhang, P.; Yan, X.; et al. Comparison of the Revised 4th (2016) and 5th (2022) Editions of the World Health Organization Classification of Myelodysplastic Neoplasms. Leukemia 2022, 36, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N Engl J Med 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N Engl J Med 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- DeZern, A.E.; Malcovati, L.; Ebert, B.L. CHIP, CCUS, and Other Acronyms: Definition, Implications, and Impact on Practice. American Society of Clinical Oncology Educational Book 2018. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. New England Journal of Medicine 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Schuermans, A.; Honigberg, M.C. Clonal Haematopoiesis in Cardiovascular Disease: Prognostic Role and Novel Therapeutic Target. Nat Rev Cardiol 2025. [Google Scholar] [CrossRef]
- Xie, Z.; Komrokji, R.; Al Ali, N.; Regelson, A.; Geyer, S.; Patel, A.; Saygin, C.; Zeidan, A.M.; Bewersdorf, J.P.; Mendez, L.; et al. Risk Prediction for Clonal Cytopenia: Multicenter Real-World Evidence. Blood 2024, 144, 2033–2044. [Google Scholar] [CrossRef]
- Huber, S.; Baer, C.; Hutter, S.; Wossidlo, N.; Hoermann, G.; Pohlkamp, C.; Walter, W.; Meggendorfer, M.; Kern, W.; Haferlach, T.; et al. Genomic Landscape of CCUS Compared to MDS and Its Implications on Risk Prediction. Leukemia 2024, 38, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Gallì, A.; Travaglino, E.; Ambaglio, I.; Rizzo, E.; Molteni, E.; Elena, C.; Ferretti, V.V.; Catricalà, S.; Bono, E.; et al. Clinical Significance of Somatic Mutation in Unexplained Blood Cytopenia. Blood 2017, 129, 3371–3378. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N Engl J Med 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical Significance of SF3B1 Mutations in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat Med 2020, 26, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Hart, S.A.; Lee, L.A.; Seegmiller, A.C.; Mason, E.F. Diagnosis of TP53 -Mutated Myeloid Disease by the ICC and WHO Fifth Edition Classifications. Blood Advances 2025, 9, 445–454. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neuberg, D.; et al. Clinical Effect of Point Mutations in Myelodysplastic Syndromes. N Engl J Med 2011, 364, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute Myeloid Leukemia Ontogeny Is Defined by Distinct Somatic Mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the Spliceosome Machinery, a Novel and Ubiquitous Pathway in Leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef]
- Visconte, V.; O. Nakashima, M.; J. Rogers, H. Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers 2019, 11, 1844. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Herzig, J.K.; Aulitzky, T.; Bullinger, L.; Spath, D.; Teleanu, V.; Kundgen, A.; Kohne, C.-H.; et al. ASXL1 Mutations in Younger Adult Patients with Acute Myeloid Leukemia: A Study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica 2015, 100, 324–330. [Google Scholar] [CrossRef]
- Stasik, S.; Middeke, J.M.; Kramer, M.; Röllig, C.; Krämer, A.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; et al. EZH2 Mutations and Impact on Clinical Outcome: An Analysis in 1,604 Patients with Newly Diagnosed Acute Myeloid Leukemia. Haematologica 2020, 105, e228–e231. [Google Scholar] [CrossRef] [PubMed]
- Ramil, G.; Pratcorona, M.; Nomdedéu, J.F. Be Aware of the X: BCOR Mutations in Myeloid Neoplasms. haematol 2024. [Google Scholar] [CrossRef]
- Eckardt, J.-N.; Stasik, S.; Röllig, C.; Sauer, T.; Scholl, S.; Hochhaus, A.; Crysandt, M.; Brümmendorf, T.H.; Naumann, R.; Steffen, B.; et al. Alterations of Cohesin Complex Genes in Acute Myeloid Leukemia: Differential Co-Mutations, Clinical Presentation and Impact on Outcome. Blood Cancer J. 2023, 13, 18. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S.; Mazzone, C.; Cesini, L.; Canichella, M.; de Fabritiis, P. CPX-351: An Old Scheme with a New Formulation in the Treatment of High-Risk AML. Cancers 2022, 14, 2843. [Google Scholar] [CrossRef]
- Gaidzik, V.I.; Bullinger, L.; Schlenk, R.F.; Zimmermann, A.S.; Röck, J.; Paschka, P.; Corbacioglu, A.; Krauter, J.; Schlegelberger, B.; Ganser, A.; et al. RUNX1 Mutations in Acute Myeloid Leukemia: Results From a Comprehensive Genetic and Clinical Analysis From the AML Study Group. JCO 2011, 29, 1364–1372. [Google Scholar] [CrossRef]
- Taube, F.; Georgi, J.A.; Kramer, M.; Stasik, S.; Middeke, J.M.; Röllig, C.; Krug, U.; Krämer, A.; Scholl, S.; Hochhaus, A.; et al. CEBPA Mutations in 4708 Patients with Acute Myeloid Leukemia: Differential Impact of bZIP and TAD Mutations on Outcome. Blood 2022, 139, 87–103. [Google Scholar] [CrossRef]
- Pan, L.; Li, Y.; Gao, H.; Cai, Y.; Lai, X.; Chen, Z.; Li, X.; Wang, S. Clinical Features and Monitoring of Germline CEBPA -Mutated Carriers. Blood 2023, 142, 1366–1366. [Google Scholar] [CrossRef]
- Falini, B.; Sorcini, D.; Perriello, V.M.; Sportoletti, P. Functions of the Native NPM1 Protein and Its Leukemic Mutant. Leukemia 2025, 39, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. NPM1-Mutated Acute Myeloid Leukemia: From Bench to Bedside. Blood 2020, 136, 1707–1721. [Google Scholar] [CrossRef] [PubMed]
- Issa, G.C.; Aldoss, I.; Thirman, M.J.; DiPersio, J.; Arellano, M.; Blachly, J.S.; Mannis, G.N.; Perl, A.; Dickens, D.S.; McMahon, C.M.; et al. Menin Inhibition With Revumenib for KMT2A-Rearranged Relapsed or Refractory Acute Leukemia (AUGMENT-101). JCO 2025, 43, 75–84. [Google Scholar] [CrossRef]
- Dali, S.A.; Al-Mashdali, A.F.; Kalfah, A.; Mohamed, S.F. Menin Inhibitors in KMT2A-Rearranged and NPM1-Mutated Acute Leukemia: A Scoping Review of Safety and Efficacy. Critical Reviews in Oncology/Hematology 2025, 213, 104783. [Google Scholar] [CrossRef] [PubMed]
- Arellano, M.L.; Thirman, M.J.; DiPersio, J.F.; Heiblig, M.; Stein, E.M.; Schuh, A.C.; Žučenka, A.; de Botton, S.; Grove, C.S.; Mannis, G.N.; et al. Menin Inhibition with Revumenib for NPM1-Mutated Relapsed or Refractory Acute Myeloid Leukemia: The AUGMENT-101 Study. Blood 2025, 146, 1065–1077. [Google Scholar] [CrossRef]
- Severina, N.; Sidorova, Y.; Risinskaya, N.; Biderman, B.; Pshenychnyy, A.; Ryzhikova, N.; Lukianova, I.; Kashlakova, A.; Sudarikov, A. PB1792: NGS VS PCR FOR THE DETECTION OF NPM1 GENE MUTATIONS IN ACUTE MYELOID LEUKEMIA. Hemasphere 2022, 6, 1672–1673. [Google Scholar] [CrossRef]
- Vonk, C.M.; Grob, T.; Rijken, M.; Kavelaars, F.G.; Konijnenburg, J.M.L.; Ossenkoppele, G.J.; Manz, M.G.; Havelange, V.; Fløisand, Y.; Löwenberg, B.; et al. Advantages of a Genomic DNA-Based next-Generation Sequencing Assay for Detection of Mutant NPM1 Measurable Residual Disease in AML. Blood Adv 2025, 9, 1069–1077. [Google Scholar] [CrossRef]
- Boluda, B.; Rodriguez-Veiga, R.; Sargas, C.; Ayala, R.; Larráyoz, M.J.; Chillón, M.C.; Soria-Saldise, E.; Bilbao, C.; Prados De La Torre, E.P.; Navarro, I.; et al. Conventional PCR Versus Next Generation Sequencing for Diagnosis of FLT3, IDH and NPM1 Mutations in Acute Myeloid Leukemia: Results of the PETHEMA PCR-LMA Study. Cancers 2025, 17, 854. [Google Scholar] [CrossRef]
- Akabane, H. How Often Do Cytogenetics and NGS Information Impact Upfront Treatment in AML? Blood 2022, 140, 11607–11608. [Google Scholar] [CrossRef]
- Shimony, S.; Stahl, M.; Stone, R.M. Acute Myeloid Leukemia: 2025 Update on Diagnosis, Risk-Stratification, and Management. American Journal of Hematology 2025, 100, 860–891. [Google Scholar] [CrossRef]
- Research, C. for D.E. and FDA Approves Ivosidenib for Myelodysplastic Syndromes. FDA 2023. [Google Scholar]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Erba, H.P.; Montesinos, P.; Kim, H.-J.; Patkowska, E.; Vrhovac, R.; Žák, P.; Wang, P.-N.; Mitov, T.; Hanyok, J.; Kamel, Y.M.; et al. Quizartinib plus Chemotherapy in Newly Diagnosed Patients with FLT3-Internal-Tandem-Duplication-Positive Acute Myeloid Leukaemia (QuANTUM-First): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; Recher, C.; Vives, S.; Zarzycka, E.; Wang, J.; Bertani, G.; Heuser, M.; Calado, R.T.; Schuh, A.C.; Yeh, S.-P.; et al. Ivosidenib and Azacitidine in IDH1-Mutated Acute Myeloid Leukemia. N Engl J Med 2022, 386, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Petermichl, V.; Fuchs, S.; Weber, M.; Gobat, K.; Micheloud, C.; Graf, L.; Gerth, Y.; Goede, J.S.; Lehmann, T.; Driessen, C.; et al. Prognostic Impact of the AML60+ Score for Elderly Patients with Acute Myeloid Leukemia Treated with Hypomethylating Agents: A Retrospective Multicentric Analysis. Cancers (Basel) 2025, 17, 2658. [Google Scholar] [CrossRef] [PubMed]
- Bérard, E.; Röllig, C.; Bertoli, S.; Pigneux, A.; Tavitian, S.; Kramer, M.; Serve, H.; Bornhäuser, M.; Platzbecker, U.; Müller-Tidow, C.; et al. A Scoring System for AML Patients Aged 70 Years or Older, Eligible for Intensive Chemotherapy: A Study Based on a Large European Data Set Using the DATAML, SAL, and PETHEMA Registries. Blood Cancer J. 2022, 12, 107. [Google Scholar] [CrossRef]
- Clichet, V.; Boyer, T. Artificial Intelligence-Based Myelodysplastic Syndromes Score, 2022 Classifications, and the Molecular International Prognostic Scoring System: A Perfect Match. haematol 2024. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and Management of AML in Adults: 2017 ELN Recommendations from an International Expert Panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Garcia-Manero, G. Myelodysplastic Syndromes: 2023 Update on Diagnosis, Risk-Stratification, and Management. Am J Hematol 2023, 98, 1307–1325. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Voso, M.T.; Fenu, S.; Latagliata, R.; Buccisano, F.; Piciocchi, A.; Aloe-Spiriti, M.A.; Breccia, M.; Criscuolo, M.; Andriani, A.; Mancini, S.; et al. Revised International Prognostic Scoring System (IPSS) Predicts Survival and Leukemic Evolution of Myelodysplastic Syndromes Significantly Better Than IPSS and WHO Prognostic Scoring System: Validation by the Gruppo Romano Mielodisplasie Italian Regional Database. JCO 2013, 31, 2671–2677. [Google Scholar] [CrossRef]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango Ossa, J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evidence 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Bacigalupo, A.; Barbui, T.; Ditschkowski, M.; Gagelmann, N.; Griesshammer, M.; Gupta, V.; Hamad, N.; Harrison, C.; Hernandez-Boluda, J.C.; et al. Indication and Management of Allogeneic Haematopoietic Stem-Cell Transplantation in Myelofibrosis: Updated Recommendations by the EBMT/ELN International Working Group. The Lancet Haematology 2024, 11, e62–e74. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Gobbi, M.; Kropf, P.L.; Issa, J.-P.J.; Roboz, G.J.; Mayer, J.; Krauter, J.; Robak, T.; Kantarjian, H.; Novak, J.; et al. Guadecitabine vs Treatment Choice in Newly Diagnosed Acute Myeloid Leukemia: A Global Phase 3 Randomized Study. Blood Adv 2023, 7, 5027–5037. [Google Scholar] [CrossRef] [PubMed]
- Blackmon, A.L.; Grunwald, M.R. Editorial: Molecular MRD Testing in Patients with Acute Myeloid Leukemia. Bone Marrow Transplant 2025, 60, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, K.D.; Mims, A.S. MRD in AML: Who, What, When, Where, and How? Blood 2024, 143, 296–298. [Google Scholar] [CrossRef]
- Heuser, M.; Freeman, S.D.; Ossenkoppele, G.J.; Buccisano, F.; Hourigan, C.S.; Ngai, L.L.; Tettero, J.M.; Bachas, C.; Baer, C.; Béné, M.-C.; et al. 2021 Update on MRD in Acute Myeloid Leukemia: A Consensus Document from the European LeukemiaNet MRD Working Party. Blood 2021, 138, 2753–2767. [Google Scholar] [CrossRef] [PubMed]
- Ivey, A.; Hills, R.K.; Simpson, M.A.; Jovanovic, J.V.; Gilkes, A.; Grech, A.; Patel, Y.; Bhudia, N.; Farah, H.; Mason, J.; et al. Assessment of Minimal Residual Disease in Standard-Risk AML. New England Journal of Medicine 2016, 374, 422–433. [Google Scholar] [CrossRef]
- Höllein, A.; Meggendorfer, M.; Dicker, F.; Jeromin, S.; Nadarajah, N.; Kern, W.; Haferlach, C.; Haferlach, T. NPM1 Mutated AML Can Relapse with Wild-Type NPM1: Persistent Clonal Hematopoiesis Can Drive Relapse. Blood Adv 2018, 2, 3118–3125. [Google Scholar] [CrossRef]
- Cocciardi, S.; Dolnik, A.; Kapp-Schwoerer, S.; Rücker, F.G.; Lux, S.; Blätte, T.J.; Skambraks, S.; Krönke, J.; Heidel, F.H.; Schnöder, T.M.; et al. Clonal Evolution Patterns in Acute Myeloid Leukemia with NPM1 Mutation. Nat Commun 2019, 10, 2031. [Google Scholar] [CrossRef]
- Marchetti, F.; Cardoso, R.; Chen, C.L.; Douglas, G.R.; Elloway, J.; Escobar, P.A.; Harper, T.; Heflich, R.H.; Kidd, D.; Lynch, A.M.; et al. Error-Corrected next Generation Sequencing – Promises and Challenges for Genotoxicity and Cancer Risk Assessment. Mutation Research - Reviews in Mutation Research 2023, 792, 108466. [Google Scholar] [CrossRef]
- Chen, H.; Yu, F.; Lu, D.; Huang, S.; Liu, S.; Zhang, B.; Shu, K.; Pu, D. Enhanced Error Suppression for Accurate Detection of Low-Frequency Variants. Electrophoresis 2025, 46, 65–75. [Google Scholar] [CrossRef]
- Romer-Seibert, J.S.; Meyer, S.E. Genetic Heterogeneity and Clonal Evolution in Acute Myeloid Leukemia. Curr Opin Hematol 2021, 28, 64–70. [Google Scholar] [CrossRef]
- Schuringa, J.J.; Bonifer, C. Dissecting Clonal Heterogeneity in AML. Cancer Cell 2020, 38, 782–784. [Google Scholar] [CrossRef]
- McMahon, C.M.; Ferng, T.; Canaani, J.; Wang, E.S.; Morrissette, J.J.D.; Eastburn, D.J.; Pellegrino, M.; Durruthy-Durruthy, R.; Watt, C.D.; Asthana, S.; et al. Clonal Selection with RAS Pathway Activation Mediates Secondary Clinical Resistance to Selective FLT3 Inhibition in Acute Myeloid Leukemia. Cancer Discovery 2019, 9, 1050–1063. [Google Scholar] [CrossRef]
- Walter, R.B. Perspective on Measurable Residual Disease Testing in Acute Myeloid Leukemia. Leukemia 2024, 38, 10–13. [Google Scholar] [CrossRef]
- Levis, M.J.; Hamadani, M.; Logan, B.R.; Jones, R.J.; Singh, A.K.; Litzow, M.R.; Wingard, J.R.; Papadopoulos, E.B.; Perl, A.E.; Soiffer, R.J.; et al. Measurable Residual Disease and Posttransplantation Gilteritinib Maintenance for Patients with FLT3-ITD-Mutated AML. Blood 2025, 145, 2138–2148. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-H.; Tang, J.-L.; Tien, F.-M.; Kuo, Y.-Y.; Wu, D.-C.; Lin, C.-C.; Tseng, M.-H.; Peng, Y.-L.; Hou, M.-F.; Chuang, Y.-K.; et al. Clinical Implications of Sequential MRD Monitoring by NGS at 2 Time Points after Chemotherapy in Patients with AML. Blood Advances 2021, 5, 2456–2466. [Google Scholar] [CrossRef] [PubMed]
- Schnittger, S.; Schoch, C.; Dugas, M.; Kern, W.; Staib, P.; Wuchter, C.; Löffler, H.; Sauerland, C.M.; Serve, H.; Büchner, T.; et al. Analysis of FLT3 Length Mutations in 1003 Patients with Acute Myeloid Leukemia: Correlation to Cytogenetics, FAB Subtype, and Prognosis in the AMLCG Study and Usefulness as a Marker for the Detection of Minimal Residual Disease. Blood 2002, 100, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.-H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S.; et al. Next-Generation Sequencing for Minimal Residual Disease Monitoring in Acute Myeloid Leukemia Patients with FLT3-ITD or NPM1 Mutations. Genes, Chromosomes and Cancer 2012, 51, 689–695. [Google Scholar] [CrossRef]
- Levis, M.J.; Hamadani, M.; Logan, B.; Jones, R.J.; Singh, A.K.; Litzow, M.; Wingard, J.R.; Papadopoulos, E.B.; Perl, A.E.; Soiffer, R.J.; et al. Gilteritinib as Post-Transplant Maintenance for AML With Internal Tandem Duplication Mutation of FLT3. JCO 2024, 42, 1766–1775. [Google Scholar] [CrossRef]
- Dillon, L.W.; Gui, G.; Page, K.M.; Ravindra, N.; Wong, Z.C.; Andrew, G.; Mukherjee, D.; Zeger, S.L.; El Chaer, F.; Spellman, S.; et al. DNA Sequencing to Detect Residual Disease in Adults With Acute Myeloid Leukemia Prior to Hematopoietic Cell Transplant. JAMA 2023, 329, 745. [Google Scholar] [CrossRef]
- Hirsch, P.; Lambert, J.; Bucci, M.; Deswarte, C.; Boudry, A.; Lambert, J.; Fenwarth, L.; Micol, J.-B.; Terré, C.; Celli-Lebras, K.; et al. Multi-Target Measurable Residual Disease Assessed by Error-Corrected Sequencing in Patients with Acute Myeloid Leukemia: An ALFA Study. Blood Cancer J. 2024, 14, 97. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A.; Shimamura, A. Genetic Predisposition to Hematologic Malignancies: Management and Surveillance. Blood 2017, 130, 424–432. [Google Scholar] [CrossRef]
- Rio-Machin, A.; Vulliamy, T.; Hug, N.; Walne, A.; Tawana, K.; Cardoso, S.; Ellison, A.; Pontikos, N.; Wang, J.; Tummala, H.; et al. The Complex Genetic Landscape of Familial MDS and AML Reveals Pathogenic Germline Variants. Nat Commun 2020, 11, 1044. [Google Scholar] [CrossRef]
- Mestre, J.; Chaparro, L.; Manzanares, A.; Xicoy, B.; Zamora, L.; Sole, F.; Calvete, O. Beyond Myeloid Neoplasms Germline Guidelines: Validation of the Thresholds Criteria in the Search of Germline Predisposition Variants. eJHaem 2024, 5, 1021–1027. [Google Scholar] [CrossRef]
- Baliakas, P.; Tesi, B.; Wartiovaara-Kautto, U.; Stray-Pedersen, A.; Friis, L.S.; Dybedal, I.; Hovland, R.; Jahnukainen, K.; Raaschou-Jensen, K.; Ljungman, P.; et al. Nordic Guidelines for Germline Predisposition to Myeloid Neoplasms in Adults: Recommendations for Genetic Diagnosis, Clinical Management and Follow-Up. HemaSphere 2019, 3, e321. [Google Scholar] [CrossRef]
- Tawana, K.; Brown, A.L.; Churpek, J.E. Integrating Germline Variant Assessment into Routine Clinical Practice for Myelodysplastic Syndrome and Acute Myeloid Leukaemia: Current Strategies and Challenges. Br J Haematol 2022, 196, 1293–1310. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A.; DiNardo, C.D.; Bolton, K. Germline Predisposition in Hematologic Malignancies: Testing, Management, and Implications. Am Soc Clin Oncol Educ Book 2024, 44, e432218. [Google Scholar] [CrossRef]
- Farina, M.; Bernardi, S.; Gandolfi, L.; Zanaglio, C.; Morello, E.; Turra, A.; Zollner, T.; Gramegna, D.; Rambaldi, B.; Cattina, F.; et al. Case Report: Late Onset of Myelodysplastic Syndrome From Donor Progenitor Cells After Allogeneic Stem Cell Transplantation. Which Lessons Can We Draw From the Reported Case? Front. Oncol. 2020, 10, 564521. [Google Scholar] [CrossRef]
- Kato, M.; Yamashita, T.; Suzuki, R.; Matsumoto, K.; Nishimori, H.; Takahashi, S.; Iwato, K.; Nakaseko, C.; Kondo, T.; Imada, K.; et al. Donor Cell-Derived Hematological Malignancy: A Survey by the Japan Society for Hematopoietic Cell Transplantation. Leukemia 2016, 30, 1742–1745. [Google Scholar] [CrossRef]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. JCO 2022, 40, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Speight, B.; Hanson, H.; Turnbull, C.; Hardy, S.; Drummond, J.; Khorashad, J.; Wragg, C.; Page, P.; Parkin, N.W.; Rio-Machin, A.; et al. Germline Predisposition to Haematological Malignancies: Best Practice Consensus Guidelines from the UK Cancer Genetics Group (UKCGG), CanGene-CanVar and the NHS England Haematological Oncology Working Group. Br J Haematol 2023, 201, 25–34. [Google Scholar] [CrossRef]
- Reilly, C.R.; Shimamura, A. Predisposition to Myeloid Malignancies in Shwachman-Diamond Syndrome: Biological Insights and Clinical Advances. Blood 2023, 141, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S. Management of Fanconi Anemia beyond Childhood. Hematology Am Soc Hematol Educ Program 2023, 2023, 556–562. [Google Scholar] [CrossRef]
- Mason, N.R.; Cahill, H.; Diamond, Y.; McCleary, K.; Kotecha, R.S.; Marshall, G.M.; Mateos, M.K. Down Syndrome-Associated Leukaemias: Current Evidence and Challenges. Ther Adv Hematol 2024, 15, 20406207241257901. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S. Molecular Targets for the Treatment of AML in the Forthcoming 5th World Health Organization Classification of Haematolymphoid Tumours. Expert Review of Hematology 2022, 15, 973–986. [Google Scholar] [CrossRef] [PubMed]
- Baliakas, P.; Tesi, B.; Cammenga, J.; Stray-Pedersen, A.; Jahnukainen, K.; Andersen, M.K.; Ågerstam, H.; Creignou, M.; Dybedal, I.; Raaschou-Jensen, K.; et al. How to Manage Patients with Germline DDX41 Variants: Recommendations from the Nordic Working Group on Germline Predisposition for Myeloid Neoplasms. HemaSphere 2024, 8, e145. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ross, S.R. Multifunctional Role of DEAD-Box Helicase 41 in Innate Immunity, Hematopoiesis and Disease. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef]
- Makishima, H.; Saiki, R.; Nannya, Y.; Korotev, S.; Gurnari, C.; Takeda, J.; Momozawa, Y.; Best, S.; Krishnamurthy, P.; Yoshizato, T.; et al. Germ Line DDX41 Mutations Define a Unique Subtype of Myeloid Neoplasms. Blood 2023, 141, 534–549. [Google Scholar] [CrossRef]
- Wang, X.; Xiao, Z.; Qin, T.; Xu, Z.; Jia, Y.; Qu, S.; Li, B.; Pan, L.; Gao, Q.; Jiao, M.; et al. Combination Therapy with Venetoclax and Azacitidine for the Treatment of Myelodysplastic Syndromes with DDX41 Mutations. Hematology 2024, 29, 2338509. [Google Scholar] [CrossRef]
- Nanaa, A.; He, R.; Foran, J.M.; Badar, T.; Gangat, N.; Pardanani, A.; Hogan, W.J.; Litzow, M.R.; Patnaik, M.; Al-Kali, A.; et al. Venetoclax plus Hypomethylating Agents in DDX41-Mutated Acute Myeloid Leukaemia and Myelodysplastic Syndrome: Mayo Clinic Series on 12 Patients. British Journal of Haematology 2024, 204, 171–176. [Google Scholar] [CrossRef]
- Alkhateeb, H.B.; Nanaa, A.; Viswanatha, D.; Foran, J.M.; Badar, T.; Sproat, L.; He, R.; Nguyen, P.; Jevremovic, D.; Salama, M.E.; et al. Genetic Features and Clinical Outcomes of Patients with Isolated and Comutated DDX41-Mutated Myeloid Neoplasms. Blood Adv 2022, 6, 528–532. [Google Scholar] [CrossRef]
- Li, P.; White, T.; Xie, W.; Cui, W.; Peker, D.; Zeng, G.; Wang, H.-Y.; Vagher, J.; Brown, S.; Williams, M.; et al. AML with Germline DDX41 Variants Is a Clinicopathologically Distinct Entity with an Indolent Clinical Course and Favorable Outcome. Leukemia 2022, 36, 664–674. [Google Scholar] [CrossRef]
- Nanaa, A.; He, R.; Viswanatha, D.; Nguyen, P.; Jevremovic, D.; Foran, J.M.; Yi, C.A.; Greipp, P.T.; Gangat, N.; Patnaik, M.; et al. Comparison between GATA2 and DDX41-Mutated Myeloid Neoplasms. Leuk Res 2022, 121, 106931. [Google Scholar] [CrossRef]
- Dimopoulos, Y.P.; Wang, W.; Wang, S.A.; Loghavi, S.; DiNardo, C.D.; Gerstein, Y.; Hu, S.; Tang, Z.; Ilagan, C.J.L.; Thakral, B.; et al. The Spectrum of Hematologic Neoplasms in Patients with Li-Fraumeni Syndrome. American J Hematol 2024, 99, 2416–2419. [Google Scholar] [CrossRef]
- Alikarami, F.; Xie, H.M.; Riedel, S.S.; Goodrow, H.T.; Barrett, D.R.; Mahdavi, L.; Lenard, A.; Chen, C.; Yamauchi, T.; Danis, E.; et al. GATA2 Links Stemness to Chemotherapy Resistance in Acute Myeloid Leukemia. Blood 2025, 145, 2179–2195. [Google Scholar] [CrossRef]
- Calvo, K.R.; Hickstein, D.D. The Spectrum of GATA2 Deficiency Syndrome. Blood 2023, 141, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef]
- Sanchez-Petitto, G.; El Boghdadly, Z.; Nicolet, D.; Cooper, J.; Eisfeld, A.-K.; Klein, V.; Walker, M.C.; Mrózek, K.; Bezerra, E.; Brammer, J.E.; et al. GATA2-Mutated AML: Clinical Outcomes and Spectrum of Infections in Patients Undergoing or Not Allogeneic Stem Cell Transplantation. Blood 2024, 144, 7309. [Google Scholar] [CrossRef]
- Santiago, M.; Liquori, A.; Such, E.; Zúñiga, Á.; Cervera, J. The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers 2023, 15, 1590. [Google Scholar] [CrossRef] [PubMed]
- de Bruijn, M.; Dzierzak, E. Runx Transcription Factors in the Development and Function of the Definitive Hematopoietic System. Blood 2017, 129, 2061–2069. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Deuitch, N.; Merguerian, M.; Cunningham, L.; Davis, J.; Bresciani, E.; Diemer, J.; Andrews, E.; Young, A.; Donovan, F.; et al. Genomic Landscape of Patients with Germline RUNX1 Variants and Familial Platelet Disorder with Myeloid Malignancy. Blood Adv 2024, 8, 497–511. [Google Scholar] [CrossRef]
- Greif, P.A.; Konstandin, N.P.; Metzeler, K.H.; Herold, T.; Pasalic, Z.; Ksienzyk, B.; Dufour, A.; Schneider, F.; Schneider, S.; Kakadia, P.M.; et al. RUNX1 Mutations in Cytogenetically Normal Acute Myeloid Leukemia Are Associated with a Poor Prognosis and Up-Regulation of Lymphoid Genes. Haematologica 2012, 97, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Homan, C.C.; Drazer, M.W.; Yu, K.; Lawrence, D.M.; Feng, J.; Arriola-Martinez, L.; Pozsgai, M.J.; McNeely, K.E.; Ha, T.; Venugopal, P.; et al. Somatic Mutational Landscape of Hereditary Hematopoietic Malignancies Caused by Germline Variants in RUNX1, GATA2, and DDX41. Blood Adv 2023, 7, 6092–6107. [Google Scholar] [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.L.; et al. Germline Mutations in ETV6 Are Associated with Thrombocytopenia, Red Cell Macrocytosis and Predisposition to Lymphoblastic Leukemia. Nat Genet 2015, 47, 535–538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 Mutations in Familial Thrombocytopenia and Hematologic Malignancy. Nat Genet 2015, 47, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.-C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-Related Thrombocytopenia and Myeloid Malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5’ UTR of ANKRD26, the Ankirin Repeat Domain 26 Gene, Cause an Autosomal-Dominant Form of Inherited Thrombocytopenia, THC2. Am J Hum Genet 2011, 88, 115–120. [Google Scholar] [CrossRef]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; De Rocco, D.; et al. Mutations in ANKRD26 Are Responsible for a Frequent Form of Inherited Thrombocytopenia: Analysis of 78 Patients from 21 Families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Potenza, L.; Borelli, E.; Bigi, S.; Giusti, D.; Longo, G.; Odejide, O.; Porro, C.A.; Zimmermann, C.; Efficace, F.; Bruera, E.; et al. Early Palliative Care in Acute Myeloid Leukemia. Cancers 2022, 14, 478. [Google Scholar] [CrossRef]
- Moore, C.G.; Stein, A.; Fathi, A.T.; Pullarkat, V. Treatment of Relapsed/Refractory AML—Novel Treatment Options Including Immunotherapy. American Journal of Hematology 2025, 100, 23–37. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S.; Andriola, C.; Rossi, M. Immunotherapy with Monoclonal Antibodies for Acute Myeloid Leukemia: A Work in Progress. Cancers 2023, 15, 5060. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Borsi, E.; Patuelli, A.; Bandini, L.; Mancini, M.; Forte, D.; Nanni, J.; Barone, M.; Grassi, A.; Cristiano, G.; et al. Tracking Response and Resistance in Acute Myeloid Leukemia through Single-Cell DNA Sequencing Helps Uncover New Therapeutic Targets. IJMS 2024, 25, 10002. [Google Scholar] [CrossRef]
- Assi, R.E.; Alfonso Pierola, A.; Kc, D.; Abaza, Y.M.; Abou Zahr, A.; Chamoun, K.; Montalban-Bravo, G.; Takahashi, K.; Jabbour, E.; Kadia, T.M.; et al. Impact of Next-Generation Sequencing (NGS) on Treatment Selection in Acute Myeloid Leukemia (AML). JCO 2018, 36, 103–103. [Google Scholar] [CrossRef]
- Upadhyay Banskota, S.; Khanal, N.; Bhatt, V.R. A Precision Medicine Approach to Management of Acute Myeloid Leukemia in Older Adults. Current Opinion in Oncology 2020, 32, 650. [Google Scholar] [CrossRef] [PubMed]
- Snaith, O.; Poveda-Rogers, C.; Laczko, D.; Yang, G.; Morrissette, J.J.D. Cytogenetics and Genomics of Acute Myeloid Leukemia. Best Pract Res Clin Haematol 2024, 37, 101533. [Google Scholar] [CrossRef]
- Dai, B.; Yu, H.; Ma, T.; Lei, Y.; Wang, J.; Zhang, Y.; Lu, J.; Yan, H.; Jiang, L.; Chen, B. The Application of Targeted RNA Sequencing for KMT2A-Partial Tandem Duplication Identification and Integrated Analysis of Molecular Characterization in Acute Myeloid Leukemia. J Mol Diagn 2021, 23, 1478–1490. [Google Scholar] [CrossRef]
- Nawas, M.T.; Kosuri, S. Utility or Futility? A Contemporary Approach to Allogeneic Hematopoietic Cell Transplantation for TP53-Mutated MDS/AML. Blood Advances 2024, 8, 553–561. [Google Scholar] [CrossRef]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, Y.; Ohanian, M.; et al. Outcomes of TP53-Mutant Acute Myeloid Leukemia with Decitabine and Venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef]
- Shimony, S.O.; Murdock, H.; Keating, J.; Reilly, C.R.; Tsai, H.K.; Gibson, C.J.; Faderl, S.; Wagner, T.; Dronamraju, N.; Lin, T.L.; et al. AML-MR Mutations Drive the Benefit of CPX-351 over 7+3 in the Pivotal Phase 3 AML Trial. Blood 2024, 144, 60. [Google Scholar] [CrossRef]
- Badar, T.; Nanaa, A.; Atallah, E.; Shallis, R.M.; Guilherme, S. de C.C.; Goldberg, A.D.; Saliba, A.N.; Patel, A.; Bewersdorf, J.P.; DuVall, A.S.; et al. Comparing Venetoclax in Combination with Hypomethylating Agents to Hypomethylating Agent-Based Therapies for Treatment Naive TP53-Mutated Acute Myeloid Leukemia: Results from the Consortium on Myeloid Malignancies and Neoplastic Diseases (COMMAND). Blood Cancer J. 2024, 14, 32. [Google Scholar] [CrossRef]
- Sallman, D.A.; Stahl, M. TP53-Mutated Acute Myeloid Leukemia: How Can We Improve Outcomes? Blood 2025, 145, 2828–2833. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging P53 in Cancer: One Protein, Many Targets. Nat Rev Drug Discov 2023, 22, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Ottone, T.; Zhdanovskaya, N.; Molica, M. How Acute Myeloid Leukemia (AML) Escapes from FMS-Related Tyrosine Kinase 3 (FLT3) Inhibitors? Still an Overrated Complication? Cancer Drug Resist 2023, 6, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Loghavi, S.; Daver, N.; Ravandi, F.; DiNardo, C.D.; Kadia, T.M.; Borthakur, G.; Jabbour, E.; Yilmaz, M.; Issa, G.C.; et al. Cytomolecular Mechanisms of Relapse and Post-Relapse Outcomes After Frontline FLT3 Inhibitor-Based Therapy in FLT3-Mutated AML. American Journal of Hematology n/a. [CrossRef]
- Sasaki, K.; Takahashi, S.; Ouchi, K.; Otsuki, Y.; Wakayama, S.; Ishioka, C. Different Impacts of TP53 Mutations on Cell Cycle-Related Gene Expression among Cancer Types. Sci Rep 2023, 13, 4868. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.; Ricker, C.; Messersmith, H.; Balmaña, J.; Domchek, S.; Stoffel, E.M.; Almhanna, K.; Arun, B.; Chavarri-Guerra, Y.; Cohen, S.A.; et al. Selection of Germline Genetic Testing Panels in Patients With Cancer: ASCO Guideline. JCO 2024, 42, 2599–2615. [Google Scholar] [CrossRef]
- Haferlach, T.; Eckardt, J.-N.; Walter, W.; Maschek, S.; Kather, J.N.; Pohlkamp, C.; Middeke, J.M. AML Diagnostics in the 21st Century: Use of AI. Seminars in Hematology 2025. [Google Scholar] [CrossRef]
- Qin, Y.; Pu, X.; Hu, D.; Yang, M. Machine Learning-Based Biomarker Screening for Acute Myeloid Leukemia Prognosis and Therapy from Diverse Cell-Death Patterns. Sci Rep 2024, 14, 17874. [Google Scholar] [CrossRef]
Figure 1.
This vignette shows the advised requirements for an effective NGS report.


Table 1.
This table shows the main genes mutated in AML or MDS, organized by functional category, with brief mechanisms of their alteration.
Table 1.
This table shows the main genes mutated in AML or MDS, organized by functional category, with brief mechanisms of their alteration.
| Functional Category | Example Genes / Alterations | Consequences of Genetic Alteration |
|---|---|---|
| Signal Transduction | FLT3, NRAS, KRAS, c-KIT, PTPN11 | Confers proliferative advantages through the hyperactivation of signaling pathways like JAK/STAT, PI3K/AKT, and RAF/MEK/ERK. |
| Myeloid Transcription Factors | RUNX1, CEBPA, or fusions like RUNX1::RUNX1T1, PML::RARA, CBFB::MYH11 | Causes transcriptional deregulation, which impairs or blocks normal hematopoietic differentiation. |
| Tumor Suppressor Genes | TP53, WT1, PHF6 | Deregulates normal transcription and alters cell cycle checkpoints and responses to cellular stress, often leading to increased proliferation. |
| Spliceosome Complex | SRSF2, SF3B1, U2AF1, ZRSR2 | Alters the proper maturation of mRNA, causing events like intron retention or exon skipping, which can lead to dysfunctional proteins. |
| Multifunctional Protein | NPM1 | Nucleophosmin, causes the protein to be abnormally located in the cytoplasm, affecting ribosome biogenesis and the stability of tumor suppressors like p53. |
| Cohesin Complex | SMC1A, SMC3, STAG2, RAD21 | Affects chromosomal segregation and gene expression by altering chromatin accessibility, leading to increased proliferation and decreased differentiation. |
| DNA Methylation | DNMT3A, TET2, IDH1/2 | Leads to global changes in the epigenetic landscape by altering DNA methylation patterns, which affects gene expression. |
| Chromatin Modifiers | ASXL1, EZH2, or fusions involving KMT2A | Perturbs epigenetic homeostasis through aberrant histone modifications, leading to widespread changes in gene transcription. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



