
Submitted:
30 October 2025
Posted:
03 November 2025
You are already at the latest version
Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by considerable heterogeneity in both its underlying biological mechanisms and clinical presentation. High-dimensional transcriptomic datasets offer an opportunity to characterise this variation at the molecular level; however, traditional statistical methods struggle with their scale and complexity. Machine learning approaches can reduce dimensionality and uncover latent patterns, enabling the identification of molecular subtypes that may refine prognosis and support patient stratification.Recent transcriptomic studies employing unsupervised machine learning have identified ALS subtypes with distinct molecular and clinical characteristics. Redefining ALS into more homogeneous molecular and clinical subtypes could transform all areas of ALS research by supporting novel experimental designs and precision medicine approaches. In this review, we summarise and critically assess these studies, discussing their findings, strengths, and limitations, and highlighting research gaps and challenges that should be addressed to enable their translation into biomedical and clinical practice.
Keywords:
ALS
; Machine Learning
; Molecular subtypes
; disease stratification
; transcriptomics
Introduction
The biological and clinical heterogeneity of amyotrophic lateral sclerosis (ALS) [1] greatly impacts our efforts in understanding its pathogenesis and developing treatments by acting as a major confounding factor in studies and limiting the effectiveness of clinical procedures. Previous research into the genetics of people with ALS has uncovered a number of genetic contributors to disease pathogenesis including rare mutations and common polymorphisms in many genes, among which SOD1, C9orf72, FUS, and TARDBP can causally explain the largest proportion of patients [2,3,4,5,6,7]. Despite these advances, large effect variants only causally explain about 20% of ALS [8,9]. As a result, a re-classification of ALS based on our current knowledge of disease-associated gene variants would not be widely applicable. This has prompted researchers to look beyond single-gene approaches and toward more comprehensive molecular profiling strategies that can capture the complex and variable processes underlying all ALS and dissect the disease heterogeneity. Technological advances in high throughput molecular profiling have led to the identification of various molecular subtypes of ALS utilising unsupervised machine learning (ML) models to analyse the ALS transcriptome. Most studies implementing such an approach identify a range of molecular ALS subgroups consistent with alterations in distinct biological processes such as transposable element (TE) expression (ALS-TE), oxidative stress (ALS-Ox), neuroinflammation (ALS-Neu), and increase in astrocyte activity (ALS-Glia) [10,11,12,13].
This literature review will explore the various methodologies and findings across recent studies on the identification of molecular subtypes of ALS and their potential clinical relevance.
Chronology of Identification of ALS Molecular Subtype in the ALS Transcriptome
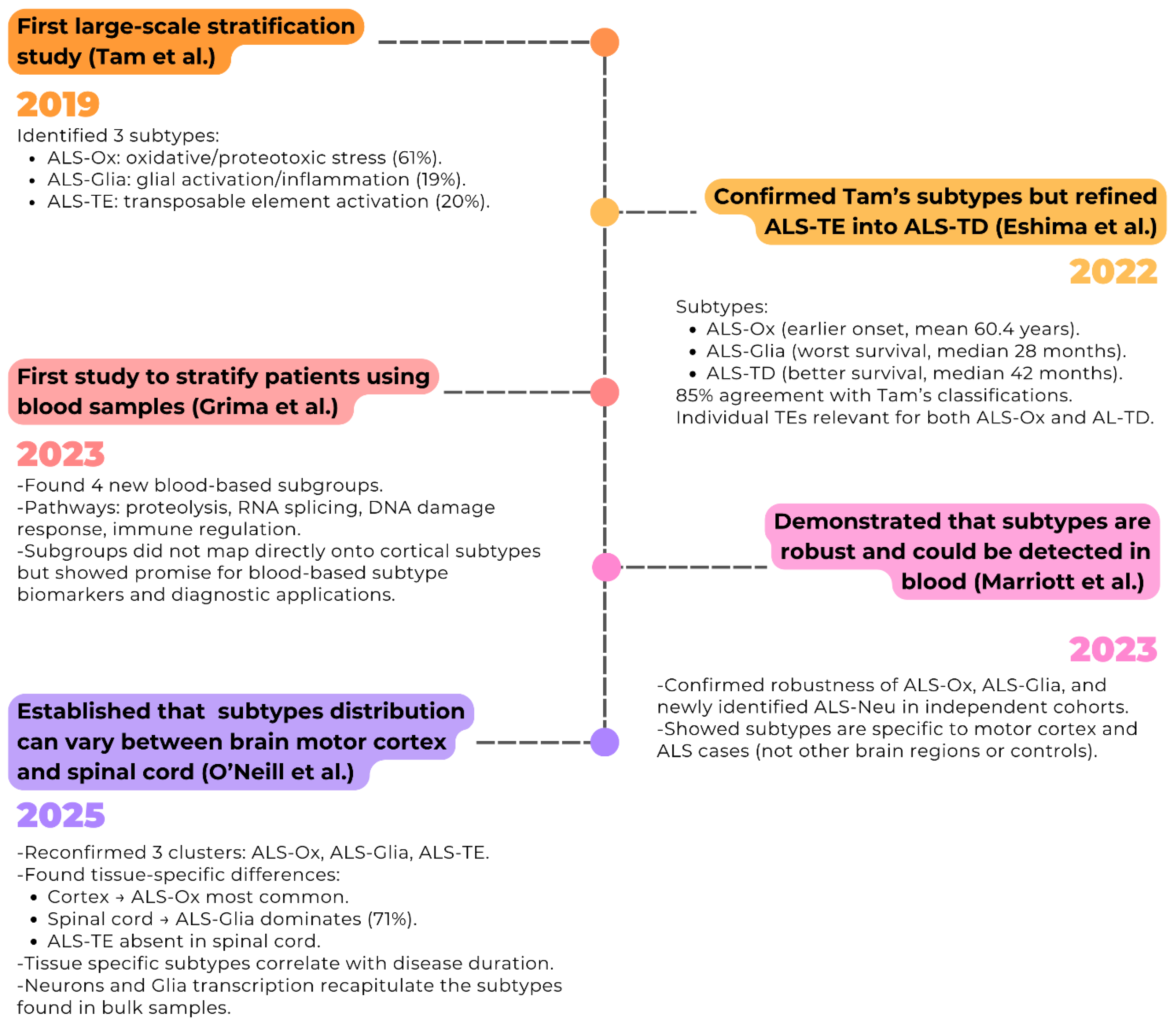
Research exploring the molecular subtypes of ALS using ML and transcriptomics is relatively recent (Figure 1), with the first large scale molecular stratification study published in 2019 [13]. Using unsupervised ML to analyse the NYGC ALS Consortium transcriptomic data of 148 post-mortem cortex samples of 77 people with ALS, they identified three distinct subtypes. The largest (61% of patients) was characterised by alterations in oxidative and proteotoxic stress processes, while a second (19%) showed predominant glial activation and inflammatory signatures. A third group (20%) displayed elevated transposable element expression and depletion of pathways normally regulated by TDP-43, thereby linking this subtype to TDP-43 dysfunction [13]. Following this initial study, several others have explored transcriptome-based stratification in ALS, not only seeking to replicate these findings but also exploring the various potential applications this method can provide, such as integrating additional datasets and tissue types, applying alternative analytical methods, or assessing the prognostic and clinical significance of these subtypes.
Utilising 451 samples from 208 pwALS, Eshima et al. [10] increased the sample size, investigating the relationship between molecular subtypes and clinical outcomes using patient survival and age of onset. They were able to find three molecular subtypes consistent with the ones found by Tam et al., despite some differences partially due to different methodologies for the characterisation of subgroups. Indeed, considering the 140 samples shared between the two studies, they observed 85% agreement in sample classification. However, the larger sample size and methodological differences contributed to the redefinition of the ALS-TE subgroup into the ALS-transcriptional dysregulation subtype (ALS-TD), and the upregulation of specific TEs was found in samples belonging to both ALS-TD and ALS-Ox groups. Notably, the ALS-TD subtype reflected many of the hallmark pathways of TDP-43 dysfunction, including impaired transcriptional regulation, cryptic exon–associated nonsense-mediated decay, and aberrant expression of transposable and non-coding RNAs, suggesting that TDP-43 pathology underlies its transcriptomic profile [10]. Clinically, individuals with ALS-TD had a better prognosis, with a median survival of 42 months (Table 1). By contrast, ALS-Ox was associated with an earlier age of onset and intermediate survival (mean age of onset 60.4 ± 1.16 years and median survival 36 months), while ALS-Glia displayed the worst prognosis, with a median survival of 28 months and an average age of onset of 63.2 ± 1.83 years (Table 1).
Following these findings, Grima et al. [14] published in 2023, a study that investigated if molecular subtypes could also be identified and distinguished de novo from whole peripheral blood RNA-seq data. Using samples from 96 people with sporadic ALS (sALS), they applied unsupervised K-means clustering and identified four patient subgroups. The gene expression profiles of these clusters were also explored; differential gene expression analysis between clusters identified 5756 sub-group defining genes with most differentially expressed in two clusters relative to all other clusters. Furthermore, the study also used GO biological pathway and KEGG pathway enrichment of these subgroup-defining genes and identified pathways involving proteolysis, RNA splicing, DNA damage response, and immune regulation, suggesting that peripheral signatures may reflect heterogeneous systemic responses to ALS. Unlike earlier studies that stratified post-mortem motor cortex tissue, this work focused on whole blood, a minimally invasive and clinically accessible biospecimen. Although the resulting subgroups cannot be directly aligned with the established cortical molecular subtypes identified in other studies, their work demonstrates the potential of blood-based transcriptomics for patient stratification and lays a foundation for developing accessible biomarkers that could be monitored longitudinally throughout disease progression.
Another study published in 2023 conducted by Marriott et al. [11] further investigated the robustness and clinical applicability of the ALS molecular subtypes across independent cohorts. Using post-mortem motor cortex ALS samples from the King’s College London Brain Bank as a discovery cohort (n=112) [15,16], they validated the three cortical molecular subtypes in an independent dataset from TargetALS (n=93 sALS samples). To test tissue and disease specificity, using linear discriminate analysis they projected the expression signatures onto occipital cortex (n=45) and cerebellum ALS samples (n=128), and motor cortex samples of controls (n=59), showing that the subtypes were largely restricted to motor cortex and people with ALS, with logistic regression-based classifiers achieving excellent discrimination between motor cortex, the other brain regions, and between cases and controls. Unlike earlier studies that incorporated both gene and TE expression counts into a combined matrix for clustering, this study derived subtypes solely from gene expression data, demonstrating that the molecular signatures are robust even without TE features. They also examined whether the subtypes could be detected in peripheral blood from people with ALS, applying the classifier to RNA sequencing (RNA-seq) data from polymorphic blood mononuclear cells (n=15) and microarray data from whole blood (n=397). While separation was less distinct than in brain tissue, they reported high assignment probabilities (80–90%) across all three groups, suggesting that the subtypes are at least partially recapitulated in blood and may provide a foundation as accessible biomarkers for patient stratification.
In 2025, O’Neill et al. [12], a team of scientists from the same lab as Tam et al. 2019, built upon their original findings by training a deep ALS neural net classifier (DANCer) on their NYGC dataset in order to generate a classifier of subtype assignment able to identify the ALS subtype in the newly released expanded cohort from the NYGC ALS Consortium. Furthermore, to determine whether the subtypes were present in this new cohort they re-ran a non-smooth non-negative matrix factorisation (nsNMF) model with the new independent samples and identified 3 clusters consistent with their original 2019 findings, i.e. ALS-Ox, ALS-Glia, and ALS-TE. Crucially, this study compared molecular subtypes between post-mortem motor cortex and spinal cord, analysing 664 ALS and control samples. Within this dataset, 267 individuals with ALS had both motor cortex and spinal cord available, allowing direct comparison of subtype distributions within the same donors. The analysis revealed noticeable tissue-specific differences, where ALS-Glia represented the smallest group in cortex (10%) but the majority in spinal cord (71%), while the ALS-TE subtype was absent from spinal cord (Figure 2B). Moreover, disease duration was found to correlate with TDP-43 dysfunction in the motor cortex and with inflammatory signatures in spinal cord, highlighting the clinical relevance of these transcriptomic subtypes.
Brain Molecular Subtypes and Clinical Relevance
Consistency of the Molecular Subtypes Across Studies
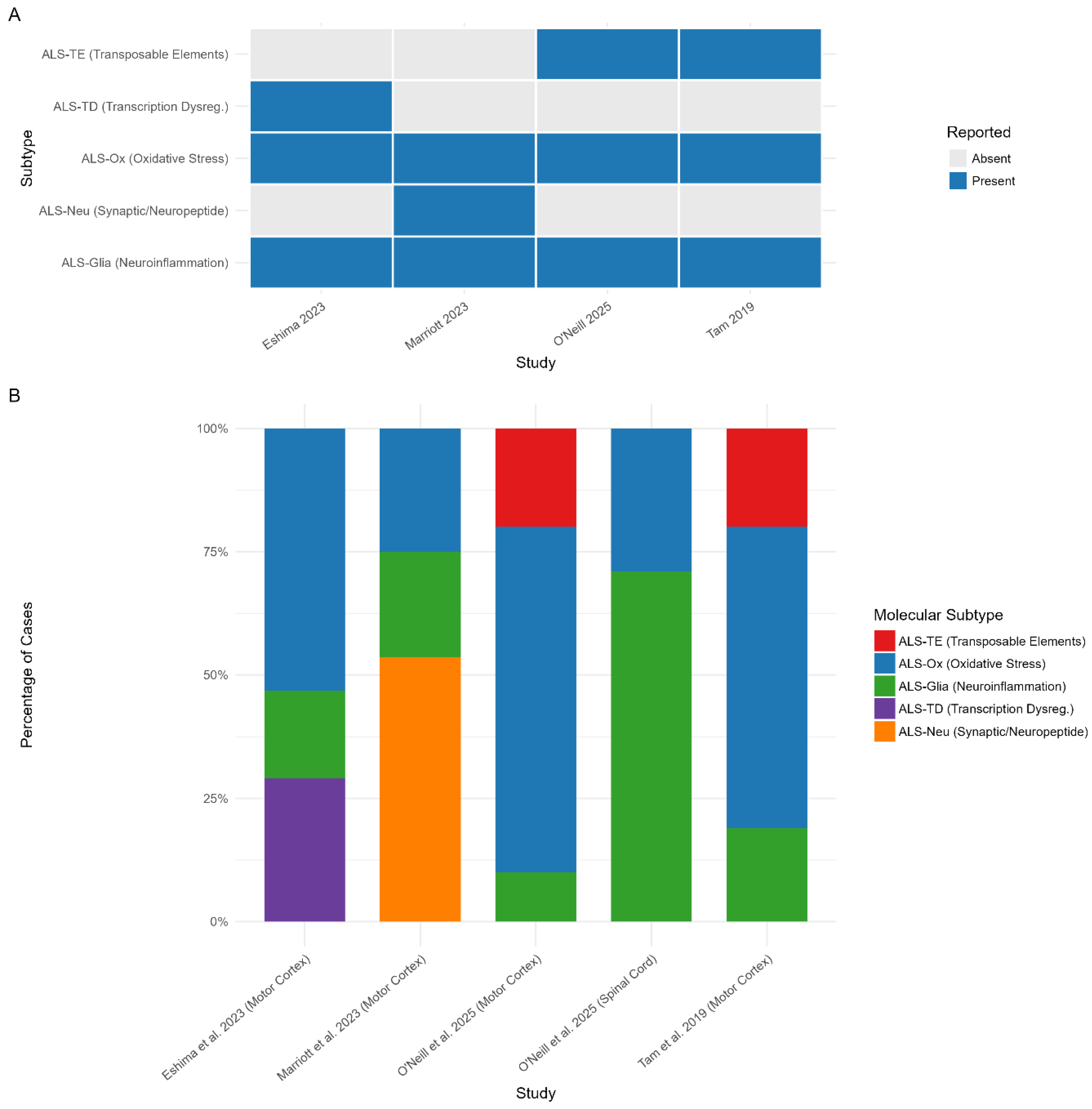
Across all studies exploring and identifying the molecular subtypes in ALS brain samples, a total of 5 have been identified across various cohorts (Figure 2A). This includes ALS-Ox, ALS-Glia, ALS-TE, ALS-TD, and ALS synaptic and neuropeptide signalling (ALS-Neu) [10,11,12,13]. Noticeably, both ALS-Ox and ALS-Glia were established in all studies and by all teams whereas ALS-TE, ALS-TD, and ALS-Neu were only identified by individual teams (Figure 2A). Eshima et al., [10] identified the ALS-TD subtype, which displays molecular pathways broadly consistent with those originally observed in ALS-TE by Tam et al., suggesting potential mechanistic overlap between transcriptional dysregulation and transposable element activation. However, while TE expression characterised only the ALS-TE subtype in Tam et al., the expression of individual TEs was a characteristic of both ALS-TD and ALS-Ox in Eshima et al. The biological relevance of most identified subtypes is supported by molecular mechanisms that are well-established in the literature exploring ALS pathogenesis. Each subtype demonstrates distinct pathological processes, including; oxidative stress (ALS-Ox) involving mitochondrial dysfunction and reactive oxygen species accumulation, consistent with decades of research demonstrating oxidative stress as a core pathogenic mechanism in ALS, particularly in SOD1-associated disease where mutant SOD1 was known to generate toxic reactive oxygen species [17,18]; glial activation and neuroinflammation (ALS-Glia) characterised by microglial and astrocytic dysfunction, validating extensive prior evidence of neuroinflammation in ALS including early PET imaging studies showing microglial activation and neuropathological findings of reactive astrogliosis in post-mortem tissues [19,20]; transcriptional disruption (ALS-TD) involving RNA-binding protein dysfunction, directly confirming the central role of RNA metabolism dysfunction established through discoveries of TDP-43, FUS, and C9orf72 mutations, all of which encode RNA-binding proteins [21]; and altered neuropeptide signalling (ALS-Neu) affecting motor neuron survival and synaptic function, aligning with emerging evidence of synaptic dysfunction and altered neurotrophic signalling in ALS, extending beyond the traditional focus on motor neuron cell death [22,23]. Transposable element dysregulation (ALS-TE) represented a novel pathogenic mechanism not previously recognized in ALS, though it had been linked to other neurodegenerative diseases and aging [24,25]. Tam et al. demonstrated in their study demonstrated that TE are linked to TDP-43 pathology and genomic instability in vitro and in vivo.
Figure 2.
Reported ALS molecular subtypes in the nervous tissue across studies. (A) Heatmap showing whether each subtype was identified in Eshima et al., [10], Marriott et al., [11], O’Neill et al., [12], and Tam et al. [13]. (B) Relative prevalence of subtypes reported in each study. Across datasets, ALS-Glia and ALS-Ox subtypes are consistently observed, while ALS-TE and ALS-TD appear more study- and tissue-specific. ALS-Neu was uniquely described by Marriott et al. [11].
Figure 2.
Reported ALS molecular subtypes in the nervous tissue across studies. (A) Heatmap showing whether each subtype was identified in Eshima et al., [10], Marriott et al., [11], O’Neill et al., [12], and Tam et al. [13]. (B) Relative prevalence of subtypes reported in each study. Across datasets, ALS-Glia and ALS-Ox subtypes are consistently observed, while ALS-TE and ALS-TD appear more study- and tissue-specific. ALS-Neu was uniquely described by Marriott et al. [11].
ALS-TE: Increase in Transposable Element Expression Subtype
The ALS-TE subtype was identified by Tam, et al., [13] and subsequently reproduced by O’Neill, et al., [12] with both studies leveraging the NYGC ALS consortium dataset and overlapping teams. The subtype represents approximately 20% of ALS across both respective studies (Table 1; Figure 2B). Using single-cell analysis of frontal and motor cortex tissue, O’Neill, et al., [12] were able to demonstrate greater evidence of TDP-43 dysfunction in motor neurons, elevated TE expression in astrocytes, and increased enrichment of microglia in homeostatic clusters for samples belonging to this phenotype. ALS-TE is also characterised by the increased expression of TE and TDP-43 dysfunction; with key molecular features including aberrant retrotransposon activation, dysregulated RNA processing, and increased pseudogene expression [12,13]. Regarding the clinical correlations of this subtype, O’Neill, et al., [12] identified that individuals within this subgroup have a significant negative correlation with disease duration (r = −0.25, p < 0.05), indicating shorter survival times, though the specific magnitude of this difference was not quantified. However, Tam, et al., [13] were unable to establish this correlation but did identify an association with limb onset.
The role of TEs in ALS is still under investigation despite being implicated in aging and other neurodegenerative disorders [18,32]. The identification of the ALS-TE subtype reinforced the previous notion of the potential association of endogenous retroviruses in ALS pathogenesis [26,27,28,29,30,31]. Tam et al.’s findings implicate a wider range of TEs in ALS beyond endogenous retroviruses and support the role TE dysregulation as a major disease mechanism in ALS, specifically relevant for a distinct subset of ALS patient samples. The discovery of the TE subtype in ALS provided the first systematic evidence that genomic instability through retrotransposon activation constitutes a distinct pathogenic pathway in a substantial subset of ALS patients [13]. The finding directly supported emerging hypotheses about TDP-43’s role in genomic instability, demonstrating that loss of TDP-43 nuclear function leads to widespread increase in transposable element expression [18,24].
ALS-Ox: Oxidative Stress Subtype
The ALS-Ox subtype was concordant across all studies and displayed the largest proportion of pwALS, representing 25–70% of cases within their respective cohorts (Table 1). This subtype is characterised by an increase in oxidative stress, proteotoxic stress and mitochondrial dysfunction. Its molecular signatures include upregulation of SOD1, OXR1 and NEFL, and altered oxidative phosphorylation [10,11,12,13]. Using single-cell analysis of frontal and motor cortex tissue, O’Neill, et al., [12] identified an upregulation in genes associated to autophagy and lysosome activity in motor neurons. Similarly, Marriott, et al., [11] identified a higher composition of astrocytes and endothelial cells within this subtype using cell deconvolution analysis, while Eshima, et al., [10] found it associated with excitatory and inhibitory neurons using the same approach. Furthermore, patients belonging to this group displayed intermediate survival outcomes (36 months) and an age of onset (60±1.16 years); as shown in Table 1 [10].
ALS-Glia: Neuroinflammation/Glial Activation Subtype
Individuals belonging to the ALS-Glia subtype represented the smallest subgroup across all studies, ranging from 10% to 21.4% [10,11,12,13]. This specific subtype is characterised by increased activity in neuroinflammatory signatures and glial cell activation. Cell composition analyses consistently revealed increased proportions of microglia, oligodendrocytes, glial progenitors, and vascular cells in samples associated with this subtype [10,11,13]. Additional evidence from single-cell data further supports microglial enrichment and activation [12]. This subtype correlates with the poorest prognosis (28 months median survival), and the latest age of onset (63.2 ± 1.83 years), suggesting an accentuated neuroinflammation may associate with an aggressive disease progression (Table 1) [10,12]. The identification of the ALS-Glia subtype supported extensive prior evidence of neuroinflammation in ALS, including early PET imaging studies demonstrating microglial activation and neuropathological findings of reactive astrogliosis in post-mortem tissues [19]. This subtype has significantly impacted the field by reinforcing the central role of neuroinflammation in ALS pathogenesis and highlighting potential therapeutic targets focused on modulating microglial activation and inflammatory cascades, establishing neuroinflammation as a key driver of disease progression alongside traditional motor neuron degeneration paradigms [20,33,34,35].
ALS-TD: Transcription Dysregulation in ALS
The ALS-TD subgroup was only identified in the study conducted by Eshima, et al., [10]; representing 29.1% of the study’s sample cohort. Individuals in this subtype are more prone to transcription dysregulation, which is characterised by the aberrant expression of pseudogenes, intronic regions, and long non-coding RNA (Table 1). However, although this subtype shares similar pathways with the subtype ALS-TE, samples in this cluster did not exhibit TDP-43 dysfunction, suggesting dysregulation of alternative underlying mechanisms that govern transcription processes. The study was unable to identify any significant differences between the ALS-TD phenotype and cell composition but were able to establish that individuals with this phenotype have a more favourable prognosis (median survival: 42 months, Table 1), suggesting the underlying pathogenic mechanisms may exhibit less aggressive features than other subtypes [10].
ALS-Neu: Synaptic and Neuropeptide signalling Subtype
The ALS-Neu subgroup was only identified in the study conducted by Marriott, et al., [11]. It represented the highest proportion of samples within the study at a rate of 53.6% (Table 1). According to Marriott, et al [11], this subtype is characterised by the enrichment of pathways associated with neuronal and synaptic signalling, specifically related to processes involved in binding and receptor interaction, cAMP and neuroactive ligand transcription pathways, and neuropeptide activity. Through deconvolution analysis, motor neurons appeared to be more enriched in this subtype when compared to the other phenotypes [11]. However, the study was unable to establish a correlation between this phenotype and age of onset or disease duration, although it did report an association with inflammatory processes. Importantly, this analysis did not incorporate TE data, and therefore the presence of subtypes such as ALS-TE could not be assessed. Future studies that integrate TE expression data may help to confirm the existence of additional subtypes beyond ALS-Neu.
Tissue Specific Differences in Molecular Signatures
The study conducted by O’Neill, et al., [12] also explored the composition of subgroups between the frontal and motor cortex tissue with spinal cord tissue. They identified significant regional differences existing in subtype distribution and features. In spinal cord tissue, ALS-Glia predominates (71% vs. 29% for ALS-Ox), contrasting with motor cortex where ALS-Ox is more common (Figure 2A). Spinal cord ALS-Glia samples show stronger TDP-43 splicing defects, while cortical ALS-TE samples exhibit more pronounced TE expression (Table 1). Importantly, these tissue-specific differences can occur even within the same individuals, with analysis of 267 patients having both motor cortex and spinal cord samples showing that the majority display different molecular subtypes between tissues [12]. This finding suggests that ALS molecular subtypes are not fixed for each patient but can vary across different tissues, highlighting the regional heterogeneity of ALS pathology and the need to consider tissue context when classifying subtypes.
Biomarkers of Molecular Subtype
Although transcriptomic subtypes were initially defined in post-mortem motor cortex tissue, some of the reviewed studies have investigated whether they can also be identified pre-mortem in peripheral samples and thus serve as biomarkers. Grima et al. [14] investigated both diagnostic and prognostic transcriptomic biomarkers, as well as molecular subtypes, in peripheral blood from individuals with sporadic ALS (sALS). For diagnostic biomarkers, the authors identified 245 genes differentially expressed between sALS cases and controls and also identified 6 genes exhibiting of differentially expressed isoforms. These features were used to train a Random Forest Classification model which was able to discriminate sALS from controls with 78% accuracy. The study also explored the potential of prognostic biomarkers in peripheral blood samples. They performed weighted correlation network analysis to determine if co-expressed genes correlated with clinical features and were able to identify an association between 3 co-expressed gene modules with age at sample collection and age at disease onset. They also identified a single module (consisting of 25 genes) associated with sex differences; these enriched genes are characterised to be involved in bacterial defence response. The study also explored two forms of patient stratification to investigate the molecular heterogeneity between subgroups and to identify relevant biomarkers. They first analysed the differences in gene expression between early and late-stage patients when compared to controls and were able to identify 279 differentially expressed genes between late-stage sALS cases and controls but were only able to identify a single differentially expressed gene (C4BPA) between early stage sALS and controls. Separately, they applied unsupervised clustering analysis, which revealed four distinct patient clusters with divergent gene-expression profiles, supporting the presence of underlying molecular heterogeneity among people with sALS. These findings imply that peripheral transcriptomic profiles could reflect disease subtypes, though further validation is required to determine whether such clustering corresponds to previously defined cortical subtypes or holds prognostic value across independent cohorts.
Marriott et al., [11] also trained subtype classifiers using motor cortex bulk RNA-seq with nsNMF and then projected these signatures onto peripheral blood datasets, including PBMC RNA-seq and whole-blood microarrays. They reported that ALS-Ox, ALS-Glia, and ALS-Neu subtypes could be detected in blood with high assignment probabilities, although the degree of separation between groups was weaker than that observed in the cortex tissue. This study provided the first evidence that molecular programmes derived from post-mortem motor cortex tissue are at least partially preserved in blood, supporting their potential as non-invasive biomarkers.
Catanese et al. [36], as part of a larger study whose primary aim was to investigate the transcriptional landscape of the main genetic subtypes of ALS (C9orf72, TARDBP, SOD1 and FUS), investigated the presence of ALS molecular expression signatures in blood and their relationship with those in central nervous system samples. They first used a deep learning model to identify an expression signature in a large, Dutch dataset of expression data from blood samples (397 ALS patients and 645 controls) [37] that was able to discriminate between case and control samples. The resulting gene set was enriched for genes related to toll-like receptor (TLR) cascade, immune response and autophagy. By overlapping and analysing their blood signature with other expression datasets of CNS samples and hiPSC-derived motor neurons from people with ALS carrying C9orf72, FUS, SOD1 and TARDBP mutations, they showed common transcriptional alterations across all tissues and samples underlying and discriminating ALS from control samples. Although this study focussed on the main genetic sub-types of ALS and the transcriptional differences between ALS and controls, it provided strong evidence of the possibility to identify ML derived signatures of ALS that are consistent, at least partially, across ante-mortem and post-mortem samples.
Eshima et al., [10] added further complexity to the cross-tissue exploration of the subtype transcriptional signatures, by investigating the consistency of the molecular subtypes across motor cortex and spinal cord samples of the same individuals. They found that subtype assignments could vary depending on the region analysed from the same individual, with ALS-Glia more prominent in spinal cord and ALS-TE more detectable in motor cortex. Although this study did not extend directly to blood, the results emphasise that although the expression of some of the genes that make up an individual subtype signature might be preserved across different tissues, the overall subtype identification is strongly tissue-dependent, raising important challenges for developing consistent peripheral biomarkers.
More recently, O’Neill et al., [12] applied single-nucleus RNA-seq across the motor cortex, spinal cord, and cerebellum and explored whether molecular subtypes defined in central nervous system tissue could be reflected in peripheral blood. They found that signatures associated with ALS-Ox and ALS-Glia could indeed be identified in blood transcriptomes, supporting the idea that these subtypes manifest systemically. However, they also reported that transcriptional programmes were often region-specific, and subtype assignment could vary depending on the tissue examined from the same donor. This observation reinforces the tissue-dependence highlighted by Eshima et al., [10] and underscores both the promise and the challenge of developing reliable blood-based biomarkers.
Across these studies, certain signatures have shown stronger potential for ante-mortem detection. The oxidative phosphorylation and mitochondrial stress pathways characteristic of ALS-Ox were reproducibly observed in both cortical and peripheral datasets, suggesting systemic mitochondrial dysfunction. Similarly, MHC-II, complement activation, and interferon-stimulated modules associated with ALS-Glia were consistently detected in blood-derived immune cells, consistent with the immune-driven nature of this subtype. In contrast, features of ALS-TE and ALS-TD, such as transposable element activation and cryptic exon inclusion, remain largely confined to cortical tissue, with no robust demonstration in blood to date. Signatures linked to ALS-Neu, including synaptic signalling and axon guidance, were strongly represented in motor cortex but were less clearly observed in peripheral samples [11]. Taken together, these studies indicate that pre-mortem detection of molecular subtypes is feasible, particularly for ALS-Ox and ALS-Glia, and that blood-derived transcriptomic data can stratify patients into biologically meaningful groups. However, reproducibility and resolution of tissue outside of central nervous system remain limited, and some subtypes, such as ALS-TE and ALS-TD, have yet to be reliably identified in blood. Prospective validation in independent, longitudinal cohorts is therefore essential before molecular subtypes can be translated into clinically actionable biomarkers, and these findings highlight their potential utility for stratified clinical trial design and patient enrichment strategies [10,11,12,36].

Table 1.
Molecular Subtypes of ALS Identified Across Studies. Note: The arrows indicate increase or decrease in activity of characteristic in question (↑ indicates increase in activity; ↓ indicates decrease in activity). Values in bold under the Clinical Feature Correlation column represent significant outcomes.
Table 1.
Molecular Subtypes of ALS Identified Across Studies. Note: The arrows indicate increase or decrease in activity of characteristic in question (↑ indicates increase in activity; ↓ indicates decrease in activity). Values in bold under the Clinical Feature Correlation column represent significant outcomes.
| Study | Tissue Type | Subtype | Cell Types | Proportion of pwALS | Replicated in Independent Samples | Tested on Pre-mortem Samples | Molecular characteristics | Clinical Feature Correlation | Code and Data Availability |
|---|---|---|---|---|---|---|---|---|---|
| Tam, et al., 2019 | Frontal and motor cortex tissue |
Increased Transposable element expression (ALS-TE) |
n/a | 20% | Yes | No | ↑ Retrotransposon activation ↑ TDP-43 dysfunction ↑ Transposable elements ↓ Spliceosome components ↓ Protein export pathways |
Associated with limb onset (56% of patients) No survival differences or significant correlations |
Data: All data is provided by the Gene Expression Omnibus database: Motor cortex RNA-seq datasets from CSHL motor cortex: Accession Numbers: GSE122649 CLIP-seq and RNA-seq datasets from SH-SY5Y cells: Accession Numbers: GSE122650 RNA-seq datasets provided by the NYGC ALS Consortium: Accession Numbers: GSE124439 Code is unavailable |
|
Oxidative stress (ALS-Ox) |
n/a | 61% | Yes | No | ↑ Oxidative stress markers ↑ Proteotoxic stress ↑ Autophagy pathways ↑ Oxidative phosphorylation |
No survival differences or significant correlations | |||
|
Glial markers (ALS-Glia) |
- Astrocytes (markers) - Microglia (markers) Oligodendrocytes (markers) |
19% | Yes | No | ↑ Glial activation markers ↑Neuroinflammation |
No survival differences or significant correlations | |||
| Eshima, et al., 2022 | Frontal and motor cortex tissue |
Dysregulation in Transcription (ALS-TD) |
n/a | 29.1% | No | No | ↑ Transcriptional dysregulation ↑ Pseudogenes ↑ lncRNAs, miRNAs ↑ Nonsense-mediated decay |
Better prognosis Median survival: 42 months Mean age of Onset: (62.7 ± 1.68 years) |
Data: Raw data from NCBI Run Selector: Accession code: PRJNA644618 The RSEM processed gene count matrix from Gene Expression Omnibus: Accession code: GSE153960 Processed RNA-seq count files: https://figshare.com/authors/Jarrett_Eshima/13813720 Code: Code for the analysis available in the Barbara Smith Lab GitHub repository: https://github.com/BSmithLab/ALSPatientStratification Scripts used for supervised Classification: https://github.com/plaisier-lab/U5_hNSC_Neural_G0 Classification models are unavailable. |
|
Oxidative stress (ALS-Ox) |
Cell Deconvolution analysis: - Excitatory neurons - Inhibitory neurons |
53.2% | No | No | ↑ Oxidative stress ↓ Oxidative phosphorylation ↑ Proteotoxic stress ↑ Synaptic alterations |
Intermediate survival: 36 months Mean age of Onset: (60.4 ± 1.16 years) |
|||
|
Glial markers (ALS-Glia) |
Cell Deconvolution analysis: - Microglial - Glial progenitor - Vascular cell - Inhibitory neurons |
17.7% | No | No | ↑ Glial activation ↑ Neuroinflammation ↑ MHC Class II ↑ Complement cascade ↓ Transposable elements |
Worse prognosis Median survival: 28 months Mean age of Onset: (63.2 ± 1.83 years) |
|||
| Grima, et al., 2023 | Peripheral blood tissue | Cluster 0 | n/a | 44% | No | n/a | ↑ Translation & Adaptive immune response ↓ Inflammatory / Innate immune response |
n/a |
Data: Raw Fastq files, raw gene counts and offset matrix are available at NCBI Gene Expression Omnibus: Accession code: GSE234297 Code: Code for the analysis is available in the Gitlab repository: https://gitlab.com/mq-mnd/grp_williams/sals_blood_rnaseq |
| Cluster 1 | n/a | ~10.5% | No | n/a |
↓ Proteolysis, Metabolic and RNA-splicing pathways |
n/a | |||
| Cluster 2 | n/a | ~10.5% | No | n/a | ↑ Proteolysis, Metabolic and RNA-splicing pathways (opposite to Cluster 1) | n/a | |||
| Cluster 3 | n/a | 35% | No | n/a | Intermediate profile between Clusters 1 and 2 with few unique genes | n/a | |||
| Marriott, et al., 2023 | Frontal and motor cortex tissue |
Synaptic and neuropeptide signalling (ALS-Neu) |
Cell Deconvolution analysis: - Motor Neurons |
53.6% | Yes | No | ↑ Synaptic signalling ↑ Neuropeptide activity ↑ Mitochondrial ATP synthesis ↑ cAMP signalling ↑ Neuroactive ligand binding |
Youngest mean age of onset: (58.8 ± 11.6) Disease duration in years (median(IQR)): 3.16 (1.96) |
Data: The RNA- seq validation set generated by Zucca et al. [38] can be found via the NCBI Sequence Read Archive: Accession numbers: PRJNA416880 PRJNA474387 The gene expression microarray data generated by Van Rheenan [37] are available via the Gene Expression Omnibus database: Accession number: GSE112681 The KCL Brain Bank data is available upon reasonable request. Target ALS dataset is available upon approval by the TargetALS Post-mortem Tissue Core. Code: The code used for the analyses performed: https://github.com/KHP-Informatics/HierarchicalClusteringALS/ Class Assignment models are available to use via the link: https://alsgeclustering.er.kcl.ac.uk |
|
Oxidative stress and apoptosis (ALS-Ox) |
Cell Deconvolution analysis: - Astrocytes - Endothelial cells |
25% | Yes | No | ↑ Oxidative stress ↑ Apoptosis signalling ↑ Muscle contraction ↑ Anti-inflammatory processes ↑ Metalloproteinase activity |
Oldest mean age of onset: (65.7 ± 12.3) Disease duration in years (median(IQR)): 2.30 (1.81) |
|||
|
Neuroinflammation (ALS-Glia) |
Cell Deconvolution analysis: - Microglial - Oligodendrocytes |
21.4% | Yes | No | ↑ Neuroinflammation ↑ MHC class II complex ↑ Complement cascade ↑ Interferon signalling ↑ M1 activated microglia |
Mean age of onset: (61.7 ± 15.7) Disease duration in years (median(IQR)): 2.38 (1.75) No significant correlations |
|||
| O’Neill, et al., 2025 | Frontal and motor cortex tissue |
Increased Transposable element expression (ALS-TE) |
Single cell Composition: - L5ET Neurons (TDP-43 dysfunction) - Excitatory neurons - Inhibitory neurons |
20% | No | No | ↑ TDP-43 pathology L5ET Neurons ↑Transposable elements expression in L5ET Neurons ↑ TDP-43 splicing defects |
↓Disease duration against Eigengene score (r=-0.25, P<0.05) |
Data: The RNA- seq and snRNA- seq data are available via the Gene Expression Omnibus: Accession Numbers: GSE271156 Post-mortem frontal/motor cortex and spinal cord RNA- seq was provided by the NYGC ALS Consortium: Accession Numbers: GSE137810 Code: The software developed that can quantify transposable elements from single-cell and single-nuclei datasets: https://github.com/mhammell-laboratory/CellRangerTE Class Assignment DANCer model is available via the study’s GitHub link: https://github.com/mhammell-laboratory/DANcer |
|
Oxidative stress (ALS-Ox) |
Single cell Composition: - L5ET Neurons |
70% | No | No | ↑Oxidative stress ↑Mitochondrial dysfunction |
No significant correlations | |||
|
Glial markers (ALS-Glia) |
Single cell Composition: - L5ET Neurons - Microglial |
10% | No | No | ↑Neuroinflammation ↑Microglial activation |
No significant correlations | |||
| Spinal Cord |
Oxidative stress (ALS-Ox) |
n/a | 29% | No | No | ↑Oxidative stress ↑Neuroactive ligands |
↓ Disease duration against Eigengene score (r=-0.29, P<3e-5) |
||
|
Glial markers (ALS-Glia) |
- Astrocytes (markers) - Microglia (markers) |
71% | No | No | ↑Inflammatory signatures ↑ TDP-43 dysfunction |
↓Disease duration against Eigengene score (r=-0.35, P<2e-10) |
Discussion and Future Directions
In this review, we focus on studies that aimed to identify the molecular subtypes of ALS utilising unsupervised clustering and transcriptomic data. Notably, the reviewed independent studies have identified largely overlapping subtype classifications. The ALS-Ox and ALS-Glia subtypes were consistently identified across all studies reviewed, while the ALS-TE, ALS-TD, and ALS-Neu subgroups were reported only by individual teams, most likely due to the technical and methodological differences implemented in their studies [10,11,12,13]. The characterisation and distribution of these subtypes amongst the ALS population highlight the heterogeneity of the disease and align with the underlying mechanisms that govern its pathology and development [17,18,19,20,21,22,24,38,39]. However, it is important to note that while these studies appear to validate findings across different research groups, three studies utilised overlapping samples from the NYGC ALS Consortium dataset and two studies were from the same group [10,12,13]. This sample overall is largely due to the limited availability of suitable datasets in ALS and may be a limiting factor for independent validation due to population bias and other generalisability concerns. However, while there is some overlap in sample sources, each study did apply their own distinct clustering frameworks while also investigating their robustness against independent studies. For instance, Eshima, et al., [10] validated their findings using an independent sample provided by Prudencio, et al., [40]. This cross-cohort consistency strengthens the evidence for reproducible and biologically meaningful ALS subtypes. Another notable observation is the different molecular subtype patterns identified between tissue samples from O’Neill, et al., [12] study. These findings also highlight regional vulnerability to specific pathological mechanisms and suggest that anatomically targeted therapeutic approaches may be necessary [12].
Furthermore, the studies also explored the clinical relevance for each subtype and found correlations with survival duration, age of onset and with some subtypes having an association with limb onset and inflammatory processes. These findings highlight their potential for prognostic use and clinical stratification. Although not yet implemented in routine clinical practice, their associations with key clinical features and molecular mechanisms suggest they could be used to stratify patients in clinical trials, guide personalised treatment approaches, or serve as targets for therapeutic intervention. To this end, some of the studies explored the possibility of identifying subtypes in transcriptomic data from blood samples of pwALS. They showed that subtype signatures derived from the CNS can be used to classify pwALS into these subtypes using blood derived data (Marriott et al.) and that the expression of key gene markers is consistent between ALS blood and brain samples (Catanese et al.). One study, Grima et al. [14], focussed on deriving the subtypes directly from whole-blood RNA-seq or pwALS and identified four patient subgroups (called cluster 0-3 by the authors) with distinct molecular characteristics. While these de novo blood clusters were not explicitly aligned with the cortical subtypes, their biology partly overlaps consistently with the findings from the other studies. Immune/inflammatory signatures in Cluster 0 displays similar characteristics to that of glial-activation or neuroinflammation that defines ALS-Glia in brain tissue, indicating the relevance of immune activity in ALS pathogenesis. In converse, the proteostasis and RNA-processing differences that distinguish Clusters 1 and 2 resemble similar overlap between oxidative or proteotoxic stress of ALS-Ox and the RNA-dysregulation seen in ALS-TD, respectively.
However, due to the lack of matching blood and brain samples from the same individuals, none of the studies could establish reliable blood biomarkers or investigate whether matching brain and blood samples correspond to the same subtype in pwALS. Investigating this issue is particularly important given the results from O’Neill et al., which demonstrated that samples from the same individual, but from different motor tissues, might belong to different subtypes.
Some of the studies also explored whether expression signatures could be used to discriminate between samples obtained from pwALS and non-affected controls. Marriott et al. showed that the same genes comprising the signature of the ALS subtype can distinguish brain samples of people with ALS from non-affected controls with high accuracy (AUC 0.88). Using RNA-seq data from blood samples of pwALS and controls, Grima et al. trained a machine learning classifier with high discriminatory power (accuracy 78%). Similarly, Catanese and colleagues developed a deep learning classifier using microarray data and demonstrated that the expression of key ALS genes underpinning the classifier is conserved across all ALS samples and correlates between blood and CNS samples. These results highlight the diagnostic potential of these approaches beyond their use in patient stratification.
In conclusion, the reviewed studies provide convincing evidence that molecular subtypes exist in ALS samples from both CNS and peripheral tissues, and that machine learning can identify their signatures in transcriptomic data. These subtypes and their expression footprints offer great potential for developing transformative approaches to patient clinical and biological stratification, as well as diagnostics. However, further investigation into their generalizability and usability using large, independent datasets, comprising multiple matching CNS and peripheral tissue samples, as well as longitudinally collected biofluids from the same individuals, is crucial to establish a definitive re-classification of ALS subtypes, identify viable biomarkers, and understand their evolution throughout disease progression and across tissue types.
Funding
A.A.-C. is an NIHR Senior Investigator. A.A.-C. receives salary support from the National Institute for Health and Care Research (NIHR) Dementia Biomedical Research Unit at South London and Maudsley NHS Foundation Trust and King’s College London. The work leading up to this publication was funded by the European Community’s Health Seventh Framework Program (FP7/2007-2013; grant agreement number 259867) and Horizon 2020 Program (H2020-PHC-2014-two-stage; grant agreement number 633413). This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement no. 772376-EScORIAL). This study represents independent research part funded by the NIHR Maudsley Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. A.I. is funded by South London and Maudsley NHS Foundation Trust, MND Scotland, Motor Neurone Disease Association, National Institute for Health and Care Research, Spastic Paraplegia Foundation, Rosetrees Trust, Darby Rimmer MND Foundation, the Medical Research Council (UKRI), Alzheimer’s Research UK and LifeArc.
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. New England Journal of Medicine 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Akçimen, F.; Lopez, E.R.; Landers, J.E.; Nath, A.; Chiò, A.; Chia, R.; Traynor, B.J. Amyotrophic lateral sclerosis: translating genetic discoveries into therapies. Nat. Rev. Genet. 2023, 24, 642–658. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic Lateral Sclerosis. Nature Reviews Disease Primers 2017, 3. [Google Scholar]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.-M.; Le Masson, G. Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Al Khleifat, A.; Sproviero, W.; Shatunov, A.; Jones, A.R.; Opie-Martin, S.; Naselli, E.; Topp, S.D.; Fogh, I.; Hodges, A.; et al. ALSgeneScanner: a pipeline for the analysis and interpretation of DNA sequencing data of ALS patients. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Lin, T.; Al Khleifat, A.; Jones, A.R.; Opie-Martin, S.; Coleman, J.R.; Shatunov, A.; Sproviero, W.; Williams, K.L.; Garton, F.; et al. Genome-wide Meta-analysis Finds the ACSL5-ZDHHC6 Locus Is Associated with ALS and Links Weight Loss to the Disease Genetics. Cell Rep. 2020, 33. [Google Scholar] [CrossRef]
- van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef]
- Iacoangeli, A.; A Dilliott, A.; Al Khleifat, A.; Andersen, P.M.; A Başak, N.; Cooper-Knock, J.; Couratier, P.; Decarvalho, M.; E Drory, V.; Glass, J.D.; et al. Oligogenic structure of amyotrophic lateral sclerosis has genetic testing, counselling and therapeutic implications. J. Neurol. Neurosurg. Psychiatry 2025, 1–9. [Google Scholar] [CrossRef]
- Mehta, P.R.; Iacoangeli, A.; Opie-Martin, S.; A van Vugt, J.J.F.; Al Khleifat, A.; Bredin, A.; Ossher, L.; Andersen, P.M.; Hardiman, O.; Mehta, A.R.; et al. The impact of age on genetic testing decisions in amyotrophic lateral sclerosis. Brain 2022, 145, 4440–4447. [Google Scholar] [CrossRef]
- Eshima, J.; O’Connor, S.A.; Marschall, E.; Bowser, R.; Plaisier, C.L.; Smith, B.S. Molecular subtypes of ALS are associated with differences in patient prognosis. Nat. Commun. 2023, 14. [Google Scholar] [CrossRef]
- Marriott, H.; Kabiljo, R.; Hunt, G.P.; Khleifat, A.A.; Jones, A.; Troakes, C.; Pfaff, A.L.; Quinn, J.P.; Koks, S.; Dobson, R.J.; Schwab, P.; Al-Chalabi, A.; Iacoangeli, A. Unsupervised machine learning identifies distinct ALS molecular subtypes in post-mortem motor cortex and blood expression data. Acta Neuropathol. Commun. 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- O’nEill, K.; Shaw, R.; Bolger, I.; Tam, O.H.; Phatnani, H.; Hammell, M.G. ALS molecular subtypes are a combination of cellular and pathological features learned by deep multiomics classifiers. Cell Rep. 2025, 44, 115402. [Google Scholar] [CrossRef]
- Tam, O.H.; Rozhkov, N.V.; Shaw, R.; Kim, D.; Hubbard, I.; Fennessey, S.; Propp, N.; Phatnani, H.; Kwan, J.; Sareen, D.; et al. Postmortem Cortex Samples Identify Distinct Molecular Subtypes of ALS: Retrotransposon Activation, Oxidative Stress, and Activated Glia. Cell Rep. 2019, 29, 1164–1177.e5. [Google Scholar] [CrossRef]
- Grima, N.; Liu, S.; Southwood, D.; Henden, L.; Smith, A.; Lee, A.; Rowe, D.B.; D’SIlva, S.; Blair, I.P.; Williams, K.L. RNA sequencing of peripheral blood in amyotrophic lateral sclerosis reveals distinct molecular subtypes: Considerations for biomarker discovery. Neuropathol. Appl. Neurobiol. 2023, 49. [Google Scholar] [CrossRef] [PubMed]
- Iacoangeli, A.; Fogh, I.; Selvackadunco, S.; Topp, S.D.; Shatunov, A.; van Rheenen, W.; Al-Khleifat, A.; Opie-Martin, S.; Ratti, A.; Calvo, A.; et al. SCFD1 expression quantitative trait loci in amyotrophic lateral sclerosis are differentially expressed. Brain Commun. 2021, 3. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.R.; Iacoangeli, A.; Adey, B.N.; Bowles, H.; Shatunov, A.; Troakes, C.; Garson, J.A.; McCormick, A.L.; Al-Chalabi, A. A HML6 endogenous retrovirus on chromosome 3 is upregulated in amyotrophic lateral sclerosis motor cortex. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef]
- Blasco, H.; Garcon, G.; Patin, F.; Veyrat-Durebex, C.; Boyer, J.; Devos, D.; Vourc’h, P.; Andres, C.R.; Corcia, P. Panel of Oxidative Stress and Inflammatory Biomarkers in ALS: A Pilot Study. Canadian Journal of Neurological Sciences 2016, 44, 90–95. [Google Scholar] [CrossRef]
- Liu, D.; Wen, J.; Liu, J.; Li, L. The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999, 13, 2318–2328. [Google Scholar] [CrossRef]
- Calma, A.D.; Pavey, N.; Menon, P.; Vucic, S. Neuroinflammation in amyotrophic lateral sclerosis: pathogenic insights and therapeutic implications. Curr. Opin. Neurol. 2024, 37, 585–592. [Google Scholar] [CrossRef]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’aMbrosi, N. The dual role of microglia in ALS: Mechanisms and therapeutic approaches. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef]
- Cao, M.C.; Scotter, E.L. Transcriptional targets of amyotrophic lateral sclerosis/frontotemporal dementia protein TDP-43 – meta-analysis and interactive graphical database. Dis. Model. Mech. 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Seki, S.; Kitaoka, Y.; Kawata, S.; Nishiura, A.; Uchihashi, T.; Hiraoka, S.-I.; Yokota, Y.; Isomura, E.T.; Kogo, M.; Tanaka, S. Characteristics of Sensory Neuron Dysfunction in Amyotrophic Lateral Sclerosis (ALS): Potential for ALS Therapy. Biomedicines 2023, 11. [Google Scholar] [CrossRef]
- Zuccaro, E.; Piol, D.; Basso, M.; Pennuto, M. Motor Neuron Diseases and Neuroprotective Peptides: A Closer Look to Neurons. Front. Aging Neurosci. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Gonzales, P.K.; Cook, C.N.; Gendron, T.F.; Daughrity, L.M.; Song, Y.; Ebbert, M.T.; van Blitterswijk, M.; Zhang, Y.-J.; Jansen-West, K.; et al. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum. Mol. Genet. 2017, 26, 3421–3431. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito-Maturana, B.; Arancibia, E.; Riadi, G.; Tapia, J.C.; Carrasco, M. Locus-specific analysis of Transposable Elements during the progression of ALS in the SOD1G93A mouse model. PLOS ONE 2021, 16. [Google Scholar] [CrossRef]
- Westarp, M.E.; Ferrante, P.; Perron, H.; Bartmann, P.; Kornhuber, H.H. Sporadic ALS/MND: a global neurodegeneration with retroviral involvement? J. Neurol. Sci. 1995, 129, 145–147. [Google Scholar] [CrossRef]
- Ferrante, P.; Westarp, M.; Mancuso, R.; Puricelli, S.; Westarp, M.; Mini, M.; Caputo, D.; Zuffolato, M. HTLV tax-rex DNA and antibodies in idiopathic amyotrophic lateral sclerosis. J. Neurol. Sci. 1995, 129, 140–144. [Google Scholar] [CrossRef]
- Westarp, M.; Bartmann, P.; Rossler, J.; Geiger, E.; Westphal, K.P.; Schreiber, H.; Fuchs, D.; Westerp, M.P.; Kornhuber, H.H. Antiretroviral therapy in sporadic adult amyotrophic lateral sclerosis. NeuroReport 1993, 4, 819–822. [Google Scholar] [CrossRef]
- Andrews, W.; Tuke, P.; Al-Chalabi, A.; Gaudin, P.; Ijaz, S.; Parton, M.; Garson, J. Detection of reverse transcriptase activity in the serum of patients with motor neurone disease. J. Med Virol. 2000, 61, 527–532. [Google Scholar] [CrossRef]
- Douville, R.; Liu, J.; Rothstein, J.; Nath, A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol. 2010, 69, 141–151. [Google Scholar] [CrossRef]
- Gold, J.; Rowe, D.B.; Kiernan, M.C.; Vucic, S.; Mathers, S.; van Eijk, R.P.A.; Nath, A.; Montojo, M.G.; Norato, G.; Santamaria, U.A.; et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: the Lighthouse trial. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 595–604. [Google Scholar] [CrossRef]
- Savage, A.L.; Schumann, G.G.; Breen, G.; Bubb, V.J.; Al-Chalabi, A.; Quinn, J.P. Retrotransposons in the development and progression of amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2018, 90, 284–293. [Google Scholar] [CrossRef]
- Bond, S.; Saxena, S.; Sierra-Delgado, J.A. Microglia in ALS: Insights into Mechanisms and Therapeutic Potential. Cells 2025, 14. [Google Scholar] [CrossRef]
- Bensimon, G.; Leigh, P.N.; Tree, T.; Malaspina, A.; Payan, C.A.; Pham, H.-P.; Klaassen, P.; Shaw, P.J.; Al Khleifat, A.; Amador, M.D.M.; et al. Efficacy and safety of low-dose IL-2 as an add-on therapy to riluzole (MIROCALS): a phase 2b, double-blind, randomised, placebo-controlled trial. Lancet 2025, 405, 1837–1850. [Google Scholar] [CrossRef]
- Camu, W.; Mickunas, M.; Veyrune, J.-L.; Payan, C.; Garlanda, C.; Locati, M.; Juntas-Morales, R.; Pageot, N.; Malaspina, A.; Andreasson, U.; et al. Repeated 5-day cycles of low dose aldesleukin in amyotrophic lateral sclerosis (IMODALS): A phase 2a randomised, double-blind, placebo-controlled trial. EBioMedicine 2020, 59. [Google Scholar] [CrossRef]
- Catanese, A.; Rajkumar, S.; Sommer, D.; Masrori, P.; Hersmus, N.; Van Damme, P.; Witzel, S.; Ludolph, A.; Ho, R.; Boeckers, T.M.; et al. Multiomics and machine-learning identify novel transcriptional and mutational signatures in amyotrophic lateral sclerosis. Brain 2023, 146, 3770–3782. [Google Scholar] [CrossRef]
- Van Rheenen, W.; Diekstra, F.P.; Harschnitz, O.; Westeneng, H.-J.; Van Eijk, K.R.; Saris, C.G.J.; Groen, E.J.N.; A Van Es, M.; Blauw, H.M.; Van Vught, P.W.J.; et al. Whole blood transcriptome analysis in amyotrophic lateral sclerosis: A biomarker study. PLOS ONE 2018, 13. [Google Scholar] [CrossRef]
- Zucca, S.; Gagliardi, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. RNA-Seq profiling in peripheral blood mononuclear cells of amyotrophic lateral sclerosis patients and controls. Sci. Data 2019, 6. [Google Scholar] [CrossRef]
- Valdebenito-Maturana, B.; Rojas-Tapia, M.I.; Carrasco, M.; Tapia, J.C. Dysregulated Expression of Transposable Elements in TDP-43M337V Human Motor Neurons That Recapitulate Amyotrophic Lateral Sclerosis In Vitro. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Humphrey, J.; Pickles, S.; Brown, A.-L.; Hill, S.E.; Kachergus, J.M.; Shi, J.; Heckman, M.G.; Spiegel, M.R.; Cook, C.; et al. Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. J. Clin. Investig. 2020, 130, 6080–6092. [Google Scholar] [CrossRef] [PubMed]
- Thakral, S.; Purohit, P.; Mishra, R.; Gupta, V.; Setia, P. The impact of RNA stability and degradation in different tissues to the determination of post-mortem interval: A systematic review. Forensic Sci. Int. 2023, 349. [Google Scholar] [CrossRef]
- H. Belyadi and A. Haghighat, “Chapter 3 - Machine learning workflows and types,” in Machine Learning Guide for Oil and Gas Using Python, Gulf Professional Publishing, 2021, pp. 97-123.
- Guo, Y.-T.; Li, Q.-Q.; Liang, C.-S. The rise of nonnegative matrix factorization: Algorithms and applications. Inf. Syst. 2024, 123. [Google Scholar] [CrossRef]
- Subramanian, P. Tamayo, V. K. Mootha, S. Mukherjee, B. L. Ebert, M. A. Gillette, A. Paulovich, S. L. Pomeroy, T. R. Golub, E. S. Lander and J. P. Mesirov, “Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles,” 2005.
- Mathur, R.; Rotroff, D.; Ma, J.; Shojaie, A.; Motsinger-Reif, A. Gene set analysis methods: a systematic comparison. BioData Min. 2018, 11. [Google Scholar] [CrossRef]
- Gaiani, A.; Martinelli, I.; Bello, L.; Querin, G.; Puthenparampil, M.; Ruggero, S.; Toffanin, E.; Cagnin, A.; Briani, C.; Pegoraro, E.; Soraru, G. Diagnostic and prognostic biomarkers in amyotrophic lateral sclerosis: Neurofilament light chain levels in definite subtypes of disease. JAMA Neurolog 2017, 74, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Heiman-Patterson, T.D.; Sher, R.B.; Blankenhorn, E.A.; Alexander, G.; Deitch, J.S.; Kunst, C.B.; Maragakis, N.; Cox, G. Effect of genetic background on phenotype variability in transgenic mouse models of amyotrophic lateral sclerosis: A window of opportunity in the search for genetic modifiers. Amyotroph. Lateral Scler. 2011, 12, 79–86. [Google Scholar] [CrossRef]
- Venneti, S.; Robinson, J.L.; Roy, S.; White, M.T.; Baccon, J.; Xie, S.X.; Trojanowski, J.Q. Simulated brain biopsy for diagnosing neurodegeneration using autopsy-confirmed cases. Acta Neuropathol. 2011, 122, 737–745. [Google Scholar] [CrossRef]
- Harvey, C.; Nowak, A.; Zhang, S.; Moll, T.; Weimer, A.K.; Barcons, A.M.; Souza, C.D.S.; Ferraiuolo, L.; Kenna, K.; Zaitlen, N.; et al. Evaluation of a biomarker for amyotrophic lateral sclerosis derived from a hypomethylated DNA signature of human motor neurons. BMC Med Genom. 2025, 18. [Google Scholar] [CrossRef]
- Gomes, L.C.; Hänzelmann, S.; Hausmann, F.; Khatri, R.; Oller, S.; Parvaz, M.; Tzeplaeff, L.; Pasetto, L.; Gebelin, M.; Ebbing, M.; et al. Multiomic ALS signatures highlight subclusters and sex differences suggesting the MAPK pathway as therapeutic target. Nat. Commun. 2024, 15. [Google Scholar] [CrossRef]
- Bild and P. George Febbo, “Application of a priori established gene sets to discover biologically important differential expression in microarray data,” 2005.
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: Current approaches and outstanding challenges. PLOS Comput. Biol. 2012, 8. [Google Scholar] [CrossRef]
- Filippi, M.; Agosta, F. Does neuroinflammation sustain neurodegeneration in ALS? Neurology 2016, 87, 2508–2509. [Google Scholar] [CrossRef]
- Jo, M.; Lee, S.; Jeon, Y.-M.; Kim, S.; Kwon, Y.; Kim, H.-J. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp. Mol. Med. 2020, 52, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Sakowski, S.A.; Schuyler, A.D.; Feldman, E.L. Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 63–73. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nature Communications 2016, 7. [Google Scholar] [CrossRef]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; Fazal, R.; Germeys, C.; Quaegebeur, A.; Fendt, S.M.; Carmeliet, P.; Verfaillie, C.; Van Damme, P.; Ghesquière, B.; De Bock, K.; Van Den Bosch, L. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nature Communications 2019, 10. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Neale, B.M.; Heskes, T.; Posthuma, D. The statistical properties of gene-set analysis. Nat. Rev. Genet. 2016, 17, 353–364. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Endogenous retroviruses in ALS: A reawakening? Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- J. A. Garson, L. Usher, A. Al-Chalabi, J. Huggett, E. F. Day and A. L. McCormick, “Quantitative analysis of human endogenous retrovirus-K transcripts in postmortem premotor cortex fails to confirm elevated expression of HERV-K RNA in amyotrophic lateral sclerosis,” Acta neuropathologica communications, vol. 7, no. 1, p. 45, 3 2019. [CrossRef]
- McCormick, A.L.; Brown, R.H.; Cudkowicz, M.E.; Al-Chalabi, A.; Garson, J.A. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 2008, 70, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.J.; Al-Chalabi, A.; Ferrante, K.; Cudkowicz, M.E.; Brown, R.H.; Garson, J.A. Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology 2005, 64, 454–458. [Google Scholar] [CrossRef]
- J. A. Garson, L. Usher, A. Al-Chalabi, J. Huggett, E. F. Day and A. L. McCormick, “Quantitative analysis of human endogenous retrovirus-K transcripts in postmortem premotor cortex fails to confirm elevated expression of HERV-K RNA in amyotrophic lateral sclerosis,” Acta neuropathologica communications, vol. 7, no. 102, 2019.
Figure 1.
Timeline summarising key studies that investigated molecular subtypes of ALS using stratification approaches. Early work by Tam et al. [13] identified three cortical subtypes (ALS-Ox, ALS-Glia, ALS-TE), which were later refined and validated by Eshima et al. [10]. Subsequent studies in 2023 extended these findings to blood-based stratification [11,14], highlighting potential biomarker applications, while also demonstrating subtype robustness in independent cohorts. Most recently, O’Neill et al. [12] revealed tissue-specific distributions between cortex and spinal cord, suggesting spatial heterogeneity in subtype prevalence.
Figure 1.
Timeline summarising key studies that investigated molecular subtypes of ALS using stratification approaches. Early work by Tam et al. [13] identified three cortical subtypes (ALS-Ox, ALS-Glia, ALS-TE), which were later refined and validated by Eshima et al. [10]. Subsequent studies in 2023 extended these findings to blood-based stratification [11,14], highlighting potential biomarker applications, while also demonstrating subtype robustness in independent cohorts. Most recently, O’Neill et al. [12] revealed tissue-specific distributions between cortex and spinal cord, suggesting spatial heterogeneity in subtype prevalence.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



