Submitted:
30 October 2025
Posted:
31 October 2025
You are already at the latest version
Abstract
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the two most common forms of dementia due to neurodegeneration. AD is characterized by extracellular amyloid‑β (Aβ) plaques and intracellular tau neurofibrillary tangles, whereas DLB is defined by α‑synuclein (α‑Syn)–containing Lewy bodies. Although AD and DLB exhibit divergent core features, the disorders frequently co‑occur and converge on shared endpoints. Co‑pathology is common and linked to more severe cognitive decline, faster progression, and clinicopathological heterogeneity. Here, we discuss the current understanding of shared and unique clinical and neuropathological features of AD and DLB. We compare genetic risk and pathological drivers (Aβ and tau in AD; α-Syn in DLB) and their overlapping co-pathology, and review downstream mechanisms—mitochondrial dysfunction, oxidative stress, neuroinflammation, and cerebrovascular contributions, including cerebral amyloid angiopathy. We highlight recent findings from state‑of‑the‑art multi‑omics (transcriptomic, proteomic, metabolomic, single‑cell/spatial) that reveal convergent and disease‑specific molecular signatures of AD and DLB. We outline a framework for emerging next‑generation biomarkers—from blood‑based and cerebrospinal fluid assays to imaging and digital measures—for diagnosis and stratification, and discuss potential translational implications. Together, these advances help to disentangle shared from disease‑specific mechanisms, which is essential for improved diagnosis and the development of precise, disease‑modifying therapies.
Keywords:
Alzheimer’s disease
; dementia with Lewy bodies
; amyloid-β
; tau
; alpha-synuclein
; biomarkers
1. Introduction
On 3 November 1906, at a German psychiatrists meeting in Tübingen, Germany, Alois Alzheimer, a psychiatrist and neuropathologist gave a talk on “A peculiar severe disease process of the cerebral cortex”, describing a woman named Auguste Deter with symptoms of presenile dementia, and reported the post-mortem autopsy of her brain showing senile plaques and neurofibrillary tangles [1,2]. A few years later in 1910, Emil Kraepelin, a German psychiatrist, named the disease condition as “Alzheimer’s disease” [3,4]. Alzheimer’s disease (AD) is the most common progressive neurodegenerative disorder caused by neuronal cell death. AD currently affects around 50 million patients globally and this number is projected to reach approximately 152 million by 2050 [5,6]. Most AD cases are prevalent particularly among adults aged 65 years or older, with a life expectancy of about 3-10 years [7] . Depending on the stage, AD patients often exhibit a severe decline in cognitive and behavioral ability, initially with memory and spatial navigation problems, and other symptoms such as executive dysfunction (planning/organisation), impaired reasoning and judgement, attention and concentration problems, personality and behaviour changes, mood changes, social withdrawal, agitation and aggression, paranoia/delusions, and sleep disturbances [8,9,10,11] .
Early stage AD exhibits histological alterations and atrophy in the entorhinal cortex within the hippocampus of the medial temporal lobe, a brain region crucial for episodic memory formation and consolidation and spatial navigation [12,13,14]. Biochemists George Glenner and Caine Wong reported the accumulation of a 4-kDa peptide, which they termed “amyloid-β protein” (Aβ), the major constituent of extracellular amyloid plaques [15,16]. Similarly, Inge Grundke-Iqbal and Khalid Iqbal identified microtubule-associated protein tau (MAPT) labeling some neurofibrillary tangles and plaque neurites, in AD brain [17,18].
Dementia with Lewy bodies (DLB) is an age-associated neurodegenerative disorder that is less common than AD [19]. DLB is a member of group of neurodegenerative diseases referred to as synucleinopathies, which also include Parkinson’s disease (PD) and Multiple system atrophy (MSA). Synucleinopathies are a group of disorders characterized by pathological accumulation of the misfolded form of the small presynaptic protein alpha-synuclein (α-Syn), which is encoded by the gene SNCA. Major synucleinopathies are Lewy body disease (LBD) [20]. LBD refers to a group of disorders comprising Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and dementia with Lewy bodies (DLB), which shows the shared neuropathological hallmark of deposition of Lewy bodies within neurons and extensive axonal Lewy neurites [21,22]. However, among synucleinopathies, MSA is characterized by accumulation of inclusions known as glial cytoplasmic inclusions containing synuclein aggregates in oligodendrocytes, while Lewy body pathology is absent and, therefore, the LBD group does not include MSA [21,23,24,25].
DLB has an incidence of 0.5–1.6 per 1,000 person-years and affects an estimated 1.4 million people in the United States [26]. It accounts for ~7.5% of all dementia cases [27]. DLB typically begins in the late fifties and constantly increases with age [28,29], and it is more prevalent in males than females [30]. Historically, in 1912 Jakob Heinrich Lewy, who was studying Parkinson’s disease at Alois Alzheimer’s laboratory, observed eosinophilic intraneuronal inclusion bodies, which were named Lewy bodies by Nikolaevich Tretiakoff in 1919 [31]. DLB gained momentum in 1961, when Okazaki et al. reported that patients with dementia who died shortly thereafter with severe extrapyramidal rigidity [32]. Later, Japanese psychiatrist Kenji Kosaka and colleagues observed Lewy bodies in the brainstem and cerebral cortex in post-mortem autopsy samples from more than 20 patients with different cognitive impairment [33,34]. In 1995, the term “dementia with Lewy bodies” was first proposed at the First International Workshop (Newcastle upon Tyne, England), with diagnostic criteria focused on three core features: impairment in cognitive function, hallucinations, and Parkinsonian features [33,35,36]. In 1997, Spillantini and colleagues identified α-Syn as a major component of Lewy bodies in the brain tissue from patients with DLB and PD [37].
AD and DLB frequently converge clinically and pathologically, complicating diagnosis, prognostication, medication selection, and eligibility for clinical trials. In this review, we briefly compare clinical and neuropathological features of AD and DLB. We integrate evidence across fluid biomarkers, genetics, and imaging to delineate shared versus disease-specific biology. Finally, we translate the evidence from the findings into therapeutic implications and outline new future research directions with potential next steps for targeted treatments.
2. Clinical and Neuropathological Overlap Between AD and DLB
2.1. Clinical Features of AD
AD usually co-exists with ageing and presents with cognitive, behavioral, and psychological symptoms. In the early stage, individuals with AD show amnestic features such as memory loss, trouble learning new information, repeating questions, misplacing items, and difficulty with spatial navigation [11,38]. As the disease progresses, the moderate stage includes memory loss; disorientation of time and place; language difficulties; and impaired executive function such as poor reasoning and planning, with changes in mood (apathy, irritability, or depression), difficulty in solving complex daily tasks, and impaired judgement. In the late stages, AD patients show global cognitive failure, dependency for activities of daily living, gait impairment, dysphagia, and other complex neurological and psychiatric symptoms such as agitation, delusions or occasional hallucinations, and extrapyramidal symptoms such as rigidity or gait disturbance [11,39]. Initially, in 1984, the criteria were set by the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA criteria for AD) as a suitable procedure for a clinical diagnosis of AD and have recently been updated [40,41,42]. The National Institute on Aging and Alzheimer’s Association (NIA–AA) updated the clinical approach guidelines, introduced the use of biomarkers, and proposed a new biological scheme: amyloid, tau, neurodegeneration (A/T(N)) for research [43]. In day-to-day practice, clinicians still diagnose the disease based on medical records, physical and neurological examination, neuropsychological evaluation, and neuroimaging [44].
2.2. Clinical Features of DLB
Individuals with DLB also show cognitive with characteristic non-amnestic features that often different from typical AD. The major symptoms of DLB are progressive cognitive or visuospatial impairment with fluctuating attention and alertness, parkinsonism (rigidity, slowness, tremor, shuffling gait, falls), recurrent visual hallucinations, REM sleep behaviour disorder (RBD), and antipsychotic sensitivity causing worse parkinsonism, confusion, and sleepiness [45,46]. In the later stages DLB patients commonly develop autonomic instability (orthostatic hypotension, urinary incontinence), repeated falls, and increased sensitivity to antipsychotics [30]. The 2017 DLB Consortium criteria diagnose probable DLB with two or more core clinical features, or one core feature plus an indicative biomarker: reduced striatal dopamine-transporter uptake (Positron Emission Tomography / Single-Photon Emission Computed Tomography, PET/SPECT), reduced myocardial iodine-123-metaiodobenzylguanidine (MIBG) uptake, or polysomnographic confirmation of rapid eye movement (REM) sleep without atonia [47,48,49]. Many symptoms of DLB overlap with PD, hence, the timing of the symptoms is diagnostically crucial. The “1-year rule”: dementia that begins before or within 1 year of the onset of parkinsonism helps separate DLB from PDD. DLB is favoured when dementia precedes or appears within 1 year of the onset of parkinsonism [35,48,50,51].
AD and DLB have some distinct early symptoms that help differentiate them. AD starts with prominent episodic memory loss and a steady decline, spatial navigation deficits, and features like hallucinations or parkinsonism usually appear late [44,52]. However, in DLB, early visual hallucinations, fluctuating cognitive features, RBD, and parkinsonism are hallmarks; autonomic failure and marked antipsychotic sensitivity increase the diagnostic confidence for DLB [53,54]. On bedside testing, DLB shows visuospatial and attentional deficits disproportionate to memory impairment, whereas AD is characterised by predominant amnestic deficits. Timing of motor signs is important: parkinsonism near or preceding cognitive symptoms supports DLB, while late parkinsonism in a prolonged amnestic course favors AD with superimposed extrapyramidal signs [55]. Recognition of these discriminators reduces misdiagnosis and avoids toxic antipsychotic exposure in DLB.
2.3. Neuropathological Features of AD
At the autopsy, AD brains often show moderate cerebral cortical atrophy primarily with narrowed gyri and widened sulci, early and significant atrophy of the medial temporal lobe (hippocampal/entorhinal atrophy), and symmetrical dilation of the lateral ventricles (hydrocephalus ex vacuo) compared with healthy age-matched controls [56,57,58]. Microscopically, AD brains show accumulation of extracellular Aβ (diffuse and neuritic) and intraneuronal neurofibrillary tangles composed of hyperphosphorylated tau [59,60,61]. In a landmark study by Braak H. and Braak E., tau pathology progresses in a predictable Braak sequence: transentorhinal (Stage I–Stage II) → limbic/hippocampal (Stage III–Stage IV) → widespread neocortical (Stage V–Stage VI), tracking clinical severity; Aβ deposition follows Thal phases from neocortex to deeper structures [62,63,64,65]. Thal phases (1–5) stage the spread of amyloid-β plaques in AD: starting in the neocortex (1), then hippocampal/allocortical regions (2), striatum and diencephalon (3), brainstem (4), and finally the cerebellum (5) [65]. Neuroinflammation such as activated microglia and astrocytosis, synaptic loss, and neuronal death accompany these lesions, and tangle burden correlates best with cognitive impairment [56]. Current NIA–AA neuropathologic guidelines report “AD neuropathologic change” on a continuum, integrating Braak neurofibrillary tangle stage, Thal Aβ phase, and neuritic-plaque scores [66].
2.4. Neuropathological Features of DLB
DLB brains also show cortical atrophy, but it is less severe than in AD [67]. Most prominently in early stages, for instance in the medial temporal lobe (hippocampus), atrophy is more profound in AD patients than in DLB. DLB patients show Lewy pathology, which is observed in the brainstem (mainly the substantia nigra), limbic, and neocortical regions [68]. DLB brains mainly exhibit depigmentation of the substantia nigra and locus coeruleus due to loss of the majority of pigmented neurons. In later stages, DLB individuals’ brains show moderate cortical atrophy [67,69,70,71]. Microscopically, DLB patients show two general types of Lewy bodies: classic brainstem Lewy bodies with a dense eosinophilic core and a pale halo (visible by haematoxylin and eosin stain), and cortical Lewy bodies, which are smaller, lack the halo, and are mainly observed by α-Syn immunohistochemistry. α-Syn is the primary component of Lewy bodies [22,72,73]. However, post-mortem studies of DLB brains suggest that microglial activation is not prominent in DLB compared to AD [74].
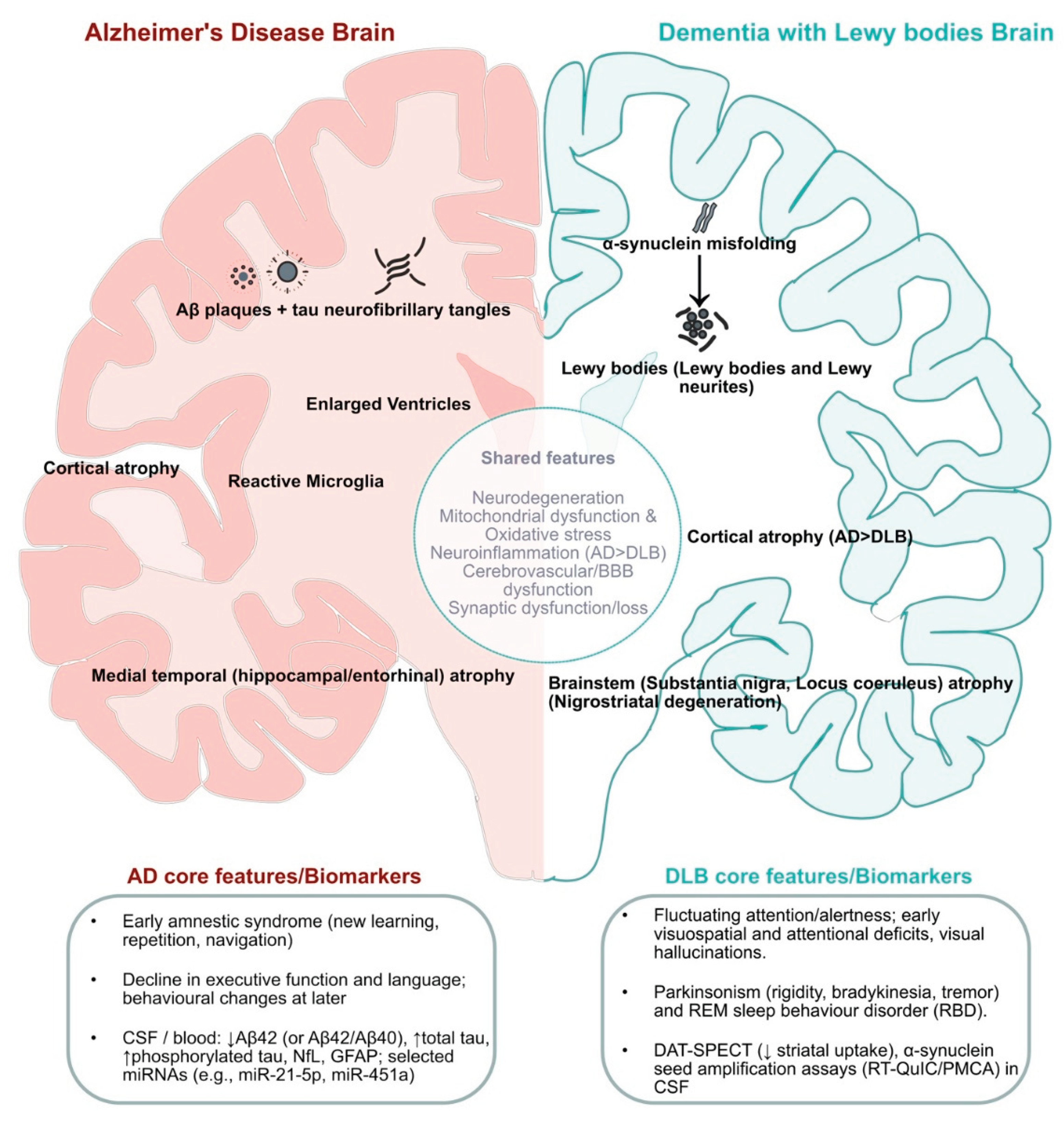
Comparatively, DLB post-mortem tissue shows widespread cortical and limbic α-Syn–containing inclusions, whereas AD shows medial temporal (hippocampal) atrophy with abundant neuritic Aβ plaques and high-stage tau tangles. Co-pathology is common, but lesion type, regional burden, and staging (Braak/Thal) usually distinguish the two [67,75,76,77]. An overview of shared versus disease-specific features is shown in Figure 1.
4. Emerging Next-Generation Biomarkers
The current well-established tools for diagnosing AD are CSF biomarkers and neuroimaging. Imaging techniques include advanced PET, such as Aβ-PET to visualise Aβ deposition and tau-PET to assess tau pathology, together with 18F-fluorodeoxyglucose (18F-FDG) PET and structural magnetic resonance imaging (MRI). The latter two serve as indicators of neurodegeneration [155,156]. Major reliable CSF and advanced blood-based biomarkers for pathological changes in AD are Aβ42 (often expressed as the Aβ42/Aβ40 ratio), total tau, phosphorylated tau, neurofilament light chain (NfL), the microRNA subtypes miR-21-5p and miR-451a, and glial fibrillary acidic protein (GFAP) [157,158].
Beyond Aβ and tau, other non-traditional and utilising minimally invasive alternative biomarkers such as salivary (e.g., lactoferrin, acetylcholinesterase), urinary (AD7c neuronal thread protein, extracellular vesicle proteins), lipidomic shifts (ceramide/sphingomyelin), synaptic/axonal proteins (neurogranin), circulating microRNAs, and gut-/fecal-derived readouts that reflects to inflammation, synaptic injury, mitochondrial stress, and membrane lipid dysregulation enhance diagnostic accuracy [159].
Interestingly, detection of Spatial navigation ability measured with the Sea Hero Quest smartphone game is emerging as a sensitive, early “cognitive fingerprint” of AD risk. In large, real-world large demographic samples, navigation performance can flag preclinical risk and has been reported to outperform some standard memory tests, positioning such early digital biomarkers [38,160].
The well-known current clinical practices for diagnosing DLB are (i) ¹²³I-FP-CIT dopamine transporter SPECT (DAT-SPECT) for nigrostriatal uptake, (ii) polysomnography for REM sleep without atonia, and (iii) ¹²³I-MIBG cardiac scintigraphy showing reduced cardiac uptake. DAT SPECT offers high specificity; however, it has only moderate sensitivity in prodromal stages [161]. A new line of research has offered detection of α-Syn in biological samples as an additional avenue for biomarker development [162]. Development of α-Syn seed amplification assays (SAAs) such as RT-QuIC (Real-Time Quaking-Induced Conversion) and PMCA (Protein Misfolding Cyclic Amplification) show high sensitivity and specificity in CSF for PD and hold promise for stratifying synucleinopathies, including DLB, using seeded-aggregation kinetics [163]. No blood test is yet clinically validated for DLB, although plasma/exosomal α-Syn are under study and some non-traditional matrices including SAAs on peripheral tissues and body fluid olfactory mucosa and urine have shown poor agreement with CSF [164].
Sjaelland and colleagues, in a systematic review that screened 4,295 records and included 20 studies, reported that 17 different portable and wearable digital health technologies have been used to capture digital biomarkers of non-cognitive symptoms in DLB, but validation and feasibility reporting remain uncertain. Future digital biomarker–related studies with standardized evaluation are needed before adoption for clinical use [165].
A multiplex approach that integrates core clinical features with neuroimaging and fluid biomarkers is needed to improve the diagnostic accuracy of DLB [166,167]. Given the complex nature of AD and DLB pathobiology, no single marker can fully capture disease progression across all stages. Combined use of fluid biomarkers with imaging biomarkers and emerging new biomarkers can provide comprehensive biological insights. Using fluid and imaging measures together provides multilevel insight that better supports precision diagnosis, prognosis and treatment monitoring, thereby offering new opportunities for diagnosis, hence, effective disease management.
5. Conclusions and Therapeutic Future Directions
Increasing cases of AD and DLB impose a steep and rising societal and economic burden such as caregiver stress and health-system costs. Despite impressive progress in the field, disease-modifying options remain limited aside from recently approved anti-amyloid monoclonal antibodies for AD [168], so there is an urgent need for therapies that halt progression. AD and DLB frequently co-occur and share pathology, complicating diagnosis, prognosis, and medication choice. Hence, it is important to investigate the contribution of co-pathology in each patient. Determining whether a patient has pure AD, pure DLB, or a mixed phenotype is crucial for clinical trial selection and interpretation.
While established imaging and CSF tests are valuable, the current biomarker toolkit for distinguishing mixed versus pure disease is still incomplete. Larger, longitudinal, and standardized studies including α-Syn SAAs, A/T(N) panels, proteomic/metabolomic signatures, peripheral or blood-based signatures and rigorously validated digital biomarkers could help in clarifying these complex questions. Leveraging patient-derived induced pluripotent stem cells (iPSCs) and advanced brain organoid models that recapitulate α-Syn, tau, and Aβ pathologies will accelerate target discovery and mechanism-driven pharmacology. High-throughput CRISPR perturbation platforms such as unbiased screens [169] should be used to reveal druggable targets. Modern drug-design toolkits: computer-aided drug design, integrative drug designing approach [170], enhancing targeted protein degradation (e.g., PROTACs), molecular chaperone modulators, and peptide/binder design platforms such as BindCraft [171] can be deployed to design and identify selective modulators of α-Syn, tau, and upstream trafficking/clearance machinery. Future studies should be directed at investigating a combinatorial treatment approach, such as pairing an anti-amyloid agent with an anti-α-Syn therapy, with or without an anti-tau component and other additional strategies for synaptic repair, mitochondrial support, glial-immune modulation, and neurovascular protection.
In conclusion, deeper investigation of the molecular mechanisms, particularly the synergistic interplay of Aβ, α-Syn, and tau, is needed to uncover overlapping pathology and points of divergence. Clarifying these networks will open new avenues for earlier and more accurate diagnostics and for rational, mechanism-based therapeutic interventions.
Author Contributions
Conceptualization, Investigation, Writing – original draft: S.N. Visualization: S.N. Supervision, review & editing: T.H. Both authors have read and approved the final version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
18F-FDG 18F-fluorodeoxyglucose
A/T(N) amyloid / tau / neurodegeneration
Aβ amyloid-β
AD Alzheimer’s disease
ADNI Alzheimer’s Disease Neuroimaging Initiative
BBB blood–brain barrier
BACE1 β-site APP cleaving enzyme 1
CAA cerebral amyloid angiopathy
CSF cerebrospinal fluid
CTF99 C-terminal fragment 99 (of APP)
DAT-SPECT dopamine transporter single-photon emission computed tomography
DLB dementia with Lewy bodies
GFAP glial fibrillary acidic protein
GWAS genome-wide association studies
iPSC induced pluripotent stem cells
LBD Lewy body disease
LBV Lewy body variant (of Alzheimer’s disease)
LMTK2 Lemur Tyrosine Kinase 2
MAPK mitogen-activated protein kinase
MAPT microtubule-associated protein tau
MIBG metaiodobenzylguanidine
miR microRNA
MRI magnetic resonance imaging
MSA multiple system atrophy
NfL neurofilament light chain
NIA–AA National Institute on Aging–Alzheimer’s Association
NINCDS–ADRDA National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association
PD Parkinson’s disease
PDD Parkinson’s disease dementia
PET positron emission tomography
PFF pre-formed fibril
PheWAS phenome-wide association studies
PMCA Protein Misfolding Cyclic Amplification
RBD REM sleep behaviour disorder
REM rapid eye movement
RNA-seq RNA sequencing
RT-QuIC real-time quaking-induced conversion
SAA(s) seed amplification assays
SPECT single-photon emission computed tomography
TDP-43 TAR DNA-binding protein 43
TMT-MS tandem mass tag mass spectrometry
α-Syn alpha-synuclein
References
- Hippius, H.; Neundörfer, G. The Discovery of Alzheimer’s Disease. Dialogues in Clinical Neuroscience 2003, 5, 101–108. [Google Scholar] [CrossRef]
- Thakor, V.S.; Tyagi, A.; Lee Jr., J. M.; Coffman, F.; Mittal, R. Alois Alzheimer (1864-1915): The Father of Modern Dementia Research and the Discovery of Alzheimer’s Disease. 2024. [Google Scholar] [CrossRef]
- Yang, H.D.; Kim, D.H.; Lee, S.B.; Young, L.D. History of Alzheimer’s Disease. Dement Neurocognitive Disord 2016, 15, 115. [Google Scholar] [CrossRef] [PubMed]
- Ciurea, V.A.; Covache-Busuioc, R.-A.; Mohan, A.G.; Costin, H.P.; Voicu, V. Alzheimer’s Disease: 120 Years of Research and Progress. JMedLife 2023, 16, 173–177. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia Prevention, Intervention, and Care: 2020 Report of the Lancet Commission. The Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef] [PubMed]
- Xiaopeng, Z.; Jing, Y.; Xia, L.; Xingsheng, W.; Juan, D.; Yan, L.; Baoshan, L. Global Burden of Alzheimer’s Disease and Other Dementias in Adults Aged 65 Years and Older, 1991–2021: Population-Based Study. Front. Public Health 2025, 13, 1585711. [Google Scholar] [CrossRef]
- Liang, C.-S.; Li, D.-J.; Yang, F.-C.; Tseng, P.-T.; Carvalho, A.F.; Stubbs, B.; Thompson, T.; Mueller, C.; Shin, J.I.; Radua, J.; et al. Mortality Rates in Alzheimer’s Disease and Non-Alzheimer’s Dementias: A Systematic Review and Meta-Analysis. The Lancet Healthy Longevity 2021, 2, e479–e488. [Google Scholar] [CrossRef]
- Barnes, J.; Dickerson, B.C.; Frost, C.; Jiskoot, L.C.; Wolk, D.; Van Der Flier, W.M. Alzheimer’s Disease First Symptoms Are Age Dependent: Evidence from the NACC Dataset. Alzheimer’s & Dementia 2015, 11, 1349–1357. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Förstl, H.; Kurz, A. Clinical Features of Alzheimer’s Disease.
- Zvěřová, M. Clinical Aspects of Alzheimer’s Disease. Clinical Biochemistry 2019, 72, 3–6. [Google Scholar] [CrossRef]
- Trejo-Lopez, J.A.; Yachnis, A.T.; Prokop, S. Neuropathology of Alzheimer’s Disease. Neurotherapeutics 2022, 19, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.M. Entorhinal Cortex Dysfunction in Alzheimer’s Disease. Trends in Neurosciences 2023, 46, 124–136. [Google Scholar] [CrossRef]
- Tacikowski, P.; Kalender, G.; Ciliberti, D.; Fried, I. Human Hippocampal and Entorhinal Neurons Encode the Temporal Structure of Experience. Nature 2024, 635, 160–167. [Google Scholar] [CrossRef]
- Tanzi, R.E.; Bertram, L. Twenty Years of the Alzheimer’s Disease Amyloid Hypothesis: A Genetic Perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochemical and Biophysical Research Communications 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.-C.; Wisniewski, H.M. Alzheimer Paired Helical Filaments: Immunochemical Identification of Polypeptides. Acta Neuropathol 1984, 62, 259–267. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Capouch, S.D.; Farlow, M.R.; Brosch, J.R. A Review of Dementia with Lewy Bodies’ Impact, Diagnostic Criteria and Treatment. Neurol Ther 2018, 7, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. Journal of Parkinson’s Disease 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy Bodies: An Update and Outlook. Mol Neurodegeneration 2019, 14, 5. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathological Spectrum of Synucleinopathies. Mov Disord. 2003, 18, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Lantos, P.L. Papp–Lantos Inclusions and the Pathogenesis of Multiple System Atrophy: An Update. Acta Neuropathol 2010, 119, 657–667. [Google Scholar] [CrossRef]
- Jellinger, K.A. Heterogeneity of Multiple System Atrophy: An Update. Biomedicines 2022, 10, 599. [Google Scholar] [CrossRef]
- Galvin, J.E.; Chrisphonte, S.; Cohen, I.; Greenfield, K.K.; Kleiman, M.J.; Moore, C.; Riccio, M.L.; Rosenfeld, A.; Shkolnik, N.; Walker, M.; et al. Characterization of Dementia with Lewy Bodies (DLB) and Mild Cognitive Impairment Using the Lewy Body Dementia Module (LBD-MOD). Alzheimer’s & Dementia 2021, 17, 1675–1686. [Google Scholar] [CrossRef]
- Vann Jones, S.A.; O’Brien, J.T. The Prevalence and Incidence of Dementia with Lewy Bodies: A Systematic Review of Population and Clinical Studies. Psychol. Med. 2014, 44, 673–683. [Google Scholar] [CrossRef]
- Wakisaka, Y.; Furuta, A.; Tanizaki, Y.; Kiyohara, Y.; Iida, M.; Iwaki, T. Age-Associated Prevalence and Risk Factors of Lewy Body Pathology in a General Population: The Hisayama Study. Acta Neuropathologica 2003, 106, 374–382. [Google Scholar] [CrossRef]
- Ratnavel, A.; Dino, F.R.; Jiang, C.; Azmy, S.; Wyman-Chick, K.A.; Bayram, E. Risk Factors and Predictors for Lewy Body Dementia: A Systematic Review. npj Dement. 2025, 1, 20. [Google Scholar] [CrossRef]
- Prasad, S.; Katta, M.R.; Abhishek, S.; Sridhar, R.; Valisekka, S.S.; Hameed, M.; Kaur, J.; Walia, N. Recent Advances in Lewy Body Dementia: A Comprehensive Review. Disease-a-Month 2023, 69, 101441. [Google Scholar] [CrossRef] [PubMed]
- Trétiakoff, K.N. Contribution à L’étude De L’anatomie Pathologique Du Locus Niger De Soemmering Avec Quelques Deductions Relatives A La Pathogenie Des Troubles Du Tonus Musculaire Et De La Maladie De Parkinson. Paris: Universtié de Paris. 1919.
- Okazaki, H.; Lipkin, L.E.; Aronson, S.M. Diffuse Intracytoplasmic Ganglionic Inclusions (Lewy Type) Associated with Progressive Dementia and Quadriparesis in Flexion. J Neuropathol Exp Neurol 1961, 20, 237–244. [Google Scholar] [CrossRef]
- Kosaka, K.; Iseki, E. Dementia with Lewy Bodies. Current Opinion in Neurology 1996, 9, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, K. Lewy Bodies in Cerebral Cortex. Report of Three Cases. Acta Neuropathol 1978, 42, 127–134. [Google Scholar] [CrossRef]
- McKeith, I.G.; Galasko, D.; Kosaka, K.; Perry, E.K.; Dickson, D.W.; Hansen, L.A.; Salmon, D.P.; Lowe, J.; Mirra, S.S.; Byrne, E.J.; et al. Consensus Guidelines for the Clinical and Pathologic Diagnosis of Dementia with Lewy Bodies (DLB): Report of the Consortium on DLB International Workshop. Neurology 1996, 47, 1113–1124. [Google Scholar] [CrossRef]
- Mueller, C.; Ballard, C.; Corbett, A.; Aarsland, D. Historical Landmarks in Dementia with Lewy Bodies. The Lancet Neurology 2017, 16, 348. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Coughlan, G.; Laczó, J.; Hort, J.; Minihane, A.-M.; Hornberger, M. Spatial Navigation Deficits — Overlooked Cognitive Marker for Preclinical Alzheimer Disease? Nat Rev Neurol 2018, 14, 496–506. [Google Scholar] [CrossRef]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer Disease. Nat Rev Dis Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS—ADRDA Work Group under the Auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 939–333. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging-Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimer’s & Dementia 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E.; et al. Revised Criteria for Diagnosis and Staging of Alzheimer’s Disease: Alzheimer’s Association Workgroup. Alzheimer’s & Dementia 2024, 20, 5143–5169. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a Biological Definition of Alzheimer’s Disease. Alzheimer’s & Dementia 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Porsteinsson, A.P.; Isaacson, R.S.; Knox, S.; Sabbagh, M.N.; Rubino, I. Diagnosis of Early Alzheimer’s Disease: Clinical Practice in 2021. The Journal of Prevention of Alzheimer’s Disease 2021, 8, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, P.C.; McKeith, I.G. The Clinical Characteristics of Dementia with Lewy Bodies and a Consideration of Prodromal Diagnosis. Alz Res Therapy 2014, 6, 46. [Google Scholar] [CrossRef]
- Yamada, M.; Komatsu, J.; Nakamura, K.; Sakai, K.; Samuraki-Yokohama, M.; Nakajima, K.; Yoshita, M. Diagnostic Criteria for Dementia with Lewy Bodies: Updates and Future Directions. JMD 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Capouch, S.D.; Farlow, M.R.; Brosch, J.R. A Review of Dementia with Lewy Bodies’ Impact, Diagnostic Criteria and Treatment. Neurol Ther 2018, 7, 249–263. [Google Scholar] [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.-P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and Management of Dementia with Lewy Bodies: Fourth Consensus Report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef]
- Bencze, J.; Seo, W.; Hye, A.; Aarsland, D.; Hortobágyi, T. Dementia with Lewy Bodies – a Clinicopathological Update. 2020. [Google Scholar] [CrossRef]
- Beach, T.G.; Serrano, G.E.; Zhang, N.; Driver-Dunckley, E.D.; Sue, L.I.; Shill, H.A.; Mehta, S.H.; Belden, C.; Tremblay, C.; Choudhury, P.; et al. Clinicopathological Heterogeneity of Lewy Body Diseases: The Profound Influence of Comorbid Alzheimer’s Disease 2024.
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and Management of Dementia with Lewy Bodies: Third Report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef]
- Laczó, M.; Svacova, Z.; Lerch, O.; Martinkovic, L.; Krejci, M.; Nedelska, Z.; Horakova, H.; Matoska, V.; Vyhnalek, M.; Hort, J.; et al. Spatial Navigation Deficits in Early Alzheimer’s Disease: The Role of Biomarkers and APOE Genotype. J Neurol 2025, 272, 438. [Google Scholar] [CrossRef]
- Liang, J.; Li, R.; Wong, G.; Huang, X. Lewy Body Dementia: Exploring Biomarkers and Pathogenic Interactions of Amyloid β, Tau, and α-Synuclein. Mol Neurodegeneration 2025, 20, 90. [Google Scholar] [CrossRef]
- Rongve, A.; Soennesyn, H.; Skogseth, R.; Oesterhus, R.; Hortobágyi, T.; Ballard, C.; Auestad, B.H.; Aarsland, D. Cognitive Decline in Dementia with Lewy Bodies: A 5-Year Prospective Cohort Study. BMJ Open 2016, 6, e010357. [Google Scholar] [CrossRef] [PubMed]
- Mirza, S.S.; Saeed, U.; Ramirez, J.; Herrmann, N.; Stuss, D.T.; Black, S.E.; Masellis, M. Effects of White Matter Hyperintensities, Neuropsychiatric Symptoms, and Cognition on Activities of Daily Living: Differences between Alzheimer’s Disease and Dementia with Lewy Bodies. Alz & Dem Diag Ass & Dis Mo 2022, 14, e12306. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol Neurodegeneration 2019, 14, 32. [Google Scholar] [CrossRef]
- Perl, D.P. Neuropathology of Alzheimer’s Disease. Mount Sinai J Medicine 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, B.C.; Bakkour, A.; Salat, D.H.; Feczko, E.; Pacheco, J.; Greve, D.N.; Grodstein, F.; Wright, C.I.; Blacker, D.; Rosas, H.D.; et al. The Cortical Signature of Alzheimer’s Disease: Regionally Specific Cortical Thinning Relates to Symptom Severity in Very Mild to Mild AD Dementia and Is Detectable in Asymptomatic Amyloid-Positive Individuals. Cerebral Cortex 2009, 19, 497–510. [Google Scholar] [CrossRef]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T. Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severiti of Azheimer’s Disease.
- Browne, D.F.; Smirnov, D.S.; Coughlin, D.G.; Peng, I.; Standke, H.G.; Kim, Y.; Pizzo, D.P.; Unapanta, A.; Andreasson, T.; Hiniker, A.; et al. Early Alzheimer’s Disease with Frequent Neuritic Plaques Harbors Neocortical Tau Seeds Distinct from Primary Age-Related Tauopathy. Nat Commun 2025, 16, 1851. [Google Scholar] [CrossRef]
- Wang, J.-Z.; Xia, Y.-Y.; Grundke-Iqbal, I.; Iqbal, K. Abnormal Hyperphosphorylation of Tau: Sites, Regulation, and Molecular Mechanism of Neurofibrillary Degeneration. JAD 2012, 33, S123–S139. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-D.; Lu, J.-Y.; Li, H.-Q.; Yang, Y.-X.; Jiang, J.-H.; Cui, M.; Zuo, C.-T.; Tan, L.; Dong, Q.; Yu, J.-T.; et al. Staging Tau Pathology with Tau PET in Alzheimer’s Disease: A Longitudinal Study. Transl Psychiatry 2021, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of Aβ-Deposition in the Human Brain and Its Relevance for the Development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging–Alzheimer’s Association Guidelines for the Neuropathologic Assessment of Alzheimer’s Disease: A Practical Approach. Acta Neuropathol 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy Bodies: An Update and Outlook. Mol Neurodegeneration 2019, 14, 5. [Google Scholar] [CrossRef]
- Bencze, J.; Szarka, M.; Bencs, V.; Szabó, R.N.; Smajda, M.; Aarsland, D.; Hortobágyi, T. Neuropathological Characterization of Lemur Tyrosine Kinase 2 (LMTK2) in Alzheimer’s Disease and Neocortical Lewy Body Disease. Sci Rep 2019, 9, 17222. [Google Scholar] [CrossRef]
- Nedelska, Z.; Ferman, T.J.; Boeve, B.F.; Przybelski, S.A.; Lesnick, T.G.; Murray, M.E.; Gunter, J.L.; Senjem, M.L.; Vemuri, P.; Smith, G.E.; et al. Pattern of Brain Atrophy Rates in Autopsy-Confirmed Dementia with Lewy Bodies. Neurobiology of Aging 2015, 36, 452–461. [Google Scholar] [CrossRef]
- Harper, L.; Fumagalli, G.G.; Barkhof, F.; Scheltens, P.; O’Brien, J.T.; Bouwman, F.; Burton, E.J.; Rohrer, J.D.; Fox, N.C.; Ridgway, G.R.; et al. MRI Visual Rating Scales in the Diagnosis of Dementia: Evaluation in 184 Post-Mortem Confirmed Cases. Brain 2016, 139, 1211–1225. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Toledo, J.B.; Walker, L.; Gelpi, E.; Gentleman, S.; Halliday, G.; Hortobagyi, T.; Jellinger, K.; Kovacs, G.G.; Lee, E.B.; et al. Neuropathological Consensus Criteria for the Evaluation of Lewy Pathology in Post-Mortem Brains: A Multi-Centre Study. Acta Neuropathol 2021, 141, 159–172. [Google Scholar] [CrossRef]
- Kanazawa, T.; Uchihara, T.; Takahashi, A.; Nakamura, A.; Orimo, S.; Mizusawa, H. Three-Layered Structure Shared Between Lewy Bodies and Lewy Neurites—Three-Dimensional Reconstruction of Triple-Labeled Sections. Brain Pathology 2008, 18, 415–422. [Google Scholar] [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and Molecular Diagnosis of Synucleinopathies. Mol Neurodegeneration 2021, 16, 83. [Google Scholar] [CrossRef]
- Amin, J.; Holmes, C.; Dorey, R.B.; Tommasino, E.; Casal, Y.R.; Williams, D.M.; Dupuy, C.; Nicoll, J.A.R.; Boche, D. Neuroinflammation in Dementia with Lewy Bodies: A Human Post-Mortem Study. Transl Psychiatry 2020, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.J.; Karas, G.; Paling, S.M.; Barber, R.; Williams, E.D.; Ballard, C.G.; McKeith, I.G.; Scheltens, P.; Barkhof, F.; O’Brien, J.T. Patterns of Cerebral Atrophy in Dementia with Lewy Bodies Using Voxel-Based Morphometry. NeuroImage 2002, 17, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.J.; Barber, R.; Mukaetova-Ladinska, E.B.; Robson, J.; Perry, R.H.; Jaros, E.; Kalaria, R.N.; O’Brien, J.T. Medial Temporal Lobe Atrophy on MRI Differentiates Alzheimer’s Disease from Dementia with Lewy Bodies and Vascular Cognitive Impairment: A Prospective Study with Pathological Verification of Diagnosis. Brain 2009, 132, 195–203. [Google Scholar] [CrossRef]
- Coughlin, D.G.; Hurtig, H.I.; Irwin, D.J. Pathological Influences on Clinical Heterogeneity in Lewy Body Diseases. Movement Disorders 2020, 35, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Röhr, S. Making Time for Brain Health: Recognising Temporal Inequity in Dementia Risk Reduction.
- Yegambaram, M.; Manivannan, B.; Beach, T.; Halden, R. Role of Environmental Contaminants in the Etiology of Alzheimer’s Disease: A Review. CAR 2015, 12, 116–146. [Google Scholar] [CrossRef]
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental Pollutants as Risk Factors for Neurodegenerative Disorders: Alzheimer and Parkinson Diseases. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Giridharan, V.V.; Masud, F.; Petronilho, F.; Dal-Pizzol, F.; Barichello, T. Infection-Induced Systemic Inflammation Is a Potential Driver of Alzheimer’s Disease Progression. Front. Aging Neurosci. 2019, 11, 122. [Google Scholar] [CrossRef]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harbor Perspectives in Medicine 2012, 2, a006296–a006296. [Google Scholar] [CrossRef]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.-C. Genetics of Alzheimer’s Disease: Where We Are, and Where We Are Going. Current Opinion in Neurobiology 2020, 61, 40–48. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias. Nat Genet 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Korologou-Linden, R.; Bhatta, L.; Brumpton, B.M.; Howe, L.D.; Millard, L.A.C.; Kolaric, K.; Ben-Shlomo, Y.; Williams, D.M.; Smith, G.D.; Anderson, E.L.; et al. The Causes and Consequences of Alzheimer’s Disease: Phenome-Wide Evidence from Mendelian Randomization. Nat Commun 2022, 13, 4726. [Google Scholar] [CrossRef]
- Mitra, S.; Bp, K.; C R, S.; Saikumar, N.V.; Philip, P.; Narayanan, M. Alzheimer’s Disease Rewires Gene Coexpression Networks Coupling Different Brain Regions. npj Syst Biol Appl 2024, 10, 50. [Google Scholar] [CrossRef]
- Bellenguez, C.; Grenier-Boley, B.; Lambert, J.-C. Genetics of Alzheimer’s Disease: Where We Are, and Where We Are Going. Current Opinion in Neurobiology 2020, 61, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Kamatham, P.T.; Shukla, R.; Khatri, D.K.; Vora, L.K. Pathogenesis, Diagnostics, and Therapeutics for Alzheimer’s Disease: Breaking the Memory Barrier. Ageing Research Reviews 2024, 101, 102481. [Google Scholar] [CrossRef] [PubMed]
- Härtig, W.; Bauer, A.; Brauer, K.; Grosche, J.; Hortobágyi, T.; Penke, B.; Schliebs, R.; Harkany, T. Functional Recovery of Cholinergic Basal Forebrain Neurons under Disease Conditions: Old Problems, New Solutions? Reviews in the Neurosciences 2002, 13. [Google Scholar] [CrossRef]
- Jellinger, K.A. Dementia with Lewy Bodies and Parkinson’s Disease-Dementia: Current Concepts and Controversies. J Neural Transm 2018, 125, 615–650. [Google Scholar] [CrossRef]
- Agin, A.; Blanc, F.; Bousiges, O.; Villette, C.; Philippi, N.; Demuynck, C.; Martin-Hunyadi, C.; Cretin, B.; Lang, S.; Zumsteg, J.; et al. Environmental Exposure to Phthalates and Dementia with Lewy Bodies: Contribution of Metabolomics. J Neurol Neurosurg Psychiatry 2020, 91, 968–974. [Google Scholar] [CrossRef]
- An, D.; Xu, Y. Environmental Risk Factors Provoke New Thinking for Prevention and Treatment of Dementia with Lewy Bodies. Heliyon 2024, 10, e30175. [Google Scholar] [CrossRef]
- Chia, R.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; Ding, J.; et al. Genome Sequencing Analysis Identifies New Loci Associated with Lewy Body Dementia and Provides Insights into Its Genetic Architecture. Nat Genet 2021, 53, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kaivola, K.; Shah, Z.; Chia, R. ; International LBD Genomics Consortium; Black, S. E.; Gan-Or, Z.; Keith, J.; Masellis, M.; Rogaeva, E.; Brice, A.; et al. Genetic Evaluation of Dementia with Lewy Bodies Implicates Distinct Disease Subgroups. Brain 2022, 145, 1757–1762. [Google Scholar] [CrossRef]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of Novel Risk Loci, Causal Insights, and Heritable Risk for Parkinson’s Disease: A Meta-Analysis of Genome-Wide Association Studies. The Lancet Neurology 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Talyansky, S.; Le Guen, Y.; Kasireddy, N.; Belloy, M.E.; Greicius, M.D. APOE-Ε4 and BIN1 Increase Risk of Alzheimer’s Disease Pathology but Not Specifically of Lewy Body Pathology. acta neuropathol commun 2023, 11, 149. [Google Scholar] [CrossRef]
- Guerreiro, R.; Ross, O.A.; Kun-Rodrigues, C.; Hernandez, D.G.; Orme, T.; Eicher, J.D.; Shepherd, C.E.; Parkkinen, L.; Darwent, L.; Heckman, M.G.; et al. Investigating the Genetic Architecture of Dementia with Lewy Bodies: A Two-Stage Genome-Wide Association Study. The Lancet Neurology 2018, 17, 64–74. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol Med 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Mobley, W.C. Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights From Alternative Hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; De Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid Appearance and Local Toxicity of Amyloid-β Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2008, 451, 720–724. [Google Scholar] [CrossRef]
- Suzuki, N.; Cheung, T.T.; Cai, X.-D.; Odaka, A.; Otvos, L.; Eckman, C.; Golde, T.E.; Younkin, S.G. An Increased Percentage of Long Amyloid β Protein Secreted by Familial Amyloid β Protein Precursor (βApp717 ) Mutants. Science 1994, 264, 1336–1340. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; LeVine, H. Alzheimer’s Disease and the Amyloid-β Peptide. JAD 2010, 19, 311–323. [Google Scholar] [CrossRef]
- Chen, X.-Q.; Mobley, W.C. Exploring the Pathogenesis of Alzheimer Disease in Basal Forebrain Cholinergic Neurons: Converging Insights From Alternative Hypotheses. Front. Neurosci. 2019, 13, 446. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The Cholinergic Hypothesis of Alzheimer’s Disease: A Review of Progress. Journal of Neurology, Neurosurgery & Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. The Journal of Prevention of Alzheimer’s Disease 2019, 6, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Lucas, J.J.; Pérez, M.; Hernández, F. Role of Tau Protein in Both Physiological and Pathological Conditions. Physiological Reviews 2004, 84, 361–384. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Grundke-Iqbal, I.; Iqbal, K. Alzheimer’s Disease Hyperphosphorylated Tau Sequesters Normal Tau into Tangles of Filaments and Disassembles Microtubules. Nat Med 1996, 2, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Moloney, C.M.; Lowe, V.J.; Murray, M.E. Visualization of Neurofibrillary Tangle Maturity in Alzheimer’s Disease: A Clinicopathologic Perspective for Biomarker Research. Alzheimer’s & Dementia 2021, 17, 1554–1574. [Google Scholar] [CrossRef]
- Aguzzi, A.; Lakkaraju, A.K.K. Cell Biology of Prions and Prionoids: A Status Report. Trends in Cell Biology 2016, 26, 40–51. [Google Scholar] [CrossRef]
- Vasconcelos, B.; Stancu, I.-C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.-N.; et al. Heterotypic Seeding of Tau Fibrillization by Pre-Aggregated Abeta Provides Potent Seeds for Prion-like Seeding and Propagation of Tau-Pathology in Vivo. Acta Neuropathol 2016, 131, 549–569. [Google Scholar] [CrossRef]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol 2014, 71, 505. [Google Scholar] [CrossRef]
- Vasconcelos, B.; Stancu, I.-C.; Buist, A.; Bird, M.; Wang, P.; Vanoosthuyse, A.; Van Kolen, K.; Verheyen, A.; Kienlen-Campard, P.; Octave, J.-N.; et al. Heterotypic Seeding of Tau Fibrillization by Pre-Aggregated Abeta Provides Potent Seeds for Prion-like Seeding and Propagation of Tau-Pathology in Vivo. Acta Neuropathol 2016, 131, 549–569. [Google Scholar] [CrossRef]
- McAleese, K.E.; Walker, L.; Erskine, D.; Thomas, A.J.; McKeith, I.G.; Attems, J. TDP-43 Pathology in Alzheimer’s Disease, Dementia with Lewy Bodies and Ageing. Brain Pathology 2017, 27, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Soga, T.; Okano, H.J.; Parhar, I. α-Synuclein-Mediated Neurodegeneration in Dementia with Lewy Bodies: The Pathobiology of a Paradox. Cell Biosci 2021, 11, 196. [Google Scholar] [CrossRef]
- Neupane, S.; De Cecco, E.; Aguzzi, A. The Hidden Cell-to-Cell Trail of α-Synuclein Aggregates. Journal of Molecular Biology 2022, 167930. [Google Scholar] [CrossRef]
- Ayers, J.I.; Lee, J.; Monteiro, O.; Woerman, A.L.; Lazar, A.A.; Condello, C.; Paras, N.A.; Prusiner, S.B. Different α-Synuclein Prion Strains Cause Dementia with Lewy Bodies and Multiple System Atrophy. Proc. Natl. Acad. Sci. U.S.A. 2022, 119, e2113489119. [Google Scholar] [CrossRef]
- Van Der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van Den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The Structural Differences between Patient-Derived α-Synuclein Strains Dictate Characteristics of Parkinson’s Disease, Multiple System Atrophy and Dementia with Lewy Bodies. Acta Neuropathol 2020, 139, 977–1000. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Leuner, K.; Schütt, T.; Kurz, C.; Eckert, S.H.; Schiller, C.; Occhipinti, A.; Mai, S.; Jendrach, M.; Eckert, G.P.; Kruse, S.E.; et al. Mitochondrion-Derived Reactive Oxygen Species Lead to Enhanced Amyloid Beta Formation. Antioxidants & Redox Signaling 2012, 16, 1421–1433. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The Alzheimer’s Disease Mitochondrial Cascade Hypothesis: A Current Overview. Journal of Alzheimer’s Disease 2023, 92, 751–768. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. α-Synuclein Promotes Mitochondrial Deficit and Oxidative Stress. The American Journal of Pathology 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Billingsley, K.J.; Barbosa, I.A.; Bandrés-Ciga, S.; Quinn, J.P.; Bubb, V.J.; Deshpande, C.; Botia, J.A.; Reynolds, R.H.; Zhang, D.; Simpson, M.A.; et al. Mitochondria Function Associated Genes Contribute to Parkinson’s Disease Risk and Later Age at Onset. npj Parkinsons Dis. 2019, 5, 8. [Google Scholar] [CrossRef]
- Spano, M. The Possible Involvement of Mitochondrial Dysfunctions in Lewy Body Dementia: A Systematic Review. Functional Neurology 2015. [CrossRef] [PubMed]
- Ye, J.; Dai, X.; Zhang, C.; Duan, Z.; Zhou, G.; Wang, J. Investigating the Causal Relationships between Mitochondrial Proteins and Dementia with Lewy Bodies. Journal of Alzheimer’s Disease 2025, 105, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Millot, P.; Pujol, C.; Paquet, C.; Mouton-Liger, F. Impaired Mitochondrial Dynamics in the Blood of Patients with Alzheimer’s Disease and Lewy Body Dementia. Alzheimer’s & Dementia 2023, 19, e075795. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. The Lancet Neurology 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of Pro-Inflammatory Cytokines Released from Microglia in Alzheimer’s Disease. Ann Transl Med 2015; 3(10): 136 2015, Ann Transl Med 2015. [Google Scholar]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835. [Google Scholar] [CrossRef]
- Heneka, M.T.; Van Der Flier, W.M.; Jessen, F.; Hoozemanns, J.; Thal, D.R.; Boche, D.; Brosseron, F.; Teunissen, C.; Zetterberg, H.; Jacobs, A.H.; et al. Neuroinflammation in Alzheimer Disease. Nat Rev Immunol 2025, 25, 321–352. [Google Scholar] [CrossRef]
- Plantone, D.; Pardini, M.; Righi, D.; Manco, C.; Colombo, B.M.; De Stefano, N. The Role of TNF-α in Alzheimer’s Disease: A Narrative Review. Cells 2023, 13, 54. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Murray, T.E.; Noyovitz, B.; Narayana, K.; Gray, T.E.; Le, J.; He, J.; Simtchouk, S.; Gibon, J.; Alcorn, J.; et al. Cardiolipin Released by Microglia Can Act on Neighboring Glial Cells to Facilitate the Uptake of Amyloid-β (1–42). Molecular and Cellular Neuroscience 2023, 124, 103804. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Attems, J. Prevalence and Impact of Vascular and Alzheimer Pathologies in Lewy Body Disease. Acta Neuropathol 2008, 115, 427–436. [Google Scholar] [CrossRef]
- Jellinger, K.A. Alzheimer Disease and Cerebrovascular Pathology: An Update. Journal of Neural Transmission 2002, 109, 813–836. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.A. Only Cerebral Capillary Amyloid Angiopathy Correlates with Alzheimer Pathology? A Pilot Study. Acta Neuropathologica 2004, 107, 83–90. [Google Scholar] [CrossRef]
- Rajeev, V.; Fann, D.Y.; Dinh, Q.N.; Kim, H.A.; De Silva, T.M.; Lai, M.K.P.; Chen, C.L.-H.; Drummond, G.R.; Sobey, C.G.; Arumugam, T.V. Pathophysiology of Blood Brain Barrier Dysfunction during Chronic Cerebral Hypoperfusion in Vascular Cognitive Impairment. Theranostics 2022, 12, 1639–1658. [Google Scholar] [CrossRef] [PubMed]
- Zenaro, E.; Piacentino, G.; Constantin, G. The Blood-Brain Barrier in Alzheimer’s Disease. Neurobiology of Disease 2017, 107, 41–56. [Google Scholar] [CrossRef]
- Candelario-Jalil, E.; Dijkhuizen, R.M.; Magnus, T. Neuroinflammation, Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke 2022, 53, 1473–1486. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, Y.; Zhao, Y.; Huang, T.Y.; Zheng, Q.; Wang, X. Peripheral and Central Neuroimmune Mechanisms in Alzheimer’s Disease Pathogenesis. Mol Neurodegeneration 2025, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Cechetto, D.F.; Hachinski, V.; Whitehead, S.N. Vascular Risk Factors and Alzheimer’s Disease. Expert Review of Neurotherapeutics 2008, 8, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Surendranathan, A.; Rowe, J.B.; O’Brien, J.T. Neuroinflammation in Lewy Body Dementia. Parkinsonism & Related Disorders 2015, 21, 1398–1406. [Google Scholar] [CrossRef]
- Rajkumar, A.P.; Bidkhori, G.; Shoaie, S.; Clarke, E.; Morrin, H.; Hye, A.; Williams, G.; Ballard, C.; Francis, P.; Aarsland, D. Postmortem Cortical Transcriptomics of Lewy Body Dementia Reveal Mitochondrial Dysfunction and Lack of Neuroinflammation. The American Journal of Geriatric Psychiatry 2020, 28, 75–86. [Google Scholar] [CrossRef]
- Jellinger, K.A. Prevalence of Vascular Lesions in Dementia with Lewy Bodies. A Postmortem Study. J Neural Transm 2003, 110, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.C.B.; Carter, E.K.; Dammer, E.B.; Duong, D.M.; Gerasimov, E.S.; Liu, Y.; Liu, J.; Betarbet, R.; Ping, L.; Yin, L.; et al. Large-Scale Deep Multi-Layer Analysis of Alzheimer’s Disease Brain Reveals Strong Proteomic Disease-Related Changes Not Observed at the RNA Level. Nat Neurosci 2022, 25, 213–225. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Knörnschild, F.; Zhang, E.J.; Ghosh Biswas, R.; Kobus, M.; Chen, J.; Zhou, J.X.; Rao, A.; Sun, J.; Wang, X.; Li, W.; et al. Correlations of Blood and Brain NMR Metabolomics with Alzheimer’s Disease Mouse Models. Transl Psychiatry 2025, 15, 87. [Google Scholar] [CrossRef]
- Oka, T.; Matsuzawa, Y.; Tsuneyoshi, M.; Nakamura, Y.; Aoshima, K.; Tsugawa, H. ; the Alzheimer’s Disease Metabolomics Consortium; Weiner, M. ; Aisen, P.; Petersen, R.; et al. Multiomics Analysis to Explore Blood Metabolite Biomarkers in an Alzheimer’s Disease Neuroimaging Initiative Cohort. Sci Rep 2024, 14, 6797. [Google Scholar] [CrossRef]
- Greally, S.; Kumar, M.; Schlaffner, C.; Van Der Heijden, H.; Lawton, E.S.; Biswas, D.; Berretta, S.; Steen, H.; Steen, J.A. Dementia with Lewy Bodies Patients with High Tau Levels Display Unique Proteome Profiles. Mol Neurodegeneration 2024, 19, 98. [Google Scholar] [CrossRef]
- Goralski, T.M.; Meyerdirk, L.; Breton, L.; Brasseur, L.; Kurgat, K.; DeWeerd, D.; Turner, L.; Becker, K.; Adams, M.; Newhouse, D.J.; et al. Spatial Transcriptomics Reveals Molecular Dysfunction Associated with Cortical Lewy Pathology. Nat Commun 2024, 15, 2642. [Google Scholar] [CrossRef]
- Pan, X.; Donaghy, P.C.; Roberts, G.; Chouliaras, L.; O’Brien, J.T.; Thomas, A.J.; Heslegrave, A.J.; Zetterberg, H.; McGuinness, B.; Passmore, A.P.; et al. Plasma Metabolites Distinguish Dementia with Lewy Bodies from Alzheimer’s Disease: A Cross-Sectional Metabolomic Analysis. Front. Aging Neurosci. 2024, 15, 1326780. [Google Scholar] [CrossRef]
- Canal-Garcia, A.; Branca, R.M.; Francis, P.T.; Ballard, C.; Winblad, B.; Lehtiö, J.; Nilsson, P.; Aarsland, D.; Pereira, J.B.; Bereczki, E. Proteomic Signatures of Alzheimer’s Disease and Lewy Body Dementias: A Comparative Analysis. Alzheimer’s & Dementia 2025, 21, e14375. [Google Scholar] [CrossRef]
- Shantaraman, A.; Dammer, E.B.; Ugochukwu, O.; Duong, D.M.; Yin, L.; Carter, E.K.; Gearing, M.; Chen-Plotkin, A.; Lee, E.B.; Trojanowski, J.Q.; et al. Network Proteomics of the Lewy Body Dementia Brain Reveals Presynaptic Signatures Distinct from Alzheimer’s Disease. Mol Neurodegeneration 2024, 19, 60. [Google Scholar] [CrossRef]
- Habbal, S.; Mian, M.; Imam, M.; Tahiri, J.; Amor, A.; Reddy, P.H. Harnessing Artificial Intelligence for Transforming Dementia Care: Innovations in Early Detection and Treatment. Brain Organoid and Systems Neuroscience Journal 2025, 3, 122–133. [Google Scholar] [CrossRef]
- Goddard, T.R.; Brookes, K.J.; Sharma, R.; Moemeni, A.; Rajkumar, A.P. Dementia with Lewy Bodies: Genomics, Transcriptomics, and Its Future with Data Science. Cells 2024, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Lian, P.; Guo, Y.; Yu, J. Biomarkers in Alzheimer’s Disease: Emerging Trends and Clinical Implications. Chinese Medical Journal 2025, 138, 1009–1012. [Google Scholar] [CrossRef]
- Jellinger, K.A. Criteria for the Neuropathological Diagnosis of Dementing Disorders: Routes out of the Swamp? Acta Neuropathol 2009, 117, 101–110. [Google Scholar] [CrossRef]
- McGettigan, S.; Nolan, Y.; Ghosh, S.; O’Mahony, D. The Emerging Role of Blood Biomarkers in Diagnosis and Treatment of Alzheimer’s Disease. Eur Geriatr Med 2023, 14, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Grande, G.; Valletta, M.; Rizzuto, D.; Xia, X.; Qiu, C.; Orsini, N.; Dale, M.; Andersson, S.; Fredolini, C.; Winblad, B.; et al. Blood-Based Biomarkers of Alzheimer’s Disease and Incident Dementia in the Community. Nat Med 2025, 31, 2027–2035. [Google Scholar] [CrossRef] [PubMed]
- Afrin, M.R.; Upadhyaya, P.G.; Hashim, A.; Bhattacharya, K.; Chanu, N.R.; Das, D.; Khanal, P.; Deka, S. Advanced Biomarkers: Beyond Amyloid and Tau: Emerging Non-Traditional Biomarkers for Alzheimer`s Diagnosis and Progression. Ageing Research Reviews 2025, 108, 102736. [Google Scholar] [CrossRef]
- Coutrot, A.; Schmidt, S.; Coutrot, L.; Pittman, J.; Hong, L.; Wiener, J.M.; Hölscher, C.; Dalton, R.C.; Hornberger, M.; Spiers, H.J. Virtual Navigation Tested on a Mobile App Is Predictive of Real-World Wayfinding Navigation Performance. PLoS ONE 2019, 14, e0213272. [Google Scholar] [CrossRef]
- Hansen, N.; Bouter, C.; Müller, S.J.; Van Riesen, C.; Khadhraoui, E.; Ernst, M.; Riedel, C.H.; Wiltfang, J.; Lange, C. New Insights into Potential Biomarkers in Patients with Mild Cognitive Impairment Occurring in the Prodromal Stage of Dementia with Lewy Bodies. Brain Sciences 2023, 13, 242. [Google Scholar] [CrossRef]
- Holden, S.K. Updates in Fluid, Tissue, and Imaging Biomarkers for Dementia with Lewy Bodies and Implications for Biologically Based Disease Definitions. Curr Treat Options Neurol 2024, 26, 189–201. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Surmeier, D.J.; Simuni, T.; Merchant, K.; Caughey, B.; Soto, C.; Fares, M.-B.; Heym, R.G.; Melki, R. Alpha-Synuclein Seed Amplification Assays: Data Sharing, Standardization Needed for Clinical Use. Science AdvAnceS 2025. [CrossRef]
- Bsoul, R.; McWilliam, O.H.; Waldemar, G.; Hasselbalch, S.G.; Simonsen, A.H.; Von Buchwald, C.; Bech, M.; Pinborg, C.H.; Pedersen, C.K.; Baungaard, S.O.; et al. Accurate Detection of Pathologic α-Synuclein in CSF, Skin, Olfactory Mucosa, and Urine with a Uniform Seeding Amplification Assay. acta neuropathol commun 2025, 13, 113. [Google Scholar] [CrossRef]
- Sjaelland, N.S.; Gramkow, M.H.; Hasselbalch, S.G.; Frederiksen, K.S. Digital Biomarkers for the Assessment of Non-Cognitive Symptoms in Patients with Dementia with Lewy Bodies: A Systematic Review. JAD 2024, 100, 431–451. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, M.; Vermunt, L.; Peeters, C.F.W.; Sieben, A.; Hok-A-Hin, Y.S.; Lleó, A.; Alcolea, D.; Van Nee, M.; Engelborghs, S.; Van Alphen, J.L.; et al. CSF Proteome Profiling Reveals Biomarkers to Discriminate Dementia with Lewy Bodies from Alzheimer´s Disease. Nat Commun 2023, 14, 5635. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Comorbid Pathologies and Their Impact on Dementia with Lewy Bodies—Current View. Int. J. Mol. Sci. 2025, 26, 7674. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Neupane, S.; Nikolić, L.; Maraio, L.; Goiran, T.; Karpilovsky, N.; Sellitto, S.; Bouris, V.; Yin, J.-A.; Melki, R.; Fon, E.A.; et al. Large-Scale Bidirectional Arrayed Genetic Screens Identify OXR1 and EMC4 as Modifiers of α-Synuclein Aggregation. bioRxiv 2025. [CrossRef]
- Neupane, S.; Khadka, J.; Rayamajhi, S.; Pandey, A.S. Binding Modes of Potential Anti-Prion Phytochemicals to PrPC Structures in Silico. Journal of Ayurveda and Integrative Medicine 2023, 14, 100750. [Google Scholar] [CrossRef] [PubMed]
- Pacesa, M.; Nickel, L.; Schellhaas, C.; Schmidt, J.; Pyatova, E.; Kissling, L.; Barendse, P.; Choudhury, J.; Kapoor, S.; Alcaraz-Serna, A.; et al. One-Shot Design of Functional Protein Binders with BindCraft. Nature 2025, 646, 483–492. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison of AD and DLB shared and discriminating features. Left hemisphere (AD-predominant): extracellular Aβ plaques and intraneuronal tau tangles accompanied by medial-temporal and cortical atrophy and ventriculomegaly. Right hemisphere (DLB-predominant): α-synuclein Lewy bodies and neurites following a brainstem (substantia nigra, locus coeruleus). The middle circular band summarizes shared features: mitochondrial dysfunction/oxidative stress, neuroinflammation, cerebrovascular/BBB, and synaptic/proteostasis–lysosome–autophagy stress. The bottom boxes summarize the core clinical profiles and indicative biomarkers for AD and DLB. AD, Alzheimer’s disease; DLB, dementia with Lewy bodies; Aβ, amyloid-β; α-syn, α-synuclein; DAT, dopamine transporter; FDG, fluorodeoxyglucose; GFAP, glial fibrillary acidic protein; LBV, Lewy body variant of AD; NfL, neurofilament light; RT-QuIC, real-time quaking-induced conversion; PMCA, protein misfolding cyclic amplification; DAT-SPECT, dopamine transporter single-photon emission computed tomography.
Figure 1.
Comparison of AD and DLB shared and discriminating features. Left hemisphere (AD-predominant): extracellular Aβ plaques and intraneuronal tau tangles accompanied by medial-temporal and cortical atrophy and ventriculomegaly. Right hemisphere (DLB-predominant): α-synuclein Lewy bodies and neurites following a brainstem (substantia nigra, locus coeruleus). The middle circular band summarizes shared features: mitochondrial dysfunction/oxidative stress, neuroinflammation, cerebrovascular/BBB, and synaptic/proteostasis–lysosome–autophagy stress. The bottom boxes summarize the core clinical profiles and indicative biomarkers for AD and DLB. AD, Alzheimer’s disease; DLB, dementia with Lewy bodies; Aβ, amyloid-β; α-syn, α-synuclein; DAT, dopamine transporter; FDG, fluorodeoxyglucose; GFAP, glial fibrillary acidic protein; LBV, Lewy body variant of AD; NfL, neurofilament light; RT-QuIC, real-time quaking-induced conversion; PMCA, protein misfolding cyclic amplification; DAT-SPECT, dopamine transporter single-photon emission computed tomography.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



