
Submitted:
30 October 2025
Posted:
31 October 2025
You are already at the latest version
Abstract
Dysregulation of CK1 isoforms is linked to various types of diseases, including neuro-degeneration and different types of neoplasia such as colon-, pancreatic-, breast-, and ovarian cancer. For CK1 isoforms, a plethora of effective small molecule inhibitors are available. However, only a few degraders of CK1α and, more recently, PROteolysis TArgeting Chimeras (PROTACs) for CK1δ/CK1ε have been reported. In this study, we applied the PROTAC concept by harnessing molecular modeling to design and synthe-size a series of candidate CK1δ-targeting PROTACs based on a highly specific and po-tent benzothiazole-based CK1δ-inhibitor that we previously developed in our lab. In the present study, we established a modular synthetic platform to systematically gen-erate a set of PROTAC degrader candidates consisting of the CK1δ-specific inhibitor scaffold, alkyl- and PEG-linker motifs with various lengths, and Cereblon (CRBN)-engaging pomalidomide and thalidomide derivatives as E3 ligase binders. We demonstrate that several PROTACs degrade CK1δ/ε in various cells. The most potent PROTAC P1d inhibits the phosphorylation of downstream substrates through CK1δ/ε degradation. We establish the requirement of CUL4ACRBN and the proteasome for P1d-mediated degradation of CK1δ/ε.
Keywords:
PROTACs
; protein degradation
; CK1δ
; kinases
; cancer therapy
1. Introduction
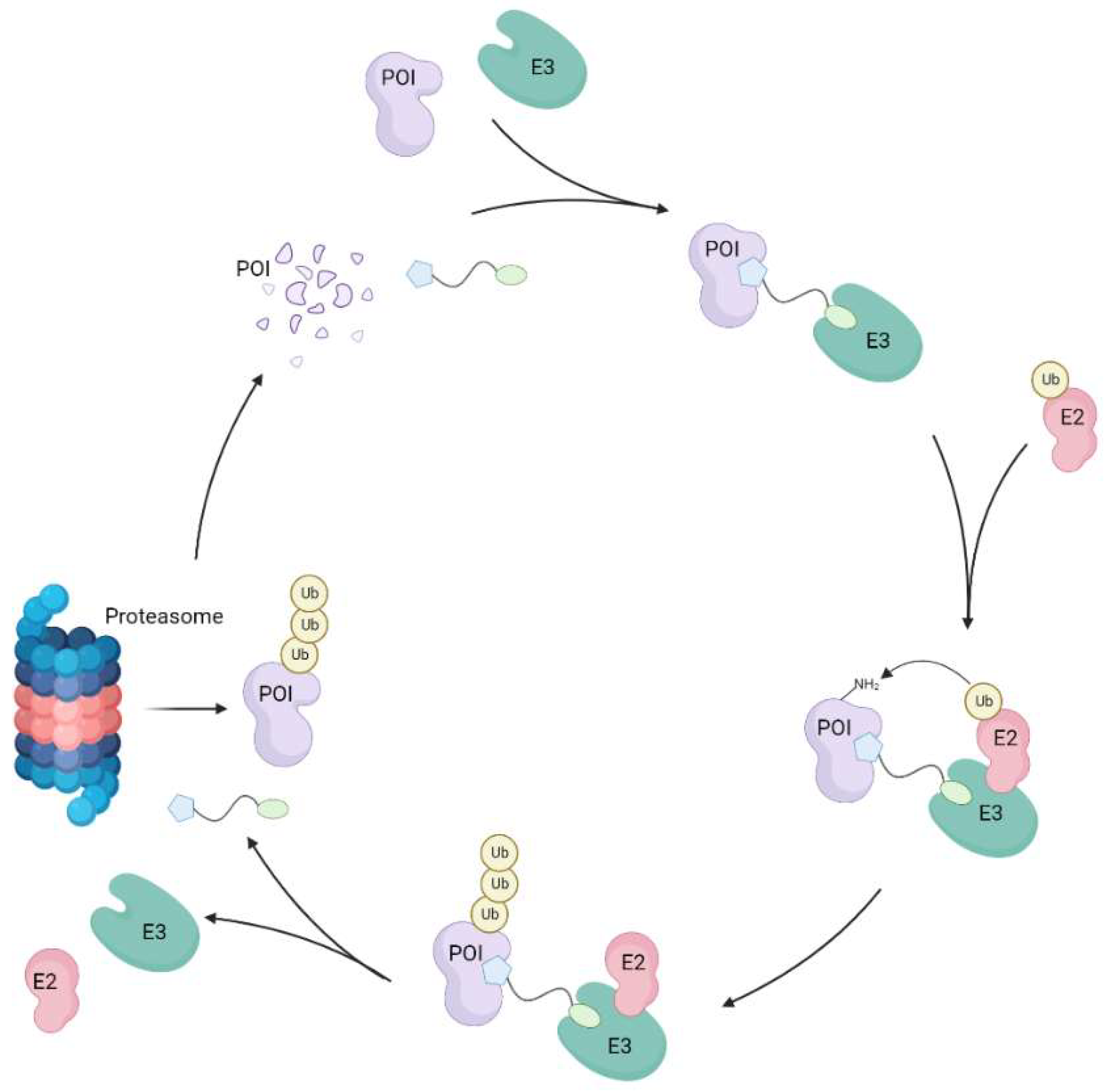
The concept of proteolysis targeting chimeras (PROTACs) represents contemporary and innovative therapeutic modalities, first established by Crews and colleagues in 2001 [1]. PROTACs’ catalytic mode of action (MOA) potentially offers several advantages over small molecule inhibitors (SMIs), such as the accessibility of previously undruggable targets, enhanced protein isoform selectivity, or reduced dosing frequency [2]. PROTACs induce the degradation of a protein of interest (POI) by hijacking the intrinsic cellular ubiquitin-proteasome system (UPS), thereby triggering a conditional POI-knockdown phenotype [1,3]. The UPS itself is an essential pathway in cells that physiologically regulates the life cycle of proteins. Hence, while regulating the degradation of damaged or misfolded proteins in general, it plays major roles, e.g., in the regulation of the cell cycle, as well as inflammatory and immune responses [4,5,6]. Dysregulation of the UPS is often associated with diseases, including different forms of cancer as well as neurodegenerative diseases such as Parkinson’s, Alzheimer’s, or Huntington’s disease [7,8,9]. Compared to SMIs, the alternative MOA of PROTACs correlates to their nature as heterobifunctional molecules. Typical PROTAC compounds, e.g., consist of both an E3 ligase ligand and a POI ligand, which are connected via different types of linkers [10]. These chimeric molecules are therefore able to simultaneously recruit the respective E3 ligase and the POI, forming ternary complexes. Consecutively, due to the induced spatial proximity, the E3 ligase is able to bind an E2 ligase that transfers ubiquitin moieties to lysine residues on the POI surface, thereby labelling that protein for cellular waste disposal [11,12]. Consequently, lysine polyubiquitination is then recognized by the 26S proteasome, and the targeted POI is degraded, while the PROTAC itself is released and able to form another ternary complex (Figure 1) [13,14]. In this regard, PROTACs are able to go through multiple cycles of complexing with POI and E3 ligase, facilitating catalytic degradation of the POI in sub-stoichiometric amounts [15].
When designing and developing novel, effective PROTAC compounds, picking the right linker motif plays a major role (“linkerology”). Since both the POI- and E3 ligase-addressing ligands are mostly set in their physicochemical properties, variation in the used linker is often key to influencing PROTAC features, such as biological activity, physicochemical parameters coding for solubility, cellular permeability, and metabolic stability [16,17,18]. Of note, empirically finding the right linker length is often key, and the absence or presence of a single atom within the linker moiety can differentiate whether a PROTAC is bioactive or not. In line with this notion, linkers too short may not allow each ligand to interact with their respective protein due to steric hindrance, which makes it impossible to form the required ternary complexes. As a result, respective compounds do not lead to polyubiquitination and subsequent degradation. If the linker is too long, ternary complexes may be formed, although POI-degradation may still not occur due to the formation of “non-ideal” proximity between POI and E3 ligase being insufficient to result in ubiquitination [14,19].
1.1. CK1 isoforms as proteins of interest for PROTAC design
The highly conserved and ubiquitously expressed CK1 family of serine/threonine kinases consists of 7 members (α, β, γ1, γ2, γ3, δ, and ε). CK1 isoforms and various splice variants in humans are known to be linked to several regulatory roles such as DNA processing and repair, proliferation, apoptosis, cell differentiation, and circadian rhythm [20,21,22,23,24]. One of the most researched and best-characterised CK1-regulated mechanisms to date is the canonical Wnt/β-catenin pathway, which mainly controls cell proliferation. If Wnt receptors are stimulated through binding of respective Wnt ligands, β-catenin is translocated to the nucleus, which ultimately leads to transcription and expression of Wnt-dependent target genes. However, if Wnt ligands are absent, a “destruction complex” consisting of adenomatous polyposis coli (APC), AXIN, CK1α, and glycogen synthase kinase 3 (GSK3) is formed in the cytoplasm to capture β-catenin and induce its degradation through phosphorylation of CK1α and GSK3 [23,25,26]. Additionally, CK1δ/ε control Wnt signalling via phosphorylation of Dishevelled (DVL1/2/3) proteins [27]. Dysregulation of the CK1δ isoform is known to be linked to various forms of diseases, e.g., cancer (i.e., colon, pancreatic, breast, and ovarian cancer), as well as neurodegenerative and metabolic diseases, rendering this isoform a highly attractive target for medicinal chemistry-based approaches [28,29,30].
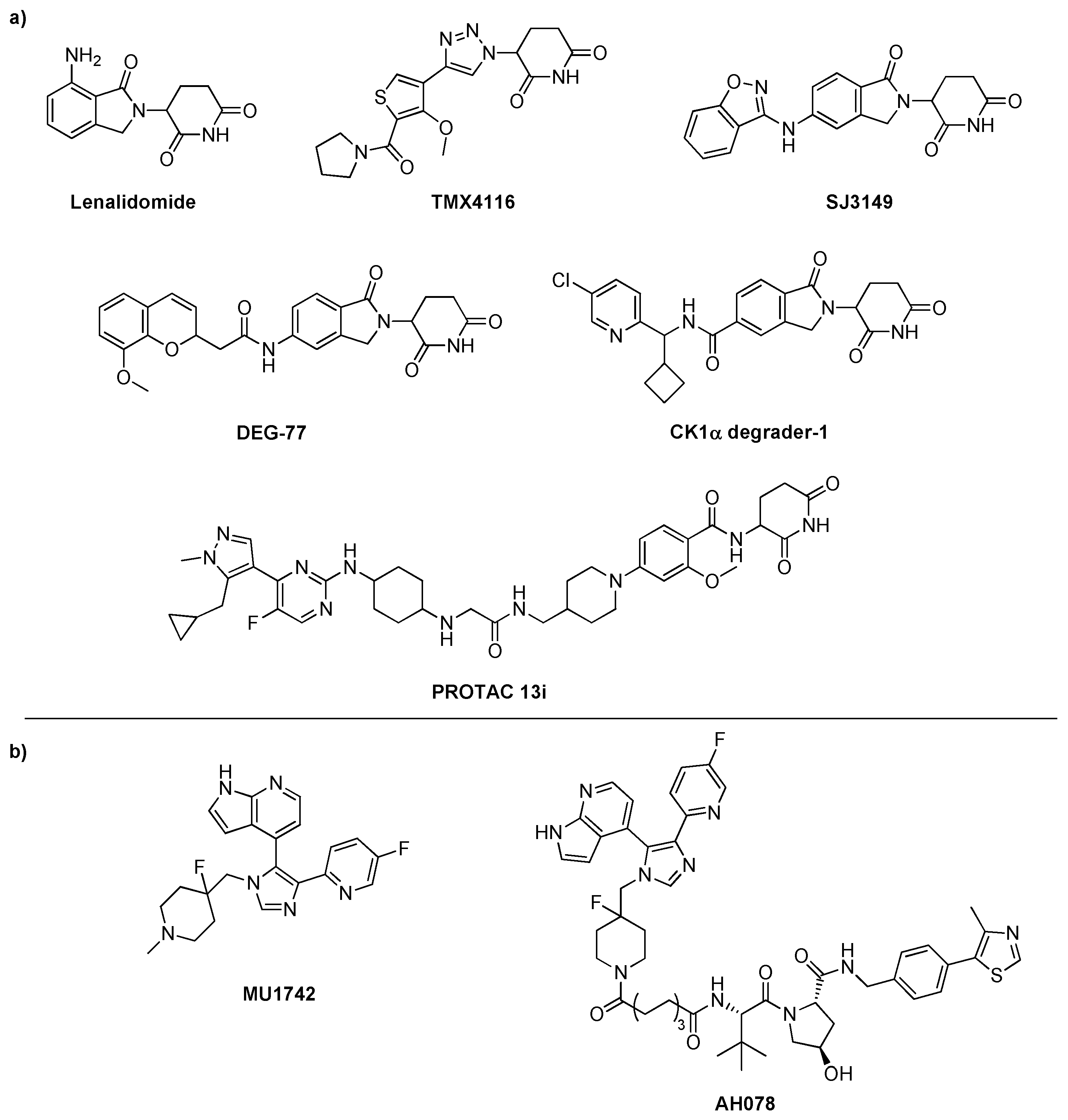
When it comes to CK1 targeted protein degradation (TPD), so far, compounds targeting CK1α have been reported, including CK1 molecular glue degraders like lenalidomide, TMX4116, SJ7095/SJ3149, DEG-77 [31], CK1α degrader-1 [32], and PROTAC 13i [33] (Scheme 1). While our work was in progress, in December 2024, Haag et al. reported their degrader compounds targeting CK1δ and CK1ε. They demonstrated that although compound AH078 showed a reported DC50 value of 0.55 μM and Dmax of 70 % towards CK1δ, all compounds lacked isoform selectivity regarding CK1δ/ε, likely due to the high homology between their respective kinase ATP-binding domains [23,28,34].
Motivated especially by the lack of CK1δ-specific degraders at the inception of our project, we started a synthetic project towards the development of a platform containing various types of linker motifs of different lengths and variable types of E3 ligase ligands. In contrast to the “tear drop” type of CK1δ inhibitor used by Haag et al [34] in their recently reported CK1 PROTACs, we set out to employ a “linear” CK1δ inhibitor moiety in our project, since former efforts towards “linear” CK1δ/ε-specific inhibitors in our group led to the discovery of SMI 1 (Scheme 2) [35,36]. The CK1δ-specific inhibitor is based on the scaffold of established IWP compounds (i.e., IWP-2) [37,38] and known CK1-specific benzimidazole-based inhibitors such as Bischof-5 [39]. Such degrader probes would be highly useful to further study CK1 isoform-dependent processes, e.g., in cancer cells. Based on strong SAR data and having structural data of a ligand-protein complex for SMI 1 in hand, we first aimed to identify a suitable linker-attachment position at the CK1δ-specific inhibitor scaffold.
2. Results and Discussion
2.1. CK1δ PROTAC design based on SMI 1
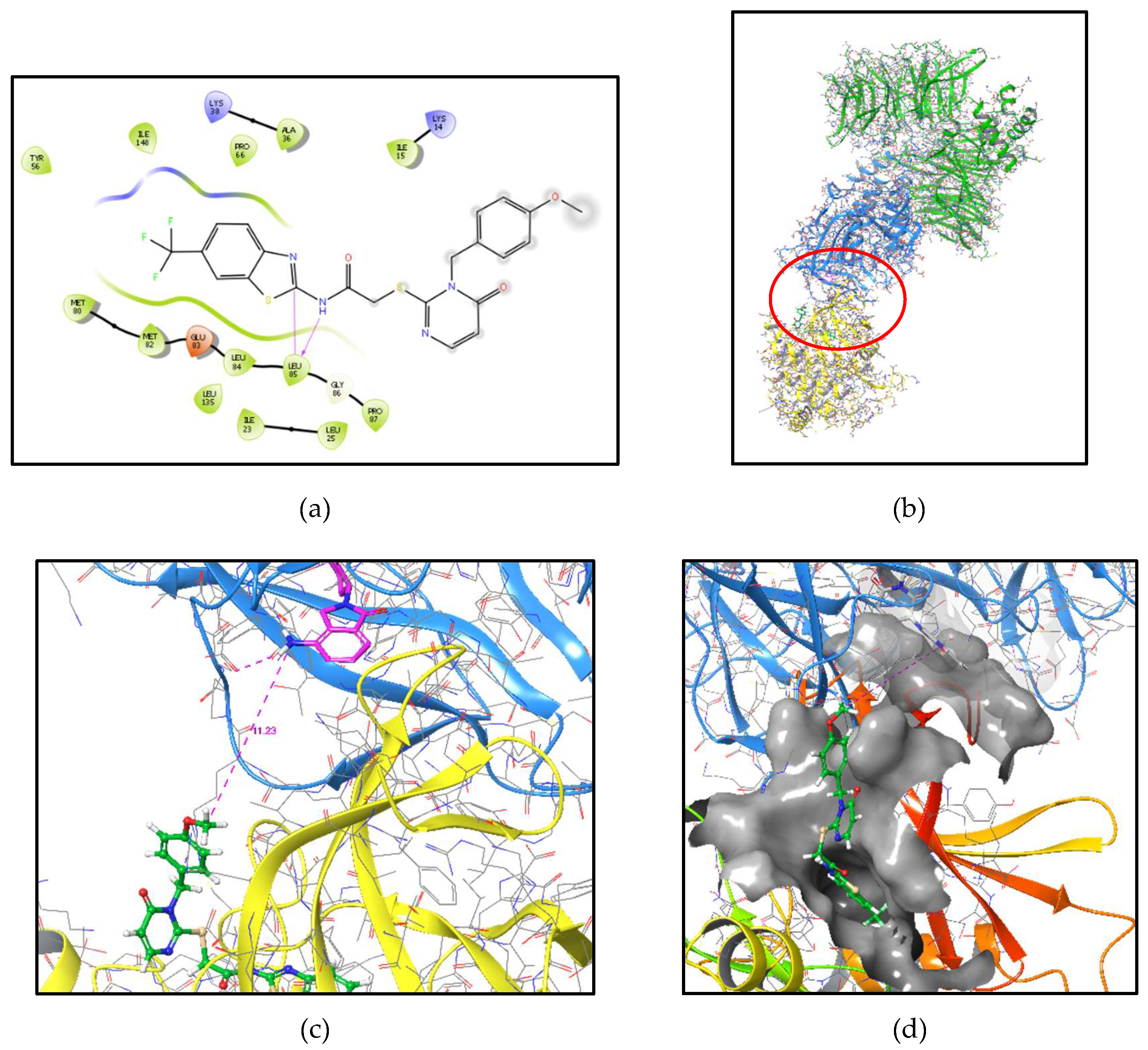
Having established SMI 1 as a highly specific and potent inhibitor towards CK1δ, we set out to integrate this scaffold into the development of the desired CK1δ-targeting PROTACs. SAR data and initial molecular modeling using our ligand-CK1δ structure complex (pdb 5OKT) [35] suggested the p-MeO benzyl position of SMI 1 as a suitable position for linker attachment. This moiety occupies the solvent-open hydrophobic region II (HRII) of the CK1-ATP binding pocket (Figure 2a), thus offering a strong rationale for effective CK1δ PROTAC design. Besides defining the p-benzyl moiety of the POI ligand as the desired function for linker-attachment, we chose well-known thalidomide derivatives to address Cereblon (CRBN) as the employed E3-ligase for the first set of potential CK1δ-PROTACs. Having defined both POI and E3 ligands, we next focused on the linker moiety itself. As previously described, the design of the linker is often crucial to the efficacy of PROTACs, due to the linker’s strong influence on both physicochemical properties as well as PROTAC functionality itself (i.e., forming of a stable ternary complex, etc.). In order to rationalize a chemical space for eligible linker moieties, we performed molecular modeling using a CK1α–CRL4(CRBN) - lenalidomide complex structure (pdb 5FQD) [40] as a model system in which we replaced the CK1α part by our CK1δ structure with SMI 1 docked into the ATP binding pocket (Figure 2b-d). Herein, we determined an approximately 11 Å distance between the p-MeO-benzyl position of SMI 1 and the nitrogen atom of lenalidomide in its CRBN-binding pose. Noteworthy, being aware of lenalidomide’s role as a molecular glue in the original ligand-protein structure, we considered the modelled linker distance in our CK1δ-PROTAC approach only as a rough estimation. Therefore, we did not focus on the modelled 11 Å distance but instead designed a variable set of linker moieties to cover a broad range for the aimed candidate compounds.
Motivated by the modeling hypothesis based on the ligand-protein-structure (pdb 5OKT), in our first attempts, we aimed toward the designed CK1δ-PROTACs on the p-MeO benzyl moiety of SMI 1 to attach the linker also via ether moieties. However, our various attempts to cleave the p-MeO position of SMI 1 did not result in sufficient yields of the corresponding phenol compound that would be needed. Therefore, we started implementing the respective aniline derivative I9 to attach the linker as amides (see Scheme 4). Noteworthy, SAR of the former project towards SMI 1 demonstrated variable substitution of the p-position of the benzyl moiety to impact CK1δ/ε-isoform selectivity. Accordingly, attaching different linker moieties to this position via amide instead of ether is anticipated to potentially influence the isoform selectivity of the corresponding PROTACs and is considered in due course of this project.
2.2. Synthesis of the key aniline CK1δ POI ligand I9
In order to generate the favoured aniline intermediate (I9), synthesis of the corresponding p-nitro group compound (I8) was performed according to the general convergent method for synthesizing SMI 1. Subsequent reduction of the said p-nitro group delivered targeted I9 (Scheme 3) [36,41,42,43]. Briefly, for the general synthesis strategy, 2-chloracetylchloride was reacted with the benzothiazole building block I2 to yield amide I3. On the other hand, precursors I4 and I5 were implemented under basic conditions with nano iron in order to provide selective NH-substitution of tautomeric I4 to generate intermediate I6. Next, the thioxo group was introduced using phenyl chlorothionoformate to afford intermediate I7. By coupling benzothiazole I3 with the substituted tetrahydrothieno-pyrimidinone I7, we were able to generate intermediate I8. Finally, the nitro group in the p-position was catalytically reduced with hydrogen to the amine function in order to afford the desired key intermediate I9. It is to be noted that the hydrogen pressure had to be increased to 3 bar to yield the intermediate I9 in sufficient yields.
2.3. Synthesis of candidate CK1δ PROTACs containing alkyl linker motifs
As previously described, most PROTACs published so far contain either PEG or alkyl linker motifs [17]. Due to their chemical accessibility and extensive literature on attaching these linkers to thalidomide derivatives, we decided on alkyl linker motifs in our first set of PROTAC compounds. The synthesis of the intermediate compounds L2a-d through L4a-d was performed based on a method reported by Schmuck et al. (Scheme 4) by N-Cbz-protection of the corresponding amino acid under basic conditions using benzyl chloroformate in the first step and generating the respective tert-butyl ester compound afterward by using tert-butyl alcohol, pyridine, and phosphoryl chloride. In the final step of the alkyl linker preparation, the Cbz protecting group is cleaved by stirring intermediates L3a-d and 10 % (m/m) Pd/C under a hydrogen atmosphere to yield intermediate compounds L4a-d [44]. Initially, the syntheses resulted in unsatisfactory yields, which we were able to increase by extending the reaction time by letting each step stir overnight. After successfully preparing the linker motifs, the compounds were coupled according to Kim et al. (Scheme 4) with the regioisomeric thalidomide derivatives 2-(2,6-dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione or 2-(2,6-dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione, respectively, under basic conditions to generate the desired intermediates L6a-d and L8a-d [45].
Having the variable linker-thalidomide compound series L6a-d and L8a-d in hand, we aimed to finally generate the first two sets of PROTAC compounds by coupling the respective intermediates I9 and L6a-d or L8a-d to the inhibitor scaffold via an amide function. Therefore, the tert-butyl ester of the linker motifs needed to be cleaved first to afford the corresponding carboxylic acid compounds. Hence, in a one-pot approach, the intermediate compounds were dissolved in CH2Cl2, and TFA was added [45]. The solvent was evaporated, and the residue was dried under high vacuum to afford the carboxylic acid compounds in quantitative yields. The first attempts of amide coupling itself were performed according to Ghosh et al. (Scheme 4) using 1.5 equiv. HATU and 5.0 equiv. DIPEA, but only with insufficient yields. We therefore decided to instead perform the amide coupling by employing EDC hydrochloride, HOBt, DMAP, and DIPEA, which led to significantly higher, though still low yields in the range of 6 – 24 %. However, we considered these yields to be sufficient in the context of initial test compounds [46].
Even though the employed amide coupling method using EDC, HOBT, and DMAP was successful for the synthesis of the initial degrader compounds P1a-d (linker lengths ranging from three through six methylene groups), we were, however, unable to generate the PROTAC compounds P2b-c (four and five methylene groups, respectively) by this method. To follow up on these molecules, we first verified the functionality of compounds L8b-c to actually undergo the desired amide coupling by utilizing p-toluidine as a dummy reactant for compound I9. Interestingly, both syntheses were performed successfully with quite good yields in the range of 85 %. Next, various conditions were tested in order to yield compounds P2b-c (Table 1), but all with no success. Since the dummy reaction yields were excellent, we hypothesized that the actual inhibitor scaffold somehow prevents the coupling, especially with these linker moieties (four and five methylene groups), probably by forming complexes, thus hindering the actual amide coupling reaction. In line with this notion, molecular modelling suggested interactions between the hinge binder of I9 and the imide structure of the employed thalidomide/pomalidomide (Figure 3), potentially complicating the formation of the desired amide bond.
Therefore, we performed the coupling in various solvents and conditions, and also added glutarimide in an attempt to saturate the hinge binder of I9 so that the amide coupling moieties may be accessible to each other. However, in our hands, all these attempts did not lead to the successful formation of the desired degrader compounds. Hence, the exact molecular background of this method delivering various lengths of alkyl-linker compounds, with the exception of compounds P2b-c, remains unclear. We next decided to focus on generating a set of CK1δ-PROTACs containing PEG linker motifs, due to their overall likely higher solubility and cell permeability. Furthermore, we favoured these especially regarding PROTAC compounds containing VHL ligands [47], which we intend to integrate into our synthetic platform in the future.
2.4. Synthesis of candidate CK1δ PROTACs containing PEG linker motifs
We sought to couple our POI ligand I9 and the thalidomide derivatives via different types of PEG moieties. First, the corresponding diol precursors were coupled with tert-butyl acrylate according to the method by Cromm et al. (Scheme 5) in the sense of a Michael-Addition under basic conditions [48]. Noteworthy, the diol precursors were used in a fivefold excess to avoid Michael-Addition to occur on both primary alcohol functions. According to the reported method, in the first attempts of attaching the PEG linker motifs to the thalidomide derivative, the remaining primary alcohol function was iodinated using iodine, triphenylphosphine, and imidazole, and then coupled with 2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione under basic SN-conditions [48]. The latter step, however, resulted only in insufficient yields of approximately 2 – 10 % for the aimed intermediates L10a-d, respectively. The main reason for this was the significant formation of a side product showing dual linker substitution of L12 at both the acidic phenol-OH as well as the imide nitrogen-NH of the thalidomide part (not shown). Therefore, we opted to first protect the primary alcohol functions of intermediates L10a-d with a tosyl moiety as a leaving group in order to avoid substitution at the imide nitrogen based on the sterically demanding tosyl moiety (Scheme 5). This strategy proved to be successful, and by coupling these tosyl-protected intermediates with 2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione, we were able to increase the yield of the corresponding intermediates L13a-d to up to 80 %, while simultaneously keeping dual substitution to a minimum (Scheme 5). Finally, the syntheses of the newly designed CK1δ-PROTACs containing PEG linker motifs were performed according to the method used for producing the first two sets, P1a-d, P2a, and P2d (Scheme 6). As formerly observed in these final steps for the alkyl series, the coupling reactions for the PEG series yielding P3a-d and P4a-d also resulted in low but still sufficient yields for the biological testing of the PROTACs.
2.5. Biological Results
2.5.1. Differential degradation of CK1δ/ε by different PROTACs
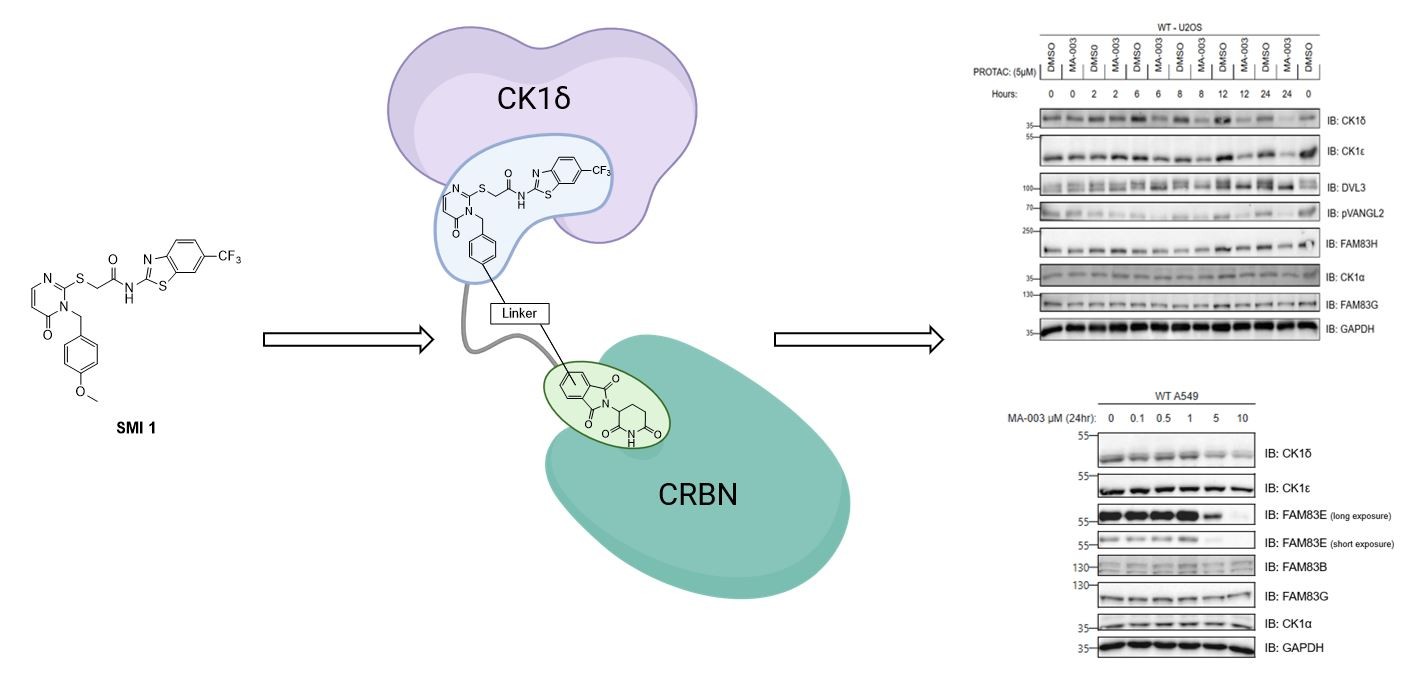
We tested the first series of PROTACs that we synthesized for their ability to degrade CK1α, CK1δ, and CK1ε in wild-type (WT) U2OS osteosarcoma cells at 0.5 and 5 µM for 24 h prior to lysis. Compared to DMSO, I9 and SMI 1-treated controls, P1d caused a reduction in levels of both CK1δ and CK1ε at 5 µM, but no change in levels of CK1α (Figure 4), demonstrating isoform selectivity. P2d caused a reduction in levels of CK1α, CK1δ, and CK1ε at 5 µM (Figure 4). In contrast, P1b and P1c did not cause any changes in levels of CK1α, CK1δ, and CK1ε at any concentrations (Figure 4). The specificity of antibodies recognizing CK1α, CK1δ, and CK1ε was confirmed by immunoblotting extracts from the respective knockout DLD-1 cells (Figure 4). The levels of GAPDH did not change with any compound treatment (Figure 4).
Encouraged by P1d causing reduced levels of CK1δ and CK1ε, once we completed the synthesis of the remaining molecules, we sought to test the remaining series of PROTACs side-by-side along with P1d in WT U2OS cells at 1 µM and 5 µM concentrations for 24 h, along with the inhibitor warhead I9 as a control, for their ability to degrade CK1δ and CK1ε. Treatment of U2OS cells with 1 µM I9 slightly increased the levels of CK1δ but did not substantially alter levels of CK1ε compared to DMSO treatment (Figure 5). P1d, P3d, and P4d led to a dose-dependent reduction in levels of CK1δ and CK1ε at 1 µM and 5 µM compared to DMSO-treated controls (Figure 5). P3c and P4b both displayed a slight dose-dependent reduction in levels of CK1δ/ε, while P3a, P3b, P1a, P4a, and P4c showed no robust dose-dependent decrease in levels of CK1δ/ε (Figure 5).
DVL3 and Vangl2 are known CK1δ/ε substrates [27,49]. Therefore, inhibition or degradation of CK1δ/ε would be expected to block the phosphorylation of these proteins. DVL3 is heavily phosphorylated by CK1δ/ε, causing an upward electrophoretic mobility shift of DVL3. No robust DVL3 mobility shift was observed with 1 µM I9 or any of the PROTACs, while a minor downward mobility shift was observed with most PROTACs at 5 µM (Figure 5), potentially suggesting inhibition of CK1δ/ε at 5 µM. A lack of robust electrophoretic mobility shift of DVL3 evident here was possibly due to the use of 4-20% polyacrylamide Bis-Tris precast gels (Invitrogen), and we have subsequently improved this for further experiments by using 8% polyacrylamide gels. Vangl2 is phosphorylated on Ser84 by CK1δ/ε [50]. Treatment of cells with 1 µM I9 caused a reduction in pVangl2 signal compared to DMSO-treated control (Figure 5), suggesting successful inhibition of CK1δ/ε. P1d, P3d, and P4d, as well as all other PROTACs, especially at 5 µM, led to a reduction of pVangl2 signal compared to DMSO-treated control (Figure 5). This data suggests that most PROTACs lead to inhibition of CK1δ/ε, even if not all of them cause degradation of CK1δ/ε (Figure 5).
CK1δ/ε co-exist in cells with SACK1A, B, E, and H (formerly known as FAM83A, B, E & H) [51]. Treatment of U2OS cells with PROTACs did not yield co-depletion of SACK1H, even under conditions where certain PROTACs led to a depletion of CK1δ/ε (Figure 5). In similar assays, CK1α degraders DEG-77 and SJ3149 have been shown to co-degrade CK1α-interactors SACK1B, SACK1D, SACK1F, and SACK1G [52]. The levels of CK1α or SACK1G, which only interacts with CK1α but not CK1δ/ε, were also not substantially affected by any of the PROTACs (Figure 5). The levels of GAPDH, employed as a loading control, were unaltered by any of the treatment conditions (Figure 5).
2.5.2. PROTACs as inhibitors of CK1δ/ε
Despite some PROTACs not causing degradation of CK1δ/ε, all PROTACs at 5 µM caused a robust reduction in levels of pVangl2 and noticeable downward mobility shift of DVL3 (Figure 6A), suggesting the PROTACs inhibited CK1δ/ε. We evaluated the potency of CK1δ inhibition in vitro by the three best degraders, P1d, P3d, and P4d, relative to warhead inhibitor compound I9. An in vitro kinase assay for CK1δ using a peptide substrate was conducted by the MRC PPU International Centre for Kinase Profiling (https://www.kinase-screen.mrc.ac.uk/). The IC50 for CK1δ inhibition in vitro for P3d was 1.96 µM, while it was ~2.2 µM for P1d and P4d, and 3.9 µM for CK1δ inhibitor I9 (Figure 6A), suggesting that the PROTACs were more potent inhibitors of CK1δ in vitro than the inhibitor warhead molecule. Without further controls, these findings make it hard to interpret whether the cellular effects of PROTACs are due to chemical inhibition of CK1δ/ε or the modest degradation of CK1δ/ε, or the combined effects of both. In similar assays in vitro, we showed that P1d and P4d did not inhibit CK1α kinase activity substantially (Figure 6B), suggesting the compounds were selective against CK1δ/ε.
2.5.3. Extended dose-response analysis of CK1δ/ε degradation by the best degrader P1d across multiple cells.
Initial tests of PROTACs in cells revealed that P1d showed the most potent CK1δ/ε degradation. Therefore, a comprehensive dose response of P1d was performed in multiple cell lines to assess CK1δ/ε degradation and downstream consequences. In U2OS cells, a robust degradation of CK1δ and CK1ε was observed at 5 and 10 µM when treated for 24 h (Figure 7A). Concomitantly, at the given concentrations, there was a clear reduction in levels of pVANGL2 (Figure 7A), suggesting inhibition of VANGL2 phosphorylation as CK1δ/ε were degraded. No substantial changes in CK1α, SACK1H, or GAPDH were observed at any concentrations of P1d (Figure 7A), suggesting selective degradation of CK1δ/ε and no co-degradation of SACK1H. In A549 adenocarcinoma cells, P1d also caused a robust degradation of CK1δ and CK1ε at 5 and 10 µM (Figure 7A). Unlike U2OS cells, A549 cells express detectable levels of CK1δ/ε-binders SACK1B and SACK1E. No co-depletion of SACK1B was observed, but a robust co-depletion of SACK1E, which exists in complex with CK1δ/ε, at 5 and 10 µM was observed (Figure 7A), potentially suggesting co-degradation of CK1δ/ε-SACK1E complex. No substantial changes in levels of CK1α and its interactor SACK1G or GAPDH were observed at any concentrations of P1d (Figure 7B).
2.5.4. Kinetic analysis of CK1δ/ε degradation by P1d
Given robust degradation of CK1δ/ε by P1d at 5 µM for 24 h, specific time points across 0-24 h were chosen to assess the kinetics of degradation. Compared to DMSO control, P1d showed a time-dependent degradation of CK1δ/ε, where degradation started at 6 h and reached maximal levels at 24 h following P1d treatment (Figure 8). In line with the degradation kinetics of CK1δ/ε, a downward mobility shift of DVL3 was observed, indicating inhibition of DVL3 phosphorylation (Figure 8). Similarly, a reduction in levels of pVANGL2 was observed at 2 h following P1d treatment and showed almost complete reduction after 6 h (Figure 8). Throughout the full 24 h with P1d treatment, there was no change in levels of CK1α, its interactor SACK1G, or GAPDH (Figure 8).
2.5.5. Establishing mode of action for P1d
PROTACs recruit a native E3 ligase to ubiquitylate the POI, which triggers the UPS system to successfully degrade the POI. We sought to explore the mode of action through which P1d led to the reduction in levels of CK1δ/ε. As P1d uses a thalidomide warhead that binds to and recruits the CUL4CRBN E3 ligase, we sought to investigate the requirement of CRBN by using CRBN-knockout (KO) U2OS cells. As shown earlier, treatment of WT U2OS cells with 5 μM and 10 μM P1d led to a robust degradation of CK1δ/ε compared to DMSO control (Figure 9A). In contrast, in CRBN-KO cells, generated through CRISPR/Cas9 genome editing (Glennie et al, 2025) and validated by an absence of signal with anti-CRBN immunoblot, no CK1δ/ε degradation was observed upon treatment of cells with 5 μM and 10 μM P1d (Figure 4-8A), confirming the requirement of CRBN for CK1δ/ε degradation. More importantly, WT and CRBN-KO U2OS cells also allowed us to determine the relative contribution of CK1δ/ε degradation vs. inhibition by 5 μM and 10 μM P1d on downstream signalling, as only inhibition would be possible in CRBN-KO cells. Excitingly, the extent of downward mobility shift of DVL3 and reduction in VANGL2 phosphorylation observed in WT U2OS cells caused by P1d at both 5 μM and 10 μM was much lower in CRBN-KO cells (Figure 9A), suggesting that the degradation of CK1δ/ε in WT cells caused a better inhibition of downstream signalling than just the inhibition of CK1δ/ε in CRBN-KO U2OS cells caused by P1d. No reduction in levels of SACK1H, CK1α, SACK1G, or GAPDH was observed by P1d in WT or CRBN-KO U2OS cells (Figure 9A). CUL4ACRBN is activated by NEDDylation, which can be inhibited by the NAE1 inhibitor MLN4924. Treatment of U2OS cells with 1 μM MLN4924 blocked degradation of CK1δ/ε caused by 5 μM P1d (Figure 9B). Similarly, the proteasome inhibitor MG132 at 20 μM also blocked degradation of CK1δ/ε caused by 5 μM P1d (Figure 9B), suggesting the requirement of the proteasome for degradation. Used as positive controls, MLN4924 blocked NEDDylation of CUL2, causing disappearance of the higher mobility species, and MG132 led to accumulation of polyubiquitylated chains compared to DMSO controls (Figure 9B). Collectively, these data suggest that P1d causes CK1δ/ε degradation through CUL4CRBN and the proteasome, and the degradation at the same PROTAC concentration appears to impact downstream signalling much more than inhibition.
2.5.6. Correlation between Computed and Experimental Linker Dimensions
To better understand the relationship between linker length and the observed efficacy in degradation of our compounds, we compared the computationally estimated linker distance with experimentally realized structures. As previously mentioned, the calculated optimal linker length was determined to be approximately 11 Å (Figure 2c). Interestingly, our best-performing compounds (P1d and P4d, Figure 5) contain shorter linkers, measuring 7.413 Å and 8.492 Å, respectively (Table 2). Compounds P3b and P4b, whose linker lengths are closest to the calculated value, exhibit minimal or no degradation under the tested conditions. Although the current modeling approach does not yet allow for a precise prediction of optimal linker dimensions, it has provided a valuable guideline that helped to orient our experimental design and interpret the observed structure–activity trends. Since the current synthesis route only allowed successful generation of compounds containing shorter alkyl linkers, the exact influence of longer alkyl chains remains to be clarified. In future studies, we intend to synthesize and evaluate compounds with extended alkyl linkers to systematically investigate how increasing linker length affects both molecular stability and functional performance.
3. Materials and Methods
3.1. Molecular Modeling
Molecular Modeling was performed using Maestro 13.8 (Schrödinger). The apoCK1α/Crl4(CRBN)/lenalidomide complex (pdb 5FQD) and CK1δ-ligand structure (pdb 5OKT) were imported and processed using the standard settings within the Protein Preparation Wizard. The CK1α part of 5FQD was used to align CK1δ (pdb 5OKT) by the structural alignment tool, followed by undisplaying of the CK1α part. Molecular structure of ligand SMI 1 was prepared by minimization (MacroModel) followed by LigPrep. Docking (SP mode) SMI 1 into the receptor grid of the ATP binding pocket of CK1δ (pdb 5OKT) revealed a binding pose that was subsequently minimized using the overall workspace entries by Prime and MacroModel. The 2D ligand-protein interaction diagram was used to illustrate the binding pose. In the 3D structure, the molecular measurement between the carbon atom of p-MeO of SMI 1 and the amino nitrogen atom of lenalidomide bound to the Crl4(Crbn)/Ck1δ engagement site revealed a distance of approximately d = 11 Å, suggesting a putative linker length between the molecules. The solvent accessible protein surfaces of the structures have been generated using the standard settings of the “surface binding site” tool.
3.2. Chemistry
3.2.1. General
Reagents and solvents were purchased from abcr, Deutero, Merck, Roth, or TCI and used as received.
Analytical RP-HPLC experiments for all intermediate compounds were performed using an Agilent 1290 (Agilent Technologies, Heilbronn, Germany) system (column: Agilent Poroshell, EC-C18, 2.7 μm, 50 x 3 mm) with 0.1 %acetic acid in water (Solvent A) and acetonitrile (Solvent B) as eluents. All experiments were performed using a gradient with an increase of 5.88 % Solvent B/min at a flow rate of 0.8 ml/min with a total run time of 16 min, injection volume of 1 μL, and detection between 210 – 400 nm.
Analytical RP-HPLC experiments for the PROTAC compounds were performed using an Agilent 1100 system (column: STAGROMA®C18 3 μm, 125 x 4 mm; Stagroma AG, Reinach, Switzerland) with 0.01 M Potassium dihydrogen phosphate buffer pH 2.3 (Solvent A) and acetonitrile (Solvent B) as eluents. All experiments were performed using a linear gradient with an increase of 3.33 % Solvent B/min at a flow rate of 1.5 ml/min with a total run time of 30 min, injection volume of 10 μL, and detection at 254 nm.
MS spectra of all intermediate compounds were measured using an AmaZon SL (Bruker) system. HR-MS characterizations of the PROTAC compounds were performed using a Q Exactive Plus Quadrupole-Orbitrap (Thermo Scientific) system. NMR spectra of all compounds were generated using a Bruker Avance III 400 system with the corresponding deuterated solvent as an internal standard.
3.2.2. Synthesis of Compound I3
2-Amino-6-(trifluoromethyl) benzothiazole (I2, 1.00 equiv) and TEA (1.10 equiv) are dissolved in 5 mL dichloromethane. Chloroacetyl chloride (I1, 1.10 equiv) is dissolved in 5 mL dichloromethane and added to the reaction mixture dropwise over an hour. The reaction mixture is left stirring at RT overnight. The solvent is evaporated to afford the product, which is dissolved in ethyl acetate, washed with water, and concentrated in vacuo again. The product is recrystallized (ethanol/water 1:1) to afford compound I3.
3.2.3 Synthesis of Compound I6
KOH (1.5 equiv) is ground, weighed in, and left under vacuum for 45 min at RT. The system is flooded with argon afterwards. KOH is dissolved in 9.5 mL DMSO while stirring at 110 °C. 4(3H)-Pyrimidinone (I4, 1.00 equiv) and 4-Nitrobenzyl chloride (I5, 1.10 equiv) are dissolved in 10 mL DMSO and added to the reaction mixture via injection. Iron (nano) (0.10 equiv) is suspended in 0.5 mL DMSO and added via injection. Afterwards, the reaction mixture is left stirring at 110 °C for 2 h. After cooling down, the reaction mixture is decanted on ice and, if needed, neutralized with a few drops of 1M HCl. The aqueous layer is extracted with CH2 Cl2 [3x 30 mL]. The organic layers are combined, washed with water [3x 30 mL], and dried over sodium sulfate. The solvent is evaporated, and the crude product is purified via flash chromatography (silica gel, petroleum ether/ethyl acetate 80/20 – 0/100) to afford Compound I6.
3.2.4. Synthesis of Compound I7
Intermediate I6 (1.00 equiv) and sodium bicarbonate (1.10 equiv) are dissolved in 30 mL EA/H2O 1:1. O-Phenyl chlorothionoformate (2.50 equiv) is added dropwise, and the reaction mixture is left stirring overnight afterwards. The aqueous layer is extracted with EA (3x 15 mL). The organic layers are combined, washed with brine (3x 15 mL), and dried over sodium sulfate. The solvent is evaporated, and the isolated intermediate stage is dissolved in 15 mL MeOH. TEA (6.00 equiv) is added, and the reaction mixture is left stirring at 80 °C for 4h. The solvent is evaporated, and the crude product is first purified via flash chromatography (dichloromethane/methyl alcohol). The crude is then suspended in ethyl acetate and filtered. The residue is dissolved in dichloromethane, and the solvent is evaporated to afford compound I7.
3.2.5. Synthesis of Compound I8
Intermediate I7 (1.00 equiv) and intermediate I8 (1.00 equiv) are dissolved in 10 mL DMF. TEA (3.00 equiv) is added via injection, and the reaction mixture is left stirring at 80 °C for 2h. After cooling down, the reaction is quenched with water and extracted with EA (3x 15 mL). The organic layers are combined, washed with brine (2x 15 mL) and water (1x 15 mL), and dried with sodium sulfate. The solvent is evaporated and the crude product purified via flash chromatography (silica gel, petroleum ether/ethyl acetate 80/20 – 0/100) to afford compound I8.
3.2.6. Synthesis of Compound I9
Intermediate I8 is dissolved in 20 mL of methyl alcohol. Palladium on activated carbon (10 %, 0.1 equiv) is added to the reaction mixture. The atmosphere is exchanged for hydrogen (3 bar). The reaction is stirred at room temperature for 48 h. Afterwards, the catalyst is filtered off through a Celite pad, which is washed with methyl alcohol (3x 20 mL). The filtrate and washings are combined, the solvent is evaporated, and the crude product is purified via flash chromatography (silica gel, dichloromethane/methyl alcohol 99/1 – 90/10) to afford compound I9.
3.2.7. General Procedure for the Synthesis of Linker Compounds L2a-d
The respective ϖ-Aminocarboxylic acid is dissolved in 2N sodium hydroxide solution (17 mL) and cooled in an ice bath to 0 °C. Once 0 °C is reached, benzyl chloroformate (1.10 equiv) and 2N sodium hydroxide solution (19 mL) are added within 2 minutes. The reaction mixture is left stirring at room temperature overnight and extracted with diethyl ether (4x 40 mL) afterwards. The aqueous layer is separated, and the pH is adjusted to 2 with 2N hydrochloric acid. The resulting emulsion is extracted with ethyl acetate (3x 30 mL). The organic phases are combined, washed with brine (3x 20 mL), and dried over sodium sulfate. The solvent is evaporated to afford compounds L2a-d.
3.2.8. General Procedure for the Synthesis of Linker Compounds L3a-d
The respective compound L2a-d is dissolved in tert-butyl alcohol (45 mL). Pyridine (27 mL) is added, and the reaction mixture is chilled to -10 °C. Once -10 °C is reached, phosphoryl chloride (1.10 equiv) is added dropwise under vigorous stirring. The reaction mixture is stirred at -10 °C for 30 minutes and then left stirring at room temperature overnight. The reaction mixture is concentrated in vacuo and diluted with water (20 mL). The reaction mixture is extracted with ethyl acetate (3x 50 mL) afterwards. The organic layers are combined, washed with water (3x 55 mL), saturated sodium sulfate solution (3x 55 mL), water again (3x 55 mL), 2N potassium bisulfate solution (3x 55 mL), brine (2x 30 mL), and dried over sodium sulfate. The solvent is evaporated to afford the corresponding compounds L3a-d.
3.2.9. General Procedure for the Synthesis of Linker Compounds L4a-d
The respective compound L3a-d is dissolved in methyl alcohol (30 mL). Palladium on activated carbon (10 %, 0.10 equiv) is added, and the atmosphere is first exchanged for nitrogen and then exchanged for hydrogen. The reaction mixture is left stirring at room temperature overnight. The catalyst is filtered off through a Celite pad, which is washed with methyl alcohol (3x 20 mL). The filtrate and washings are combined, and the solvent is evaporated to afford the corresponding compounds L4a-d.
3.2.10. General Procedure for the Synthesis of Compounds L6a-d
The respective compound L4a-d and 2-(2,6-Dioxopiperidin-3-yl)-4-fluoroisoindoline-1,3-dione (1.00 equiv) are dissolved in DMSO (10 mL). N, N-Diisopropylethylamine (DIPEA, 3.0 equiv) is added, and the reaction mixture is stirred at 90 °C for 12 hours. After cooling down, the reaction mixture is diluted with water (30 mL) and extracted with ethyl acetate (3x 30 mL). The organic layers are combined, washed with brine (3x 30 mL), and dried over sodium sulfate. The solvent is evaporated and the corresponding crude product is purified by flash column chromatography (C18-reversed phase silica gel, water/acetonitrile = 95/5 – 0/100). The solvent is evaporated to afford the corresponding compounds L6a-d.
3.2.11. General Procedure for the Synthesis of Compounds L8a-d
The respective compound L4a-d and 2-(2,6-Dioxopiperidin-3-yl)-5-fluoroisoindoline-1,3-dione (1.00 equiv) are dissolved in DMSO (10 mL). N, N-Diisopropylethylamine (DIPEA, 3.0 equiv) is added, and the reaction mixture is stirred at 90 °C for 12 hours. After cooling down, the reaction mixture is diluted with water (30 mL) and extracted with ethyl acetate (3x 30 mL). The organic layers are combined, washed with brine (3x 30 mL), and dried over sodium sulfate. The reaction mixture is concentrated in vacuo, and the corresponding crude product is purified by flash column chromatography (C18-reversed phase silica gel, water/acetonitrile = 95/5 – 0/100). The solvent is evaporated to afford the corresponding compounds L8a-d.
3.2.12. General Procedure for the Synthesis of Linker Compounds L10a-d
The respective glycol and tert-butyl acrylate (1.00 equiv) are dissolved in acetonitrile (20 mL). Benzyltrimethylammonium hydroxide (Triton B) solution (40 % in water, 0.04 equiv.) is added, and the reaction mixture is stirred at room temperature for 120 hours. The solvent is evaporated and the corresponding crude product is purified by flash column chromatography (silica gel, petroleum ether/ethyl acetate = 100/0 – 0/100). The solvent is evaporated to afford the corresponding compounds L10a-d.
3.2.13. General Procedure for the Synthesis of Linker Compounds L11a-d
The respective compound L10a-d and Tosyl chloride (1.50 equiv) are dissolved in dichloromethane (5 mL). Triethylamine (3.00 equiv) and 4-dimethylaminopyridine (DMAP, 0.10 equiv) are added, and the reaction mixture is stirred at room temperature overnight. The solvent is evaporated, and the corresponding crude product is purified by flash column chromatography (silica gel, petroleum ether/ethyl acetate = 80/20 – 50/50). The solvent is evaporated to afford the corresponding compounds L11a-d.
3.2.14. General Procedure for the Synthesis of Compounds L13a-d
The respective compound L11a-d and 2-(2,6-dioxopiperidin-3-yl)-4-hydroxyisoindoline-1,3-dione (1.00 equiv) are dissolved in DMF (5 mL). Potassium bicarbonate (1.50 equiv) and sodium iodide (0.10 equiv) are added, and the reaction mixture is stirred at 80 °C for 12 hours. After cooling down, the reaction mixture is quenched with water (10 mL) and extracted with ethyl acetate (3x 20 mL). The organic layers are combined and dried over sodium sulfate. The solvent is evaporated and the corresponding crude product is purified by flash column chromatography (C18-reversed phase silica gel, water/acetonitrile = 95/5 – 0/100). The solvent is evaporated to afford the corresponding compounds L13a-d.
3.2.15. General Procedure for the Synthesis of Compounds L15a-d
The respective compound L11a-d and 2-(2,6-dioxopiperidin-3-yl)-5-hydroxyisoindoline-1,3-dione (1.00 equiv) are dissolved in DMF (5 mL). Potassium bicarbonate (1.50 equiv) and sodium iodide (0.10 equiv) are added, and the reaction mixture is stirred at 80 °C for 12 hours. After cooling down, the reaction mixture is quenched with water (10 mL) and extracted with ethyl acetate (3x 20 mL). The organic layers are combined and dried over sodium sulfate. The solvent is evaporated and the corresponding crude product is purified by flash column chromatography (C18-reversed phase silica gel, water/acetonitrile = 95/5 – 0/100). The solvent is evaporated to afford the corresponding compounds L15a-d.
3.2.16. General Procedure for the Synthesis of PROTAC Compounds P1a-d, P2a, P2d, P3a-d and P4a-d
First, the respective compounds L6a-d, L8a, L8d, L13a-d, or L15a-d are dissolved in dichloromethane (1 mL). 1 mL of trifluoroacetic acid is added, and the reaction mixture is stirred at room temperature for 1 h. The solvent is evaporated, and the product is dried under high vacuum to afford the corresponding carboxylic acid in a quantitative yield. The deprotection of the tert-butyl ester is verified via LC-MS, and the intermediate product is used without further purification.
The intermediate is dissolved in dichloromethane (10 mL), and intermediate I9 (1.00 equiv) is added. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC, 1.00 equiv), Hydroxybenzotriazole (HOBt, 0.10 equiv), DMAP (1.00 equiv), and DIPEA (5.00 equiv) are added, and the reaction mixture is stirred at room temperature for 48 h. The solvent is removed, the residue is dissolved in ethyl acetate (10 mL), and quenched with saturated sodium bicarbonate solution (30 mL). The aqueous layer is extracted with ethyl acetate (3x 30 mL). The organic layers are combined, dried over sodium sulfate, and the solvent is evaporated. The crude product is purified by flash column chromatography twice (1: silica gel, petroleum ether/ethyl acetate 80/20 – 0/100, elution with dichloromethane/methyl alcohol 90/10; 2: c18-reversed phase silica gel, water/acetonitrile 5/95 – 0/100) to yield the corresponding PROTAC compounds P1a-d, P2a, P2d, P3a-d, and P4a-d.
3.3. Biology
3.3.1. Cell culture, drug treatment, and lysis
U2OS osteosarcoma, A549 adenocarcinoma, and DLD1 colorectal cancer cells were cultured in Dulbecco’s modified Eagle Medium (DMEM) supplemented with 10% (v/v) foetal bovine serum (FBS), 2 mM L-glutamine (Lonza), 100 U/mL penicillin (Lonza), and 0.1 mg/mL streptomycin (Lonza). Cells were incubated in a humidified incubator at 37˚C with 5% CO2 and handled under aseptic conditions within a laminar flow hood. For passaging, cells were washed in PBS and incubated with trypsin/EDTA at 37˚C until detached and transferred to fresh plates. CRBN-/- U2OS cells were generated using a CRISPR/Cas9 genome editing approach as described previously [53]. CSNK1A1-/- [52], CSNK1D-/- and CSNKE-/- DLD-1 cells were generated by K. Dunbar (Sapkota lab). All knockout cells were validated by immunoblotting (Figure 4) and DNA sequencing. All PROTAC compounds and small molecule inhibitors were dissolved in DMSO and were added directly to culture medium at the indicated concentrations and incubated with cells for the specified times prior to cell lysis. An equivalent volume of DMSO was added to cells as a treatment control. MG132 (Abcam; Ab141003) and MLN4294 (Sigma-Aldrich; 5054770001) were also diluted in DMSO and applied to cells as above. Cells were lysed by removing culture media, washing twice with ice-cold PBS and then scraping directly into ice-cold NP-40 cell lysis buffer (50 mM Tris-HCl pH 7.5, 0.27 M sucrose, 150 mM NaCl, 1 mM EGTA, 1 mM EDTA, 1 mM sodium orthovanadate, 10 mM sodium b-glycerophosphate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate and 1% NP-40) supplemented with 1x cOmplete protease inhibitor cocktail (Roche) and 1 mM DTT. Cell extracts were transferred to Eppendorf tubes and clarified by centrifugation at 14000 rpm for 15 min at 4˚C. Protein concentration was measured by Bradford assay. For SDS-PAGE analysis, samples were prepared to an equal protein concentration by normalising with NP-40 lysis buffer and 1X LDS sample buffer supplemented with 2.5% β-mercaptoethanol. Samples were boiled at 95˚C for 5 min before SDS-PAGE. 3.3.2 SDS-PAGE and immunoblotting
Cell extracts (20 µg protein) were loaded into 8% polyacrylamide gels or precast 4-20% NuPAGE™ Midi Bis-Tris gels (Invitrogen) and resolved at 120V for 90-120 min. Protein was then transferred to a nitrocellulose membrane. Membranes were blocked in 5% (w/v) non-fat milk (Marvel) in TBS-T (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.2% Tween 20) for 1 h and incubated overnight at 4˚C with the appropriate primary antibodies, also diluted in 5 ml TBS-T/5% non-fat milk. Membranes were then washed 3x10 min using TBS-T in a shaking platform. They were then incubated with HRP-conjugated secondary antibodies diluted in 5% non-fat milk + TBS-T for 1 h at room temperature, after which they were washed 3x10 min in TBST-T. Signal detection was performed using an enhanced chemiluminescence (ECL) system and data obtained using the ChemiDoc Imaging System (Bio-Rad). Primary antibodies used were: CK1δ (MRC PPU; SA609); CK1α (MRC PPU; SA527); CK1ε (MRC PPU; SA610); DVL3 (ProteinTech; 13444 1-AP); pVANGL2 (AbClonal Technology; AP1206) Vinculin (CST; 4650); GAPDH (ProteinTech; 60004-1Ig); GAPDH-HRP (ProteinTech; HRP-60004); SACK1A (MRC PPU; S911D); SACK1B (MRC PPU; SA270); SACK1E (MRC PPU; SA272); SACK1H (MRC PPU; SA273); SACK1G (Abcam; Ab121750); CUL2 (Invitrogen; 51-8100); Ubiquitin (Abcam; Ab19247); Cereblon (CST; 60312/71810); Goat anti-rabbit IgG (7074, CST, 1:2500) and Rabbit anti-sheep IgG (31480, Thermo Fisher Scientific, 1:2500).
3.3.3. In vitro kinase assays for IC50 determination
The in vitro kinase assays were performed by MRC PPU International Centre for Kinase Profiling (https://www.kinase-screen.mrc.ac.uk/services/ic50). CK1α or δ (5 mU diluted in 20 mM HEPES pH7.5, 0.15 M NaCl, 0.1 mM EGTA, 0.1% Triton X-100, 5 mM DTT, 50% glycerol) was assayed against CK1 substrate peptide (RRKDLHDDEEDEAMSITA) in a final volume of 25.5 µl containing 20 mM HEPES pH 7.5, 0.15 M NaCl, 0.1 mM EDTA, 5 mM DTT, 0.1% Triton-X 100, CK1 peptide (0.5 mM), 10 mM magnesium acetate, indicated concentrations of compounds or DMSO control and 0.02 mM [33P-γATP](500 -1000 cpm/pmole) and incubated for 30 min at room temperature. Assays are stopped by the addition of 5 µl of 0.5 M (3%) orthophosphoric acid and then harvested onto P81 Unifilter plates with a wash buffer of 50 mM orthophosphoric acid. 33P-radioactivity (cpm) for the kinase assay conducted in the presence of DMSO control was assigned 100 % CK1 activity.
4. Conclusions
In this study, we established a flexible synthetic platform for the modular preparation of designed CK1δ-PROTACs following the ternary scheme CK1δ-ligand - linker - E3 ligase ligand. We applied this platform to generate two different sets of projected PROTAC degrader compounds towards CK1δ. The synthetic route previously published by our group in 2019 was applied and extended to generate an inhibitor compound (I9) containing an aniline moiety, facilitating linker attachment via amide coupling. All dimeric intermediate compounds (L6a-d, L8a-d) were generated in sufficient quantities to be employed in the final amide coupling in order to generate the PROTAC compounds.
While alkyl-linker PROTAC compounds P1a-d (three through six methylene groups respectively) were synthesized successfully with yields in the range from 6 to 22 %, compounds P2b and P2c (four and five methylene groups) of this series could not be generated by the method. Despite our efforts towards expanded variations regarding solvent, reaction time, temperature, and coupling reagents, all attempts were ineffective in yielding the desired compounds P2b and P2c.
The set of PROTACs containing PEG linker motifs, however, were synthesized successfully, possibly due to their length and higher conformational flexibility compared to alkyl linkers. Overall, we were successful in establishing a versatile and effective synthetic platform for the systematic synthesis of various PROTAC candidate compounds.
We were able to apply all our PROTAC candidate molecules in various cells to assess the potential degradation of CK1 isoforms, their interacting partners, and assess the impact on phosphorylation of downstream substrates. A few PROTACs induced a dose-dependent degradation of CK1δ and CK1ε but not the related kinase CK1α. P1d was the most potent CK1δ/ε degrader, robustly degrading these kinases at concentrations of 5 μM and 10 μM within 6 h in multiple cell lines, including U2OS and A549. We established that P1d degrades CK1δ/ε via the CUL4CRBN E3 ligase complex and the proteasome.
A key challenge in utilizing kinase inhibitors as warheads for PROTACs is that the PROTACs could still act as inhibitors even in the absence of degradation, and the effects of PROTACs on downstream biology might be due to inhibition, without the added benefit of degradation. Therefore, it is important to evaluate the relative contribution of inhibition vs degradation on downstream biology, as well as evaluate the potency of the kinase inhibition in vitro. For our top PROTAC P1d, an in vitro kinase inhibition assay against CK1δ carried out in parallel with the inhibitor I9, showed IC50 values of 2.16 μM compared to 3.43 μM for I9. These data suggested the potential of P1d to be a more potent cellular inhibitor of CK1δ/ε than I9. However, the superiority of P1d PROTAC at inhibiting downstream CK1δ/ε via degradation compared to inhibition comes from the observations when WT and CRBN-KO U2OS cells were treated with P1d. Here, P1d degrades CK1δ/ε in WT cells but not in CRBN-KO U2OS cells; however, it should still be able to inhibit CK1δ/ε activity in CRBN-KO cells. Excitingly, P1d-induced inhibition of both DVL3 and VANGL2 phosphorylation at both 5 μM and 10 μM in WT U2OS cells is much more potent than in CRBN-KO U2OS cells, suggesting that it is indeed the degradation of CK1δ/ε that accounts for the potent inhibition of downstream signalling. Nonetheless, 10 μM of P1d still potently inhibits DVL3 and VANGL2 phosphorylation in CRBN-KO U2OS cells, implying the compound enters the cells and inhibits CK1δ/ε, as no CK1δ/ε degradation is seen in CRBN-KO U2OS cells. Another exciting observation we made is that in A549 cells, P1d induced degradation of SACK1E (formerly FAM83E) but no other SACK1 proteins, although the functional significance of this needs further investigation. In cells it is known that CK1δ/ε exist in complex with SACK1A/FAM83A, SACK1B/FAM83B, SACK1E/FAM83E and SACK1H/FAM83H, but the function of SACK1E- CK1δ/ε complexes are unknown [23].
The current minimum effective dose of P1d of 5 μM required to degrade CK1δ/ε does not compare favourably with more potent PROTACs against other targets that work at low nanomolar concentrations. This limitation is most likely due to the utilized CK1δ inhibitor moiety, which inhibits CK1δ in vitro with an IC50 of 3.43 μM. We aim to develop new series of compounds with a more potent inhibitor warhead, and alternative linkers as well as other E3 ligands, including those recruiting VHL, KLHDC2, and cIAP2. Based on the established procedures providing dimers containing E3 ligase ligands and linkers of varying motifs and lengths readily available, we aim to apply the modular platform for the preparation of further PROTAC sets towards various proteins of interest.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figures S1-S14: 1H- and 13C-NMR spectra for the PROTAC compounds; Figures S15-S28: HPLC and MS spectra for the PROTAC compounds.
Author Contributions
Conceptualization, M.A., G.P.S. and C.P.; methodology, M.A., M.H., L.G. and T.T.; software, C.P.; validation, M.A., G.P.S. and C.P.; formal analysis, M.A., M.H., L.G. and T.T.; investigation, M.A., L.G. and T.T.; resources, M.A., G.P.S. and C.P.; data curation, M.A., M.H. L.G. and T.T.; writing—original draft preparation, M.A., G.P.S. and C.P; writing—review and editing, M.H., T.T., G.P.S. and C.P; visualization, M.A., G.P.S. and C.P.; supervision, G.P.S. and C.P.; project administration, M.A., G.P.S. and C.P.; funding acquisition, G.P.S. and C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research in G.P.S.’s laboratory was funded by the UKRI Medical Research Council (MC_UU_00038/6). T.T. was funded by the University of Dundee’s Black British Scholarship Programme. L.G. was funded by MRC PPU Prize PhD studentship. The research in C.P.’s laboratory received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author(s).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| CK1 | Casein Kinase 1 |
| PROTAC | Proteolysis Targeting Chimera |
| PEG | Polyethylene Glycol |
| MOA | Mode of Action |
| SMI | Small Molecule Inhibitor |
| POI | Protein of Interest |
| UPS | Ubiquitin Proteasome System |
| APC | Adenomatous Polyposis Coli |
| AXIN | Axis Inhibition Protein |
| GSK3 protein | Glycogen Synthase Kinase 3 Protein |
| HRII | Hydrophobic Region II |
| ATP | Adenosine triphosphate |
| TPD | Targeted Protein Degradation |
| IWP | Inhibitor of Wnt Production |
| CRBN | Cereblon |
| p-MeO | Para-Methoxy |
| SAR | Structure Activity Relationship |
| TEA | Triethylamine |
| DMSO | Dimethylsulfoxide |
| DMF | Dimethylformamide |
| equiv. | Equivalent |
| TFA | Trifluororacetic acid |
| HATU | Hexafluorophosphate Azabenzotriazole Tetramethyl Uronium |
| DIPEA | N, N-Diisopropylethylamine |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| HOBt | Hydroxybenzotriazole |
| DMAP | 4-Dimethylaminopyridine |
| MeCN | Acetonitrile |
| RP-HPLC | Reversed Phase High Performance Liquid Chromatography |
| MS | Mass Spectrometry |
| HR-MS | High resolution Mass Spectrometry |
| NMR | Nuclear Magnetic Resonance |
| EA | Ethyl acetate |
| MeOH | Methanol |
References
- Békés, M.; Langley, D. R.; Crews, C. M. Nature reviews. Drug discovery 2022, 3, 181. [CrossRef]
- Li, K.; Crews, C. M. Chemical Society reviews 2022, 12, 5214. [CrossRef]
- Nalawansha, D. A.; Crews, C. M. Cell Chemical Biology 2020, 8, 998. [CrossRef]
- Hershko, A.; Ciechanover, A. Annual review of biochemistry 1998, 425. [CrossRef]
- Kleiger, G.; Mayor, T. Trends in cell biology 2014, 6, 352. [CrossRef]
- Nandi, D.; Tahiliani, P.; Kumar, A.; Chandu, D. Journal of biosciences 2006, 1, 137. [CrossRef]
- Paiva, S.-L.; Da Silva, S. R.; Araujo, E. D. de; Gunning, P. T. Journal of medicinal chemistry 2018, 2, 405. [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Nature medicine 2014, 11, 1242. [CrossRef]
- Jiang, Y.; Beaudet, A. L. Current opinion in pediatrics 2004, 4, 419. [CrossRef]
- Paiva, S.-L.; Crews, C. M. Current Opinion in Chemical Biology 2019, 111. [CrossRef]
- An, S.; Fu, L. EBioMedicine 2018, 553. [CrossRef]
- Berndsen, C. E.; Wolberger, C. Nature Structural & Molecular Biology 2014, 4, 301. [CrossRef]
- Schreiber, A.; Peter, M. Biochimica et biophysica acta 2014, 1, 163. [CrossRef]
- Alabi, S. B.; Crews, C. M. The Journal of biological chemistry 2021, 100647. [CrossRef]
- Bondeson, D. P.; Mares, A.; Smith, I. E. D.; Ko, E.; Campos, S.; Miah, A. H.; Mulholland, K. E.; Routly, N.; Buckley, D. L.; Gustafson, J. L.; Zinn, N.; Grandi, P.; Shimamura, S.; Bergamini, G.; Faelth-Savitski, M.; Bantscheff, M.; Cox, C.; Gordon, D. A.; Willard, R. R.; Flanagan, J. J.; Casillas, L. N.; Votta, B. J.; Besten, W. den; Famm, K.; Kruidenier, L.; Carter, P. S.; Harling, J. D.; Churcher, I.; Crews, C. M. Nature chemical biology 2015, 8, 611. [CrossRef]
- Zagidullin, A.; Milyukov, V.; Rizvanov, A.; Bulatov, E. Exploration of targeted anti-tumor therapy 2020, 5, 381. [CrossRef]
- Troup, R. I.; Fallan, C.; Baud, M. G. J. Exploration of targeted anti-tumor therapy 2020, 5, 273. [CrossRef]
- Poongavanam, V.; Peintner, S.; Abeje, Y.; Kölling, F.; Meibom, D.; Erdelyi, M.; Kihlberg, J. ACS medicinal chemistry letters 2025. [CrossRef]
- Cyrus, K.; Wehenkel, M.; Choi, E.-Y.; Han, H.-J.; Lee, H.; Swanson, H.; Kim, K.-B. Molecular bioSystems 2011, 2, 359. [CrossRef]
- Knippschild, U.; Krüger, M.; Richter, J.; Xu, P.; García-Reyes, B.; Peifer, C.; Halekotte, J.; Bakulev, V.; Bischof, J. Frontiers in oncology 2014, 96. [CrossRef]
- Narasimamurthy, R.; Hunt, S. R.; Lu, Y.; Fustin, J.-M.; Okamura, H.; Partch, C. L.; Forger, D. B.; Kim, J. K.; Virshup, D. M. Proceedings of the National Academy of Sciences 2018, 23, 5986. [CrossRef]
- Sinnberg, T.; Wang, J.; Sauer, B.; Schittek, B. BMC cancer 2016, 594. [CrossRef]
- Fulcher, L. J.; Sapkota, G. P. The Biochemical journal 2020, 23, 4603. [CrossRef]
- Gybeľ, T.; Čada, Š.; Klementová, D.; Schwalm, M. P.; Berger, B.-T.; Šebesta, M.; Knapp, S.; Bryja, V. The Journal of biological chemistry 2024, 7, 107407. [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Signal transduction and targeted therapy 2022, 1, 3. [CrossRef]
- MacDonald, B. T.; Tamai, K.; He, X. Developmental cell 2009, 1, 9. [CrossRef]
- Bryja, V.; Bernatík, O. 2014, 207. [CrossRef]
- Xu, P.; Ianes, C.; Gärtner, F.; Liu, C.; Burster, T.; Bakulev, V.; Rachidi, N.; Knippschild, U.; Bischof, J. Gene 2019, 144005. [CrossRef]
- Mazzoldi, E. L.; Pastò, A.; Ceppelli, E.; Pilotto, G.; Barbieri, V.; Amadori, A.; Pavan, S. Frontiers in oncology 2019, 1211. [CrossRef]
- Cheong, J. K.; Virshup, D. M. The international journal of biochemistry & cell biology 2011, 4, 465. [CrossRef]
- Nishiguchi, G.; Caine, E. A.; McGowan, K.; Shi, Z.; Aggarwal, A.; Mayasundari, A.; Price, J.; Yang, L.; Li, Y.; Fu, X.; Mascibroda, L. G.; Das, S.; Daniels, D. L.; Urh, M.; Klco, J. M.; Riching, K. M.; Rankovic, Z. ACS medicinal chemistry letters 2024. [CrossRef]
- MedchemExpress.com. Available online: https://www.medchemexpress.com/ck1%CE%B1-degrader-1.html (accessed on 6 March 2025).
- Wang, K.; Jiang, M.; Liu, H.; Meng, C.; Li, M.; Lu, H. Bioorganic chemistry 2024, 107319. [CrossRef]
- Haag, A.; Němec, V.; Janovská, P.; Bartošíková, J.; Adhikari, B.; Müller, J.; Schwalm, M. P.; Čada, Š.; Ohmayer, U.; Daub, H.; Kim, Y.; Born, F.; Wolf, E.; Bryja, V.; Knapp, S. Journal of medicinal chemistry 2025, 1, 506. [CrossRef]
- García-Reyes, B.; Witt, L.; Jansen, B.; Karasu, E.; Gehring, T.; Leban, J.; Henne-Bruns, D.; Pichlo, C.; Brunstein, E.; Baumann, U.; Wesseler, F.; Rathmer, B.; Schade, D.; Peifer, C.; Knippschild, U. Journal of medicinal chemistry 2018, 9, 4087. [CrossRef]
- Liu, C.; Witt, L.; Ianes, C.; Bischof, J.; Bammert, M.-T.; Baier, J.; Kirschner, S.; Henne-Bruns, D.; Xu, P.; Kornmann, M.; Peifer, C.; Knippschild, U. International journal of molecular sciences 2019, 24. [CrossRef]
- Schade, D.; Plowright, A. T. Journal of medicinal chemistry 2015, 24, 9451. [CrossRef]
- Chen, B.; Dodge, M. E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.-W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N. S.; Roth, M. G.; Amatruda, J. F.; Chen, C.; Lum, L. Nature chemical biology 2009, 2, 100. [CrossRef]
- Bischof, J.; Leban, J.; Zaja, M.; Grothey, A.; Radunsky, B.; Othersen, O.; Strobl, S.; Vitt, D.; Knippschild, U. Amino acids 2012, 4, 1577. [CrossRef]
- Petzold, G.; Fischer, E. S.; Thomä, N. H. Nature 2016, 7597, 127. [CrossRef]
- Hernández-Núñez, E.; Tlahuext, H.; Moo-Puc, R.; Torres-Gómez, H.; Reyes-Martínez, R.; Cedillo-Rivera, R.; Nava-Zuazo, C.; Navarrete-Vazquez, G. European journal of medicinal chemistry 2009, 7, 2975. [CrossRef]
- Roopan, S. M.; Khan, F. R. N.; Mandal, B. K. Tetrahedron letters 2010, 17, 2309. [CrossRef]
- Xu, J.; Yadan, J.-C. Synlett 1995, 03, 239. [CrossRef]
- Schmuck, C.; Rehm, T.; Geiger, L.; Schäfer, M. The Journal of organic chemistry 2007, 16, 6162. [CrossRef]
- Kim, K.; Lee, D. H.; Park, S.; Jo, S.-H.; Ku, B.; Park, S. G.; Park, B. C.; Jeon, Y. U.; Ahn, S.; Kang, C. H.; Hwang, D.; Chae, S.; Du Ha, J.; Kim, S.; Hwang, J. Y.; Kim, J.-H. Scientific reports 2019, 1, 19654. [CrossRef]
- Ghosh, A. K.; Shahabi, D. Tetrahedron letters 2021.
- Abeje, Y. E.; Wieske, L. H. E.; Poongavanam, V.; Maassen, S.; Atilaw, Y.; Cromm, P.; Lehmann, L.; Erdelyi, M.; Meibom, D.; Kihlberg, J. Journal of medicinal chemistry 2025, 1, 638. [CrossRef]
- Cromm, P. M.; Samarasinghe, K. T. G.; Hines, J.; Crews, C. M. Journal of the American Chemical Society 2018, 49, 17019. [CrossRef]
- Kaucká, M.; Petersen, J.; Janovská, P.; Radaszkiewicz, T.; Smyčková, L.; Daulat, A. M.; Borg, J.-P.; Schulte, G.; Bryja, V. Cell communication and signalling : CCS 2015, 2. [CrossRef]
- Yang, W.; Garrett, L.; Di Feng; Elliott, G.; Liu, X.; Wang, N.; Wong, Y. M.; Choi, N. T.; Yang, Y.; Gao, B. Cell research 2017, 12, 1466. [CrossRef]
- Fulcher, L. J.; Bozatzi, P.; Tachie-Menson, T.; Wu, K. Z. L.; Cummins, T. D.; Bufton, J. C.; Pinkas, D. M.; Dunbar, K.; Shrestha, S.; Wood, N. T.; Weidlich, S.; Macartney, T. J.; Varghese, J.; Gourlay, R.; Campbell, D. G.; Dingwell, K. S.; Smith, J. C.; Bullock, A. N.; Sapkota, G. P. Science signalling 2018, 531. [CrossRef]
- Glennie, L.; Curnutt, N.; Cartwright, T.; Dunbar, K.; Le Chatelier, B.; Wood, N. T.; Macartney, T. J.; Woo, C. M.; Sapkota, G. P. bioRxiv : the preprint server for biology 2025. [CrossRef]
- Dunbar, K.; Jones, R. A.; Dingwell, K.; Macartney, T. J.; Smith, J. C.; Sapkota, G. P. Life science alliance 2021, 2. [CrossRef]
Figure 1.
Schematic representation of the mechanism of action of PROTAC compounds. PROTACs hijack the UPS by forming ternary complexes with the POI and an E3 ligase, and therefore artificially inducing proximity between these proteins. The latter then recruits an E2 ligase, which transfers its ubiquitin molecules onto the POI. The polyubiquitinated POI is then marked for degradation by the 26S proteasome. Due to its catalytic nature, PROTACs can undergo this cycle of inducing polyubiquitination and degradation of a targeted POI in sub-stoichiometric amounts. Figure created with BioRender.com.
Figure 1.
Schematic representation of the mechanism of action of PROTAC compounds. PROTACs hijack the UPS by forming ternary complexes with the POI and an E3 ligase, and therefore artificially inducing proximity between these proteins. The latter then recruits an E2 ligase, which transfers its ubiquitin molecules onto the POI. The polyubiquitinated POI is then marked for degradation by the 26S proteasome. Due to its catalytic nature, PROTACs can undergo this cycle of inducing polyubiquitination and degradation of a targeted POI in sub-stoichiometric amounts. Figure created with BioRender.com.
Scheme 1.
a) Chemical structures of CK1α degrader probes, including both molecular glue (lenalidomide, TMX4116, SJ3149, DEG-77, CK1α degrader-1) and PROTAC (PROTAC 13i) compounds. b) Chemical structure of the CK1-“tear drop” inhibitor (MU1742) utilized to synthesize degrader compound AH078 containing an alkyl linker as well as a VHL ligand.
Scheme 1.
a) Chemical structures of CK1α degrader probes, including both molecular glue (lenalidomide, TMX4116, SJ3149, DEG-77, CK1α degrader-1) and PROTAC (PROTAC 13i) compounds. b) Chemical structure of the CK1-“tear drop” inhibitor (MU1742) utilized to synthesize degrader compound AH078 containing an alkyl linker as well as a VHL ligand.
SCHEME 2.
Combining relevant moieties from the chemical structures of IWP-2 and Bischof-5 led to the discovery of SMI 1, a highly CK1δ-specific and cellular effective inhibitor with an CK1δ IC50 value of 0.09 ± 0.01 μM.
SCHEME 2.
Combining relevant moieties from the chemical structures of IWP-2 and Bischof-5 led to the discovery of SMI 1, a highly CK1δ-specific and cellular effective inhibitor with an CK1δ IC50 value of 0.09 ± 0.01 μM.
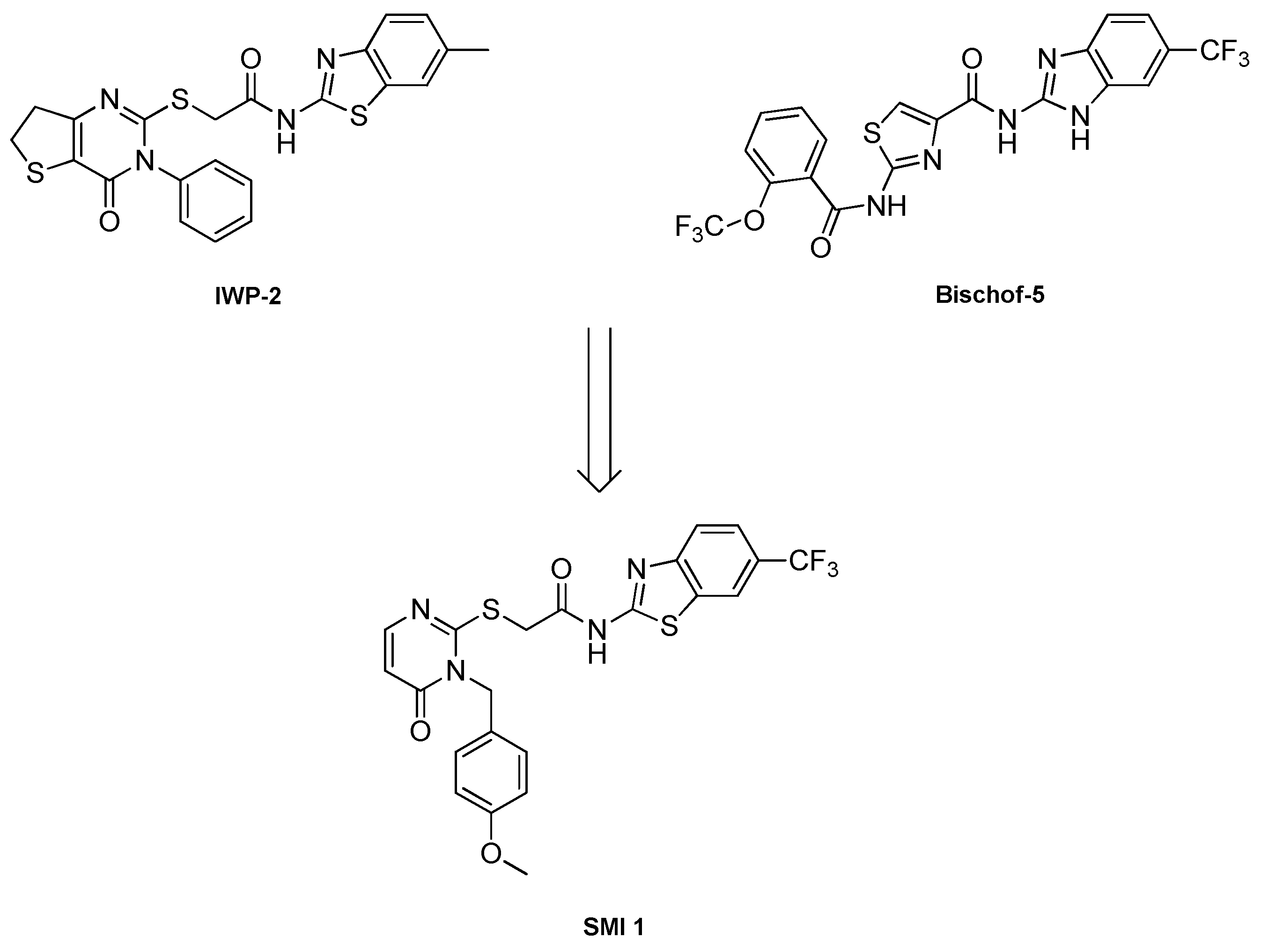
FIGURE 2.
Molecular modelling to guide the design of candidate CK1δ-PROTAC compounds. a) SMI 1 docked in the ATP binding pocket of CK1δ (pdb 5OKT) [35] illustrated as a 2D-Ligand-interaction-diagram showing key interactions of the binding pose of SMI 1 within the ATP pocket of CK1δ (bidental hinge-binding motif to Leu85 is highlighted). The benzyl moiety of SMI 1 with the p-MeO position is sitting in the solvent open area (grey dots), indicating this position to be suitable for linker attachment. b) Molecular modelling based on a x-ray defined apoCK1α/CRL4(CRBN)/lenalidomide complex (pdb 5FQD) [40]. We aligned CK1δ (pdb 5OKT, yellow) with docked SMI 1 (green) in the ATP binding pocket on the original CK1α structure (not shown anymore) to generate a CK1δ (yellow) - SMI 1 (green)/Crl4(CRBN) (blue/green) - lenalidomide (magenta) complex as a model system. The protein-interaction site of CK1δ-SMI 1/CRBN-lenalidomide is highlighted c) Close-up view of the highlighted area with the measured distance (dotted purple line) between the methyl group of the p-MeO function of SMI 1 (green) and the amino nitrogen atom of lenalidomide bound to CRL4(CRBN) (blue) revealed a linker length of approximately d = 11 Å used for estimating the linker length of the designed CK1δ PROTACs in this study. d) Close-up view of the highlighted area (side-perspective) also showing the solvent accessible protein surface.
FIGURE 2.
Molecular modelling to guide the design of candidate CK1δ-PROTAC compounds. a) SMI 1 docked in the ATP binding pocket of CK1δ (pdb 5OKT) [35] illustrated as a 2D-Ligand-interaction-diagram showing key interactions of the binding pose of SMI 1 within the ATP pocket of CK1δ (bidental hinge-binding motif to Leu85 is highlighted). The benzyl moiety of SMI 1 with the p-MeO position is sitting in the solvent open area (grey dots), indicating this position to be suitable for linker attachment. b) Molecular modelling based on a x-ray defined apoCK1α/CRL4(CRBN)/lenalidomide complex (pdb 5FQD) [40]. We aligned CK1δ (pdb 5OKT, yellow) with docked SMI 1 (green) in the ATP binding pocket on the original CK1α structure (not shown anymore) to generate a CK1δ (yellow) - SMI 1 (green)/Crl4(CRBN) (blue/green) - lenalidomide (magenta) complex as a model system. The protein-interaction site of CK1δ-SMI 1/CRBN-lenalidomide is highlighted c) Close-up view of the highlighted area with the measured distance (dotted purple line) between the methyl group of the p-MeO function of SMI 1 (green) and the amino nitrogen atom of lenalidomide bound to CRL4(CRBN) (blue) revealed a linker length of approximately d = 11 Å used for estimating the linker length of the designed CK1δ PROTACs in this study. d) Close-up view of the highlighted area (side-perspective) also showing the solvent accessible protein surface.
SCHEME 3.
Synthesis of POI ligand and intermediate I9. (a) 1.1 equiv. TEA in CH2Cl2, rt, 24 h; (b) 1.5 equiv. KOH and nano iron (cat.) in abs. DMSO under inert atmosphere, 110 °C, 2 h; (c) (i) 2.5 equiv. phenyl chlorothionoformate and 1.1 equiv. sodium bicarbonate in ethyl acetate/water (1:1), rt, overnight; (ii) 6.0 equiv. TEA in methanol, 80 °C, 4 h; (d) 3.0 equiv. TEA in abs. DMF, 80 °C, 2 h; (e) H2 (3 bar) and 10 % (m/m) Pd/C in methanol, rt, 24 h.
SCHEME 3.
Synthesis of POI ligand and intermediate I9. (a) 1.1 equiv. TEA in CH2Cl2, rt, 24 h; (b) 1.5 equiv. KOH and nano iron (cat.) in abs. DMSO under inert atmosphere, 110 °C, 2 h; (c) (i) 2.5 equiv. phenyl chlorothionoformate and 1.1 equiv. sodium bicarbonate in ethyl acetate/water (1:1), rt, overnight; (ii) 6.0 equiv. TEA in methanol, 80 °C, 4 h; (d) 3.0 equiv. TEA in abs. DMF, 80 °C, 2 h; (e) H2 (3 bar) and 10 % (m/m) Pd/C in methanol, rt, 24 h.
SCHEME 4.
Synthesis of the first set of PROTAC compounds P1a-d, as well as P2a and P2d containing alkyl linker motifs. (a) 1.1 equiv. benzyl chloroformate in 2 N sodium hydroxide solution, rt, overnight; (b) 1.1 equiv. phosphoryl chloride in tBuOH/Pyridine (3:2), rt, overnight; (c) H2 and 10 % (m/m) Pd/C in methanol, rt, overnight; (d) 3.0 equiv. DIPEA in DMSO, 90 °C, 14 h; (e) (i) 1 mL TFA in CH2Cl2, rt, 1 h; (ii)1.0 equiv. EDC x HCl, 0.1 equiv. HOBt, 1.0 equiv. DMAP and 5.0 equiv. DIPEA in MeCN, rt, 48 h.
SCHEME 4.
Synthesis of the first set of PROTAC compounds P1a-d, as well as P2a and P2d containing alkyl linker motifs. (a) 1.1 equiv. benzyl chloroformate in 2 N sodium hydroxide solution, rt, overnight; (b) 1.1 equiv. phosphoryl chloride in tBuOH/Pyridine (3:2), rt, overnight; (c) H2 and 10 % (m/m) Pd/C in methanol, rt, overnight; (d) 3.0 equiv. DIPEA in DMSO, 90 °C, 14 h; (e) (i) 1 mL TFA in CH2Cl2, rt, 1 h; (ii)1.0 equiv. EDC x HCl, 0.1 equiv. HOBt, 1.0 equiv. DMAP and 5.0 equiv. DIPEA in MeCN, rt, 48 h.
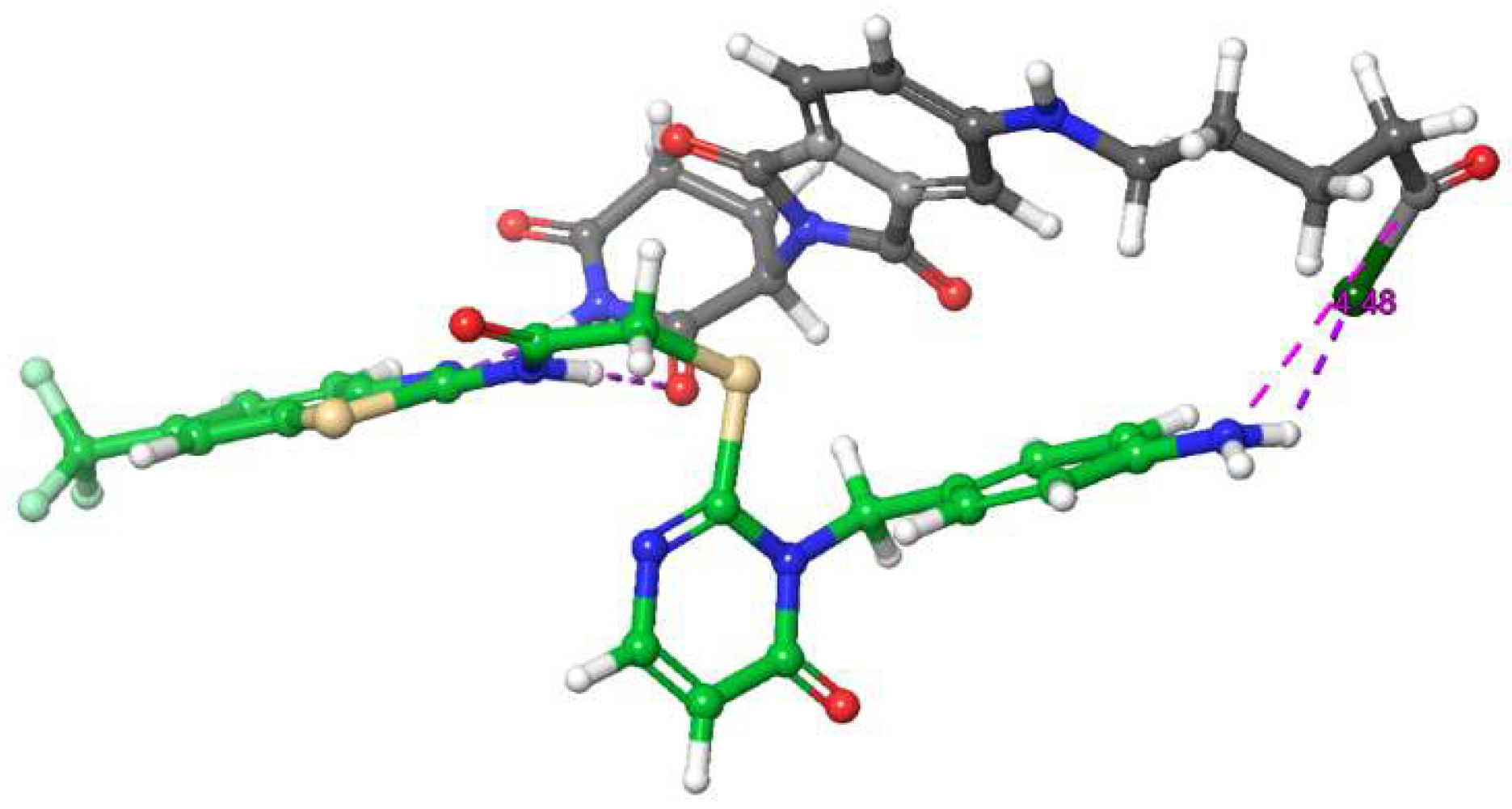
FIGURE 3.
Exemplary molecular modelling suggesting complex-forming interactions between I9 and L8c, thereby potentially preventing the amide coupling reaction.
FIGURE 3.
Exemplary molecular modelling suggesting complex-forming interactions between I9 and L8c, thereby potentially preventing the amide coupling reaction.
Scheme 5.
Synthesis for compounds L13a-d and L15a-d containing PEG linker motifs. (a) 1.0 equiv. tert-butyl acrylate and 0.04 equiv. Triton B solution (40 % in water), rt, 120 h; (b) 1.5 equiv. 3.0 equiv. TEA and 0.1 equiv. DMAP in CH2Cl2, rt. 24 h; (c) 1.5 equiv. potassium bicarbonate and 0.1 equiv. sodium iodide in DMF, 80 °C, 16 h.
Scheme 5.
Synthesis for compounds L13a-d and L15a-d containing PEG linker motifs. (a) 1.0 equiv. tert-butyl acrylate and 0.04 equiv. Triton B solution (40 % in water), rt, 120 h; (b) 1.5 equiv. 3.0 equiv. TEA and 0.1 equiv. DMAP in CH2Cl2, rt. 24 h; (c) 1.5 equiv. potassium bicarbonate and 0.1 equiv. sodium iodide in DMF, 80 °C, 16 h.
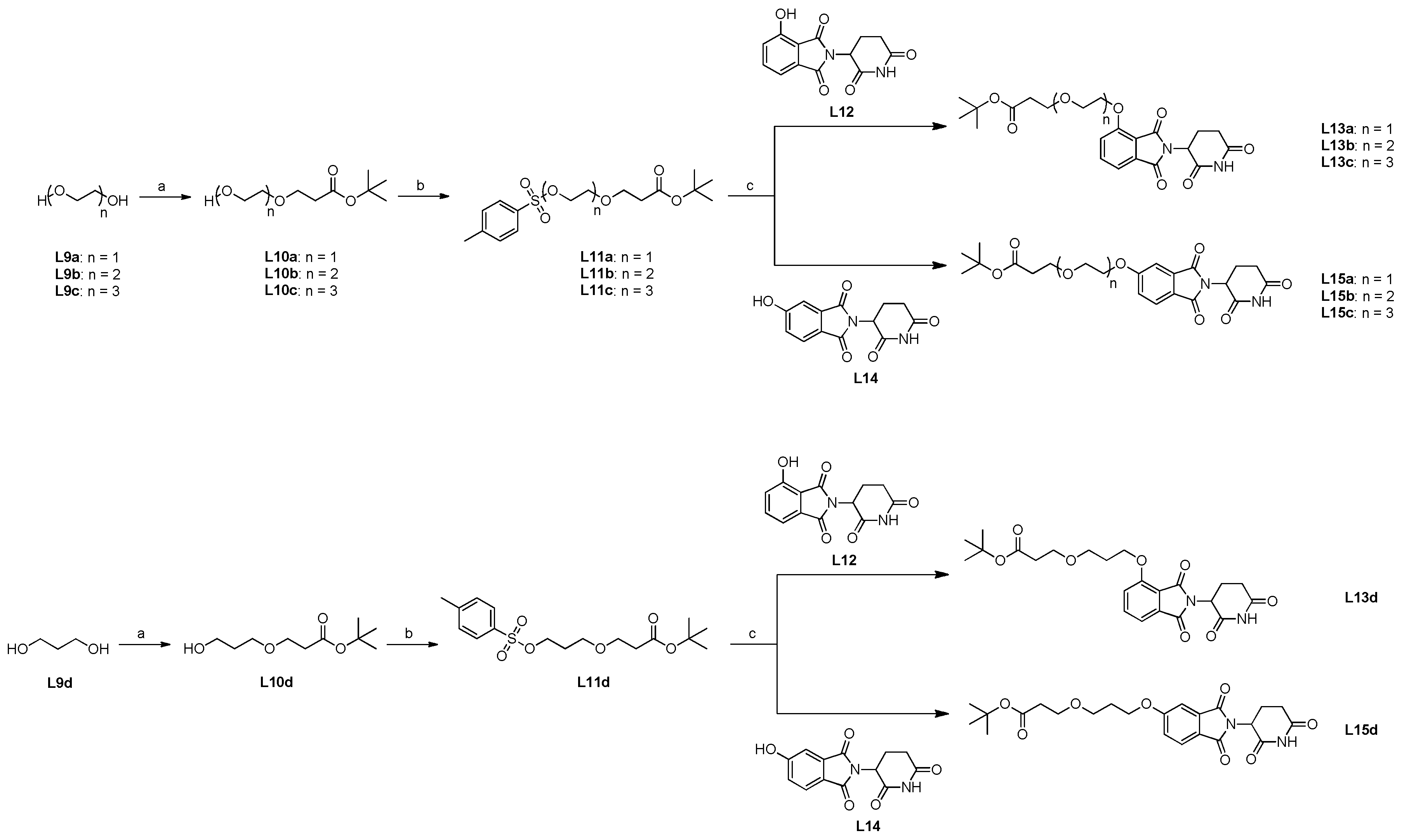
SCHEME 6.
Synthesis for the second and third set of PROTAC compounds P3a-d and P4a-d containing PEG linker motifs. (a) (i) 1 mL TFA in CH2Cl2, rt, 1 h; (ii)1.0 equiv. EDC x HCl, 0.1 equiv. HOBt, 1.0 equiv. DMAP and 5.0 equiv. DIPEA in MeCN, rt, 48 h.
SCHEME 6.
Synthesis for the second and third set of PROTAC compounds P3a-d and P4a-d containing PEG linker motifs. (a) (i) 1 mL TFA in CH2Cl2, rt, 1 h; (ii)1.0 equiv. EDC x HCl, 0.1 equiv. HOBt, 1.0 equiv. DMAP and 5.0 equiv. DIPEA in MeCN, rt, 48 h.
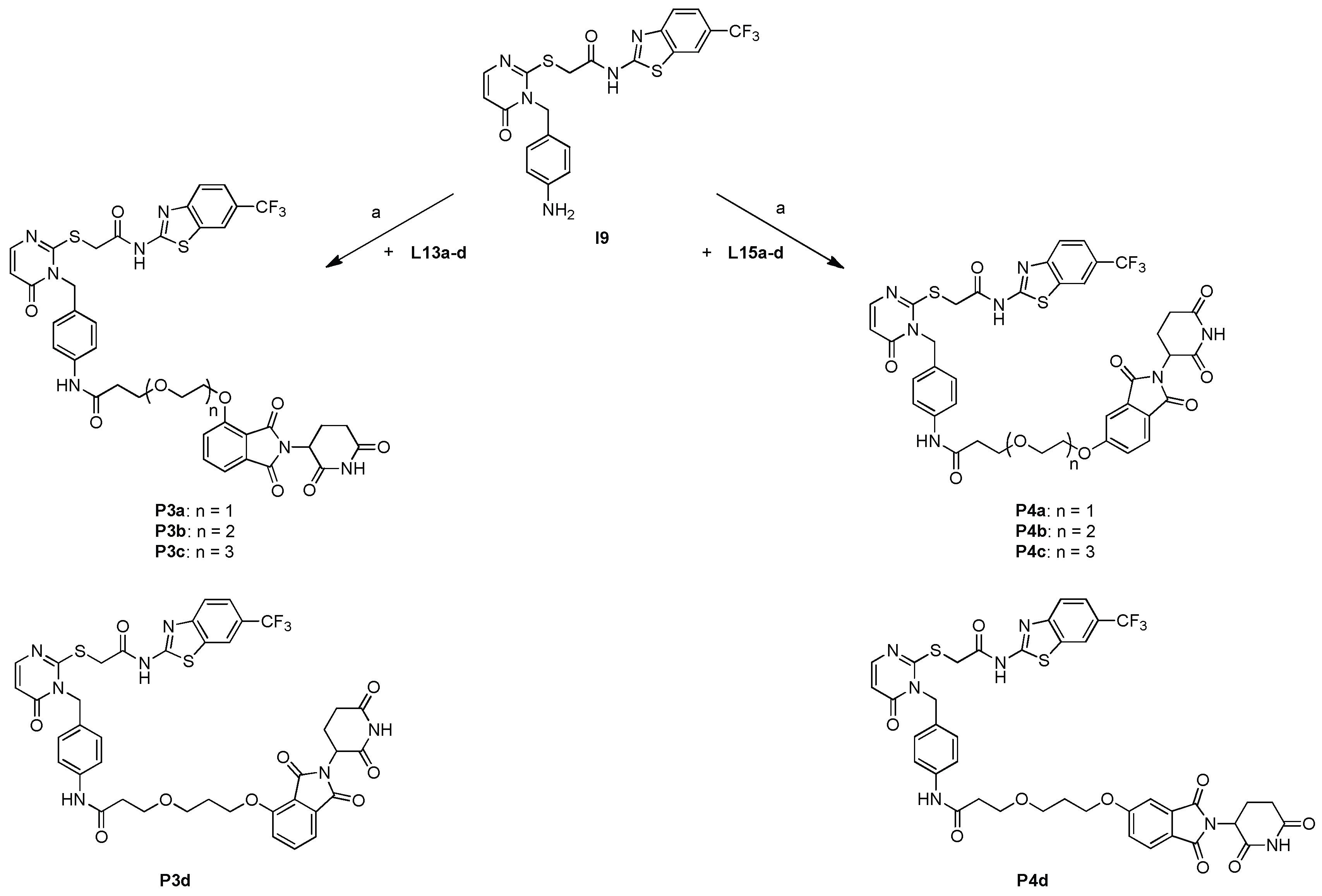
FIGURE 4.
Dose response of PROTAC compounds P1b, P1c, P1d & P2d at 0.5 and 5 μM. Wildtype (WT) U2OS cells were treated with DMSO, I9, SMI 1, or the indicated PROTACs at 0.5 and 5 μM for 24 h prior to lysis. Extracts (20 µg) were resolved by 4-20% Bis-Tris pre-cast SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. GAPDH was employed as a loading control. Extracts from DLD-1 CSNK1A1-/-, CSNK1D-/- and CSNK1E-/- cells were included to validate CK1α, CK1δ and CK1ε antibodies respectively. All blots are representative of three independent biological repeats.
FIGURE 4.
Dose response of PROTAC compounds P1b, P1c, P1d & P2d at 0.5 and 5 μM. Wildtype (WT) U2OS cells were treated with DMSO, I9, SMI 1, or the indicated PROTACs at 0.5 and 5 μM for 24 h prior to lysis. Extracts (20 µg) were resolved by 4-20% Bis-Tris pre-cast SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. GAPDH was employed as a loading control. Extracts from DLD-1 CSNK1A1-/-, CSNK1D-/- and CSNK1E-/- cells were included to validate CK1α, CK1δ and CK1ε antibodies respectively. All blots are representative of three independent biological repeats.
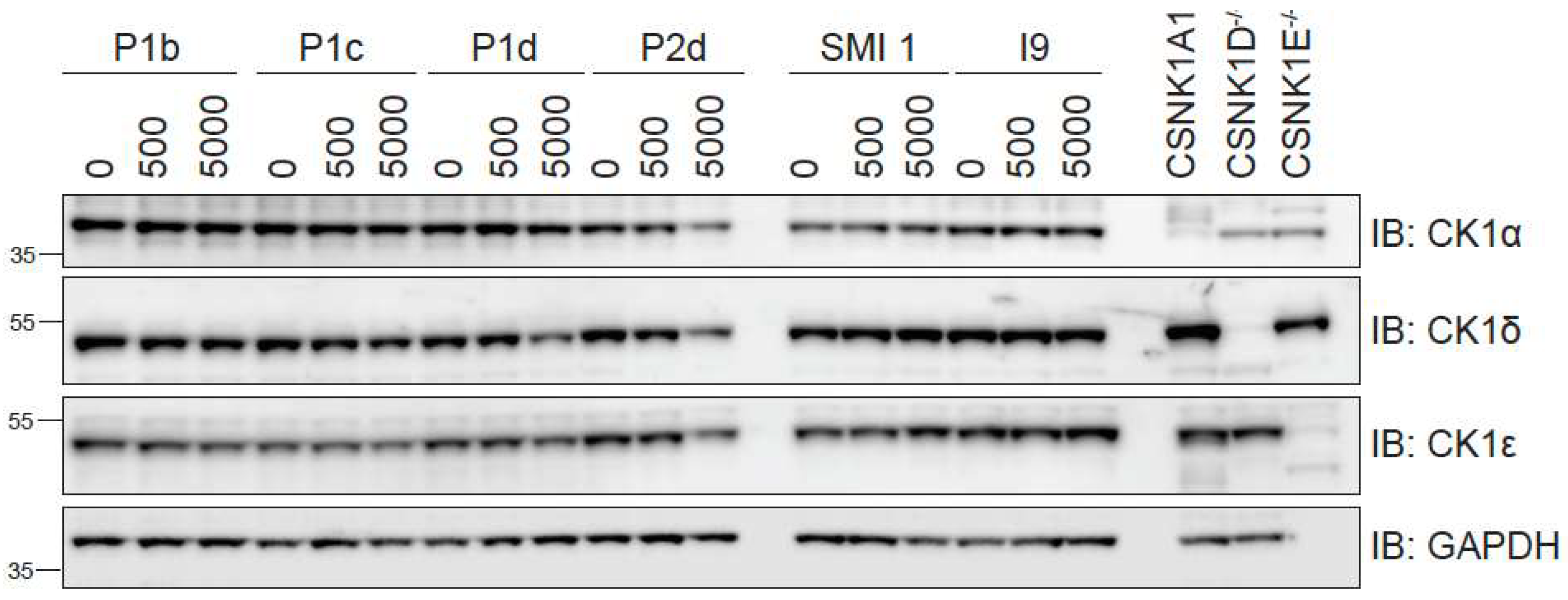
FIGURE 5.
Dose response of all PROTAC compounds at 1 and 5 μM. Wildtype (WT) U2OS cells were treated with DMSO, reference inhibitor I9 at 1 μM, or indicated PROTACs (P1d to P4d) at 1 and 5 μM for 24 h prior to lysis. Extracts (20 µg) were resolved by 4-20% Bis-Tris pre-cast SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. GAPDH was employed as a loading control. All blots are representative of three independent biological repeats.
FIGURE 5.
Dose response of all PROTAC compounds at 1 and 5 μM. Wildtype (WT) U2OS cells were treated with DMSO, reference inhibitor I9 at 1 μM, or indicated PROTACs (P1d to P4d) at 1 and 5 μM for 24 h prior to lysis. Extracts (20 µg) were resolved by 4-20% Bis-Tris pre-cast SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. GAPDH was employed as a loading control. All blots are representative of three independent biological repeats.
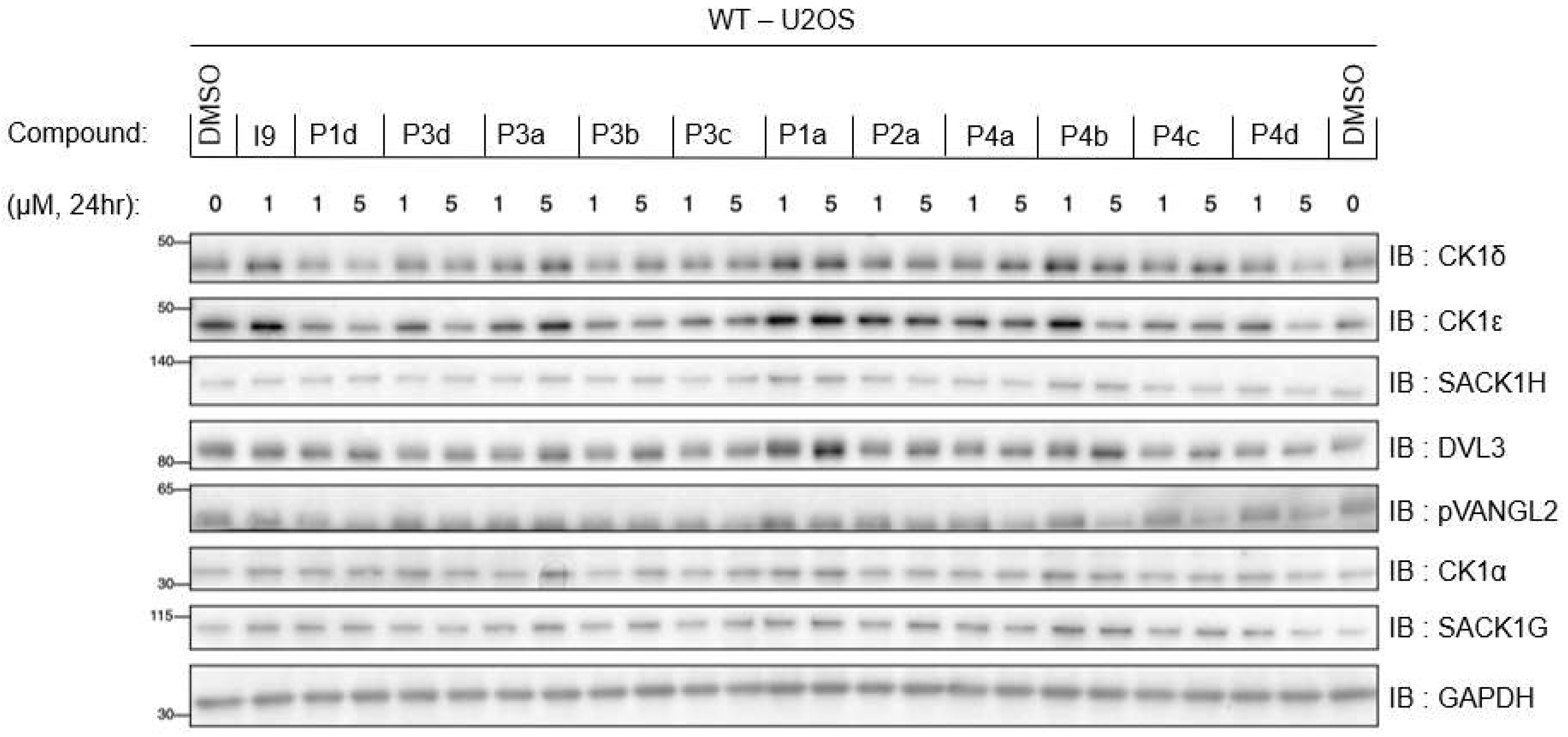
FIGURE 6.
In vitro IC50 determination for the indicated compounds in vitro against CK1δ (A) and CK1α (B) kinase activity using a peptide substrate. All assays performed by the MRC PPU International Centre for Kinase Profiling.
FIGURE 6.
In vitro IC50 determination for the indicated compounds in vitro against CK1δ (A) and CK1α (B) kinase activity using a peptide substrate. All assays performed by the MRC PPU International Centre for Kinase Profiling.
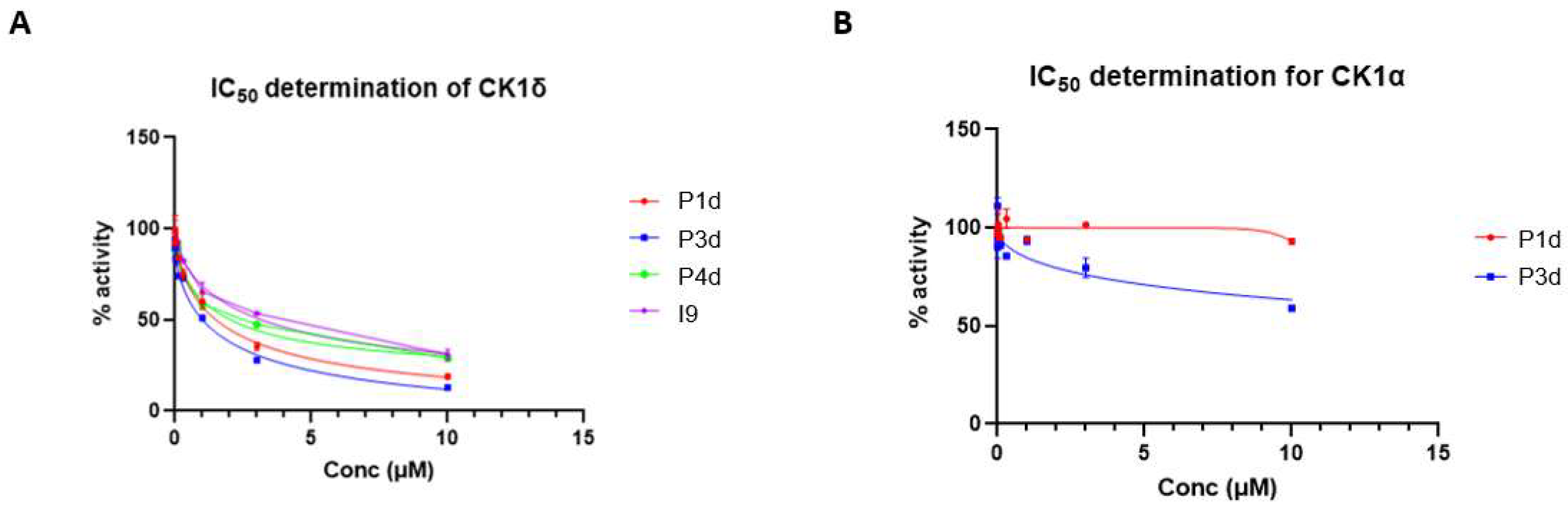
FIGURE 7.
Dose response of compound P1d at 0-10μM in various cell lines. (A) Wildtype (WT) U2OS, and (B) A549 adenocarcinoma cells were treated with DMSO or P1d (from 0.1-10 µM) for 24 h prior to lysis. Extracts (20 µg) were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. All blots are representative of three independent biological repeats.
FIGURE 7.
Dose response of compound P1d at 0-10μM in various cell lines. (A) Wildtype (WT) U2OS, and (B) A549 adenocarcinoma cells were treated with DMSO or P1d (from 0.1-10 µM) for 24 h prior to lysis. Extracts (20 µg) were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. All blots are representative of three independent biological repeats.
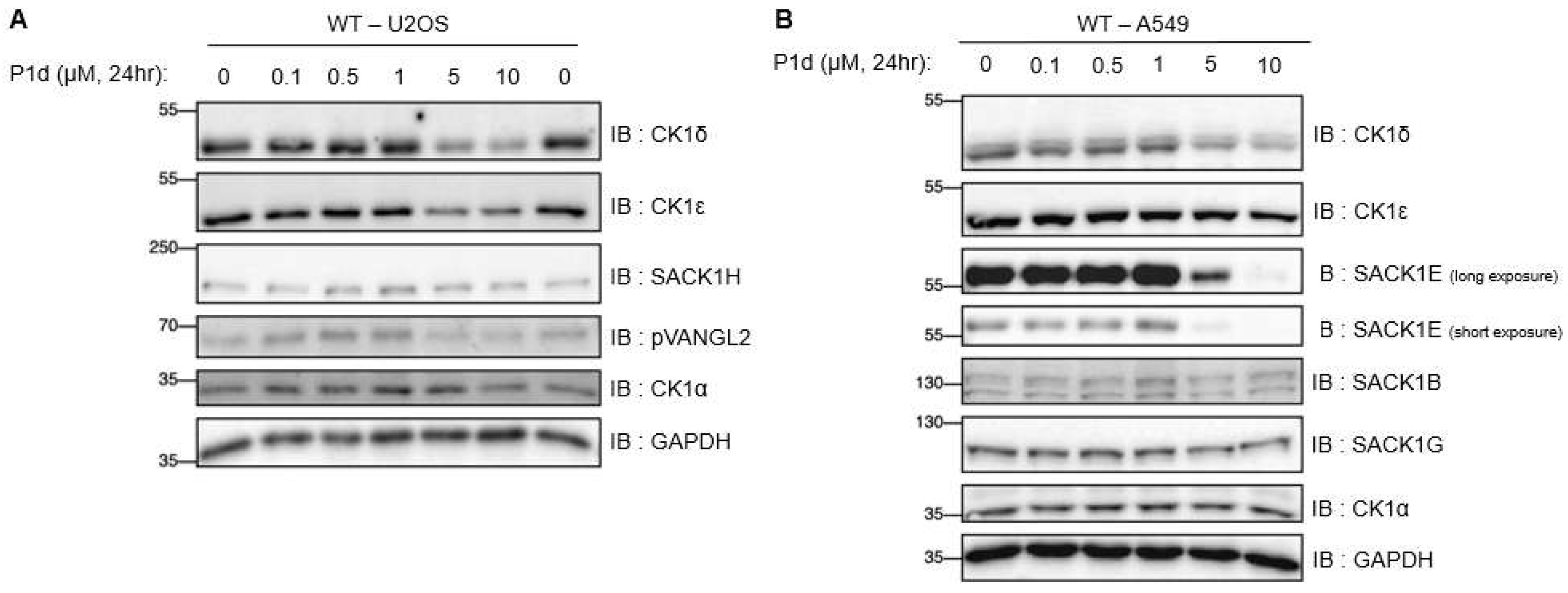
FIGURE 8.
Kinetic analysis of CK1δ/ε degradation by compound P1d at 5 μM. Wildtype (WT) U2OS cells were treated with DMSO, or 5 μM P1d for the indicated times prior to lysis. Extracts (20 µg) were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. All blots are representative of three independent biological repeats.
FIGURE 8.
Kinetic analysis of CK1δ/ε degradation by compound P1d at 5 μM. Wildtype (WT) U2OS cells were treated with DMSO, or 5 μM P1d for the indicated times prior to lysis. Extracts (20 µg) were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. All blots are representative of three independent biological repeats.
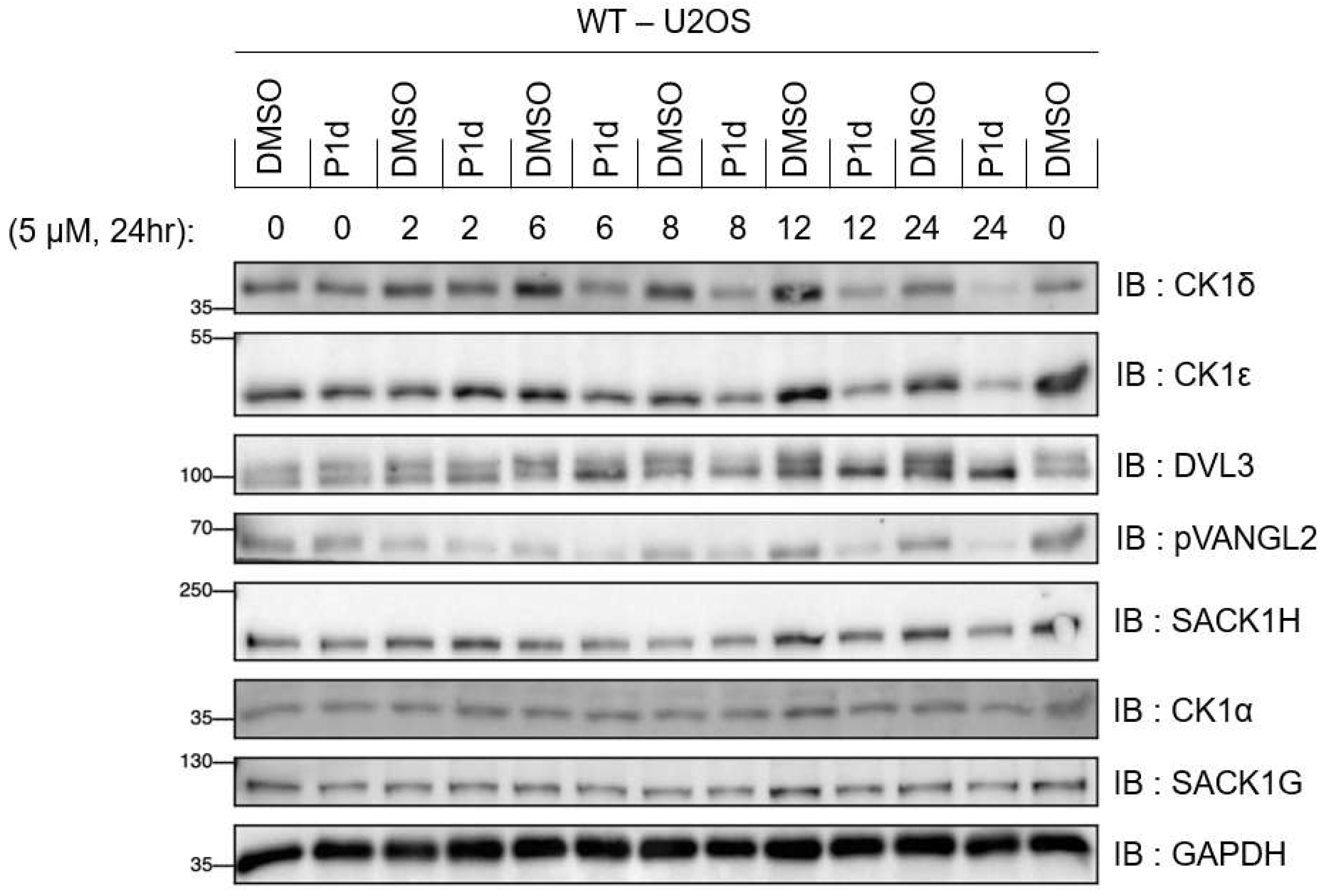
FIGURE 9.
Mode of E3 ligase action compound P1d at 5 and 10μM. A) Wildtype (WT) or CRBN-knock out (CRBN - KO) U2OS cells were treated with DMSO, or P1d at 5 and 10 µM for 24 h prior to lysis. (B) WT U2OS cells were treated with DMSO, or P1d at 5 µM without or together with 1 µM MLN4924 (NAE1 inhibitor) or 20 µM of MG132 (proteasome inhibitor). For A & B, extracts (20 µg) were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. All blots are representative of three independent biological repeats.
FIGURE 9.
Mode of E3 ligase action compound P1d at 5 and 10μM. A) Wildtype (WT) or CRBN-knock out (CRBN - KO) U2OS cells were treated with DMSO, or P1d at 5 and 10 µM for 24 h prior to lysis. (B) WT U2OS cells were treated with DMSO, or P1d at 5 µM without or together with 1 µM MLN4924 (NAE1 inhibitor) or 20 µM of MG132 (proteasome inhibitor). For A & B, extracts (20 µg) were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and immunoblots were performed with the indicated antibodies. All blots are representative of three independent biological repeats.

Table 1.
Employed conditions in order to try to generate compounds P2b-c.
| I9 | L8b-c | Solvent | Base (5 equiv.) |
Coupling Reagents | Temperature [° C] | Reaction Time | Additive |
|---|---|---|---|---|---|---|---|
| 1 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | DMSO | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | DMF | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | THF | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 25 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | Potassium carbonate | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | TEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | DBU | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | Mole sieve (3 Å) |
| 2 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 2 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | - |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | Glutarimide (5 equiv.) |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | EDC, HOBT, DMAP | 40 °C | 48 h | Glutarimide (10 equiv.) |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | HATU | 40 °C | 5 h | - |
| 1 equiv. | 1 equiv. | MeCN | Potassium carbonate | HATU | 40 °C | 5 h | - |
| 1 equiv. | 1 equiv. | MeCN | TEA | HATU | 40 °C | 5 h | - |
| 1 equiv. | 1 equiv. | MeCN | DBU | HATU | 40 °C | 5 h | - |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | HATU | 40 °C | 5 h | Mole sieve (3 Å) |
| 1 equiv. | 1 equiv. | MeCN | DIPEA | HATU | 70 °C | 5 h | - |
Table 1.
Overview of Compounds and Corresponding Linker Lengths.
| PROTAC | Linker length (Å)1 |
|---|---|
| P1a | 4.179 |
| P1b | 4.991 |
| P1c | 6.428 |
| P1d | 7.413 |
| P2a | 4.179 |
| P2d | 7.413 |
| P3a | 7.103 |
| P3b | 10.712 |
| P3c | 14.062 |
| P3d | 8.492 |
| P4a | 7.103 |
| P4b | 10.712 |
| P4c | 14.062 |
| P4d | 8.492 |
1 Measured linker lengths between the amide nitrogen connecting the linker to the inhibitor and the nitrogen or oxygen atom linking the linker to the thalidomide derivative. Linker lengths were measured based on 3D molecular models of the corresponding PROTACs, constructed and energy-minimized using Chem3D Pro 12.0.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



