Submitted:
27 October 2025
Posted:
05 November 2025
You are already at the latest version
Abstract
Iron is essential in all cells, but paradoxically accumulates in various regions across neurodegenerative diseases including Multiple Sclerosis, Alzheimer’s disease, traumatic brain injuries, frontotemporal dementia, Amyotrophic Lateral Sclerosis, stroke, and Lewy bodies associated with Parkinson’s. Furthermore, neurodegenerative pathologies frequently co-exist or compound; when such pathologies threshold into clinical progression across (overlapping) neurodegenerative diseases remain unexplained. We propose a new unifying theory: localized iron accumulation facilitates chronic neurodegeneration, wherein location-specific and temporal iron accumulation result in individual and concurrent neurodegenerative diseases. Excess ferrous iron can trigger ferroptosis, while ferric iron stored in ferritin involves cellular senescence: complementary and self-sustaining processes promoting oxidative stress, mitochondrial and endolysosomal dysfunction, and other hallmarks involved in chronic neurodegeneration. Finally, our proposed ferroptotic-senescence axis novelly accounts for both singular and compound proteinopathies. Our theory highlights important, novel priorities for diagnostics to screen for concurrent neurodegenerative diseases, and therapeutics for targeting iron-driven cell death and senescence.
Keywords:
Iron
; Chronic Neurodegeneration
; Neurodegenerative diseases
; Compound pathologies
; Mixed Diseases
; Ferroptosis
; Cellular senescence
Main
Despite neurodegenerative diseases resulting in millions suffering worldwide, treatments remain elusive. Extensive work has described many clinical, imaging, and neuropathological hallmarks of individual neurodegenerative diseases, but concurrent pathological findings shared between entities have received relatively little attention. Yet, patient investigations and post-mortem samples mostly present with mixed pathologies that co-exist more frequently than random chance[1,2]. This causes difficulties disentangling and diagnosing neurodegenerative diseases. Lewy body diseases (LBD) frequently demonstrate mixed aggregates of Alzheimer’s disease (AD)-associated amyloid-beta (Ab), neurofibrillary pathology (i.e., phosphorylated tau (p-tau) aggregates), and Parkinson’s disease (PD) associated phosphorylated alpha-synuclein (p-a-syn)[1,2,3].
Vascular contributions to cognitive impairment and dementia (VCID) involve cortical and subcortical infarction, lacunes, cerebral microbleeds and superficial siderosis, perivascular space enlargement, cerebral amyloid angiopathy (CAA), and white matter hyperintensities (WMH) visible on MRI; frequently resulting vascular dementia is difficult to disentangle from AD-associated proteinopathies[4]. Cerebrovascular pathology significantly acts as a risk and contribution to other neurodegenerative diseases[5]. WMH also arise in multiple sclerosis (MS)[6,7], and p-tau accumulation occurs in non-AD tauopathies including traumatic brain injuries (TBI), chronic traumatic encephalopathy (CTE), and progressive supranuclear palsy (PSP)[8]. How can these overlapping findings be reconciled mechanistically, yet differentiable? These problems extend to symptomology. Cognitive impairment is associated with AD. Yet, cognitive impairment frequently occurs in diseases without significant Ab burden including MS, Huntington’s disease (HD), and multiple system atrophy (MSA)[9,10,11].
Empirically studying these questions remains difficult. Still, understanding why disease etiologies inadvertently compound critically highlights gaps towards how and why these diseases (co-)occur. If a theoretical framework better parsimoniously reconciles these questions, downstream explanations and predictions can further improve treatments for neurodegenerative diseases. Notably, localized iron accumulation within the brain occurs in disease-associated cells and numerous neurodegenerative pathologies (Figure 1)[12,13,14,15]. This localized iron is explicitly different than systemic iron dysregulation disorders (ex. transfusion related iron overload or hereditary hemochromatosis); systemic iron dysregulation proportionally damages more brain-peripheral organ including the liver, wherein peripheral immune cells and the blood-brain barrier can act as ‘first-pass’ factors in absorbing and weakening potential significant effects of iron toxicity in chronic neurodegeneration.
Although not every MRI protocol is equally sensitive in assessing iron, chronic neurodegenerative diseases correlate well with increased localized iron accumulation[12,13,16,17,18,19,20]. Localized iron accumulation also independently predicts cognitive impairment and other symptomology[13,21,22,23], and localizes within the aforementioned neurodegenerative pathologies (even in other neurological diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), epilepsy, spinal cord injuries (SCI), and rarer genetic neuroferritinopathies)[24,25,26,27,28,29,30,31]. Notably, iron accumulation in the nervous system does not consistently correlate with plasma iron measures; systemic iron accumulation in disorders of iron-handling also do not reflect brain iron stores[32].
Localized iron accumulation is not the sole determinant of disease-specific pathologies. But does it correlate with disease progression, or does it also causally contribute to (co-existing) progression in these diseases? If so, how does this precisely operate? Does localized iron accumulation explain synergistic and concurrent disease pathologies? Finally, how does localized iron accumulation discern specific neurodegenerative diseases, while also being a significant driver of chronic neurodegeneration overall?
We propose that local iron accumulation causally facilitates chronic neurodegeneration. The spatiotemporal manner (i.e., location-specific, temporal spread from one or more regions to subsequent areas) of localized iron accumulation is necessarily involved in chronic neurodegeneration; spatiotemporal progression and combinations of localized iron accumulation result in progression for (concurrent) neurodegenerative diseases. We novelly integrate that localized iron accumulation inevitably facilitates numerous neurodegenerative hallmarks via (i) accelerating oxidative stress and ferroptosis cell death, and (ii) ferritin-mediated senescence that initially delays ferroptosis, but ultimately exacerbates disease progression. We then establish how localized iron accumulation amplifies frequent, mixed pathologies (including compound proteinopathies).
Neurodegenerative pathologies chronically converge into oxidative stress, ferroptosis, and ferrous iron accumulation
Numerous studies pinpoint ferroptosis (i.e., oxidized, ferrous iron (Fe2+)-involved cell death) as significantly contributing to chronic neurodegeneration (Figure 2). As interventional studies and reviews supporting that iron accumulation drives neurodegenerative pathologies (rather than only being a consequence) are established, we will cite more comprehensive literature here[13,16,33,34,35,36]. Our efforts will focus on theory streamlining iron imbalances occurring within the cell, beginning with how excessive, Fe2+-catalyzed oxidative stress causes ferroptosis.
Regardless of specific neurodegenerative pathology, chronic neurodegeneration converges into reactive oxygen and nitrogen species (ROS) formation. When ROS creation overwhelms anti-oxidant systems, oxidative stress arises leading to cellular damage; this occurs in chronic neurodegeneration, due to disease-associated signalling (ex. pro-inflammatory cytokines, cGAS-STING), endogenous creation and/or increasing burden of disease pathologies[3,37]. Such pathologies include glutamatergic excitotoxicity, myelin debris, protein aggregates (including those of Ab, p-tau, and/or p-a-syn), DNA-damaging toxins (ex. manganese), and/or overwhelming iron exposure (whether created by cerebrovascular pathology and/or pre-existing ferroptosis)[3,8,38,39,40,41,42]. Finally, prominent TDP-43 deposition correlates with iron burden across FTD variants, LATE, and AD[43,44,45].
Phagocytosing cells and external (protein) aggregates is metabolically exhausting, and also induces oxidative stress[46]. Additionally, oxidative stress induces mitochondrial dysfunction[41,47,48]. As mitochondria metabolize iron for healthy cell functioning, mitochondrial damage results in further Fe2+ release; oxidative stress and mitochondrial dysfunction engage in positive feedback loops[47,48]. Here, exacerbated mitochondrial dysfunction acts as a critical determinant towards whether cells undergo ferroptosis[41,42].
Excessive intracellular Fe2+ levels comprise the Fenton reaction, wherein Fe2+ reacts with H2O2 to generate potent ROS[33]. Although mitochondria damage significantly contributes to the LIP and sufficient Fe2+ for ferroptosis[41,42], other mechanisms also contribute to increased Fe2+. DMT1 imports Fe2+ from lysosomes or externally at the cell membrane, and cytosolic/lysosomal STEAP3 converts Fe3+ into Fe2+ available for the LIP[16,33]. Fe3+ is also internalized by transferrin and other receptors (ex. CD163) into a cell or as ferritin-bound iron, and can be later converted into Fe2+ for the LIP (ex. via STEAP3 and/or ferritinophagy). Oxidative stress only arises when ROS creation outweighs anti-oxidant systems; while further signalling mechanisms are detailed comprehensively elsewhere[16,33], these include primary System Xc- involving the glutathione-GPX4 system and FSP1-mediated CoQ10 redox reactions (Figure 2), alongside exogenous iron chelators.
Available iron for metabolic use (i.e., labile iron pool (LIP)) vs iron efflux must also be considered for ferroptosis. Transmembrane ferroportin exports Fe2+; Fe2+ can be converted and stored as ferric iron (Fe3+) via ferritin (made of heavy (FTH) and light chain (FTL) subunits)[33,49]. In iron overload, excess ferritin-bound iron can be secreted via lysosomal processing and extracellular vesicles (EV)[33,50,51] (Figure 3). However, ferroptosis requires ferritinophagy to occur, wherein NCOA4 facilitates ferritin degradation; this releases Fe3+ back into the cell to eventually be converted into more oxidizing Fe2+ (Figure 2). This is due to ROS also inducing autophagy[52], activating downstream NCOA4, subsequent autophagy-mediated ferritin degradation, and further Fe2+ creation to sufficiently induce ferroptosis[33,53]. If iron-dependent oxidative stress outweighs anti-ferroptosis systems (via LIP-involving Fe2+, ferritinophagy, and overwhelming ROS levels), overwhelming lipid peroxidation disintegrates the cell membrane to induce ferroptosis[16,33].
Finally, Fe2+ directly facilitates protein aggregates including Aβ, p-tau, SOD1, and α-syn[34]. Even if pathology aggregation precedes significant iron accumulation, iron accumulation significantly aggravates greater pathology formation and seeding[13,16,33,34]. As organisms cannot be completely depleted of iron and survive, localized iron accumulation is argued here as one inevitable, causal progressor in chronic neurodegenerative diseases. Altogether, Fe2+ buildup promotes multiple neurodegenerative pathologies and mitochondrial dysfunction[12,18,19,20,34,36,48,54].
Because all cells require iron to function, oxidative stress, ferroptosis, and Fe2+ accumulation inevitably occurs within any neurodegenerative disease. We further propose that excess ferroptosis is a causal progressor in these neurodegenerative diseases (Figure 2). Beyond theoretical rationales, studies support that excessive ferroptosis facilitates or exacerbates neurodegeneration caused by disease pathologies[16,33,35,36]. Ferroptosis inhibitors do not completely test if iron accumulation drives chronic neurodegeneration, as they do not address detrimental effects of ferritin-involved senescence. Still, ferroptosis inhibition has been shown to slow both the underlying pathology and the resulting symptom decline across numerous neurodegenerative disease models, particularly when implemented early in disease trajectories[16,19,35,55,56,57].
Chronic neurodegenerative pathologies induce senescence when averting ferroptosis and ferritinophagy
Fe2+ accumulation involves ferroptosis. Yet, a percentage of cells with accumulated iron can resist cell death, exhibit enhanced survival in-vitro without exposure to potent ferroptosis inducers (ex. erastin or liproxstatin-1), and persist to late-stage disease progression[36,56,58,59,60]. Cellular iron overload can induce cellular senescence instead of ferroptosis, with senescent cells showing ferroptosis resistance[14,54,58,61,62,63]. What differentiates these outcomes is currently not well explained[16], nor the relation between senescent cells, iron accumulation, and chronic neurodegeneration. We novelly reconcile these paradoxes by addressing Fe2+ (favouring ferroptosis) versus Fe3+ (favouring cellular senescence) accumulation. We first overview how senescence induction is initiated to avert ferroptosis, before explaining that Fe3+-based accumulation (including via senescent phenotypes) causally facilitates multiple neurodegenerative hallmarks and compound pathology progression.
Senescence is a functional state characterized by (i) cell cycle arrest and death resistance, with (ii) accompanying organelle dysfunction[3,64]. Senescence induction and particular markers vary across cell types and disease context; nonetheless, all senescent cells arise from sublethal stressors[64,65,66]. Cell cycle arrest (often assessed by p16INK4a or p21 positivity[3,64]) in senescent cells also present with organelle impairment. This includes buildup of endolysosomal products such as lipofuscin, DNA damage (marked by γH2A.X), and mitochondrial damage leading into mitochondrial DNA (mtDNA) leakage; this leads to a functional senescence-associated secretory phenotype (SASP)[67,68].
The SASP involves increased cGAS-STING signalling and downstream type I IFN secretion, MMP release, and other mediators varying across cell types[64]. While more disease-context pathologies and explanations will be given regarding specific cell types below, SASP regulators also include C/EBP, NF-kb, and HMGB1[3,64]. Finally, although senescence initially acts likely both an anti-death and anti-cancer mechanism[3,64], senescence buildup is increased over time; senolytic drugs (i.e., selectively killing senescent cells) increase healthspan and inhibit chronic disease conditions in mouse models[3,69,70].
Sublethal levels of cell stressors induce senescence[58,66,68]. These stressors include neurodegenerative pathologies observed in patients (Figure 1, 3): internalized creation and/or increased oxidative stress, myelin debris, proteinopathies (including Ab, p-tau, and/or p-a-syn), DNA damage, mitochondrial damage, and/or internalized Fe2+ accumulation irrespective of other confirmed pathologies[3,54,55,58,61,62,68,71,72,73,74,75,76,77,78,79,80,81,82]. Phagocytosing cells and external (protein) aggregates generates oxidative stress[46], which induces sublethal stress and subsequent senescence through mitochondrial damage[79]. Finally, while iron overload can induce mitochondrial damage and senescence, oxidative stress (even when not due to initial, external iron overload) can induce mitochondrial-dependent damage and intracellular Fe2+ accumulation[47,48].
How do cells avert ferroptosis arising out of neurodegenerative pathologies, and instead enter senescence to survive sublethal stressors? In in-vitro conditions without lethal iron overload or exposure to potent ferroptosis inductors, (i) converting Fe2+ into Fe3+, and (ii) sequestering Fe3+ into ferritin allows cells to become senescent, yet resistant to ferroptosis[14,36,54,58,63]. Explicitly, sufficiently inhibiting ferritinophagy allows Fe2+ sequestering into Fe3+ accumulation through ferritin. This averts ferroptosis and sustains cell survival[14,54,83] (Figure 3). While ferritinophagy inhibition occurs, mitochondrial damage and mtDNA release via oxidative stress activates signalling pathways inducing senescence (to avert cell death). Specific signalling cascade likely involves upstream, ROS-mediated, DNA damage activating p21 (into cell cycle arrest) and BNIP3, which alongside other ROS creation (Figure 2); BNIP3 leads into mitochondrial fatty acid oxidation, eventually activating p16INK4a, Rb, and downstream cell cycle arrest inducing senescence[79] (Figure 3). Finally, cell cycle arrest induces lipid droplet formation to resist ferroptosis and limit excessive lipid peroxidation[84].
Further explaining ferritinophagy inhibition, oxidative stress also oxidizes cholesterol (i.e., oxysterols) recognized by LXR and subsequent LXR-induced, APOE upregulation[85,86]. APOE is not only upregulated in response to oxidative stress (including Fe2+-catalyzed ROS generation), but also inhibits ferritinophagy via the PI3K/AKT/mTOR pathway[83]. APOE also facilitates senescence[87,88,89], and is often secreted and uptaken by nearby cells[3,90]. This further explains APOE involvement broadly in chronic neurodegeneration and across numerous neurodegenerative diseases[91]: excessive APOE responds to oxidative stress (via at least oxysterols), inhibits ferritinophagy, and promotes senescence induction[3,83,87,88,89,90].
Cells possess IRE-IRP (iron response element – iron regulatory protein) systems detecting and responding to Fe2+. When exposed to non-lethal levels of ferroptosis-based stressors (Figure 2), IRP can bind to ferritin to inhibit ferritinophagy as a response to high LIP and available Fe2+ (to indirectly promote Fe2+ conversion into Fe3+-sequestered ferritin)[49,51,54]. As senescent cells sequester more iron as Fe3+ bound-ferritin[14,54,58,62], is ferritin-Fe3+ considered a marker enriched in senescence? Although explanations will follow, briefly yes; senescent nervous system cell types that are functionally senescent (and not dead) and accumulate endolysosomal products should also accumulate iron. Ferritin enrichment is likely difficult to precisely discern in nervous system cell types with homeostatic iron abundance (ex. oligodendrocytes for myelin synthesis), but should be more distinguishable in neuronal and CNS immune cell populations that do not healthily accumulate iron.
Senescent cells with inhibited ferritinophagy and impaired endolysosomal systems have upregulated ferritin (leading into sequestered and unavailable Fe3+ that the IRE-IRP does not detect); this leads to these senescent cells absorbing greater free Fe2+ and sequestering more Fe3-bound ferritin than non-senescent cells[14,54,61,63]. In environments containing neurodegenerative disease pathologies and ferroptosis, the IRE-IRP system constantly detects uptaken and/or intracellular Fe2+ (thereby synthesizing further ferritin to sequester said Fe2+ into stored Fe3+)[49,51]; as senescent cells feature endolysosomal impairment, ferritin is unable to be effectively degraded and sustains positive feedback loops accumulating iron[14,54,62]. This ferritin synthesis can occur even when Fe2+ is not effectively converted into ferritin, resulting in senescent cells with (i) decreased ferritin-iron binding, albeit (ii) still with upregulated ferritin and total iron accumulation[58].
Finally, iron is also accumulated in lipofuscin; lipofuscin localizes to lysosomes, and lipofuscin-bound iron is an enriched marker in senescent cells[92]. This provides a complementary, yet synergistic paradigm synergizing with ferroptosis, addressing how localized iron accumulation can create chronically maladaptive senescent phenotypes and dysfunction beyond the LIP and oxidative stress (Figure 3).
Ferric iron-rich, senescent cells maladaptively accelerate neurodegenerative pathology burden
As senescent cells can uptake Fe2+ and accumulate Fe3-bound ferritin[54,61,63], these cells can act as a better, albeit short-term buffer against further ferroptosis than non-senescent cells. Yet, pathologies arising in neurodegenerative diseases require clearance. Nervous system cell types have differential ferritin and ferritinophagy activity under equivalent ferroptosis-based stress[93]. What roles do these cells play in chronic neurodegeneration? We explain here that (i) maladaptive responses by iron-accumulated, senescent cells causally progress neurodegenerative pathologies greater than that of either homeostatic or dead cells; further explanations will overview (ii) senescent neurons and CNS macrophages, particularly iron-rich microglia.
Senescent cells become endolysomally dysfunctional and contain impaired mitochondria[3,68,79]. Although genetic and sporadic factors vary per disease, a convergent feature observed across neurodegenerative disease progression involves aggregate buildup. These aggregates include myelin debris, proteinopathies (ex. Ab, p-tau, and/or p-a-syn), DNA damage markers, and mitochondrial damage; if endolysosomal performance were sufficiently functional, these aggregates should be cleared over time and neurodegenerative disease progression stopped. This does not occur in chronic neurodegeneration, and senescent cells further contribute to disease pathology propagation[3,54,55,58,61,62,68,71,72,73,74,75,76,77,78,79,80,81,82,94,95,96,97,98].
When senescent cells avert ferroptosis, Fe3+-bound ferritin accumulation can occur either in cytoplasm or within endolysosomal compartments. Although endolysosomal, Fe3+-ferritin accumulation has not been mechanistically elucidated within senescent cells, ongoing oxidative stress and lysosomal damage has been shown to induce ferritin accumulation[99]; overall increased ferritin levels (due to decreased ferritinophagy) may still involve increased lysosomal ferritin targeting, albeit without significant ferritin degradation due to IRP, APOE, and/or senescence-decreased lysosomal acidity[3,64,83,100]. Regardless, endolysosomal compartments also act as convergent or targeted hotspots for neurodegenerative pathologies. When lysosomal compartments become faulty (such as occurring within senescent cells), pH alterations favour singular and compound neurodegenerative pathology aggregation instead of healthy degradation[3,101].
Oxidative stress occurs in senescent cells, even after averting cell death[3,64,79]. Internalized neurodegenerative pathologies and surrounding disease environments (containing said pathologies) do not disappear when a senescent cell becomes endolysosomally impaired. Thus, two pathways may deal with overwhelming neurodegenerative pathology aggregates: (i) endolysosomal-facilitated degradation, which is poorly performed within senescent cells, or (ii) secretory release of internalized aggregates. (ii) is favoured in senescent cells, wherein internalized components (including mitochondrial components, type I IFNs, and protein aggregates) are secreted to nearby cells and induce dysfunction in non-senescent cells[3,68,70,71,72,74,76,77,78,88,102,103]. Sustained cGAS-STING signalling induces TFEB-mediated lysosomal biogenesis[104,105], whereby TFEB-upregulated lysosomes become dysfunctional in senescence[105]. Therefore, in senescent cells, increased and dysfunctional lysosomal numbers facilitate neurodegenerative pathologies to co-aggregate with iron (Figure 3), before being secreted by senescent cells to maladaptively induce chronic neurodegeneration. Furthermore, alongside contributions to oxidative stress via the Fenton reaction[33], Fe2+ also directly engages with and accelerates proteinopathy aggregation across multiple neurodegenerative diseases[12,34].
Although healthy neurons likely demonstrate less iron accumulation relative to glia[93], selectively-vulnerable neuronal populations are likely susceptible to both Fe2+-involved ferroptosis and Fe3+-involved senescence. Senescent iPSC-derived neurons (harbouring ferritin mutations) have been shown to aggregate both iron and ferritin in genetic neuroferritinopathy models, increased LIP, decreased NCOA4 (indicating reduced ferritin breakdown), and increased ferritin-iron storages despite poorer ferritin functionality; this resulted in these transgenic cells displaying greater iron accumulation, ferroptosis, and senescence[58]. Neurofibrillary tau tangles (NFT) with both p-tau and Lewy bodies (LB) are enriched in iron[20,59,60]; selectively vulnerable (i) excitatory neuronal populations (in tauopathies) and (ii) dopaminergic neurons (in α-synucleinopathies) responsible for creating these aggregates are enriched in senescent markers[3,96,98,106].
Iron-rich, immunosenescent cells exacerbate multiple neurodegenerative hallmarks and seed compound pathologies
Among cell types and different capacities to store iron, most evidence currently supports that iron-rich, CNS macrophages are neurodegeneration-associated. Across neurodegenerative diseases, CNS iron-rich immune cell enrichment has been detected via MRI, histology, and transcriptomics[6,7,19,43,44,107,108,109,110,111,112,113,114,115,116,117,118]. Ferritin-enriched microglia and CNS-infiltrating macrophages are senescence-enriched across both in-vitro, rodent, and post-mortem patient studies. While studies should further clarify their roles in chronic neurodegeneration, we will overview how CNS iron-rich immune cells (including microglia and CNS-infiltrating macrophages) are enriched in senescent phenotypes.
Single-nucleus RNA sequencing likely underrepresent non-nuclear FTL/FTH transcripts[119], and Fe3+-ferritin accumulation may involve greater buildup through ferritinophagy inhibition rather than increased FTH/FTL transcription. Regardless, patient microglia (or CNS-infiltrating macrophages) displaying hypertrophic morphologies and/or dystrophy (i.e., reduced morphology complexity with ferritin upregulation[3,118]) are ferritin-enriched[55,108,110,117,118], and ferritin-enriched, human microglia states are enriched in multiple senescence-associated genes[71,120,121,122]. Senescence markers and ferritin enrichment in human microglia have been confirmed across multiple diseases and studies; these include y.H2AX, p16INK4a+ and p21[55,71,121,122]. Combined staining has also been performed, wherein post-mortem MS brains, chronic active lesions are enriched with CDKN2A, FTL/FTH, and APOE microglia and CNS macrophages supporting senescent, iron-rich immunosenescent cells; the authors also confirmed protein (p16INK4a+, FTL+, APOE+) and lipofuscin staining at these sites[122]. Mouse microglial states with upregulated CDKN2A, FTL/FTH, and APOE are increased in older non-transgenic mice, supporting existence of a p16INK4a+ and Fe3+-ferritin enriched, senescent microglia population[70,97]; increased p16INK4a+ microglia proportions correlate with upregulated SA-β-gal+, FTL, and lowered Lamin B1 in mice chronically exposed to low-dose arsenic and resulting in cognitive impairment[123].
Human SV40 microglial cultures upregulate p16INK4a+ and ferritin, upon becoming senescent after sirtuin 5 knockdown[124]. Primary mouse and SV40 human microglial cultures exposed to iron overload display dystrophy with impaired organelle dysfunction, via increased endoplasmic reticulum stress and dysfunctional autophagy[125,126,127], additional senescence-associated markers[3,128]. Human iPSC-derived microglial cultures overloaded with iron exhibit several senescence-associated markers in increased oxidative stress, dysfunctional mitochondria accompanied by increased glycolysis, and impaired phagocytic clearance of extracellular aggregates indicating dysfunctional endolysosomal systems[129] (and serving as another hallmark in senescent microglia[72,130]).
Senescent CNS microglia (and likely other, infiltrating immune cells) are iron-enriched [3,55,70,71,97,120,121,123,131]. Microglia are also particularly susceptible towards becoming dysfunctional under iron overload[36,132]. Theoretically, CNS macrophages (particularly in microglia) accumulating iron can only have undergone (i) ferroptosis, (ii) senescence, or (iii) transient internalization of external, iron-rich objects (ex. phagocytosing erythrocytes). As these cells do not homeostatically accumulate Fe3+-ferritin (unlike oligodendrocytes)[36,132], CNS iron-rich macrophages should not display such ferritin enrichment if they completed ferroptosis (due to ferritinophagy[33,54,63]), nor have such enriched ferritin levels if they were non-maladaptively senescent. Excess iron-rich phagocytosis also facilitates chronic neurodegeneration (ex. blood vessel leakage leading into CNS erythrocyte infiltration and/or engulfing iron-rich materials); additionally, sustained phagocytosis of iron-rich debris, including myelin, results in increased senescent microglia populations[55,76,82,97]. As patient microglia across numerous neurodegenerative diseases have been shown to display Fe3+-ferritin enrichment[6,7,107,108,110,112,113,114,115,117,118,133], this completes a complimentary, theoretical explanation that iron-rich, CNS macrophages are enriched in senescence.
Immune cells phagocytose senescent cells[134]. Speculatively, even if other iron-rich, senescent cell types are created via sublethal pathology exposure, such cells may not excessively accumulate until nearby immune cells also become exhausted and senescent[3]. We additionally propose that iron-rich CNS immunosenescent buildup integrates multiple neurodegenerative hallmarks, and likely contributes to iron buildup correlating with disease progression[13,21,22,23]. Furthermore, as senescent CNS macrophages are iron-enriched[3,55,70,71,97,120,121,123,131], their functions in driving chronic neurodegeneration includes detrimental effects mediated by empirical studies researching both iron-rich and senescent microglia (or CNS infiltrating macrophage) populations.
Although senescent populations comprise a smaller percentage of all CNS macrophages, senescent cells disproportionally drive numerous neurodegenerative pathologies and consequent symptomatic progression[3,70,71,97,102,103,116,127,130,135,136,137,138,139,140,141,142,143] (Figure 3). In primary mouse cultures, (i) exposure to p-tau aggregates or neurons with p-tau aggregates induced microglial senescence, with impaired phagocytosis of further p-tau aggregates and increased MMP-3 secretions[72]; (ii) extended Aβ treatment induced senescence, with significantly greater MMP-2 and MMP-12 secretion[144]. In Aβ-overexpressing mice approximating AD, both MMP-3 and MMP-12 levels were also increased in treatment conditions preserving higher levels of senescent microglia[130,139]. TBI increased senescence microglial populations in aged mice, correlating with behavioural deficits (decreased motor functioning and increased anxiety-like behaviour) and increased MMP-9 levels[145]. As increased MMP levels are upregulated across multiple neurodegenerative diseases[146], and the SASP often enriches MMP release[64,94], iron-rich, CNS immunosenescent cells likely contribute to chronic neurodegeneration via exacerbated MMP release (Figure 3).
As iron-rich microglia display increased oxidative stress and impaired mitochondria[124,127,129], mitochondrial oxidation leads into cGAS-STING signalling[68], and senescent microglia sustain cGAS-STING signalling[71,103,140], iron-rich, immunosenescent cells should sustain cGAS-STING signalling and downstream type I IFN release. Although cGAS-STING signalling is beneficial in anti-viral responses[104], in chronic neurodegeneration, upregulated type I IFN release facilitates exacerbated synaptic loss (i.e., a classical neurodegenerative hallmark) in models with (i) significant Aβ or p-tau proteinopathies and (ii) simultaneously increased senescent microglial populations[71,102,130,147,148]. Additionally, sustained cGAS-STING signalling facilitates increased CNS pro-inflammatory states, synaptic loss (including via neuronal death), and spatial memory performance in aged, non-transgenic mice[103]. This outlines CNS iron-rich, immunosenescent cells as also causally facilitating cGAS-STING-mediated, chronic neurodegeneration.
Although senescent and iron-rich microglia displaying impaired endolysosomal systems[124,125,126,127,129], a primary role of CNS macrophages is to be competent phagocytes; i.e., to efficiently clear and degrade neurodegenerative pathologies[3,101]. As senescence impairs such clearance capabilities, iron-rich, CNS macrophages are predicted to not only (i) fail at degrading internalized neurodegenerative pathologies (i.e., contributing further to sustaining disease environments and chronic neurodegeneration), but to also instead (ii) maladaptively secrete aggregates. cGAS-STING signalling induces TFEB-mediated lysosomal biogenesis[104,105]; in combination with senescence-involved, endolysosomal impairment and immune cell roles in phagocytosing to a greater extent than other cell types, this (iii) unintendedly transforms iron-rich, CNS immunosenescent cells into initiators and accelerators of compound neurodegenerative pathologies. As explained below, compound pathologies are proposed to be secreted by iron-rich senescent cells. However, the reverse is not true; not every senescent cell must necessarily seed and secrete compound pathologies. Nonetheless, default patient presentation of concurrent neurodegenerative pathologies can now be theoretically explained[1,2], particularly via iron-rich, progressive CNS senescence.
Dystrophic microglia enrichment in patient brains likely occurs via initial p-tau pathology, and both patient correlations and preclinical models support that dystrophic microglia drive chronic neurodegeneration via accelerating the spread of both Aβ and p-tau aggregates[71,111]. Iron-rich, neuritic amyloid plaques also spread p-tau pathology faster than p-tau spread mediated via NFT, even in patient brains[31,149,150]. Finally, microglia directly secrete aggregates forming amyloid plaques[3,151]. As CNS immunosenescent cells should maladaptively secrete endolysosomal aggregates (due to failed degradation of such aggregates[3]), iron deposits within neuritic plaques can now be explained via lysosomal Fe3+-ferritin from these incompetent cells. Alternatively, these iron-rich cells and iron-positive neuritic plaques seed and drive compound proteinopathies facilitating chronic neurodegeneration.
These explanations further account for patient brains with LBD, containing amyloid plaques also with both p-α-syn and p-tau inclusions[152]. Although not yet investigated, we predict that iron-rich CNS immunosenescent cells were unable to fully degrade singular or mixed Aβ, p-tau, and/or p-α-syn; instead, these cells secreted these partially-digested proteins (forming amyloid plaques containing all three aggregates). Reinforcing these predictions, after non-transgenic mice were injected with primary mouse microglia pre-treated with Aβ, p16INK4a+ senescent microglia numbers were increased and accompanied multiple hallmarks of chronic neurodegeneration, including increased synaptic loss, increased pro-inflammatory cytokine levels of TNF-α, and aggregate buildups in sporadic p-tau and p-α-syn inclusions[141].
As iron-rich CNS macrophages comprise hypertrophic and/or dystrophic morphologies[3,110,112,116], this also explains why patients commonly display dystrophic and/or “activated” microglia around neurodegenerative pathologies[6,7,43,44,71,109,110,111,112,113,114,115,116]. If these iron-rich cells become senescent after sublethal exposures to these pathologies and/or exacerbate such pathologies (via seeding and secreting, partially-digested aggregates), this rationalizes their increased presence around neurodegenerative pathologies[3,55,110,111,117,120,143]. Dystrophic, ferritin-rich, and γH2A.X-positive (senescence-associated) microglia have also been found to surround both cerebrovascular injuries and WMH; these cells also contained both lipid droplets and myelin debris[55], likely as a consequence of becoming senescent via environment-dependent, overwhelming myelin phagocytosis. Notably, iron-rich, CNS immunosenescent cells and their proximity to damaging, neurodegenerative pathologies may differ from protective non-iron-rich states also localized to these pathologies. For example, homeostatic, non-senescent microglia creating protective amyloid plaques should not be iron-rich[151]. Creating such plaques limit Aβ aggregates from causing further damage to nearby cells, in contrast to iron-rich neuritic plaques that accelerate the spread of proteinopathy[31,149,150].
Senescence can be induced by different sublethal stressors. We predict that iron-rich, immunosenescent cells drive chronic neurodegeneration with further substates; the particular neurodegenerative pathologies they accelerate and spread will depend on the neurodegenerative pathologies present in their environment. This is supported via rodent models, wherein disease-associated microglia have been facilitated accelerating context-specific spreading of Aβ, p-tau, and/or p-α-syn (mixed) aggregates[3,71,111,125,126,141,143]. Finally, iron-rich microglia also may likely contribute to chronic neurodegeneration via promoting calcium-mediated, neuronal death. SV40 human microglial and BV2 mouse microglial cultures overloaded with iron release significantly higher levels of glutamate[126,135]; iPSC-derived, senescent microglial cultures secrete DLK1 ligands that initiate higher intracellular calcium peaks within iPSC-derived neurons[138].
Spatiotemporal iron accumulation is predicted to discern specific and concurrent diseases with clinical progression in chronic neurodegeneration
If localized Fe2+ and Fe3+ accumulation together drive singular and compound neurodegenerative pathologies, how can this theory discern specific diseases and subsequent clinical progression? Although localized iron accumulation is predicted to drive chronic neurodegeneration, each specific disease should follow a unique combination of spatiotemporally affected locations and consequent clinical etiology. In AD, significant p-tau pathology follows Braak staging. This is tracked as earlier burden in the entorhinal cortex and later significantly damaging the cerebral cortex, parallelling the cognitive decline trajectory of the disease[3,153]; this specific, spatiotemporal disease progression contrasts other forms of dementia significantly involving different CNS regions at different timepoints especially in early-mid disease progression[4]. PD may be isolated out via initial autonomic nervous system entry routes involving p-α-syn pathology (ex. via the olfactory bulb or vagus nerve), before involving the SNpc and motor symptoms[154,155]. This spatiotemporal specificity can further differentiate other disease trajectories including ALS, which involves earlier and specific iron accumulation (and specifically enriched iron microglia burden) in deeper motor cortex cortical layers[109].
We resolve these paradoxes by a novel postulate: Localized iron accumulation drives chronic neurodegeneration, with the spatiotemporal manner of this iron accumulation determining both specific vs concurrent disease etiologies and emergent, clinical progression (Figure 4). As ferroptosis, neurodegenerative pathologies, and ferritin-involved senescence all require local propagation to cause damage, localized iron accumulation can happen within one region and also start independently in a distal location. For example, vascular events inducing WMH can occur earlier in a person’s prefrontal cortex to induce both ferroptosis and iron-rich, senescent microglia[55]; while this lead into subtler cognitive deficits and VCID, such events can happen independently or in synergy with p-tau and CNS iron-rich senescence abundance occurring in the temporal cortex in their senior years[3,71,111]. Although further studies are required, we can apply a gross, anatomical simplification of this theory to a patient (i) beginning with WMH that goes to (ii) develop mixed AD and PD pathologies.
In AD, greater iron accumulation follows Braak staging and disease severity[113,114,115,153]. In PD, greater iron accumulation should occur in the autonomic nervous system prior to the SNpc[20,26,30]. Take a patient with mixed LBD, (iia) beginning with dementia and later gaining motor impairment vs (iib) evolving past initial parkinsonian symptoms with additional cognitive decline. This theory would predict that patient (iia) has now evolved to accumulate significantly greater localized iron accumulation in regions following AD Braak staging, while also accumulating more CNS iron in the SNpc; this constitutes AD evolving into DLB. Patient (iib) would present with PD with dementia or DLB (depending on disease progression speed), and should also involve greater iron accumulation across both the SNpc and AD Braak staging regions. Biologically, (iib) would involve earlier, greater iron accumulation in the SNpc (vs healthy, age-adjusted SNpc iron levels) relative to regions involved later in AD Braak staging (ex. parietal lobe).
Comparisons of greater vs lesser iron accumulation should be compared for regions between disease vs healthy individuals, not between disease-specific regions; certain locations may have higher homeostatic capacities for localized iron accumulation vs other regions before clinical emergence. Why certain regions are selectively vulnerable is not understood well, but may result from ratios of glia:neurons within each region[156], Speculatively, how much more iron glia and or neurons can tolerate neurodegenerative pathology occurrence in any region before undergoing localized iron accumulation (by ferroptosis or ferritin-enriched senescence) may dictate this selective vulnerability. Potential contributing factors include blood-brain barrier permeability, metabolic rate and susceptibility (involving greater ROS generation and/or iron usage in cells), resilience against neurodegenerative pathology exposure, and differential expression of iron transport proteins. Further insights into these aspects may better improve understanding localized iron accumulation as a causal contributor and therapeutic in chronic and concurrent neurodegeneration.
Overall, concurrent clinical progression can now be explained via concurrent iron accumulation across multiple locations (Figure 4). Explicitly, behavioural deficits may arise out of combined iron-accumulated regions working together; therefore, localized iron accumulation may be required in multiple regions before becoming clinically significant.
Discussion
Our paradigm-shift theory proposes that localized iron accumulation causally progresses chronic neurodegeneration, due to conserved, universal systems governing cellular oxidative stress, ferroptosis, and ferritin-involved senescence. We have explained and integrated how localized iron accumulation is not only involved in (i) contributing to specific neurodegenerative pathologies, but also importantly drives (ii) compound proteinopathies. Localized iron accumulation ultimately serves as a causal contributor to (iii) singular and co-existing chronic neurodegenerative diseases. Strengths of our theories include explaining such iron burden as a causal rather correlational driving factor in these diseases, explaining how and why compound pathologies frequently exist in a complex patient brain, and explain iron contributions to chronic neurodegeneration beyond ferroptosis (including account for ferroptosis-resistant senescence that still involves accumulating iron).
Our theory does not address if (i) acute symptomatic appearance is due to localized iron accumulation, nor cases where (ii) insufficient iron accumulation may contribute to clinical decline in diseases. These deserve future investigation. Explicitly, localized iron accumulation is not the only contributory factor towards chronic neurodegeneration. For instance, p-tau pathology still causally contributes to AD and TBI, APOE4/4 homozygote carriers contribute towards multiple neurodegenerative diseases including AD[90]. Even with such shared risk factors, comprising genetic and/or environmental causes, localized iron accumulation still simultaneously functions as an inevitable, causal driver in chronic, neurodegenerative diseases. Findings that could contradict our theory and current literature include decreased iron burden in regions with increased neurodegenerative pathologies, CNS senescent cells accumulating less iron than non-senescent cells, or comprehensive iron-incorporated treatments failing to improve clinical decline. However, the methods in these papers would not only need to have adequate sensitivity in detecting such iron (ex. magnet strength and post-processing analysis after MRI scans), but also adequately address all the different mechanisms (i.e., ferroptosis and ferritin-involved senescence) facilitated by localized iron accumulation.
Regarding future experiments, it is unassessed if other CNS-residing, iron-rich cell populations are senescence-enriched. This includes glia (ex. astrocytes and oligodendrocyte lineage cells), other infiltrating immune cell populations (ex. CD8+ T cells), and BAMs. Although future studies should investigate these other cells, whether they are iron-rich and senescent do not invalidate the theory’s postulates (i.e., ferroptosis and Fe3+-accumulated, senescent neurons and CNS immune cells significantly contribute to neurodegenerative disease progression). As amygdalar neurons can contain NFT and LB[157], iron-rich senescent neurons may also contribute towards initiating and secreting compound pathologies.
MRI studies already point towards iron-rich microglia and neurodegenerative pathologies as biological indicators to differentiate healthy vs disease populations. Why do some individuals develop neurodegenerative diseases while others do not, despite iron accumulation increasing over time universally? Although more precise MRI measures should be developed, neurodegenerative diseases occur over time and akin to a progressive spectrum, rather than binary absolutes; similarly, older individuals with increased iron accumulation should display more subtler symptomatic deficits, but may not reach a level classified as sufficiently “disease-clinical” until they accumulate sufficient, localized iron accumulation.
This framework will hopefully support further detection of singular and concurrent neurodegenerative disease progression. Although iron loss can occur over demyelination within neurodegenerative diseases, pathological localized iron accumulation should outweigh iron loss occurring through demyelination (i.e., via buildup of myelin debris, ferroptosis, blood brain barrier leakage, erythrocyte infiltration, and/or iron-rich immunosenescent cells). Further diagnostic methods and accuracy should be targeted to enhance iron accumulation detection in chronic neurodegeneration; nonetheless, net increases to iron accumulation, even with accompanying demyelination, have already been observed[158,159]. Finally, drugs potentially targeting mechanisms involving iron accumulation, via both ferroptosis and senescent cells, may better treat compound pathologies across neurodegenerative diseases.
Author Contributions
V.Z.L. conceptualized article ideas, wrote the first manuscript draft, and provided figures. All authors critically discussed manuscript content, with subsequent editing, revising, and improving the manuscript.
Acknowledgements
V.Z.L. is grateful to be supported by graduate doctoral Vascular Training (VAST) Platform and Ontario Graduate Scholarship (OGS) awards.
Competing Interests
E.H.M. is a speaker for Siemens Healthineers and consultant for Boston Scientific Corp. All other authors declare no competing interests.
List of Abbreviations
ALS: Amyotrophic lateral sclerosis
APP/Aβ: Amyloid precursor protein; Amyloid-beta
AD: Alzheimer’s disease
APOE: Apolipoprotein E
ATG7: Autophagy related 7
BAM: Border-associated macrophages
CAA: Cerebral amyloid angiopathy
CDKN2A / p16INK4A: Cyclin-dependent kinase inhibitor 2A
cGAS-STING: Cyclic GMP-AMP synthase; Stimulator of interferon genes
CoQ10: Coenzyme Q10
CNS: Central nervous system
CTE: Chronic traumatic encephalopathy
C/EBP: CCAAT-enhancer-binding protein
DLK1: Delta-like non-canonical Notch ligand 1
DMT1: Divalent metal transporter 1
EV: Extracellular vesicle
Fe2+: Ferrous iron
Fe3+: Ferric iron
FSP1: Ferroptosis suppressor protein 1
FTD: Frontotemporal dementia
FTH1/FTL: Ferritin heavy chain 1 / Ferritin light chain
GCL: Glutamate–cysteine ligase
GLS: Glutaminase
GPX-4: Glutathione peroxidase 4
GSH: Glutathione (reduced form)
GSK3: Glycogen synthase kinase-3
GSS: Glutathione Disulfide (oxidized glutathione)
HD: Huntington’s disease
HMGB1: High mobility group box protein 1
H2O2: Hydrogen peroxide
IFN: Interferon
iPSC: Induced pluripotent stem cell
IRE-IRP: Iron responsive element – Iron regulatory protein system
LATE: Limbic-predominant age-related TDP-43 encephalopathy
LB: Lewy bodies
LBD: Lewy body diseases
LIP: Labile iron pool
LXR: Liver X receptor
MMP: Matrix metalloprotease
MRI: Magnetic resonance imaging
MS: Multiple sclerosis
MSA: Multiple system atrophy
NCOA4: Nuclear receptor coactivator 4
NF-κβ: Nuclear factor kappa-light-chain-enhancer of activated B cells
NFT: Neurofibrillary tangle
NLRP3: NLR family pyrin domain containing 3
PD: Parkinson’s disease
PI3K/AKT/mTOR: Phosphoinositide 3-kinase/Protein kinase B/Mechanistic target of rapamycin pathway
pATM: Phosphorylated ataxia telangiectasia mutated
PSP: Progressive supranuclear palsy
p-α-syn: Phosphorylated alpha-synuclein
p21: Cyclin-dependent kinase inhibitor 1
p38 MAPK: p38 mitogen-activated protein kinases
p53: Transformation-related protein 53 (TRP53)
Rb: Retinoblastoma protein
ROS: Reactive oxygen and nitrogen species
SA-β-gal: Senescence-associated beta-galactosidase
SASP: Senescence-associated secretory phenotype
SCI: Spinal cord injury
SNpc: Substantia nigra pars compacta
System Xc- / SLC7A11: Cysteine/glutamate transporter (sodium-independent, chloride-dependent cysteine-glutamate antiporter)
STEAP3: Six-transmembrane epithelial antigen of the prostate 3
SV40: Simian virus 40 (polyoma virus)
TBI: Traumatic brain injury
TNF-α: Tumour necrosis factor alpha
TFEB: Transcription factor EB
VCID: Vascular contributions to cognitive impairment and dementia
WMH: White matter injuries and hyperintensities
γH2A.X: Phosphorylated serine 139 on histone family member X
References
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: principles and practice. BMJ 2019, 72. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; et al. Pathological combinations in neurodegenerative disease are heterogeneous and disease-associated. Brain 2023, 146, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- Lau, V.; Ramer, L.; Tremblay, M.-È. An aging, pathology burden, and glial senescence build-up hypothesis for late onset Alzheimer’s disease. Nat. Commun. 2023, 14, 1670. [Google Scholar] [CrossRef] [PubMed]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease - lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef]
- Paolini Paoletti, F.; Simoni, S.; Parnetti, L.; Gaetani, L. The Contribution of Small Vessel Disease to Neurodegeneration: Focus on Alzheimer’s Disease, Parkinson’s Disease and Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 4958. [Google Scholar] [CrossRef]
- Lerma-Martin, C. Cell type mapping reveals tissue niches and interactions in subcortical multiple sclerosis lesions. Nat. Neurosci. 2024, 27. [Google Scholar] [CrossRef]
- Alsema, A.M.; et al. Spatially resolved gene signatures of white matter lesion progression in multiple sclerosis. Nat. Neurosci. 2024, 27, 1–13. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef]
- Paulsen, J.S. Cognitive Impairment in Huntington Disease: Diagnosis and Treatment. Curr. Neurol. Neurosci. Rep. 2011, 11, 474–483. [Google Scholar] [CrossRef]
- Stankovic, I.; et al. Cognitive impairment in multiple system atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 857–867. [Google Scholar] [CrossRef]
- DeLuca, G.C.; Yates, R.L.; Beale, H.; Morrow, S.A. Cognitive Impairment in Multiple Sclerosis: Clinical, Radiologic and Pathologic Insights. Brain Pathol. 2014, 25, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Ripamonti, M.; Moro, A.S.; Cozzi, A. Iron imbalance in neurodegeneration. Mol. Psychiatry 2024, 29, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Berggren, K.L.; Marks, E.; Fox, J.H. Impact of high iron intake on cognition and neurodegeneration in humans and in animal models: a systematic review. Nutr. Rev. 2017, 75, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; et al. Iron retardation in lysosomes protects senescent cells from ferroptosis. Aging 2024, 16, 7683–7703. [Google Scholar] [CrossRef]
- Andersen, H.H.; Johnsen, K.B.; Moos, T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell. Mol. Life Sci. 2014, 71, 1607–1622. [Google Scholar] [CrossRef]
- Leon-Oliva, D.D.; et al. Improving understanding of ferroptosis: molecular mechanisms, connection with cellular senescence and implications for aging. 2024, 10.
- Ravanfar, P.; et al. Systematic Review: Quantitative Susceptibility Mapping (QSM) of Brain Iron Profile in Neurodegenerative Diseases. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.M.; Aaseth, J.O. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef]
- Guo, J.; Tuo, Q.; Lei, P. Iron, Ferroptosis, and Ischemic Stroke. J. Neurochem. 2023, 165. [Google Scholar] [CrossRef]
- Riederer, P.; et al. Lewy bodies, iron, inflammation and neuromelanin: pathological aspects underlying Parkinson’s disease. J. Neural Transm. 2023, 130, 627–646. [Google Scholar] [CrossRef]
- Spence, H.; McNeil, C.J.; Waiter, G.D. The impact of brain iron accumulation on cognition: A systematic review. 2020, 15. [CrossRef]
- Chen, L.; et al. Quantitative Susceptibility Mapping of Brain Iron and β-Amyloid in MRI and PET Relating to Cognitive Performance in Cognitively Normal Older Adults. Radiology 2021, 298, 353–362. [Google Scholar] [CrossRef]
- Zachariou, V.; Pappas, C.; Bauer, C.E.; Seago, E.R.; Gold, B.T. Exploring the links among brain iron accumulation, cognitive performance, and dietary intake in older adults: A longitudinal MRI study. Neurobiol. Aging 2024, 145. [Google Scholar] [CrossRef]
- Li, Z.; et al. Paramagnetic susceptibility measured by magnetic resonance imaging as an in vivo biomarker for iron pathology in epilepsy. Sci. Adv. 2025, 11, eads8149. [Google Scholar] [CrossRef]
- Lu, W.; et al. Relaxometry network based on MRI R2* mapping revealing brain iron accumulation patterns in Parkinson’s disease. NeuroImage 2024, 303, 120943. [Google Scholar] [CrossRef]
- Alushaj, E.; et al. Increased iron in the substantia nigra pars compacta identifies patients with early Parkinson’s disease: A 3T and 7T MRI study. NeuroImage Clin. 2024, 41, 103577. [Google Scholar] [CrossRef]
- Yao, S.; et al. Quantitative Susceptibility Mapping Reveals an Association between Brain Iron Load and Depression Severity. Front. Hum. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Yan, S.; Sun, J.; Chen, Y.; Selim, M.; Lou, M. Brain iron deposition in white matter hyperintensities: a 3-T MRI study. AGE 2013, 35, 1927–1936. [Google Scholar] [CrossRef]
- Valdés Hernández, M.; et al. Do white matter hyperintensities mediate the association between brain iron deposition and cognitive abilities in older people? Eur. J. Neurol. 2016, 23, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, M.-S. Brain Iron Accumulation in Atypical Parkinsonian Syndromes: in vivo MRI Evidences for Distinctive Patterns. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; et al. Ferritin is a component of the neuritic (senile) plaque in Alzheimer dementia. Acta Neuropathol. (Berl.) 1990, 81, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Xu, W.; Wang, X.; Peng, D.; Jiang, Z. Exploring the Causal Relationship Between Inflammatory Cytokines and MRI-Derived Brain Iron: A Mendelian Randomization Study. Brain Behav. 2024, 14, e70181. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; et al. Ferroptosis: past, present and future. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Joppe, K.; Roser, A.-E.; Maass, F.; Lingor, P. The Contribution of Iron to Protein Aggregation Disorders in the Central Nervous System. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef]
- Jhelum, P.; et al. Ferroptosis induces detrimental effects in chronic EAE and its implications for progressive MS. Acta Neuropathol. Commun. 2023, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.K.; et al. Microglia ferroptosis is regulated by SEC24B and contributes to neurodegeneration. Nat. Neurosci. 2023, 26, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of TNF. Oxid. Med. Cell. Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Stys, P. White Matter Injury Mechanisms. Curr. Mol. Med. 2004, 4, 113–130. [Google Scholar] [CrossRef]
- Emerit, J.; Edeas, M.; Bricaire, F. Neurodegenerative diseases and oxidative stress. Biomed. Pharmacother. 2004, 58, 39–46. [Google Scholar] [CrossRef]
- Goldman, S.M. Environmental Toxins and Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 141–164. [Google Scholar] [CrossRef]
- Javadov, S. Mitochondria and ferroptosis. Curr. Opin. Physiol. 2022, 25, 100483. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Zhao, Y.; Gao, G. Mitochondria regulation in ferroptosis. Eur. J. Cell Biol. 2020, 99, 151058. [Google Scholar] [CrossRef]
- Tisdall, M.D.; et al. Ex vivo MRI and histopathology detect novel iron-rich cortical inflammation in frontotemporal lobar degeneration with tau versus TDP-43 pathology. NeuroImage Clin. 2022, 33, 102913. [Google Scholar] [CrossRef] [PubMed]
- Giannini, L.A.A.; et al. Cortical iron accumulation in MAPT- and C9orf 72-associated frontotemporal lobar degeneration. Brain Pathol. 2023, 33, e13158. [Google Scholar] [CrossRef] [PubMed]
- Waldron, F.M.; et al. Brain Iron as a Surrogate Biomarker of Pathological TDP-43 Identifies Brain Region-Specific Signatures in Ageing, Alzheimer’s Disease and Amyotrophic Lateral Sclerosis. bioRxiv 2025. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Seo, A.Y.; et al. Mitochondrial iron accumulation with age and functional consequences. Aging Cell 2008, 7, 706–716. [Google Scholar] [CrossRef]
- Chen, Y.; et al. Oxidative stress induces mitochondrial iron overload and ferroptotic cell death. Sci. Rep. 2023, 13, 15515. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef]
- Truman-Rosentsvit, M.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Dhekne, H.S.; Toyokuni, S.; Kishi, F. CD63 is regulated by iron via the IRE-IRP system and is important for ferritin secretion by extracellular vesicles. Blood 2021, 138, 1490–1503. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Hou, W.; et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Masaldan, S.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, P.A.; et al. Ferroptosis of Microglia in Aging Human White Matter Injury. Ann. Neurol. 2023, 94, 1048–1066. [Google Scholar] [CrossRef] [PubMed]
- da Costa Caiado, M.J.; Dolga, A.M.; den Dunnen, W.F.A. Iron(ing) out parkinsonisms: The interplay of proteinopathy and ferroptosis in Parkinson’s disease and tau-related parkinsonisms. Redox Biol. 2025, 79, 103478. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; et al. The Emerging Roles of Ferroptosis in Huntington’s Disease. NeuroMolecular Med. 2019, 21, 110–119. [Google Scholar] [CrossRef]
- Cozzi, A.; et al. Stem Cell Modeling of Neuroferritinopathy Reveals Iron as a Determinant of Senescence and Ferroptosis during Neuronal Aging. Stem Cell Rep. 2019, 13, 832–846. [Google Scholar] [CrossRef]
- Good, P.F.; Perl, D.P.; Bierer, L.M.; Schmeidler, J. Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer’s disease: A laser microprobe (LAMMA) study. Ann. Neurol. 1992, 31, 286–292. [Google Scholar] [CrossRef]
- Smith, M.A.; Harris, P.L.R.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. 1997, 94, 9866–9868. [Google Scholar] [CrossRef]
- Killilea, D.W.; Wong, S.L.; Cahaya, H.S.; Atamna, H.; Ames, B.N. Iron accumulation during cellular senescence. Ann. N. Y. Acad. Sci. 2004, 1019, 365–367. [Google Scholar] [CrossRef]
- Maus, M.; et al. Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat. Metab. 2023, 5, 1–20. [Google Scholar] [CrossRef]
- Admasu, T.D.; et al. Selective ablation of primary and paracrine senescent cells by targeting iron dyshomeostasis. Cell Rep. 2023, 42. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.G.; et al. In vivo DNA replication dynamics unveil aging-dependent replication stress. Cell 2024, 187. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Cell senescence, rapamycin and hyperfunction theory of aging. Cell Cycle 2022, 21, 1456–1467. [Google Scholar] [CrossRef]
- Rovira, M.; et al. The lysosomal proteome of senescent cells contributes to the senescence secretome. Aging Cell 2022, 21, e13707. [Google Scholar] [CrossRef]
- Victorelli, S.; et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature 2023, 622, 627–636. [Google Scholar] [CrossRef]
- Xu, M.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246. [Google Scholar] [CrossRef]
- Zhang, X.; et al. Rejuvenation of the aged brain immune cell landscape in mice through p16-positive senescent cell clearance. Nat. Commun. 2022, 13, 5671. [Google Scholar] [CrossRef]
- Jin, M.; et al. Type I interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer’s disease. Cell Stem Cell 2022, 29, 1135–1153.e8. [Google Scholar] [CrossRef]
- Brelstaff, J.H.; et al. Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Shen, Q.; et al. Cell senescence induced by toxic interaction between α-synuclein and iron precedes nigral dopaminergic neuron loss in a mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 2024, 45, 268–281. [Google Scholar] [CrossRef]
- Hong, B.; Ohtake, Y.; Itokazu, T.; Yamashita, T. Glial senescence enhances α-synuclein pathology owing to its insufficient clearance caused by autophagy dysfunction. Cell Death Discov. 2024, 10, 1–12. [Google Scholar] [CrossRef]
- Safaiyan, S.; et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Matsudaira, T.; et al. Cellular senescence in white matter microglia is induced during ageing in mice and exacerbates the neuroinflammatory phenotype. Commun. Biol. 2023, 6, 665. [Google Scholar] [CrossRef]
- Zhang, X.; et al. Exposure to Manganese Induces Autophagy–Lysosomal Pathway Dysfunction-Mediated Tauopathy by Activating the cGAS–STING Pathway in the Brain. Environ. Health 2024, 3. [Google Scholar] [CrossRef] [PubMed]
- López-Polo, V.; et al. Release of mitochondrial dsRNA into the cytosol is a key driver of the inflammatory phenotype of senescent cells. Nat. Commun. 2024, 15, 7378. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; et al. Mitochondrial fatty acid oxidation drives senescence. Sci. Adv. 2024, 10, eado5887. [Google Scholar] [CrossRef] [PubMed]
- Torres-Querol, C.; et al. Acute ischemic stroke triggers a cellular senescence-associated secretory phenotype. Sci. Rep. 2021, 11, 15752. [Google Scholar] [CrossRef]
- Baixauli-Martín, J.; et al. Spatio-Temporal Characterization of Cellular Senescence Hallmarks in Experimental Ischemic Stroke. Int. J. Mol. Sci. 2025, 26, 2364. [Google Scholar] [CrossRef]
- Gross, P.S.; et al. Senescent-like microglia limit remyelination through the senescence associated secretory phenotype. Nat. Commun. 2025, 16, 2283. [Google Scholar] [CrossRef]
- Belaidi, A.A.; et al. Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy. Mol. Psychiatry 2024, 29, 211–220. [Google Scholar] [CrossRef]
- Lee, H.; et al. Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance. Nat. Commun. 2024, 15, 79. [Google Scholar] [CrossRef]
- Courtney, R.; Landreth, G.E. LXR regulation of brain cholesterol: from development to disease. Trends Endocrinol. Metab. TEM 2016, 27, 404–414. [Google Scholar] [CrossRef]
- Jiang, Q.; et al. ApoE promotes the proteolytic degradation of Aβ. Neuron 2008, 58, 681. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; et al. Destabilizing heterochromatin by APOE mediates senescence. Nat. Aging 2022, 2, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; et al. Microglial apolipoprotein E particles contribute to neuronal senescence and synaptotoxicity. iScience 2024, 27, 110006. [Google Scholar] [CrossRef] [PubMed]
- Bancaro, N.; et al. Apolipoprotein E induces pathogenic senescent-like myeloid cells in prostate cancer. Cancer Cell 2023, 41, 602–619.e11. [Google Scholar] [CrossRef]
- Windham, I.A.; Cohen, S. The cell biology of APOE in the brain. Trends Cell Biol. 2024, 34, 338–348. [Google Scholar] [CrossRef]
- Giau, V.V.; Bagyinszky, E.; An, S.S.A.; Kim, S.Y. Role of apolipoprotein E in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2015, 11, 1723–1737. [Google Scholar] [CrossRef]
- Höhn, A.; Jung, T.; Grimm, S.; Grune, T. Lipofuscin-bound iron is a major intracellular source of oxidants: Role in senescent cells. Free Radic. Biol. Med. 2010, 48, 1100–1108. [Google Scholar] [CrossRef]
- Reinert, A.; Morawski, M.; Seeger, J.; Arendt, T.; Reinert, T. Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci. 2019, 20, 25. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Ren, J.; Chen, Q.; Chen, Z.J. cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 2017, 114, E4612. [Google Scholar] [CrossRef]
- Karabag, D.; et al. Characterizing microglial senescence: Tau as a key player. J. Neurochem. 2023, 166. [Google Scholar] [CrossRef] [PubMed]
- Du, T.; et al. Nuclear alpha-synuclein accelerates cell senescence and neurodegeneration. Immun. Ageing 2024, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Carver, C.M.; et al. Senescent and disease-associated microglia are modifiable features of aged brain white matter. Res. Sq. rs.3.rs-3467812 (2023). [CrossRef]
- Yoon, Y.-S.; et al. Senescence and impaired DNA damage responses in alpha-synucleinopathy models. Exp. Mol. Med. 2022, 54, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Carlsson, F.; Laskar, A.; Yuan, X.-M.; Li, W. Lysosomal membrane permeabilization causes oxidative stress and ferritin induction in macrophages. FEBS Lett. 2011, 585, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Kaji, S.; et al. Apolipoprotein E aggregation in microglia initiates Alzheimer’s disease pathology by seeding β-amyloidosis. Immunity 2024, 57. [Google Scholar] [CrossRef]
- Quick, J.D.; et al. Lysosomal acidification dysfunction in microglia: an emerging pathogenic mechanism of neuroinflammation and neurodegeneration. J. Neuroinflammation 2023, 20, 185. [Google Scholar] [CrossRef]
- Jin, M.; et al. Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nat. Commun. 2021, 12, 6565. [Google Scholar] [CrossRef]
- Gulen, M.F.; et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 1–7. [Google Scholar] [CrossRef]
- Lv, B.; et al. A TBK1-independent primordial function of STING in lysosomal biogenesis. Mol. Cell 2024, 84. [Google Scholar] [CrossRef] [PubMed]
- Curnock, R.; et al. TFEB-dependent lysosome biogenesis is required for senescence. EMBO J. 2023, 42, e111241. [Google Scholar] [CrossRef] [PubMed]
- Musi, N.; et al. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Martinez-Valbuena, I.; Lang, A.E.; Kovacs, G.G. Cellular iron deposition patterns predict clinical subtypes of multiple system atrophy. Neurobiol. Dis. 2024, 197, 106535. [Google Scholar] [CrossRef]
- Simmons, D.A.; et al. Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 2007, 55, 1074–1084. [Google Scholar] [CrossRef]
- Kwan, J.Y.; et al. Iron Accumulation in Deep Cortical Layers Accounts for MRI Signal Abnormalities in ALS: Correlating 7 Tesla MRI and Pathology. PLOS ONE 2012, 7, e35241. [Google Scholar] [CrossRef]
- Shahidehpour, R.K.; et al. Dystrophic microglia are associated with neurodegenerative disease and not healthy aging in the human brain. Neurobiol. Aging 2021, 99, 19–27. [Google Scholar] [CrossRef]
- Shahidehpour, R.K.; Nelson, P.T.; Katsumata, Y.; Bachstetter, A.D. Exploring the link between dystrophic microglia and the spread of Alzheimer’s neuropathology. Brain 2024, awae258. [Google Scholar] [CrossRef]
- Swanson, M.E.V.; et al. Quantitative immunohistochemical analysis of myeloid cell marker expression in human cortex captures microglia heterogeneity with anatomical context. Sci. Rep. 2020, 10, 11693. [Google Scholar] [CrossRef]
- Kenkhuis, B.; et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 2021, 9, 27. [Google Scholar] [CrossRef]
- Zeineh, M.M.; et al. Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol. Aging 2015, 36, 2483–2500. [Google Scholar] [CrossRef] [PubMed]
- van Duijn, S.; et al. Cortical Iron Reflects Severity of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Pisa, M.; et al. Aberrant iron deposition in the multiple sclerosis spinal cord relates to neurodegeneration. Preprint 2024. [Google Scholar] [CrossRef]
- Tsering, W.; et al. Preferential clustering of microglia and astrocytes around neuritic plaques during progression of Alzheimer’s disease neuropathological changes. J. Neurochem. 2025, 169, e16275. [Google Scholar] [CrossRef]
- Swanson, M.E.V.; et al. Identification of a dysfunctional microglial population in human Alzheimer’s disease cortex using novel single-cell histology image analysis. Acta Neuropathol. Commun. 2020, 8, 170. [Google Scholar] [CrossRef]
- Thrupp, N.; et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020, 32, 108189. [Google Scholar] [CrossRef]
- Fancy, N.N.; et al. Characterisation of premature cell senescence in Alzheimer’s disease using single nuclear transcriptomics. Acta Neuropathol. (Berl.) 2024, 147, 78. [Google Scholar] [CrossRef]
- Wang, Q.; et al. Single cell transcriptomes and multiscale networks from persons with and without Alzheimer’s disease. Nat. Commun. 2024, 15, 5815. [Google Scholar] [CrossRef]
- Fagiani, F.; et al. Spatially-restricted inflammation-induced senescent-like glia in multiple sclerosis and patient-derived organoids. Nat. Commun. 2025, 16, 8477. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, J.; Wang, J.; Pan, X. Arsenic exposure induces neural cells senescence and abnormal lipid droplet accumulation leading to social memory impairment in mice. Environ. Pollut. 2025, 368, 125779. [Google Scholar] [CrossRef]
- Zhao, X.; et al. Up-regulated succinylation modifications induce a senescence phenotype in microglia by altering mitochondrial energy metabolism. J. Neuroinflammation 2024, 21, 296. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Model Senescent Microglia Induce Disease Related Changes in α-Synuclein Expression and Activity. Biomolecules 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Angelova, D.M.; Brown, D.R. Altered Processing of β-Amyloid in SH-SY5Y Cells Induced by Model Senescent Microglia. ACS Chem. Neurosci. 2018, 9, 3137–3152. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, A.; et al. Iron accumulation in microglia triggers a cascade of events that leads to altered metabolism and compromised function in APP/PS1 mice. Brain Pathol. 2019, 29, 606–621. [Google Scholar] [CrossRef]
- Wei, L.; et al. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J. Neuroinflammation 2023, 20, 208. [Google Scholar] [CrossRef]
- Kenkhuis, B.; et al. Iron accumulation induces oxidative stress, while depressing inflammatory polarization in human iPSC-derived microglia. Stem Cell Rep. 2022. [Google Scholar] [CrossRef]
- Choi, I.; et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat. Cell Biol. 2023, 1–12. [Google Scholar] [CrossRef]
- Malvaso, A.; et al. Microglial Senescence and Activation in Healthy Aging and Alzheimer’s Disease: Systematic Review and Neuropathological Scoring. Cells 2023, 12, 2824. [Google Scholar] [CrossRef]
- Jiao, L.; et al. Iron metabolism mediates microglia susceptibility in ferroptosis. Front. Cell. Neurosci. 2022, 16, 995084. [Google Scholar] [CrossRef]
- Roe, M.T.; Dawson, D.V.; Hulette, C.M.; Einstein, G.; Crain, B.J. Microglia Are Not Exclusively Associated with Plaque-rich Regions of the Dentate Gyrus in Alzheimer’s Disease. J. Neuropathol. Exp. Neurol. 1996, 55, 366–371. [Google Scholar] [CrossRef]
- Langhi Prata, L.G.P.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent cell clearance by the immune system: Emerging therapeutic opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; et al. Soluble iron accumulation makes microglia to overproduce and release glutamate via aconitase 1, tace and glutaminase c in ALS spinal cords. J. Neurol. Sci. 2017, 381, 710. [Google Scholar] [CrossRef]
- Ogrodnik, M.; et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell 2021, 20, e13296. [Google Scholar] [CrossRef] [PubMed]
- Liddell, J.R.; et al. Microglial ferroptotic stress causes non-cell autonomous neuronal death. Mol. Neurodegener. 2024, 19, 14. [Google Scholar] [CrossRef]
- Gan, L.; et al. Soluble DLK1 secreted by telomere-shortening-induced senescent microglia impairs myelination and alters neuronal activity. Preprint 2024. [Google Scholar] [CrossRef]
- Shin, H.J.; et al. Rejuvenating aged microglia by p16ink4a-siRNA-loaded nanoparticles increases amyloid-β clearance in animal models of Alzheimer’s disease. Mol. Neurodegener. 2024, 19, 25. [Google Scholar] [CrossRef]
- Carling, G.K.; et al. Alzheimer’s disease-linked risk alleles elevate microglial cGAS-associated senescence and neurodegeneration in a tauopathy model. Neuron 2024, 112, 3877–3896.e8. [Google Scholar] [CrossRef]
- Lee, S.H.; et al. Amyloid-β-activated microglia can induce compound proteinopathies. Brain 2024, 147. [Google Scholar] [CrossRef]
- Chung, S.; et al. Blockade of STING activation alleviates microglial dysfunction and a broad spectrum of Alzheimer’s disease pathologies. Exp. Mol. Med. 2024, 1–16. [Google Scholar] [CrossRef]
- Hu, Y.; et al. Replicative senescence dictates the emergence of disease-associated microglia and contributes to Aβ pathology. Cell Rep. 2021, 35, 109228. [Google Scholar] [CrossRef]
- Caldeira, C.; et al. Key Aging-Associated Alterations in Primary Microglia Response to Beta-Amyloid Stimulation. Front. Aging Neurosci. 2017, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.M.; et al. Old age increases microglial senescence, exacerbates secondary neuroinflammation, and worsens neurological outcomes after acute traumatic brain injury in mice. Neurobiol. Aging 2019, 77, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or Foes: Matrix Metalloproteinases and Their Multifaceted Roles in Neurodegenerative Diseases. Mediators Inflamm. 2015, 2015, 620581. [Google Scholar] [CrossRef] [PubMed]
- Roy, E.R.; et al. Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity 2022, 55, 879–894.e6. [Google Scholar] [CrossRef]
- Roy, E.R.; et al. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 2020, 130, 1912. [Google Scholar] [CrossRef]
- He, Z.; et al. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef]
- Li, T.; et al. The neuritic plaque facilitates pathological conversion of tau in an Alzheimer’s disease mouse model. Nat. Commun. 2016, 7, 12082. [Google Scholar] [CrossRef]
- Baligács, N.; et al. Homeostatic microglia initially seed and activated microglia later reshape amyloid plaques in Alzheimer’s Disease. Nat. Commun. 2024, 15, 10634. [Google Scholar] [CrossRef]
- Bassil, F.; et al. Amyloid-Beta (Aβ) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Aβ Pathology. Neuron 2020, 111. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. (Berl.) 2006, 112, 389–404. [Google Scholar] [CrossRef]
- Borghammer, P. The α-Synuclein Origin and Connectome Model (SOC Model) of Parkinson’s Disease: Explaining Motor Asymmetry, Non-Motor Phenotypes, and Cognitive Decline. J. Park. Dis. 2021, 11, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Adler, C.H.; et al. Unified Staging System for Lewy Body Disorders: Clinicopathologic Correlations and Comparison to Braak Staging. J. Neuropathol. Exp. Neurol. 2019, 78, 891. [Google Scholar] [CrossRef] [PubMed]
- Reinert, A.; Morawski, M.; Seeger, J.; Arendt, T.; Reinert, T. Iron concentrations in neurons and glial cells with estimates on ferritin concentrations. BMC Neurosci. 2019, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.L.; Martin, J.A.; Lee, V.M.-Y.; Trojanowski, J.Q. Convergence of Lewy bodies and neurofibrillary tangles in amygdala neurons of Alzheimer’s disease and Lewy body disorders. Acta Neuropathol. (Berl.) 1996, 91, 475–481. [Google Scholar] [CrossRef]
- Wang, C.; et al. Methods for quantitative susceptibility and R2* mapping in whole post-mortem brains at 7T applied to amyotrophic lateral sclerosis. Neuroimage 2020, 222, 117216. [Google Scholar] [CrossRef]
- Bagnato, F.; et al. Untangling the R2* contrast in multiple sclerosis: A combined MRI-histology study at 7.0 Tesla. PLoS ONE 2018, 13, e0193839. [Google Scholar] [CrossRef]
- Zhang, W.; et al. Brain Iron Deposits in Thalamus Is an Independent Factor for Depressive Symptoms Based on Quantitative Susceptibility Mapping in an Older Adults Community Population. Front. Psychiatry 2019, 10. [Google Scholar] [CrossRef]
Figure 1.
Increased Localized Iron Across Disease Pathologies and Chronic Neurodegeneration. A visual snapshot of localized iron accumulation and its presence across numerous neurodegenerative diseases are included, alongside hallmarks of chronic neurodegeneration. The hourglass at the top denotes increased localized iron accumulation over disease progression, alongside the clinical severity resulting from the extent of chronic neurodegeneration. Although other chronic neurodegenerative and neuropsychiatric diseases involve increased localized iron over disease progression and clinical decline, they are not included here due to insufficient amounts of current studies. The outermost circle of text involves specific neurodegenerative diseases; the middle ring comprises iron-rich pathologies or aggregates appearing in these diseases, and the center includes iron-rich CNS macrophages that have been noted to be present in neurodegenerative settings across both singular and mixed disease presentation. At the bottom, a panel of key disease pathologies and/or maladaptive responses (ex. cGAS-STING signalling) involved with chronic neurodegeneration are included. Finally, studies and reviews linking localized iron accumulation to each disease or pathology are listed for reference[6,7,12,18,19,20,24,25,26,27,28,29,30,31,34,57,107,108,110,112,113,114,115,116,117,118,133,135,158,160]. Figure created with BioRender.com.
Figure 1.
Increased Localized Iron Across Disease Pathologies and Chronic Neurodegeneration. A visual snapshot of localized iron accumulation and its presence across numerous neurodegenerative diseases are included, alongside hallmarks of chronic neurodegeneration. The hourglass at the top denotes increased localized iron accumulation over disease progression, alongside the clinical severity resulting from the extent of chronic neurodegeneration. Although other chronic neurodegenerative and neuropsychiatric diseases involve increased localized iron over disease progression and clinical decline, they are not included here due to insufficient amounts of current studies. The outermost circle of text involves specific neurodegenerative diseases; the middle ring comprises iron-rich pathologies or aggregates appearing in these diseases, and the center includes iron-rich CNS macrophages that have been noted to be present in neurodegenerative settings across both singular and mixed disease presentation. At the bottom, a panel of key disease pathologies and/or maladaptive responses (ex. cGAS-STING signalling) involved with chronic neurodegeneration are included. Finally, studies and reviews linking localized iron accumulation to each disease or pathology are listed for reference[6,7,12,18,19,20,24,25,26,27,28,29,30,31,34,57,107,108,110,112,113,114,115,116,117,118,133,135,158,160]. Figure created with BioRender.com.
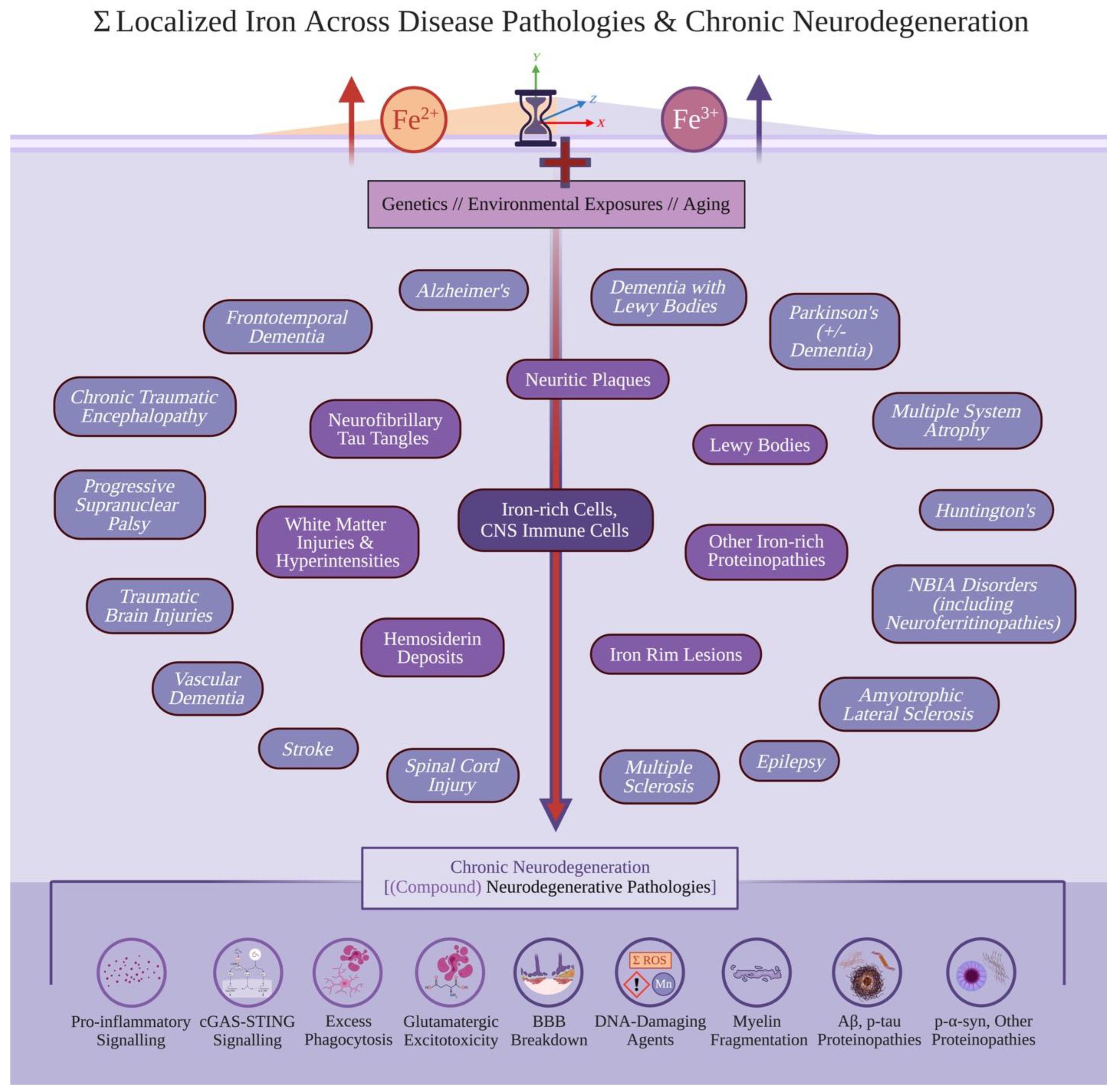
Figure 2.
Ferrous Iron Accumulation, Oxidative Stress, and Ferroptosis in Chronic Neurodegeneration. To explain how localized iron accumulation causally drives chronic neurodegeneration, a schematic of intracellular iron buildup and transport systems is provided. When excessive Fe2+ accumulates intracellularly, this increases the LIP leading into causally downstream Fenton reaction, mitochondrial dysfunction, and ROS creation outweighing anti-oxidant systems; continued persistence of excess Fe2+, when combined with ferritinophagy degrading ferritin (stabilizing Fe2+ into Fe3+), leads into lipid peroxidation and cell death in ferroptosis. As oxidative stress and mitochondrial dysfunction also induce intracellular Fe2+ accumulation, these three factors are involved to engage in positive feedback loops to drive singular and compound neurodegenerative pathologies involved in chronic neurodegeneration. Figure created with BioRender.com.
Figure 2.
Ferrous Iron Accumulation, Oxidative Stress, and Ferroptosis in Chronic Neurodegeneration. To explain how localized iron accumulation causally drives chronic neurodegeneration, a schematic of intracellular iron buildup and transport systems is provided. When excessive Fe2+ accumulates intracellularly, this increases the LIP leading into causally downstream Fenton reaction, mitochondrial dysfunction, and ROS creation outweighing anti-oxidant systems; continued persistence of excess Fe2+, when combined with ferritinophagy degrading ferritin (stabilizing Fe2+ into Fe3+), leads into lipid peroxidation and cell death in ferroptosis. As oxidative stress and mitochondrial dysfunction also induce intracellular Fe2+ accumulation, these three factors are involved to engage in positive feedback loops to drive singular and compound neurodegenerative pathologies involved in chronic neurodegeneration. Figure created with BioRender.com.
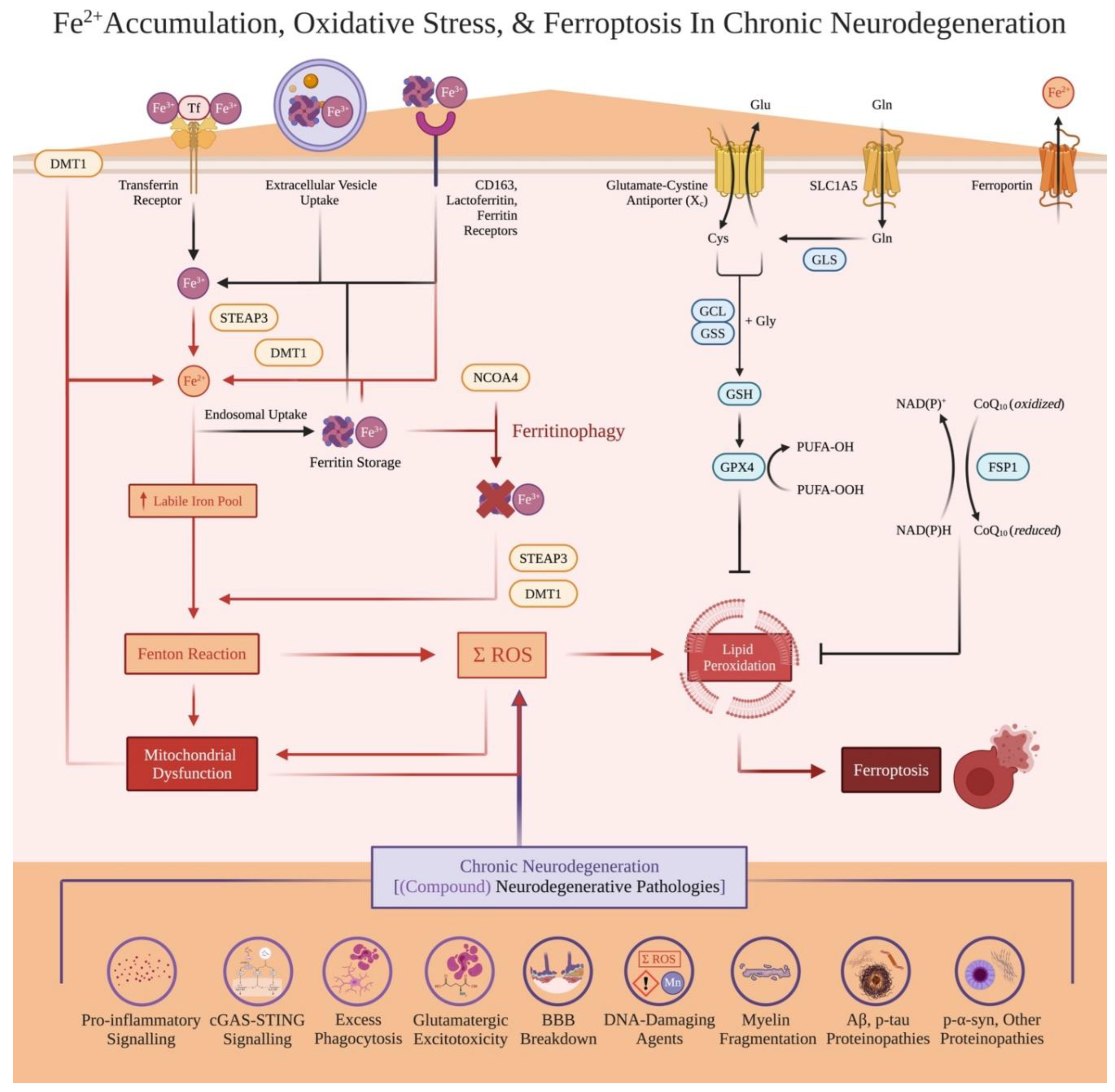
Figure 3.
Ferroptosis-Averted Senescence, Ferric Iron Accumulation, and Chronic Neurodegeneration. To explain how localized iron accumulation causally drives chronic neurodegeneration, alternative survival mechanisms exempting ferroptosis are provided. When ferritinophagy is inhibited and cells survive sublethal stressors (which converge into iron accumulation, ROS creation, and mitochondrial dysfunction), these cells avert ferroptosis but become senescent instead; such cells feature cell cycle arrest (often enacted through p16 or p21) and organelle impairment, wherein their internalized aggregates reflect their cell type, disease environment, and surrounding compound (or possibly, singular) neurodegenerative pathologies. These cells also build up ferritin binding to Fe3+, due to their ability to buffer and survive via converting massive amounts of intracellular Fe2+. Due to exposure to sublethal oxidative stress, some lipid peroxidation is predicted to occur in the cell resulting in decreased cytoplasmic integrity, but not to extents observed in complete ferroptosis. Iron-rich aggregate-producing, senescent neurons have been observed in the literature likely due to selective vulnerability; iron-rich CNS macrophages in infiltrating macrophages and microglia are senescent, and are proposed to maladaptively (i) exacerbate cGAS-STING mediated neurodegeneration, alongside (ii) seed iron-containing neurodegenerative pathologies. These pathologies may be of a singular nature (ex. only p-tau), but can also comprise compound aggregates resulting in and explaining mixed disease presentation). Iron-rich, CNS immunosenescent cells also (iii) further induce dysfunction in non-senescent cells via inducing paracrine senescence and not clearing out localized neurodegenerative pathologies. These behaviours unfortunately lead to these iron-rich, senescent cells being accelerators and involved contributors towards chronic neurodegeneration, rather than being bystanders or tangential to (compound) disease progression. Figure created with BioRender.com.
Figure 3.
Ferroptosis-Averted Senescence, Ferric Iron Accumulation, and Chronic Neurodegeneration. To explain how localized iron accumulation causally drives chronic neurodegeneration, alternative survival mechanisms exempting ferroptosis are provided. When ferritinophagy is inhibited and cells survive sublethal stressors (which converge into iron accumulation, ROS creation, and mitochondrial dysfunction), these cells avert ferroptosis but become senescent instead; such cells feature cell cycle arrest (often enacted through p16 or p21) and organelle impairment, wherein their internalized aggregates reflect their cell type, disease environment, and surrounding compound (or possibly, singular) neurodegenerative pathologies. These cells also build up ferritin binding to Fe3+, due to their ability to buffer and survive via converting massive amounts of intracellular Fe2+. Due to exposure to sublethal oxidative stress, some lipid peroxidation is predicted to occur in the cell resulting in decreased cytoplasmic integrity, but not to extents observed in complete ferroptosis. Iron-rich aggregate-producing, senescent neurons have been observed in the literature likely due to selective vulnerability; iron-rich CNS macrophages in infiltrating macrophages and microglia are senescent, and are proposed to maladaptively (i) exacerbate cGAS-STING mediated neurodegeneration, alongside (ii) seed iron-containing neurodegenerative pathologies. These pathologies may be of a singular nature (ex. only p-tau), but can also comprise compound aggregates resulting in and explaining mixed disease presentation). Iron-rich, CNS immunosenescent cells also (iii) further induce dysfunction in non-senescent cells via inducing paracrine senescence and not clearing out localized neurodegenerative pathologies. These behaviours unfortunately lead to these iron-rich, senescent cells being accelerators and involved contributors towards chronic neurodegeneration, rather than being bystanders or tangential to (compound) disease progression. Figure created with BioRender.com.
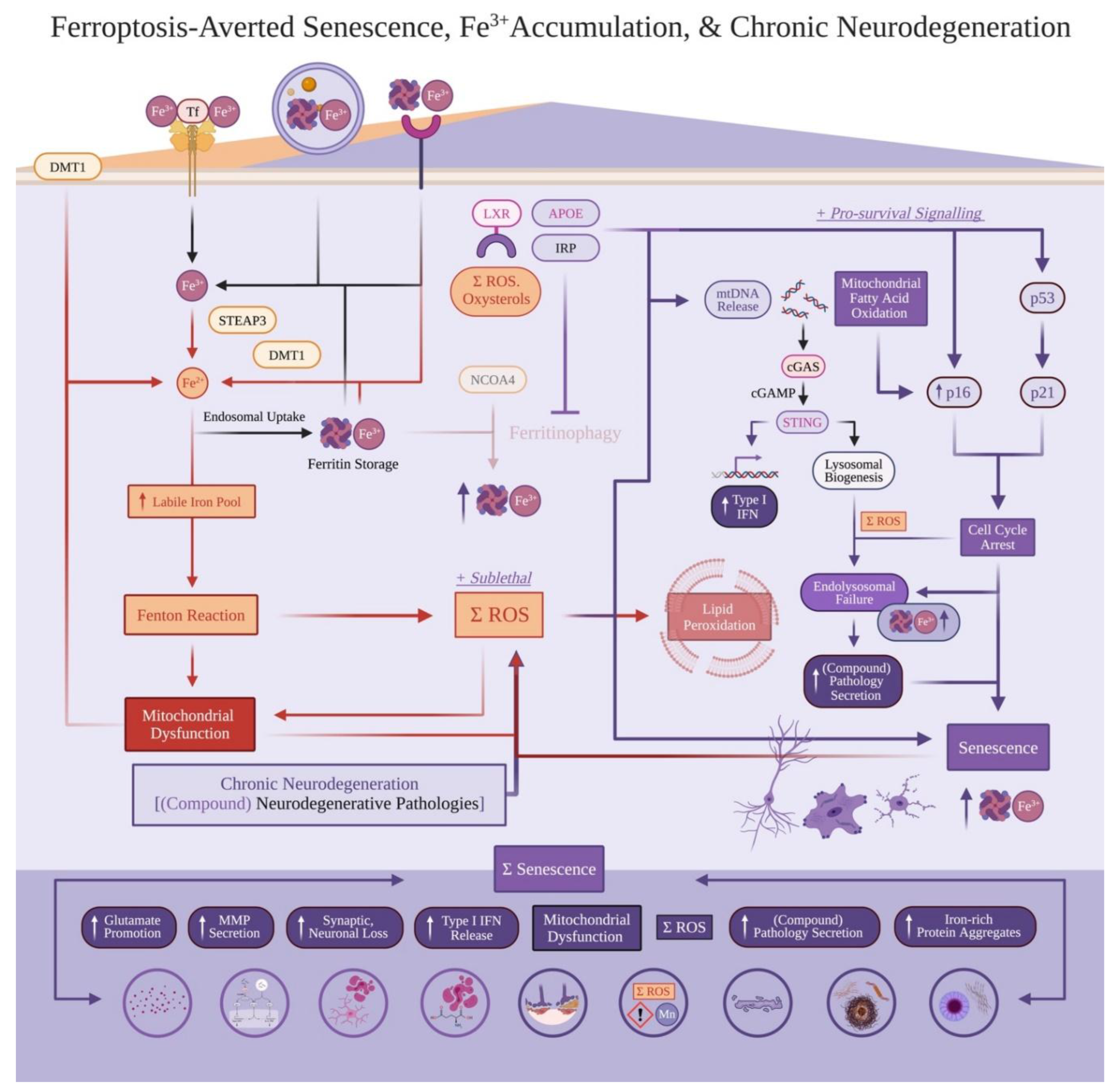
Figure 4.
Spatiotemporal Iron Accumulation Progresses Specific and Concurrent Neurodegeneration. While ferroptosis and senescence occur even in healthy neurodevelopment, their buildup and accompanying localized iron accumulation is predicted to drive chronic neurodegeneration in a spatiotemporal manner. Aligning with post-mortem studies, most patients are predicted to present with mixed and synergizing neurodegenerative diseases; this is proposed to be due to localized iron accumulation (via ferroptosis and senescence buildup). However, localized iron accumulation can still occur and drive isolated or co-existing, unrelated neurodegenerative diseases; a potential instance may include a rare, co-existing confirmation of cerebellar strokes in a patient confirmed with AD. In the bottom half, an example is given as to how cerebrovascular contributions (VCID) add to chronic neurodegeneration earlier in life; when localized iron accumulation sufficiently increases in the temporal lobe, increased global iron burden (due to VCID) leads into a patient with subsequent dementia diagnosis involving mixed p-tau spreading, Aβ burden, and cerebrovascular pathology (VCID + AD). Near late-life disease progression, this example patient later accumulates more iron leading into additional LB aggregation and motor impairment (VCID + AD + PD). The hourglass represents increasing total burden of chronic neurodegeneration and severity of clinical progression, wherein spatial CNS regions accumulate more iron over time leading into mixed neurodegenerative diseases. Finally, this framework of spatiotemporal, localized iron accumulation novelly preserves specificity in (i) predicting singular neurodegenerative diseases, while also (ii) explaining and accounting for compound disease progression. Figure created with BioRender.com.
Figure 4.
Spatiotemporal Iron Accumulation Progresses Specific and Concurrent Neurodegeneration. While ferroptosis and senescence occur even in healthy neurodevelopment, their buildup and accompanying localized iron accumulation is predicted to drive chronic neurodegeneration in a spatiotemporal manner. Aligning with post-mortem studies, most patients are predicted to present with mixed and synergizing neurodegenerative diseases; this is proposed to be due to localized iron accumulation (via ferroptosis and senescence buildup). However, localized iron accumulation can still occur and drive isolated or co-existing, unrelated neurodegenerative diseases; a potential instance may include a rare, co-existing confirmation of cerebellar strokes in a patient confirmed with AD. In the bottom half, an example is given as to how cerebrovascular contributions (VCID) add to chronic neurodegeneration earlier in life; when localized iron accumulation sufficiently increases in the temporal lobe, increased global iron burden (due to VCID) leads into a patient with subsequent dementia diagnosis involving mixed p-tau spreading, Aβ burden, and cerebrovascular pathology (VCID + AD). Near late-life disease progression, this example patient later accumulates more iron leading into additional LB aggregation and motor impairment (VCID + AD + PD). The hourglass represents increasing total burden of chronic neurodegeneration and severity of clinical progression, wherein spatial CNS regions accumulate more iron over time leading into mixed neurodegenerative diseases. Finally, this framework of spatiotemporal, localized iron accumulation novelly preserves specificity in (i) predicting singular neurodegenerative diseases, while also (ii) explaining and accounting for compound disease progression. Figure created with BioRender.com.
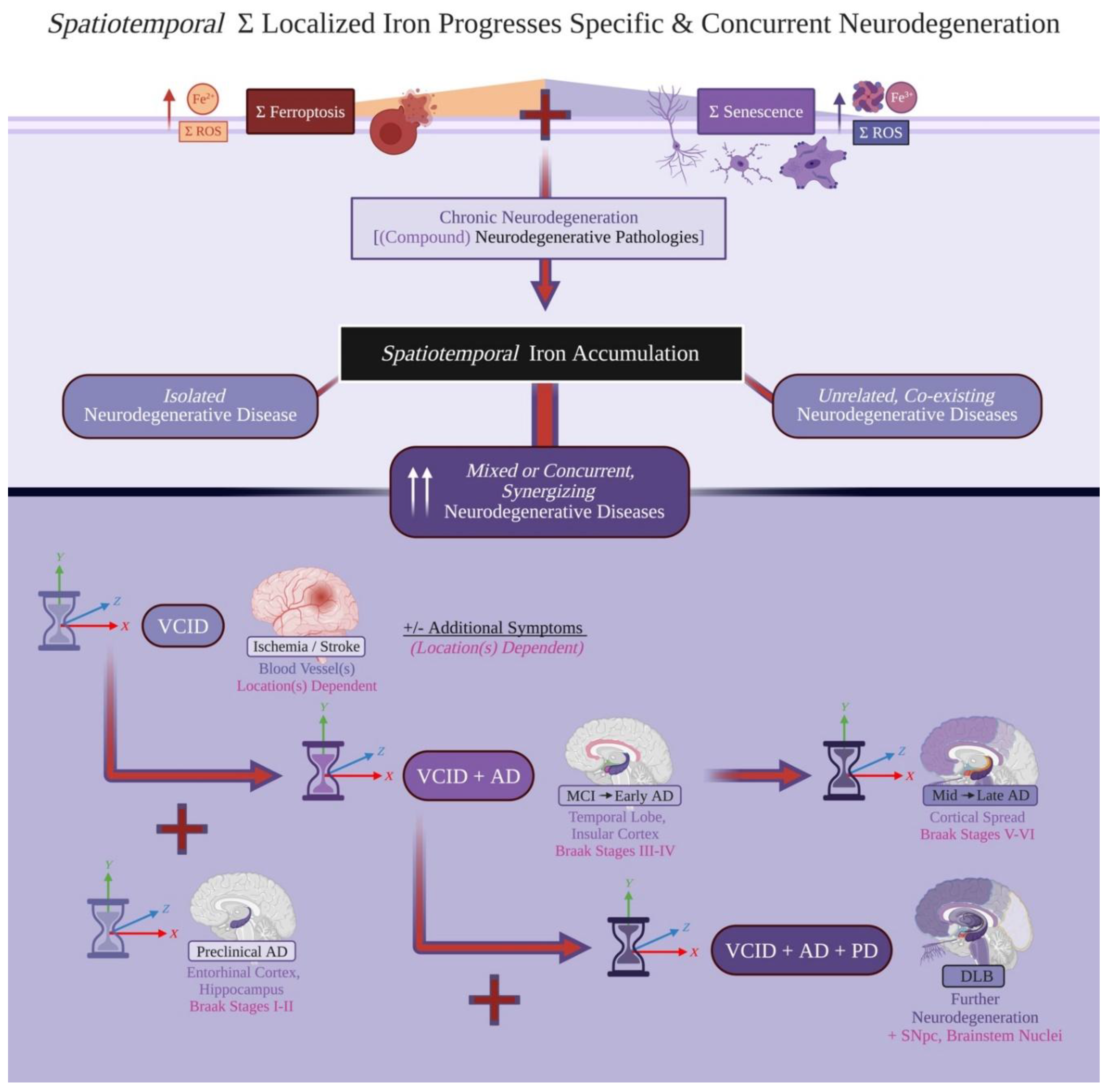
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



