
Submitted:
31 October 2025
Posted:
31 October 2025
You are already at the latest version
Abstract
Chemical machining serves essential applications throughout the electronics industry in the manufacture of diverse metallic components. Etchants of transition metal salts are especially favored for their ability to be regenerated by simple oxidizing agents, including atmospheric oxygen, allowing steady-state processing. Despite their relevance, elementary steps in the reaction mechanisms of copper chemical machining have not yet been fully characterized. This review draws from supporting literature to assert hypotheses of intermediate coordination states in the heterogeneous electron transfer mechanism of the surface dissolution reaction by each of three common etchants of transition metal salts: ferric chloride, acidic cupric chloride, and alkaline cupric ammine chloride. Primary evidence is evaluated as well for mechanisms asserted of their aerobic regeneration pathways. Additionally, the reader is directed to relevant mathematical models cited throughout the article, and areas for further research are identified in each section.
Keywords:
etching
; etchant
; anodic dissolution
; corrosion
; cupric chloride
; ferric chloride
; copper ammine
1. Introduction
The dissolution process where complimentary redox reactions occur spontaneously and simultaneously on the same surface of the electrode is known as chemical machining and was heavily advanced during the 1900s by the electronics industry for application in the manufacture of conductors on printed circuit boards, lead frames of semiconductor devices, and other electronic components [1,2]. In these modern applications, both photographic and chemical techniques are employed sequentially, a process termed photochemical machining, to selectively remove the area to be machined (Figure 1): the workpiece is coated with a photoactive polymer, areas to be retained are exposed and developed, then the undesired material is chemically machined, leaving material retained only in areas protected beneath the photopolymer [1,2,3]. In general, this process is known by many names including etching, wet etching, photoetching, photochemical milling, photoengraving, and chemical micromachining among others depending on context [1,2,3,4].
Chemical machining of Group 11 elements (Cu, Ag, Au) by this solution method has the comparative advantages of lower capital investment costs for tools, independence from physical properties of the workpiece, high selectivity regarding other films and materials in device structures, and high precision on micrometer scales [2,5]. These methods are not suitable, however, on nanometer scales where controlled profiles are required for advanced microelectronics, due to the isotropic nature of chemical machining [1,2,3,4,5,6]. Machining by liquid, as well as by gaseous and vapor-phase etchants, is generally isotropic, having a non-zero etching rate in all directions [2,5]. As microcomponents are advanced toward smaller dimensions, the thickness of the material becomes comparable to the lateral pattern dimensions to the extent which the lateral etching imparted by isotropy of the etchant may become intolerable or difficult to control [4,7,8]. This has been noted as the most influential factor in fabricating three-dimensional microcomponents of micro-electromechanical systems [4]. For this reason, chemical machining equipment is usually designed with pressurized sprays impacting the workpiece with etchant perpendicularly, which has been shown to give some preference to vertical etching over the lateral direction [3,6,9]. Several mathematical models have been developed which simulate the isotropic nature of chemical machining [10,11,12].
Alternative process have been developed toward anisotropic machining at nanometer scales with nearly vertical side walls, using machining agents such as ion beams, laser and high-intensity photon sources, and plasmas [1,5]. Although, these methods pose a much greater expense which keeps chemical machining a more viable option when pattern dimensions would tolerate its degree of isotropy. Additionally, chemical machining generally provides greater etching rates and selectivity [1]. Common etchants used for photochemical machining are transition metal salts: acidic ferric chloride, acidic cupric chloride, and alkaline cupric ammine chloride; others include chromic-sulfuric acid, ammonium persulfate, and hydrogen peroxide with sulfuric acid [1,4]. The transition metal salts have the comparative advantages of greater reaction rates and the capability to be regenerated for a steady-state system [1,2,13]. This is especially true for cupric chloride, where reaction products maintain the bath composition so long as chloride is kept in excess, but less true for ferric chloride, where reaction products change the bath composition from its starting condition as dissolved copper species accumulate [1,2,13].
In production of multi-layered printed circuit boards, conductors are connected between layers by a through-hole plating process typically utilizing metallic resist during machining, such as a tin-lead alloy, rather than a photopolymer [1]. Selectivity is therefore required of the etchant to remove copper while preserving the metallic resist. Among the three noted etchants of transition metal salts, alkaline cupric ammine chloride provides the greatest etch rate and capacity for dissolved copper with the selectivity needed to preserve tin and tin-lead alloy resists. Although, these benefits come by greater operation and copper recovery costs [2,4,13]. In contrast, acidic cupric chloride is favored due to its lower costs and chemical simplicity in machining and regeneration, but it exhibits the slowest etch rate of the three without selectivity for copper, limiting its typical application in printed circuit board manufacturing to chemical machining of individual layers [2,4,13,14,15]. Ferric chloride is a universal metal etchant which has decreased in popularity for high-volume machining of copper due to its lower capacity for dissolved copper than acidic cupric chloride, despite usually having greater etch rates [2,4,13,15,16].
Despite a long-standing history and industrial usage, elementary steps in the reaction mechanisms of copper chemical machining have yet to be fully characterized. The machining principle, however, is known to involve two sequential stages: oxidation of the metal surface followed by solvation of the oxidation products [5,17]. Therefore, the overall rate of etching is not only dependent on the intrinsic redox reaction but also on the dissolution of oxidation products from the metal surface.
While reduction of the Cu(II) metal center of reacting etchant is observed to occur exclusively through two successive, one-electron transitions between Cu(II) and Cu(0) [18,19], oxidation of the solid copper workpiece is more intricate. At sufficiently low applied potentials in acidic chloride solutions, which applies to both ferric chloride and cupric chloride, electrodissolution of copper may proceed by a single step involving termolecular addition of two chlorides to solid copper [20]. Although, this is relevant only to electrochemical methods which apply an external bias rather than to industrial chemical machining usually operating at open-circuit potentials. Otherwise, two-step oxidation is seen near the open-circuit potential in acidic chloride solutions [18], while solid copper in alkaline ammonia solutions exhibits two-step oxidation only for thin layers of electroplated copper electrodes tested from 0.03-µm thickness [19]. Indeed, the bidirectional diffusion limitation at the copper surface imposed by its passivation in conjunction with viscosity arising from high specific gravity and ionic strength of industrial etchants makes industrial chemical machining reactions generally mass transfer-limited and diffusion-controlled [3,7,21,22,23].
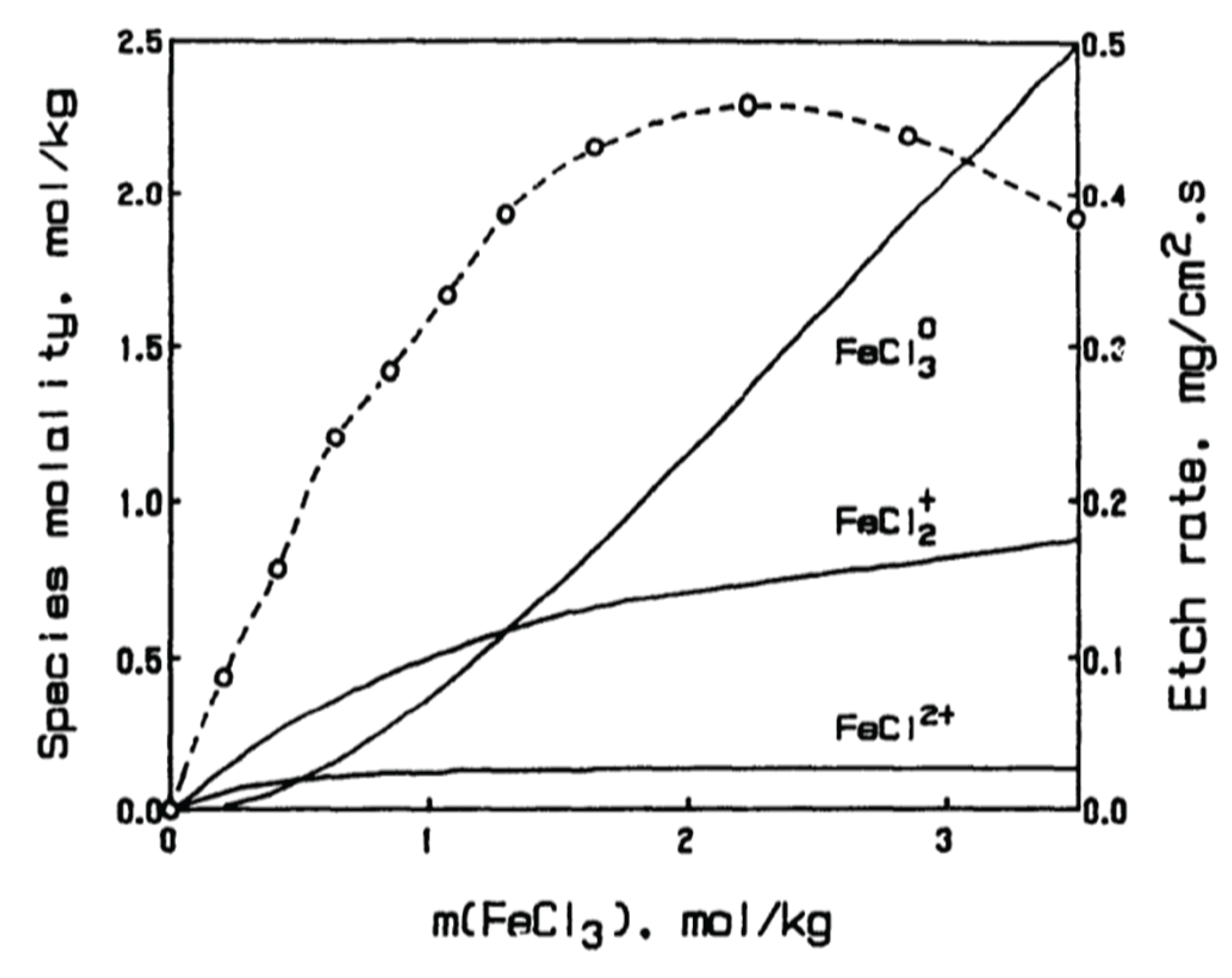
In the example of ferric chloride, the actual dissolution rate of copper increases nearly linearly with ferric chloride molarity, as expected by the mass action law, but then reverses after a maximum [3,24]. This reversal is seen only in the actual dissolution rate, where effects of mass transfer are neglected. Correcting for mass transfer shows the rate intrinsic to the dissolution reaction instead continues to increase asymptotically at greater concentrations, demonstrating the limitations of the diffusion-controlled process [3,24].
Essential to the goal of designing and modeling reactors for anisotropic chemical machining is to establish an understanding of the mechanisms driving the reactions as well as of those which are limiting. This review draws from supporting literature to assert hypotheses of the heterogeneous electron transfer (ET) mechanism involved in the surface machining reaction of each of the three case etchants and evaluates primary evidence for their aerobic regeneration mechanisms as well. Passivation and stability of the dissolved metals will be examined in another review.
2. Chemical Machining
While the etching mechanisms of ferric chloride, cupric chloride, and alkaline cupric ammine chloride are all commonly explained using their neutral salts (FeCl3, CuCl2, and Cu(NH3)4Cl2,, respectively) as the active oxidizing agents, this supposition is not well-supported by literature. Rather, machining mechanisms have been found to proceed by intermediate coordination states [25]; or, as in the case of ferric chloride, studies correlating etch rate with speciation have found diminishing effects on etch rate with prevalence of the neutral salt, hypothesizing FeCl3 to be nonreactive [3] or even to act as an inhibitor [24]. Although, the indication of inhibition rather than inactivity seems confounded due to diffusion limitations at high ionic strengths. When corrected for mass transfer, the etch rate is seen to increase asymptotically up to 3.5 M rather than reversing, exemplifying the importance of examining intrinsic kinetics in addition to those observed [3].
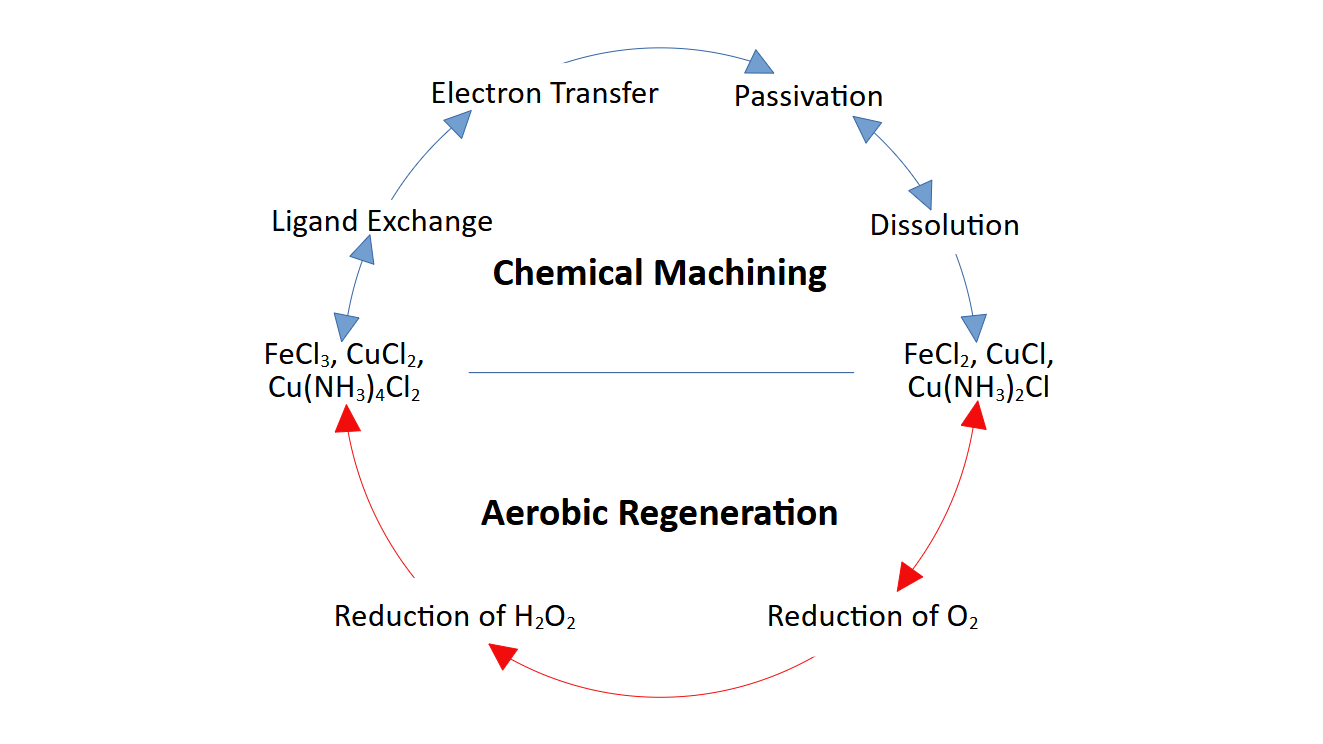
For each of the three case etchants, equilibrium in known between coordination states of the metal center [25,26,27]. The coordination numbers of reported dominant species at industrial concentrations overlap between oxidized and reduced forms, indicating a probable ET pathway with reduced reorganization energy consistent with adiabatic transfer. Given the speciation distributions and ligand exchange equilibria discussed in this section, it is hypothesized here that ET for each of the three etchants is mediated by ligand exchange which equalizes inner-sphere coordination of the oxidized and reduced forms of the metal center to facilitate ET. Although, this pathway is dependent on both, rapid ligand exchange rates on the order of the intrinsic ET timescale to constitute a pre-equilibrium condition in addition to sufficient electronic coupling between the donor and acceptor. Pre-equilibrium seems likely in most cases when considering the small ligands involved (chloride and ammonia) and the effect on lability by Jahn-Teller distortion brought by the d9 configuration of the copper(II) reactants [28]. Electronic coupling may also be benefited, in the absence of passivation, by the heterogeneous nature of chemical machining reactions: solid-phase metal reacting with liquid-phase etchant. Literature related to this hypothesis is reviewed within this section.
2.1. Ferric Chloride
The ferric chloride etchant is unique in that its salt serves two purposes in the chemical machining of copper, reacting twice as an oxidizing agent. The solid copper is first oxidized to copper(I), which forms a passive copper(I) chloride film adsorbed onto the copper surface that dissolves in the presence of free chloride [15,17]. Then, dissolved copper(I) products are again oxidized by ferric chloride to copper(II) chloride, which acts independently as cupric chloride etchant in the same bath volume [15]. This result brings complexity to the use of ferric chloride etchant on copper. The bath composition changes as the reaction proceeds and the overall etchant behavior is altered [29].
Throughout a range of ionic strengths encompassing industrial etchant concentrations, the predominant complexes of iron(III) are FeCl3 and FeCl2+, accounting for well over 50% of all species [24,26,30,31] (Figure 2). Lesser concentrations of FeCl2+ and hydrated Fe3+ are equilibrated in solution, and at high chloride concentrations, FeCl4- is observed as well [24,32]. As for the reduced form, iron(II) is known only to form the stable complexes FeCl2 and FeCl– in aqueous chloride solutions, with the dichloro complex most prevalent [26,30,33,34,35]. These dichloro complexes of iron(III) and iron(II) prefer identical structures of their inner coordination sphere, with chloride ligands in trans positions of the otherwise hydrated octahedron [30,35,36]. Thus, there may be a viable ET pathway with reduced reorganization energy, Reactions (1) and (2).
FeCl3(aq) ⇌ FeCl2+(aq) + Cl–(aq)
FeCl2+(aq) + Cu(s) + Cl–(aq) → FeCl2(aq) + CuCl(s)
Net Reaction:
FeCl3(aq) → FeCl2(aq) + CuCl(s)
The literature search did not yield rate constants for the ligand exchange from FeCl3 to FeCl2+, which is necessary to evaluate the premise of ligand exchange fast enough to constitute a pre-equilibrium condition to ET. However, other exchange rates for the ferric complex are reported on the order of 103 M–1s–1 [31].
The oxidation of dissolved copper(I) to copper(II) by ferric chloride has been proposed to proceed by simultaneous pathways of free Fe3+ and FeOH2+ as the oxidizing agents [37]. This does not seem applicable under industrial conditions, however, given the pH range of 0.0-1.3 and the micromolar FeCl3 concentrations at which the experiment was performed, considering the known concentration-dependence of iron chloride speciation (Figure 2). These proposed species do not comprise a significant portion of iron(III) species at greater concentrations [24,26]. Under industrial conditions, this homogeneous ET instead likely proceeds by some non-adiabatic mechanism which may involve the trichloro or again the dichloro ferric complex with a bridging ligand between the two solvated metal complexes. A complete conjecture will not be proposed in this article but must be addressed by further research.
2.2. Acidic Cupric Chloride
Copper(II) complexes are extremely labile with typical ligand exchange rate constants reported on the order of 106−109 M−1s−1 [38]. The trichloro complex, CuCl3–, has been reported as the dominant species in reacting solutions [7,39,40]. Although, speciation distributions describing coordination of more than two chlorides as a function of copper(II) chloride concentration seem to be lacking [7,26], despite their known formation in solution [27,38,41]. Otherwise, the dichloro complex has been reported in majority concentrations [26]. As for copper(I), it is well-agreed that dichloro and trichloro states are also most prevalent, with the trichloro complex, CuCl32–, being the major species in concentrated solutions [23,26,42]. Considering the potential for fast lability to provide sufficient pre-equilibrium, it may be the case that ET proceeds with significant quantity by reduction of both, the trichloro and dichloro cupric complex, Reactions (4)–(7).
CuCl2(aq) + Cl–(aq) ⇌ CuCl3–(aq)
CuCl3–(aq) + Cu(s) + Cl–(aq) → CuCl32–(aq) + CuCl(s)
And simultaneously,
CuCl2(aq) + Cu(s) + Cl–(aq) → CuCl2–(aq) + CuCl(s)
CuCl2–(aq) + Cl–(aq) ⇌ CuCl32–(aq)
Both resulting in the net reaction:
CuCl2(aq) + Cu(s) + 2Cl–(aq) → CuCl32–(aq) + CuCl(s)
The viability of these mechanisms to proceed with such quantity would still depend on the specific ligand exchange rates relative to the intrinsic ET timescale as well as on the prevalence of dichloro copper(II). Reduction of the trichloro cupric complex is also the most thermodynamically favored, indicated by its greater standard reduction potential among other cupric chloro complexes [40].
2.3. Alkaline Cupric Ammine Chloride
In molar aqueous ammonia, copper(II) tetrammine and copper(I) diammine are the predominant species, preferring square-planar and linear configurations of ammine ligands, respectively [43,44]. This difference in preferred coordination geometry poses a reorganization energy barrier to direct reduction of the tetrammine, suggesting there may be an intermediate configuration which allows ET to proceed with reduced inner sphere reorganization. Indeed, Butler-Volmer analysis of the Cu(II)/Cu(I) redox couple in aqueous ammonium sulfate solutions shows the tetrammine complex is not the electroactive species [25]. Rather, a triammine intermediate was determined to be the active species in reduction of Cu(II), independent of electrode material, by a mechanistic analysis with impedance spectroscopy, Reactions (9)–(11). It should be noted that cuprous oxide, Cu2O, is also known to form as a passive species in addition to cuprous chloride, CuCl [45]; however, the associated formation mechanism is omitted here and deferred to a future review.
Cu(NH3)42+(aq) ⇌ Cu(NH3)32+(aq) + NH3(aq)
Cu(NH3)32+(aq) + Cu(s) + Cl–(aq) → Cu(NH3)3+(aq) + CuCl(s)
Cu(NH3)3+(aq) ⇌ Cu(NH3)2+(aq) + NH3(aq)
Net Reaction:
Cu(NH3)42+(aq) + Cu(s) + Cl–(aq) → Cu(NH3)2+(aq) + 2NH3(aq) + CuCl(s)
The mechanistic analysis itself is based on the supposition of the oxidized and reduced forms having equal coordination numbers. This premise seems reasonable considering likely fast exchange equilibrium of the small ammonia ligand in conjunction with the greater energetic barrier of an otherwise direct ET [25]. Equal coordination numbers of both species then promotes adiabatic ET following fast ligand exchange pre-equilibrium, which is supported by the finding of the transfer step being rate-limiting [25]. The triammine species has also been identified spectrophotometrically in ammonia solutions at unit ionic strength [46] as well as in liquid ammonia [47].
3. Aerobic Regeneration
A primary benefit of chemical machining with etchants of transition metal salts is their ability to be regenerated for continuous, steady-state machining [1,13]. Common oxidizing agents are used to regenerate the reduced reaction products, including sodium chlorate, hydrogen peroxide, gaseous chlorine, gaseous oxygen, and electrochemical methods [13,48,49,50]. Although, regeneration by oxygen from atmospheric air has long been considered the most appealing option, on account of its comparative advantages in cost and safety [13,48,51,52].
Being an instance of mass transfer with chemical reaction [53], considerations also have long been made for machine designs which sparge air through the etchant bath for increased regeneration efficiency, increasing the reaction rate by the mass-action law as reactant concentration increases [48,52]. As a non-polar gas, oxygen exhibits very low solubility in water which decreases further with electrolyte concentration [54,55]. Solubility of oxygen in water also decreases as temperature rises from 0–100 °C but then increases above 100 °C as temperature continues to rise [55]. pH alone does not have a direct effect on solubility [56]. Rather, its main influence would be in changing protonation states of dissolved species which may then exert some effect on oxygen solubility. Mathematical models have been developed to determine oxygen solubility in inorganic solutions at a given temperature [57]; although, the assumption of negligible undissociated ionic salts is likely to lead to underestimations in the case of etchants at industrial ionic strengths. Otherwise, simple estimations exist for oxygen solubility in electrolyte solutions relative to pure water [54].
Mechanisms have been proposed for oxidation by oxygen of iron(II) and copper(I), relating to the acidic etchants, ferric and cupric chloride. An outer-sphere ET is described [58], Reactions (13)–(15), where ML and ML+ are generalized as the reduced and oxidized metal center coordination complex, respectively. Ligands and phase labels are neglected, as kinetically active complexes in these paths have not yet been elucidated and all species other than water are aqueous.
M + O2 + H+ ⇌ M+ + HO2
M + HO2 → M+ • HO2–
M+ • HO2– → M+ + HO2–
Alternatively, an inner-sphere mechanism is proposed for the first ET to oxygen, Reactions (16) and (17) [58,59].
M + O2 ⇌ M • O2
M + M • O2 + H+ → 2M+ + HO2–
Following dissociation of the perhydroxyl anion, HO2–, from both mechanisms, either from the transient outer-sphere complex or the inner-sphere adduct, reduction continues to form hydrogen peroxide and follows the standard reduction mechanism for hydrogen peroxide in acidic solution to form water, Reactions (18)–(21) [58,60].
HO2– + H+ ⇌ H2O2
M + H2O2 → M+ + OH– + OH
M + OH → M+ + OH–
2OH– + 2H+ → 2H2O
In either case, the net reaction is achieved:
4M + O2 + 4H+ → 4M+ + 2H2O
The application of these mechanisms to each of the three case etchants is discussed within this section.
3.1. Ferric Chloride
Oxidation of iron(II) by oxygen is a slow process on the order of 10-2 M–1s–1 [58], which seems unsuitable for the purpose of maintaining stable machining conditions for most production demands. Independent of acid concentration, oxygen is not observed to regenerate iron(III) on the necessary timescale of live industrial processing [3,52]. Early research even suggested oxygen to act as a surface poison reducing active centers on the copper surface available for reaction, as etching rates in solutions near 1.5-3.5 M FeCl3 are decreased when saturated with oxygen compared to inert gas [3]. Most success was found with up to 89% conversion after 5 hours of oxygen sparged into boiling etchant under reflux [61]. The limitation is not largely due to mass transfer control, as sparging with reduced oxygen bubble surface area by a static mixing design was also ineffective in significantly reducing timescale [48]. Only slow regeneration was observed overnight when the surface area of the etchant droplets were reduced and exposed to ambient air, which appears to have sufficed for limited production demand [48]. However, other gaseous oxidizers are known to be effective, including chlorine and ozone [48,52], thus indicating a mechanism with activation control, rather than diffusion control, for oxidation by molecular oxygen.
The activation energy has been calculated in the range of 71-78 kJ mol–1, decreasing with additional chloride [59]. It has been proposed that the activation limitation lies in the formation of the probable hydrogen peroxide intermediate [3], stating the reduction potential of hydrogen peroxide under standard operating conditions is insufficient to oxidize the ferrous ion. Hydrogen peroxide is, however, a known, effective oxidizer for ferric chloride etchant and is commonly used among other options including sodium chlorate and electrochemical methods [48,62,63]. The limiting step of the mechanism must then be before the formation of a hydrogen peroxide intermediate.
Considering the reduction potentials of involved species [58], the equilibrium of Reaction (13), the first ET to oxygen of the outer-sphere mechanism, is the most thermodynamically unfavorable. The reduction potential of the Fe(II)/Fe(III) couple when the reduced form is in excess is consistently greater by at least 0.1 V than the reduction of O2 to form the perhydroxyl radical, HO2, except at high acidity [58]. The addition of chloride would be expected to promote the reduction of O2 by favoring the forward reaction of equilibrium Reaction (13), since Fe(III) is better stabilized by Cl– than Fe(II), as given by its greater stability constants at various ionic strengths [26] and greater oxidation state with cationic charge density as the harder acid [64]. Indeed, the difference between the reduction potentials diminishes as chloride concentration is increased, which, in conjunction with the finding that rate constants for the oxidation by hydrogen peroxide increases similarly, is aligned with the observation of overall faster oxidation of iron(II) by molecular oxygen with lower activation energy as chloride concentration increases [58,59,65].
Given the unfavorable thermodynamics of Reaction (13), the outer-sphere ET to O2 is not likely for iron(II) unless stronger ligands for Fe(III) are present to further promote the forward reaction of the equilibrium, such as phosphate and fluoride [58]. The derived rate equation of the outer-sphere mechanism predicts an inverse dependence on Fe(II) concentration, which is not observed experimentally, and disagreement also exists between theoretical kinetics and observed rate data of the elementary steps [58]. The inner-sphere mechanism is in better agreement with observed dependence on Fe(II) concentration and observed kinetics, being first order with respect to oxygen and either first or second order with respect to Fe(II), depending on chloride concentration, with concentrations greater than about 3 M shifting to first order kinetics [58,59,66]. Thus, the inner-sphere mechanism, Reactions (16)–(17), for the first ET from molecular oxygen is a more likely contributor to the oxidation of iron(II) [58].
3.2. Acidic Cupric Chloride
The rate of oxidation of copper(I) by molecular oxygen is on the order of 101 M–1s–1, nearly 500 times greater than that of iron(II), whereas the corresponding rates of oxidation by hydrogen peroxide are very similar and much faster still than oxidation by oxygen [58]. This is true even in the presence of excess copper(II), which is known to retard oxidation of copper(I) by oxygen while the rate of oxidation by hydrogen peroxide is unaffected [58,60,67]. Arising from these faster rates, the reaction is controlled primarily by mass transfer, specifically by the diffusion of oxygen on the solution-side of the boundary layer [68]. Mathematical models have been developed to describe oxidation of copper(I) by air in packed, co-current column reactors [12,69], as well as to describe the position of the reaction plane with accounts for diffusion and fluid flow generated by a rotating disk electrode [40].
Opposite the case of ferric chloride, reduction potentials [58] show thermodynamic favor of the first ET to oxygen by the outer-sphere mechanism, Reaction (13). The reduction potential of O2 to form HO2 is consistently greater than that of the Cu(I)/Cu(II) couple, except at pH greater than about 1.0, but the potential difference diminishes quickly with increasing chloride concentration up to 4.5 M [58]. Under this equilibrium, additional chloride would promote the reverse reaction, thereby inhibiting oxidation, since Cu(I) is better stabilized by Cl– than Cu(II), as given by its greater stability constants at various ionic strengths [26] and being a more polarizable, softer acid for the borderline ligand [64]. This effect has been observed experimentally [58,67]. Activation energy for the net reaction was calculated as 12.5 kJ mol–1 at 4 M total chloride and 15 mM CuCl, but this barrier is likely to increase directly with chloride, considering its noted retarding effect [70]. Oxidation is thus more likely to proceed by the outer-sphere mechanism for the first ET, as it shows better alignment of observed kinetic data with theoretical kinetics, and the derived rate equation correctly predicts an inverse dependence on copper(II) [58,67].
There is some disagreement regarding reaction orders. Most studies concur first order with respect to oxygen, while dependence on cuprous species is either determined also as first order or to display second order kinetics when tested at greater concentrations, indicating a possible shift or differences in data analysis [67,68,70]. Also disputed is the effect of speciation. Investigations have disagreed among propositions of which cuprous chloride complex is kinetically active [67,70]. This question should be investigated further, and insight may be gained by measuring rate constants as copper concentration varies from millimolar to several molar under fixed and variable chloride concentration with reference to known speciation distributions.
3.3. Alkaline Cupric Ammine Chloride
Claimed in industry to regenerate by dissolved oxygen from ambient air [50], this conjecture for alkaline cupric ammine chloride etchant apparently has not been proven experimentally. In fact, oxygen solubility in both ammonium chloride and ammonium sulfate decreases rapidly with ionic strength, reaching millimolar solubility less than that of chloride solution by factors near 57–71 at equivalent ionic strength [55]. The literature search did not yield any studies specifically examining homogeneous oxidation of copper(I) by air or oxygen in alkaline media of ammonia and ammonium chloride, indicating this essential process is severely under-studied.
Inferences can be made of the mechanism, however, similarly to that of ferric and acidic cupric chloride. Proton-coupled electron transfer steps proposed by the outer-sphere and inner-sphere mechanisms would be disfavored by alkaline pH, as speciation of reduction intermediates shifts toward their unprotonated conjugates, such as the superoxide anion radical, O2–. Differences in preferred ammine coordination number by Cu(I) and Cu(II) suggests also intermediate ammine configurations in the oxidation pathway, as described similarly in a previous section for the mechanism of the machining reaction. A related study has detected significant presence of [Cu2II(NH3)4O2]2+ as an intermediate in the aerobic oxidation of Cu(I) by extended X-ray absorption fine structure (EXAFS) spectroscopy and supported by density functional theory (DFT), indicating a mechanism of inner-sphere adducts [71]. Although, oxidation was conducted at 200 °C, so lower temperatures may not be able to maintain the steric strain of such an intermediate, and more stable intermediates are instead likely to become more viable closer to industrial conditions.
Equilibrium of the first ET to oxygen would depend on the effect of relative ligand stabilization between the oxidized and reduced forms to favor either the forward or reverse reaction [58]. DFT calculations of ligand field stabilization energy for an array of hydrated ammine configurations of both Cu(I) and Cu(II) show significantly greater stabilization energy for Cu(II) in all cases [43]. Therefore, favor would be expected for the forward reaction, the oxidation of Cu(I). The effect of varying ligand concentration and mixed ligand speciation [72] on rate constants and reduction potentials should be investigated for mechanistic insight as well, although kinetic results would need correction for mass transfer to account for the noted simultaneous result of reduced solubility.
4. Conclusions
While the mechanisms of copper chemical machining by the transition metal salts of ferric chloride, acidic cupric chloride, and alkaline cupric ammine chloride are commonly simplified as proceeding with their neutral salts as the acting oxidizing agents, literature has been presented in support of a hypothesis that these etchants instead proceed through intermediate coordination complexes. It is then recommended, to further investigate the hypothesis and proposed mechanisms, that further research include a mechanistic analysis by electrochemical impedance spectroscopy [73], or some other comprehensive method, in conjunction with DFT calculations of free energies and electronic coupling. Any kinetic investigations should certainly include corrections for mass transfer to avoid confounding results with diffusion control at high ionic strengths. Such investigations may finally provide a comprehensive understanding of the machining mechanisms with detailed intrinsic (mass transfer-corrected) kinetics and actual kinetics, relative rates between ligand exchange and ET, and reaction coordinate diagrams showing potential energy along the complete pathway. Although the mathematical rigor predicating mechanistic analysis by electrochemical impedance spectroscopy seems to discourage its use [73], the technique has already been applied successfully to the case of alkaline cupric ammine chloride etchant [25].
Aerobic oxidation of the reduced forms of these etchants has been discussed since the mid-1900s, but mechanisms of such regeneration have not been well-characterized until recently [58], likewise to their machining mechanisms. However, it seems aerobic regeneration of alkaline cupric ammine chloride still has yet to be elucidated, despite its industrial usage. Establishing a detailed description of the mechanisms driving these fundamental, industrial processes remains essential to the goal of designing and modeling reactors for anisotropic chemical machining of copper surfaces. This article provides a review of the contributing knowledge base along with novel insight and further direction for further research.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The author recently ended employment with a printed circuit board manufacturer engaged in photochemical machining. The entirety of this research was conducted independently by the author only after employment had ceased with neither any affiliation nor funding.
Abbreviations
The following abbreviations are used in this manuscript:
| ET | Electron Transfer |
| DFT | Density Functional Theory |
| EXAFS | X-ray absorption fine structure |
References
- Datta, M.; Romankiw, L.T. Application of Chemical and Electrochemical Micromachining in the Electronics Industry. J. Electrochem. Soc. 1989, 136, 285C. [Google Scholar] [CrossRef]
- Cakir, O. Photochemical Machining of Engineering Materials. In Proceedings of the Trends in the Development of Machinery and Associated Technology; TMT: Istanbul, Turkey, 2008; pp. 109–112. [Google Scholar]
- Burrows, W.H.; Lewis, C.T.Jr.; Saëre, D.E.; Brooks, R.E. Kinetics of the Copper-Ferric Chloride Reaction and the Effects of Certain Inhibitors. Ind. Eng. Chem. Proc. Des. Dev. 1964, 3, 149–159. [Google Scholar] [CrossRef]
- Çakır, O. Review of Etchants for Copper and Its Alloys in Wet Etching Processes. KEM 2007, 364–366, 460–465. [Google Scholar] [CrossRef]
- Choi, T.-S.; Hess, D.W. Chemical Etching and Patterning of Copper, Silver, and Gold Films at Low Temperatures. ECS J. Solid State Sci. Technol. 2015, 4, N3084–N3093. [Google Scholar] [CrossRef]
- Swartzell, J.C. Etching Parameters: Their Individual and Collective Impact on High Density Circuit Production. Circuit World 1981, 8, 21–23. [Google Scholar] [CrossRef]
- Georgiadou, M.; Alkire, R. Anisotropic Chemical Etching of Copper Foil: I. Electrochemical Studies in Acidic Solutions. J. Electrochem. Soc. 1993, 140, 1340–1347. [Google Scholar] [CrossRef]
- Georgiadou, M.; Alkire, R. Anisotropic Chemical Etching of Copper Foil: II. Experimental Studies on Shape Evolution. J. Electrochem. Soc. 1993, 140, 1348–1355. [Google Scholar] [CrossRef]
- Atta, R.M. Effect of Applying Air Pressure during Wet Etching of Micro Copper PCB Tracks with Ferric Chloride. International Journal of Materials Research 2022, 113, 795–808. [Google Scholar] [CrossRef]
- Bruzzone, A.A.G.; Reverberi, A.P. An Experimental Evaluation of an Etching Simulation Model for Photochemical Machining. CIRP Annals 2010, 59, 255–258. [Google Scholar] [CrossRef]
- Georgiadou, M.; Alkire, R. Anisotropic Chemical Pattern Etching of Copper Foil: III. Mathematical Model. J. Electrochem. Soc. 1994, 141, 679–689. [Google Scholar] [CrossRef]
- Kao, A.S.; Stenger, H.G.; Georgakis, C.; Covert, K.L.; Kurowski, J.A. Etch Profile Development in Spray Etching Processes. J. Electrochem. Soc. 1992, 139, 2202–2211. [Google Scholar] [CrossRef]
- Cakir, O. Copper Etching with Cupric Chloride and Regeneration of Waste Etchant. Journal of Materials Processing Technology 2006, 175, 63–68. [Google Scholar] [CrossRef]
- Choi, J.-C.; Lee, Y.-S.; Lee, J.; Kwon, H.-W.; Lee, J.-H. Etching Behaviors of Galvanic Coupled Metals in PCB Applications. In Proceedings of the 2018 Pan Pacific Microelectronics Symposium (Pan Pacific); February 2018; pp. 1–4. [Google Scholar]
- Çakır, O.; Temel, H.; Kiyak, M. Chemical Etching of Cu-ETP Copper. Journal of Materials Processing Technology 2005, 162–163, 275–279. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, Q.; Zhang, X.; Liu, F.; Li, C.; Yang, L.; Xu, C.; Song, Z. Influence of Etchants on Etched Surfaces of High-Strength and High-Conductivity Cu Alloy of Different Processing States. Materials 2024, 17, 1966. [Google Scholar] [CrossRef]
- Jin, L.; Li, K.; Wang, Z.-Y.; Chen, X. Electrochemical and Theoretical Studies for Forward Understanding the Mechanism and the Synergistic Effects of Accelerating Ions in Copper Foil Etching. Materials Today Communications 2025, 42, 111483. [Google Scholar] [CrossRef]
- Low, C.T.J.; Ponce De Leon, C.; Walsh, F.C. Copper Deposition and Dissolution in Mixed Chloride–Sulphate Acidic Electrolytes: Cyclic Voltammetry at Static Disc Electrode. Transactions of the IMF 2015, 93, 74–81. [Google Scholar] [CrossRef]
- DARCHEN, A.; DRISSI-DAOUDI, R.; IRZHO, A. Electrochemical Investigations of Copper Etching by Cu(NH3)4Cl2 in Ammoniacal Solutions. Journal of Applied Electrochemistry 1997, 27, 448–454. [Google Scholar] [CrossRef]
- Cooper, R.S.; Bartlett, J.H. Convection and Film Instability Copper Anodes in Hydrochloric Acid. J. Electrochem. Soc. 1958, 105, 109. [Google Scholar] [CrossRef]
- Kear, G.; Barker, B.D.; Walsh, F.C. Electrochemical Corrosion of Unalloyed Copper in Chloride Media––a Critical Review. Corrosion Science 2004, 46, 109–135. [Google Scholar] [CrossRef]
- Allen, D.M.; Almond, H.J.A. Characterisation of Aqueous Ferric Chloride Etchants Used in Industrial Photochemical Machining. Journal of Materials Processing Technology 2004, 149, 238–245. [Google Scholar] [CrossRef]
- Braun, M.; Nobe, K. Electrodissolution Kinetics of Copper in Acidic Chloride Solutions. J. Electrochem. Soc. 1979, 126, 1666. [Google Scholar] [CrossRef]
- Bryce, C. The Kinetics of Copper Etching in Ferric Chloride-Hydrochloric Acid Solutions, McGill University, Department of Chemical Engineering, 1992.
- Brown, O.R.; Wilmott, M.J. A Kinetic Study of the Cu(NH3)4II/Cu(NH3)21 Redox Couple at Carbon Electrodes. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1985, 191, 191–199. [Google Scholar] [CrossRef]
- Kimura, R.T.; Haunschild, P.A.; Liddell, K.C. A Mathematical Model for Calculation of Equilibrium Solution Speciations for the FeCl3-FeCl2-CuCl2-CuCl-HCl-NaCl-H2O System at 25 ‡C. Metall Trans B 1984, 15, 213–219. [Google Scholar] [CrossRef]
- Ramette, R.W. Copper(II) Complexes with Chloride Ion. Inorg. Chem. 1986, 25, 2481–2482. [Google Scholar] [CrossRef]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. 11. In Advanced Inorganic Chemistry; A Wiley-Interscience publication; Wiley: New York, 1999; ISBN 978-0-471-19957-1. [Google Scholar]
- Saubestre, E.B. Copper Etching in Ferric Chloride. Ind. Eng. Chem. 1959, 51, 288–290. [Google Scholar] [CrossRef]
- Apted, M.J.; Waychunas, G.A.; Brown, G.E. Structure and Specification of Iron Complexes in Aqueous Solutions Determined by X-Ray Absorption Spectroscopy. Geochimica et Cosmochimica Acta 1985, 49, 2081–2089. [Google Scholar] [CrossRef]
- Strahm, Ulrich. ; Patel, R.C.; Matijevic, Egon. Thermodynamics and Kinetics of Aqueous Iron(III) Chloride Complexes Formation. J. Phys. Chem. 1979, 83, 1689–1695. [Google Scholar] [CrossRef]
- Marston, A.L.; Bush, S.F. Raman Spectral Investigation of the Complex Species of Ferric Chloride in Concentrated Aqueous Solution. Appl Spectrosc 1972, 26, 579–584. [Google Scholar] [CrossRef]
- Heinrich, C.A.; Seward, T.M. A Spectrophotometric Study of Aqueous Iron (II) Chloride Complexing from 25 to 200 °C. Geochimica et Cosmochimica Acta 1990, 54, 2207–2221. [Google Scholar] [CrossRef]
- Lee, M.-S. Chemical Equilibria in Ferrous Chloride Acid Solution. Met. Mater. Int. 2004, 10, 387–392. [Google Scholar] [CrossRef]
- Luin, U.; Arčon, I.; Valant, M. Structure and Population of Complex Ionic Species in FeCl2 Aqueous Solution by X-Ray Absorption Spectroscopy. Molecules 2022, 27, 642. [Google Scholar] [CrossRef]
- Persson, I. Ferric Chloride Complexes in Aqueous Solution: An EXAFS Study. J Solution Chem 2018, 47, 797–805. [Google Scholar] [CrossRef]
- Orth, R.J.; Liddell, K.C. Rate Law and Mechanism for the Oxidation of Copper(I) by Iron(III) in Hydrochloric Acid Solutions. Ind. Eng. Chem. Res. 1990, 29, 1178–1183. [Google Scholar] [CrossRef]
- Mereshchenko, A.S.; Olshin, P.K.; Karabaeva, K.E.; Panov, M.S.; Wilson, R.M.; Kochemirovsky, V.A.; Skripkin, M.Y.; Tveryanovich, Y.S.; Tarnovsky, A.N. Mechanism of Formation of Copper(II) Chloro Complexes Revealed by Transient Absorption Spectroscopy and DFT/TDDFT Calculations. J Phys Chem B 2015, 119, 8754–8763. [Google Scholar] [CrossRef]
- Ramette, R.W.; Fan, G. Copper(II) Chloride Complex Equilibrium Constants. Inorg. Chem. 1983, 22, 3323–3326. [Google Scholar] [CrossRef]
- Georgiadou, M.; Alkire, R. Modelling of Copper Etching in Aerated Chloride Solutions. Journal of Applied Electrochemistry 1998, 28, 127–134. [Google Scholar] [CrossRef]
- Hathaway, B.J. A New Look at the Stereochemistry and Electronic Properties of Complexes of the Copper(II) Ion. In Proceedings of the Complex Chemistry; Springer Berlin Heidelberg: Berlin, Heidelberg, 1984; pp. 55–118. [Google Scholar]
- Fritz, J.J. Chloride Complexes of Copper(I) Chloride in Aqueous Solution. J. Phys. Chem. 1980, 84, 2241–2246. [Google Scholar] [CrossRef]
- Pavelka, M.; Burda, J.V. Theoretical Description of Copper Cu(I)/Cu(II) Complexes in Mixed Ammine-Aqua Environment. DFT and Ab Initio Quantum Chemical Study. Chemical Physics 2005, 312, 193–204. [Google Scholar] [CrossRef]
- Noguchi, D. The CCDC Database of Crystal Structures of Tetraamminecopper (II) [Cu(NH3)4]2+: Complicated Geometry of a Well-Known Complex Ion. Journal of the Korean Chemical Society 2022, 66, 61–66. [Google Scholar] [CrossRef]
- Larin, V.I.; Khobotova, E.B.; Datsenko, V.V.; Dobriyan, M.A. The Chemical Passivation of Copper in Ammonia Solutions Containing Chlorine Ions. Russ. J. Phys. Chem. A 2008, 82, 1490–1494. [Google Scholar] [CrossRef]
- Braish, T.F.; Duncan, R.E.; Harber, J.J.; Steffen, R.L.; Stevenson, K.L. Equilibria, Spectra, and Photochemistry of Copper(I)-Ammonia Complexes in Aqueous Solution. Inorg. Chem. 1984, 23, 4072–4075. [Google Scholar] [CrossRef]
- Nilsson, K.B.; Persson, I. The Coordination Chemistry of Copper(I) in Liquid Ammonia, Trialkyl and Triphenyl Phosphite, and Tri-n-Butylphosphine Solution. Dalton Trans. 2004, 1312–1319. [Google Scholar] [CrossRef]
- Allen, D.M.; Jefferies, P. An Economic, Environment-Friendly Oxygen-Hydrochloric Acid Regeneration System for Ferric Chloride Etchants Used in Photochemical Machining. CIRP Annals 2006, 55, 205–208. [Google Scholar] [CrossRef]
- Chemcut Corporation Process Guidelines for Cupric Chloride Etching 2015.
- Chemcut Corporation Process Guidlines for Alkaline Etching 2002.
- Fadhil, B.H. COPPER ETCHING IN AIR REGENERATED CUPRIC CHLORIDE SOLUTION. jcoeng 2010, 16, 5771–5777. [Google Scholar] [CrossRef]
- Black, O.D.; Cutler, L.H. New Method for Etching Copper. Ind. Eng. Chem. 1958, 50, 1539–1540. [Google Scholar] [CrossRef]
- McCabe, W.L.; Smith, J.C.; Harriott, P. Gas Absorption. In UNIT OPERATIONS OF CHEMICAL ENGINEERING; McGraw-Hill, 2005; pp. 607–610 ISBN 0-07-124710-6.
- Narita, E.; Lawson, F.; Han, K.N. Solubility of Oxygen in Aqueous Electrolyte Solutions. Hydrometallurgy 1983, 10, 21–37. [Google Scholar] [CrossRef]
- Miyamoto, H.; Yampolski, Y.; Young, C.L. IUPAC-NIST Solubility Data Series. 103. Oxygen and Ozone in Water, Aqueous Solutions, and Organic Liquids (Supplement to Solubility Data Series Volume 7). Journal of Physical and Chemical Reference Data 2014, 43, 033102. [Google Scholar] [CrossRef]
- Groisman, A.S.; Khomutov, Ν.Ε. Solubility of Oxygen in Electrolyte Solutions. Russian Chemical Reviews 1990, 59, 707–727. [Google Scholar] [CrossRef]
- Tromans, D. Oxygen Solubility Modeling in Inorganic Solutions: Concentration, Temperature and Pressure Effects. Hydrometallurgy 1998, 50, 279–296. [Google Scholar] [CrossRef]
- Akilan, C.; Nicol, M.J. Kinetics of the Oxidation of Iron(II) by Oxygen and Hydrogen Peroxide in Concentrated Chloride Solutions – A Re-Evaluation and Comparison with the Oxidation of Copper(I). Hydrometallurgy 2016, 166, 123–129. [Google Scholar] [CrossRef]
- Iwai, M.; Majima, H.; Izaki, T. A Kinetic Study on the Oxidation of Ferrous Ion with Dissolved Molecular Oxygen. Denki Kagaku1961 1979, 47, 409–414. [Google Scholar] [CrossRef]
- Nicol, M.J. Kinetics of the Oxidation of Copper(I) by Hydrogen Peroxide in Acidic Chloride Solutions. South African Journal of Chemistry 1982, 35, 77–79. [Google Scholar]
- Hock, S.J. Precipitation of Hematite and Recovery of Hydrochloric Acid from Aqueous Iron(II, III) Chloride Solutions by Hydrothermal Processing, McGill University, Department of Mining and Materials Engineering, 2009.
- Kim, Dae-Weon; Park, Il-Jeong; Kim, Geon-Hong; Chae, Byung-man; Lee, Sang-Woo; Choi, Hee-Lack; Jung, Hang-Chul Oxidation Process for the Etching Solution Regeneration of Ferric Chloride Using Liquid and Solid Oxidizing Agent. Clean Technology 2017, 23, 158–162. [CrossRef]
- Qiang, Z.; Chang, J.-H.; Huang, C.-P. Electrochemical Regeneration of Fe2+ in Fenton Oxidation Processes. Water Research 2003, 37, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Colborn, R.; Nicol, M. An Investigation into the Kinetics and Mechanism of the Oxidation of Iron(II) by Oxygen in Aqueous Chloride Solutions. Journal of the South African Institute of Mining and Metallurgy 1973, 73, 281–289. [Google Scholar]
- Posner, A.M. The Kinetics of Autoxidation of Ferrous Ions in Concentrated HCl Solutions. Trans. Faraday Soc. 1953, 49, 382. [Google Scholar] [CrossRef]
- Nicol, M.J. Kinetics of the Oxidation of Copper(I) by Oxygen in Acidic Chloride Solutions. South African Journal of Chemistry 1984, 37, 77–80. [Google Scholar]
- Hine, F.; Yamakawa, K. Mechanism of Oxidation of Cuprous Ion in Hydrochloric Acid Solution by Oxygen. Electrochimica Acta 1970, 15, 769–781. [Google Scholar] [CrossRef]
- Deront, M.; Samb, F.M.; Adler, N.; Péringer, P. Volumetric Oxygen Mass Transfer Coefficient in an Upflow Cocurrent Packed-Bed Bioreactor. Chemical Engineering Science 1998, 53, 1321–1330. [Google Scholar] [CrossRef]
- Tran, T.; Swinkels, D.A.J. THE KINETICS OF OXIDATION OF Cu(I) CHLORIDE BY OXYGEN IN NaCl-HCl SOLUTIONS. Hydrometallurgy 1986, 15, 281–295. [Google Scholar] [CrossRef]
- Negri, C.; Selleri, T.; Borfecchia, E.; Martini, A.; Lomachenko, K.A.; Janssens, T.V.W.; Cutini, M.; Bordiga, S.; Berlier, G. Structure and Reactivity of Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-Ion-Exchanged Chabazite Catalyst for NH3-Mediated Selective Catalytic Reduction. J Am Chem Soc 2020, 142, 15884–15896. [Google Scholar] [CrossRef] [PubMed]
- Velásquez-Yévenes, L.; Ram, R. The Aqueous Chemistry of the Copper-Ammonia System and Its Implications for the Sustainable Recovery of Copper. Cleaner Engineering and Technology 2022, 9, 100515. [Google Scholar] [CrossRef]
- Macdonald, D.D. Review of Mechanistic Analysis by Electrochemical Impedance Spectroscopy. Electrochimica Acta 1990, 35, 1509–1525. [Google Scholar] [CrossRef]
Figure 1.
The photochemical machining process: (a) A copper workpiece coated with a photopolymer; (b) The photopolymer imaged and developed, leaving undesired copper areas exposed for machining; (c) Undesired copper chemically machined with visible isotropy; (d) The photopolymer removed, leaving only the intended copper pattern on the workpiece.
Figure 1.
The photochemical machining process: (a) A copper workpiece coated with a photopolymer; (b) The photopolymer imaged and developed, leaving undesired copper areas exposed for machining; (c) Undesired copper chemically machined with visible isotropy; (d) The photopolymer removed, leaving only the intended copper pattern on the workpiece.
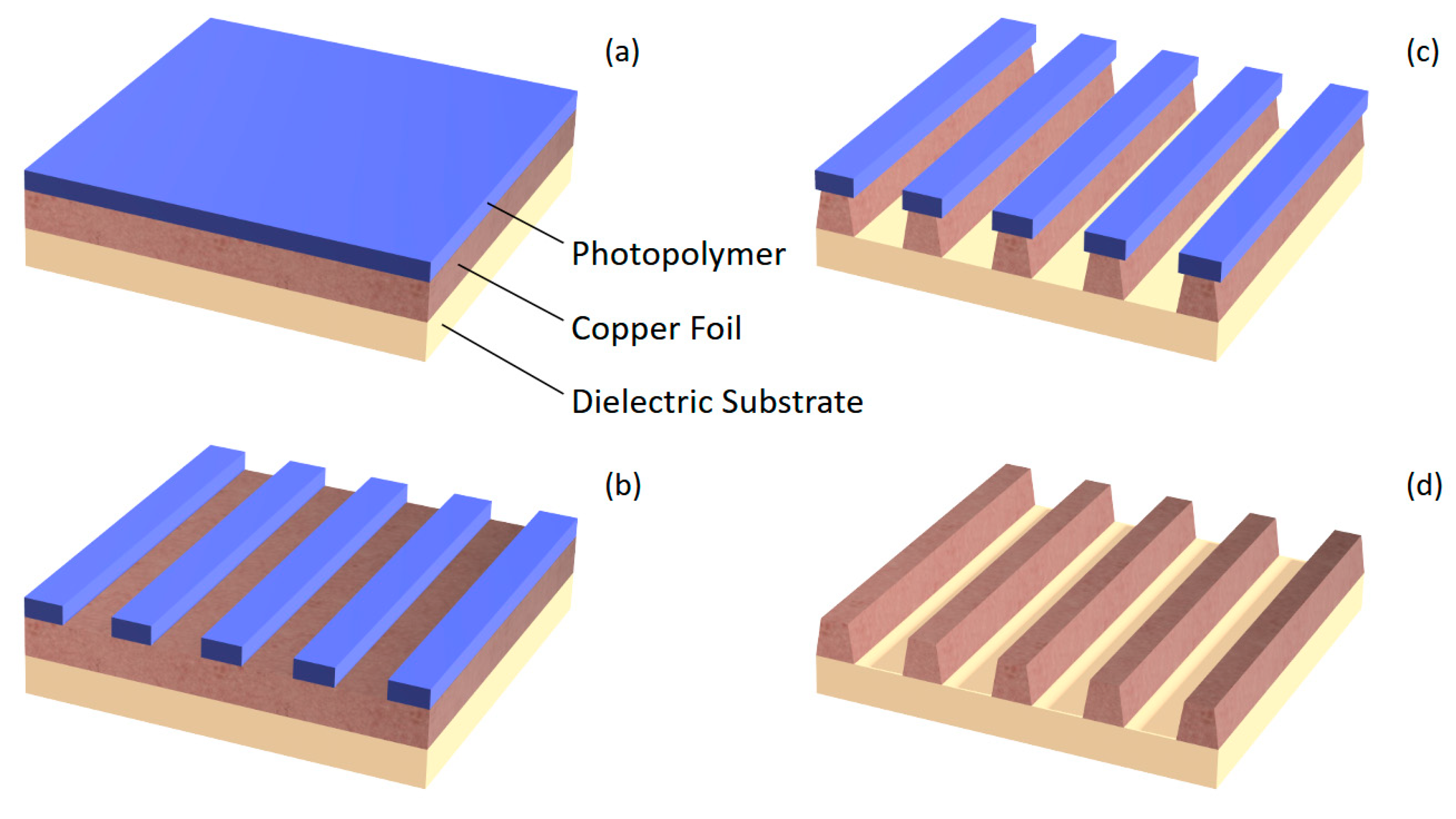
Figure 2.
Ferric chloride speciation distribution and observed etch rate with FeCl3 molality at constant acid concentration (0.84 M HCl) at 50 °C. Adapted from Bryce, 1992 [24].
Figure 2.
Ferric chloride speciation distribution and observed etch rate with FeCl3 molality at constant acid concentration (0.84 M HCl) at 50 °C. Adapted from Bryce, 1992 [24].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



