
Submitted:
21 October 2025
Posted:
22 October 2025
You are already at the latest version
Abstract
Transition metal cyanides form a diverse family of coordination compounds, exhibiting a range of interesting physical and functional properties. Such features are determined by the ability of that ligand to serve as an electron density bridge between the involved transition metal centers and the diversity of coordination modes for the cyanide ligand (CN¯). In fact, such an ability results in a coupling and overlapping of metals electron clouds. In that context, X-ray photoelectron spectroscopy (XPS) appears as an excellent tool for probing the interaction of the CN bridge with the bridged metal centers, the electron density on these last ones, their effective valence, the electron density redistribution effects, and many other features related to the electronic structure of these solids. This review discusses the scope of XPS for probing the electronic structure of the titled family of coordination polymers. Understanding the scope of using this spectroscopic technique for studying these materials opens up new opportunities for engineering their potential applications.
Keywords:
transition metal cyanides
; emerging wide band gap semiconductors
; spin crossover materials
1. Introduction
X-Ray Photoelectron Spectroscopy (XPS) is a multi-element analytical technique that provides information on the nature of the elements present in the sample under study, their valence, electronic configuration, and bonding interactions with the atoms in their coordination sphere. Such structural features are probed by measuring the kinetic energy (KE) of emitted photoelectrons to obtain their binding energy according to Ebinding = Ephoton –(Ekinetic + ϕ), where ϕ is the work function. The emitted photoelectrons that can escape from the solid without energy loss due to inelastic interactions with the solid surface under study typically originate from a depth of up to 10 nm (equivalent to approximately 60 atomic layers) from the very topmost surface atoms. This spectroscopic technique is mainly a surface technique; nevertheless, the relatively small region of a solid sample that is probed provides valuable information on the electronic structure of materials, including those based on coordination compounds. The fundamentals of XPS remain well documented [1,2].
For the CN¯ ligand, strong bonding interactions and different coordination geometries are distinctive features [3]. In coordination polymers based on transition metal cyanides, the simple bridge M-C≡N-T between the metal centers (M, T) dominates [4,5]. The metals (M, T) coordination geometries at the C and N ends of the CN ligand depend on their nature [6]. The CN ligand forms a strong coordination bond at its C end and, in consequence, has a particular preference for transition metals with depopulated eg orbitals. The physical properties of cyanide-based coordination polymers are mainly determined by the subtraction of electron density from the metal M and its relocation at the CN 5σ orbital at the N end. This results in an overlapping of the electron cloud of the M and T metals when the coordination polymer is formed. Such a charge density redistribution explains many of the physical properties of cyanide-based coordination polymers [3,4,7,8]. The study of cyanide-based transition metal coordination polymers has attracted significant attention over several decades [9,10,11,12,13,14,15,16]. However, such interest has been renewed in more recent studies [17,18,19,20,21,22,23], which is the revised subject of this contribution. In the family of coordination polymers known as Prussian blue analogs (PBAs), the two metal centers (M and T) are found to have an octahedral coordination, resulting in MC6 and TN6 octahedra that form three-dimensional (3D) frameworks. The most common combinations of M and T metals in PBAs are M = Ti, V, Cr, Mn, Fe, Ru, Os, Co, Rh, Ir, Pd, and Pt; and T = Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Ag, Zn, and Cd [13,18,19,20,21,22,23]. With T = Zn, the tetrahedral ZnN4 coordination is also usual [19,24].
For M = Ni(II), Pd(II), and Pt(II), related to their strong bonding interaction with the C end of the CN ligand, the formation of a planar M(CN)4 building block results, originating in layered (2D) coordination polymers, T(L)n[M(CN)4], where L is a pillar molecule which occupies the axial coordination sites for the metal T. That pillar molecule may be water or an organic molecule capable of forming a coordination bond with the T metal [25,26,27,28,29,30]. The resulting family of coordination polymers is known as Hofmann-like solids. XPS spectrum provides valuable information on the electronic structure of that family of 2D coordination polymers [20]. The pillar molecule has the potential to modulate the crystal field around the T metal, making the spin transition possible. This is a common thermal-induced effect observed with T = Fe(II) [28,29,30,31,32,33,34].
With Cu(I), Ag(I), and Au(I), the CN ligand forms linear (1D) coordination polymers, M(CN), where adjacent chains remain packed through physical interactions to form 3D solids [35,36]. This subseries is characterized by a specific structural disorder for the metal coordination to the ligand C or N end, relative to the ∙∙∙M−N≡C−M−N≡C−M∙∙∙ configuration, which is the most stable one, according to thermodynamic considerations. The XPS spectrum provides valuable information on such a structural disorder [21,37]. For these three metals (M), the formation of dicyanide-based coordination polymers, T[M(CN)2]2, with a 1D structure, is also possible, but such coordination polymers have not been studied by XPS. For Zn, Cd, and Hg, which also have an nd10 electronic configuration, metal dicyanides are formed: Zn(CN)2, Cd(CN)2, and Hg(CN)2. In this subseries, with a tetrahedral coordination of the metal to the ligand, a particular disorder has been reported for Zn and Cd. In contrast, for Hg, an ordered ligand configuration corresponds [38,39,40]. Except for Hg, the remaining metals in the series form metal cyanides that show a pronounced negative thermal expansion behavior. The experimental XPS results show that this surface technique is an excellent probe for the nature of the structural disorder in this subseries of nd10 metal cyanides [21].
The CN ligand forms octacyanide coordination polymers, T2[M(CN)8] with M = Nb(IV), Mo(IV), and W(IV). Analog solids with these metals in other valence states are also possible. Related to the accommodation of eight CN ligands in their coordination sphere, repulsive interactions between neighboring C atoms appear. This has implications in the π-back-bonding interaction between the CN ligand and the inner metal (M) and in the physical and functional properties of that series of coordination polymers [41].
Transition metal nitroprussides, T[Fe(CN)5NO], are another series of transition metal cyanide-based materials to be considered in this review. This series shares with the remaining coordination polymers herein considered, the π-back-bonding ability of the involved ligands (CN, NO) [42]. Such a π-back-bonding interaction is particularly pronounced for the NO ligand. In fact, according to the XPS spectrum, the effective valence of the iron atom is close to Fe(IV) (discussed below) [22]. The electron density removed from the iron atom is relocated to the O atom of the NO group. This charge relocation has pronounced implications for the physical and functional properties of this series of coordination polymers (discussed below). For dicyanides, T[M(CN)2]2 with M = Cu(+), Ag(+), and Au(+) subseries, and octacyanide, T2[M(CN)8] subseries, no XPS studies have been reported. As mentioned, the remaining cyanide-based coordination polymers have been studied from XPS data [18,19,20,21,22,23,37]. Regarding the physical and functional properties of these materials, such features will be discussed in the context of their XPS spectra (see below). This contribution aims to shed light on the potential of XPS as a tool for probing the electronic structure and related physical and functional properties of cyanide-based coordination polymers.
2. Transition Metal Hexacyanometallates (Prussian Blue Analogs)
This family of coordination polymers is typically formed through a precipitation reaction from aqueous solutions of an appropriate salt of the T metal and a complex anion, such as K4[Fe(CN)6]. For T = Fe(3+), the formed coordination polymer has the formula unit Fe4[Fe(CN)6]3, corresponding to the so-called Prussian blue, the oldest documented coordination polymer [43]. Because the CN ligand behaves as a strong ligand at its C end, such a complex ion is formed for a metal with a maximum of six nd electrons, e.g., Fe(II), Fe(III), Ru(II), Os(II), Co(III), Rh(III), Ir(III), Pd(IV), and Pt(IV) [3]. In the following, the metal in the complex ion will be labeled as the inner metal (M), while the metal bound at the N end is referred to as the outer metal (T). In such coordination polymers, the antibonding π *-orbitals of the CN ligand are found overlapping the t2g orbitals of the metal (M), subtracting electron density from this last one (Figure 1a). The electron density removed from the inner metal is relocated to the CN5σ orbital. In the recorded XPS spectrum, such an electron density accumulation at the CN5σ orbital is appreciated as a decrease in the N 1s core-level binding energy (BE) (Figure 1b). When the coordination polymer is formed, (Tn)(6-m)/n[Mm(CN)6], the electron density accumulated at the CN5σ orbital is partially donated to the outer metal (T), (Figure 1c). This is probed as an increase for the N 1s core-level binding energy (Figure 1d). Such an electron density redistribution during the formation of coordination polymers has relevant implications in the physical and functional properties of PBAs.
The charge density overlapping between neighboring paramagnetic metal centers determines the superexchange integral in molecular magnets and, consequently, the appearance of a magnetic ordering, which is characterized by a critical temperature (Tc). Transition metal cyanide-based coordination polymers are the prototypes of materials for studying molecular magnets. Within PBAs, the highest Tc values are observed for the combination of V2+ and [CrIII(CN)6] [44] (Figure 2). These two metal centers, characterized by a relatively low nuclear charge, possess extended t2g orbitals, which facilitate the overlapping of their electron clouds through the π-back-bonding mechanism. The plot shown in Figure 2 illustrates the dependence of the Tc value on the electron density accumulated at the CN5σ orbital, sensed through the N 1s core-level binding energy for different combinations of paramagnetic T metals with [Cr(CN)6]3− and [Fe(CN)6]3-. In the subseries of molecular magnets based on the [Cr(CN)6]3- ion, the highest values of Tc have been reported [3], which is congruent with the accumulation of the electron density at the CN5σ orbital. The values of Tc for the coordination polymers formed with the [Fe(CN)6]3- complex anion are relatively low, in the 10 to 25 K range [45], which corresponds to the relatively minor electron density accumulated at the CN5σ orbital. For [Mn(CN)6]4-, [Mn(CN)6]3-, and [Mn(CN)6]2−, also paramagnetic complex ions, the formation of PBA-type coordination polymers with magnetic behavior has been reported [46,47], but, for these compositions, no XPS data are available. For this reason, the correlation shown in Figure 2 was limited to [Cr(CN)6]3− and [Fe(CN)6]3− paramagnetic complex ions. Within the coordination polymers resulting from these two complex ions, the highest Tc values correspond to Cr as the inner metal (Figure 2). The mentioned nuclear charge effect explains this behavior. The dependence of the Tc value on the outer metal (T) results from its ability to subtract electron density from the CN5σ orbital, which is determined by the metal’s polarizing power.
An interesting effect in PBAs is the possibility of inducing an electron transfer between the metal centers by the incidence of light, e.g. Co(III)K[Fe(II)(CN)6] (diamagnetic) to form Co(2+)K[Fe(III)(CN)6] (paramagnetic) with a magnetic ordering at low temperature [8] (Figure 2, Inset). This is an exciting effect that has captured the interest of the research community due to its potential applications for information storage devices, spintronics, and sensors. The photoinduced electron transfer between neighboring metal centers in other members of the PBA family of coordination polymers is well-known [3]. This is possible due to a decrease in the energy barrier height between the metal centers, because of their electron clouds overlapping through the π-back donation mechanism. The deep blue color of Prussian blue is also a consequence of such a decrease in the energy barrier for the electron transfer between the metal center through the CN bridge. Prussian blue exhibits broad light absorption in the 600 to 800 nm spectral region, which is why this coordination polymer appears with a deep blue color [48].
XPS is an excellent probe for the valence, electronic configuration, and coordination geometry of the involved metal centers. A change in the number of valence electrons and in their electronic configuration, e.g., low or high spin configuration, modifies the energy levels of the core electrons of the atoms and the binding energy of these electrons. Figure 3a shows the N 1s peak and the corresponding binding energy (BE) for the nitrogen atom in the CN ligand for KCN, K4[Fe(CN)6], and K3[Fe(CN)6]. The formation of the complex ion is characterized by a negative shift in the N 1s BE value of about -0.7 eV relative to KCN, which is related to an accumulation of electron density on the N atom, particularly at the CN5σ orbital. Such an electron density is subtracted from the iron atom via π-back bonding interaction. For K3[Fe(CN)6], the analog BE shift is lower, of about -0.5 eV, due to a minor number of valence electrons in the metal center, in this case the Fe(III) atom. In these two potassium hexacyanoferrates, the iron atom is found with a low-spin electronic configuration. The difference in BE for the N 1s core-level electrons between low-spin Fe(II) and Fe(III) is -0.2 eV [20]. When the iron atom is found with a high-spin electronic configuration, the difference in BE between Fe(2+) and Fe(3+) is above 0.5 eV. Such a difference is a consequence of a stronger charge subtraction from the Fe(II), via back donation, when compared with the Fe(III) species. This indicates that the XPS can be used to discriminate between π and σ bonding interactions.
XPS also helps to discriminate between different coordination numbers or coordination geometries for a given metal center, because these structural features modify the binding energy of its core-level electrons. Figure 3b illustrates this fact for the Zn atom in PBAs, which could be found with octahedral or tetrahedral coordination. The difference in BE value for the Zn 2p3/2 for these two coordination numbers is relatively small, without possibilities to be used to discriminate between them. For the case of Zn, due to its 3d10 electronic configuration, a better probe for the coordination geometry is the Zn LMM Auger peaks (see Figure S1). For the Zn atom in tetrahedral coordination, the most intense Zn LMM Auger peak appears with a kinetic energy shift of +0.3 eV, relative to the octahedral coordination.
In PBAs, the CN ligands are found coordinated to only a metal center at their C and N ends. But within the transition and post-transition metal cyanides, the coordination of the N end to two metal centers is also possible [49,50,51]. This is the case, for instance, of the T2[M(CN)6] series with T = Mn(2+) and Cd; and M = Fe(II), Ru(II) and Os(II) [49], Ni(2+) with M = Ru(II) and Os(II) [50], and Sn2[Fe(CN)6], and Pb2[Fe(CN)6] [51]. In this subseries of metal hexacyanometallates, the CN ligand has a bifurcated coordination to the outer metal (T). This supposes a larger charge subtraction from the N end, which induces a stronger charge subtraction from the inner metal (M) via π-back bonding. At the same time, a more positive N atom must result. The observed BE value for the N 1s core-level electrons shows a slight increase. A larger energy shift is observed for Cd 3d5/2 core-level electrons (Figure 3c). For iron as the inner metal (M), the 57Fe Mossbauer spectrum identifies a more negative shift isomer (δ) value when the N end changes from linear to bifurcated coordination [49]. This informs us of a stronger π back-bonding interaction for the bifurcated coordination, when compared with a single coordination to the outer metal [49].
In addition to the mentioned magnetic and magneto-optical properties, PBAS have also demonstrated potential applications for gas storage and separation, as negative and zero thermal expansion materials, energy conversion and storage, heterogeneous catalysis, health care, in environmental remediation, and as wide band gap semiconductors [3], among others. The physical properties that support these functional applications are determined by their electronic structure, and in consequence, XPS could be a proper probe technique to provide valuable information, but such studies and derived information are limited.
2.1. Transition Metal Tetracyanometallates (Hofmann-Like Coordination Polymers)
For Ni, Pd, and Pt as inner metal (M), the CN ligand forms a square-planar complex ion, [M(CN)4]2¯. The coordination of the N end of their four CN ligands to divalent transition metals (T) leads to the formation of layered (2D) coordination polymers, T[M(CN)4]. When these coordination polymers are prepared by the precipitation method from aqueous solutions, the interlayer region is occupied by water molecules. Coordinated water molecules occupy the axial coordination sites for the T metal. These coordinated water molecules stabilize additional water molecules in the interlayer region through hydrogen bonding interactions. Depending on the number of weakly bonded water molecules per formula unit, three different phases are formed: L1: T(H2O)2[M(CN)4]•1H2O; K: T(H2O)2[M(CN)4]•2H2O; L0: T(H2O)2[M(CN)4]•4H2O [25]. In the presence of an organic ligand in the solution during the precipitation reaction, it could be found occupying the axial coordination sites of the metal (T). Such an organic ligand could be imidazole and its derivatives [26], 1-methyl-2-pyrrolidone [27], pyridine and its derivatives [28,29,30,31,32,33,34], among other ligands [52,53]. If the coordinated organic ligand is unable to form hydrogen bonds with the water molecules, an anhydrous solid is obtained. Such a series of 2D solids is known as Hofmann-like coordination polymers.
Figure 4 shows the N 1s and K 2p core-level spectral regions for the K2[M(CN)4] series. These three metals (Ni(2+), Pd(2+), and Pt(2+)) have diamagnetic character, with relatively simple XPS spectra [20]. Relative to KCN, the N 1s BE value decreases in the order Pt <Pd <Ni (Figure 4, Inset). This order is sensing the accumulation of electron density at the CN5 orbital due to the π-back-bonding interaction. The shift order in the BE for the N 1s core-level electrons peak, according to the involved metal (Pt, Pd, Ni), reflects the ability of the latter metals to donate charge density to the CN ligand. Pd and Pt are heavier metals, with extended t2g orbitals, which facilitate the charge retro-donation process. A surprising fact from these spectra is the bonding interaction between the K atom and the [M(CN)4]2− complex ion via the CN5σ molecular orbital. It seems that the interaction of the potassium ion with the complex anion is not merely electrostatic. This suggests that the complex ion is transferring electron density to the K+ atom through the N end of the CN ligand. According to the electronic configuration for the potassium ion, 1s2 2s2 2p6 3s2 3p6 4s0, it has a low energy available empty 4s orbital, which can receive electron density from the CN ligand, but preserving the fundamentally ionic character for the K–NC interaction, which is evidenced by the salt solubility in water [20].
When the T[M(CN)4] coordination polymer is formed, the electron density accumulated at the CN5σ orbital is at least partially donated to the outer metal (T). This is appreciated as an increase in the N 1s core-level binding energy (Figure 5, Inset b), an effect like that already discussed for PBAs. That charge donation, from the CN5σ molecular orbital to the T metal, reinforces the stability of the coordination polymer. Through this mechanism, the electron clouds of the metals (M, T) in the layer remain strongly coupled. When the T metal has a diamagnetic character, e.g., Zn, Cd, the layer behaves as a diamagnetic material. For T = Mn, Fe, Co, Ni, and Cu, the layer shows a weak antiferromagnetic character at room and medium temperatures, except for a weak cooperative magnetic interaction between the T paramagnetic metals from neighboring layers through the molecules coordinated to these T metals [53]. Such a behavior is observed when the sample is cooled at low temperatures [54,55]. Such a cooperative magnetic interaction has a weak ferromagnetic character for water, pyridine, and imidazole and their derivatives occupying the interlayer region. These molecules are usually referred to as pillar molecules. For water, the ferromagnetic interaction is mediated through a network of hydrogen bonds [56]. For the organic pillars, such a ferromagnetic ordering is possible by the π−π coupling between neighboring aromatic rings [54,55,56].
For T = Fe with pyridine and its derivatives as pillar molecules (L), a spin-crossover transition is usually observed upon sample cooling [28,29,30,31,32,33,34]. The energy separation between the t2g and eg levels of the iron atom, traditionally labeled as 10Dq, is close to kT. When the sample is cooled, reducing the kinetic energy related to the atoms’ vibration and, in consequence, the entropic contribution (∆S) to ∆G (∆G = ∆H-∆S) where ∆H is the enthalpy change, the electrons found at the eg orbitals of the iron atom migrate to its t2g orbital, corresponding to a low spin (LS) electronic configuration. Such a high spin (HS) to low spin (LS) transition induced by the sample cooling can be reversed by the sample warming. The temperature range where that reversible effect is observed depends on the bonding interaction of the iron atom with its ligands, the four CN ligands at their N end within the layer, and the pyridinic N atoms from the axial ligand. An increase in the electron density found at the CN5σ orbital favors the HS to LS transition. This is particularly evident in all the Fe(L)2[M(CN)4] series, where L represents the pillar molecule. Within that series of Hofmann-like coordination polymers, the higher electron density at the CN5σ orbital corresponds to M = Pt, and, in consequence, the highest temperature where the HS−LS is observed for Fe(L)2[P(CN)4] (Figure 5). The value of that transition temperature follows a linear dependence on the BE for N 1s core-level electrons.
3. nd10 Metal Cyanides.
The nd10 metal cyanide series, Zn(CN)2, Cd(CN)2, Hg(CN)2, CuCN, AgCN, and AuCN, can be divided into two subseries. In the dicyanides of Zn, Cd, and Hg, the metal is found with a tetrahedral (Zn, Cd) or pseudo-tetrahedral coordination (Hg) when forming coordination polymers with the CN ligand. These coordination polymers have a 3D structure. For the remaining three metals (Cu, Ag, Au), the coordination is linear to form chains (1D), which remain packed through dispersive forces and metal-metal interactions to create 3D solids.
The structural disorder in terms of the CN orientation characterizes the coordination environment for the metal in CuCN, AgCN, AuCN, Zn(CN)2, and Cd(CN)2, as appreciated in their XPS spectra (Figure 6). In thermodynamic terms, the most stable ligand configuration in the metal (M) coordination sphere must be −C≡N−M−C≡N−, which for Zn and Cd corresponds to a M(CN)2(NC)2 coordination environment. The deviation from this ideal ligand configuration has been established from structural NMR studies [57]. The peak width in their XPS N 1s core level (Figure 6), of about 2.5 eV, relative to the one observed for Hg(CN)2, 2.1 eV, is a consequence of such structural disorder. For Hg(CN)2, without such a structural feature, the peak width and metal binding energy show values corresponding to a material with a well-defined coordination environment.
For the CuCN, AgCN, and AuCN subseries, the N 1s, Cu 2p3/2, Ag 3d5/2, and Au 4f7/2 binding energy values shed light on the electronic structure of these metal cyanides. The C 1s core level in metal cyanides appears overlapped with the peak from adventitious carbon at 284.8 eV. From this fact, the C 1s peak has a limited scope on the electronic structure of metal cyanide. From this fact, the C 1s Relative to KCN, for Cu and Ag, the N 1s core-level spectrum senses a more positive character for the N atom. On the T-NC coordination bond formation, electron density is removed from the N end of the CN ligand, which results in a slight shift of the N 1s spectra to the high binding energy side (Figure 6). The metal’s capability to subtract electron density from the CN5σ orbital is determined by its polarizing power, Ze/r2, which for these three metals follows the order Cu >Ag >Au [58]. The deviation of the N 1s BE for AuCN relative to KCN, -0.1 eV, is probing the presence of a metal-metal interaction. This is congruent with the usually high frequency value for the CN stretching, 2238 cm-1 (Figure 6, Inset).
AuCN samples have been submitted to mechanical milling (grinding) to induce structural disorder to be studied by XPS [37]. XPS, complemented with Raman spectra, reveals the presence of N≡C−Au−N≡C, C≡N−Au−N≡C, and N≡C−Au−C≡N, in the grounded samples. The induced disorder in AuCN modifies its physical and functional properties, among them, its optical features. This simple metal cyanide has relevant industrial applications, e.g., the extractive gold industry [59,60].
4. Transition Metal Nitroprussides
Transition metal nitroprussides comprise a series of coordination polymers formed by the assembling of the pseudoctahedral anionic block pentacyanonitrosylferrate(II), [Fe(CN)5NO]2−, through a second transition metal (T) linked at the N end of the CN ligands. The nitroprusside ion has C4v symmetry, where the degenerated metal t2g are split into b2(dxy) and e(dxz, dyz) orbitals. The electronic structure for the nitroprusside ion, [Fe(CN)5NO]2-, was calculated and reported by Manoharan and Gray about 60 years ago [60], which has been used to shed light on the photochemistry of this ion and its behavior in aqueous solutions [61].
Figure 7 shows the reported high-resolution XPS spectra for the elements present in sodium nitroprusside, Na2[Fe(CN)5NO]•2H2O, recorded at 77 K [22]. The XPS study of metal nitroprussides at room temperature leads to a specific sample degradation by interaction with the X-ray beam, local sample heating, and excitation of transversal vibration modes for the NO+ group that induce the Fe-NO bond rupture. That figure reveals that the iron atom in the nitroprusside ion has a relatively low electron density, at least compared with the one observed for the ferrocyanide ion, included in that figure for comparison. The difference in 2p3/2 binding energy between the iron atom in the nitroprusside and the ferrocyanide ion is 2 eV. The difference between ferricyanide and ferrocyanide ions is 0.3 eV (Figure 3). A higher value for the binding energy indicates a lower electron density on the considered atom and vice versa. Such a difference of binding energy, of 2 eV, gives an idea of the low electron density that resides on the iron atom due to the strong charge-withdrawing ability of the NO group in the nitroprusside ion. An analog result can be inferred from Mossbauer spectroscopy, where the isomer shift value is -0.27 mm/s relative to metallic iron [62].
The electron density withdrawn by the NO+ group from the iron atom is found, at least, partially located at its O atom, due to its higher electronegative character. The XPS spectrum confirms this fact: the O 1s core-level peak appears 0.9 eV below the corresponding peak for the oxygen atom in the water molecules of sodium nitroprusside (Figure 7, Inset) [22]. At the same time, the N 1s core level binding energy for the nitrogen atom in the NO+ group has a relatively high value; its peak appears at 402.5 eV, which confirms a high accumulation of electron density on the O atom. The strong π-back-bonding interaction of the NO+ group with the iron atom reduces the capability of this last one to donate charge density to the CN ligands via π-back donation. The difference for N 1s BE between the CN ligand in sodium nitroprusside and sodium ferrocyanide is + 0.3 eV, confirming a weaker π-back-bonding interaction of the CN ligand with the n atom in the nitroprusside ion. That relatively low electron density found at the N end of the CN ligands in the nitroprusside ion has relevant implications on its reactivity with transition metal ions, and on the stability of the formed coordination polymers.
The strong electron density subtraction from the iron atom by the NO group weakens the Fe-NCaxial coordination bond. This opens the possibility of inducing a selective rupture of that bond to form 2D transition metal nitroprussides. Such a possibility was experimentally realized by sonicating a suspension of transition metal nitroprussides in a methanolic solution containing 1-methyl-2-pyrrolydone (1m2p). In the formed 2D solid, the organic molecule (1m2p) appears to occupy the axial coordination sites for the outer metal (T), playing the role of a bimolecular pillar [63]. After that first evidence on a route to obtain 2D transition metal nitroprusside, the preparative route was repeated with many other molecules, among them, pyridine and its derivatives, and imidazole and its derivatives [64] (Figure 8). In this family of pillared 2D transition metal nitroprussides, the axial CN at its N end and the NO group at its O end accumulated electron density. In the interlayer region, between these charge centers, a repulsive interaction between adjacent layers could appear. This fact has relevant importance in the physical properties of pillared 2D transition metal nitroprusides. That repulsive interaction could confer certain structural rigidity for the volume contraction in the formed pillared 2D solids. Figure 8 shows the rupture of the axial T-NCaxial coordination bond in 3D transition metal nitroprussides by an appropriate ligand, which behaves as a scissor, to form a 2D transition metal nitroprusside. The scissor molecule is then found occupying the axial coordination sites for the outer metal (T), in the resulting 2D coordination polymers.
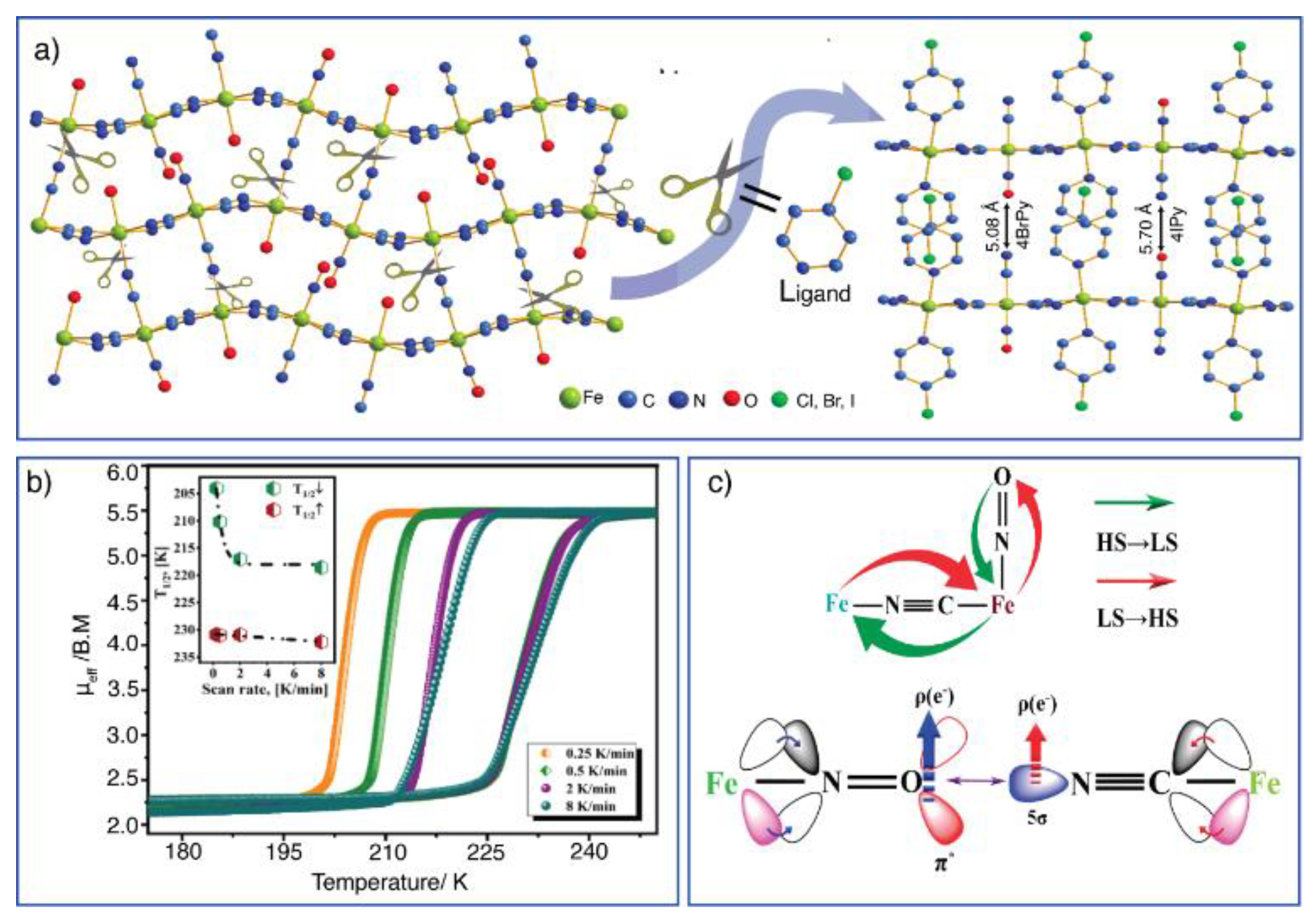
For T= Fe, like the already discussed for pillared ferrous tetracyanometallates, a thermal-induced spin crossover effect is possible in ferrous 2D nitroprusides. Figure 8b shows the SCO effect in the Fe(4X-pyridine)2[Fe(CN)5NO] series with X = Cl, Br, and I [65]. The hysteresis loops show a pronounced dependence on the sample cooling rates used during the magnetic data recording (kinetic effects). This effect is a consequence of the repulsive electrostatic interaction between CN∙∙∙ON axial ligands between adjacent layers. Such a repulsive interaction favors the layer separation during the sample warming, and no kinetic effect is observed. The observed kinetic effect in the SCO transition in ferrous 2D nitroprussides on the sample cooling is observed for the axial CN∙∙∙ON distance below 8 Å [66]. This is possible for small to medium-length pillar molecules. But the SCO observed is possible for such pillar molecules. The explanation of that behavior is found in the electron density redistribution during the sample cooling, when electron density accumulated at the O end of the NO ligand migrates to the CN5σ orbital, and then, on the sample warming, returns to the NO ligand (Figure 8c). The SCO has been reported for several ferrous 2D nitroprussides [67,68,69] where that electron density migration must be present.
5. Conclusions
XPS is a surface spectroscopy that provides information up to 10 nm in depth, equivalent to about 60 atomic layers from the very topmost surface atoms. This contribution discusses the potential of this technique to shed light on the electronic structure and bonding interactions in coordination polymers based on transition metal cyanides, a family of solids with interesting physical and functional properties, among them, as semiconductors, molecular magnets, spin-crossover materials, environmental protection and remediation solids, and for gas storage and separation, etc. XPS provides a conclusive clue on the nature of many of these physical and functional properties. Herein, information on the bonding properties derived from XPS for the following series of that family of materials is discussed, particularly Prussian blue analogs, transition metal tetracyanides (Hofmann-like coordination polymers), d10 metal cyanides, including those with 1D structure, and transition metal nitroprussides. No similar volume of information has been previously summarized in a single contribution.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Arely Cano: Writing- review & editing, writing original draft, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization.
Funding
This study was supported by a Cinvestav (Mexico) project in the context of a research program on emergent semiconducting materials, among them those with 1D and 2D dimensions.
Data Availability Statement
This contribution summarizes the author’s experience with the application of XPS spectroscopy to structural studies in materials. All additional data on this subject are available from the author.
Acknowledgments
The author thanks the Cinvestav (Mexico) for a research contract for the study of emerging semiconductor materials.
Conflicts of Interest
The author declares no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| XPS | X-ray Photoelectron Spectroscopy |
| PBAs | Prussian blue analogs (PBAs) |
| KE | Kinetic energy |
| BE | Binding energy |
| Tc | Critical temperature |
| HS | High spin |
| LS | Low spin |
| SCO | Spin Crossover |
References
- Watts, J.F.; Wolstenholme, J. An Introduction to Surface Analysis by XPS and AES; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2020. [Google Scholar]
- van der Heide, P. X-Ray Photoelectron Spectroscopy—An Introduction to Principles and Practices; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012. [Google Scholar]
- Avila, Y.; Acevedo-Peña, P.; Reguera, L.; Reguera, E. Recent progress in transition metal hexacyanometallates: From structure to properties and functionality. Co-ord. Chem. Rev. 2022, 453. [Google Scholar] [CrossRef]
- Dunbar, K.R.; Heintz, R.A. Chemistry of Transition Metal Cyanide Compounds: Modern Perspectives. Prog. Inorg. Chem. 1997, 45, 283. [Google Scholar]
- Sharpe, A.G. The Chemistry of Cyano Complexes of the Transition Metals; Academic; Academic Press: London, UK, 1976. [Google Scholar]
- Pike, R.D. Structure and Bonding in Copper(I) Carbonyl and Cyanide Complexes. Organometallics 2012, 31, 7647–7660. [Google Scholar] [CrossRef]
- Ferlay, S.; Mallah, T.; Ouahès, R.; Veillet, P.; Verdaguer, M. A room-temperature organometallic magnet based on Prussian blue. Nature 1995, 378, 701–703. [Google Scholar] [CrossRef]
- Sato, O.; Iyoda, T.; Fujishima, A.; Hashimoto, K. Photoinduced Magnetization of a Cobalt-Iron Cyanide. Science 1996, 272, 704–705. [Google Scholar] [CrossRef] [PubMed]
- Vannerberg, N.G. The ESCA-spectra of sodium and potassium cyanide and of the sodium and potassium salts of the hexacyanometallates of the first transition metal series. Chem. Scr. 1976, 9, 122–126. [Google Scholar]
- Fluck, E.; Inoue, H.; Yanagisawa, S. Mössbauer and X-ray photoelectron spectroscopic studies of prussian blue and its related compounds. Z. Anorg. Allg. Chem. 1977, 430, 241–249. [Google Scholar] [CrossRef]
- Fluck, E.; Inoue, H.; Nagao, M.; Yanagisawa, S. Bonding properties in Prussian blue analogues of the type Fe[Fe(CN)5X]·xH2O. J. Inorg. Nucl. Chem. 1979, 41, 287–292. [Google Scholar] [CrossRef]
- Adams, I.; Thomas, J.M.; Bancroft, G.M.; Butler, K.D.; Barber, M. Correlation between core-electron binding energies and Mössbauer chemical isomer shifts for inorganic complexes containing iron(II) low spin. J. Chem. Soc. Chem. Commun. 1972, 751–752. [Google Scholar] [CrossRef]
- Wertheim, G.K.; Rosencwaig, A. Characterization of Inequivalent Iron Sites in Prussian Blue by Photoelectron Spectroscopy. J. Chem. Phys. 1971, 54, 3235–3237. [Google Scholar] [CrossRef]
- Koster, A. X-ray spectroscopy of the valence band in simple and complex cyanides. Chem. Phys. Lett. 1973, 23, 18–20. [Google Scholar] [CrossRef]
- Oku, M. X-ray photoelectron spectroscopic studies on the kinetics of photoreduction of Fe III in single-crystal K3(Fe,Co)(CN)6 surfaces cleaved in situ. J. Chem. Soc. Faraday Trans. 1993, 89, 743–747. [Google Scholar] [CrossRef]
- Lummen, T.T.A.; Gengler, R.Y.N.; Rudolf, P.; Lusitani, F.; Vertelman, E.J.M.; van Koningsbruggen, P.J.; Knupfer, M.; Molodtsova, O.; Pireaux, J.-J.; van Loosdrecht, P.H.M. Bulk and Surface Switching in Mn−Fe-Based Prussian Blue Analogues. J. Phys. Chem. C 2008, 112, 14158–14167. [Google Scholar] [CrossRef]
- Gerber, S.J.; Erasmus, E. Electronic effects of metal hexacyanoferrates: An XPS and FTIR study. Mater. Chem. Phys. 2018, 203, 73–81. [Google Scholar] [CrossRef]
- Cano, A.; Rodríguez-Hernández, J.; Shchukarev, A.; Reguera, E. Intercalation of pyrazine in layered copper nitroprusside: Synthesis, crystal structure and XPS study. J. Solid State Chem. 2019, 273, 1–10. [Google Scholar] [CrossRef]
- Cano, A.; Rodríguez-Hernández, J.; Reguera, L.; Rodríguez-Castellón, E.; Reguera, E. On the Scope of XPS as Sensor in Coordination Chemistry of Transition Metal Hexacyanometallates. Eur. J. Inorg. Chem. 2019, 2019, 1724–1732. [Google Scholar] [CrossRef]
- Cano, A.; Monroy, I.; Ávila, M.; Velasco-Arias, D.; Rodríguez-Hernández, J.; Reguera, E. Relevant electronic interactions related to the coordination chemistry of tetracyanometallates. An XPS study. New J. Chem. 2019, 43, 18384–18393. [Google Scholar] [CrossRef]
- Cano, A.; Avila, Y.; Avila, M.; Reguera, E. Structural information contained in the XPS spectra of nd10 metal cyanides. J. Solid State Chem. 2019, 276, 339–344. [Google Scholar] [CrossRef]
- Cano, A.; Lartundo-Rojas, L.; Shchukarev, A.; Reguera, E. Contribution to the coordination chemistry of transition metal nitroprussides: a cryo-XPS study. New J. Chem. 2019, 43, 4835–4848. [Google Scholar] [CrossRef]
- Cano, A.; Reguera, L.; Avila, M.; Velasco-Arias, D.; Reguera, E. Charge Redistribution Effects in Hexacyanometallates Evaluated from XPS Data. Eur. J. Inorg. Chem. 2019, 2020, 137–145. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, J.; Reguera, E.; Lima, E.; Balmaseda, J.; Martínez-García, R.; Yee-Madeira, H. An atypical coordination in hexacyanometallates: Structure and properties of hexagonal zinc phases. J. Phys. Chem. Solids 2007, 68, 1630–1642. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, J.; Lemus-Santana, A.; Vargas, C.; Reguera, E. Three structural modifications in the series of layered solids T(H2O)2[Ni(CN)4]·xH2O with T = Mn, Co, Ni: Their nature and crystal structures. Comptes Rendus Chim. 2011, 15, 350–355. [Google Scholar] [CrossRef]
- González, M.; Lemus-Santana, A.A.; Rodríguez-Hernández, J.; Knobel, M.; Reguera, E. π–π Interactions and magnetic properties in a series of hybrid inorganic–organic crystals. J. Solid State Chem. 2013, 197, 317–322. [Google Scholar] [CrossRef]
- Lemus-Santana, A.A.; González, M.; Rodríguez-Hernández, J.; Knobel, M.; Reguera, E. 1-Methyl-2-Pyrrolidone: From Exfoliating Solvent to a Paramagnetic Ligand. J. Phys. Chem. A 2013, 117, 2400–2407. [Google Scholar] [CrossRef]
- Kitazawa, T.; Gomi, Y.; Takahashi, M.; Takeda, M.; Enomoto, M.; Miyazaki, A.; Enoki, T. Spin-crossover behaviour of the coordination polymer FeII(C5H5N)2NiII(CN)4. J. Mater. Chem. 1996, 6, 119–121. [Google Scholar] [CrossRef]
- Kitazawa, T.; Hosoya, K.; Takahashi, M.; Takeda, M.; Marchuk, I.; Filipek, S.M. 57Fe Mössbauer spectroscopic study for Fe(pyridine)2Ni(CN)4spin-crossover compound. J. Radioanal. Nucl. Chem. 2003, 255, 509–512. [Google Scholar] [CrossRef]
- Hosoya, K.; Kitazawa, T.; Takahashi, M.; Takeda, M.; Meunier, J.F.; Molna´r, G.; Bousseksou, A. Unexpected isotope effect on the spin transition of the coordination polymer Fe (C 5 H 5 N) 2 [Ni (CN) 4]. J. Phys. Chem. Chem. Phys. 2003, 5, 1682–1688. [Google Scholar] [CrossRef]
- Avila, Y.; Pérez, O.; Sánchez, L.; Vázquez, M.C.; Mojica, R.; González, M.; Ávila, M.; Rodríguez-Hernández, J.; Reguera, E. Spin crossover in Hofmann-like coordination polymers. Effect of the axial ligand substituent and its position. New J. Chem. 2023, 47, 10781–10795. [Google Scholar] [CrossRef]
- Terrero, R.; Avila, Y.; Mojica, R.; Cano, A.; Gonzalez, M.; Avila, M.; Reguera, E. Thermally induced spin-crossover in the Fe(3-ethynylpyridine)2[M(CN)4] series with M = Ni, Pd, and Pt. The role of the electron density found at the CN 5σ orbital. New J. Chem. 2022, 46, 9618–9628. [Google Scholar] [CrossRef]
- Muñoz, M.C.; Real, J.A. Thermo-, piezo-, photo- and chemo-switchable spin crossover iron(II)-metallocyanate based coordination polymers. Co-ord. Chem. Rev. 2011, 255, 2068–2093. [Google Scholar] [CrossRef]
- Martínez, V.; Gaspar, A.B.; Muñoz, M.C.; Bukin, G.V.; Levchenko, G.; Real, J.A. Synthesis and Characterisation of a New Series of Bistable Iron(II) Spin-Crossover 2D Metal–Organic Frameworks. Chem. – A Eur. J. 2009, 15, 10960–10971. [Google Scholar] [CrossRef]
- Hibble, S.J.; Eversfield, S.G.; Cowley, A.R.; Chippindale, A.M. Copper(I) Cyanide: A Simple Compound with a Complicated Structure and Surprising Room-Temperature Reactivity. Angew. Chem. Int. Ed. Engl. 2004, 43, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Hibble, S.J.; Cheyne, S.M.; Hannon, A.C.; Eversfield, S.G. CuCN: A Polymorphic Material. Structure of One Form Determined from Total Neutron Diffraction. Inorg. Chem. 2002, 41, 4990–4992. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Arias, D.; Mojica, R.; Zumeta-Dubé, I.; Ruíz-Ruíz, F.; Puente-Lee, I.; Reguera, E. New Understanding on an Old Compound: Insights on the Origin of Chain Sequence Defects and Their Impact on the Electronic Structure of AuCN. Eur. J. Inorg. Chem. 2021, 2021, 3742–3751. [Google Scholar] [CrossRef]
- Ding, P.; Liang, E.J.; Jia, Y.; Du, Z.Y. Electronic structure, bonding and phonon modes in the negative thermal expansion materials of Cd(CN)2and Zn(CN)2. J. Physics: Condens. Matter 2008, 20, 275224. [Google Scholar] [CrossRef]
- Hibble, S.J.; Chippindale, A.M.; Marelli, E.; Kroeker, S.; Michaelis, V.K.; Greer, B.J.; Aguiar, P.M.; Bilbé, E.J.; Barney, E.R.; Hannon, A.C. Local and Average Structure in Zinc Cyanide: Toward an Understanding of the Atomistic Origin of Negative Thermal Expansion. J. Am. Chem. Soc. 2013, 135, 16478–16489. [Google Scholar] [CrossRef]
- Korčok, J.L.; Leznoff, D.B. Thermal expansion of mercury(II) cyanide and HgCN(NO3). Polyhedron 2013, 52, 72–77. [Google Scholar] [CrossRef]
- Avila, Y.; Vázquez, M.; Piedra, O.; Rodríguez-Hernández, J.; Reguera, L.; Ávila, M.; Reguera, E. Structural features of the Zn2[M(CN)8] series with M = Nb, Mo, and W and their negative thermal expansion behavior. J. Phys. Chem. Solids 2025. [Google Scholar] [CrossRef]
- Reguera, L.; Avila, Y.; Reguera, E. Transition metal nitroprussides: Crystal and electronic structure, and related properties. Co-ord. Chem. Rev. 2021, 434. [Google Scholar] [CrossRef]
- Ware, M. Prussian Blue: Artists’ Pigment and Chemists’ Sponge. J. Chem. Educ. 2008, 85, 614. [Google Scholar] [CrossRef]
- Holmes, S.M.; Girolami, G.S. Sol−Gel Synthesis of KVII[CrIII(CN)6]·2H2O: A Crystalline Molecule-Based Magnet with a Magnetic Ordering Temperature above 100 °CCl. J. Am. Chem. Soc. 1999, 121, 5593. [Google Scholar] [CrossRef]
- Martínez-Garcia, R.; Knobel, M.; Reguera, E. Thermal-Induced Changes in Molecular Magnets Based on Prussian Blue Analogues. J. Phys. Chem. B 2006, 110, 7296–7303. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, W.E.; Miller, J.S. Magnetic Ordering and Spin-Glass Behavior in First-Row Transition Metal Hexacyanomanganate(IV) Prussian Blue Analogues. Inorg. Chem. 2000, 39, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Her, J.-H.; Stephens, P.W.; Kareis, C.M.; Moore, J.G.; Min, K.S.; Park, J.-W.; Bali, G.; Kennon, B.S.; Miller, J.S. Anomalous Non-Prussian Blue Structures and Magnetic Ordering of K2MnII[MnII(CN)6] and Rb2MnII[MnII(CN)6]. Inorg. Chem. 2010, 49, 1524. [Google Scholar] [CrossRef] [PubMed]
- Reguera, E.; Marín, E.; Calderón, A.; Rodríguez-Hernández, J. Photo-induced charge transfer in Prussian blue analogues as detected by photoacoustic spectroscopy. Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 2007, 68, 191–197. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, J.; Reguera, E.; Lima, E.; Balmaseda, J.; Martínez-García, R.; Yee-Madeira, H. An atypical coordination in hexacyanometallates: Structure and properties of hexagonal zinc phases. J. Phys. Chem. Solids 2007, 68, 1630–1642. [Google Scholar] [CrossRef]
- Reguera, L.; López, N.L.; Rodríguez-Hernández, J.; Martínez-García, R.; Reguera, E. Hydrothermal Recrystallization as a Strategy to Reveal the Structural Diversity in Hexacyanometallates: Nickel and Copper Hexacyanoosmates(II). Eur. J. Inorg. Chem. 2020, 2020, 1763–1774. [Google Scholar] [CrossRef]
- Zubkov, V.; Tyutyunnik, A.; Berger, I.; Maksimova, L.; Denisova, T.; Polyakov, E.; Kaplan, I.; Voronin, V. Anhydrous tin and lead hexacyanoferrates (II). Solid State Sci. 2001, 3, 361–367. [Google Scholar] [CrossRef]
- Echevarría, F.; Lemus-Santana, A.; González, M.; Rodríguez-Hernández, J.; Reguera, E. Intercalation of thiazole in layered solids. A 3D framework supported in dipolar and quadrupolar intermolecular interactions. Polyhedron 2015, 95, 75–80. [Google Scholar] [CrossRef]
- Torres, L.; Ávila-Santos, M.; Vega-Moreno, J.; Portales-Martínez, B.; Vera, M.A.; Lemus-Santana, A.A. Crystallographic and Spectroscopic Footprints of Hydrophobic and Hydrophilic Hydrogen Bonding within 2D Coordination Polymers. Cryst. Growth Des. 2025, 25, 924–935. [Google Scholar] [CrossRef]
- González, M.; Lemus-Santana, A.; Rodríguez-Hernández, J.; Aguirre-Velez, C.; Knobel, M.; Reguera, E. Intermolecular interactions between imidazole derivatives intercalated in layered solids. Substituent group effect. J. Solid State Chem. 2013, 204, 128–135. [Google Scholar] [CrossRef]
- Di Santo, A.; Osiry, H.; Reguera, E.; Alborés, P.; Carbonio, R.E.; Ben Altabef, A.; Gil, D.M. New coordination polymers based on 2-methylimidazole and transition metal nitroprusside containing building blocks: synthesis, structure and magnetic properties. New J. Chem. 2017, 42, 1347–1355. [Google Scholar] [CrossRef]
- Gonzalez, M.; Osiry, H.; Martínez, M.; Rodríguez-Hernández, J.; Lemus-Santana, A.; Reguera, E. Magnetic interaction in a 2D solid through hydrogen bonds and π-π stacking. J. Magn. Magn. Mater. 2019, 471, 70–76. [Google Scholar] [CrossRef]
- Hibble, S.J.; Chippindale, A.M.; Marelli, E.; Kroeker, S.; Michaelis, V.K.; Greer, B.J.; Aguiar, P.M.; Bilbé, E.J.; Barney, E.R.; Hannon, A.C. Local and Average Structure in Zinc Cyanide: Toward an Understanding of the Atomistic Origin of Negative Thermal Expansion. J. Am. Chem. Soc. 2013, 135, 16478–16489. [Google Scholar] [CrossRef]
- Zhang, X.; Xi, R.; Yin, S.; Cao, X.; Zhang, Y.; Lin, L.; Chen, R.; Wu, H. Three inorganic-organic hybrid complexes based on isopolymolybdate and derivatives of 1H-4-nitroimidazole. J. Solid State Chem. 2018, 258, 737–743. [Google Scholar] [CrossRef]
- Rees, K.; van Deventer, J. The mechanism of enhanced gold extraction from ores in the presence of activated carbon. Hydrometallurgy 2000, 58, 151–167. [Google Scholar] [CrossRef]
- Manoharan, P.T.; Gray, H.B. Electronic Structure of Nitroprusside Ion. J. Am. Chem. Soc. 1965, 87, 3340–3348. [Google Scholar] [CrossRef]
- Crespo, P.; Odio, O.; Ávila, Y.; Perez-Cappe, E.; Reguera, E. Effect of water and light on the stability of transition metal nitroprussides. J. Photochem. Photobiol. A: Chem. 2021, 412. [Google Scholar] [CrossRef]
- Ávila, Y.; Osiry, H.; Plasencia, Y.; E Torres, A.; González, M.; A Lemus-Santana, A.; Reguera, E. From 3D to 2D Transition Metal Nitroprussides by Selective Rupture of Axial Bonds. Chem. – A Eur. J. 2019, 25, 11327–11336. [Google Scholar] [CrossRef]
- Avila, Y.; Vázquez, M.; Rodríguez-Hernández, J.; Mojica, R.; Lemus-Santana, A.; Avila, M.; González, M.; Reguera, E. Coexistence of two magnetic sublattices in 2D transition metal nitroprussides, T(L)2[Fe(CN)5NO] with T = Mn, Co, Ni; L = 2-ethylimidazole, Imidazo [1,2-a]pyridine. J. Solid State Chem. 2025, 352. [Google Scholar] [CrossRef]
- Avila, Y.; Mojica, R.; Vázquez, M.C.; Sánchez, L.; González, M.; Rodríguez-Hernández, J.; Reguera, E. Spin-crossover in the Fe(4X-pyridine)2[Fe(CN)5NO] series with X = Cl, Br, and I. Role of the distortion for the iron atom coordination environment. New J. Chem. 2022, 47, 238–249. [Google Scholar] [CrossRef]
- Avila, Y.; Scanda, K.; Díaz-Paneque, L.A.; Cruz-Santiago, L.A.; González, M.; Reguera, E. The Nature of the Atypical Kinetic Effects Observed for the Thermally Induced Spin Transition in Ferrous Nitroprussides with Short Organic Pillars. Eur. J. Inorg. Chem. 2022, 24, e202200252. [Google Scholar] [CrossRef]
- Avila, Y.; Plasencia, Y.; Osiry, H.; Martínez-dlCruz, L.; González Montiel, M.; Reguera, E. Thermally Induced Spin Transition in a 2D Ferrous Nitroprusside. Eur. J. Inorg. Chem. 2019, 47, 4966–4973. [Google Scholar] [CrossRef]
- Avila, Y.; Crespo, P.M.; Plasencia, Y.; Mojica, H.R.; Rodríguez-Hernández, J.; Reguera, E. Intercalation of 3X-pyridine with X= F, Cl, Br, I, in 2D ferrous nitroprusside. Thermal induced spin transition in Fe (3F-pyridine) 2 [Fe (CN) 5NO]. J. Solid State Chem. 2020, 286, 121293. [Google Scholar] [CrossRef]
- Avila, Y.; Crespo, P.M.; Plasencia, Y.; Mojica, H.R.; Rodríguez-Hernández, J.; Reguera, E. Thermally induced spin crossover in Fe(PyrDer)2[Fe(CN)5NO] with PyrDer = 4-substituted pyridine derivatives. New J. Chem. 2020, 44, 5937–5946. [Google Scholar] [CrossRef]
Figure 1.
(a): Accumulation of electron density at the CN5σ orbital, probed by a decrease in the BE value for the N 1s core-level electrons (b); (c): the coordination of the CN ligand at its N end to a T metal involves the donation of charge density to this last one, which is detected as an increase in the value of BE for the N 1s core-level electrons (d). This figure was prepared with the author’s data [23].
Figure 1.
(a): Accumulation of electron density at the CN5σ orbital, probed by a decrease in the BE value for the N 1s core-level electrons (b); (c): the coordination of the CN ligand at its N end to a T metal involves the donation of charge density to this last one, which is detected as an increase in the value of BE for the N 1s core-level electrons (d). This figure was prepared with the author’s data [23].
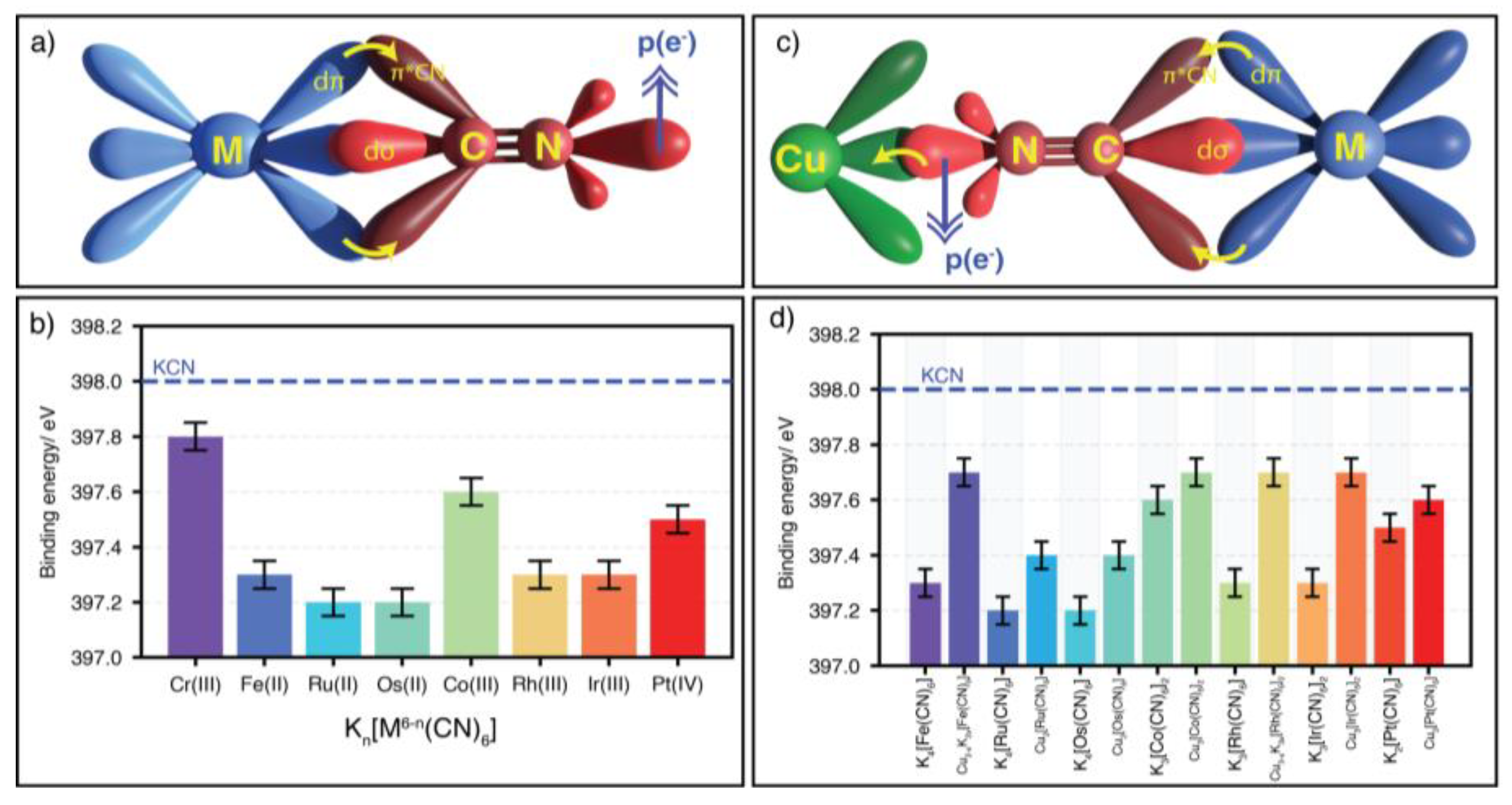
Figure 2.
Correlation between the critical temperature of magnetic ordering (Tc) and the N 1s BE values for the (T)n[CrIII(CN)6] and T3[FeIII(CN)6]2 subseries of PBAs. Inset: Photoinduced electron transfer in Co(III)K[Fe(II)(CN)6] when it is irradiated with UV-vis light.
Figure 2.
Correlation between the critical temperature of magnetic ordering (Tc) and the N 1s BE values for the (T)n[CrIII(CN)6] and T3[FeIII(CN)6]2 subseries of PBAs. Inset: Photoinduced electron transfer in Co(III)K[Fe(II)(CN)6] when it is irradiated with UV-vis light.
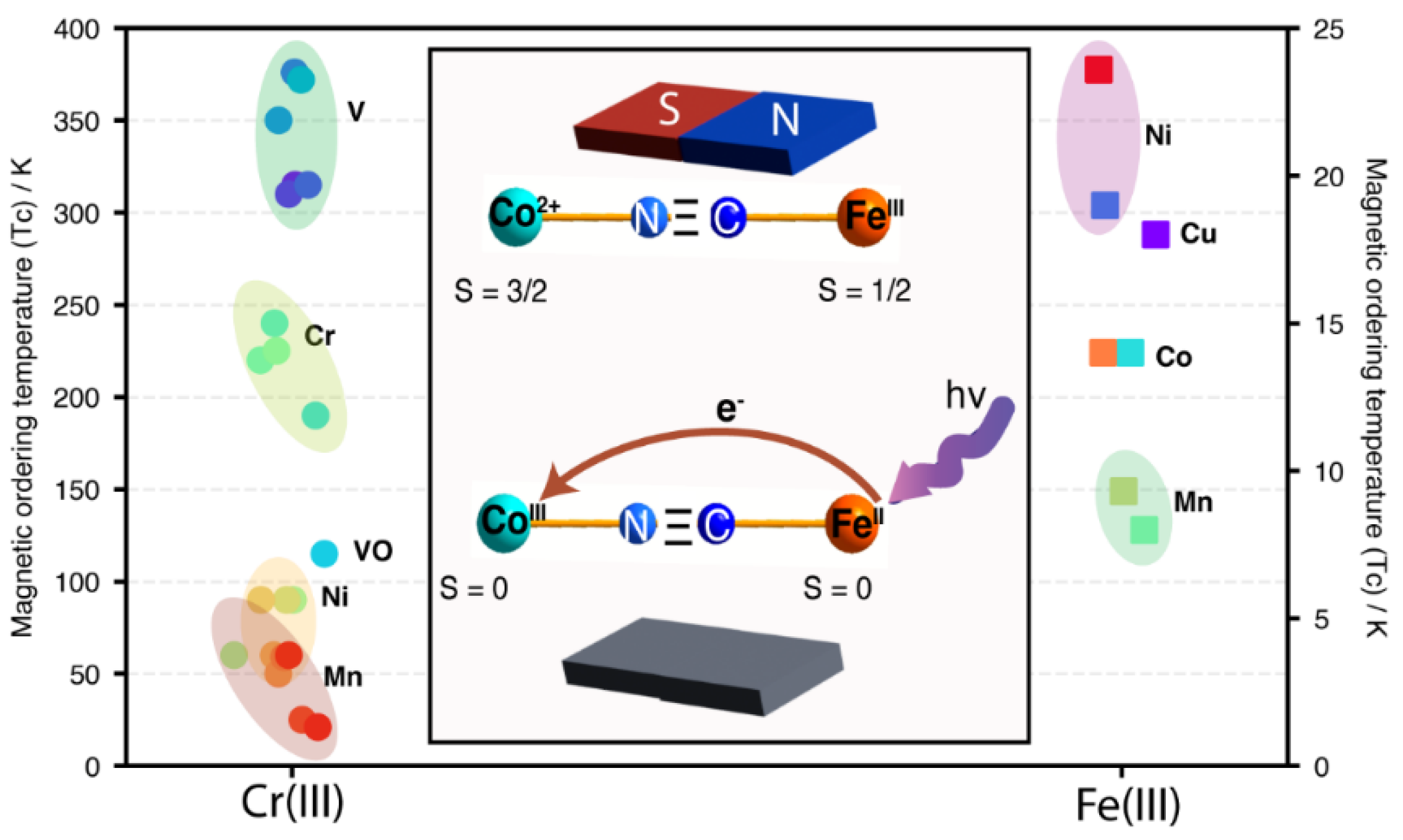
Figure 3.
XPS spectrum as a sensor for the valence and coordination geometry of the involved metals in PBAs and related solids, and the coordination mode for the CN ligand. A) N 1s core-level peak of KCN, K4[Fe(CN)6] and K3[Fe(CN)6]; B) Zn 2p3/2 core-level peak for Zn3[Co(CN)6]2•xH2O (cubic phase) and Zn3[Co(CN)6]2 (hexagonal phase); C) Cd 3d5/2 core-level peak in cadmium hexacyanometallates with the N end of the CN ligand coordinated to one and two cadmium atoms.
Figure 3.
XPS spectrum as a sensor for the valence and coordination geometry of the involved metals in PBAs and related solids, and the coordination mode for the CN ligand. A) N 1s core-level peak of KCN, K4[Fe(CN)6] and K3[Fe(CN)6]; B) Zn 2p3/2 core-level peak for Zn3[Co(CN)6]2•xH2O (cubic phase) and Zn3[Co(CN)6]2 (hexagonal phase); C) Cd 3d5/2 core-level peak in cadmium hexacyanometallates with the N end of the CN ligand coordinated to one and two cadmium atoms.
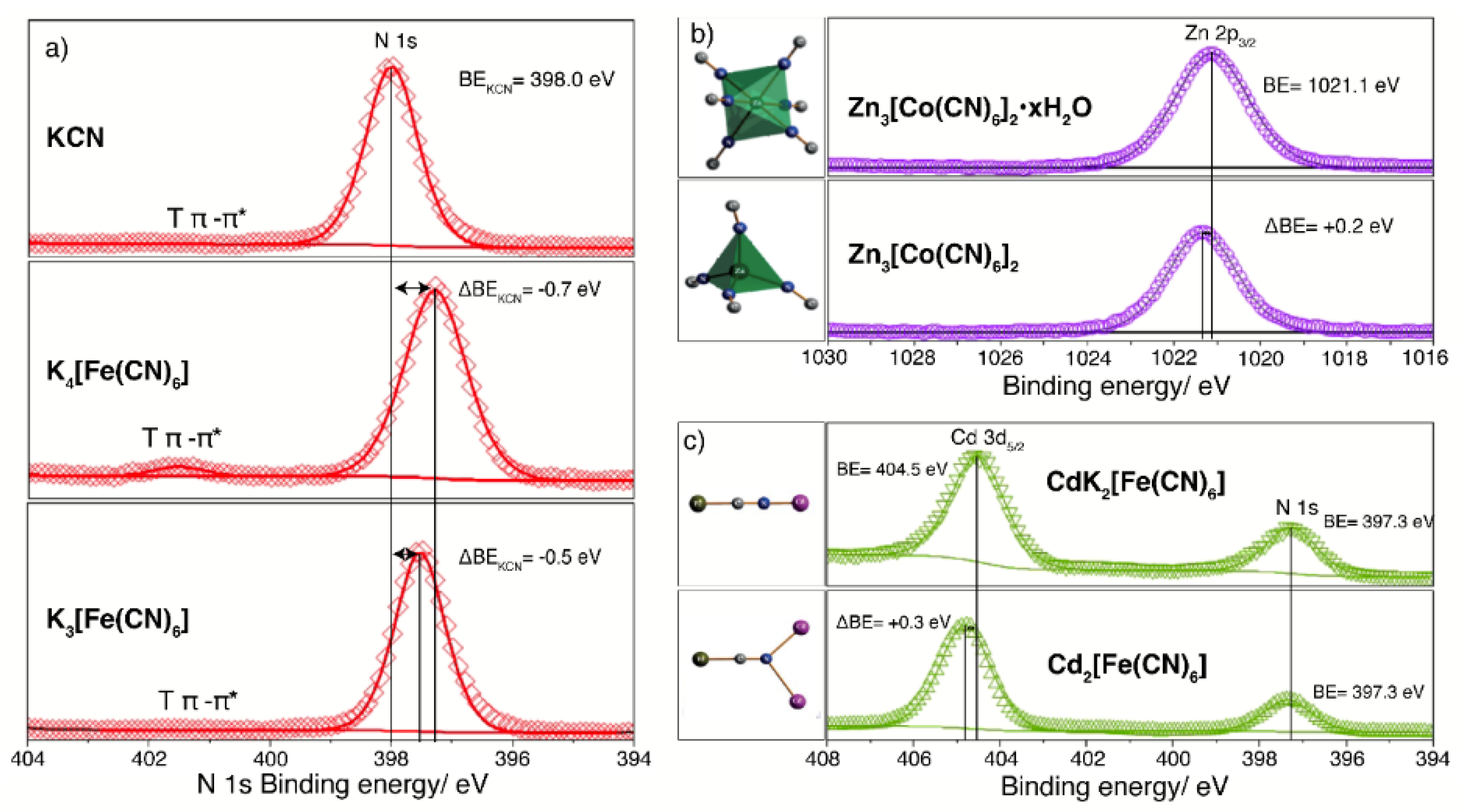
Figure 4.
XPS spectra for the K2[M(CN)4] series, for M = Ni, Pd, and Pt. a) N 1s core-level peak and its BE value; b) K 2p core-level peaks and the BE value for K 2p3/2. For comparison, the spectrum for KCN was included.
Figure 4.
XPS spectra for the K2[M(CN)4] series, for M = Ni, Pd, and Pt. a) N 1s core-level peak and its BE value; b) K 2p core-level peaks and the BE value for K 2p3/2. For comparison, the spectrum for KCN was included.
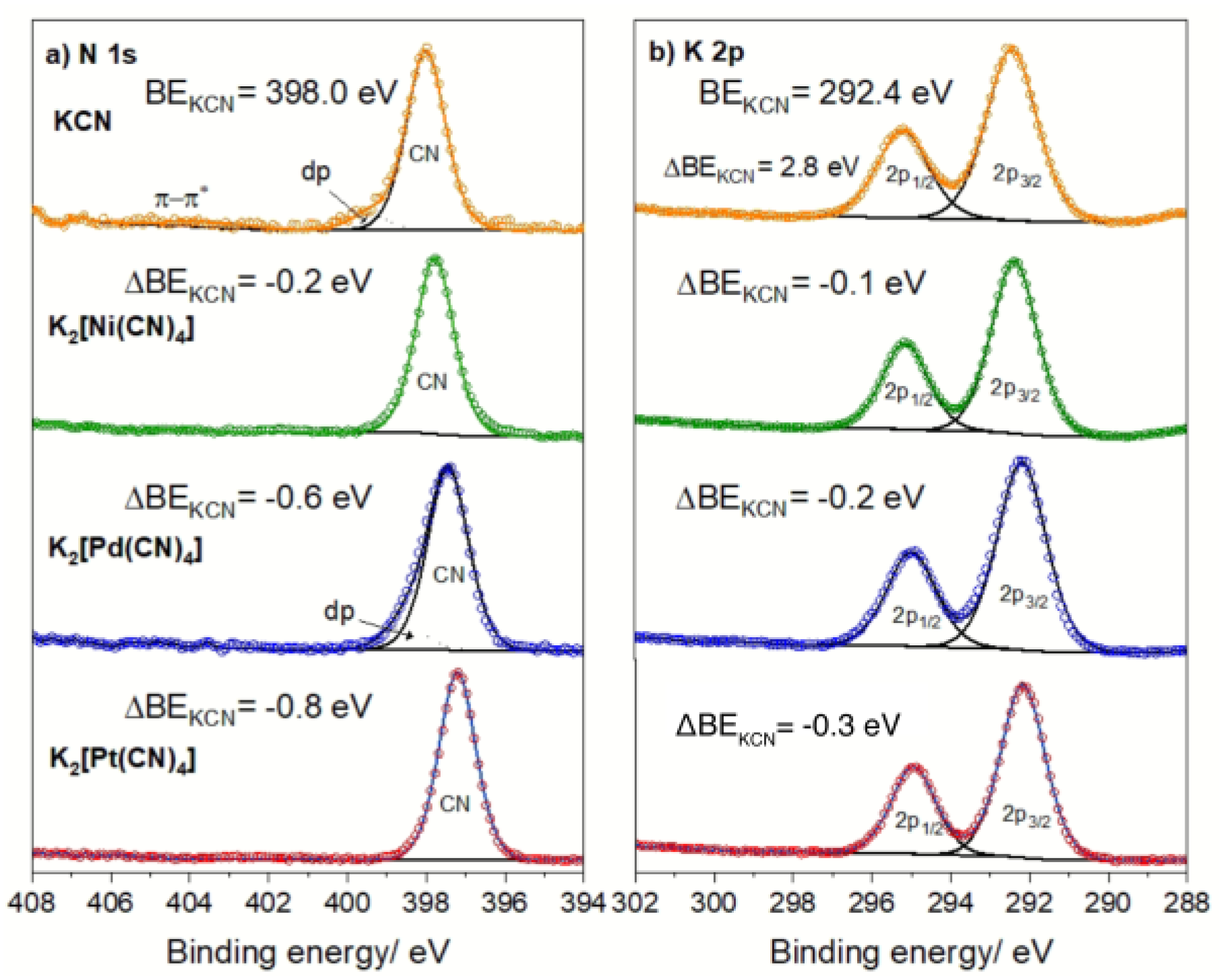
Figure 5.
Dependence of HS−LS transition temperature on the electron density accumulated at the CN5σ orbital, herein illustrated for the Fe(3-ethynylpyridine)2[M(CN)4] series versus N 1s core-level binding energy for the K2[M(CN)4] series. Insets: a) Spin crossover hysteresis loop for Fe(3-ethynylpyridine)2[Pt(CN)4]; b) N1s core-level binding energy for the K2[M(CN)4] and Ni[M(CN)4]•xH2O series; c) N 1s core-level peak for K2[Pt(CN)4] salt.
Figure 5.
Dependence of HS−LS transition temperature on the electron density accumulated at the CN5σ orbital, herein illustrated for the Fe(3-ethynylpyridine)2[M(CN)4] series versus N 1s core-level binding energy for the K2[M(CN)4] series. Insets: a) Spin crossover hysteresis loop for Fe(3-ethynylpyridine)2[Pt(CN)4]; b) N1s core-level binding energy for the K2[M(CN)4] and Ni[M(CN)4]•xH2O series; c) N 1s core-level peak for K2[Pt(CN)4] salt.
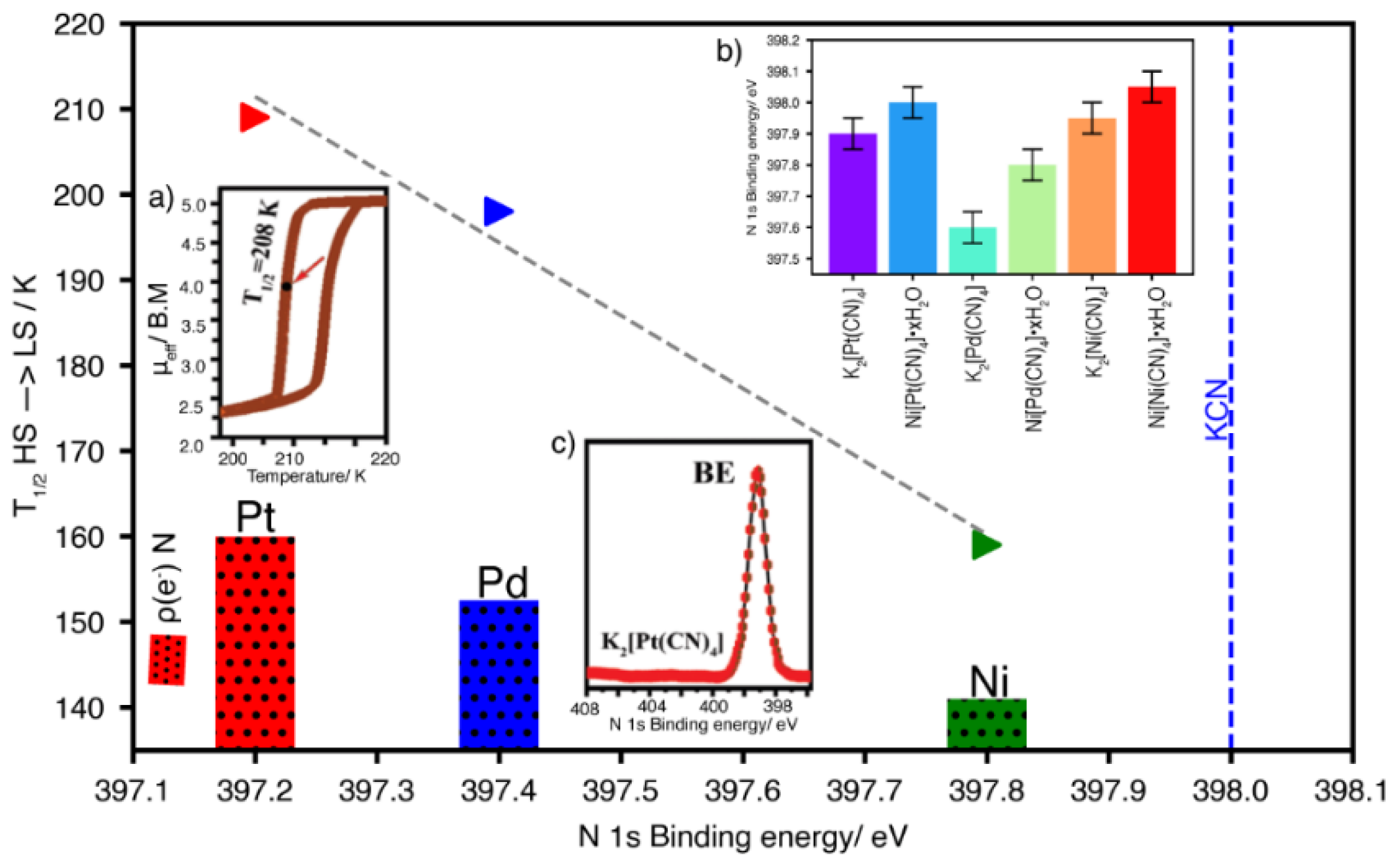
Figure 6.
XPS N 1s core-level peak of the nd10 cyanide series. a) MCN subseries with M = Cu(+), Ag(+) and Au(+); b) M(CN)2 subseries, with M = Zn, Cd, Hg. Insets: υ (CN) stretching Raman peak and atomic packing for the two subseries.
Figure 6.
XPS N 1s core-level peak of the nd10 cyanide series. a) MCN subseries with M = Cu(+), Ag(+) and Au(+); b) M(CN)2 subseries, with M = Zn, Cd, Hg. Insets: υ (CN) stretching Raman peak and atomic packing for the two subseries.
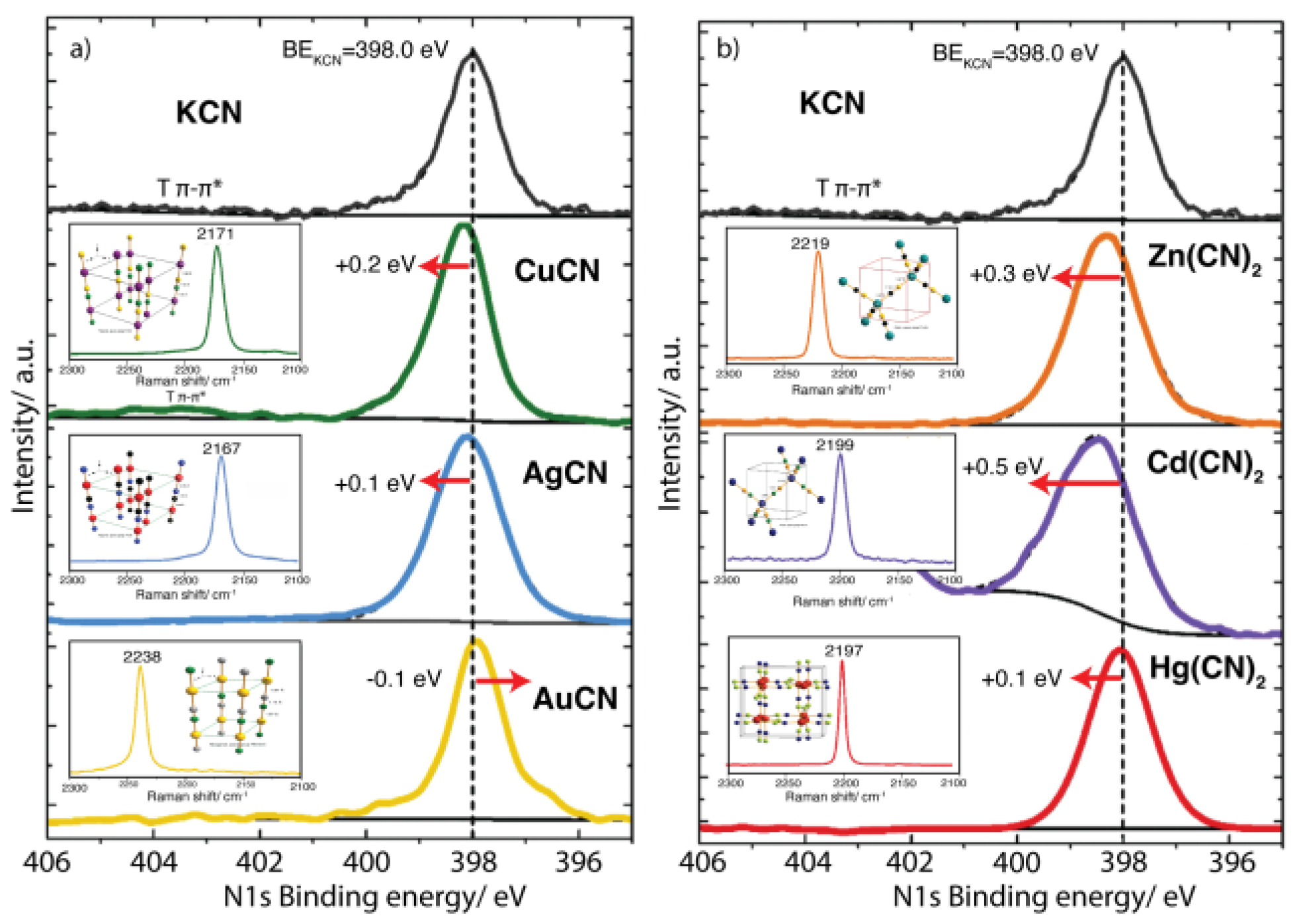
Figure 7.
XPS spectrum for sodium nitroprusside. For comparison, the spectrum for sodium hexacyanoferrate (II) was included: Left) Fe 2p3/2 spectral region where weak peaks from decomposition byproducts (Fe2+ and Fe0 species) also appear; Right) N 1s spectral region; Inset i) O 1s charge distribution difference for NO and H2O. This figure was prepared using original data from the author [11].
Figure 7.
XPS spectrum for sodium nitroprusside. For comparison, the spectrum for sodium hexacyanoferrate (II) was included: Left) Fe 2p3/2 spectral region where weak peaks from decomposition byproducts (Fe2+ and Fe0 species) also appear; Right) N 1s spectral region; Inset i) O 1s charge distribution difference for NO and H2O. This figure was prepared using original data from the author [11].
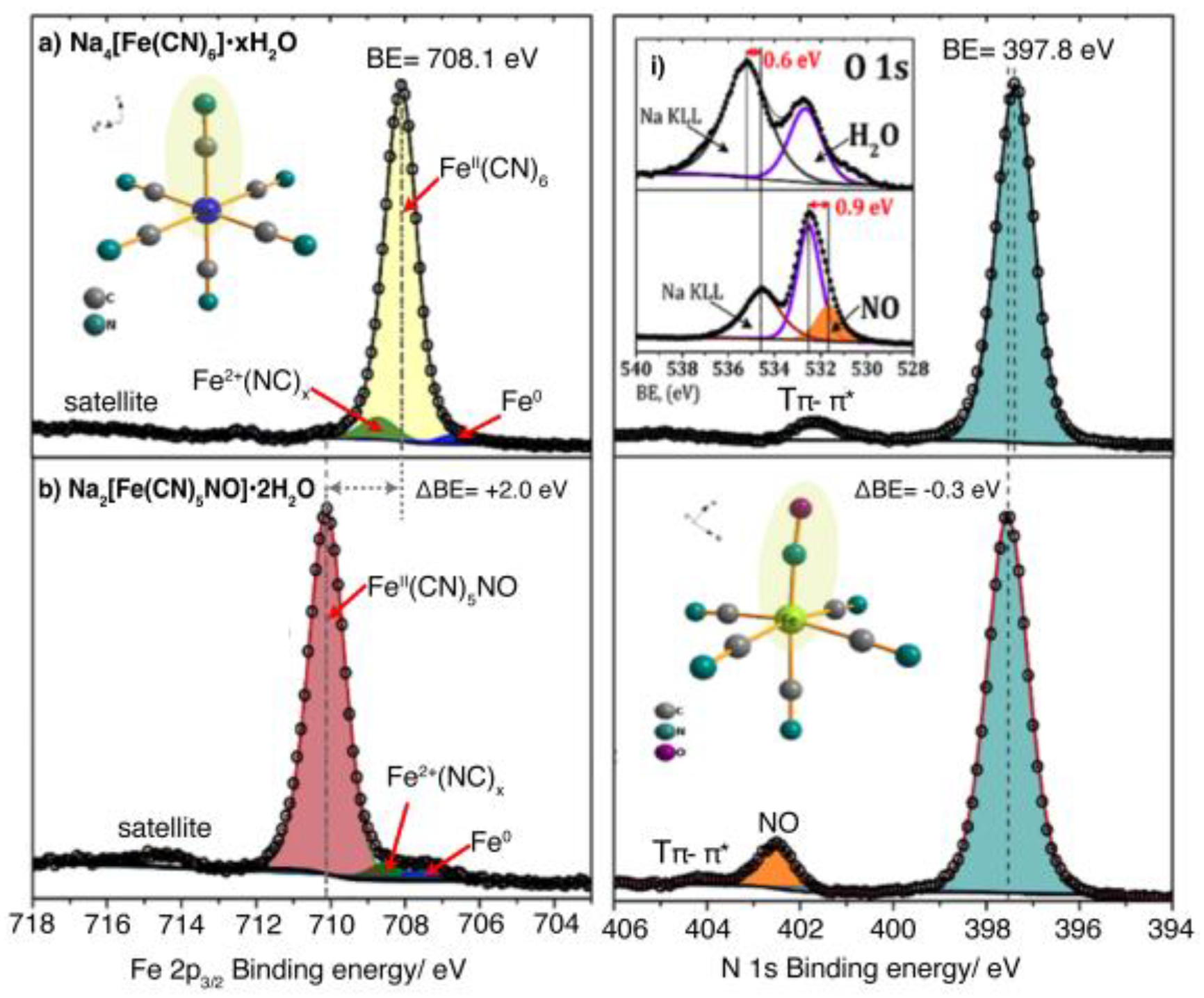
Figure 8.
a) Formation of 2D transition metal nitroprussides from their 3D analogs, inducing the rupture of the axial Fe-NCaxial coordination bond using an appropriate ligand (scissors). In the formed 2D solid, the ligand is found occupying the axial coordination sites for the metal (T). b) For T = Fe, in the resulting hybrid inorganic-organic solid, with pyridine and its derivatives as a pillar, thermal-induced spin crossover (SCO) can be observed. For a short pillar molecule, the SCO is an unexpected effect due to the repulsive NO∙∙∙NC interaction since the HS to LS transition involves a unit cell volume contraction; c) The SCO effect in this series of Hofmann-like coordination polymers involves an electron density redistribution in the solid.
Figure 8.
a) Formation of 2D transition metal nitroprussides from their 3D analogs, inducing the rupture of the axial Fe-NCaxial coordination bond using an appropriate ligand (scissors). In the formed 2D solid, the ligand is found occupying the axial coordination sites for the metal (T). b) For T = Fe, in the resulting hybrid inorganic-organic solid, with pyridine and its derivatives as a pillar, thermal-induced spin crossover (SCO) can be observed. For a short pillar molecule, the SCO is an unexpected effect due to the repulsive NO∙∙∙NC interaction since the HS to LS transition involves a unit cell volume contraction; c) The SCO effect in this series of Hofmann-like coordination polymers involves an electron density redistribution in the solid.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



