Submitted:
30 October 2024
Posted:
30 October 2024
You are already at the latest version
Abstract
Endohedral metallo-borospherenes M@B40 have received considerable attention since the discovery of B40 in 2014. However, the coordination bonding nature of most of the actinide-doped endohedral An@B40 still remains in disputes or unexplored. Extensive first-principles theory calculations performed herein unveil the ground states of triplet U@B40 (1, C2v, 3A2), quartet U@B40- (2, C2v, 4B1), quintet Np@B40+ (3, C2v, 5A1), sextet Np@B40 (4, C2, 6A), septet Pu@B40 (5, C2v, 7A2), octet Am@B40 (6, C2v, 8A2), and octet Cm@B40+ (7, C2v, 8A2) at the coupled-cluster with triple excitations CCSD(T) level. Detailed principal interacting spin orbital (PISO) and adaptive natural density partitioning (AdNDP) analyses reveal their coordination bonding patterns and show that, with the numbers of unpaired α-electrons in parallel spins varying from nα = 2, 3, 4, 5, 6, 7, to 7 in these complexes, the percentage contribution of the An 5f-involved PISO pairs to overall coordination bonding interactions decreases monotonously from 41% to 1%, the contribution of An 6d-involved PISO pairs increases monotonously from 47% to 72%, while the marginal contribution of An 7s-involved PISO pairs remains basically unchanged (4~7%). The IR, Raman, and photoelectron spectra of the most concerned species are computationally simulated to facilitate their characterizations in future experiments.
Keywords:
Actinides
; Metallo-Borospherenes
; First-Principles Theory
; Structures
; Coordination Bonding Patterns
1. Introduction
The discovery of the first all-boron fullerenes D2d B40-/0 in 2014[1] and C3/C2 B39- in 2015[2] paves the way for borospherene chemistry, with special attention paid to the structures and bonding of metallo-borospherenes.[3] Our group predicted at density functional theory (DFT) level the first endohedral metallo-borospherenes C2v Ca@B40 and D2d Sr@B40 and exohedral metallo-borospherenes Cs M&B40 (M = Be, Mg) in 2015.[4] Similar endohedral rare-earth-metal-doped Cs Sc@B40, C2v Y@B40, and C2v La@B40 have also been proposed at DFT.[5] Dong et al. proposed a B40 fullerene decorated with six Ti atoms as a promising candidate for hydrogen storage.[6] Fa et al. studied the structural stability of endohedral C2v Na@B40 and D2d Ba@B40 and exohedral Cs M&B40 (M = Li, K and Tl) at DFT.[7] Sr-doping was found to increase the conductance of B40 fullerene due to the decreased energy gap in D2d Sr@B40.[8] The Ti atom in Ti@B40 is found to reside very close to the boron framework, while the doubly doped Ti2@B40 possesses a singlet cube-like structure with Cs symmetry.[9] The exohedral Nin∈B40 complex series (n = 1-4) feature quasi-planar hepta-coordinate Ni centers on the cage surfaces in η7-B7 heptagons.[10] Li et al. predicted that Cu, Ag and Au atoms in MB40 (M = Cu, Ag and Au) favor the exohedral configuration.[11] Wang et al. predicted in 2017 the first singlet endohedral actinide-metal-doped D2d U@B40 at the pure DFT Perdew-Burke-Ernzerhof (DFT-PBE) level which, with the U atom located exactly at the center of the B40 cage, satisfies the 32-electron principle of 1S21P61D101F14.[12] However, at the hybrid PBE0 level, a slightly distorted triplet C1 U@B40 appears to be the ground state of the neutral species which lies 0.70 eV more stable than its singlet counterpart D2d U@B40.[13] Shi et al. systematically explored actinide-doped AnBm series (An = Ac, Th, Pa, U, Np, Pu, Am, Cm; m = 7, 20, 24, 36, 38, 39, 40) and suggested that doping with the right actinides may stabilize Bn clusters.[14,15,16,17,18] The lanthanide-doped octet D2d Eu@B40 (8B2) and septet Cs Gd@B40 (7A”) have also been predicted in theory.[19] Li et al. explored the ThB40 which revealed obvious covalent characters between the Th center and the B40 cage.[20]
In this work, we systematically investigated the coordination bonding nature of actinide-doped endohedral borospherenes An@B400/+/- (An = U, Np, Pu, Am, Cm) at the first-principles theory level. Extensive coupled-cluster calculations with triple excitations (CCSD(T)) reveal the ground states of U@B40 (1, C2v, 3A2), U@B40- (2, C2v, 4B1), Np@B40+ (3, C2v, 5A1), Np@B40 (4, C2, 6A), Pu@B40 (5, C2v, 7A2), Am@B40 (6, C2v, 8A2), and Cm@B40+ (7, C2v, 8A2) with the numbers of unpaired α-electrons of nα = 2, 3, 4, 5, 6, 7, and 7, respectively. Detailed Principal Interacting Spin Orbital (PISO) and adaptive natural density partitioning (AdNDP) analyses unveil the coordination bonding patterns of the complex series and quantitatively evaluated the variation trends of percentage contributions of An 5f-, 6d-, and 7s-involved PISO pairs to the overall coordination bonding energies with the numbers of unpaired α-electrons (nα) in the complex systems.
2. Results and Discussion
2.1. Structures and Stabilities
The optimized three lowest-lying isomers with different spin multiplicities of U@B40 and U@B40- are shown in Figure 1(a) and Figure 1(b), respectively. The optimized ground-state structures of the An@B400/+/- series (An = U, Np, Pu, Am, Cm) are collectively shown in Figure 1(c), with their alternative low-lying isomers with different spin multiplicities depicted according to their relative energies in Figure S1. As shown in Figure 1 and Figure S1, the calculated CCSD(T) relative energies at the most accurate theoretical level implemented in this work provide strong evidence to support both the hybrid PBE0 and TPSSh approaches.
Interestingly, as clearly shown in Figure 1(a), the triplet C2v U@B40 (1, 3A2) with two unpaired 5f α-electrons proves to be the well-defined ground state of the neutral complex which lies 0.93 and 1.01 eV more stable than the quintet C2 U@B40 (5B) and singlet D2d U@B40 (1A1) at the most accurate CCSD(T) level achieved in this work, respectively. Such a relative energy order qualitatively agrees with that obtained at both the hybrid DFT-PBE0 and DFT-TPSSh levels, but totally differs from that of previously reported results at pure DFT-PBE.[21] We believe the hybrid DFT and CCSD(T) relative energies are more reliable than that obtained at the pure DFT-PBE. As expected, with one extra electron attached, the U@B40- monoanion with three unpaired 5f α-electrons appears to have a quartet ground state of C2v U@B40- (2, 4B1) which lies 0.25 and 0.89 eV more stable than the sextet C2v U@B40- (6A2) and doublet C2v U@B40- (2A2) at CCSD(T), respectively. Detailed BOMD simulations collectively shown in Figure S2 indicate that both U@B40 (1) and U@B40- (2) are dynamically stable at 300K, with the small calculated root-mean-square-deviations of RMSD = 0.12, 0.08 Å and maximum bond length deviations of MAXD = 0.41, 27 Å, respectively.
Substituting the U coordination center in U@B40 (1) with heavier actinide metals Np, Pu, Am, and Cm, the quintet C2v Np@B40+ (3, 5A1), sextet C2 Np@B40 (4, 6A), septet C2v Pu@B40 (5, 7A2), octet C2v Am@B40 (6, 8A2), and C2v Cm@B40+ (7, 8A2) are obtained systematically which prove to be the ground states of the systems, as shown in Figure 1c and Figure S1, with the second lowest-lying C1 Np@B40+ (7A), C2v Np@B40 (4B2), C2v Pu@B40 (5B2), C2v Am@B40 (10B1), and Cs Cm@B40+ (6A′) being 0.41, 0.19, 0.07, 0.04, and 0.77 eV less stable than their corresponding ground states at CCSD(T), respectively. As discussed in details below, the ground states triplet U@B40 (1), quartet U@B40- (2), quintet Np@B40+ (3), sextet Np@B40 (4), septet Pu@B40 (5), octet Am@B40 (6), and octet Cm@B40+ (7) possess increasing spin multiplicities, with the numbers of unpaired α-electrons in parallel spins varying from nα = 2, 3, 4, 5, 6, 7, to 7, indicating that the increased valence electrons in complexes 1-7 are consecutively distributed in unpaired α-orbitals of the systems.
2.2. Bonding Pattern Analyses
As demonstrations, detailed AdNDP and PISO bonding patterns of the triplet C2v U@B40 (1) are presented in Figure 2. As shown in Figure 2(a), U@B40 (1) contains 1 1c-1e fxz2-type bond and 1 1c-1e fyz2-type bond on the U coordination center with the occupation numbers of ON = 0.92 and 0.93, respectively, 40 3c-2e and 8 6c-2e σ bonds on the B40 ligand with ON = 1.76-1.94, 4 6c-2e, 4 7c-2e, and 4 8c-2e π coordination bonds between the B40 ligand and U center with ON = 1.72-1.86, and 2 41c-2e σ coordination bonds between B40 and U with ON = 2.00. It is the two unpaired 5f α-electrons in parallel spins that determine the triplet ground state of the system (3A2).
Detailed PISO analyses on U@B40 (1) in Figure 2(b) with the B40 ligand and U coordination center as interacting fragments help to unveil a precise description of the coordination bonding pattern in the complex. As clearly shown in Figure 2(b), U@B40 (1) has two unpaired α-PISO 5f orbitals with the PISO populations of 0.921 and 0.933, respectively, as the singly occupied molecular orbitals (SOMOs) of the complex which have no corresponding β-PISO counterparts to correlate with, while all the remaining α-PISO and β-PISO pairs in exact one-to-one corresponding relationships are fully paired in couples, rendering the system a triplet ground state (3A2). The two unpaired α-PISO 5f orbitals turn out to well correspond to the 1c-1e fxz2-type bond and 1c-1e fyz2-type bond obtained by AdNDP analyses in Figure 2(a), respectively. Their small PISO-based bond indexes (PBI) of 0.146 and 0.125 indicate that the interactions between the two unpaired α-PISO U 5f orbitals and the corresponding nearly empty α-molecular orbitals of B40 ligand with the small PISO populations of 0.079 and 0.067 make marginable contributions (1.9% and 1.6%, respectively) to the overall coordination energy in the complex, with the remaining PISO pairs with PISO 5f populations between 0.084~0.294 and PBI values between 0.154~0.451 dominate the overall coordination interactions between the B40 ligand and U center. In combinations, these PISO pairs result in the 4 6c-2e, 4 7c-2e, and 4 8c-2e π coordination bonds and 2 41c-2e σ coordination bonds obtained in AdNDP analyses discussed above.
As shown in Figure 3, similar AdNDP and PISO bonding patterns exist for quartet U@B40- (2, 4B1), quintet Np@B40+ (3, 5A1), septet Pu@B40 (5, 7A2), octet C2v Am@B40 (6, 8A2), and octet C2v Cm@B40+ (7, 8A2) which possess three, four, six, seven, and seven unpaired α-PISO 5f electrons with the PISO populations between 0.87~0.91, 0.93-0.99, 0.79~0.98, 0.91~0.99, and 0.99~1.00 and PISO-based bond indexes between PBI = 0.16~0.23, 0.03~0.12, 0.04~0.34, 0.01~0.17, 0.01~0.02, respectively. Interestingly, the α-SOMO of the sextet Np@B40 (4) which contributes 7.8% to the overall coordination interaction turns out to be a typical α-bond with the comparable Np 5f α-PISO population of 0.432 and B40 β-PISO population of 0.568 and the PISO-based bond index of PBI = 0.491, respectively. Such an α-bond with nonnegligible contributions from both the Np coordination center and B40 ligand corresponds to a 41c-1e bond in AdNDP bonding analyses, as clearly shown in Figure 3.
2.3. Percentage Contributions of An 5f-, 6d-, and 7s-Involved PISO Pairs to the Overall Coordination Interactions
To compare the percentage contributions of An 5f-, 6d-, and 7s-involved PISO pairs to the overall An--B40 coordination interaction energies, we categorized the orbital types of An atoms involved in the PISO bonding patterns by their orbital shapes and consider the contributions of the corresponding PISO pairs separately. As shown in Figure 4, with the numbers of unpaired α-electrons in parallel spins varying from nα = 2, 3, 4, 5, 6, 7, to 7 in the complex series, the calculated overall An--B40 coordination interaction energies decrease generally from U@B40 (1, 3A2), U@B40- (2, 4B1), Np@B40+ (3, 5A1), Np@B40 (4, 6A), Pu@B40 (5, 7A2), to Am@B40 (6, 8A2) and increase slightly at Cm@B40+ (7, 8A2), with the percentage contributions of An 5f-involved PISO pairs to the overall coordination bonding interactions decreasing monotonously from 41% to 1%, the dominating contributions of An 6d-involved PISO pairs increasing monotonously from 47% to 72%, and the marginal contributions of An 7s-involved PISO pairs remaining basically unchanged (4~7%). The slight increase in overall coordination interaction energy at Cm@B40+ (7) mainly originates from the obvious increased contribution of the Cm 6d orbitals. These results show that with the metal center varying from U, Np, Pu, Am, to Cm, the tendency of the An-5f orbitals to participate in coordination bonding interactions with the B40 ligand weakens gradually from left to right in the periodic table, with the seven unpaired 5f α-electrons (5f7) in Cm@B40+ (7) contribute only about 1% to the overall coordination interaction energy, indicating an obvious actinide contraction in atomic radii from left to right in the periodic table. Figure 4 indicates that the An 6d atomic orbitals dominate the coordination interaction between the An centers and B40 ligand in the concerned An@B40 species, while An 5f and 7s make only minor contributions.
2.4. Simulated IR, Raman and PE Spectra
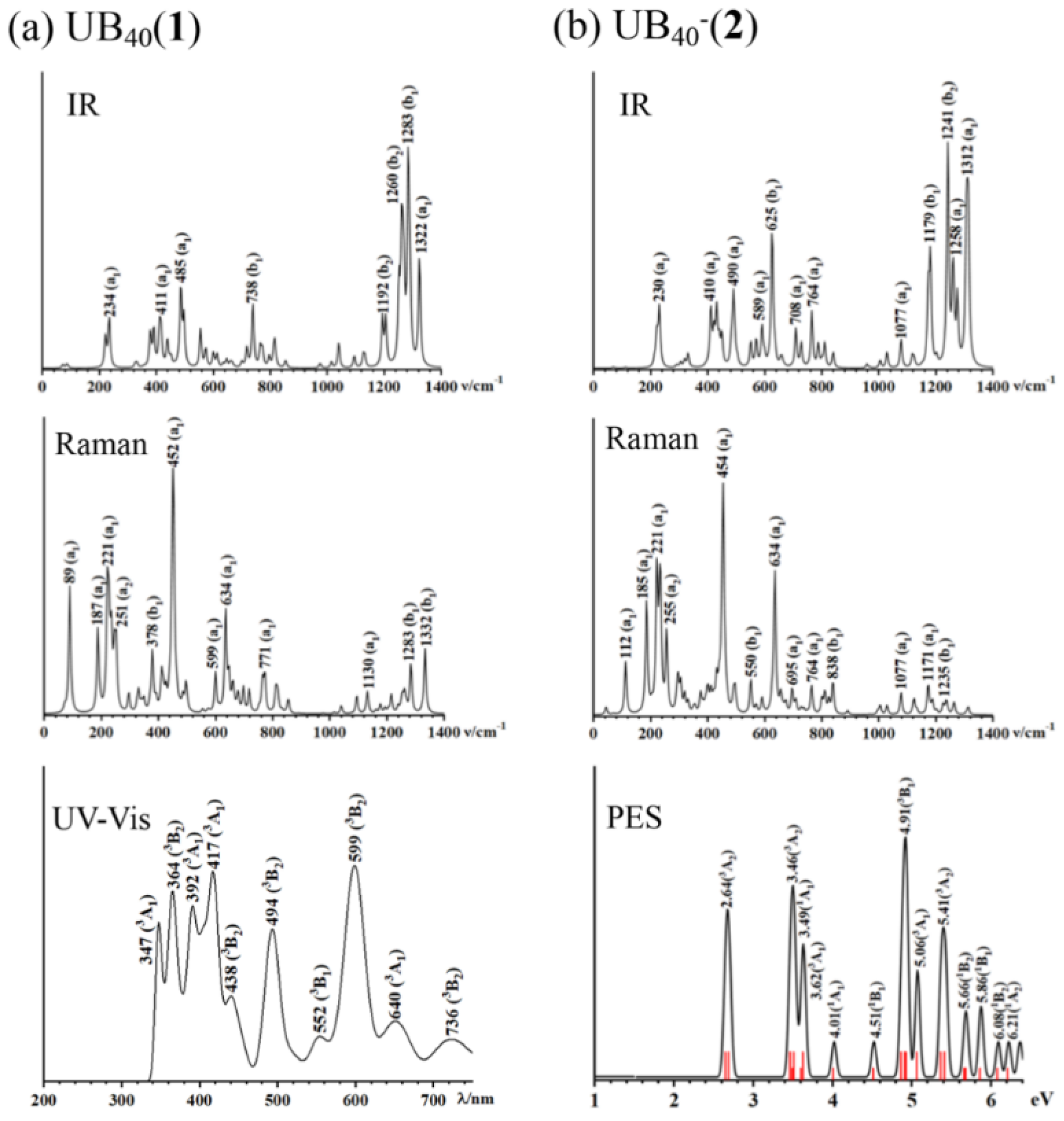
Infrared (IR) and photoelectron spectra (PES) measurements have proven to be the powerful approaches to characterize boron nanoclusters in gas phases.[1,2,3] We depict the simulated IR, Raman, and UV-Vis spectra of U@B40 (1) in Figure 5(a) and the calculated IR, Raman, and PES of U@B40- (2) in Figure 5(b) at PBE0 level to facilitate their future experimental characterizations. U@B40 (1) exhibits strong IR peaks at 234 (a1), 411 (a1), 485 (a1), 738 (b1), and 1283 (b1) cm-1, while its Raman spectrum features strong vibrational modes at 89 (a1), 221 (a1), 452 (a1), 634 (a1), and 1332 (b1). It is noticed that U@B40 (1) and U@B40- (2) possess the radial breathing modes (RBMs) at 452 cm-1 (a1) and 454 cm-1 (a1), respectively, which turns out to be slightly blue-shifted from that (428 cm-1 (a1)) of the empty D2d B40 borospherene at the same theoretical level. Similar IR and Raman spectra exist for U@B40- (2). The UV-vis spectrum of U@B40 (1) and PES spectrum of U@B40- (2) were calculated using the time-dependent DFT approach (TD-DFT) at PBE0 level. Since U@B40- has a quartet state, one-electron detachment from the anion could lead to triplet or singlet final states in the neutral. The first vertical detachment energy at VDE1 = 2.64 eV (3A2) for U@B40- was calculated as the energy difference between the anionic ground state and the neutral ground state at the optimized anion geometry. Higher vertical detachment energies at VDE = 3.46 (3A2), 4.91 (3B1), and 5.41 (3A2) eV correspond to vertical detachment transitions to the excited states of the neutral.
4. Theoretical Methods
The structures of endohedral actinide-metal-doped An@B400/+/- (An = U, Np, Pu, Am, Cm) were fully optimized at both the hybrid DFT-PBE0[22] and DFT-TPSSh[23] levels, with the 6-311+G(d)[24] basis set used for B and the scalar-relativistic Stuttgart energy-consistent pseudopotential with the 32-valence-electron and associated ECP60MWB_SEG valence basis set[25,26] employed for An. The single-point relative energies were further refined at the more accurate domain based local pair-natural orbital based singles and doubles coupled cluster method (DLPNO-CCSD(T))[27] implemented in the ORCA program[28], with the segmented all-electron relativistically contracted basis sets with the DKH2 Hamiltonians (SARC-DHK-TZVP) used for An and DKH-def2-SVP basis set chosen for B.[29] Vibrational frequency and wavefunction stability checks were performed at PBE0 level to make sure that all the lowest-lying isomers obtained are true minima of the systems without imaginary frequencies. All the PBE0 and TPSSh computations were performed using the Gaussian09[30] program package. Detailed Born-Oppenheimer molecular dynamic (BOMD) simulations were performed on both U@B40 (1) and U@B40- (2) at 300 K for 30 ps. BOMD simulation was implemented employing the CP2K[31] code with the GTH-PBE pseudopotentials and the TZVP-MOLOPTSR-GTH basis sets. The infrared and Raman spectra of C2v U@B40 (1) and C2v U@B40- (2) were simulated at PBE0/6-311+G(d). The UV-vis absorption spectra of U@B40 (1) and PE spectrum of U@B40- (2) were simulated using the time-dependent DFT method (TD-DFT-PBE0) approach.[32,33]
Chemical bonding patterns were analyzed employing both the AdNDP [34,35] method and principal interacting orbital (PIO) [36] approach based on the natural population analyses using the NBO 6.0[37] program. In this work, PISO [38] analyses based on PIO calculations were performed on the open-shell An@B40-/0/+ series. The PIO analyses were also carried out using the Gaussian 09 program with 6-31G* basis set used for B atoms and ECP60MWB_SEG employed for An. The VMD[39] program was used for the visualization of structures and molecular orbitals.
5. Conclusions
In summary, we have predicted in this work the ground states of triplet U@B40 (1), quartet U@B40- (2), quintet Np@B40+ (3), sextet Np@B40 (4), septet Pu@B40 (5), octet Am@B40 (6), and octet Cm@B40+ (7) at CCSD(T) level, revealed their coordination bonding patterns using both the PISO and AdNDP approaches, and calculated the percentage contributions of An 5f-, 6d-, and 7s-involved PISO pairs to the overall coordination interaction energies at PBE0 level, respectively, unveiling the coordination bonding nature of these actinide-doped endohedral metallo-borospherenes both qualitatively and quantitatively. Such high spin-multiplicity actinide-doped endohedral metallo-borospherenes could be expanded to all the actinides in the periodic table to form various magnetic complexes and crystals with potential applications in digital device.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Relative energies of the low-lying isomers of An@B400/+/- (An = Np to Cm) with different spin multiplicities at PBE0/B/6-311+G*/An/ECP60MWB, TPSSh/B/6-311+G*/An/ECP60MWB, and CCSD(T) levels, Figure S2: Molecular dynamics simulations of U@B40 (1) and U@B40- (2) at 300 K, with the calculated root-mean-square-deviations (RMSD) and maximum bond length deviations (MAXD) indicated, respectively, Table S1: Optimized coordinates (x, y, z) of C2v U@B40 (1), C2v U@B40- (2), C2v Np@B40+ (3), C2 Np@B40 (4), C2v Pu@B40 (5), C2v Am@B40 (6) and C2v Cm@B40+ (7) at PBE0 level.
Author Contributions
S.L. conceived the project and finalized the manuscript. X.Z. performed the DFT calculations. Z.W. and S.L. provided valuable discussion. X.Z wrote the manuscript, and all authors participated in the revision of the manuscript and agreed to the published version of the manuscript.
Data Availability Statement
All the data are available online.
Acknowledgments
The work was supported by the National Natural Science Foundation of China (22373061 and 21973057 to S.-D. Li).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Zhai, H.J.; Zhao, Y.F.; Li, W.L.; Chen, Q.; Bai, H.; Hu, H.S.; Piazza, Z.A.; Tian, W.J.; Lu, H.G.; Wu, Y.B.; et al. Observation of an All-Boron Fullerene. Nat Chem 2014, 6, 727–731. [CrossRef]
- Chen, Q.; Li, W.L.; Zhao, Y.F.; Zhang, S.Y.; Hu, H.S.; Bai, H.; Li, H.R.; Tian, W.J.; Lu, H.G.; Zhai, H.J.; et al. Experimental and Theoretical Evidence of an Axially Chiral Borospherene. ACS Nano 2015, 9, 754–760. [CrossRef]
- Jian, T.; Chen, X.; Li, S.D.; Boldyrev, A.I.; Li, J.; Wang, L.S. Probing the Structures and Bonding of Size-Selected Boron and Doped-Boron Clusters. Chem Soc Rev 2019, 48, 3550–3591. [CrossRef]
- Bai, H.; Chen, Q.; Zhai, H.J.; Li, S.D. Endohedral and Exohedral Metalloborospherenes: M@B40 (M = Ca, Sr) and M&B40 (M = Be, Mg). Angewandte Chemie—International Edition 2015, 54, 941–945. [CrossRef]
- Jin, P.; Hou, Q.; Tang, C.; Chen, Z. Computational Investigation on the Endohedral Borofullerenes M@B40 (M = Sc, Y, La). Theor Chem Acc 2015, 134, 1–10. [CrossRef]
- Dong, H.; Hou, T.; Lee, S.T.; Li, Y. New Ti-Decorated B 40 Fullerene as a Promising Hydrogen Storage Material. Sci Rep 2015, 5, 1–8. [CrossRef]
- Fa, W.; Chen, S.; Pande, S.; Zeng, X.C. Stability of Metal-Encapsulating Boron Fullerene B40. Journal of Physical Chemistry A 2015, 119, 11208–11214. [CrossRef]
- An, Y.; Zhang, M.; Wu, D.; Fu, Z.; Wang, T.; Xia, C. Electronic Transport Properties of the First All-Boron Fullerene B40 and Its Metallofullerene Sr@B40. Physical Chemistry Chemical Physics 2016, 18, 12024–12028. [CrossRef]
- Jin, P.; Yang, L.; Liu, C.; Hou, Q.; Li, L. Computational Prediction of the Endohedral Metalloborofullerenes Tin@B40 (n = 1, 2). Theor Chem Acc 2017, 136, 1–12. [CrossRef]
- Li, H.R.; Tian, X.X.; Luo, X.M.; Yan, M.; Mu, Y.W.; Lu, H.G.; Li, S.D. Heteroborospherene Clusters Nin € B40 (n = 1-4) and Heteroborophene Monolayers Ni2 € B14 with Planar Heptacoordinate Transition-Metal Centers in H7-B7 Heptagons. Sci Rep 2017, 7, 1–7. [CrossRef]
- Li, S.X.; Zhang, Z.P.; Long, Z.W.; Qin, S.J. Structures, Stabilities and Spectral Properties of Metalloborospherenes MB 0/- 40 (M = Cu, Ag, and Au). RSC Adv 2017, 7, 38526–38537. [CrossRef]
- Yu, T.; Gao, Y.; Xu, D.; Wang, Z. Actinide Endohedral Boron Clusters: A Closed-Shell Electronic Structure of U@B40. Nano Res 2018, 11, 354–359. [CrossRef]
- Wang, J.; Xie, W.; Jiang, W.; Wu, X.; Wang, Z. The Reliability of the Density-Functional Theory in Actinide Endohedral Systems. Adv Theory Simul 2019, 2, 1–7. [CrossRef]
- Zhang, N.; Li, A.; Wang, C.; Wu, Q.; Lan, J.; Chai, Z.; Zhao, Y.; Shi, W. Theoretical Prediction of Chiral Actinide Endohedral Borospherenes. New Journal of Chemistry 2021, 45, 6803–6810. [CrossRef]
- Zhang, N.; Wang, C.; Wu, Q.; Lan, J.; Chai, Z.; Shi, W. Highly Stable Actinide(Iii) Complexes Supported by Doubly Aromatic Ligands†. Physical Chemistry Chemical Physics 2022, 24, 5921–5928. [CrossRef]
- Wang, C.Z.; Bo, T.; Lan, J.H.; Wu, Q.Y.; Chai, Z.F.; Gibson, J.K.; Shi, W.Q. Ultrastable Actinide Endohedral Borospherenes. Chemical Communications 2018, 54, 2248–2251. [CrossRef]
- Wang, J.; Wang, C.Z.; Wu, Q.Y.; Lan, J.H.; Chai, Z.F.; Nie, C.M.; Shi, W.Q. Construction of the Largest Metal-Centered Double-Ring Tubular Boron Clusters Based on Actinide Metal Doping. Journal of Physical Chemistry A 2022, 126, 3445–3451. [CrossRef]
- Zhang, N.; Wang, C.; Wu, Q.; Lan, J.; Chai, Z.; Shi, W. Highly Stable Actinide(Iii) Complexes Supported by Doubly Aromatic Ligands†. Physical Chemistry Chemical Physics 2022, 24, 5921–5928. [CrossRef]
- Xi, C.; Yang, L.; Liu, C.; You, P.; Li, L.; Jin, P. Lanthanide Metals in the Boron Cages: Computational Prediction of M@Bn (M = Eu, Gd; n = 38, 40). Int J Quantum Chem 2018, 118, 1–11. [CrossRef]
- Li, Y.; Wang, Y.; Zhou, Z.; Gao, Y.; Chen, Y.; Zhang, G.; Ma, C. Insights into ThB40: Stability, Electronic Structure, and Interaction. Molecules 2024, 29. [CrossRef]
- Wang, J.; Xie, W.; Jiang, W.; Wu, X.; Wang, Z. The Reliability of the Density-Functional Theory in Actinide Endohedral Systems. Adv Theory Simul 2019, 2, 1–7. [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. Journal of Chemical Physics 1999, 110, 6158–6170. [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. Journal of Chemical Physics 2003, 119, 12129–12137. [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J Chem Phys 1980, 72, 650–654. [CrossRef]
- Cao, X.; Dolg, M. Segmented Contraction Scheme for Small-Core Actinide Pseudopotential Basis Sets. Journal of Molecular Structure: THEOCHEM 2004, 673, 203–209. [CrossRef]
- Cao, X.; Dolg, M.; Stoll, H. Valence Basis Sets for Relativistic Energy-Consistent Small-Core Actinide Pseudopotentials. Journal of Chemical Physics 2003, 118, 487–496. [CrossRef]
- Guo, Y.; Riplinger, C.; Becker, U.; Liakos, D.G.; Minenkov, Y.; Cavallo, L.; Neese, F. Communication: An Improved Linear Scaling Perturbative Triples Correction for the Domain Based Local Pair-Natural Orbital Based Singles and Doubles Coupled Cluster Method [DLPNO-CCSD(T)]. Journal of Chemical Physics 2018, 148. [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip Rev Comput Mol Sci 2012, 2, 73–78. [CrossRef]
- Pantazis, D.A.; Neese, F. All-Electron Scalar Relativistic Basis Sets for the Actinides. J Chem Theory Comput 2011, 7, 677–684. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.; et al. Gaussian 09, Revision D.01, Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Vandevondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput Phys Commun 2005, 167, 103–128. [CrossRef]
- Bauernschmitt, R.; Ahlrichs, R. Treatment of Electronic Excitations within the Adiabatic Approximation of Time Dependent Density Functional Theory. Chem Phys Lett 1996, 256, 454–464. [CrossRef]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular Excitation Energies to High-Lying Bound States from Time-Dependent Density-Functional Response Theory: Characterization and Correction of the Time-Dependent Local Density Approximation Ionization Threshold. Journal of Chemical Physics 1998, 108, 4439–4449. [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Physical Chemistry Chemical Physics 2008, 10, 5207–5217. [CrossRef]
- Tkachenko, N. V.; Boldyrev, A.I. Chemical Bonding Analysis of Excited States Using the Adaptive Natural Density Partitioning Method. Physical Chemistry Chemical Physics 2019, 21, 9590–9596. [CrossRef]
- Zhang, J.X.; Sheong, F.K.; Lin, Z. Unravelling Chemical Interactions with Principal Interacting Orbital Analysis. Chemistry—A European Journal 2018, 24, 9639–9650. [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J Comput Chem 2013, 34, 1429–1437. [CrossRef]
- Sheong, F.K.; Zhang, J.X.; Lin, Z. Principal Interacting Spin Orbital: Understanding the Fragment Interactions in Open-Shell Systems. Physical Chemistry Chemical Physics 2020, 22, 10076–10086. [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J Mol Graph 1996, 14, 33–38. [CrossRef]
Figure 1.
Optimized three low-lying isomers of (a) U@B40 and (b) U@B40- with their relative energies indicated in eV at PBE0, TPSSh (parentheses), and CCSD(T)/PBE0 (square brackets) levels, respectively, and optimized ground-state structures of (c) C2v U@B40 (1,3A2), C2v U@B40- (2, 4B1), C2v Np@B40+ (3, 5A1), C2 Np@B40 (4, 6A), C2v Pu@B40 (5, 7A2), C2v Am@B40 (6, 8A2), and C2v Cm@B40+ (7, 8A2) at PBE0.
Figure 1.
Optimized three low-lying isomers of (a) U@B40 and (b) U@B40- with their relative energies indicated in eV at PBE0, TPSSh (parentheses), and CCSD(T)/PBE0 (square brackets) levels, respectively, and optimized ground-state structures of (c) C2v U@B40 (1,3A2), C2v U@B40- (2, 4B1), C2v Np@B40+ (3, 5A1), C2 Np@B40 (4, 6A), C2v Pu@B40 (5, 7A2), C2v Am@B40 (6, 8A2), and C2v Cm@B40+ (7, 8A2) at PBE0.
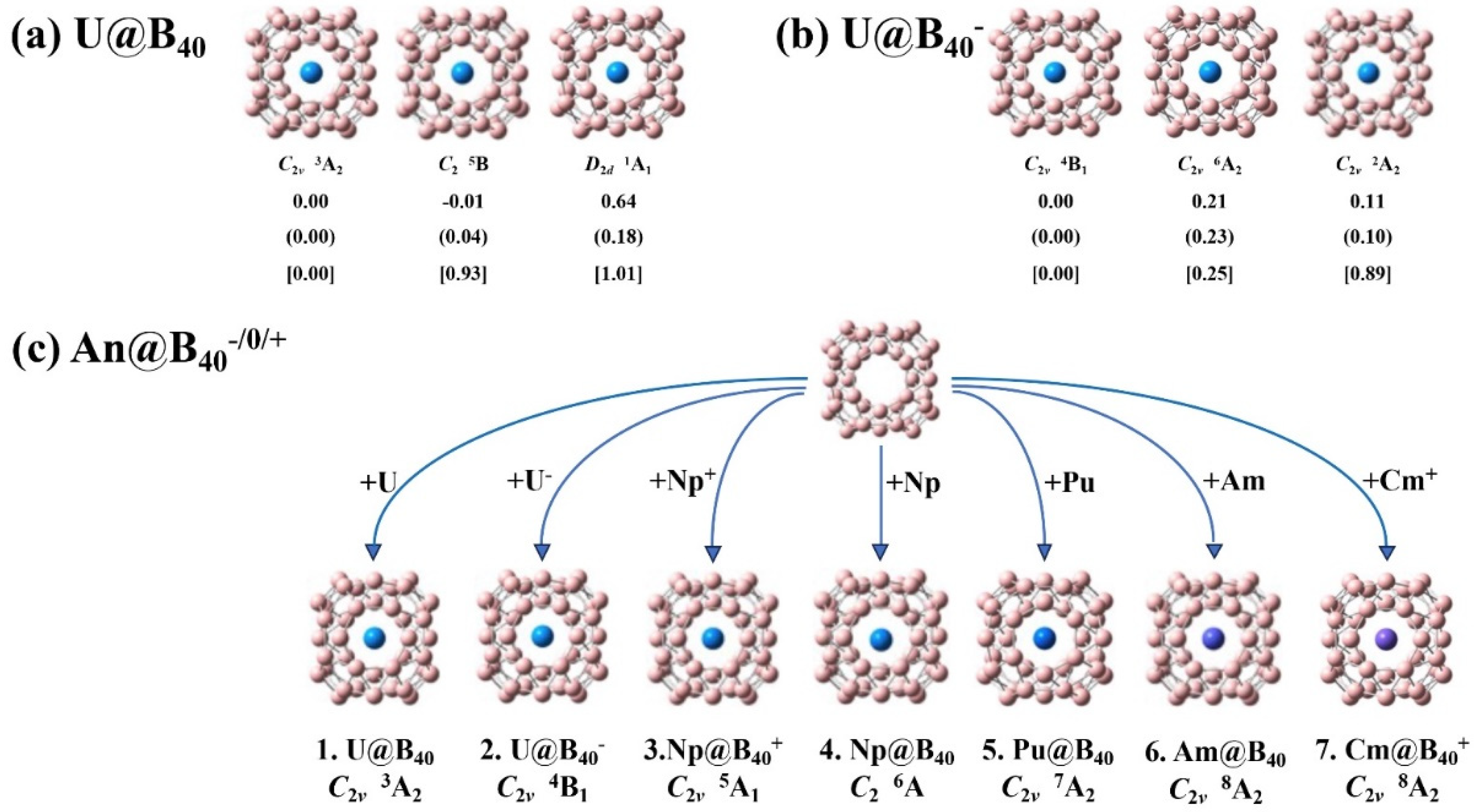
Figure 2.
(a) AdNDP bonding pattern of triplet C2v U@B40 (1), with the occupation numbers (ON) indicated. (b) PISO bonding pattern of C2v U@B40 (1) with the U coordination center and B40 ligand as interacting fragments, with the corresponding occupation numbers (PISO Pop.), PIO-based bond indexes (PBI), and percentage contributions (contrib/%) to the overall coordination interactions indicated.
Figure 2.
(a) AdNDP bonding pattern of triplet C2v U@B40 (1), with the occupation numbers (ON) indicated. (b) PISO bonding pattern of C2v U@B40 (1) with the U coordination center and B40 ligand as interacting fragments, with the corresponding occupation numbers (PISO Pop.), PIO-based bond indexes (PBI), and percentage contributions (contrib/%) to the overall coordination interactions indicated.
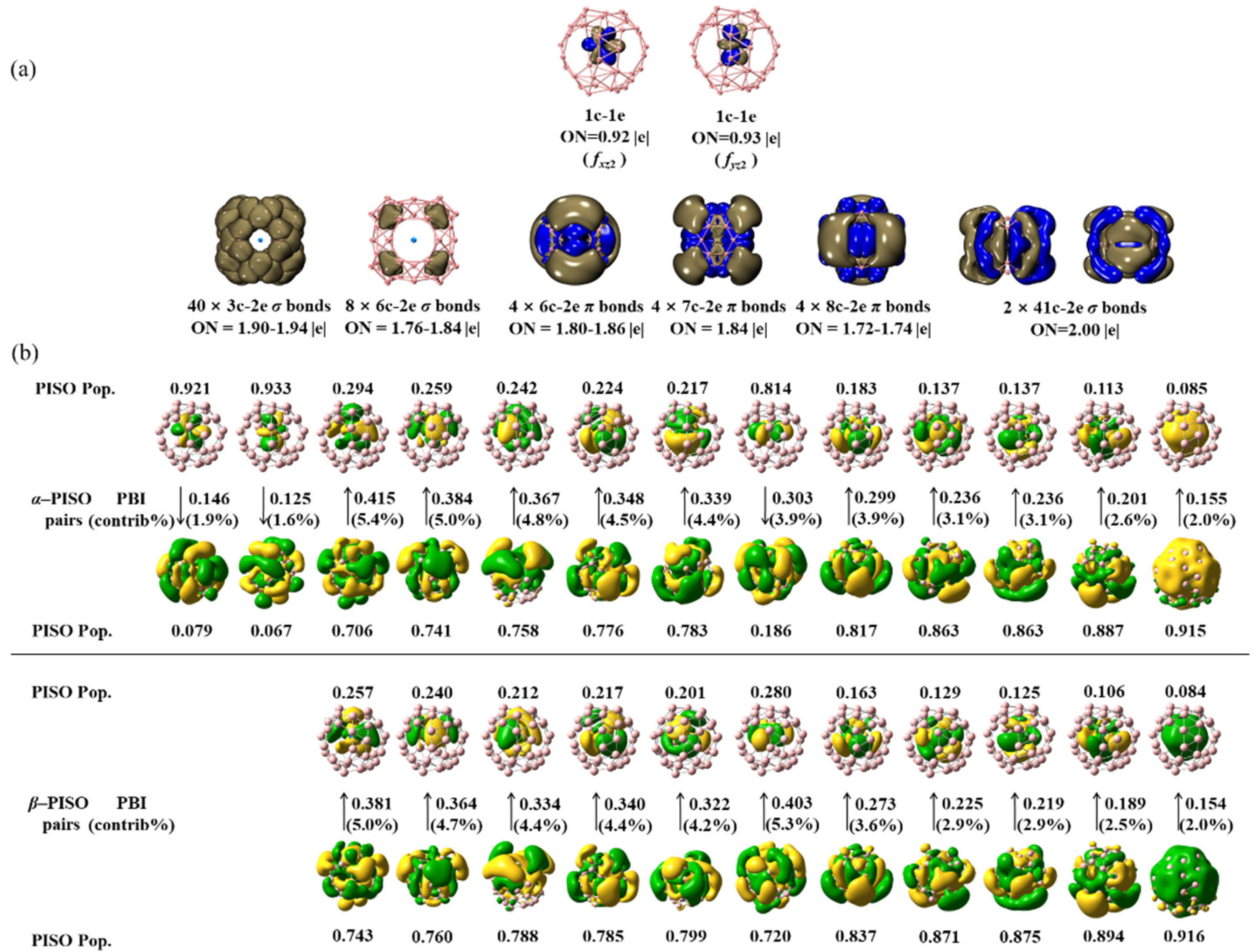
Figure 3.
Unpaired PISO α-orbitals of U@B40 (1, 3A2), U@B40- (2, 4B1), Np@B40+ (3, 5A1), Np@B40 (4, 6A), Pu@B40 (5, 7A2), Am@B40 (6, 8A2), and Cm@B40+ (7, 8A2), with the α-spin occupation numbers (PISO Pop.) associated with the principal interacting spin orbitals, PISO-based bond indexes (PBI), and their percentage contributions (contrib/%) to the overall coordination interactions between the An coordination center and B40 ligand indicated. The corresponding AdNDP analyses of the singly occupied 1c-1e α-5f orbitals in 1, 2, 3, 5, 6, and 7 and 41c-1e α-bond in Np@B40 (4) are compared at the bottom, with the occupation numbers (ON) indicated.
Figure 3.
Unpaired PISO α-orbitals of U@B40 (1, 3A2), U@B40- (2, 4B1), Np@B40+ (3, 5A1), Np@B40 (4, 6A), Pu@B40 (5, 7A2), Am@B40 (6, 8A2), and Cm@B40+ (7, 8A2), with the α-spin occupation numbers (PISO Pop.) associated with the principal interacting spin orbitals, PISO-based bond indexes (PBI), and their percentage contributions (contrib/%) to the overall coordination interactions between the An coordination center and B40 ligand indicated. The corresponding AdNDP analyses of the singly occupied 1c-1e α-5f orbitals in 1, 2, 3, 5, 6, and 7 and 41c-1e α-bond in Np@B40 (4) are compared at the bottom, with the occupation numbers (ON) indicated.

Figure 4.
Variation of the calculated overall An--B40 coordination interaction energies highlighted in blue and the corresponding PISO percentage contributions of 5f-, 6d-, and 7s-orbital-involved pair interactions highlighted in red in U@B40 (1, 3A2), U@B40- (2, 4B1), Np@B40+ (3, 5A1), Np@B40 (4, 6A), Pu@B40 (5, 7A2), Am@B40 (6, 8A2), and Cm@B40+ (7, 8A2) with the numbers of singly occupied 5f electrons (nα) at PBE0 level.
Figure 4.
Variation of the calculated overall An--B40 coordination interaction energies highlighted in blue and the corresponding PISO percentage contributions of 5f-, 6d-, and 7s-orbital-involved pair interactions highlighted in red in U@B40 (1, 3A2), U@B40- (2, 4B1), Np@B40+ (3, 5A1), Np@B40 (4, 6A), Pu@B40 (5, 7A2), Am@B40 (6, 8A2), and Cm@B40+ (7, 8A2) with the numbers of singly occupied 5f electrons (nα) at PBE0 level.
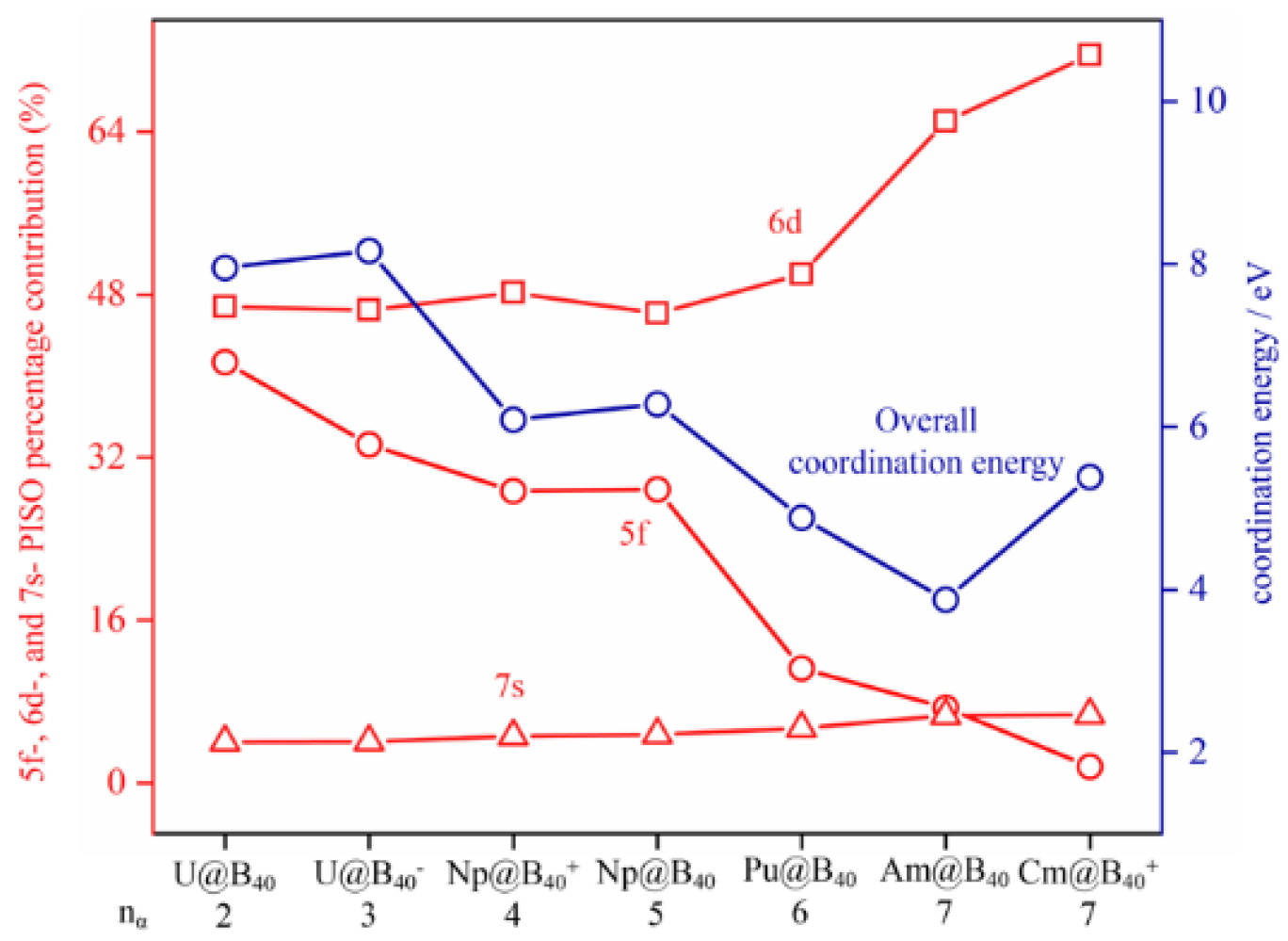
Figure 5.
Simulated IR, Raman and UV-Vis spectra of (a) C2v U@B40 (1, 3A2) and IR, Raman, photoelectron spectrum (PES) of (b) C2v U@B40- (2, 4B1) at PBE0 level.
Figure 5.
Simulated IR, Raman and UV-Vis spectra of (a) C2v U@B40 (1, 3A2) and IR, Raman, photoelectron spectrum (PES) of (b) C2v U@B40- (2, 4B1) at PBE0 level.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



