Submitted:
19 October 2025
Posted:
20 October 2025
You are already at the latest version
Abstract
Polyploid crops such as wheat, Brassica and cotton are critical in global agricultural and economic system. However, their productivity is threatened increasingly by biotic stresses such as disease, and abiotic stresses such as heat, both exacerbated by climate change. Understanding the molecular basis of stress responses in these crops is crucial but remains challenging due to their complex genetic makeup—characterized by large sizes, multiple genomes and limited annotation resources. Proteomics is a powerful approach to elucidate molecular mechanisms, enabling the identification of stress-responsive proteins, cellular localization, physiological, biochemical and meta-bolic pathways, protein-protein interactions, and post-translational modifications. It also sheds light on the evolutionary consequences of genome duplication and hybridi-zation. Breeders can improve stress tolerance and yield traits by characterizing the proteome of polyploid crops. Functional and subcellular proteomics, and the identifi-cation and introgression of stress-responsive protein biomarkers, are promising for crop improvement. Nonetheless, several challenges remain, including inefficient pro-tein extraction method, limited organelle-specific data, insufficient protein annota-tions, low proteoform coverage, reproducibility and lack of target-specific antibodies. This review explores the genomic complexity of three key allopolyploid crops—wheat, oilseed Brassica, and cotton—summarizes recent proteomic insights into heat stress and pathogen response and discusses current challenges and future directions for ad-vancing proteomics in polyploid crop improvement through proteomics.
Keywords:
wheat
; cotton
; oilseed brassica
; climate-resilient
; disease
; breeding
; yield
; quality
1. Introduction
Polyploid crops—with multiple sets of chromosomes—play an important role in global agriculture. They are the key drivers of crop evolution and contribute to global food security [1]. Polyploids exhibit greater vigor and yield potential than their diploid relatives. Whether natural or induced, polyploidization enhances heterozygosity, reduces inbreeding depression and mitigates deleterious mutations. Polyploids are classified into two major types: autoploids, with duplicated identical genomes [2], and alloploids formed by hybridization of more than two copies of divergent genomes [3].
Examples of autoploid crops include potato (Solanum tuberosum; 2n = 4x = 48 [4]) and sugarcane (Saccharum spp.; 2n = 62, 80, 96, 112, or 128 [5]). In contrast, alloploids include wheat (Triticum aestivum; AABBDD; 2n = 6x = 42 [6]), Brassica species (Brassica spp.; 2n = 2x = 34 (BBCC), 36 (AABB) and 38 (AACC) [7]) and cotton (Gossypium hirsitum; AADD; 2n = 2x = 52 [8]). Other notable alloploids include oat (Avena sativa; AACCDD; 2n = 6x = 42), different millets (e.g., finger millet, porso millete, fonio millet, and Indian- and Japanese-barnyard millet) and quinoa (Chenopodium quinoa; AABB; 2n = 4x = 36) [8]. Due to their dietary, industrial, and economic value, this review focuses on three globally significant allopolyploid crops—wheat, oilseed Brassica, and cotton.
Wheat (Triticum aestivum) is the most widely grown cereal crop, critical for food, animal feed and trade. In 2023–2024, global wheat production was estimated 788 million tons (MT), with 209.6 MT traded [9]. Despite this, production has declined while demand continues to rise, highlighting the urgent need for yield improvement.
Oilseed Brassica, a member of the Brassicaceae family, includes 4,000 of species, many of which are cultivated for edible oil [10]. Among them, B. napus (rapeseed/canola), B. campestris, and B. juncea are the most important [11]. However, B. napus is widely grown globally due to its low erucic acid and glucosinolate levels and high oleic and linolenic acid content. In contrast, B. campestris and B. juncea, which have higher erucic acid, are more regionally cultivated—B. campestris in temperate countries such as Australia, Germany, Sweden, France and Canada, and B. juncea in Asia [12]. Global canola production reached 89.39 MT in 2023–2024, with a projected decline for 2024–2025 [13].
Cotton has industrial value in textiles, paper, oil, chipboard and livestock feed industries [14]. It is valued at around $600 billion to the global textile industry economy [15], with an estimated production of 24.12 MT in 2023–2024 [16].
The demand for these crops is rising due to global population growth—projected to increase by 25% by 2050 [17]. Wheat production must double by 2050 [18,19], cotton production must reach 28.1 MT by 2032 [20], and canola production in Canada is projected to increase by 3 MT by 2030 [21]. However, achieving these targets is hampered by biotic (e.g., pathogens and pests) and abiotic (e.g., heat, drought and salinity) stresses, which are becoming more severe with climate change. For instance, drought can reduce yields by 32% in wheat [22], ~60% in canola [23], and 31% in cotton [24]. A 1°C increase above 15 ℃ can reduce global wheat yields by 6% [25], and a similar increase above 30 ℃ can reduce canola and cotton yields by 7% [26] and 6.1% [27], respectively. These stresses disrupt plant phenotypes, physiology, biochemical, and metabolism, ultimately reducing yield and product quality.
For example, an individual heat stress (HS) and combined HS and drought stresses reduce canola yield and oil concentration significantly [28], and impair cotton ball development [29]. In wheat, HS stress at the reproductive stage can cause pollen malformation and male sterility, [30,31,32] and a combined HS and drought stresses can cause mitotic and meiotic arrest, ovule abortion and reduce stigma receptivity [33], drastically reducing grain number and quality. Biotic stresses are equally harmful, with powdery mildew in wheat causing up to 33% yield loss [34], and clubroot disease in canola [35] and cotton leafroll dwarf virus [36] causing up to 100% yield loss in severe cases.
Understanding the molecular basis of stress responses is essential to combat these threats. During stress, plant nucleus receives signals to alter gene expressions—exportation of mRNA from nucleus to cytoplasm and biosynthesis of stress-induced novel proteins [37,38,39]. To acclimatize stress, plant phenotype changes due to the biological function of the novel proteins. As biotic and abiotic stress response, the biological function of a protein is governed by proteoforms—the basic molecular forms of proteins produced from a single gene including genetic variants, alternative splice variants and post-translational modifications (PTMs) [40]—, their cellular localization, and protein-protein and protein-other non-protein compound’s interactions [38,39]. Therefore, while genomics, transcriptomics, and phenomics have been useful to study changes in DNA, RNA [41], and phenotypes [42], respectively, proteomics provides more direct insights into stress adaptation mechanisms [43]. Unlike transcriptomics, which may not correlate with actual protein levels [44]. For example, transcriptomics techniques such as microarray and RNA-seq covers 48–57% of phenotypic variation and has moderate correlation with mRNA and protein (identified by label-free proteomics) expression [45]. Proteomics can identify proteins and proteoforms actively involved in stress-related physiological, biochemical, and metabolic processes [46], and protein–protein interactions [47]. Proteomics is also a valuable tool for understanding the consequences of polyploidization [48].
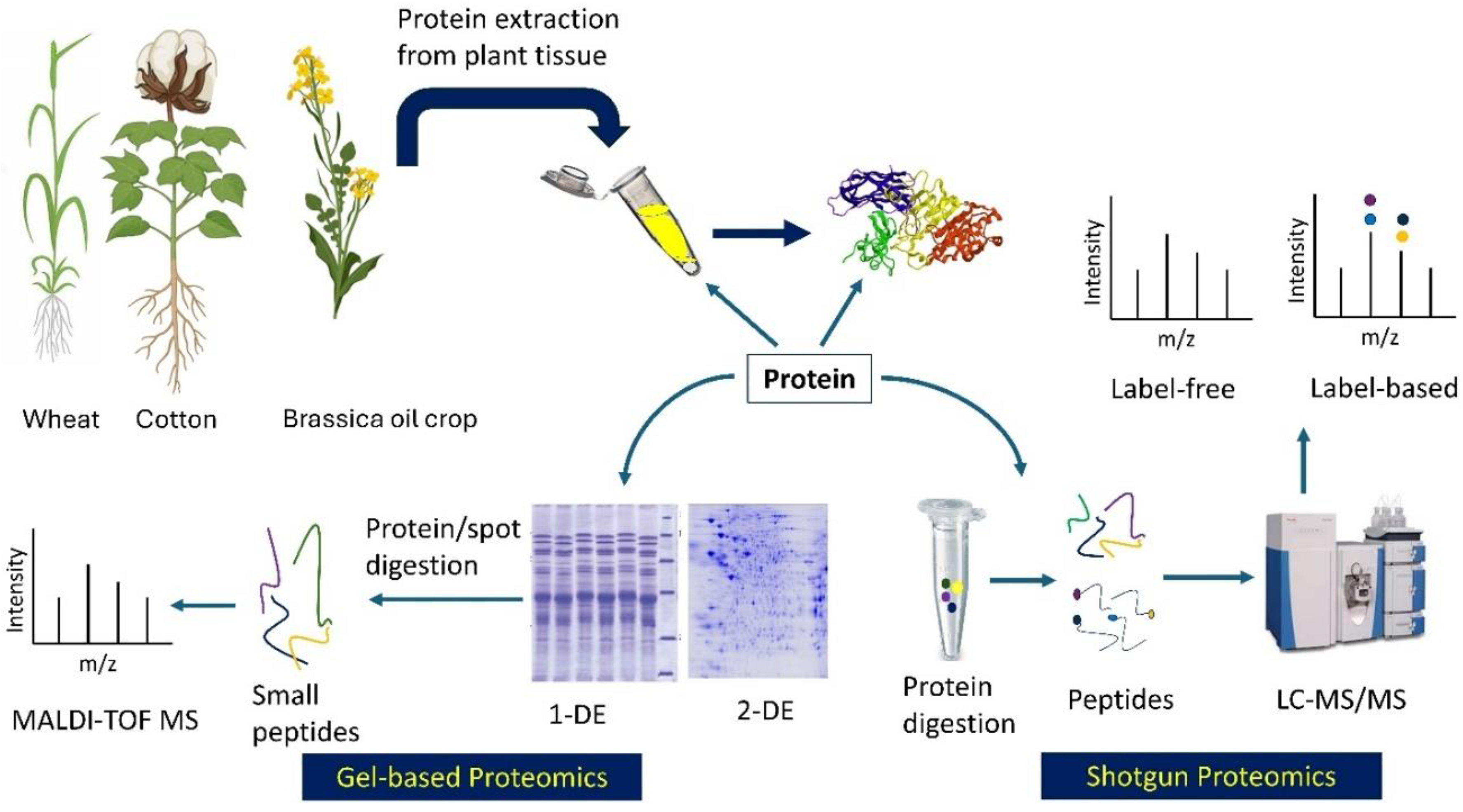
Several proteomic tools are popularly used in polyploid crops to get stress-responsive molecular insights (Figure 1). Mass spectrometry (MS) is the most extensively used tool in proteomics since its discovery in 1913 [49]. Techniques such as 2-dimensional polyacrylamide gel electrophoresis-MS (2DE-PAGE/MS) and its improved version, 2D-DIGE (2-dimensional difference gel electrophoresis), have evolved into advanced methods such as quadrupole (Q), time-of-flight (TOF), orbitrap, matrix-assisted laser desorption/ionization couple with TOF (MALDI-TOF), and shotgun proteomics [43,48]. Shotgun methods such as label-based method, isobaric tags for relative and absolute quantitation (iTRAQ), and label-free quantitative proteomics are widely used. While label-free approaches offer precise quantification of all peptides of a sample, they are data-dependent acquisition (DDA) method and therefore, often fail to identify extensive low abundant protein or peptide [50]. To overcome the challenge, sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) ), a data-independent acquisition (DIA) method, is being applied in crops like, including wheat [51,52] and cotton [53].
Despite these advances, proteomic research in polyploid crops remains challenging due to genome complexity, inefficient protein extraction methods, lack of organelle-specific databases, insufficient protein annotations, and limited protein validation [43,54,55,56]. The genomic complexity of allopolyploids arises from the genomic shocks due to hybridization causing genome rearrangement and altered gene expression than their diploid parents [57]. Compared to diploids, the proteomic data interpretation in the polyploids can be challenged by the “homeologs collapse”—wrong assembly of two homeologs into a single gene [58]—and “homeologe expression bias”—unequal expressions between homeologs caused by the duplicated gene actions [59]. For the first time, identification of “homeologe expression bias” in the protein data between the A and B subgenomes in Arachis hypogaea provides insights into the biological processes in the homeologous and paralogous protein expression [60].
However, recent advancement in genome sequencing, functional and subcellular proteomics improved extraction methods, and identification of stress-related protein markers highlight the growing potential of proteomics for enhancing stress resistance in polyploid crops. Though proteomics identifies the gene response for environmental stimuli, multi-omics—integration of other omics such as genomics, transcriptomics, epigenomics and metabolomics with proteomics—could provide more comprehensive understanding stress tolerance in polyploids than the single approach [61].
Figure 1.
Proteomic approaches used in allopolyploid crops: Gel-based (bottom left) and shotgun (bottom right). The gel photos were modified from Gris and Baldoni (2013) and Nadeem et al. (2016). Crops were generated from BioRender (https://app.biorender.com/); 1-DE and 2-DE = one- and two-dimensional gel electrophoresis; MALDI-TOF = matrix assisted laser desorption ionization - time of flight; MS = Mass spectrophotometry; LC = liquid chromatography.
Figure 1.
Proteomic approaches used in allopolyploid crops: Gel-based (bottom left) and shotgun (bottom right). The gel photos were modified from Gris and Baldoni (2013) and Nadeem et al. (2016). Crops were generated from BioRender (https://app.biorender.com/); 1-DE and 2-DE = one- and two-dimensional gel electrophoresis; MALDI-TOF = matrix assisted laser desorption ionization - time of flight; MS = Mass spectrophotometry; LC = liquid chromatography.
This review highlights the genomic complexity of polyploid crops—wheat, oilseed Brassica and cotton—progress of their proteomics for HS and biotic stress (disease) focusing on comparative proteomics, challenges of proteomics in identifying functional proteins and proteoforms of polyploids, and the prospects of proteomics, particularly subcellular proteomics, in polyploid breeding.
2. Genome Complexity of Polyploidy
2.1. Wheat
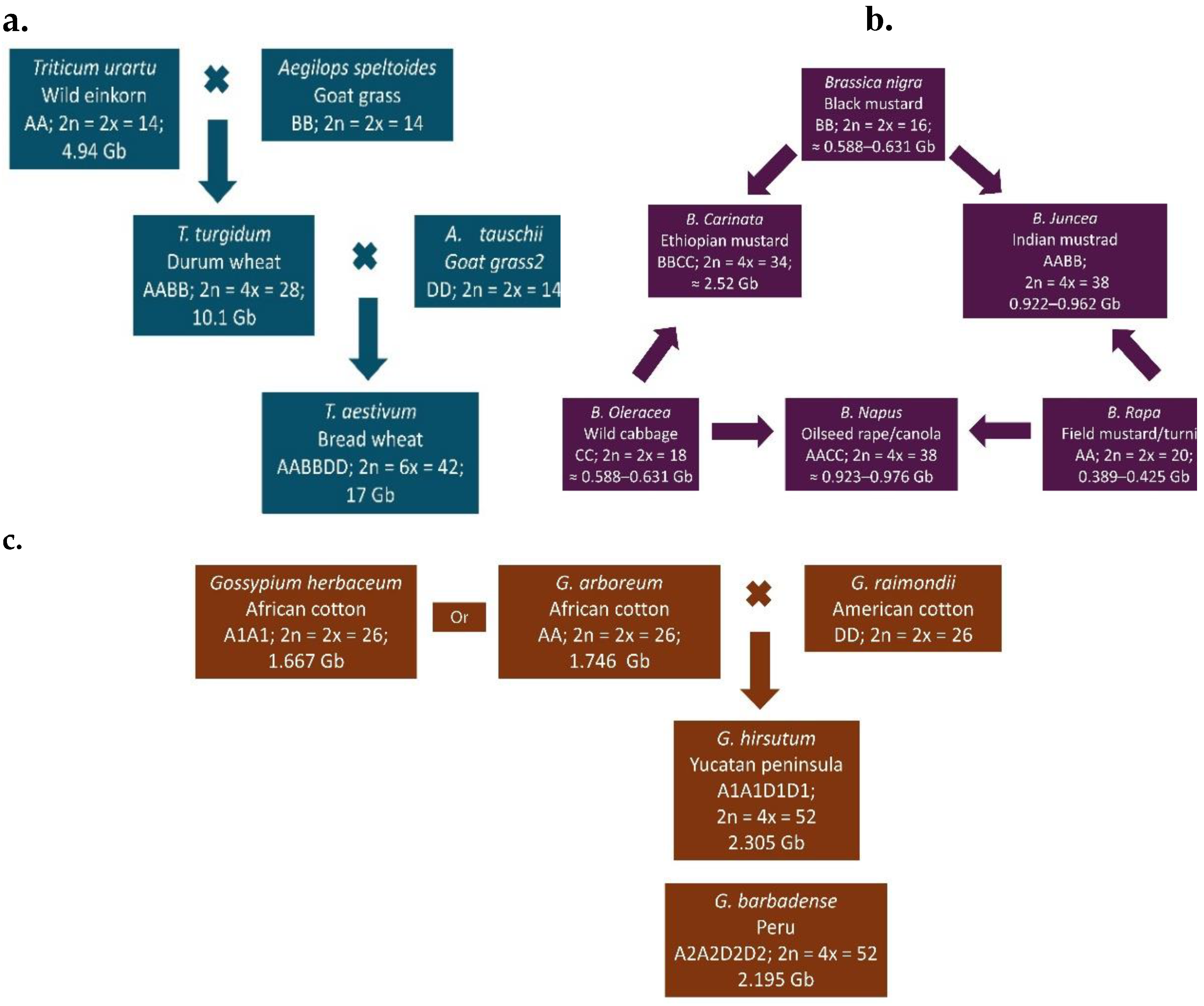
Bread wheat (T. aestivum; AABBDD; 2n = 6x = 42), a hexaploidy species in the Poaceae family, is one of the world’s most important staple cereal crops. It evolved naturally through two successive hybridization events. First, a diploid wheat (T. urartu; AA; 2n = 2× = 14) crossed with a wild goat grass (Aegilops speltoides; BB; 2n = 2× = 14), producing a tetraploid wild emmer wheat (T. turgidum spp. diccoides; AABB; 2n = 4× = 28). This tetraploid then hybridized with another goat grass (A. tauschii; DD; 2n = 2× = 14), resulting in modern hexaploidy bread wheat (Figure 2a) [6,62]. The estimated genome sizes of hexaploid, tetraploid and diploid wheat are approximately 17 Gb [63], 10.1 Gb [64] and 4.94 Gb [65], respectively.
2.2. Oilseed Brassica Crops
The genus Brassica, within the Brassicaceae family, includes economically important species cultivated for oilseeds, vegetables and condiments [66]. The Brassica genus has three diploid species—B. nigra (BB; 2n = 16), B. oleracea (CC; 2n = 18) and B. rapa (AA; 2n = 20). They evolved, by intercrossing into three amphidiploid species, B. carinata (BBCC; 2n = 34), B. juncea (AABB; 2n = 36) and B. napus (AACC; 2n = 38) (Figure 2b) [7]. Among the three diploids, B. rapa is the oldest and most widely distributed [67], with a genome size ranging from ~389.2–424.59 Mb [68,69]. The genome sizes of B. oleracea and B. nigra are estimated at ~587.7–630.7 Mb [70,71] and ~446.5–534.2 Mb [72,73], respectively. The genome sizes of the amhidiploids are approximately 922–961.72 Mb for B. juncea [74,75], 921.5–975.8 Mb for B. acarinata [76].
Figure 2.
Evolution and genome structure of three major allopolyploid crops: (a) wheat (Triticum aestivum L.), (b) oilseed Brassica crops, and (c) cotton (Gossypium hirsutum and G. barbadense); adapted from [77]).
Figure 2.
Evolution and genome structure of three major allopolyploid crops: (a) wheat (Triticum aestivum L.), (b) oilseed Brassica crops, and (c) cotton (Gossypium hirsutum and G. barbadense); adapted from [77]).
2.3. Cotton
Cotton belonging to the genus Gossypium and Malvaceae family has been cultivated for more than 7000 years [78] for fibres, medicine and animal feed [79]. Although there are around 50 species, only four are widely cultivated: two American species—G. hirsutum (A1A1D1D1; 2n = 4x = 52) and G. barbadense (A2A2D2D2 ;2n = 4x = 52)—and two Asian and African species—G. arboreum (AA; 2n = 2x = 26) and G. herbaceum (A1A1; 2n = 2x = 26) (Figure 2c) [77,79,80,81]. Forty-three Gossypium species are diploid (n = 13), classified into genome groups A–G and K while seven are tetraploid (AADD) [82,83]. The A-genome originated in Africa and the D-genome in Mexico [82]. Allotetraploid or amphidiploid cottons species—G. hirsutum (2,305.24 Mbp), G. barbadense (2,195.8 Mbp), G. tomentosum (2,193.56 Mbp), G. mustelinum (2,315.09 Mbp), G. darwinii (2,182.96 Mbp) [84], G. ekmanianum (2,341.87 Mbp), and G. stephensii (2,291.84 Mbp) [85]—are the result of polyploidization between G. arboreum (A genome; 1,746 Mb; [86] or G. herbaceum (1,667 Mb;[87]) and G. raimondii (D genome; 885 Mb; [88,89,90] 2015). Among these cotton species, G. hirsutum is widely cultivated species globally [91].
3. Proteomic Studies on Heat Stress Tolerance in Polyploid Crops
Heat stress significantly impairs crop growth, development, yield, grain, seed or fibre quality. Although HS can adversely affect plants’ all developmental stages, the anthesis and post-anthesis phases are particularly sensitive in crops like wheat, oilseed brassica, and cotton. Protein—functional gene expression products—play key roles in a plant’s HS response by regulating various metabolic pathways. For example, HS increases biomolecule kinetics resulting protein degradation or misfolding risk; protective proteins or proteoforms accumulate to reduce the harmful effect of HS on the cells [38]. Different HS-induced proteins, particularly heat shock proteins (HSPs), and proteoforms are strongly associated with HS tolerance across different developmental stages. This section discusses how HS affects crops, and the molecular mechanisms of the HS response revealed by proteomics approaches.
3.1. Wheat
In wheat, HS adversely impacts every stage, from seedling emergence to maturity, ultimately reducing grain yield and quality [92]. Terminal HS—occurring during the reproductive stage—is detrimental to yield [93]. Heat stress during tillering disrupts pollen viability, spike formation, seed set, and embryo development [94,95]. Post anthesis HS inhibits the translocation of photosynthates and starch accumulation into grains and reduces stem carbohydrate [95]. It also leads to increased reactive oxygen species (ROS), osmolyte accumulation, decreased RuBisCo activity, and degradation of seed proteins[96]. However, the accumulation of HSPs, antioxidant enzymes (e.g., catalase (CAT), peroxidase (H2O2), superoxide dismutase (SOD) and glutathione (GSH)) and proline, along with the suppression of anabolic and catabolic process, are critical for HS tolerance [43,95]. Numerous proteomic studies in recent years have examined HS responses at different growth stages in wheat (Table 1). Our previous review summarized recent progress in wheat proteomics under HS [43].
Heat stress causes wheat seedling leaves to roll and turn yellow [113]. During early stress stages, H2O2 levels rise while peroxidase drops; at later stages, antioxidant enzymes like ascorbate peroxidase (APX), glutathione reductase (GR) and SOD increase. Cytoplasmic proteins such as 2-cysteine peroxiredoxin (Ta2CP) contribute to HS tolerance and chlorophyl metabolism. Arabidopsis with overexpressed Ta2CP had normal green leaves with high chlorophyll a and b content, low H2O2 and ROS leading to thermotolerance. A 2-DE and MALDI TOF/TOF proteomics identified that upregulation of photosynthesis associated proteins, including RuBisCO activase A and PEP carboxylase, and signal transduction pathway in wheat seedling contributes to HS tolerance by increasing CO2 assimilation [114]. A small HSP gene, TaHSP23.9 (a member of the HSP20/ACD family) significantly upregulated in wheat leaves and developing grain under HS at both the transcription (mRNA) and translation levels, making it a promising marker for HS tolerance [102]. Phosphorylation, a major PTMs, also play important roles in HS tolerance. Due to the amino acid substitution in TaSG-D1 allele, TaSG-D1E286K gene undergoes phosphorylation and stabilizes the expression of TraesCS5B02G380200 (TaPIF4), resulting improved HS tolerance in seedling and mature stages of T. sphaerococcum [115].
Heat stress disrupts photosynthesis and pollination leading to male sterility and reduced grain yield and quality in wheat. Sowing time plays a crucial role in mitigating HS effects. For example, delayed sowing during 2018–2019 resulted in 11.16% and 10.10% increases in grain weight and yield in wheat, respectively, due to higher photosynthetic rates in flag leaves [98]. This improvement was linked to increased energy flow into the electron transport system and upregulation of differentially abundant proteins (DAPs) involved in photosynthetic electron transport (e.g., PsbH and PsbR), the Calvin cycle, and chlorophyll biosynthesis. Conversely, terminal HS under standard sowing dates significantly reduces photosynthesis and starch accumulation, diminishing grain yield and quality, respectively (Kumar, Dubey et al. 2021). Combined 2D-PAGE and transcriptomic analyses revealed that mitogen-activated protein kinase (MAPK) contributes to terminal HS tolerance by promoting proline, starch, and H2O2 accumulation, enhancing granule integrity, positively regulating stress-related gene expression and amylolytic activities while suppressing photosynthesis-related genes. Moreover, MAPK improves oxidative stress tolerance through its positive correlations with SOD and HSP17. MAPK signaling also involved in HS-induced male sterility in wheat [30]. Downregulation of MAPK related genes (e.g., TraesCS3A02G149800.1) under HS negatively affects ROS scavenging and pollen development. Further, DAPs related with phenylpropanoid biosynthesis, and starch and sucrose metabolism cause anther indehiscence and starch deficiency in it. Excess ROS accumulation in anthers inhibits starch and sucrose synthesis through phenylpropanoid biosynthesis. Upregulation of the HS-pathway thermosensitive male sterile 1 gene, TraesCS4B02G193500.2, causes male sterility under HS.
Heat stress also affects spike differentiation in wheat [51]. A comparative transcriptomic and proteomic study across three spike differentiation stages (pistil and primordium differentiation, anther separation and tetrad formation) identified 210 HS-associated transcripts/proteins, including key genes in abscisic acid (ABA) and proline biosynthesis pathways. Downregulation of 9-cis-epoxy carotenoid dioxygenases and upregulation of zeaxanthin epoxidase and ascorbate oxidation modulate ABA biosynthesis. Similarly, ornithine aminotransferase and pyrroline-5-carboxylate reductase enhance proline accumulation via ornithine and glutamate pathways. Phosphorylation, a major PTM, regulates reproductive organ development and photosynthetic apparatus in leaves in wheat under HS [116]. A DDA LC-MS/MS identified 14 phosphoproteins under HS, which activated phosphatases and kinases and generated their phosphoforms. A protein kinase, TraesCS6B01G377500.3, was associated with upregulated S711 and S762 phosphosites. Downregulated splicing variant of S12, TraesCS2D01G281200.1, was found in wheat spike under HS. Serine or threonine dominated phosphosites—S3236 and T3238—maintained protein functions by preserving phosphorylation pool under HS. However, identification of proteins and proteoforms is recommended to understand the co-regulation of these phosphosites to combat HS.
While recent proteomic studies have focused on heat response during various growth stages, earlier work concentrated on HS effects on grain quality. For example, proteomic study using 2-DE and MALDI-TOF showed that HS reduced grain weight by decreasing starch synthesis proteins such as, glucose-1-phosphate adenyl transferase, although catalase protected cells from H2O2 [117]. Heat stress also weakens dough properties due to reductions in glutenin and gliadins, and increase in HSPs, although sHSPs improve thermotolerance during grain filling stage [118].
Combined day–night temperatures HS conditions are more detrimental than high daytime temperatures alone with 9% more grain weight lost under these conditions due to increased proline and H2O2 levels in flag leaves [99]. Using 2D-PAGE and MALDI-TOF, 153 and 95 DAPs were identified in wheat grain under day and day-night HS conditions, respectively. Proteins associated with starch biosynthesis, cell wall synthesis, defense, and storage were altered—particularly downregulation of starch and cell wall-related proteins—resulting in poor grain development and premature senescence in both conditions. Day-night temperatures reduced thermotolerance, grain yield and quality by restricting the expression of transcription factors (TFs), genes associated with signaling pathway and abiotic stress, and function of protein folding machinery
Heat stress also affects flour quality and by altering protein abundance in wheat grain [104]. Using an iTRAQ, 116 and 91 up- and down-regulated DPEs were identified in Gaocheng 8901, respectively. A decreased starch accumulation and increased gluten, gliadin, globulin and albumin content resulted in a reduced thousand grain yield in wheat. Downregulation of HSP90, low-molecular-weight glutenin subunits (LMW-GSs) and starch branching enzyme IIb reduced both grain yield and quality. Five DAPs—elicitor responsive gene 3, brassinosteroid-insensitive 1, histone cell cycle regulator, chaperone protein and splicing arginine serine-rich 7—were also linked to protein–protein interactions affecting grain yield and quality.
Finally, HS reduces dough quality by modulating the abundance of storage proteins. Approximately 57.5% of high-molecular-weight glutenin subunits, LMW-GSs, α- and γ-gliadins, and avenin-like proteins were upregulated, while 79.3% were downregulated under HS [97].
Moreover, peroxidase, RuBisCo activase, sHSPs and photosynthetic electron transport are crucial for thermotolerance during seedling stage, while proteins related to antioxidants, MAPK, proline and H2O2 accumulation, and phosphofroms are vital for during grain filling stage. Globulin, gliadin and albumin are essential in determining grain quality under HS.
3.2. Oilseed Brassica Crops
Heat stress affects all the growth stages of the oilseed Brassica, but flowering stage is the most sensitive [119]. It reduces gametophyte fertility, pollen viability, germination and causes seedless pod resulting in a decreased thousand grain weight, reduced yield and poor oil quality [119,120]. Among the cultivated species, B. napus is more sensitive to HS than B. juncea and B. campestris [120]. Proteins, such as HSPs (e.g., HSP101, HSP70 and HSP17.6) [119], gucosinolates, including glucobrassican, glucocarphanin and glucoraphasatin, gluconapin, sinigrin, and progoitrin [121] and glyoxalase I [122] contribute to thermotolerance. Phytohormone signaling pathways also play critical role in HS-tolerance.
Although proteomic study on oilseed Brassica started earlier [123,124], proteomic study in HS on Brassica started only a decade ago. The first proteomic study on HS in B. napus found ascorbate peroxidase as a key protein for thermotolerance in seedlings using 2-DE [125]. Proteins related to energy and metabolism reduced growth under HS through consuming major nutritive reserves. Additionally, HS severely impaired seed germination [122]. However, elevated levels of glyoxalase-I enhanced seed thermotolerance by detoxifying methylglyoxal, maintaining redox balance, and increasing SOD activity.
Beyond canola, proteomic studies have also been conducted on other oilseed Brassica species. Over the last five years, advancement of proteomic techniques has accelerated progress in HS responses in oilseed Brassica (Table 1). Role of protease accumulation in dead pericarps in B. juncea was identified by label-free proteomics [126]. Dead organs such as seed coat and pericarp affect yield [127]. A combined proteomic and metabolomic study on B. juncea under HS found the high abundance of several proteins, including HSP70-5, HSP104, and multiple sHSPs, with proteases being the most abundant in the pericarp [126]. Heat primarily induced the accumulation of cysteine proteases, 25 HS-induced proteins and sugars on its dead pericarp.
Heat stress also disrupts biochemical and metabolic pathways in B. juncea [110]. Increased antioxidants and flavonoid, with minimal impacts on photosynthetic performance, contribute to thermotolerance in mustard seedling. Phenylalanine ammonia lyase and respiratory burst oxidase homolog protein-A improves thermotolerance by synthesizing metabolites (e.g., flavonoids and lignins) and producing ROS, respectively. A combined proteomics and peptidomics—a comprehensive analysis of all peptides of an organism [128])—in oilseed Brassica under HS for 4 and 8 h found 25 and 24 upregulated, and 41 and 23 downregulated DAPs and 532 and 570 peptide sequences, respectively. Key thermotolerance associated proteins included HSPs (21.7, 14.7 and 17.5 kDa HSP groups), sHSPs and TFs, including HS transcription factor A-4a, Myb family Atlg14600, and WRKY—related to signal transductions, regulating transcriptions, repairing DNA and processing RNA.
In B. campestris seedling, thermotolerance under both heat and cold stress involves several molecular pathways, including redox homeostasis, photosynthesis, chaperons, HSPs, carbohydrate metabolism, and signal transduction [112]. Under both stresses, 1022 DAPs (172 upregulated and 324 downregulated) and 1784 DAPs under HS were found using a combined TMT and LC-MS. The critical role of redox homeostasis in thermotolerance was validated through the reduced glutathione-to-oxidized glutathione ratio and decreased oxidative damage in transgenic Arabidopsis overexpressing GLU1, a gene encoding a redox-associated protein.
3.3. Cotton
Heat stress critically affects all the developmental stages of cotton, with the reproductive stage the most sensitive [129]. It impairs seed germination, shoot and root growth, and induces male sterility, ultimately reducing yield and fiber quality. Increased leaf temperature due to HS disrupts photosynthesis by inhibiting RuBisCO activation [130]. In G. barbadense, HS restricts photosynthesis by limiting the regeneration capacity of ribulose-1,5-bisphosphate and impeding electron transport [131]. Traits such as membrane thermostability, high proline and soluble sugar can improve thermotolerance in cotton [132]. Additionally, regulation of microRNAs, signaling pathways, including calcium, ROS, carbohydrate, transcription factors, phytohormones and gene regulation pathway, and HSPs play critical role in its thermotolerance [129]. Despite numerous studies on the physiological impacts of HS on cotton, proteomic studies remain limited. However, recent proteomic studies have provided valuable insights on thermotolerance in cotton (Table 1).
In the leaves of cotton seedlings, expression of the mitochondrial protein GhHSP24.7 increased HS-sensitivity due to increased ROS, mitochondrial acetylation activity and decreased ATP levels [107]. However, suppressed GhHSP24.7 enhanced thermotolerance by decreasing stomatal conductance and H2O2, activating GhHDA14, maintaining chloroplast structure and improving ROS scavenging.
The reproductive stage is particularly vulnerable, as HS disrupts pollen development at tetrad, uninucleate and binucleate stages, pollen maturation, pollination and embryo formation [133]. Pollen at the tetrad stage is most susceptible to damage at 38 ℃, but recovery is possible through the upregulation protective proteins such as HSP (15.7 kDa) and peptidylprolyl isomerase [53]. However, under 40 ℃ at tetrad stage, mature pollen grain size and pollen tube reduced by 36% and 120 µm, respectively, resulting two-thirds of yield reduction [107]. Under these HS conditions, HSP70, and HSP70-90 organizing protein 3-like upregulated, while late embryogenesis abundant protein (LEA) 2 downregulated. HSP70, heat shock cognate 70 kDa protein 2-like, luminal-binding protein 5-like and luminal-binding protein 5-like protect pollen from HS through protein export, folding and processing pathways.
A recent proteomic study identified over 8000 HS-induced DAPs at the flowering stage [105]. Increased abundance of β-glucosidase, different photosynthetic proteins, NDH subunit of subcomplex B1, NADH ubiquinone oxidoreductase (Complex I), stress responsive proteins, 1-aminocyclopropane-1-carboxylate oxidase and HSPs (e.g., Hsp70, Hsp90 and Hsp80) are crucial for thermotolerance. These proteins contribute to HS-tolerance by maintaining membrane and cell wall integrity, producing energy, maintaining osmotic balance, enhancing proline accumulation and ROS scavenging, and regulating electron transport in photosynthesis channels, carbohydrate metabolism and protein folding, and preventing protein aggregation.
Heat stress also reduces insecticidal proteins in Bt cotton’s bolls [108]. Under a combined heat (38 ℃) and drought (40% field capacity) stress, the insecticidal proteins decreased by 38.3 and 30.7 ng/g fresh weight of boll shell in 2026 and 2017, respectively. That reduction may be the result of the downregulation of signal recognition particles (SRPs) and SRP receptors, restriction of peptide chain translocation into the endoplasmic reticulum and increase of ubiquitin-mediated proteolysis.
Overall, proteins associated with photosynthesis, carbohydrate metabolism, stress response, ROS regulation, protein folding and aggregation and electron transport play pivotal roles in cotton growth and HS tolerance.
4. Proteomic studies for biotic stress
Biotic stress—caused by living organisms such as bacteria, fungi, viruses, and insects—significantly threatens on global food security [134]. Emerging pathogenic strains further intensify this challenge, continuously threatening crop productivity worldwide. Plant resistance to pathogens is a complex trait governed by a multifaceted interplay of morphological, genetic, biochemical, and molecular processes, with proteomic insights playing pivotal role [135,136,137]. Therefore, a comprehensive understanding of these mechanisms for essential to addressing biotic stress in complex plant genomes. Proteins are crucial in interpreting multiple signals derived from biotic stress and activating stress-associated defense responses [138]. Proteins and PTMs contribute directly to plant immunity, while subcellular protein localization provides key insights into how plants respond to biotic stress. Therefore, proteomics—a powerful and evolving analytical tool—offers substantial potential for developing disease resistance, particularly in polyploid crops [139,140].
4.1. Wheat
Wheat production is challenged by different biotic stresses, including fungi, virus, nematodes and aphids [141]. Among these, fungal pathogens such as rusts, mildew, and blights are the most destructive. Numerous proteomic studies have explored the mechanisms underlying wheat–pathogen interactions to understand both compatible and incompatible responses [142,143,144,145,146]. For example, a comprehensive proteomic revealed key virulence and host defense proteins—including PR1, PR4, and GST—associated with early resistance mechanisms against wheat rust [146]. In the wheat apoplast, increased level of protein-modifying enzymes, particularly serine proteases, along with other stress associated proteins mitigate infection-induced damage by Magnaporthe oryzae [147].
Another proteomic study found the upregulation of 4100 and 447 DAPs in wheat infected by Tilletia controversa and T. foetida, respectively [148]. Antioxidant-related proteins were upregulated in both cases, while those involved in plant–pathogen interactions and glutathione metabolism were downregulated. However, thaumatin like proteins and HSPs contributed to increase the resistance against these pathogens.
Separately, 366 DAPs for wheat infected by Fusarium pseudograminearum (causal agent of Fusarium crown rot) were identified using TMT tool [149]. Resistant cultivars exhibited significant enrichment of proteins linked to different cellular activities—glucoside, electron transport, cellulose synthase and oxidoreductase—metabolic processes, plant-pathogen interactions, and antioxidant activities [149,150]. In the resistant variety Xinong 538, unique proteins encoded by Chitinase IV, Thaumatin-like 1, PR1.1, and PR1.2 genes were strongly associated with resistance to Fusarium head blight (FHB) [150]. Further, an FHB resistant network—Ca2+, phytohormone- and phenylalanine-signaling pathways for stomatal closure, disease resistance and thickening cell wall, respectively—was proposed. Although wheat cultivars vary in FHB susceptibility, they share a core set of defense-related proteins [151]. During FHB infection, pectin-derived oligogalacturonides (produced via homogalacturonan degradation) act as damage-associated molecular patterns and are recognized by wall-associated kinase 1 in Arabidopsis, a plant receptor kinase functioning as a pattern recognition receptor. Similarly, in wheat, the Stb6 gene encodes a wall-associated kinase -like protein that recognizes AvrStb6, the fungal avirulence effector, thereby conferring resistance to Septoria tritici blotch [142]. Under biotic stress, proteomic investigations have identified key proteins, including type II metacaspase and wheat cold shock domain protein family 1, in regulating carbohydrate metabolism and photosynthesis [152]. In the resilient wheat cultivar “Xingmin318,” 741 DAPs were identified in response to Puccinia striiformis and Blumeria graminis infections, indicating distinct defense strategies against these pathogens [153].
A comparative proteomic study in a resistant wheat line “N9134” identified 394 DAPs in response to Blumeria graminis f. sp. tritici infection [154]. Among these, few DAPs were primarily associated with pathogenesis-related processes, oxidative stress responses, and primary metabolism. Key metabolic pathways for plant's defense response includs phenylpropanoid biosynthesis, phenylalanine metabolism, and photosynthesis-antenna proteins. The identification of protein motifs rich in leucine repeats and histidine sites, along with eight predicted PPI networks, provides insights into potential molecular mechanisms underlying resistance to powdery mildew.
Although proteomics has significantly advanced our understanding of plant–pathogen interactions at the molecular level, the narrow genetic diversity of modern wheat varieties remains a major challenge. Continuous research and breeding programs are essential to enhance wheat’s resistance to evolving pathogens [141,155].
In summary, proteins involved with antioxidant defense, glutathione metabolism, carbohydrate metabolism, photosynthesis, calcium-, phytohormone- and phenylalanine-mediated signaling pathways play vital roles in wheat’s resistance to biotic stress.
4.2. Oilseed Brassica Crops
Proteomic studies have revealed that biotic stress activates detoxification, redox homeostasis, and defense-related pathways in oilseed Brassica. Black rot, caused by Xanthomonas campestris pv. campestris (Xcc) severely affects cruciferous vegetables, including Brassica napus [156]. Label-free proteomic analysis identified 158 and 163 Xcc-responsive DAPs in the susceptible cultivar Mosa and the resistant cultivar Capitol, respectively, which were categorized into five major functional groups: antioxidative systems, proteolysis, photosynthesis, redox, and innate immunity. Mosa showed increased protein degradation and oxidative stress markers, while Capitol exhibited high redox potential and an enhanced innate immune response. Interestingly, reduced photosynthesis due to the downregulation of photosystem II-related proteins improved disease resistance.
Clubroot, caused by Plasmodiophora brassicae, has threatened Brassica productivity for over a century [157]. Researchers are employing advanced tools, including proteomics, to develop clubroot-resistant cultivars. Using LC with tandem MS, 73 putative proteins were identified on the quantitative trait loci in chromosomes A3 and A8 in the clubroot resistant B. napus cultivar [158]. A total of 937 proteins had significant changes in abundance, which were associated with pathways related to calcium signalling, reactive oxygen species, dehydrins, lignin, and phytohormones. Additionally, 73 orthologous proteins linked to clubroot reistant loci were identified, highlighting potential genetic targets for developing clubroot-resistant cultivars. Among them, BnaA02T0335400WE, BnaA03T0374600WE, BnaA03T0262200WE, and BnaA03T0464700WE associated with calcium signaling—play crucial role in defense against Plasmodiophora brassicae. In B. oleracea, proteins associated with ABA and glucose signaling, and fructose-bisphosphate aldolase are critical for clubroot defence [159]. Most of the abundant proteins were localized to outside the nucleus, particularly in the chloroplast, thylakoid, stroma, and membranes. Additionally, restriction of energy metabolism enhances resistance.
Stem rot, caused by Sclerotinia sclerotiorum is another major disease of Brassica [160]. The first proteomic study on S. sclerotiorum infection in B. napus, identified upregulated antioxidant enzymes peroxidase and SOD as contributors to the disease resistance [161]. Additional proteins related to photosynthesis, metabolism, hormone signaling and protein folding were also important for defense mechanisms. Several proteomic studies have expanded the understanding of stem rot resistance mechanisms in B. napus [160,162,163,164]. A TMT-based analysis identified 221 unique proteins and 173 DAPs in response to stem rot infection, including proteins involved in ROS homeostasis, lipid signaling, DNA methylation, histone modification, defense responses, and cyanate lyase activity [164].
In B. juncea, 20–22 DAPs were identified for stem rot using SDS-PAGE [165]. Further, nano LC-MS revealed peptidyl-prolyl cis-trans isomerase as the key protein for stem rot defense, interacting with other proteins related with protein importation into nucleus and RNA exportation, including GTP binding nuclear proteins, 60S ribosomal protein, and protein kinase. Investigation into alternative splicing in S. sclerotiorum-infected B. napus across five infection stages identified a total of 130 genes with 98 differentially expressed genes for defense mechanism through alternate splicing [166]. Most dominant alternative splicing sites were on intron or alternative 3′ splice sites. However, further proteomic investigations focusing on PTMs are needed to deepen our understanding of host–pathogen interactions.
Overall, proteins associated with photosynthesis, ROS homeostasis, lignin synthesis, lipid- and Ca2+- signaling, DNA methylation and histone modification, contribute to the biotic stress resistance in oilseed Brassica.
4.3. Cotton
Phytopathogens—fungi, bacteria and viruses, cause significant yield loss in cotton [167]. Among the fungi, Verticilium dahlia, Fusarium oxysporum, Thielaviopsis basicola [168] and Rhizoctonia solani [169] are major causal agents for cotton diseases. Bacterial blight by Xanthomonas campestris also cause significant yield loss in cotton [170]. Cotton proteomics has advanced significantly over the past decade, improving the understanding of pathogenicity and plant defense responses at the molecular level [54].
Numerous proteomic studies have focused on unravelling the molecular mechanism of Verticillium wilt (VW), a devastating soil borne disease caused by V. dahlia that severely affects cotton yield and quality [171]. The first root proteomic study on VW in cotton (G. barbadense) identified 51 upregulated and 17 downregulated proteins using 2-DE [172]. This study highlighted the roles of the ethylene signaling pathway, pentose phosphate pathway, and Bet v 1 family proteins in defense. Further investigation found that one of the proteins of Bet v 1 family proteins, PR10 (GbPR10.5D1), contibuted to resistance by modulating lipid metabolism and defense signaling pathways [173]. Moreover, the ethylene response factor, GbERF1-like enhanced VW resistance by promoting lignin synthesis [174]. More recently, a DIA-based proteomic comparison between two contrasting G. barbadense cultivars—XH21 (resistant) and XH7 (susceptible)—identified 885 DAPs associated with disease resistance [175]. Further, weighted gene co-expression network analysis revealed oxidoreductase and peroxidase activities significantly enriched pathways for VW resistance. Silencing ascorbate peroxidase proteins reduced VW resistance, underscoring the importance of APX-mediated redox metabolism in plant defense.
The nuclear protein, GthGAPC2, was identified in Gossypium thurberi via proteomic analysis as the key to VW resistance [176]. Silencing GthGAPC2 increased susceptibility by reducing lignin content and disrupting redox balance, while its overexpression in tobacco and Arabidopsis confirmed its role in disease resistance, highlighting its potential as a promising candidate for breeding programs. Mi et al. (2024) [177] demonstrated that the GhMPK9-GhRAF39_1-GhWRKY40a signaling pathway enhanced cotton disease resistance by regulating the GhERF1b- and GhABF2-mediated pathways. GhMAC3e, a homologous gene of a key component of MOS4-associated complex, was implicated in growth and defense responses against VW [178]. A phosphoproteomic study identified 359 and 287 differential phosphoproteins in one and three days post-VW inoculation in resistant line, respectively [179]. Analysis of PTMs revealed 37 enriched serine (Ser) and four threonine (Thr) motifs, of which 19 Ser and two Thr were novel. Moreover, the upregulation of burst oxidase homolog D (GhRbohD) increased phosphorylation after VW infection, increasing resistance by decreasing ROS, H2O2, nitric oxide and calcium levels while promoting lignin and callose accumulation [180].
Fusarium wilt (FW), caused by F. oxysporum, also results in considerable yield losses in cotton [181], though limited proteomic studies have explored this disease. Using an SDS-PAGE, it was found that proteins associated with polyketides, cellular process (e.g., lipid biosynthesis, protein and nucleotide metabolism, protein folding, carbohydrate metabolism and oxidative stress response), HSP70 and protease are significantly enriched in the F. oxysporum f.sp. vasinfectum [182]. These proteins are crucial in mediating host–pathogen interactions via extracellular vesicles. In contrast MAPK cascade involving GhMPK20, GhMKK4 and GhWRKY40 negatively regulated FW resistance [183]. However, GhMPK20 plays key role in defence signal transduction pathway to improve FW resistance in cotton. Further, a phrosphoproteomics identified GhMORG1 (a MAPK scaffold protein of the GhMKK6–GhMPK4 cascade) that enhances FW resistance in cotton [184]. Overexpression of GhMORG1 in cotton protoplasts significantly increased the activity of this pathway.
In response to Rhizoctonia solani infection, iTRAQ analysis identified 174 DAPs, with proteins involved in ROS homeostasis—such as glutathione S-transferases and glutathione peroxidase—and those linked to lignin and phenylpropanoid biosynthesis, DNA methylation, and histone modification significantly upregulated [185]. Particularly, the crucial role of secondary metabolic pathways in fungal pathogen defense was confirmed by the altered expression of phenylpropanoid biosynthesis-related proteins.
Bacterial blight, caused by Xanthomonas campestris pv. Malvacearum, defense mechanisms include the downregulation of photosynthesis-related proteins, such as ribulose-biphosphate carboxylase activase, ATP synthase β subunit, RuBisCo large subunit-binding protein, and glyceraldehyde-3-phosphate dehydrogenase B [186]. Further, ROS-regulating proteins, including peroxidase, peroxiredoxins, and Prx type 2-Cys, play important roles in enhancing resistance.
Overall, proteins associated with singnaling pathways—ethylene, calcium and defense—, lipid and carbohydrate metabolism, DNA-methylation, histone modification and peroxidase activity contribute to the biotic stress resistance in cotton.
5. Challenges of Proteomics in Polyploid Crops
Proteomics have been used in allopolyploids, including wheat, oilseed Brassica and cotton, to harness the molecular insights of abiotic and biotic stress tolerance. However, these studies face several challenges, primarily due to the complex nature of polyploid genomes, difficulties in protein identification, reproducibility and the lack of comprehensive protein databases.
The genomic complexity of allopolyploid is the result of the combination of divergent genomes through interspecific hybridization. Gene loss during interspecific hybridization or further polyploidization (e.g., in wheat), continued recombination between the homoeologous chromosomes (e.g., Brassica), and chromosomal translocations between subgenomes increase the complexity of the genomic landscape [187,188]. While proteomics analysis can detect chromosomal rearrangements by comparing DAPs or proteoforms between progenitor lines, the specific parental contributions often remain unresolved [48].
For example, bread wheat has a large and complex genome (~17 Gb across three sets of chromosomes) [63], with 55–63% comprising repetitive sequences [189]. Paralogous genes in allopolyploids often share > 90% amino acid sequence identity. This high redundancy makes it difficult to identify novel proteins and protein–protein interactions associated with distinct phenotypes [48,190]. Studies have shown that 65–70% of transcriptomic data does not correspond to proteomic outcomes [210], potentially due to its amino acid composition and alternative splicing or PTMs. Protein isoforms, altered protein sequences by alternate splicing, and PTMs, biochemical modification of amino acid residues of proteins, are two major proteoforms for determining biological functions of proteins in plants [39]. Therefore, proteoforms vary in developmental stages of crops, tissue types and external stimulus [191].
Besides genetically functional proteins, identification of proteofroms is crucial due to their important role in allopolyploid evolution and stress tolerance. Homologous genes—orthologous and paralogous—and homeologous genes are a significant part of polyploid evolution. Homeologous genes or homeologs are the homologous gene pairs originated from different species but merged to an allopolyploid genome by alloplolyploidization [192], while protein isoforms are formed due to their expression [193]. For example, 18.7% of expressed genes of allotetraploid wheat (AADD) have similar type alternate splicing in two subgenomes [193], while 90 novel protein isoforms of a HSP,TaHSP90, developed from three homeologs were found in hexaploid wheat [194]. Protein isoform of TaYRG1.1 (TaYRG1.6), increases susceptible to yellow rust disease in wheat [195], while TaHSFA6e originated form alternative splicing of a HS transcription factor gene increases HS-tolerance [196].
Similarly, PTMs also play an important role in stress tolerance in polyploids. Recently, the model of controlling cellular functions by PTM proposed as: enzymes such as kinases, “writer”, catalyze the PTM addition to amino acid residues, other enzymes such as phosphatases de-ubiquitinases and -acetylases, “erasers”, play role to deletion of PTMs from target amino acid residues, while a protein domains, “readers”, play role for interacting between the PTMs and the other proteins [197]. However, this PTM crosstalk is underexplored in plant proteomes, which can be addressed by incorporating of artificial intelligence (AI). Critical PTMs will be identified using AI in the PTM analysis to edit bases using CRISPR-Cas9 for altering the target traits [198]. Under different external stimuli, a single protein can develop the same or different kinds of PTMs, interacting with different target motifs [39]. For example, different temperature stresses activated phosphatase and kinase and produced different phosphofroms of a target protein in wheat [116]. However, progress in allopolyploid proteomics for stress-related proteoform identification lags far behind compared with other plants and human proteomics due to the inefficiency of the current proteomic approaches.
Protein enrichment protocol has been recommended to identify 1 to 5% proteoforms of the total protein in wheat [37]. One hundred and eighty-four proteoforms were identified in blood samples, using a nano-particle enrichment MS proteomics [199], which technique could be explored in alloploids. Further, despite having poor reproducibility, and limited ability to detect low-abundance and low-molecular-weight proteins precisely by 2-DE [207], it can identify proteoforms coupled with other proteomic approaches, such as LC-MS/MS [200]. 2-DE can identify the isoelectric point and relative mass of proteoforms combining with other approaches, such as Orbitrap MS, iTRAQ, TMT, and stable isotope labelling of amino acid in cell-culture (SILAC) [200,201], limitedly used in plant proteomics. Advanced proteomic technique, DIA, can quantify proteins and PTMs precisely, but it requires sophisticated software to overcome the data complexity challenges [202]. Recently DIA has been advanced due to its incorporation into Orbitrap (a high-resolution mass analyzer) [202]—mostly used for DDA—, trapped ion mobility spectrometry [203] and high-field asymmetric waveform ion mobility spectrometry [204], but limitedly applied in plant proteomics. With further optimization, this method can be adapted for improved protein quantification from the complex polyploid tissues.
In addition, sample preparation methods and different proteomic approaches influence true proteoform identification. Several recent review articles discussed the technical challenges of using top-down and bottom-up approaches for proteoform identifications [205,206,207,208,209]. Though top-down proteomics with MS identifies intact proteoforms [205], it has several major challenges, including proteoform mass limit of 30 kDa [210], and requirements of compulsory reproducible samples and high level of sample fractions [211], resulting in difficulties in finding exact localization [212]. However, improved liquid/gas-phase separation of MS instruments and bioinformatic tools [212]—e.g., TDPortal, Proteoform Suite and MetaMorpheus—, false-discovery rate determination, proteoform annotations and databases, and proteome validation [211] are required to overcome the challenges. “Super charging”—forcefully bringing a single charge state of all the molecules of a proteoform within the mass range of the MS instrument—and modified 2DE-PAGE improve the sensitivity of the top-down approach for higher proteoform coverage [206]. In contrast, though bottom-up proteomics is a sensitive, faster and easier approach than top-down proteomics to identify high and low abundant proteoforms [200,208], it has limitations in identifying complete proteform information and distinguishing between multiple proteoforms originating from a single protein due to protein digestion and a lack of proteoform database [211,213]. SWATH-MS, and a combination of bottom-up and 2DE approaches can be useful for high-resolution proteome identification in bottom-up proteomics [208].
Despite significant advancement in proteomics techniques, data reproducibility is still a key challenge for proteomics. Though addressing the challenges in human proteomics has been initiated, it remains under-represented in plant proteomics. In human proteomics, reproducibility and data accuracy in DIA-MS has been improved by short-time data acquisition from different instruments in triplicates [214]. Furthermore, a pipeline (ProNorM), developed using computational modules, mitigates instrumental variations, resulting in high reproducibility and statistical analysis potential. On the other hand, DDA’s reproducibility can be improved by dual-search approach—a spectral library is built using the searched sequence data, and further same data is searched in the library [215]. This method also minimizes the requirement of large database size, but two different sizes libraries result significant differences in protein and peptide identification. Improved software tools are required for the practical use of the proposed approach. Though the progress of top-down and bottom-up proteomics for proteform identification techniques is mostly explored in animal proteomics, they can be explored in plant proteomics, including polyploids, with an improved protein extraction protocol, complete protein and proteoform databases and bioinformatic tools.
Protein extraction protocol from allopolyploid tissues is still being optimized. The complex cell wall composition and presence of secondary metabolites can hinder complete protein extraction [43,216]. For example, wheat tissues have polysaccharides, lipids, polyphenols, and other secondary metabolites [217], while cotton fibers contain polyphenols and lipids in the cell wall [54]. An improved TCA-B (trichloroacetic acid/acetone-borax/PVPP/phenol) protocol of protein extraction from polysachharide and polyphenol rich tissues for further 2-DE and LC-MS/MS has been proposed [218]. Combination of two different protein extraction methods with an additional phenol extraction yielded high- quality protein from cotton leaves. A combination of multiple protein extraction methods also facilitates getting more protein coverage. Using a combination of two protein extraction buffers, isoelectric focusing and phenol, pairing pressure cycling technology for further label-free quantification yielded 66 higher proteins than the commonly used phenol extraction method in cotton fibre [219]. Protein extraction buffer and digestion enzymes can be modified according to the crops and tissues used for proteomics. Testing seven different protein extraction protocols in polyploid grains, including wheat and barley, Tris-HCL or urea-based extraction buffer coupled with trypsin-based membrane digestion with filter-aided sample preparation (FASP) found effective for a higher number of protein and peptide identifications than the conventional method [220]. However, trypsin digestion is limited to a shortage of lysine and arginine cleavage sites, often restricting precise proteoform identification, while multiple enzymatic digestions—trypsin and glutamyl endoproteinase—improve grain and metabolic proteins identification in wheat [221]. High quality single seed storage protein extraction from oilseed Brassica—B. rapa, B. nigra, B. juncea and B. fruticulosa—for 1D-SDS-PAGE based proteomics was improved by using a ball grinder with stainless steel beads in 2.0 mL Safe-Lock tubes with rounded bottoms and hinged lock lids with methanol for 1.2–5.5 mg of single seed grinding [222]. Tris-Glycine and tris-tricine based extraction buffers were found efficient for high (25–120 kDa) and low (2–25 kDa) molecular weight protein separation. Thus, optimized protein extraction protocols are crucial for successful proteomic studies in allopolyploids [43,54,222], while combination of different protein extraction strategies could be more efficient than a single method.
Another associated challenge is less precise and incomplete protein annotations. Like other diploids, annotation databases of allopolyploids are often incomplete. Although several genomic databases are available for allopolyploid crops—such as Wheat@URGI [223], Bolbase [224], BRAD [225], and CottonGen [226]—they often fall short of providing a comprehensive match between transcriptomic and proteomic data [44,48]. Inadequate annotation further complicates the identification of protein localization, PTMs, and protein variants in allopolyploids like wheat [216]. Multiomics databases—Wheatomics, BnIR and COTTONOMICS for wheat [227], B. napus [228] and cotton [229], respectively—offer better annotation. However, individual high-quality protein databases remain essential. For example, the wheat proteome database remains incomplete [43,230], limiting our ability to investigate complex, polygenic traits like abiotic stress tolerance [55], organelle-specific proteomics [chloroplast [231], mitochondria [232] and transmembrane [233] ], and single-cell proteomics [234]].
Difficulties in pathogenic protein identification, and lack of optimized in-vitro mimicking media as well as in-vivo models [235] often limit proteomics to understand the molecular mechanism of biotic stress tolerance of crops. Pathogenic protein extraction complexity from the host cell [236], MS-based instrument’s limitations, including ionization suppression [237] and lack of complete genome sequence of the pathogens [238] are the major challenges of pathogenic protein identification. Consequently, protein abundance comparison becomes difficult due to the disproportionate host and pathogenic proteins [238], leading to challenges in proteoforms and PTMs identification. The challenges could be intensified if new pathogenic strains evolved. Use of advanced proteomic approaches, such as DIA, reducing heterogeneity among the infected cells through improving pathogen inoculation methods, using improved mimicking media, and improving pathogenic annotation databases, can improve the understanding of host-pathogen interactions. For example, a recently optimized in vitro-mimicking media improved the protein dynamics in Botrytis cinerea [239], a major pathogen of many crops, including cotton [240]. Protein-protein interactions (PPI) also play a crucial role in host-pathogen interactions through controlling cellular mechanisms. Recently developed TurboID-based proximity labeling method overcomes the limitations of low efficiency of PPI identification, using the conventional method [241]. Recently, a TurboID-based proximity protocol labeling has been proposed for a polyploid, potato [242], indicating the prospect of using this method for other polyploids with developing protocols. However, the requirement of wild variety as control, and optimization of biotin addition time and enzyme (Turbo) [241] needs to be considered during protocol development.
Proteomics often fail to contribute to breeding due to a lack of validation of the protein or proteoform biomarkers. Popular validation approach using antibody-based methods such as Western blotting remains extremely challenging [243] due to lack of strong affinity to the target protein or amino acid residues and the ability to mark only the target, selectivity [244]. While researchers are searching for an efficient protein validation method, using CRISPR/Cas knockdown/knockout of the target-protein encoding functional genes is a promising approach [245]. For proteoform validation, immunoassays [246] and Proteoform-predictor [247] can be explored in polyploids.
6. Prospects of Proteomics in Polyploid Breeding
Rapid advances of genome sequencing technologies have increased the availability of gene sequences for most of the crops. However, identification of protein coding regions in the genome and their functions in crop performances is essential. With large sequence data in databases, geneticists face the challenge of deciphering the protein products of gene expression. Proteomics, the large-scale study of proteins, holds great promise for revolutionizing plant breeding by enhancing our understanding of the genetic factors underlying complex traits in polyploid crops [48]. Despite major progress over the last two decades in understanding genetic and genomic consequences of polyploidy, research into proteomic-level changes in polyploids remains in its early stages. Protein isoforms and PTMs are key regulators of protein function, activity, localization, and interactions. Current research efforts focus increasingly on linking genotype to proteome to phenotype, and proteomics is expected to emerge as a critical field in polyploid breeding over the coming decade.
Polyploid crops are sensitive to abiotic stresses during flowering and grain filling, during which ROS causes oxidative stress and reduce yield [248]. These stresses affect photosynthesis and increase photorespiration by altering cellular homeostasis. However, the complex interactions among gene copy numbers, stress-induced expression changes, and the resulting impacts on the transcriptome, translatome, proteome, metabolome, phenotype, and plant fitness are not fully understood. Investigating the effect of gene content and expression changes in the proteome of in auto- and allopolyploids is crucial. Identification of biologically important proteins is also important. To get better understanding of the biologically significant low-abundance proteins, researchers often reduce sample complexity or remove high-abundance proteins.
Polyploidy involves whole-genome duplications, which enables, newly formed angiosperm polyploids to tolerate extensive genomic restructuring [188]. These large-scale genome alternations within just one or a few generations result in significant changes to the transcriptome, metabolome, and proteome, ultimately leading to altered phenotypes. High-throughput DNA sequencing technologies offer to determine the parental origin of sequences in both the genomic DNA and the transcriptome and to facilitate comparative analysis between natural and synthetic polyploids and their diploid relatives [188,249]. Synthetic allopolyploid studies have proven invaluable for investigating genome plasticity in angiosperms; the genomic response to polyploidy can be rapid, targeted, and highly species specific [250]. For example, synthetic Brassica allopolyploids have 25 to 38% of tissue-specific proteins exhibit deviate from the expected additive pattern of parent protein expression. Thus, rapid genomic changes resulting from interspecific hybridization drive adaptation and speciation [251].
Beyond the evolutionary research challenge, subcellular proteins or multiplex proteins, which are often located in two or more locations, are important for stress tolerance, identification remains challenging [252]. In wheat, isolating specific organelles from whole tissue complicates subcellular proteomic analyses. As a result, many subcellular proteins—particularly stress-related and housekeeping proteins—remain unclassified. A recent artificial intelligence (AI)-based predictor, pLoc-mPlant, has been advanced to pLoc bal-mPlant to identify subcellular proteins in plants using Gene Ontology (GO) annotations and balanced training datasets [253]. The subcellular location resources in The Crop Proteins of Annotated Location database (https://croppal.org/) can be used to improve the model and develop crop-specific datasets for wheat and canola, but need to include other polyploids, including cotton. To understand the host-pathogen interactions, couple with crop predictors, pathogen specific subcellular protein predictors such as Gram-LocEN [254] and FunSecKB2 [255], can be considered. Furthermore, recently proposed dual-perspective protein profiling in clinical proteomics [256] coupled with bioinformatic tools can be applied in crops, including polyploids, to identify novel resistance mechanism, host-pathogen relationships, and disease protection strategies [198]. Therefore, AI-based predictors can help identify organelle-specific proteins, improving our understanding of stress responses and cellular functions in polyploids.
Understanding polyploidization serves as a complementary tool in crop improvement, particularly in identifying differential proteins and their roles in functional expression and evolution. Studying proteome alterations caused by polyploidization gives breeders insights into developing crops with improved traits, boosting productivity and resilience. For example, proteomic studies on hexaploid Brassica identified 452 DAPs with a significant expression bias toward the tetraploid progenitor [257]. In wheat, comparative proteomic analyses across diploid, tetraploid, and hexaploid genotypes revealed that protein expression in hexaploid wheat is influenced not just by individual donor genomes but also by complex interactions among these genomes [258]. Similarly, in cotton, allopolyploidization has led to altered gene expression patterns and diversified proteomic profiles. Hu et al. (2015) [259] reported that 4.4–12.8% of differential protein expression between allopolyploids and their diploid progenitors with A and D genome while over 80% of them were additive in nature. Additionally, proteogenomics—combination of proteomics, genomics and transcriptomics—is highly promising to refine gene annotations of these complex polyploids, discover protein coding regions in the genes and identify novel peptides [260]. For example, using proteogenomics, tissue specific proteomes including novel proteins and their subcellular locations were identified in the 24 organs in T. aestivum [230]. Thus, proteomics combining with other omics, multi-omics, can provide better insights into the molecular mechanisms of the stress response of the polyploids for improving their stress-resilience.
Comparative proteomic studies have significantly contributed to identifying and validating candidate genes for crop improvement. These analyses provide molecular insights into grain yield increase and stress tolerance mechanisms, helping breeders to develop high-yielding varieties with improved stress resilience. For example, in wheat, Daba et al. (2020) [261] identified 3,182 DAPs involved in tillering, spike initiation, and kernel development after anthesis using quantitative proteomics. These proteins are promising targets for boosting yield potential. Moreover, DAPs have been identified in relation to various stress tolerances, including heat [110], salinity [262], cold [263], drought [264], and disease resistance [158,265]. In cotton, proteomic research revealed key proteins associated with high-quality fiber development [54,266], and DAPs linked to drought [267,268], cold [269] and salinity tolerance [270].
Overall, proteomics helps elucidate how environmental stresses modify protein structure in polyploid crops. Future advancements in functional and subcellular proteomics will enhance our understanding of stress response pathways and facilitate the development of resilient crop varieties. Notably, breakthroughs in wheat genomics have greatly supported breeding efforts, and the integration of new stress-tolerant genes into breeding pipelines will accelerate the development of homozygous lines with high genetic gains. As proteomic separation techniques improve, proteomics will become increasingly central to modern crop breeding programs.
7. Conclusion
Allopolyploid crops such as wheat, oilseed Brassica, and cotton are vital for global food security and economic growth. However, HS and pathogenic infections drastically reduce grain yield, highlighting the need to improve climate resilience and yield stability. Advancement of proteomic approaches have been utilized for unravelling the molecular mechanisms underpinning stress responses in these crops. Among the three allopolyploids, proteomic research in wheat progressed most. Across these allopolyploid crops, several proteins—particularly those involved in photosynthesis, ROS scavenging, carbohydrate metabolism, peroxidase activity, and HSPs—and protein isoforms have been commonly associated with HS tolerance without grain yield penalty. On the other hand, the proteins involved in responses to biotic stress vary according to the disease specific pathogens. Nonetheless, proteins linked to photosynthesis, defense mechanisms, calcium signaling, lipid metabolism, and lignin biosynthesis commonly contribute to pathogen resistance in the allopolyploids.
Understanding stress-induced proteins at different growth stages is critical for breeding climate-resistant cultivars. In wheat, for example, upregulated proteins such as RuBisCo activase A and PEP carboxylase, along with antioxidant enzymes like SOD, CAT, and GSH, contribute to HS-tolerance in seedling. Additionally, proteins related to starch biosynthesis, low-molecular-weight glutenin subunits, and gliadin help maintain grain yield and quality under HS. Despite the promising potential of proteomics to enhance our understanding of HS responses and host–pathogen interactions in allopolyploid crops, several limitations remain. Lack of improved protein extraction methodologies, comprehensive protein annotation databases, investigation into protein isoforms, PTMs, and PPI, reproducibility, functional validation of candidate protein biomarkers and subcellular proteomics are major challenges of proteomics. Combinations of multiple protein extraction methods can offer better protein coverage than the single extraction method. Advanced DIA or double-search DDA used in other biological systems can be adapted for improved proteoform identification and reproducibility. An improved TurboID-based proximity labeling method can decipher the PPI to understand the cellular functions of the crops during stress conditions. CRISPR/Cas, immunoassays, and Proteoform-predictor can be potential proteoform validation methods.
Despite challenges, proteomics has great prospects in polyploid breeding. Proteomics can not only be successfully used in discovering the evolution of the crops and stress associated genes but also identify the subcellular localization of the candidate proteins. Integration of AI into the existing MS-based proteomic approaches and other predictive tools are promising for identifying stress-induced proteoforms and their subcellular locations. Proteomics coupled with other omics can be also useful for clear understanding of the stress-resilience mechanisms of the allopolyploids. Moreover, addressing the current challenges and gaps of proteomics will be the key to translating molecular insights into practical tools for stress-resilient breeding programs in allopolyploid crops.
Author Contributions
T. H conceptualized, wrote and reviewed majority sections of the original draft through interpretating literatures. R. B. wrote a section, reviewed, and edited the original draft. M. N. S. and M. O. K. wrote another section. K. H. M. S. critically reviewed, commented on and edited the manuscript. G. Y. reviewed and provided ideas for generating figures, and S. I. commented on and edited the draft. All the authors read and approved the final manuscript.
Data Availability Statement
No new data were created or analyzed in this study.
Acknowledgments
The author is cordially thankful to Samalka Wijeweera, Postdoctoral Research Associate at the University of Western Australia, for helping search for relevant literatures.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| HS | Heat stress |
| HSPs | Heat shock proteins |
| ROS | Reactive oxygen species |
| MAPK | Mitogen-activated protein kinase |
| SOD | Superoxide dismutase |
| PPI | Protein-protein interaction |
| AI | Artificial intelligence |
References
- Sattler, M.C.; Carvalho, C.R.; Clarindo, W.R. The polyploidy and its key role in plant breeding. Planta 2015, 243, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Rédei, G.P. Encyclopedia of genetics, genomics, proteomics, and informatics; Springer Science & Business Media: 2008.
- Tossi, V.E.; Tosar, L.J.M.; Laino, L.E.; Iannicelli, J.; Regalado, J.J.; Escandón, A.S.; Baroli, I.; Causin, H.F.; Pitta-Álvarez, S.I. Impact of polyploidy on plant tolerance to abiotic and biotic stresses. Front. Plant Sci. 2022, 13, 869423. [Google Scholar] [CrossRef]
- Kui, L.; Majeed, A.; Ahmed, S.; Khan, M.S.S.; Islam, F.; Chen, J.; Dong, Y. Solanum tuberosum (potato). Trends Genet. 2022, 38, 1193–1195. [Google Scholar] [CrossRef]
- Sforça, D.A.; Vautrin, S.; Cardoso-Silva, C.B.; Mancini, M.C.; Cruz, M.V.R.-D.; Pereira, G.d.S.; Conte, M.; Bellec, A.; Dahmer, N.; Fourment, J.; et al. Gene Duplication in the Sugarcane Genome: A Case Study of Allele Interactions and Evolutionary Patterns in Two Genic Regions. Front. Plant Sci. 2019, 10, 553. [Google Scholar] [CrossRef]
- El Baidouri, M.; Murat, F.; Veyssiere, M.; Molinier, M.; Flores, R.; Burlot, L.; Alaux, M.; Quesneville, H.; Pont, C.; Salse, J. Reconciling the evolutionary origin of bread wheat (Triticum aestivum). New Phytol. 2016, 213, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Branca, F.; Cartea, E. Brassica. In Wild crop relatives: genomic and breeding resources: oilseeds; Springer: 2010; pp. 17-36.
- Akagi, T.; Jung, K.; Masuda, K.; Shimizu, K.K. Polyploidy before and after domestication of crop species. Curr. Opin. Plant Biol. 2022, 69, 102255. [Google Scholar] [CrossRef]
- FAO. World food situation: FAO Cereal Supply and Demand Brief; FAO (Food and Agriculture Organization of the United States): Roam, Italy, 2/12/2022 2025. [Google Scholar]
- Friedt, W.; Tu, J.; Fu, T. Academic and economic importance of Brassica napus rapeseed. In The Brassica Napus Genome, Shengyi Liu, Rod Snowdon, Chalhoub, B., Eds.; Springer Cham: 2018; pp. 1-20.
- Rathore, S.S.; Babu, S.; Shekhawat, K.; Singh, V.K.; Upadhyay, P.K.; Singh, R.K.; Raj, R.; Singh, H.; Zaki, F.M. Oilseed Brassica Species Diversification and Crop Geometry Influence the Productivity, Economics, and Environmental Footprints under Semi-Arid Regions. Sustainability 2022, 14, 2230. [Google Scholar] [CrossRef]
- Rai, P.K.; Yadav, P.; Kumar, A.; Sharma, A.; Kumar, V.; Rai, P. Brassica juncea: A crop for food and health. In The Brassica juncea Genome; Springer: 2022; pp. 1-13.
- USDA. Oilseeds: world markets and trade; United States Department of Agriculture Foreign Agricultural Service: 2025; pp. 13-21.
- Akın, S. The strategic importance of cotton production for the world and Türkiye. In Best Crop Management and Processing Practices for Sustainable Cotton Production, Gürsoy, S., and, Akın, S., Eds.; IntechOpen: 2024.
- Khan, M.A.; Wahid, A.; Ahmad, M.; Tahir, M.T.; Ahmed, M.; Ahmad, S.; Hasanuzzaman, M. World cotton production and consumption: An overview. In Cotton production and uses: Agronomy, crop protection, and postharvest technologies, Ahmad, S., Hasanuzzaman, M., Eds.; Springer: Singapore, 2020; pp. 1–7. [Google Scholar]
- ICAC. Current Global Cotton Market Outlook for 2024/2025; Washington, DC, 2025.
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754. [Google Scholar] [CrossRef] [PubMed]
- FAO. Cereal production, utilization, and trade reaching record levels in 2021/22; Food and Agriculture Organization: Rome, Italy, 3/02/2022 2022. [Google Scholar]
- CIMMYT. Wheat research. Available online: https://www.cimmyt.
- OECD/FAO. OECD-FAO Agricultural outlook 2023-2032. Cotton 2023, 236–247. [Google Scholar] [CrossRef]
- OECD-FAO. OECD-FAO Agricultural Outlook 2021-2030; Organisation for Economic Co-operation and Development (OECD) and and the Food and Agriculture Organization (FAO): Paris, 2021. [Google Scholar]
- Zhang, J.; Zhang, S.; Cheng, M.; Jiang, H.; Zhang, X.; Peng, C.; Lu, X.; Zhang, M.; Jin, J. Effect of Drought on Agronomic Traits of Rice and Wheat: A Meta-Analysis. Int. J. Environ. Res. Public Heal. 2018, 15, 839. [Google Scholar] [CrossRef]
- Elferjani, R.; Soolanayakanahally, R. Canola Responses to Drought, Heat, and Combined Stress: Shared and Specific Effects on Carbon Assimilation, Seed Yield, and Oil Composition. Front. Plant Sci. 2018, 9, 1224. [Google Scholar] [CrossRef]
- Wang, R.; Ji, S.; Zhang, P.; Meng, Y.; Wang, Y.; Chen, B.; Zhou, Z. Drought Effects on Cotton Yield and Fiber Quality on Different Fruiting Branches. Crop. Sci. 2016, 56, 1265–1276. [Google Scholar] [CrossRef]
- Asseng, S.; Ewert, F.; Martre, P.; Rotter, R.P.; Lobell, D.B.; Cammarano, D.; Kimball, B.A.; Ottman, M.J.; Wall, G.W.; White, J.W.; et al. Rising temperatures reduce global wheat production. Nat. Clim. Change 2015, 5, 143–147. [Google Scholar] [CrossRef]
- Kutcher, H.; Warland, J.; Brandt, S. Temperature and precipitation effects on canola yields in Saskatchewan, Canada. Agric. For. Meteorol. 2010, 150, 161–165. [Google Scholar] [CrossRef]
- Sharma, R.K.; Dhillon, J.; Kumar, P.; Reddy, K.R.; Reed, V.; Dodds, D.M.; Reddy, K.N. Modelling the climate change and cotton yield relationship in Mississippi: Autoregressive distributed lag approach. Ecol. Indic. 2024, 166. [Google Scholar] [CrossRef]
- Secchi, M.A.; Fernandez, J.A.; Stamm, M.J.; Durrett, T.; Prasad, P.V.; Messina, C.D.; Ciampitti, I.A. Effects of heat and drought on canola (Brassica napus L.) yield, oil, and protein: A meta-analysis. Field Crop. Res. 2023, 293. [Google Scholar] [CrossRef]
- Bista, M.K.; Adhikari, B.; Sankarapillai, L.V.; Pieralisi, B.; Reddy, K.R.; Jenkins, J.; Bheemanahalli, R. Drought and heat stress induce differential physiological and agronomic trait responses in cotton. Ind. Crop. Prod. 2024, 222. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Xie, L.; Wu, H.; Han, S.; Hu, L.; Zhang, F.; Wang, H. Quantitative proteomic analysis reveals hub proteins for high temperature-induced male sterility in bread wheat (Triticum aestivum L.). Front. Plant Sci. 2024, 15, 1426832. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Yan, G.; Siddique, K.H. ; &; Cowling, W. A. Heat stress during meiosis has lasting impacts on plant growth and reproduction in wheat (Triticum aestivum L.). Agronomy 2022, 12, 987. [Google Scholar]
- Onyemaobi, I.; Liu, H.; Siddique, K.H.M.; Yan, G. Both Male and Female Malfunction Contributes to Yield Reduction under Water Stress during Meiosis in Bread Wheat. Front. Plant Sci. 2017, 7, 2071. [Google Scholar] [CrossRef] [PubMed]
- Zahra, N.; Wahid, A.; Hafeez, M.B.; Ullah, A.; Siddique, K.H.; Farooq, M. Grain development in wheat under combined heat and drought stress: Plant responses and management. Environ. Exp. Bot. 2021, 188. [Google Scholar] [CrossRef]
- Bregaglio, S.; Willocquet, L.; Kersebaum, K.C.; Ferrise, R.; Stella, T.; Ferreira, T.B.; Pavan, W.; Asseng, S.; Savary, S. Comparing process-based wheat growth models in their simulation of yield losses caused by plant diseases. Field Crop. Res. 2021, 265. [Google Scholar] [CrossRef]
- Strehlow, B.; de Mol, F.; Struck, C. Risk Potential of Clubroot Disease on Winter Oilseed Rape. Plant Dis. 2015, 99, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Parkash, V.; Sharma, D.B.; Snider, J.; Bag, S.; Roberts, P.; Tabassum, A.; West, D.; Khanal, S.; Suassuna, N.; Chee, P. Effect of Cotton Leafroll Dwarf Virus on Physiological Processes and Yield of Individual Cotton Plants. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Prášil, I.T.; Klíma, M.; Nesvadba, Z.; Vítámvás, P.; Ovesná, J. Proteomics of wheat and barley cereals in response to environmental stresses: Current state and future challenges. J. Proteom. 2023, 282, 104923. [Google Scholar] [CrossRef] [PubMed]
- Kosová, K.; Vítámvás, P.; Urban, M.O.; Prášil, I.T.; Renaut, J. Plant Abiotic Stress Proteomics: The Major Factors Determining Alterations in Cellular Proteome. Front. Plant Sci. 2018, 9, 122. [Google Scholar] [CrossRef]
- Kosová, K.; Vítámvás, P.; Prášil, I.T.; Klíma, M.; Renaut, J. Plant Proteoforms Under Environmental Stress: Functional Proteins Arising From a Single Gene. Front. Plant Sci. 2021, 12, 793113. [Google Scholar] [CrossRef]
- Smith, L.M.; The Consortium for Top Down Proteomics; Kelleher, N. L. Proteoform: a single term describing protein complexity. Nat. Methods 2013, 10, 186–187. [Google Scholar] [CrossRef]
- Xicoy, H.; Vila, M.; Laguna, A. Systems Medicine in Parkinson׳ s Disease: Joining Efforts to Change History. In Systems Medicine Integrative, Qualitative and Computational Approaches, Wolkenhauer, O., Ed.; 2021; Volume 2, pp. 1-14.
- Singh, B.; Mishra, S.; Bohra, A.; Joshi, R.; Siddique, K.H. Crop phenomics for abiotic stress tolerance in crop plants. In Biochemical, physiological and molecular avenues for combating abiotic stress tolerance in plants, Wani, S.H., Ed.; Elsevier: 2018; pp. 277-296.
- Halder, T.; Choudhary, M.; Liu, H.; Chen, Y.; Yan, G.; Siddique, K.H.M. Wheat Proteomics for Abiotic Stress Tolerance and Root System Architecture: Current Status and Future Prospects. Proteomes 2022, 10, 17. [Google Scholar] [CrossRef]
- Halder, T.; Stroeher, E.; Liu, H.; Chen, Y.; Yan, G.; Siddique, K.H. Protein biomarkers for root length and root dry mass on chromosomes 4A and 7A in wheat. J. Proteom. 2023, 291, 105044. [Google Scholar] [CrossRef]
- Fu, X.; Fu, N.; Guo, S.; Yan, Z.; Xu, Y.; Hu, H.; Menzel, C.; Chen, W.; Li, Y.; Zeng, R.; et al. Estimating accuracy of RNA-Seq and microarrays with proteomics. BMC Genom. 2009, 10, 1–9. [Google Scholar] [CrossRef]
- Ahmad, P.; Latef, A.A.H.A.; Rasool, S.; Akram, N.A.; Ashraf, M.; Gucel, S. Role of Proteomics in Crop Stress Tolerance. Front. Plant Sci. 2016, 7, 1336. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Gayen, D. Decoding post-translational modification for understanding stress tolerance in plant. Crop. Des. 2024, 3. [Google Scholar] [CrossRef]
- Soltis, D.E.; Misra, B.B.; Shan, S.; Chen, S.; Soltis, P.S. Polyploidy and the proteome. Biochim. et Biophys. Acta (BBA) - Proteins Proteom. 2016, 1864, 896–907. [Google Scholar] [CrossRef]
- Thomson, J.J. Bakerian Lecture:—Rays of positive electricity. In Proceedings of the Proceedings of The Royal Society, London, 01 August, 1913.
- Müller, F.; Kolbowski, L.; Bernhardt, O.M.; Reiter, L.; Rappsilber, J. Data-independent Acquisition Improves Quantitative Cross-linking Mass Spectrometry. Mol. Cell. Proteom. 2019, 18, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, W.; Liu, Y.; Shen, Y.; Li, W. Unlocking Wheat's Heat Stress Survival Secrets: A Comprehensive Study of Spike Development’s Metabolic Responses. J. Plant Growth Regul. 2024, 43, 1875–1890. [Google Scholar] [CrossRef]
- Shen, C.-C.; Chen, M.-X.; Xiao, T.; Zhang, C.; Shang, J.; Zhang, K.-L.; Zhu, F.-Y. Global proteome response to Pb(II) toxicity in poplar using SWATH-MS-based quantitative proteomics investigation. Ecotoxicol. Environ. Saf. 2021, 220, 112410. [Google Scholar] [CrossRef]
- Masoomi-Aladizgeh, F.; McKay, M.J.; Asar, Y.; Haynes, P.A.; Atwell, B.J. Patterns of gene expression in pollen of cotton (Gossypium hirsutum) indicate downregulation as a feature of thermotolerance. Plant J. 2021, 109, 965–979. [Google Scholar] [CrossRef]
- Zhou, M.; Sun, G.; Sun, Z.; Tang, Y.; Wu, Y. Cotton proteomics for deciphering the mechanism of environment stress response and fiber development. J. Proteom. 2014, 105, 74–84. [Google Scholar] [CrossRef]
- Barkla, B.J.; Vera-Estrella, R.; Pantoja, O. Progress and challenges for abiotic stress proteomics of crop plants. Proteomics 2013, 13, 1801–1815. [Google Scholar] [CrossRef] [PubMed]
- Sharif, I.; Aleem, S.; Junaid, J.A.; Ali, Z.; Aleem, M.; Shahzad, R.; Farooq, J.; Khan, M.I.; Arshad, W.; Ellahi, F. Multiomics approaches to explore drought tolerance in cotton. J. Cotton Res. 2024, 7, 1–13. [Google Scholar] [CrossRef]
- D’agostino, N.; Fasano, C. Editorial: Genetics and Genomics of Polyploid Plants. Genes 2024, 15, 1377. [Google Scholar] [CrossRef] [PubMed]
- Aufiero, G.; Fruggiero, C.; D’angelo, D.; D’agostino, N. Homoeologs in Allopolyploids: Navigating Redundancy as Both an Evolutionary Opportunity and a Technical Challenge—A Transcriptomics Perspective. Genes 2024, 15, 977. [Google Scholar] [CrossRef]
- Grover, C.E.; Gallagher, J.P.; Szadkowski, E.P.; Yoo, M.J.; Flagel, L.E.; Wendel, J.F. Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol. 2012, 196, 966–971. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, Y.; Zhang, T.; Chen, M.; Song, H. Proteome evaluation of homolog abundance patterns in Arachis hypogaea cv. Tifrunner. Plant Methods 2022, 18, 1–11. [Google Scholar] [CrossRef]
- Zhang, B.; Kuster, B. Proteomics Is Not an Island: Multi-omics Integration Is the Key to Understanding Biological Systems. Mol. Cell. Proteom. 2019, 18, S1–S4. [Google Scholar] [CrossRef]
- Peng, J.H.; Sun, D.; Nevo, E. Domestication evolution, genetics and genomics in wheat. Mol. Breed. 2011, 28, 281–301. [Google Scholar] [CrossRef]
- Borrill, P.; Harrington, S.A.; Uauy, C. Applying the latest advances in genomics and phenomics for trait discovery in polyploid wheat. Plant J. 2018, 97, 56–72. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.-Q.; Zhao, S.; Liu, D.; Wang, J.; Sun, H.; Zhang, C.; Fan, H.; Li, D.; Dong, L.; Tao, Y.; et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature 2013, 496, 87–90. [Google Scholar] [CrossRef]
- Cheng, F.; Wu, J.; Wang, X. Genome triplication drove the diversification of Brassica plants. Hortic. Res. 2014, 1, 14024. [Google Scholar] [CrossRef]
- Vélez-Gavilán, J. Brassica rapa (field mustard). CABI Compendium. CABI International. doi 2022, 10. [Google Scholar]
- Cai, C.; Wang, X.; Liu, B.; Wu, J.; Liang, J.; Cui, Y.; Cheng, F.; Wang, X. Brassica rapa Genome 2.0: A Reference Upgrade through Sequence Re-assembly and Gene Re-annotation. Mol. Plant 2016, 10, 649–651. [Google Scholar] [CrossRef]
- Zhang, L.; Liang, J.; Chen, H.; Zhang, Z.; Wu, J.; Wang, X. A near-complete genome assembly ofBrassica rapaprovides new insights into the evolution of centromeres. Plant Biotechnol. J. 2023, 21, 1022–1032. [Google Scholar] [CrossRef]
- Guo, N.; Wang, S.; Gao, L.; Liu, Y.; Wang, X.; Lai, E.; Duan, M.; Wang, G.; Li, J.; Yang, M.; et al. Genome sequencing sheds light on the contribution of structural variants to Brassica oleracea diversification. BMC Biol. 2021, 19, 1–15. [Google Scholar] [CrossRef]
- Ji, G.; Long, Y.; Cai, G.; Wang, A.; Yan, G.; Li, H.; Gao, G.; Xu, K.; Huang, Q.; Chen, B.; et al. A new chromosome-scale genome of wild Brassica oleracea provides insights into the domestication of Brassica crops. J. Exp. Bot. 2024, 75, 2882–2899. [Google Scholar] [CrossRef] [PubMed]
- Perumal, S.; Koh, C.S.; Jin, L.; Buchwaldt, M.; Higgins, E.E.; Zheng, C.; Sankoff, D.; Robinson, S.J.; Kagale, S.; Navabi, Z.-K.; et al. A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 2020, 6, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Paritosh, K.; Pradhan, A.K.; Pental, D. A highly contiguous genome assembly of Brassica nigra (BB) and revised nomenclature for the pseudochromosomes. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef]
- Heng, S.; Cui, M.; Li, X.; Zhang, S.; Mao, G.; Xing, F.; Wan, Z.; Wen, J.; Shen, J.; Fu, T. The high quality genome of potherb mustard Xuecai (Brassica juncea var. multiceps) provides new insights into leaf shape variation. J. Integr. Agric. 2024, 24, 1461–1476. [Google Scholar] [CrossRef]
- Niu, Y.; Liu, Q.; He, Z.; Raman, R.; Wang, H.; Long, X.; Qin, H.; Raman, H.; Parkin, I.A.; Bancroft, I.; et al. A Brassica carinata pan-genome platform for Brassica crop improvement. Plant Commun. 2023, 5, 100725. [Google Scholar] [CrossRef]
- Emani, C. Gossypium. Wild Crop Relatives: Genomic and Breeding Resources: Industrial Crops 2011, 109-122.
- Egbuta, M.A.; McIntosh, S.; Waters, D.L.E.; Vancov, T.; Liu, L. Biological Importance of Cotton By-Products Relative to Chemical Constituents of the Cotton Plant. Molecules 2017, 22, 93. [Google Scholar] [CrossRef] [PubMed]
- Wegier, A.; Alavez, V.; Piñero, D. Cotton: traditional and modern uses. Ethnobotany of Mexico: interactions of people and plants in Mesoamerica 2016, 439-456.
- Adams, K.L.; Wendel, J.F. Exploring the genomic mysteries of polyploidy in cotton. Biol. J. Linn. Soc. 2004, 82, 573–581. [Google Scholar] [CrossRef]
- Zhang, X.; Zhai, C.; He, L.; Guo, Q.; Zhang, X.; Xu, P.; Su, H.; Gong, Y.; Ni, W.; Shen, X. Morphological, cytological and molecular analyses of a synthetic hexaploid derived from an interspecific hybrid between Gossypium hirsutum and Gossypium anomalum. Crop. J. 2014, 2, 272–277. [Google Scholar] [CrossRef]
- Percival, A.; Wendel, J.; Stewart, J. Taxonomy and germplasm resources. Cotton: origin, history, technology, and production 1999, 33, 63. [Google Scholar]
- Wendel, J.F.; Clark Cronn, R.; Clark, R.; Cronn, R. Polyploidy and the Evolutionary History of Cotton. Advances in Agronomy 2003, 78. [Google Scholar]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago, L.M.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020, 52, 525–533. [Google Scholar] [CrossRef]
- Peng, R.; Xu, Y.; Tian, S.; Unver, T.; Liu, Z.; Zhou, Z.; Cai, X.; Wang, K.; Wei, Y.; Liu, Y.; et al. Evolutionary divergence of duplicated genomes in newly described allotetraploid cottons. Proc. Natl. Acad. Sci. 2022, 119. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, G.; Wang, K.; Sun, F.; Yuan, Y.; Song, G.; Li, Q.; Ma, Z.; Lu, C.; Zou, C.; et al. Genome sequence of the cultivated cotton Gossypium arboreum. Nat. Genet. 2014, 46, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Ramaraj, T.; E Grover, C.; Mendoza, A.C.; A Arick, M.; Jareczek, J.J.; Leach, A.G.; Peterson, D.G.; Wendel, J.F.; A Udall, J. The Gossypium herbaceum L. Wagad genome as a resource for understanding cotton domestication. G3 Genes|Genomes|Genetics 2022, 13. [Google Scholar] [CrossRef]
- Hendrix, B.; STEWART, J.M. Estimation of the Nuclear DNA Content of Gossypium Species. Ann. Bot. 2005, 95, 789–797. [Google Scholar] [CrossRef]
- Grover, C.E.; Gallagher, J.P.; Jareczek, J.J.; Page, J.T.; Udall, J.A.; Gore, M.A.; Wendel, J.F. Re-evaluating the phylogeny of allopolyploid Gossypium L. Mol. Phylogenetics Evol. 2015, 92, 45–52. [Google Scholar] [CrossRef]
- Wendel, J.F.; Grover, C.E. Taxonomy and evolution of the cotton genus, Gossypium. Cotton 2015, 57, 25–44. [Google Scholar]
- Meng, Q.; Gu, J.; Xu, Z.; Zhang, J.; Tang, J.; Wang, A.; Wang, P.; Liu, Z.; Rong, Y.; Xie, P.; et al. Comparative analysis of genome sequences of the two cultivated tetraploid cottons, Gossypium hirsutum (L.) and G. barbadense (L.). Ind. Crop. Prod. 2023, 196. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Nahar, K.; Alam, M.M.; Roychowdhury, R.; Fujita, M. Physiological, Biochemical, and Molecular Mechanisms of Heat Stress Tolerance in Plants. Int. J. Mol. Sci. 2013, 14, 9643–9684. [Google Scholar] [CrossRef]
- Sarkar, S.; Islam, A.A.; Barma, N.C.D. ; &; Ahmed, J. U. Tolerance mechanisms for breeding wheat against heat stress: A review. South African Journal of Botany 2021, 138, 262–277. [Google Scholar]
- Xu, J.; Lowe, C.; Hernandez-Leon, S.G.; Dreisigacker, S.; Reynolds, M.P.; Valenzuela-Soto, E.M.; Paul, M.J.; Heuer, S. The Effects of Brief Heat During Early Booting on Reproductive, Developmental, and Chlorophyll Physiological Performance in Common Wheat (Triticum aestivum L.). Front. Plant Sci. 2022, 13, 886541. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ahmad, M.; Ahmed, M.; Hussain, M.I. Rising Atmospheric Temperature Impact on Wheat and Thermotolerance Strategies. Plants 2020, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.R.; Choudhary, M.; Singh, J.; Lal, M.K.; Jha, P.K.; Udawat, P.; Gupta, N.K.; Rajput, V.D.; Garg, N.K.; Maheshwari, C.; et al. Impacts, Tolerance, Adaptation, and Mitigation of Heat Stress on Wheat under Changing Climates. Int. J. Mol. Sci. 2022, 23, 2838. [Google Scholar] [CrossRef]
- Feng, J.; Jia, Y.; Xu, B.; Bi, X.; Ge, Z.; Ma, G.; Xie, Y.; Wang, C.; Ma, D. Quantitative proteomic analysis for characterization of protein components related to dough quality and celiac disease in wheat flour, dough, and heat-treated dough. Food Chem. 2024, 461, 140924. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Chu, J.; Zhang, X.; Dong, S.; Dai, X.; He, M. Physiological and Proteomic Analyses Indicate Delayed Sowing Improves Photosynthetic Capacity in Wheat Flag Leaves Under Heat Stress. Front. Plant Sci. 2022, 13, 848464. [Google Scholar] [CrossRef] [PubMed]
- Chunduri, V.; Kaur, A.; Kaur, S.; Kumar, A.; Sharma, S.; Sharma, N.; Singh, P.; Kapoor, P.; Kaur, S.; Kumari, A.; et al. Gene Expression and Proteomics Studies Suggest an Involvement of Multiple Pathways Under Day and Day–Night Combined Heat Stresses During Grain Filling in Wheat. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef]
- Kumar, R.R.; Dubey, K.; Arora, K.; Dalal, M.; Rai, G.K.; Mishra, D.; Chaturvedi, K.K.; Rai, A.; Kumar, S.N.; Singh, B.; et al. Characterizing the putative mitogen-activated protein kinase (MAPK) and their protective role in oxidative stress tolerance and carbon assimilation in wheat under terminal heat stress. Biotechnol. Rep. 2021, 29, e00597. [Google Scholar] [CrossRef]
- Wu, B.; Qiao, J.; Wang, X.; Liu, M.; Xu, S.; Sun, D. Factors affecting the rapid changes of protein under short-term heat stress. BMC Genom. 2021, 22, 1–11. [Google Scholar] [CrossRef]
- Wang, J.; Gao, X.; Dong, J.; Tian, X.; Wang, J.; Palta, J.A.; Xu, S.; Fang, Y.; Wang, Z. Over-Expression of the Heat-Responsive Wheat Gene TaHSP23.9 in Transgenic Arabidopsis Conferred Tolerance to Heat and Salt Stress. Front. Plant Sci. 2020, 11, 243. [Google Scholar] [CrossRef]
- Kumar, R.R.; Singh, K.; Ahuja, S.; Tasleem, M.; Singh, I.; Kumar, S.; Grover, M.; Mishra, D.; Rai, G.K.; Goswami, S.; et al. Quantitative proteomic analysis reveals novel stress-associated active proteins (SAAPs) and pathways involved in modulating tolerance of wheat under terminal heat. Funct. Integr. Genom. 2018, 19, 329–348. [Google Scholar] [CrossRef]
- Zhang, Y.; Lou, H.; Guo, D.; Zhang, R.; Su, M.; Hou, Z.; Zhou, H.; Liang, R.; Xie, C.; You, M.; et al. Identifying changes in the wheat kernel proteome under heat stress using iTRAQ. Crop. J. 2018, 6, 600–610. [Google Scholar] [CrossRef]
- Perveen, A.; Sheheryar, S.; Ahmad, F.; Mustafa, G.; Moura, A.A.; Campos, F.A.P.; Domont, G.B.; Nishan, U.; Ullah, R.; Ibrahim, M.A.; et al. Integrative physiological, biochemical, and proteomic analysis of the leaves of two cotton genotypes under heat stress. PLOS ONE 2025, 20, e0316630. [Google Scholar] [CrossRef]
- Ma, W.; Li, J.; Liu, F.; Zhang, T.; Guan, X. GhHSP24.7 mediates mitochondrial protein acetylation to regulate stomatal conductance in response to abiotic stress in cotton. Crop. J. 2022, 11, 1128–1139. [Google Scholar] [CrossRef]
- Masoomi-Aladizgeh, F.; Najeeb, U.; Hamzelou, S.; Pascovici, D.; Amirkhani, A.; Tan, D.K.Y.; Mirzaei, M.; Haynes, P.A.; Atwell, B.J. Pollen development in cotton (Gossypium hirsutum) is highly sensitive to heat exposure during the tetrad stage. Plant, Cell Environ. 2020, 44, 2150–2166. [Google Scholar] [CrossRef]
- Zhang, X.; Tian, Q.; Zhao, Z.; Dong, Z.; Chen, Y.; Chen, D. Analysis of differentially expressed proteins affecting insecticidal protein content in Bt cotton under high-temperature and water deficit stress using label-free quantitation. J. Agron. Crop. Sci. 2020, 207, 1–11. [Google Scholar] [CrossRef]
- Masoomi-Aladizgeh, F.; Kamath, K.S.; Haynes, P.A.; Atwell, B.J. Genome survey sequencing of wild cotton (Gossypium robinsonii) reveals insights into proteomic responses of pollen to extreme heat. Plant, Cell Environ. 2022, 45, 1242–1256. [Google Scholar] [CrossRef]
- Rani, R.; Mawlong, I.; Balbeer, B.; Kumar, M.S.; Rai, P.K.; Singh, V.V. Proteomic, biochemical and peptidomics based analysis reveals heat responsive changes in the seedlings of Brassica juncea. J. Plant Biochem. Biotechnol. 2024, 33, 570–589. [Google Scholar] [CrossRef]
- Singiri, J.R.; Swetha, B.; Sikron-Persi, N.; Grafi, G. Differential Response to Single and Combined Salt and Heat Stresses: Impact on Accumulation of Proteins and Metabolites in Dead Pericarps of Brassica juncea. Int. J. Mol. Sci. 2021, 22, 7076. [Google Scholar] [CrossRef]
- Yuan, L.; Wang, J.; Xie, S.; Zhao, M.; Nie, L.; Zheng, Y.; Zhu, S.; Hou, J.; Chen, G.; Wang, C. Comparative Proteomics Indicates That Redox Homeostasis Is Involved in High- and Low-Temperature Stress Tolerance in a Novel Wucai (Brassica campestris L.) Genotype. Int. J. Mol. Sci. 2019, 20, 3760. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.; Shekhar, S.; Chakraborty, S.; Chakraborty, N. Wheat 2-Cys peroxiredoxin plays a dual role in chlorophyll biosynthesis and adaptation to high temperature. Plant J. 2020, 105, 1374–1389. [Google Scholar] [CrossRef] [PubMed]
- Gupta, O.P.; Mishra, V.; Singh, N.K.; Tiwari, R.; Sharma, P.; Gupta, R.K.; Sharma, I. Deciphering the dynamics of changing proteins of tolerant and intolerant wheat seedlings subjected to heat stress. Mol. Biol. Rep. 2014, 42, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Qin, Z.; Cui, G.; Chen, Z.; Cheng, X.; Peng, H.; Yao, Y.; Hu, Z.; Guo, W.; Ni, Z.; et al. Natural variation of STKc_GSK3 kinase TaSG-D1 contributes to heat stress tolerance in Indian dwarf wheat. Nat. Commun. 2024, 15, 1–12. [Google Scholar] [CrossRef]
- Vu, L.D.; Zhu, T.; Verstraeten, I.; Van De Cotte, B.; Consortium, I.W.G.S.; Gevaert, K.; De Smet, I. Temperature-induced changes in the wheat phosphoproteome reveal temperature-regulated interconversion of phosphoforms. Journal of Experimental Botany 2018, 69, 4609–4624. [Google Scholar] [CrossRef]
- Majoul, T.; Bancel, E.; Triboï, E.; Ben Hamida, J.; Branlard, G. Proteomic analysis of the effect of heat stress on hexaploid wheat grain: Characterization of heat-responsive proteins from total endosperm. Proteomics 2003, 3, 175–183. [Google Scholar] [CrossRef]
- Skylas, D.; Cordwell, S.; Hains, P.; Larsen, M.; Basseal, D.; Walsh, B.; C. Blumenthalbd, C.; Rathmell, W.; Copeland, L.; Wrigley, C. Heat Shock of Wheat During Grain Filling: Proteins Associated with Heat-tolerance. J. Cereal Sci. 2002, 35, 175–188. [Google Scholar] [CrossRef]
- Young, L.W.; Wilen, R.W.; Bonham-Smith, P.C. High temperature stress of Brassica napus during flowering reduces micro- and megagametophyte fertility, induces fruit abortion, and disrupts seed production. J. Exp. Bot. 2004, 55, 485–495. [Google Scholar] [CrossRef]
- Angadi, S.V.; Cutforth, H.W.; Miller, P.R.; McConkey, B.G.; Entz, M.H.; Brandt, S.A.; Volkmar, K.M. Response of three Brassica species to high temperature stress during reproductive growth. Can. J. Plant Sci. 2000, 80, 693–701. [Google Scholar] [CrossRef]
- Ahmad, M.; Waraich, E.A.; Skalicky, M.; Hussain, S.; Zulfiqar, U.; Anjum, M.Z.; Rahman, M.H.U.; Brestic, M.; Ratnasekera, D.; Lamilla-Tamayo, L.; et al. Adaptation Strategies to Improve the Resistance of Oilseed Crops to Heat Stress Under a Changing Climate: An Overview. Front. Plant Sci. 2021, 12, 767150. [Google Scholar] [CrossRef]
- Yan, G.; Lv, X.; Gao, G.; Li, F.; Li, J.; Qiao, J.; Xu, K.; Chen, B.; Wang, L.; Xiao, X.; et al. Identification and Characterization of a Glyoxalase I Gene in a Rapeseed Cultivar with Seed Thermotolerance. Front. Plant Sci. 2016, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Bansal, V.K.; Kav, N.N.V. Proteome-Level Investigation of Brassica carinata-Derived Resistance to Leptosphaeria maculans. J. Agric. Food Chem. 2004, 53, 313–324. [Google Scholar] [CrossRef]
- Hajduch, M.; Casteel, J.E.; Hurrelmeyer, K.E.; Song, Z.; Agrawal, G.K.; Thelen, J.J. Proteomic Analysis of Seed Filling in Brassica napus. Developmental Characterization of Metabolic Isozymes Using High-Resolution Two-Dimensional Gel Electrophoresis. Plant Physiol. 2006, 141, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Ismaili, A.; Salavati, A.; Mohammadi, P. A Comparative Proteomic Analysis of Responses to High Temperature Stress in Hypocotyl of Canola (Brassica napus L.). Protein Pept. Lett. 2015, 22, 285–299. [Google Scholar] [CrossRef]
- Swetha, B.; Singiri, J.R.; Novoplansky, N.; Grandhi, R.; Srinivasan, J.; Khadka, J.; Galis, I.; Grafi, G. Single and Combined Salinity and Heat Stresses Impact Yield and Dead Pericarp Priming Activity. Plants 2021, 10, 1627. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Li, Y.; Li, Y.; Ma, Y.; Zhao, Y.; Wang, C.; Chi, H.; Chen, M.; Ding, Y.; Guo, X. Proteomic analysis reveals that sugar and fatty acid metabolisms play a central role in sterility of the male-sterile line 1355A of cotton. Journal of Biological Chemistry 2019, 294, 7057–7067. [Google Scholar] [CrossRef]
- Schulte, I.; Tammen, H.; Selle, H.; Schulz-Knappe, P. Peptides in body fluids and tissues as markers of disease. Expert Rev. Mol. Diagn. 2005, 5, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zahid, K.R.; Ali, F.; Shah, F.; Younas, M.; Shah, T.; Shahwar, D.; Hassan, W.; Ahmad, Z.; Qi, C.; Lu, Y.; et al. Response and Tolerance Mechanism of Cotton Gossypium hirsutum L. to Elevated Temperature Stress: A Review. Front. Plant Sci. 2016, 7, 937. [Google Scholar] [CrossRef]
- Salvucci, M.E.; Crafts-Brandner, S.J. Relationship between the Heat Tolerance of Photosynthesis and the Thermal Stability of Rubisco Activase in Plants from Contrasting Thermal Environments. Plant Physiol. 2004, 134, 1460–1470. [Google Scholar] [CrossRef]
- Wise, R.R.; Olson, A.J.; Schrader, S.M.; Sharkey, T.D. Electron transport is the functional limitation of photosynthesis in field-grown Pima cotton plants at high temperature. Plant, Cell Environ. 2004, 27, 717–724. [Google Scholar] [CrossRef]
- Ashraf, M.; Saeed, M.; Qureshi, M. Tolerance to high temperature in cotton (gossypium hirsutum L.) at initial growth stages. Environ. Exp. Bot. 1994, 34, 275–283. [Google Scholar] [CrossRef]
- Majeed, S.; Rana, I.A.; Mubarik, M.S.; Atif, R.M.; Yang, S.-H.; Chung, G.; Jia, Y.; Du, X.; Hinze, L.; Azhar, M.T. Heat Stress in Cotton: A Review on Predicted and Unpredicted Growth-Yield Anomalies and Mitigating Breeding Strategies. Agronomy 2021, 11, 1825. [Google Scholar] [CrossRef]
- Sahu, B.; Choudhary, V.K.; Sahu, M.; Kumar, K.K.; Sujayanand, G.; Gopi, R.; Prakasam, V.; Sridhar, J.; Mallikarjuna, J.; Singh, H. Biotic Stress Management. In Trajectory of 75 years of Indian Agriculture after Independence; Springer: 2023; pp. 619-653.
- Dixit, S.; Sivalingam, P.N.; Baskaran, R.K.M.; Senthil-Kumar, M.; Ghosh, P.K. Plant responses to concurrent abiotic and biotic stress: unravelling physiological and morphological mechanisms. Plant Physiol. Rep. 2023, 29, 6–17. [Google Scholar] [CrossRef]
- Kayess, O.; Ashrafuzzaman, M.; Khan, M.A.R.; Siddiqui, M.N. Functional phenomics and genomics: Unravelling heat stress responses in wheat. Plant Stress 2024, 100601. [Google Scholar] [CrossRef]
- Singh, B.K.; Delgado-Baquerizo, M.; Egidi, E.; Guirado, E.; Leach, J.E.; Liu, H.; Trivedi, P. Climate change impacts on plant pathogens, food security and paths forward. Nat. Rev. Microbiol. 2023, 21, 640–656. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, S.; Liu, K.; Wang, S.; Huang, L.; Guo, L. Proteomics: a powerful tool to study plant responses to biotic stress. Plant Methods 2019, 15, 1–20. [Google Scholar] [CrossRef]
- Mmbando, G.S. Omics: A new, promising technologies for boosting crop yield and stress resilience in African agriculture. Plant Stress 2024, 11. [Google Scholar] [CrossRef]
- Haq, S.A.U.; Bashir, T.; Roberts, T.H.; Husaini, A.M. Ameliorating the effects of multiple stresses on agronomic traits in crops: modern biotechnological and omics approaches. Mol. Biol. Rep. 2023, 51, 1–35. [Google Scholar] [CrossRef]
- Bakala, H.S.; Mandahal, K.S.; Sarao, L.K.; Srivastava, P. Breeding wheat for biotic stress resistance: Achievements, challenges and prospects. Current trends in Wheat Research 2021, 12, 11–34. [Google Scholar]
- Kema, G.H.J.; Mirzadi Gohari, A.; Aouini, L.; Gibriel, H.A.Y.; Ware, S.B.; van den Bosch, F.; Manning-Smith, R.; Alonso-Chavez, V.; Helps, J.; Ben M’Barek, S.; et al. Stress and sexual reproduction affect the dynamics of the wheat pathogen effector AvrStb6 and strobilurin resistance. Nat. Genet. 2018, 50, 375–380. [Google Scholar] [CrossRef]
- Yang, F.; Melo-Braga, M.N.; Larsen, M.R.; Jørgensen, H.J.L.; Palmisano, G. Battle through Signaling between Wheat and the Fungal Pathogen Septoria tritici Revealed by Proteomics and Phosphoproteomics. Mol. Cell. Proteom. 2013, 12, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Yin, Q. Comprehensive proteomic analysis of the wheat pathogenic fungus Zymoseptoria tritici. Proteomics 2015, 16, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Vannini, C.; Domingo, G.; Fiorilli, V.; Seco, D.G.; Novero, M.; Marsoni, M.; Wisniewski-Dye, F.; Bracale, M.; Moulin, L.; Bonfante, P. Proteomic analysis reveals how pairing of a Mycorrhizal fungus with plant growth-promoting bacteria modulates growth and defense in wheat. Plant, Cell Environ. 2021, 44, 1946–1960. [Google Scholar] [CrossRef]
- Maytalman, D.; Mert, Z.; Baykal, A.; Inan, C.; Gunel, A.; Hasançebi, S. Proteomic analysis of early responsive resistance proteins of wheat (Triticum aestivum) to yellow rust (Puccinia striiformis f. sp. tritici) using ProteomeLab PF2D. Plant Omics 2013, 6, 24–35. [Google Scholar]
- Wang, J.; Diaz, J.; Hua, K.; Bellizzi, M.; Qi, L.; Zhu, L.; Qu, M.; Wang, G.-L. Proteomic identification of apoplastic proteins from rice, wheat, and barley after Magnaporthe oryzae infection. Phytopathol. Res. 2024, 6, 1–13. [Google Scholar] [CrossRef]
- He, T.; Xu, T.; Muhae-Ud-Din, G.; Guo, Q.; Liu, T.; Chen, W.; Gao, L. iTRAQ-based proteomic analysis of wheat (Triticum aestivum) spikes in response to Tilletia controversa Kühn and Tilletia foetida Kühn infection, causal organisms of dwarf bunt and common bunt of wheat. Biology 2022, 11, 865. [Google Scholar] [CrossRef]
- Qiao, F.; Yang, X.; Xu, F.; Huang, Y.; Zhang, J.; Song, M.; Zhou, S.; Zhang, M.; He, D. TMT-based quantitative proteomic analysis reveals defense mechanism of wheat against the crown rot pathogen Fusarium pseudograminearum. BMC Plant Biol. 2021, 21, 1–17. [Google Scholar] [CrossRef]
- Yang, M.; Wang, X.; Dong, J.; Zhao, W.; Alam, T.; Thomashow, L.S.; Weller, D.M.; Gao, X.; Rustgi, S.; Wen, S. Proteomics Reveals the Changes that Contribute to Fusarium Head Blight Resistance in Wheat. Phytopathology® 2021, 111, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Fabre, F.; Urbach, S.; Roche, S.; Langin, T.; Bonhomme, L. Proteomics-Based Data Integration of Wheat Cultivars Facing Fusarium graminearum Strains Revealed a Core-Responsive Pattern Controlling Fusarium Head Blight. Front. Plant Sci. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, Y.; Agarwal, D.; Ilyas, A.; Sharma, S.; Myskova, A.; Poddar, N.K. Patterns of protein expression in wheat under stress conditions and its identification by proteomics tools. In Abiotic Stresses in Wheat; Elsevier: 2023; pp. 247-258.
- Wang, Q.; Guo, J.; Jin, P.; Guo, M.; Guo, J.; Cheng, P.; Li, Q.; Wang, B. GlutathioneS-transferase interactions enhance wheat resistance to powdery mildew but not wheat stripe rust. Plant Physiol. 2022, 190, 1418–1439. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Zhang, H.; Mandal, S.N.; Wang, C.; Chen, C.; Ji, W. Quantitative proteomics reveals the central changes of wheat in response to powdery mildew. J. Proteom. 2016, 130, 108–119. [Google Scholar] [CrossRef]
- Bhatta, M.; Morgounov, A.; Belamkar, V.; Wegulo, S.N.; Dababat, A.A.; Erginbas-Orakci, G.; El Bouhssini, M.; Gautam, P.; Poland, J.; Akci, N.; et al. Genome-Wide Association Study for Multiple Biotic Stress Resistance in Synthetic Hexaploid Wheat. Int. J. Mol. Sci. 2019, 20, 3667. [Google Scholar] [CrossRef]
- Islam, T.; Lee, B.-R.; La, V.H.; Bae, D.-W.; Jung, W.-J.; Kim, T.-H. Label-Free Quantitative Proteomics Analysis in Susceptible and Resistant Brassica napus Cultivars Infected with Xanthomonas campestris pv. campestris. Microorganisms 2021, 9, 253. [Google Scholar] [CrossRef]
- Struck, C.; Rüsch, S.; Strehlow, B. Control Strategies of Clubroot Disease Caused by Plasmodiophora brassicae. Microorganisms 2022, 10, 620. [Google Scholar] [CrossRef]
- Adhikary, D.; Mehta, D.; Uhrig, R.G.; Rahman, H.; Kav, N.N.V. A Proteome-Level Investigation Into Plasmodiophora brassicae Resistance in Brassica napus Canola. Front. Plant Sci. 2022, 13, 860393. [Google Scholar] [CrossRef]
- Moon, J.Y.; Kim, S.T.; Choi, G.J.; Kwon, S.-Y.; Cho, H.S.; Kim, H.-S.; Moon, J.S.; Park, J.M. Comparative proteomic analysis of host responses to Plasmodiophora brassicae infection in susceptible and resistant Brassica oleracea. Plant Biotechnol. Rep. 2020, 14, 263–274. [Google Scholar] [CrossRef]
- Singh, J.; Yadav, P.; Budhlakoti, N.; Mishra, D.C.; Bhardwaj, N.R.; Rao, M.; Sharma, P.; Gupta, N.C. Exploration of the Sclerotinia sclerotiorum-Brassica pathosystem: advances and perspectives in omics studies. Mol. Biol. Rep. 2024, 51, 1–15. [Google Scholar] [CrossRef]
- Liang, Y.; Srivastava, S.; Rahman, M.H.; Strelkov, S.E.; Kav, N.N.V. Proteome Changes in Leaves of Brassica napus L. as a Result of Sclerotinia sclerotiorum Challenge. J. Agric. Food Chem. 2008, 56, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Garg, H.; Li, H.; Sivasithamparam, K.; Barbetti, M.J. Differentially Expressed Proteins and Associated Histological and Disease Progression Changes in Cotyledon Tissue of a Resistant and Susceptible Genotype of Brassica napus Infected with Sclerotinia sclerotiorum. PLOS ONE 2013, 8, e65205. [Google Scholar] [CrossRef]
- Liang, Y.; Rahman, M.H.; Strelkov, S.E.; Kav, N.N. Developmentally induced changes in the sclerotial proteome of Sclerotinia sclerotiorum. Fungal Biol. 2010, 114, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.-Y.; Xu, Y.-P.; Cai, X.-Z. TMT-based quantitative proteomics analyses reveal novel defense mechanisms of Brassica napus against the devastating necrotrophic pathogen Sclerotinia sclerotiorum. J. Proteom. 2016, 143, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Avtar, R.; Lakra, N.; Hooda, E.; Singh, V.K.; Bishnoi, M.; Kumari, N.; Punia, R.; Kumar, N.; Choudhary, R.R. Genetic and Proteomic Basis of Sclerotinia Stem Rot Resistance in Indian Mustard [Brassica juncea (L.) Czern & Coss.]. Genes 2021, 12, 1784. [Google Scholar] [CrossRef]
- Cheng, X.; Zhao, C.; Gao, L.; Zeng, L.; Xu, Y.; Liu, F.; Huang, J.; Liu, L.; Liu, S.; Zhang, X. Alternative splicing reprogramming in fungal pathogen Sclerotinia sclerotiorum at different infection stages on Brassica napus. Front. Plant Sci. 2022, 13, 1008665. [Google Scholar] [CrossRef]
- Kamburova, V.; Salakhutdinov, I.; Abdurakhmonov, I.Y. Cotton breeding in the view of abiotic and biotic stresses: challenges and perspectives. In Cotton, Abdurakhmonov, I.Y., Ed.; IntechOpen: 2022.
- Beckman, C. Cell irritability and localization of vascular infections in plants. Phytopathology 1966, 56, 821–824. [Google Scholar]
- Aydin, RM.H. Rhizoctonia solani and its biological control. Türkiye Tarımsal Araştırmalar Dergisi 2022, 9, 118-135. [CrossRef]
- Jalloul, A.; Sayegh, M.; Champion, A.; Nicole, M. Bacterial blight of cotton. Phytopathologia Mediterranea 2015, 54, 3–20. [Google Scholar]
- Palanga, K.K.; Liu, R.; Ge, Q.; Gong, J.; Li, J.; Lu, Q.; Li, P.; Yuan, Y.; Gong, W. Current advances in pathogen-plant interaction between Verticillium dahliae and cotton provide new insight in the disease management. J. Cotton Res. 2021, 4, 1–13. [Google Scholar] [CrossRef]
- Wang, F.-X.; Ma, Y.-P.; Yang, C.-L.; Zhao, P.-M.; Yao, Y.; Jian, G.-L.; Luo, Y.-M.; Xia, G. Proteomic analysis of the sea-island cotton roots infected by wilt pathogen Verticillium dahliae. Proteomics 2011, 11, 4296–4309. [Google Scholar] [CrossRef]
- Guo, J.; Cao, P.; Yuan, L.; Xia, G.; Zhang, H.; Li, J.; Wang, F. Revealing the contribution of GbPR10.5D1 to resistance against Verticillium dahliae and its regulation for structural defense and immune signaling. Plant Genome 2022, 15, e20271. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Jin, L.; Miao, Y.; He, X.; Hu, Q.; Guo, K.; Zhu, L.; Zhang, X. An ethylene response-related factor, GbERF1-like, from Gossypium barbadense improves resistance to Verticillium dahliae via activating lignin synthesis. Plant Mol. Biol. 2016, 91, 305–318. [Google Scholar] [CrossRef]
- Lu, T.; Zhu, L.; Liang, Y.; Wang, F.; Cao, A.; Xie, S.; Chen, X.; Shen, H.; Wang, B.; Hu, M.; et al. Comparative Proteomic Analysis Reveals the Ascorbate Peroxidase-Mediated Plant Resistance to Verticillium dahliae in Gossypium barbadense. Front. Plant Sci. 2022, 13, 877146. [Google Scholar] [CrossRef]
- Umer, M.J.; Batool, R.; Yang, M.; Zheng, J.; Nazir, M.F.; Wang, H.; Cai, X.; Hou, Y.; Xu, Y.; Wang, Y.; et al. Unravelling the Functional Role of GthGAPC2 in Cotton's Defense Against Verticillium dahliae through Proteome. Physiol. Plant. 2023, 176. [Google Scholar] [CrossRef]
- Mi, X.; Li, W.; Chen, C.; Xu, H.; Wang, G.; Jin, X.; Zhang, D.; Guo, W. GhMPK9-GhRAF39_1-GhWRKY40a regulates the GhERF1b-and GhABF2-mediated pathways to increase cotton disease resistance. Advanced Science 2024, 2404400. [Google Scholar] [CrossRef]
- Han, Z.; Qiu, Y.; Pan, T.; Wang, L.; Wang, J.; Liu, K. GhMAC3e is involved in plant growth and defense response to Verticillium dahliae. Plant Cell Rep. 2024, 43, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, Y.; Zhao, L.; Wei, F.; Feng, Z.; Feng, H. Phosphoproteomics Profiling of Cotton (Gossypium hirsutum L.) Roots in Response to Verticillium dahliae Inoculation. ACS Omega 2019, 4, 18434–18443. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, Y.; Zhou, J.; Wei, F.; Feng, Z.; Zhao, L.; Shi, Y.; Feng, H.; Zhu, H. The Respiratory Burst Oxidase Homolog Protein D (GhRbohD) Positively Regulates the Cotton Resistance to Verticillium dahliae. Int. J. Mol. Sci. 2021, 22, 13041. [Google Scholar] [CrossRef]
- Khan, M.A.; Khan, S.A.; Waheed, U.; Raheel, M.; Khan, Z.; Alrefaei, A.F.; Alkhamis, H.H. Morphological and genetic characterization of Fusarium oxysporum and its management using weed extracts in cotton. J. King Saud Univ. - Sci. 2021, 33. [Google Scholar] [CrossRef]
- Bleackley, M.R.; Samuel, M.; Garcia-Ceron, D.; McKenna, J.A.; Lowe, R.G.T.; Pathan, M.; Zhao, K.; Ang, C.-S.; Mathivanan, S.; Anderson, M.A. Extracellular Vesicles From the Cotton Pathogen Fusarium oxysporum f. sp. vasinfectum Induce a Phytotoxic Response in Plants. Front. Plant Sci. 2020, 10, 1610. [Google Scholar] [CrossRef]
- Wang, C.; He, X.; Li, Y.; Wang, L.; Guo, X.; Guo, X. The cotton MAPK kinase GhMPK20 negatively regulates resistance to Fusarium oxysporum by mediating the MKK4–MPK20–WRKY40 cascade. Mol. Plant Pathol. 2017, 19, 1624–1638. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Guo, H.; He, X.; Zhang, S.; Wang, J.; Wang, L.; Guo, D.; Guo, X. Scaffold protein GhMORG1 enhances the resistance of cotton to Fusarium oxysporum by facilitating the MKK6-MPK4 cascade. Plant Biotechnol. J. 2019, 18, 1421–1433. [Google Scholar] [CrossRef]
- Zhang, M.; Cheng, S.-T.; Wang, H.-Y.; Wu, J.-H.; Luo, Y.-M.; Wang, Q.; Wang, F.-X.; Xia, G.-X. iTRAQ-based proteomic analysis of defence responses triggered by the necrotrophic pathogen Rhizoctonia solani in cotton. J. Proteom. 2017, 152, 226–235. [Google Scholar] [CrossRef]
- Santos, I.R.; Rios, T.B.; Maximiano, M.R.; Coutinho, W.M.; De Lima, L.M.; Silva, L.P.; Oliveira-Neto, O.B.; Mehta, A. Proteomic screening for the identification of proteins involved in resistance to Xanthomonas campestris pv. malvacearum in cotton. Physiol. Mol. Plant Pathol. 2021, 113. [Google Scholar] [CrossRef]
- Hegarty, M.J.; Hiscock, S.J. The complex nature of allopolyploid plant genomes. Heredity 2009, 103, 100–101. [Google Scholar] [CrossRef]
- Lim, K.Y.; Kovarik, A.; Matyasek, R.; Chase, M.W.; Clarkson, J.J.; Grandbastien, M.A.; Leitch, A.R. Sequence of events leading to near-complete genome turnover in allopolyploid Nicotiana within five million years. New Phytol. 2007, 175, 756–763. [Google Scholar] [CrossRef]
- Garbus, I.; Romero, J.R.; Valarik, M.; Vanžurová, H.; Karafiátová, M.; Cáccamo, M.; Doležel, J.; Tranquilli, G.; Helguera, M.; Echenique, V. Characterization of repetitive DNA landscape in wheat homeologous group 4 chromosomes. BMC Genom. 2015, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Vasupalli, N.; Bhat, J.A.; Jain, P.; Sri, T.; Islam, A.; Shivaraj, S.; Singh, S.K.; Deshmukh, R.; Sonah, H.; Lin, X. Omics big data for crop improvement: Opportunities and challenges. Crop. J. 2024, 12, 1517–1532. [Google Scholar] [CrossRef]
- Yadav, B.G. ; Aakanksha; Kumar, R. ; Yadava, S.K.; Kumar, A.; Ramchiary, N. Understanding the proteomes of plant development and stress responses in Brassica crops. Journal of Proteome Research 2023, 22, 660–680. [Google Scholar]
- Glover, N.M.; Redestig, H.; Dessimoz, C. Homoeologs: What Are They and How Do We Infer Them? Trends Plant Sci. 2016, 21, 609–621. [Google Scholar] [CrossRef]
- Zhang, Z.; Xun, H.; Lv, R.; Gou, X.; Ma, X.; Li, J.; Zhao, J.; Li, N.; Gong, L.; Liu, B. Effects of homoeologous exchange on gene expression and alternative splicing in a newly formed allotetraploid wheat. Plant J. 2022, 111, 1267–1282. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, P.; Zhang, A.; Ma, L.; Xu, S.; Wang, X. Alternative Splicing Diversified the Heat Response and Evolutionary Strategy of Conserved Heat Shock Protein 90s in Hexaploid Wheat (Triticum aestivum L.). Front. Genet. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, Y.; Wang, Q.; Wang, C.; Lv, S.; Wang, Y.; Wang, J.; Wang, Y.; Yuan, J.; Zhang, H.; et al. An alternative splicing isoform of wheat TaYRG1 resistance protein activates immunity by interacting with dynamin-related proteins. J. Exp. Bot. 2022, 73, 5474–5489. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Qin, Z.; Sun, L.; Zhang, Y.; Wang, D.; Peng, H.; Yao, Y.; Hu, Z.; Ni, Z.; Sun, Q.; et al. Alternative splicing of TaHSFA6e modulates heat shock protein–mediated translational regulation in response to heat stress in wheat. New Phytol. 2023, 239, 2235–2247. [Google Scholar] [CrossRef]
- Leutert, M.; Entwisle, S.W.; Villén, J. Decoding Post-Translational Modification Crosstalk With Proteomics. Mol. Cell. Proteom. 2021, 20, 100129. [Google Scholar] [CrossRef]
- Geddes-McAlister, J.; Uhrig, R.G. The plant proteome delivers from discovery to innovation. Trends Plant Sci. 2025, 30, 837–845. [Google Scholar] [CrossRef]
- Suhre, K.; Venkataraman, G.R.; Guturu, H.; Halama, A.; Stephan, N.; Thareja, G.; Sarwath, H.; Motamedchaboki, K.; Donovan, M.K.R.; Siddiqui, A.; et al. Nanoparticle enrichment mass-spectrometry proteomics identifies protein-altering variants for precise pQTL mapping. Nat. Commun. 2024, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, B.; Zhan, X.; Schlüter, H.; Jungblut, P.R.; Coorssen, J.R. Innovating the Concept and Practice of Two-Dimensional Gel Electrophoresis in the Analysis of Proteomes at the Proteoform Level. Proteomes 2019, 7, 36. [Google Scholar] [CrossRef]
- Zhan, X.; Li, N.; Zhan, X.; Qian, S. Revival of 2DE-LC/MS in proteomics and its potential for large-scale study of human proteoforms. Med One 2018, 3. [Google Scholar]
- Fröhlich, K.; Fahrner, M.; Brombacher, E.; Seredynska, A.; Maldacker, M.; Kreutz, C.; Schmidt, A.; Schilling, O. Data-Independent Acquisition: A Milestone and Prospect in Clinical Mass Spectrometry–Based Proteomics. Mol. Cell. Proteom. 2024, 23, 100800. [Google Scholar] [CrossRef] [PubMed]
- Charkow, J.; Röst, H.L. Trapped Ion Mobility Spectrometry Reduces Spectral Complexity in Mass Spectrometry-Based Proteomics. Anal. Chem. 2021, 93, 16751–16758. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Binek, A.; Kreimer, S.; Ayres, M.; Stotland, A.; Van Eyk, J.E. High-Field Asymmetric Waveform Ion Mobility Spectrometry: Practical Alternative for Cardiac Proteome Sample Processing. J. Proteome Res. 2023, 22, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Kaulich, P.T.; Jeong, K.; Kohlbacher, O.; Tholey, A. Influence of different sample preparation approaches on proteoform identification by top-down proteomics. Nat. Methods 2024, 21, 2397–2407. [Google Scholar] [CrossRef]
- Padula, M.P.; Berry, I.J.; O′Rourke, M.B.; Raymond, B.B.; Santos, J.; Djordjevic, S.P. A Comprehensive Guide for Performing Sample Preparation and Top-Down Protein Analysis. Proteomes 2017, 5, 11. [Google Scholar] [CrossRef]
- Duong, V.-A.; Lee, H. Bottom-Up Proteomics: Advancements in Sample Preparation. Int. J. Mol. Sci. 2023, 24, 5350. [Google Scholar] [CrossRef]
- Carbonara, K.; Andonovski, M.; Coorssen, J.R. Proteomes Are of Proteoforms: Embracing the Complexity. Proteomes 2021, 9, 38. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Padula, M.P. Proteomics—The State of the Field: The Definition and Analysis of Proteomes Should Be Based in Reality, Not Convenience. Proteomes 2024, 12, 14. [Google Scholar] [CrossRef]
- Fornelli, L.; Toby, T.K. Characterization of large intact protein ions by mass spectrometry: What directions should we follow? Biochim. et Biophys. Acta (BBA) - Proteins Proteom. 2022, 1870, 140758. [Google Scholar] [CrossRef]
- Schaffer, L.V.; Millikin, R.J.; Miller, R.M.; Anderson, L.C.; Fellers, R.T.; Ge, Y.; Kelleher, N.L.; LeDuc, R.D.; Liu, X.; Payne, S.H.; et al. Identification and Quantification of Proteoforms by Mass Spectrometry. Proteomics 2019, 19, e1800361–e1800361. [Google Scholar] [CrossRef]
- Kaulich, P.T.; Tholey, A. Top-Down Proteomics: Why and When? Proteomics 2025, e202400338. [Google Scholar] [CrossRef]
- Cassidy, L.; Kaulich, P.T.; Tholey, A. Proteoforms expand the world of microproteins and short open reading frame-encoded peptides. iScience 2023, 26, 106069. [Google Scholar] [CrossRef]
- Poulos, R.C.; Hains, P.G.; Shah, R.; Lucas, N.; Xavier, D.; Manda, S.S.; Anees, A.; Koh, J.M.S.; Mahboob, S.; Wittman, M.; et al. Strategies to enable large-scale proteomics for reproducible research. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Costa, C.; Martínez-Bartolomé, S.; McClatchy, D.; Yates III, J.R. Improving proteomics data reproducibility with a dual-search strategy. Analytical Chemistry 2019, 92, 1697–1701. [Google Scholar] [CrossRef] [PubMed]
- Bacala, R.; Hatcher, D.W.; Perreault, H.; Fu, B.X. Challenges and opportunities for proteomics and the improvement of bread wheat quality. J. Plant Physiol. 2022, 275, 153743. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Yuan, H.; Gong, F.; Wu, X.; Wang, W. Protein Extraction Methods Shape Much of the Extracted Proteomes. Front. Plant Sci. 2018, 9, 802. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhu, L.; Tao, C.; Xie, Q.; Xu, X.; Chang, L.; Tan, Y.; Ding, G.; Li, H.; Wang, X. An improved protein extraction method applied to cotton leaves is compatible with 2-DE and LC-MS. BMC Genom. 2019, 20, 1–11. [Google Scholar] [CrossRef]
- Mujahid, H.; Pendarvis, K.; Reddy, J.S.; Nallamilli, B.R.R.; Reddy, K.R.; Nanduri, B.; Peng, Z. Comparative Proteomic Analysis of Cotton Fiber Development and Protein Extraction Method Comparison in Late Stage Fibers. Proteomes 2016, 4, 7. [Google Scholar] [CrossRef]
- Bose, U.; Broadbent, J.A.; Byrne, K.; Hasan, S.; Howitt, C.A.; Colgrave, M.L. Optimisation of protein extraction for in-depth profiling of the cereal grain proteome. J. Proteom. 2019, 197, 23–33. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Z.; Mao, J.; Zhang, Y.; Zhou, L.; Wei, J.; Liu, Y.; Wang, L.; Liu, F.; Cui, F. Accelerating wheat (Triticum aestivum) proteomics using a tandem digestion approach. J. Cereal Sci. 2025, 123. [Google Scholar] [CrossRef]
- Rahman, M.; Liu, L.; Barkla, B.J. A Single Seed Protein Extraction Protocol for Characterizing Brassica Seed Storage Proteins. Agronomy 2021, 11, 107. [Google Scholar] [CrossRef]
- International Wheat Genome Sequencing Consortium; Alaux, M. ; Rogers, J.; Letellier, T.; Flores, R.; Alfama, F.; Pommier, C.; Mohellibi, N.; Durand, S.; Kimmel, E.; et al. Linking the International Wheat Genome Sequencing Consortium bread wheat reference genome sequence to wheat genetic and phenomic data. Genome Biol. 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Yu, J.; Zhao, M.; Wang, X.; Tong, C.; Huang, S.; Tehrim, S.; Liu, Y.; Hua, W.; Liu, S. Bolbase: a comprehensive genomics database for Brassica oleracea. BMC Genom. 2013, 14, 664–664. [Google Scholar] [CrossRef]
- Chen, H.; Wang, T.; He, X.; Cai, X.; Lin, R.; Liang, J.; Wu, J.; King, G.; Wang, X. BRAD V3.0: an upgraded Brassicaceae database. Nucleic Acids Res. 2021, 50, D1432–D1441. [Google Scholar] [CrossRef]
- Yu, J.; Jung, S.; Cheng, C.-H.; Lee, T.; Zheng, P.; Buble, K.; Crabb, J.; Humann, J.; Hough, H.; Jones, D.; et al. CottonGen: The Community Database for Cotton Genomics, Genetics, and Breeding Research. Plants 2021, 10, 2805. [Google Scholar] [CrossRef]
- Ma, S.; Wang, M.; Wu, J.; Guo, W.; Chen, Y.; Li, G.; Wang, Y.; Shi, W.; Xia, G.; Fu, D.; et al. WheatOmics: A platform combining multiple omics data to accelerate functional genomics studies in wheat. Mol. Plant 2021, 14, 1965–1968. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, S.; Wei, L.; Huang, Y.; Liu, D.; Jia, Y.; Luo, C.; Lin, Y.; Liang, C.; Hu, Y.; et al. BnIR: A multi-omics database with various tools for Brassica napus research and breeding. Mol. Plant 2023, 16, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Dai, F.; Chen, J.; Zhang, Z.; Liu, F.; Li, J.; Zhao, T.; Hu, Y.; Zhang, T.; Fang, L. COTTONOMICS: a comprehensive cotton multi-omics database. Database 2022, 2022. [Google Scholar] [CrossRef]
- Duncan, O.; Trösch, J.; Fenske, R.; Taylor, N.L.; Millar, A.H. Resource: Mapping the Triticum aestivum proteome. Plant J. 2017, 89, 601–616. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.H.M.; Cho, K.; Choi, J.-S.; Bae, K.-H.; Komatsu, S.; Uozumi, N.; Woo, S.H. The wheat chloroplastic proteome. J. Proteom. 2013, 93, 326–342. [Google Scholar] [CrossRef]
- Jacoby, R.P.; Millar, A.H.; Taylor, N.L. Opportunities for wheat proteomics to discover the biomarkers for respiration-dependent biomass production, stress tolerance and cytoplasmic male sterility. J. Proteom. 2016, 143, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Takemori, N.; Takemori, A.; Matsuoka, K.; Morishita, R.; Matsushita, N.; Aoshima, M.; Takeda, H.; Sawasaki, T.; Endo, Y.; Higashiyama, S. High-throughput synthesis of stable isotope-labeled transmembrane proteins for targeted transmembrane proteomics using a wheat germ cell-free protein synthesis system. Mol. Biosyst. 2014, 11, 361–365. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwängler, B.; Üresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Keller, U.A.D.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Fels, U.; Gevaert, K.; Van Damme, P. Proteogenomics in Aid of Host–Pathogen Interaction Studies: A Bacterial Perspective. Proteomes 2017, 5, 26. [Google Scholar] [CrossRef] [PubMed]
- Balotf, S.; Wilson, R.; Tegg, R.S.; Nichols, D.S.; Wilson, C.R. Shotgun Proteomics as a Powerful Tool for the Study of the Proteomes of Plants, Their Pathogens, and Plant–Pathogen Interactions. Proteomes 2022, 10, 5. [Google Scholar] [CrossRef]
- Elmore, J.M.; Griffin, B.D. ; &; Walley, J.W. Advances in functional proteomics to study plant-pathogen interactions. Current Opinion in Plant Biology 2021, 63.
- Ravikumar, V.; Jers, C.; Mijakovic, I. Elucidating Host–Pathogen Interactions Based on Post-Translational Modifications Using Proteomics Approaches. Front. Microbiol. 2015, 6, 1313. [Google Scholar] [CrossRef]
- Singh, S.; Hegde, M.; Kaur, I.; Adlakha, N. Temporal proteome profiling of Botrytis cinerea reveals proteins involved in plant invasion and survival. Sci. Rep. 2025, 15, 1–11. [Google Scholar] [CrossRef]
- Gao, W.; Long, L.; Xu, L.; Lindsey, K.; Zhang, X.; Zhu, L. Suppression of the homeobox gene HDTF1 enhances resistance to Verticillium dahliae and Botrytis cinerea in cotton. J. Integr. Plant Biol. 2015, 58, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhou, J.; Zhu, D.; Gao, C. TurboID-based proximity labeling accelerates discovery of neighboring proteins in plants. Trends Plant Sci. 2023, 29, 383–384. [Google Scholar] [CrossRef]
- Shi, L.; Ferrando, T.M.; Villanueva, S.L.; Joosten, M.H.; Vleeshouwers, V.G.; Bachem, C.W. Protocol to identify protein-protein interaction networks in Solanum tuberosum using transient TurboID-based proximity labeling. STAR Protoc. 2023, 4, 102577. [Google Scholar] [CrossRef]
- Stevenson, S.E.; Chu, Y.; Ozias-Akins, P.; Thelen, J.J. Validation of gel-free, label-free quantitative proteomics approaches: Applications for seed allergen profiling. J. Proteom. 2009, 72, 555–566. [Google Scholar] [CrossRef]
- Kwon, D. The antibodies don’t work! The race to rid labs of molecules that ruin experiments. Nature 2024, 635, 26–28. [Google Scholar] [CrossRef]
- FR, T.; MOLEK, O. When antibodies mislead: the quest for validation. Nature 2020, 585, 313. [Google Scholar] [CrossRef]
- A Nierves, L.; Lin, T.-T.; Moradian, A.; Shen, Q.; Sechi, S.; MacCoss, M.J.; Qu, J.; E van Eyk, J.; Hoofnagle, A.N.; Qian, W.-J. Biomarkers, Proteoforms, and Mass Spectrometry–Based Assays for Diabetes Clinical Research. J. Clin. Endocrinol. Metab. 2025, 110, 1514–1523. [Google Scholar] [CrossRef]
- Su, T.; Fellers, R.T.; Greer, J.B.; LeDuc, R.D.; Thomas, P.M.; Kelleher, N.L. Proteoform-predictor: Increasing the Phylogenetic Reach of Top-Down Proteomics. J. Proteome Res. 2025, 24, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, F.; Alharbi, S.; Alotaibi, M.; Al Mosallam, M.; Motawei, M.; Alrajhi, A. Wheat omics: Classical breeding to new breeding technologies. Saudi J. Biol. Sci. 2021, 28, 1433–1444. [Google Scholar] [CrossRef]
- Ma, X.-F.; Fang, P.; Gustafson, J.P. Polyploidization-induced genome variation in triticale. Genome 2004, 47, 839–848. [Google Scholar] [CrossRef]
- Adams, K.; Wendel, J. Novel patterns of gene expression in polyploid plants. Trends Genet. 2005, 21, 539–543. [Google Scholar] [CrossRef]
- Leitch, A.R.; Leitch, I.J. Genomic Plasticity and the Diversity of Polyploid Plants. Science 2008, 320, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Xiao, X.; Chou, K.-C. pLoc_bal-mPlant: Predict Subcellular Localization of Plant Proteins by General PseAAC and Balancing Training Dataset. Curr. Pharm. Des. 2019, 24, 4013–4022. [Google Scholar] [CrossRef]
- Wan, S.; Mak, M.-W.; Kung, S.-Y. Gram-LocEN: Interpretable prediction of subcellular multi-localization of Gram-positive and Gram-negative bacterial proteins. Chemom. Intell. Lab. Syst. 2017, 162, 1–9. [Google Scholar] [CrossRef]
- Meinken, J.; Asch, D.K.; Neizer-Ashun, K.A.; Chang, G.H.; Cooper JR, C.R. ; &; Min, X. Computational Molecular Biology 2014, 4. [Google Scholar]
- Sukumaran, A.; Woroszchuk, E.; Ross, T.; Geddes-McAlister, J. Proteomics of host–bacterial interactions: new insights from dual perspectives. Can. J. Microbiol. 2021, 67, 213–225. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, Y.; Zou, J.; Meng, J.; Wang, J. Comparative proteomic study on Brassica hexaploid and its parents provides new insights into the effects of polyploidization. J. Proteom. 2015, 112, 274–284. [Google Scholar] [CrossRef]
- Islam, N.; Tsujimoto, H.; Hirano, H. Proteome analysis of diploid, tetraploid and hexaploid wheat: Towards understanding genome interaction in protein expression. Proteomics 2003, 3, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Rampitsch, C.; Bykova, N.V. Advances in plant proteomics toward improvement of crop productivity and stress resistancex. Front. Plant Sci. 2015, 6, 209–209. [Google Scholar] [CrossRef]
- Song, Y.-C.; Das, D.; Zhang, Y.; Chen, M.-X.; Fernie, A.R.; Zhu, F.-Y.; Han, J. Proteogenomics-based functional genome research: approaches, applications, and perspectives in plants. Trends Biotechnol. 2023, 41, 1532–1548. [Google Scholar] [CrossRef] [PubMed]
- Daba, S.D.; Liu, X.; Aryal, U.; Mohammadi, M. A proteomic analysis of grain yield-related traits in wheat. AoB PLANTS 2020, 12, plaa042. [Google Scholar] [CrossRef]
- Shu, J.; Ma, X.; Ma, H.; Huang, Q.; Zhang, Y.; Guan, M.; Guan, C. Transcriptomic, proteomic, metabolomic, and functional genomic approaches of Brassica napus L. during salt stress. PLOS ONE 2022, 17, e0262587. [Google Scholar] [CrossRef]
- Mi, W.; Liu, Z.; Jin, J.; Dong, X.; Xu, C.; Zou, Y.; Xu, M.; Zheng, G.; Cao, X.; Fang, X.; et al. Comparative proteomics analysis reveals the molecular mechanism of enhanced cold tolerance through ROS scavenging in winter rapeseed (Brassica napus L.). PLOS ONE 2021, 16, e0243292. [Google Scholar] [CrossRef]
- Koh, J.; Chen, G.; Yoo, M.-J.; Zhu, N.; Dufresne, D.; Erickson, J.E.; Shao, H.; Chen, S. Comparative Proteomic Analysis ofBrassica napusin Response to Drought Stress. J. Proteome Res. 2015, 14, 3068–3081. [Google Scholar] [CrossRef] [PubMed]
- Lan, M.; Li, G.; Hu, J.; Yang, H.; Zhang, L.; Xu, X.; Liu, J.; He, J.; Sun, R. iTRAQ-based quantitative analysis reveals proteomic changes in Chinese cabbage (Brassica rapa L.) in response to Plasmodiophora brassicae infection. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Jiang, J.; Shi, Z.; Ma, F.; Liu, K. Identification of key proteins related to high-quality fiber in Upland cotton via proteomics analysis. Plant Cell Rep. 2022, 41, 893–904. [Google Scholar] [CrossRef]
- Xiao, S.; Liu, L.; Zhang, Y.; Sun, H.; Zhang, K.; Bai, Z.; Dong, H.; Liu, Y.; Li, C. Tandem mass tag-based (TMT) quantitative proteomics analysis reveals the response of fine roots to drought stress in cotton (Gossypium hirsutum L.). BMC Plant Biol. 2020, 20, 1–18. [Google Scholar] [CrossRef]
- Zheng, M.; Meng, Y.; Yang, C.; Zhou, Z.; Wang, Y.; Chen, B. Protein expression changes during cotton fiber elongation in response to drought stress and recovery. Proteomics 2014, 14, 1776–1795. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, Y.; Khan, A.; Ullah, N.; Li, Z.; Zaheer, S.; Zhou, R.; Zhang, Z. Quantitative Proteomics-Based Analysis Reveals Molecular Mechanisms of Chilling Tolerance in Grafted Cotton Seedlings. Agronomy 2022, 12, 1152. [Google Scholar] [CrossRef]
- Peng, Z.; He, S.; Gong, W.; Xu, F.; Pan, Z.; Jia, Y.; Geng, X.; Du, X. Integration of proteomic and transcriptomic profiles reveals multiple levels of genetic regulation of salt tolerance in cotton. BMC Plant Biol. 2018, 18, 128. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Summary of proteomic studies on heat stress tolerance in wheat, cotton and oilseed Brassica during the last five years (2019–2025).
Table 1.
Summary of proteomic studies on heat stress tolerance in wheat, cotton and oilseed Brassica during the last five years (2019–2025).
| Crop | Species | Genotypes/ varieties/ lines |
Growth stages | Treatment condition | Tissue/organ | Techniques | Validation status | Total no. of DAPs/ protein groups | Unique DAPs/peptides | Common proteins | Summary | Key genes/proteins/Enzymes | Reference |
| Wheat | Triticum aestivum L. | Zhoumai 36 | Tetrad stage, binuclear stage, and trinuclear stage | Field condition | Anther | TMT; Peptide identification by nano UPLC–MS/MS | qRT-PCR | 2532 DAPs | Twenty-seven, 157, and 2348 DAPs in tetrad, binuclear, and trinuclear stages, respectively | Under HS, in the tetrad, binuclear and trinuclear stages 27, 157 and 2348 DAPs were identified, respectively. HS-disrupts different signaling pathways, including MAPK signaling pathway, phosphatidylinositol biosynthesis pathway, starch and sucrose synthesis pathway and flavinoid biosynthesis pathway, due to downregulation of related proteins resulting male sterility. | TraesCS4B02G193500.2 (thermosensitive male sterile 1), TraesCS7A02G272400.3, TraesCS4B02G193500.2 (thermosensitive male sterile 1) | [30] | |
| KX3302 (high gluten) and BN 208 (medium gluten | Maturity | Steam at 100–120 °C | Wheat flour | 4D label-free | 3783 proteins (2124 DAPs) | 2861 | Storage protein in wheat grain is associated with celiac disease, an autoimmune disease. Wheat genotypes with high- and medium-gluten have α-gliadin and HMW-GSs, and γ-gliadin and LMW-GSs as main DAPs related to the disease, respectively. Heat stress during dough stage reduces the abundance of these proteins in both genotypes resulting in improved diseases. | N/A | [97] |
||||
| Xinchun 9 | Maturity | 38 °C for 1, 2, 8, and 24 h. | Spike | Data Independent Acquisition (DIA) | qRT-PCR | 78,885 peptides | 19,503 peptides | Proteins associated with proline synthesis, ABA signaling, HSPs, and MADS TFs play an important role in thermotolerance in wheat. |
TraesCS7D02G483200 |
[51] | |||
| Tainong 18 | Maturity | Field condition | Leaf | iTRAQ | 4411 (139 DAPs) | Grain weight and grain yield increased due to delayed sowing due to improved electron transport in photosynthetic channel by upregulation of proteins-related to photosynthesis, antioxidant activities and stress. Delayed sowing increases photosynthetic electron transport associated with proteins such as PsbH, PsbR, and PetB under HS, which supports high photosynthesis to increase HS tolerance. | [98] |
||||||
| BWL4444 (HD2967+ Yr10; tolerant) | Anthesis | 35/24 ℃ | Grain | 2D-PAGE; MALDI-TOF/MS | qRT-PCR | 153 DAPs | Day-night HS suppresses stress tolerance gene expression due to activation of multiple pathways for proteins associated with protein translation, gliadins, low-and high- molecular weight glutenin, glycolysis, photosynthesis and defense. Downregulation of proteins associated with photosynthesis, glycolysis, metabolic pathways and improper protein folding increase HS-sensitivity and decreases grain weight, yield and quality. | [99] | |||||
| HD3086 (tolerant) and BT-Schomburgk (sensitive) | Flowering and grain development stages | 32 ℃ and 38 ℃ for 2 h |
Leaf | 2D-PAGE; MALDI-TOF/MS | N/A | Approximately 23,000 DAPs | Twenty-two and 11 unique DAPs in tolerant and sensitive varieties, respectively | Twenty | Mitogen-activated protein kinase (MAPK) positively increases HS tolerance by increasing proline, H2O2, gene expression of antioxidant enzymes, defence and osmolytes and HSPs. MAPK also regulates grain quality under HS. | MAPK genes | [100] | ||
| Chinese Spring | Grain filling | 30 °C and 40 °C |
Grains | Tandem mass tags (TMT) | N/A | 3742 (297 and 461 DAPs at 30 and 40 ℃, respectively) | 150 | Posttranscriptional regulation plays important role in mild HS (30 ℃), while amino acid frequency is important for protein expression during severe HS (40 ℃). Under HS, higher frequency of AAG codon was found to alter HS-induced protein expression. Further, though ribosomal protein influenced HS induced proteins, but no transcriptional regulation was evident under 30 ℃. | [101] | ||||
| Unnat Halna | Seedling | 38 °C for 4 h | Leaf | 2-DE | Vacuum-assisted Agrobacterium transformation method | 55 DAPs (10 and 29, upregulated and downregulated, respectively) | Heat stress induced 2-cysteine peroxiredoxin (2CP) increases HS-tolerance by increasing chlorophyll a and b content and decreasing H2O2 and ROX concentrations. The 2CP in wheat also interacts with protochlorophyllide reductase b,TaPORB, and play important role in chlorophyl biosynthesis. | Ta2CP | [102] | ||||
| HD2985 (tolerant) and HD2329 (sensitive) | Pollination and grain filling | 38 °C for 2 h | Leaf, stem, and spike | iTRAQ | qRT-PCR | 318 groups | 17 DAPs associated with stress associated active proteins | 9,425 (3,600 and 5825 upregulated and downregulated, respectively) | Heat shock proteins, including HSP20, HSP26, and HSC70, signaling molecules including MAPKs and CDPKs, and antioxidant enzymes, including SOD, CAT, and APX, play an important role in HS-tolerance. On the other hand, upregulated oleosin, globulin 3A, 3B, and gamma gliadin, and α/β amylases regulate grain quality of wheat under HS. | Carboxylase enzyme | [103] |
||
| Gaocheng 8901 | Flowering to maturity | 40 °C for 2 h | Milled grain | iTRAQ; LC-ESI/MS | qRT-PCR; immunobloting | 207 DAPs | Kernel weight and starch content in wheat decreases due to protein-protein interaction of HS-induced proteins, including elicitor responsive gene 3, brassinosteroid-insensitive 1, chaperone protein, histone cell cycle regulator and pre-mRNA processing factor. | [104] | |||||
| Cotton | Gossypium hirsutum | GH-Hamaliya (tolerant) and GH-Hamaliya (susceptible) | Flowering | 45/32 °C | Leaf | Label-free | N/A | 8005 | 494 | 13 | During HS, photosynthesis decreases due to decrease of intercellular CO2 concentration, stomatal conduction and water status, chlorophyll reduces due to decrease lipid peroxidation of thylakoid and chloroplast membrane. However, increased proline content supports HS-tolerance. More than 8000 DAPs were identified under HS. Upregulation of proteins associated with ROS scavenging, ATP synthesis, and major latex-like proteins, beta-glucosidase and HSP play an important role in HS-tolerance. | [105] | |
| TM-1 and W0 | Seedling | 42 ℃ for 24 h | Leaf | SDS-PAGE | Yeast two-hybrid assay for protein-protein interaction | 2426 | 728 | Three | Reduction of expression of GhHSP24.7 plays an important role in heat and drought stress tolerance through increasing ROS scavenging, decreasing acetylation, stomatal conductance and H2O2 content. | GhHSP24.7 | [106] | ||
| Sicot 71 | Reproductive | 40/30 °C for five days | Leaf | SWATH-MS | 880 | 728 | Two | In mature pollen, HS suppresses translational processes by disturbing metabolic processes, such as starch biosynthesis, glycogen catabolism, and xyloglucon processes. However, transcription remains active. In all pollen development stages, HSPs and peptidylprolyl isomerase were common. | [53] | ||||
| Gossypium hirsutum | Sicot 71 | Tetrads and binucleate stage | 36/25 ℃ and 40/30 ℃ for five days | Pollen grain | Label-free | 876 | 106 DAPs | Heat stress reduces the reproductive capacity of pollen grain of cotton decreases by decreasing the pollen grain size at the tetrad stage, increasing sugar percentage in the pollen grain and decreasing pollen viability. Upregulation of HSPs, uncluding, HSP70 kDa, proteins related to starch and sucrose metabolism, and downregulation of late embryogenesis abundant proteins contribute to the HS tolerance in cotton's pollen. | [107] | ||||
| Gossypium hirsutum | Sikang 3 | Bolling stage | 38 °C and 40% field capacity | Boll shell | 2D-PAGE; label-free quantitation | 3,094 and 3,049 | 2,927 | Under drought and HS, Bt gene expression in cotton caused increased starch hydrolysis ability and decreased fructose and glucose content. Proteins associated with protein export pathways, particularly signal recognition particles, play a crucial role in restricting the transportation of nascent peptide chain into the endoplasmic reticulum due to HS, resulting in decrease of Bt-insecticidal protein in cotton. | [108] | ||||
| Gossypium robinsonii | Australian wild cotton | Pollen development stages (tetrad, uninucleate and binucleate) | 36/25 ℃ and 40/30 ℃ for five days | Pollen grain | Nano LC-MS/MS; IDA-DIA SWATH-MS | 2704 | 422, 489 and 94 DAPs at 36 ℃ and 297, 157 and 61 DAPs at 40 ℃ in tetrad, uninucleate and binucleate, respectively | 196 DAPs | 36 and 11 at 36 and 40 °C, respectively | Heat stress at the late stage of pollen development reduces translational response and protein abundance. Plasmodesma related proteins play an important role in protein transport and cell-cell communication at the tetrad stage of pollen under HS. Downregulation of Rab proteins play crucial role in thermotolerance by inhibiting protein transport. | [109] | ||
| Brassica | Brassica juncea | Seedling | 45 °C for 4 and 8 h | Leaf | MALDI-TOF-MS | 22954 DAPs | 81 DAPs | Differentially accumulated proteins: 77 at 4h, 81 at 8h, 74 at both 4 and 8h | Under 4h and 8h of HS, 119 and 81 DAPs, 532 and 570 peptide sequences were identified, respectively. During 8h HS, accumulation of HSP, including HSPs and HS-induced TF A-4a increased. On the other hand, during 4h, MuTL like-protein-1, Cytochrome P450 85A1, CDT1_C domain-containing protein and B3 domain containing proteins, putative calcium-transporting ATPase 11 and an uncharacterized protein accumulation increased, and ribosome inactive protein accumulation decreased. | [110] | |||
| Brassica juncea | BPR 543-2 | 37 ℃, 3 h interval for five days | Pericarp | Label-free | 1078 | 209 DEP | 78 | N/A | Under HS and combination of salt-stress and HS, 367 and 209 DAPs, including HSPs, proteins related to cell division, protein disulfide isomerase, calcium binding proteins and protease, were highly accumulated, respectively. | [111] | |||
| Brassica campestris | WS-1 | Reproductive stage | 40/30 ℃ | Leaf | TMT | qRT-PCR | 2806 DAPs | 1787 DAPs | 1022 | Glucose transporter gene, BccrGLU1, improves HS-tolerance by increasing ROS scavenging, and reducing glutathione content and ratio of glutathione/oxidized glutathione. | BccrGLU1 | [112] |
N/A = not applicable; TMT = tandem mass tag, UPLC = ultra-performance liquid chromatography, MS = mass spectrometry, 4D = four dimensional, iTRAQ = isobaric tags for relative and absolute quantitation, 2-DE = two- dimensional gel electrophoresis, SDS–PAGE = sodium dodecyl sulfate–polyacrylamide gel electrophoresis, LC = liquid chromatography, ESI = electrospray ionization, SWATH = sequential window acquisition of all theoretical mass spectra, MALDI = matrix assisted laser desorption/ionization, TOF = time-of-fight, qRT-PCR = quantitative real-time reverse-transcription-polymerase chain reaction, DAPs = differentially abundant proteins; TF = transcription factors, and HS = heat stress.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



