
Submitted:
14 October 2025
Posted:
15 October 2025
You are already at the latest version
Abstract
Dementia, characterized by a progressive decline in cognitive function, is a growing global health concern due to the aging population. It affects memory, executive function, language, and other cognitive capacities, significantly impairing daily life. Despite the growing burden, effective preventive and therapeutic strategies remain elusive, emphasizing the urgent need for novel interventions. Recent advances underscore the pivotal role of neuroinflammation in dementia pathogenesis, particularly in Alzheimer's disease (AD). Chronic activation of immune cells within the central nervous system, such as microglia transition from a homeostatic to a pro-inflammatory state called Disease-associated microglia status 1 and 2 (DAM1 or DAM2), exacerbates neurodegeneration, creating a self-perpetuating cycle of inflammation and cognitive decline. This review focuses on emerging research exploring the cGAS-STING pathway’s role in dementia, examining its potential as a diagnostic and therapeutic target. The cGAS-STING pathway, integral to innate immune responses, may contribute to the chronic neuroinflammation seen in neurodegenerative diseases. By targeting this pathway, new strategies could mitigate the inflammatory processes that drive neuronal loss, offering a promising avenue for therapeutic development in dementia.
Keywords:
neurodegeneration
; dementia
; Alzheimer’s Disease
; immune response
; Disease Associated Microglia
; cGAS-STING pathway
1. Introduction
Dementia is a syndrome characterized by the progressive decline in cognitive capacities such as memory, executive function, language, and visuospatial skills, ultimately impairing daily functioning without affecting alertness or consciousness. At its core, dementia is driven by the loss of neurons and synaptic connections within the brain, which disrupts neural circuits necessary for cognitive integrity (Spires-Jones and Hyman, 2014). The incidence of dementia has risen significantly due to global aging trends, with an increase in cases from 20.2 million in 1990 to 43.8 million in 2016 (Collaborators, 2019). Projections estimate that by 2050, the number of people living with dementia will escalate to over 152.8 million globally, reflecting both increased life expectancy and the persistent lack of effective preventive measures (2020, International, 2024).
Recent advances in our understanding of dementia pathogenesis have highlighted the significant contribution of neuroinflammation to disease progression, particularly in Alzheimer’s disease (AD) and other neurodegenerative conditions (Heneka et al., 2015). Neuroinflammation is characterized by the activation of immune cells within the central nervous system (CNS), notably microglia and astrocytes, which can release pro-inflammatory cytokines, chemokines, and reactive oxygen species (ROS). While acute inflammation is necessary for CNS homeostasis and repair, chronic neuroinflammation is implicated in the exacerbation of neuronal damage, ultimately leading to cognitive decline (Ransohoff, 2016a). Microglia, the resident immune cells of the CNS, play a central role in this process. In response to pathogenic stimuli such as amyloid-β (Aβ) plaques or tau aggregates, microglia undergo a phenotypic transformation from a homeostatic to a pro-inflammatory state, releasing a cascade of inflammatory mediators (Salter and Stevens, 2017). Emerging evidence suggests that sustained activation of microglia in dementia can lead to a detrimental cycle, whereby neuroinflammation promotes further neurodegeneration, creating a self-amplifying loop (Heppner et al., 2015).
2. Dementia Classification and Neuroinflammation
Dementia is a complex disorder with various etiologies, often distinguished by the type of protein aggregation and its localization in the brain. However, the emerging role of neuroinflammation as a common thread across dementia types has garnered increasing attention. Inflammatory processes, including activation of microglia and astrocytes, have been implicated in the progression of these disorders, suggesting that targeting neuroinflammation could offer therapeutic potential (Heneka et al., 2015). The most prevalent forms of dementia—Alzheimer’s Disease (AD), Vascular Dementia (VaD), Lewy Body Dementia (LBD), and Frontotemporal Dementia (FTD)—are associated with distinct pathophysiological mechanisms, yet all are linked to chronic neuroinflammatory processes, which exacerbate neuronal damage and cognitive (Moyse et al., 2022, Zhang et al., 2023).
2.1. Vascular Dementia and Neuroinflammation
Vascular Dementia (VaD) is the second most common type of dementia and is characterized by cognitive deficits resulting from cerebrovascular disease, such as cortical infarctions and white matter lesions. The subtypes of VaD include post-stroke dementia (PSD), multi-infarct dementia (MID), and subcortical ischemic vascular dementia (SIVD) (Venkat et al., 2015). While the primary mechanism of VaD is thought to be ischemia-induced neuronal damage, recent evidence suggests that neuroinflammation plays a pivotal role in VaD progression. In particular, pro-inflammatory cytokines such as IL-6 and TNF-α, as well asactivated microglia, are known to disrupt the blood-brain barrier (BBB) exacerbate cerebral ischemia and contribute to the progressive loss of white matter integrity (Tian et al., 2022, Love and Miners, 2016). Agents targeting microglial activation such as colony-stimulating factor-1 receptor (CSF1R) inhibitor has shown promise in cerebrovascular disease model and postmortem brain tissue (Askew et al., 2024).
Moreover, advanced neuroimaging techniques, such as magnetic resonance imaging (MRI) have provided insights into the link between vascular pathology and neuroinflammation. For instance, MRI of VaD patients often reveals perivascular lesions and leukoaraiosis, which are closely associated with elevated levels of microglial activation and inflammatory marker expression (Custodero et al., 2022, Kalaria, 2016). Chronic neuroinflammation in VaD has also been associated with systemic risk factors such as hypertension and diabetes, further implicating the immune system in the progress of vascular brain damage (Kalaria, 2016). These findings underscore the potential of therapies targeting inflammatory pathways in VaD to prevent further cognitive impairment and brain atrophy.
2.2. Lewy Body Dementia and Neuroinflammation
Lewy body dementia (LBD) is characterized by the aggregation of α-synuclein into Lewy bodies, predominantly affecting the brainstem, cortex, and limbic regions. While α-synuclein aggregates are central to the pathology of LBD, mounting evidence suggests that neuroinflammation accelerates disease progression. Activated microglia have been observed surrounding Lewy bodies, where they release pro-inflammatory cytokines such as IL-1β and TNF-α, which may contribute to neuronal death and synaptic dysfunction (Loveland et al., 2023). Moreover, clinical studies reveal a correlation between the elevated neuroinflammatory markers and the severity of cognitive and motor impairments in LBD patients (Surendranathan et al., 2018, Vrillon et al., 2024), suggesting that inflammation exacerbates the neurodegenerative progress. Therapeutic approaches targeting pathways that regulate innate immune response, for instance, cGAS-STING pathway may be considered as potential treatments for LBD as the correlation with cGAS-STING pathway and synuclein pathology has been reported (Hinkle et al., 2022, Stoll and Sortwell, 2022).
2.3. Frontotemporal Dementia and Neuroinflammation
Frontotemporal dementia (FTD) involves progressive degeneration of the frontal and temporal lobes, with marked neuronal atrophy linked to proteinopathies such as tau or TDP-43 aggregates. Inflammatory processes are implicated in the neurodegeneration observed in FTD. Studies using PET imaging have revealed that increased microglial activation in patients with FTD and correlates with disease progression and severity of symptoms (Bright et al., 2019, Bevan-Jones et al., 2020, Zhang, 2015).
Moreover, genetic evidence further supports the link between inflammation and FTD. For instance, mutations in the progranulin gene (GRN), a known risk factor for familial FTD, impair anti-inflammatory processes and lead to enhanced neuroinflammation (Baker et al., 2006). Similary, C9orf72 gene disrupt autophagy and immune regulation, resulting in increased neuroinflammation and microglial activation in FTD (O’Rourke et al., 2016). Mutations in the microtubule- associated protein tau (MAPT) gene have been shown to drive neuroiniflammation by promoting tau pathology and ninducing a reactive microglial state (Leyns et al., 2017). A critical gene for microglial function, TREM2, trigerring receptor expressed on myeloid cells 2 gene is known to exacerbate neurodegeneration by imparing the microglial response to inflammatory stimuli and phagocytosis (Ulland and Colonna, 2018) These findings have proved the exploration of therapeutic strategies aiming to mitigate neuroinflammatory responses by enhancing genes activity or modulating microglial states as potential interventions (Bright et al., 2019).
3. Distinctions Between Dementia and Alzheimer’s Disease in Pathophysiology and Inflammation
Dementia is an umbrella term encompassing a range of cognitive impairments that significantly affect memory, thinking, and behavior. Alzheimer’s disease (AD), the most common form of dementia, accounts for 60-80% of all dementia cases (Association, 2024). While all individuals with Alzheimer’s have dementia, not all individuals with dementia have AD, as dementia can arise from various etiologies, including vascular causes, Lewy body formations, or frontotemporal degeneration (Jack et al., 2018).
3.1. Pathophysiology of Alzheimer’s Disease Versus Other Dementia
In the case of AD, the classical hallmark features include the accumulation of amyloid-β (Aβ) plaques and tau tangles. These aggregates primarily affect the medial temporal lobe and neocortical regions, leading to synaptic dysfunction, neuronal death, and eventual brain atrophy (Hardy and Selkoe, 2002, Jack et al., 2018). Aβ and tau accumulation is believed to trigger a neuroinflammatory cascade of events, including the activation of microglia, the resident immune cells of the central nervous system, and the release of pro-inflammatory cytokines and chemokines, which further exacerbate neuronal damage (De Strooper and Karran, 2016, Heneka et al., 2015, Heppner et al., 2015).
In contrast, other forms of dementia have distinct etiological mechanisms. VaD is primarily caused by cerebrovascular damage, leading to ischemic events, small-vessel disease, and white matter lesions. This vascular damage disrupts blood-brain barrier integrity and function, trigerring inflammatory responses and neuronal injury that contribute to cognitive decline (Gorelick et al., 2011). LBD, on the other hand, is marked by the aggregation of α-synuclein into Lewy bodies, which are often surrounded by activated microglia releasing pro-inflammatory mediators (Surendranathan et al., 2018). FTD involves the frontal and temporal lobes atrophy and shows tau or TDP-43 aggregates, which also stimulate neuroinflammatory pathway (Bright et al., 2019).
3.2. Neuroinflammation as a Shared Mechanism Across Dementias and Implications
Although these forms of dementia have distinct etiologies, neuroinflammation is increasingly recognized as a shared pathophysiological mechanism across various types of dementia. In each case, microglial activation contributes to the release of pro-inflammatory cytokines, oxidative stress and chronic neurodegeneration. Understanding these shared and distinct mechanisms is essential for developing targeted interventions. AD progresses through well-defined stages, beginning with a preclinical phase, during which mild cognitive impairment (MCI) is often noted, characterized by subtle memory loss. As the disease advances, moderate Alzheimer’s results in significant impairments in reasoning, language, and recognition. The neuroinflammatory response, particularly through the activation of the cGAS-STING pathway, is implicated in driving this disease progression from early amyloid deposition to extensive tau pathology and brain atrophy (Mathur et al., 2017).
In the severe stages of AD, widespread neuroinflammation, sustained microglial activation, and the production of pro-inflammatory mediators contribute to a self-perpetuating cycle of neuronal death and cognitive decline (Cunningham, 2013, Tejera and Heneka, 2019). Interestingly, emerging evidence suggests that chronic systemic inflammation, stemming from infections, lifestyle factors, or aging itself, may influence microglial priming and exacerbate neurodegenerative processes (Ransohoff, 2016b, Bachiller et al., 2018).
Although no cure exists for AD or other forms of dementia, targeting neuroinflammatory pathways has emerged as a promising therapeutic strategy. Interventions aimed at modulating microglial activation, inhibiting pro-inflammatory cytokines, or suppressing the cGAS-STING pathway are under investigation for their potential to slow disease progression and preserve cognitive function (Huang et al., 2023, Paul et al., 2021).
4. Current Research and Findings in Dementia and Neuroinflammation
With an aging global population and the significant socio-economic burden dementia imposes on families and healthcare systems, research in dementia has expanded rapidly, especially in identifying early-stage risk factors and diagnostic methods. Among various pathological contributors, neuroinflammation has emerged as a central process in the pathogenesis of dementia. It is now recognized that chronic and excessive activation of the brain’s immune system contributes to neuronal damage and cognitive decline (Zhang et al., 2023). Microglial dysfunction, coupled with the activation of inflammatory signaling pathwyas, exacerbates the progression of AD and other dementias. Additionally, the cGAS-STING pathway is increrasinly seen as a major player in mediating these inflammatory responses, linking cytosolic DNA sensing to neuroinflammatory cascades (Huang et al., 2023, Guo et al., 2024, Liu et al., 2024, Sharma et al., 2024, Zhang et al., 2024).
4.1. Genetic Research and Inflammatory Pathways
Recent advances in genetic research have yielded significant insights into the genetic basis of dementia,. highlighting the role of genetic variants in inflammatory pathways. Genome-wide association studies (GWAS) continue to identify novel loci associated with dementia subtypes, highlighting the complex interplay between genetic predispositions, immune function and neuroinflammatory processes in dementia (Kunkle et al., 2019, Chia et al., 2021).
One of the most well-known genetic risk factors for AD is the apolipoprotein E ε4 allele (APOE-ε4), which not only influences amyloid-β deposition but also alters microglial reactivity. APOE-ε4 carriers exhibit increased expression ofpro-inflammatory cytokines in the presence of amyloid plaques, exacerbating neurodegeneration (Yin et al., 2023, Lee et al., 2023). Variants in the triggering receptor expressed on myeloid cells 2 (TREM2) gene, which modulates microglial activation, have been identified in populations with high dementia risk and associated with an increased risk for late-onset AD. TREM2 mutations impair the microglial response to amyloid-β, resulting in inefficient clearance of toxic aggregates and heightened neuroinflammation (Ulland and Colonna, 2018).The sialic acid-binding immunoglobulin-like lectins (CD33) gene and the membrane-spanning 4-domains subfamily A (MS4A) gene families suggest additional connections to microglial signaling. CD33 variants particularly regulate the immune inhibitory receptor functions of microglia, linking them to increased neuroinflammation (Siddiqui et al., 2019, Griciuc et al., 2013). ATP-binding cassette transporter subfamily A member 7 (ABCA7) is also known to alter the response of inflammation, leading to amyloid accumulation (Dib et al., 2021). Variants in the MS4A gene cluster, which regulate immune signaling, are associated with AD risk (Kunkle et al., 2019, Naj et al., 2011). Furthermore, mutations in genes such as MAPT (tau protein) and GRN (progranulin) in frontotemporal dementia (FTD) have been shown to modulate microglial activation and the production of pro-inflammatory cytokines, amplifying neuroinflammatory responses (Baker et al., 2006, Bright et al., 2019). In LBD, genes involved in α-synuclein processing, such as SNCA, also have indirect ties to neuroinflammation through their effects on microglial activation (Chia et al., 2021) (Table 1).
4.2. Biomarkers of Neuroinflammation
The detection of biomarkers that reflect neuroinflammatory processes is an evolving area of research. While traditional biomarkers for dementia, such as amyloid-β and tau, remain pivotal, the recognition of inflammation-related markers in cerebrospinal fluid (CSF) and blood holds promise for identifying early-stage dementia and tracking disease progression. In AD, elevated levels of pro-inflammatory cytokines, including interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), have been detected in both the CSF and plasma (Daniilidou et al., 2024). Additionally, soluble TREM2 (sTREM2) is being explored as a marker of microglial activation (Ewers et al., 2020). Elevated sTREM2 levels correlate with neuroinflammatory activity and may predict the progression of AD (Nabizadeh et al., 2024, Ewers et al., 2020). In FTD, increased levels of progranulin and inflammatory cytokines, such as IL-8, are being investigated as potential biomarkers of microglial dysfunction (Bright et al., 2019). However, similar to traditional biomarkers, inflammatory biomarkers in the CSF remain difficult to utilize clinically due to the invasive nature of lumbar punctures. As a result, efforts are being made to identify reliable blood-based biomarkers. Plasma levels of sTREM2 and cytokines, as well as microRNA profiles, have shown potential in reflecting neuroinflammatory processes in both AD and FTD.
4.3. Neuroimaging and Neuroinflammation
Neuroimaging techniques have evolved beyond structural assessments and now allow the visualization of neuroinflammation in vivo. Positron Emission Tomography (PET) using ligands that target the translocator protein (TSPO), which is upregulated in activated microglia, has emerged as a powerful tool for mapping neuroinflammation in various forms of dementia (Pan et al., 2024). In AD, TSPO-PET imaging has shown increased microglial activation in regions corresponding to amyloid-β deposition, particularly in the hippocampus and frontal cortex (Pan et al., 2024, Gouilly et al., 2022). Similarly, in FTD, TSPO binding correlates with microglial activation in regions undergoing neurodegeneration, such as the frontal and temporal lobes (Zhang, 2015). This technique also holds promise in VaD, where chronic neuroinflammation in response to cerebrovascular damage has been observed using TSPO-PET. Furthermore, advanced MRI techniques such as diffusion tensor imaging (DTI) and arterial spin labeling (ASL) are being used to assess white matter integrity and cerebral blood flow, respectively, in the context of neuroinflammatory damage. In LBD, neuroinflammation visualized through these techniques may help differentiate the disease from other neurodegenerative disorders (Loveland et al., 2023).
5. Current Treatment Methods
Current therapeutic approaches for dementia primarily aim at symptom management, offering only transient relief without addressing the underlying pathophysiology. The predominant treatments, including cholinesterase inhibitors and NMDA receptor antagonists, provide modest symptomatic benefits but do little to alter the disease progression. This limitation stems from the complex etiology of dementia, wherein neuroinflammation plays a pivotal role in driving neurodegeneration (Frozza et al., 2018).
5.1. cGAS-STING Pathway and Neuroinflammation in Dementia: A Potential Therapeutic Target
The cGAS-STING pathway represents a crucial component of the body’s innate immune system, functioning primarily as a sensor for cytosolic DNA. Cyclic GMP-AMP synthase (cGAS) detects aberrant DNA within the cytoplasm—whether of exogenous origin, such as viral or bacterial DNA, or endogenous, such as mitochondrial or nuclear DNA released during cellular stress or damage. Upon binding to this DNA, cGAS catalyzes the synthesis of cyclic GMP-AMP (cGAMP), a second messenger that subsequently binds to and activates STING (Stimulator of Interferon Genes) (Ablasser and Chen, 2019). This activation triggers downstream signaling through the TANK-binding kinase 1 (TBK1), leading to the phosphorylation of the transcription factor interferon regulatory factor 3 (IRF3) and the subsequent induction of Type I interferon (IFN) and other pro-inflammatory cytokines (Mathur et al., 2017) (Figure 1). In the context of neurodegenerative diseases, such as AD and other types of dementia, the activation of the cGAS-STING pathway has gained increasing attention as a key mediator of neuroinflammation (Huang et al., 2023, Paul et al., 2021). Mitochondrial dysfunction, which is well-established in neurodegenerative conditions, can lead to the release of mitochondrial DNA (mtDNA) into the cytoplasm, where it serves as a potent activator of cGAS (Mathur et al., 2017). This activation may exacerbate chronic neuroinflammation, a hallmark of neurodegenerative diseases. Additionally, nuclear DNA instability, which may occur during cellular senescence or in the presence of oxidative stress, can also trigger the cGAS-STING pathway, further promoting neuroinflammatory responses (Figure 1).
Double-stranded DNA (dsDNA) from fragmented genome or mitochondria by DNA damage, or viral genomes activates the cytosolic sensor cGAS. This leads to the production of cyclic GMP-AMP (cGAMP), which binds to the ER membrane protein STING. Upon activation, STING translocates to the Golgi, where it triggers a signaling cascade involving TANK-binding kinase 1 (TBK1), leading to the phosphorylation of interferon regulatory factor 3 (IRF-3). Additionally, nuclear factor-kappa B (NF-κB) signaling is activated through the phosphorylation and degradation of IκB1. Both pathways induce the transcriptional activation of pro-inflammatory genes and interferons, amplifying the inflammatory response and contributing to neuronal damage in dementia by the activation of disease-associated microglia (DAMs). This feedback loop between DAMs and neuroinflammation potentially exacerbates the progression of dementia.
Emerging evidence further suggests that cGAS-STING activation is involved in the progression of several neurodegenerative diseases. In Parkinson’s disease (PD), mutations in PINK1 and Parkin genes impair mitochondrial autophagy (mitophagy), leading to the accumulation of damaged mitochondria and the release of mtDNA, which activates the cGAS-STING pathway (Sliter et al., 2018). Mouse models of α-synucleinopathies, which mimic the neuropathology of PD, show specific activation of cGAS-STING in the nigrostriatal regions, accompanied by elevated cytokine levels and enhanced neuroinflammation (Hinkle et al., 2022). In amyotrophic lateral sclerosis (ALS), TDP-43 protein aggregates have been found to disrupt mitochondrial integrity, leading to mtDNA release and subsequent cGAS-STING activation (Bright et al., 2021). A similar phenomenon has been observed in Huntington’s disease (HD), where cGAS upregulation has been identified in both murine models and human tissues (Jauhari et al., 2020, Paul et al., 2021).
Beyond these specific diseases, the cGAS-STING pathway also appears to play a significant role in broader neuroinflammatory processes. Elevated type I interferon levels, a downstream consequence of STING activation, have been documented in models of prion disease, promoting microglial activation and perpetuating neuroinflammatory cascades (Mathur et al., 2017). Post-mortem analyses of human CNS tissues from patients with AD, PD, ALS, and multiple sclerosis (MS) reveal increased STING protein levels in neurons and brain endothelial cells (Ferecskó et al., 2023, Marques et al., 2024, Govindarajulu et al., 2023). In vitro studies further demonstrate that mitochondrial stress induced by factors like palmitic acid leads to cytosolic DNA leakage and robust activation of the cGAS-STING axis, suggesting that metabolic dysfunction may be a common trigger of neuroinflammation (Bryant et al., 2022, Picca et al., 2020).
5.2. Therapeutic Targeting of cGAS-STING in Neurodegeneration
Given its pivotal role in promoting neuroinflammation, the cGAS-STING pathway is a promising therapeutic target for neurodegenerative diseases. Current strategies to inhibit this pathway focus on both cGAS and STING proteins. cGAS inhibitors aim to block the synthesis of cGAMP, thereby reducing downstream interferon production (Decout et al., 2021). These inhibitors typically target the active site of cGAS or disrupt its interaction with dsDNA. Although several small-molecule inhibitors have shown promise in preclinical models, none have progressed to clinical trials yet (Yu et al., 2024).
STING inhibition has also attracted attention, particularly in the context of cancer immunotherapy; however, its potential in neurodegenerative diseases remains largely unexplored. Most STING inhibitors aim to block the ligand-binding domain or interfere with post-translational modifications that enhance STING activity (Decout et al., 2021). Despite the lack of clinically approved inhibitors, research in this area continues to evolve, offering hope for future therapies targeting neuroinflammation in dementia and related disorders.
6. Conclusion
In summary, the cGAS-STING pathway plays a crucial role in mediating neuroinflammation in various neurodegenerative diseases, including dementia. Activation of this pathway, often triggered by mitochondrial or nuclear DNA leakage, initiates a pro-inflammatory cascade that accelerates neuronal damage and disease progression. While much remains to be understood about the precise mechanisms by which cGAS-STING contributes to neurodegeneration, the development of inhibitors targeting this pathway holds significant promise and various genetic prepositions known to be involved in neuroinflammation and neurodegeneration are worthwhile to be considered in the future research.
Acknowledgments
This work was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI22C2064). S.U.K was supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) R01 NS123456.
References
- 2020. 2020 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 16, 391-460. [CrossRef]
- ABLASSER, A. & CHEN, Z. J. 2019. cGAS in action: Expanding roles in immunity and inflammation. Science, 363, eaat8657. [CrossRef]
- ASKEW, K. E., BEVERLEY, J., SIGFRIDSSON, E., SZYMKOWIAK, S., EMELIANOVA, K., DANDO, O., HARDINGHAM, G. E., DUNCOMBE, J., HENNESSY, E., KOUDELKA, J., SAMARASEKERA, N., SALMAN, R. A., SMITH, C., TAVARES, A. A. S., GOMEZ-NICOLA, D., KALARIA, R. N., MCCOLL, B. W. & HORSBURGH, K. 2024. Inhibiting CSF1R alleviates cerebrovascular white matter disease and cognitive impairment. Glia, 72, 375-395. [CrossRef]
- ASSOCIATION, A. S. 2024. “What Is Dementia?” [Online]. Alzheimer’s Association. Available: https://www.alz.org/alzheimers-dementia/what-is-dementia [Accessed September 10 2024].
- BACHILLER, S., JIMÉNEZ-FERRER, I., PAULUS, A., YANG, Y., SWANBERG, M., DEIERBORG, T. & BOZA-SERRANO, A. 2018. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Frontiers in Cellular Neuroscience, 12. [CrossRef]
- BAKER, M., MACKENZIE, I. R., PICKERING-BROWN, S. M., GASS, J., RADEMAKERS, R., LINDHOLM, C., SNOWDEN, J., ADAMSON, J., SADOVNICK, A. D. & ROLLINSON, S. 2006a. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature, 442, 916-919. [CrossRef]
- BAKER, M., MACKENZIE, I. R., PICKERING-BROWN, S. M., GASS, J., RADEMAKERS, R., LINDHOLM, C., SNOWDEN, J., ADAMSON, J., SADOVNICK, A. D., ROLLINSON, S., CANNON, A., DWOSH, E., NEARY, D., MELQUIST, S., RICHARDSON, A., DICKSON, D., BERGER, Z., ERIKSEN, J., ROBINSON, T., ZEHR, C., DICKEY, C. A., CROOK, R., MCGOWAN, E., MANN, D., BOEVE, B., FELDMAN, H. & HUTTON, M. 2006b. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature, 442, 916-9. [CrossRef]
- BEVAN-JONES, W. R., COPE, T. E., JONES, P. S., KAALUND, S. S., PASSAMONTI, L., ALLINSON, K., GREEN, O., HONG, Y. T., FRYER, T. D., ARNOLD, R., COLES, J. P., AIGBIRHIO, F. I., LARNER, A. J., PATTERSON, K., O’BRIEN, J. T. & ROWE, J. B. 2020. Neuroinflammation and protein aggregation co-localize across the frontotemporal dementia spectrum. Brain, 143, 1010-1026.
- BRIGHT, F., CHAN, G., VAN HUMMEL, A., ITTNER, L. M. & KE, Y. D. 2021. TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Int J Mol Sci, 22. [CrossRef]
- BRIGHT, F., WERRY, E. L., DOBSON-STONE, C., PIGUET, O., ITTNER, L. M., HALLIDAY, G. M., HODGES, J. R., KIERNAN, M. C., LOY, C. T., KASSIOU, M. & KRIL, J. J. 2019. Neuroinflammation in frontotemporal dementia. Nat Rev Neurol, 15, 540-555. [CrossRef]
- BRYANT, J. D., LEI, Y., VANPORTFLIET, J. J., WINTERS, A. D. & WEST, A. P. 2022. Assessing Mitochondrial DNA Release into the Cytosol and Subsequent Activation of Innate Immune-related Pathways in Mammalian Cells. Curr Protoc, 2, e372. [CrossRef]
- CALSOLARO, V. & EDISON, P. 2016. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s & Dementia, 12, 719-732. [CrossRef]
- CHAPUIS, J., HANSMANNEL, F., GISTELINCK, M., MOUNIER, A., VAN CAUWENBERGHE, C., KOLEN, K. V., GELLER, F., SOTTEJEAU, Y., HAROLD, D., DOURLEN, P., GRENIER-BOLEY, B., KAMATANI, Y., DELEPINE, B., DEMIAUTTE, F., ZELENIKA, D., ZOMMER, N., HAMDANE, M., BELLENGUEZ, C., DARTIGUES, J. F., HAUW, J. J., LETRONNE, F., AYRAL, A. M., SLEEGERS, K., SCHELLENS, A., BROECK, L. V., ENGELBORGHS, S., DE DEYN, P. P., VANDENBERGHE, R., O’DONOVAN, M., OWEN, M., EPELBAUM, J., MERCKEN, M., KARRAN, E., BANTSCHEFF, M., DREWES, G., JOBERTY, G., CAMPION, D., OCTAVE, J. N., BERR, C., LATHROP, M., CALLAERTS, P., MANN, D., WILLIAMS, J., BUÉE, L., DEWACHTER, I., VAN BROECKHOVEN, C., AMOUYEL, P., MOECHARS, D., DERMAUT, B. & LAMBERT, J. C. 2013. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry, 18, 1225-34. [CrossRef]
- CHIA, R., SABIR, M. S., BANDRES-CIGA, S., SAEZ-ATIENZAR, S., REYNOLDS, R. H., GUSTAVSSON, E., WALTON, R. L., AHMED, S., VIOLLET, C., DING, J., MAKARIOUS, M. B., DIEZ-FAIREN, M., PORTLEY, M. K., SHAH, Z., ABRAMZON, Y., HERNANDEZ, D. G., BLAUWENDRAAT, C., STONE, D. J., EICHER, J., PARKKINEN, L., ANSORGE, O., CLARK, L., HONIG, L. S., MARDER, K., LEMSTRA, A., ST GEORGE-HYSLOP, P., LONDOS, E., MORGAN, K., LASHLEY, T., WARNER, T. T., JAUNMUKTANE, Z., GALASKO, D., SANTANA, I., TIENARI, P. J., MYLLYKANGAS, L., OINAS, M., CAIRNS, N. J., MORRIS, J. C., HALLIDAY, G. M., VAN DEERLIN, V. M., TROJANOWSKI, J. Q., GRASSANO, M., CALVO, A., MORA, G., CANOSA, A., FLORIS, G., BOHANNAN, R. C., BRETT, F., GAN-OR, Z., GEIGER, J. T., MOORE, A., MAY, P., KRÜGER, R., GOLDSTEIN, D. S., LOPEZ, G., TAYEBI, N., SIDRANSKY, E., SOTIS, A. R., SUKUMAR, G., ALBA, C., LOTT, N., MARTINEZ, E. M., TUCK, M., SINGH, J., BACIKOVA, D., ZHANG, X., HUPALO, D. N., ADELEYE, A., WILKERSON, M. D., POLLARD, H. B., NORCLIFFE-KAUFMANN, L., PALMA, J.-A., KAUFMANN, H., SHAKKOTTAI, V. G., PERKINS, M., NEWELL, K. L., GASSER, T., SCHULTE, C., LANDI, F., SALVI, E., CUSI, D., MASLIAH, E., KIM, R. C., CARAWAY, C. A., MONUKI, E. S., BRUNETTI, M., DAWSON, T. M., ROSENTHAL, L. S., ALBERT, M. S., PLETNIKOVA, O., TRONCOSO, J. C., FLANAGAN, M. E., MAO, Q., BIGIO, E. H., RODRÍGUEZ-RODRÍGUEZ, E., INFANTE, J., LAGE, C., GONZÁLEZ-ARAMBURU, I., SANCHEZ-JUAN, P., GHETTI, B., et al. 2021. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nature Genetics, 53, 294-303. [CrossRef]
- COLLABORATORS, G. D. 2019. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol, 18, 88-106. [CrossRef]
- CUNNINGHAM, C. 2013. Microglia and neurodegeneration: the role of systemic inflammation. Glia, 61, 71-90. [CrossRef]
- CUSTODERO, C., CIAVARELLA, A., PANZA, F., GNOCCHI, D., LENATO, G. M., LEE, J., MAZZOCCA, A., SABBÀ, C. & SOLFRIZZI, V. 2022. Role of inflammatory markers in the diagnosis of vascular contributions to cognitive impairment and dementia: a systematic review and meta-analysis. Geroscience, 44, 1373-1392. [CrossRef]
- DANIILIDOU, M., HOLLEMAN, J., HAGMAN, G., KÅREHOLT, I., ASPÖ, M., BRINKMALM, A., ZETTERBERG, H., BLENNOW, K., SOLOMON, A., KIVIPELTO, M., SINDI, S. & MATTON, A. 2024. Neuroinflammation, cerebrovascular dysfunction and diurnal cortisol biomarkers in a memory clinic cohort: Findings from the Co-STAR study. Translational Psychiatry, 14, 364. [CrossRef]
- DE STROOPER, B. & KARRAN, E. 2016. The Cellular Phase of Alzheimer’s Disease. Cell, 164, 603-15. [CrossRef]
- DECOUT, A., KATZ, J. D., VENKATRAMAN, S. & ABLASSER, A. 2021. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nature Reviews Immunology, 21, 548-569. [CrossRef]
- DIB, S., PAHNKE, J. & GOSSELET, F. 2021. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int J Mol Sci, 22. [CrossRef]
- EWERS, M., BIECHELE, G., SUÁREZ-CALVET, M., SACHER, C., BLUME, T., MORENAS-RODRIGUEZ, E., DEMING, Y., PICCIO, L., CRUCHAGA, C., KLEINBERGER, G., SHAW, L., TROJANOWSKI, J. Q., HERMS, J., DICHGANS, M., BRENDEL, M., HAASS, C. & FRANZMEIER, N. 2020. Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. EMBO Mol Med, 12, e12308. [CrossRef]
- FENG, T., MAI, S., ROSCOE, J. M., SHENG, R. R., ULLAH, M., ZHANG, J., KATZ, II, YU, H., XIONG, W. & HU, F. 2020. Loss of TMEM106B and PGRN leads to severe lysosomal abnormalities and neurodegeneration in mice. EMBO Rep, 21, e50219. [CrossRef]
- FERECSKÓ, A. S., SMALLWOOD, M. J., MOORE, A., LIDDLE, C., NEWCOMBE, J., HOLLEY, J., WHATMORE, J., GUTOWSKI, N. J. & EGGLETON, P. 2023. STING-Triggered CNS Inflammation in Human Neurodegenerative Diseases. Biomedicines, 11. [CrossRef]
- FROZZA, R. L., LOURENCO, M. V. & DE FELICE, F. G. 2018. Challenges for Alzheimer’s Disease Therapy: Insights from Novel Mechanisms Beyond Memory Defects. Frontiers in Neuroscience, 12. [CrossRef]
- GORELICK, P. B., SCUTERI, A., BLACK, S. E., DECARLI, C., GREENBERG, S. M., IADECOLA, C., LAUNER, L. J., LAURENT, S., LOPEZ, O. L., NYENHUIS, D., PETERSEN, R. C., SCHNEIDER, J. A., TZOURIO, C., ARNETT, D. K., BENNETT, D. A., CHUI, H. C., HIGASHIDA, R. T., LINDQUIST, R., NILSSON, P. M., ROMAN, G. C., SELLKE, F. W. & SESHADRI, S. 2011. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke, 42, 2672-713. [CrossRef]
- GOUILLY, D., SAINT-AUBERT, L., RIBEIRO, M. J., SALABERT, A. S., TAUBER, C., PÉRAN, P., ARLICOT, N., PARIENTE, J. & PAYOUX, P. 2022. Neuroinflammation PET imaging of the translocator protein (TSPO) in Alzheimer’s disease: an update. European Journal of Neuroscience, 55, 1322-1343. [CrossRef]
- GOVINDARAJULU, M., RAMESH, S., BEASLEY, M., LYNN, G., WALLACE, C., LABEAU, S., PATHAK, S., NADAR, R., MOORE, T. & DHANASEKARAN, M. 2023. Role of cGAS-Sting Signaling in Alzheimer’s Disease. Int J Mol Sci, 24. [CrossRef]
- GRICIUC, A., SERRANO-POZO, A., PARRADO, A. R., LESINSKI, A. N., ASSELIN, C. N., MULLIN, K., HOOLI, B., CHOI, S. H., HYMAN, B. T. & TANZI, R. E. 2013. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron, 78, 631-43. [CrossRef]
- GUERREIRO, R., WOJTAS, A., BRAS, J., CARRASQUILLO, M., ROGAEVA, E., MAJOUNIE, E., CRUCHAGA, C., SASSI, C., KAUWE, J. S., YOUNKIN, S., HAZRATI, L., COLLINGE, J., POCOCK, J., LASHLEY, T., WILLIAMS, J., LAMBERT, J. C., AMOUYEL, P., GOATE, A., RADEMAKERS, R., MORGAN, K., POWELL, J., ST GEORGE-HYSLOP, P., SINGLETON, A. & HARDY, J. 2013. TREM2 variants in Alzheimer’s disease. N Engl J Med, 368, 117-27. [CrossRef]
- GUO, X., YANG, L., WANG, J., WU, Y., LI, Y., DU, L., LI, L., FANG, Z. & ZHANG, X. 2024. The cytosolic DNA-sensing cGAS-STING pathway in neurodegenerative diseases. CNS Neurosci Ther, 30, e14671. [CrossRef]
- HARDY, J. & SELKOE, D. J. 2002. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science, 297, 353-356. [CrossRef]
- HENEKA, M. T., CARSON, M. J., EL KHOURY, J., LANDRETH, G. E., BROSSERON, F., FEINSTEIN, D. L., JACOBS, A. H., WYSS-CORAY, T., VITORICA, J., RANSOHOFF, R. M., HERRUP, K., FRAUTSCHY, S. A., FINSEN, B., BROWN, G. C., VERKHRATSKY, A., YAMANAKA, K., KOISTINAHO, J., LATZ, E., HALLE, A., PETZOLD, G. C., TOWN, T., MORGAN, D., SHINOHARA, M. L., PERRY, V. H., HOLMES, C., BAZAN, N. G., BROOKS, D. J., HUNOT, S., JOSEPH, B., DEIGENDESCH, N., GARASCHUK, O., BODDEKE, E., DINARELLO, C. A., BREITNER, J. C., COLE, G. M., GOLENBOCK, D. T. & KUMMER, M. P. 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol, 14, 388-405. [CrossRef]
- HEPPNER, F. L., RANSOHOFF, R. M. & BECHER, B. 2015. Immune attack: the role of inflammation in Alzheimer disease. Nature Reviews Neuroscience, 16, 358-372. [CrossRef]
- HINKLE, J. T., PATEL, J., PANICKER, N., KARUPPAGOUNDER, S. S., BISWAS, D., BELINGON, B., CHEN, R., BRAHMACHARI, S., PLETNIKOVA, O., TRONCOSO, J. C., DAWSON, V. L. & DAWSON, T. M. 2022. STING mediates neurodegeneration and neuroinflammation in nigrostriatal α-synucleinopathy. Proceedings of the National Academy of Sciences of the United States of America, 119. [CrossRef]
- HUANG, Y., LIU, B., SINHA, S. C., AMIN, S. & GAN, L. 2023. Mechanism and therapeutic potential of targeting cGAS-STING signaling in neurological disorders. Molecular Neurodegeneration, 18, 79. [CrossRef]
- INTERNATIONAL, A. S. D. 2024. Dementia statistics. Alzheimer’s Disease International.
- JACK, C. R., JR., BENNETT, D. A., BLENNOW, K., CARRILLO, M. C., DUNN, B., HAEBERLEIN, S. B., HOLTZMAN, D. M., JAGUST, W., JESSEN, F., KARLAWISH, J., LIU, E., MOLINUEVO, J. L., MONTINE, T., PHELPS, C., RANKIN, K. P., ROWE, C. C., SCHELTENS, P., SIEMERS, E., SNYDER, H. M. & SPERLING, R. 2018. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement, 14, 535-562. [CrossRef]
- JAUHARI, A., BARANOV, S. V., SUOFU, Y., KIM, J., SINGH, T., YABLONSKA, S., LI, F., WANG, X., OBERLY, P., MINNIGH, M. B., POLOYAC, S. M., CARLISLE, D. L. & FRIEDLANDER, R. M. 2020. Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J Clin Invest, 130, 3124-3136. [CrossRef]
- JONSSON, T., STEFANSSON, H., STEINBERG, S., JONSDOTTIR, I., JONSSON, P. V., SNAEDAL, J., BJORNSSON, S., HUTTENLOCHER, J., LEVEY, A. I., LAH, J. J., RUJESCU, D., HAMPEL, H., GIEGLING, I., ANDREASSEN, O. A., ENGEDAL, K., ULSTEIN, I., DJUROVIC, S., IBRAHIM-VERBAAS, C., HOFMAN, A., IKRAM, M. A., VAN DUIJN, C. M., THORSTEINSDOTTIR, U., KONG, A. & STEFANSSON, K. 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med, 368, 107-16. [CrossRef]
- KALARIA, R. N. 2016. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol, 131, 659-85. [CrossRef]
- KARCH, C. M. & GOATE, A. M. 2015. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry, 77, 43-51. [CrossRef]
- KUNKLE, B. W., GRENIER-BOLEY, B., SIMS, R., BIS, J. C., DAMOTTE, V., NAJ, A. C., BOLAND, A., VRONSKAYA, M. V., VAN DER LEE, S. J., AMLIE-WOLF, A., BELLENGUEZ, C., FRIZATTI, A., CHOURAKI, V., MARTIN, E. R., SLEEGERS, K., BADARINARAYAN, N., JAKOBSDÓTTIR, J., HAMILTON-NELSON, K. L., MORENO-GRAU, S., OLASO, R., RAYBOULD, R., CHEN, Y., KUZMA, A. B., HILTUNEN, M., MORGAN, T., AHMAD, S., VARDARAJAN, B. N., EPELBAUM, J., HOFFMANN, P., BOADA, M., BEECHAM, G. W., GARNIER, J.-G., HAROLD, D. H., FITZPATRICK, A. L., VALLADARES, O., MOUTET, M.-L., GERRISH, A., SMITH, A. V., QU, L., BACQ, D., DENNING, N., JIAN, X., ZHAO, Y., DEL ZOMPO, M., FOX, N. C., CHOI, S. H., MATEO, I., HUGHES, J. T., ADAMS, H. H. H., MALAMON, J. S., SÁNCHEZ-GARCÍA, F., PATEL, Y., BRODY, J. A., DOMBROSKI, B. A., NARANJO, M. C. D., DANIILIDOU, M., EIRIKSDOTTIR, G., MUKHERJEE, S., WALLON, D., UPHILL, J. B., ASPELUND, T., CANTWELL, L., GARZIA, F., GALIMBERTI, D., HOFER, E., BUTKIEWICZ, M., FIN, B., SCARPINI, E., SARNOWSKI, C., BUSH, W. S., MESLAGE, S., KORNHUBER, J., WHITE, C. C., SONG, Y., BARBER, R. C., ENGELBORGHS, S., SORDON, S., VOIJNOVIC, D., ADAMS, P. M., VANDENBERGHE, R., MAYHAUS, M., CUPPLES, L. A., ALBERT, M. S., DE DEYN, P. P., GU, W., HIMALI, J. J., BEEKLY, D. L., SQUASSINA, A., HARTMANN, A. M., ORELLANA, A., BLACKER, D., RODRÍGUEZ-RODRÍGUEZ, E., LOVESTONE, S., GARCIA, M. E., DOODY, R., MUNOZ-FERNADEZ, C., SUSSAMS, R., LIN, H., FAIRCHILD, T. J., BENITO, Y. A., et al. 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nature Genetics, 51, 414–430. [CrossRef]
- LEDO, J. H., LIEBMANN, T., ZHANG, R., CHANG, J. C., AZEVEDO, E. P., WONG, E., SILVA, H. M., TROYANSKAYA, O. G., BUSTOS, V. & GREENGARD, P. 2021. Presenilin 1 phosphorylation regulates amyloid-β degradation by microglia. Molecular Psychiatry, 26, 5620-5635. [CrossRef]
- LEE, S., DEVANNEY, N. A., GOLDEN, L. R., SMITH, C. T., SCHWARTZ, J. L., WALSH, A. E., CLARKE, H. A., GOULDING, D. S., ALLENGER, E. J., MORILLO-SEGOVIA, G., FRIDAY, C. M., GORMAN, A. A., HAWKINSON, T. R., MACLEAN, S. M., WILLIAMS, H. C., SUN, R. C., MORGANTI, J. M. & JOHNSON, L. A. 2023. APOE modulates microglial immunometabolism in response to age, amyloid pathology, and inflammatory challenge. Cell Rep, 42, 112196. [CrossRef]
- LEYNS, C. E. G., ULRICH, J. D., FINN, M. B., STEWART, F. R., KOSCAL, L. J., REMOLINA SERRANO, J., ROBINSON, G. O., ANDERSON, E., COLONNA, M. & HOLTZMAN, D. M. 2017. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proceedings of the National Academy of Sciences, 114, 11524-11529. [CrossRef]
- LIU, Y., ZHANG, B., DUAN, R. & LIU, Y. 2024. Mitochondrial DNA Leakage and cGas/STING Pathway in Microglia: Crosstalk Between Neuroinflammation and Neurodegeneration. Neuroscience, 548, 1-8. [CrossRef]
- LOVE, S. & MINERS, J. S. 2016. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathologica, 131, 645-658. [CrossRef]
- LOVELAND, P. M., YU, J. J., CHURILOV, L., YASSI, N. & WATSON, R. 2023. Investigation of Inflammation in Lewy Body Dementia: A Systematic Scoping Review. International Journal of Molecular Sciences, 24, 12116. [CrossRef]
- MARQUES, C., HELD, A., DORFMAN, K., SUNG, J., SONG, C., KAVUTURU, A. S., AGUILAR, C., RUSSO, T., OAKLEY, D. H., ALBERS, M. W., HYMAN, B. T., PETRUCELLI, L., LAGIER-TOURENNE, C. & WAINGER, B. J. 2024. Neuronal STING activation in amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol, 147, 56. [CrossRef]
- MATHUR, V., BURAI, R., VEST, R. T., BONANNO, L. N., LEHALLIER, B., ZARDENETA, M. E., MISTRY, K. N., DO, D., MARSH, S. E., ABUD, E. M., BLURTON-JONES, M., LI, L., LASHUEL, H. A. & WYSS-CORAY, T. 2017. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron, 96, 1290-1302.e6. [CrossRef]
- MISHRA, S., KNUPP, A., YOUNG, J. E. & JAYADEV, S. 2022. Depletion of the AD risk gene SORL1 causes endo-lysosomal dysfunction in human microglia. Alzheimer’s & Dementia, 18, e068943. [CrossRef]
- MOYSE, E., KRANTIC, S., DJELLOULI, N., ROGER, S., ANGOULVANT, D., DEBACQ, C., LEROY, V., FOUGERE, B. & AIDOUD, A. 2022. Neuroinflammation: A Possible Link Between Chronic Vascular Disorders and Neurodegenerative Diseases. Frontiers in Aging Neuroscience, 14. [CrossRef]
- NABIZADEH, F., SEYEDMIRZAEI, H. & KARAMI, S. 2024. Neuroimaging biomarkers and CSF sTREM2 levels in Alzheimer’s disease: a longitudinal study. Scientific Reports, 14, 15318. [CrossRef]
- NAJ, A. C., JUN, G., BEECHAM, G. W., WANG, L. S., VARDARAJAN, B. N., BUROS, J., GALLINS, P. J., BUXBAUM, J. D., JARVIK, G. P., CRANE, P. K., LARSON, E. B., BIRD, T. D., BOEVE, B. F., GRAFF-RADFORD, N. R., DE JAGER, P. L., EVANS, D., SCHNEIDER, J. A., CARRASQUILLO, M. M., ERTEKIN-TANER, N., YOUNKIN, S. G., CRUCHAGA, C., KAUWE, J. S., NOWOTNY, P., KRAMER, P., HARDY, J., HUENTELMAN, M. J., MYERS, A. J., BARMADA, M. M., DEMIRCI, F. Y., BALDWIN, C. T., GREEN, R. C., ROGAEVA, E., ST GEORGE-HYSLOP, P., ARNOLD, S. E., BARBER, R., BEACH, T., BIGIO, E. H., BOWEN, J. D., BOXER, A., BURKE, J. R., CAIRNS, N. J., CARLSON, C. S., CARNEY, R. M., CARROLL, S. L., CHUI, H. C., CLARK, D. G., CORNEVEAUX, J., COTMAN, C. W., CUMMINGS, J. L., DECARLI, C., DEKOSKY, S. T., DIAZ-ARRASTIA, R., DICK, M., DICKSON, D. W., ELLIS, W. G., FABER, K. M., FALLON, K. B., FARLOW, M. R., FERRIS, S., FROSCH, M. P., GALASKO, D. R., GANGULI, M., GEARING, M., GESCHWIND, D. H., GHETTI, B., GILBERT, J. R., GILMAN, S., GIORDANI, B., GLASS, J. D., GROWDON, J. H., HAMILTON, R. L., HARRELL, L. E., HEAD, E., HONIG, L. S., HULETTE, C. M., HYMAN, B. T., JICHA, G. A., JIN, L. W., JOHNSON, N., KARLAWISH, J., KARYDAS, A., KAYE, J. A., KIM, R., KOO, E. H., KOWALL, N. W., LAH, J. J., LEVEY, A. I., LIEBERMAN, A. P., LOPEZ, O. L., MACK, W. J., MARSON, D. C., MARTINIUK, F., MASH, D. C., MASLIAH, E., MCCORMICK, W. C., MCCURRY, S. M., MCDAVID, A. N., MCKEE, A. C., MESULAM, M., MILLER, B. L., et al. 2011. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet, 43, 436-41. [CrossRef]
- O’ROURKE, J. G., BOGDANIK, L., YÁÑEZ, A., LALL, D., WOLF, A. J., MUHAMMAD, A. K., HO, R., CARMONA, S., VIT, J. P., ZARROW, J., KIM, K. J., BELL, S., HARMS, M. B., MILLER, T. M., DANGLER, C. A., UNDERHILL, D. M., GOODRIDGE, H. S., LUTZ, C. M. & BALOH, R. H. 2016. C9orf72 is required for proper macrophage and microglial function in mice. Science, 351, 1324-9. [CrossRef]
- PAN, J., HU, J., MENG, D., CHEN, L. & WEI, X. 2024. Neuroinflammation in dementia: A meta-analysis of PET imaging studies. Medicine (Baltimore), 103, e38086. [CrossRef]
- PASINELLI, P. & BROWN, R. H. 2006. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature Reviews Neuroscience, 7, 710-723. [CrossRef]
- PAUL, B. D., SNYDER, S. H. & BOHR, V. A. 2021. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci, 44, 83-96. [CrossRef]
- PICCA, A., CALVANI, R., COELHO-JUNIOR, H. J., LANDI, F., BERNABEI, R. & MARZETTI, E. 2020. Mitochondrial Dysfunction, Oxidative Stress, and Neuroinflammation: Intertwined Roads to Neurodegeneration. Antioxidants (Basel), 9. [CrossRef]
- RANSOHOFF, R. M. 2016a. How neuroinflammation contributes to neurodegeneration. Science, 353, 777-783. [CrossRef]
- RANSOHOFF, R. M. 2016b. How neuroinflammation contributes to neurodegeneration. Science, 353, 777-83. [CrossRef]
- SALTER, M. W. & STEVENS, B. 2017. Microglia emerge as central players in brain disease. Nature Medicine, 23, 1018-1027. [CrossRef]
- SHARMA, O., KAUR GREWAL, A., KHAN, H. & GURJEET SINGH, T. 2024. Exploring the nexus of cGAS STING pathway in neurodegenerative terrain: A therapeutic odyssey. Int Immunopharmacol, 142, 113205. [CrossRef]
- SHI, Y., YAMADA, K., LIDDELOW, S. A., SMITH, S. T., ZHAO, L., LUO, W., TSAI, R. M., SPINA, S., GRINBERG, L. T., ROJAS, J. C., GALLARDO, G., WANG, K., ROH, J., ROBINSON, G., FINN, M. B., JIANG, H., SULLIVAN, P. M., BAUFELD, C., WOOD, M. W., SUTPHEN, C., MCCUE, L., XIONG, C., DEL-AGUILA, J. L., MORRIS, J. C., CRUCHAGA, C., FAGAN, A. M., MILLER, B. L., BOXER, A. L., SEELEY, W. W., BUTOVSKY, O., BARRES, B. A., PAUL, S. M. & HOLTZMAN, D. M. 2017. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature, 549, 523-527. [CrossRef]
- SIDDIQUI, S. S., MATAR, R., MERHEB, M., HODEIFY, R., VAZHAPPILLY, C. G., MARTON, J., SHAMSUDDIN, S. A. & AL ZOUABI, H. 2019. Siglecs in Brain Function and Neurological Disorders. Cells, 8, 1125. [CrossRef]
- SIERKSMA, A., LU, A., MANCUSO, R., FATTORELLI, N., THRUPP, N., SALTA, E., ZOCO, J., BLUM, D., BUÉE, L., DE STROOPER, B. & FIERS, M. 2020. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med, 12, e10606. [CrossRef]
- SLITER, D. A., MARTINEZ, J., HAO, L., CHEN, X., SUN, N., FISCHER, T. D., BURMAN, J. L., LI, Y., ZHANG, Z., NARENDRA, D. P., CAI, H., BORSCHE, M., KLEIN, C. & YOULE, R. J. 2018. Parkin and PINK1 mitigate STING-induced inflammation. Nature, 561, 258-262. [CrossRef]
- SPIRES-JONES, TARA L. & HYMAN, BRADLEY T. 2014. The Intersection of Amyloid Beta and Tau at Synapses in Alzheimer’s Disease. Neuron, 82, 756-771. [CrossRef]
- STOLL, A. C. & SORTWELL, C. E. 2022. Leveraging the preformed fibril model to distinguish between alpha-synuclein inclusion- and nigrostriatal degeneration-associated immunogenicity. Neurobiology of Disease, 171, 105804. [CrossRef]
- SURENDRANATHAN, A., SU, L., MAK, E., PASSAMONTI, L., HONG, Y. T., ARNOLD, R., VÁZQUEZ RODRÍGUEZ, P., BEVAN-JONES, W. R., BRAIN, S. A. E., FRYER, T. D., AIGBIRHIO, F. I., ROWE, J. B. & O’BRIEN, J. T. 2018. Early microglial activation and peripheral inflammation in dementia with Lewy bodies. Brain, 141, 3415-3427. [CrossRef]
- TEJERA, D. & HENEKA, M. T. 2019. Microglia in Neurodegenerative Disorders. Methods Mol Biol, 2034, 57-67. [CrossRef]
- TIAN, Z., JI, X. & LIU, J. 2022. Neuroinflammation in Vascular Cognitive Impairment and Dementia: Current Evidence, Advances, and Prospects. Int J Mol Sci, 23. [CrossRef]
- ULLAND, T. K. & COLONNA, M. 2018. TREM2—a key player in microglial biology and Alzheimer disease. Nat Rev Neurol, 14, 667-675. [CrossRef]
- VENKAT, P., CHOPP, M. & CHEN, J. 2015. Models and mechanisms of vascular dementia. Experimental neurology, 272, 97-108. [CrossRef]
- VRILLON, A., BOUSIGES, O., GÖTZE, K., DEMUYNCK, C., MULLER, C., RAVIER, A., SCHORR, B., PHILIPPI, N., HOURREGUE, C., COGNAT, E., DUMURGIER, J., LILAMAND, M., CRETIN, B., BLANC, F. & PAQUET, C. 2024. Plasma biomarkers of amyloid, tau, axonal, and neuroinflammation pathologies in dementia with Lewy bodies. Alzheimer’s Research & Therapy, 16, 146. [CrossRef]
- YIN, Z., ROSENZWEIG, N., KLEEMANN, K. L., ZHANG, X., BRANDÃO, W., MARGETA, M. A., SCHROEDER, C., SIVANATHAN, K. N., SILVEIRA, S., GAUTHIER, C., MALLAH, D., PITTS, K. M., DURAO, A., HERRON, S., SHOREY, H., CHENG, Y., BARRY, J. L., KRISHNAN, R. K., WAKELIN, S., RHEE, J., YUNG, A., ARONCHIK, M., WANG, C., JAIN, N., BAO, X., GERRITS, E., BROUWER, N., DEIK, A., TENEN, D. G., IKEZU, T., SANTANDER, N. G., MCKINSEY, G. L., BAUFELD, C., SHEPPARD, D., KRASEMANN, S., NOWARSKI, R., EGGEN, B. J. L., CLISH, C., TANZI, R. E., MADORE, C., ARNOLD, T. D., HOLTZMAN, D. M. & BUTOVSKY, O. 2023. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints. Nat Immunol, 24, 1839-1853. [CrossRef]
- YU, X., CAI, L., YAO, J., LI, C. & WANG, X. 2024. Agonists and Inhibitors of the cGAS-STING Pathway. Molecules, 29. [CrossRef]
- ZHANG, J. 2015. Mapping neuroinflammation in frontotemporal dementia with molecular PET imaging. J Neuroinflammation, 12, 108. [CrossRef]
- ZHANG, W., XIAO, D., MAO, Q. & XIA, H. 2023. Role of neuroinflammation in neurodegeneration development. Signal Transduction and Targeted Therapy, 8, 267. [CrossRef]
- ZHANG, Y., ZOU, M., WU, H., ZHU, J. & JIN, T. 2024. The cGAS-STING pathway drives neuroinflammation and neurodegeneration via cellular and molecular mechanisms in neurodegenerative diseases. Neurobiology of Disease, 202, 106710. [CrossRef]
- ZHANG, Z. & ZHAO, Y. 2022. Progress on the roles of MEF2C in neuropsychiatric diseases. Molecular Brain, 15, 8. [CrossRef]
- ZHAO, P., XU, Y., JIANG, L. L., FAN, X., KU, Z., LI, L., LIU, X., DENG, M., ARASE, H., ZHU, J. J., HUANG, T. Y., ZHAO, Y., ZHANG, C., XU, H., TONG, Q., ZHANG, N. & AN, Z. 2022. LILRB2-mediated TREM2 signaling inhibition suppresses microglia functions. Mol Neurodegener, 17, 44. [CrossRef]
Figure 1.
The cGAS-STING pathway in dementia progression and microglial activation.
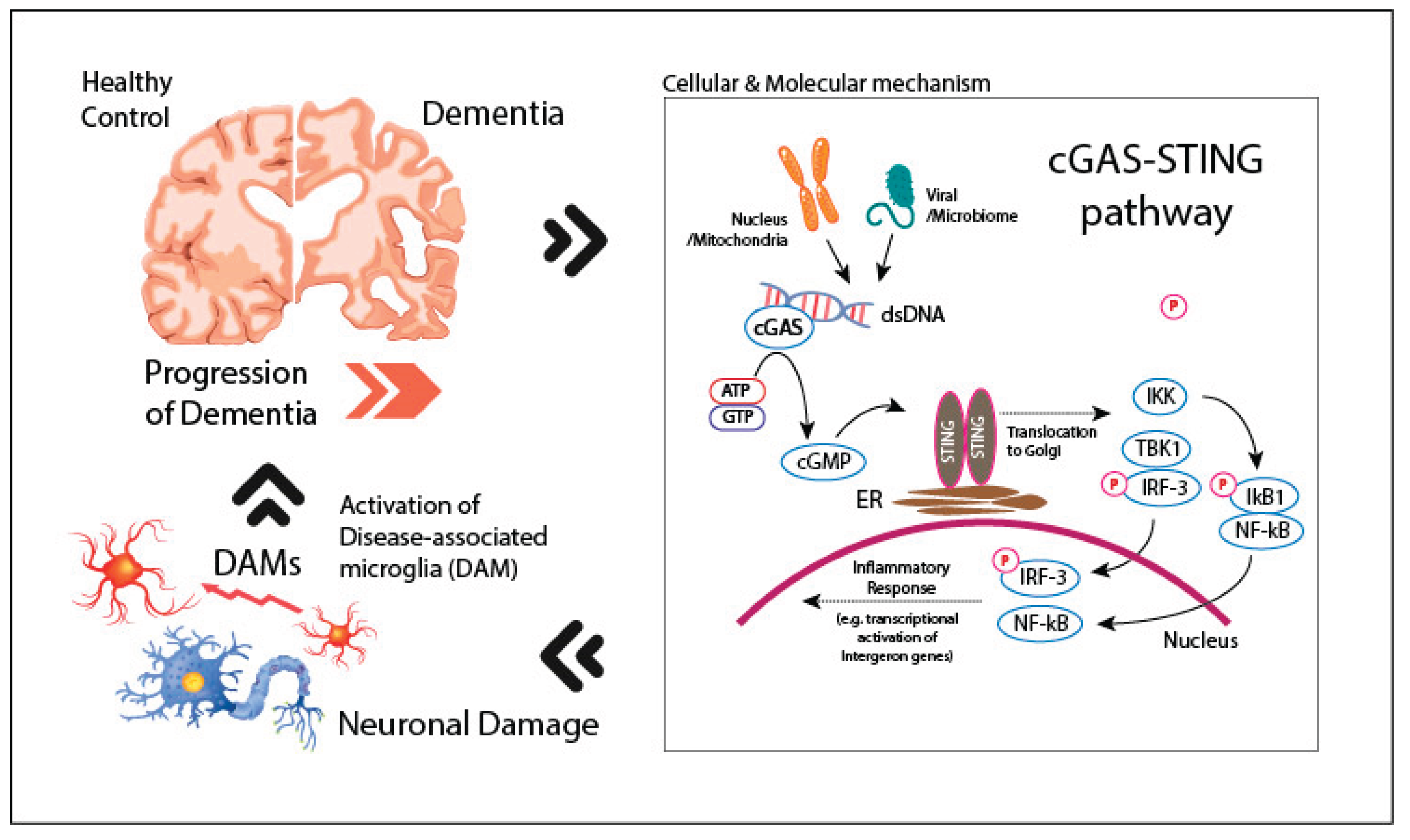

Table 1.
Genetic mutations associated with dementia that are also linked to microglial activation.
| Gene | Chromosom Location |
Role in Microglia Activation | Associated Dementia |
References |
|---|---|---|---|---|
| CR1 | 1q32.2 | Variants in CR1 impair complement regulation and enhance microglial activation via the complement cascade. | AD | (Karch and Goate, 2015) |
| BIN1 | 2q14.3 | BIN1 variants may regulate microglial activity through interaction with the immune system, including modulation of pro-inflammatory responses. | AD | (Chapuis et al., 2013) |
| MEF2C | 5q14.3 | MEF2C regulates microglial homeostasis and suppresses pro-inflammatory gene expression, thereby protecting neurons from excessive neuroinflammation. | AD | (Zhang and Zhao, 2022) |
| TREM2 | 6p21.1 | TREM2 mutations impair microglial phagocytic function and lead to chronic neuroinflammation, enhancing microglial activation. | AD, FTD | (Jonsson et al., 2013, Guerreiro et al., 2013) |
| MS4A | 11q12.2 | Modulates microglial inflammatory signaling by altering membrane protein interactions and intracellular communication pathways. | Alzheimer’s Disease | (Naj et al., 2011, Kunkle et al., 2019) |
| SORL1 | 11q24.2 | SORL1 mutations altered lysosome function of microglia, leading to amyloid ϐ accumulation. | AD | (Mishra et al., 2022) |
| PS1 | 14q24.2 | PS1 modulates microglial response to amyloid ϐ and to maintain brain homeostasis by regulating lysosomal calcium signaling and autophagy. | AD | (Ledo et al., 2021) |
| PLCG2 | 16q23.3 | Activating mutations in PLCG2 promote microglial activation and phagocytosis, improving immune response in Alzheimer’s disease. | AD | (Sierksma et al., 2020) |
| APOE ε4 | 19q13.32 | APOE ε4 induces pro-inflammatory microglial activation through various pathways, including C1q complement activation. | AD | (Shi et al., 2017) |
| CD33 | 19q13.41 | Loss of function of CD33 reduces microglial activation and enhances amyloid clearance, suggesting CD33 inhibits microglial activation in dementia. | AD | (Griciuc et al., 2013, Siddiqui et al., 2019) |
| ABCA7 | 19p13.3 | ABCA7 regulates microglial activation by modulating cholesterol efflux and lipid metabolism, which influences the microglial inflammatory response and phagocytic activity. | AD | (Dib et al., 2021) |
| LILRB2 | 19q13.42 | LILRB2 is co-expressed with TREM2 in microglia and associated with microglial activation by regulating the inflammatory response and the phagocytic activity of microglia. | AD | (Zhao et al., 2022) |
| TMEM106B | 7p21.3 | Loss of TMEM106B along with PGRN triggers the lysosomal dysfunction in microglia and exacerbates microglial activation, leading to neurodegeneration. | FTD | (Feng et al., 2020) |
| C9orf72 | 9p21.2 | C9orf72 hexanucleotide repeat expansions lead to neuroinflammation and dysregulated microglial activation, contributing to neurodegeneration. | FTD, ALS | (O’Rourke et al., 2016) |
| GRN | 17q21.31 | GRN mutations reduce progranulin levels, leading to increased neuroinflammation and excessive microglial activation. | FTD | (Baker et al., 2006b) |
| SOD1 | 21q22.11 | Mutant SOD1 increases oxidative stress and promotes neuroinflammation through enhanced microglial activation in ALS. | ALS | (Pasinelli and Brown, 2006) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



