
Submitted:
14 October 2025
Posted:
14 October 2025
You are already at the latest version
Abstract
Background/Objectives: Atrial fibrillation (AF), characteristic of chaotic atrial electrical activity along with ineffective atrial systole, remains the most frequent sustained cardiac dysrhythmia, with an overall lifetime risk for AF being approximately 15% to 40% in the global population. AF is associated with substantially enhanced risks for multiple adverse clinical outcomes, including thromboembolic cerebral stroke, dementia, chronic kidney disease, myocardial infarction, cardiac failure, and even premature cardiac demise. Although tremendous progress has been achieved toward unravelling the complex hereditary etiopathogenesis underpinning AF, it has become increasingly clear that inherited determinants predisposing to AF in a vast majority of individuals are still uncertain. Methods: A Chinese pedigree with idiopathic AF and another group of 236 cases suffering idiopathic AF along with 312 unrelated healthy volunteers were prospectively recruited. Exome-wide sequencing and Sanger sequencing assays were implemented in research participants. The functional effects of the discovered variations in the SOX5 gene were explored through dual-luciferase reporter analysis. Results: Two novel SOX5 mutants, NM_006940.6: c.355C>T; p.(Gln119*) and NM_006940.6: c.640G>T; p.(Glu214*), were identified in the AF pedigree and one of the 236 unrelated patients affected with AF, respectively. These two heterozygous truncating SOX5 variations were absent from the 624 control chromosomes. Quantitatively biochemical explorations unraveled that both Gln119*- and Glu214*-mutant SOX5 lost the ability to transactivate GJA1. Additionally, the two variations abolished the synergistic transactivation of SCN5A by SOX5 and SHOX2. Conclusions: The current findings indicate SOX5 as a novel gene contributing to AF, which adds more insight to the molecular pathogenesis of AF, and provides a potential target for personalized precision medicine.
Keywords:
arrhythmia
; atrial fibrillation
; molecular genetics
; SOX5
; transcriptional regulation
1. Introduction
As the third most prevent cardiovascular disorder next to hypertension and coronary artery disease [1], atrial fibrillation (AF), characterized by irregular atrial electrical activity resulting in disorganized atrial contractions, is the most frequently sustained cardiac dysrhythmia, affecting about 1%–2% of the general population worldwide [2,3]. According to a systematic scientific assessment of the global burden of diseases in 2019, over 43 million persons suffered from AF globally [4], and in the United States alone, AF was estimated to affect more than five million of people in 2010, with a projection of roughly 12 million in 2030 [2]. It has been reported that the total lifetime risk for the development of AF is ~30%–40% in White folks, ~20% in African Americans, and ~15% in Chinese subjects [2]. Notably, nearly one-third of patients suffering from AF are asymptomatic (so-called silent/subclinical AF) and remain undetected until the first thromboembolic complication occurs; hence, the actual incidence of AF is evidently underestimated, given that intermittent screening with a conventional electrocardiogram may miss some episodes of AF because of its paroxysmal nature [5,6]. Although the factual prevalence of undiagnosed AF is unclear in the community, a retrospective cohort investigation has revealed that about 11% of the AF patients were undiagnosed in the United States in 2015 [7]. Recently, an assessment of asymptomatic AF in the general population shows that the true prevalence of AF is at a minimum of 3%-4% if asymptomatic/device-detected AF is encompassed [8]. By disruption of atrial coordinated contraction [9], AF confers strikingly enhanced risks for a wide range of adverse clinical sequelae, encompassing reduced exercise capacity along with degraded health-correlated quality of life [10,11,12,13,14], atrial thrombosis [15,16,17,18] and thromboembolic cerebral stroke/systemic thromboembolism [19,20,21,22,23,24], cognitive impairment and early-onset dementia [25,26,27], acute renal injury/chronic kidney disease [28,29,30], atrial cardiomyopathy [31,32,33,34], myocardial infarction [35,36,37,38], congestive/chronic heart failure [39,40,41,42], malicious/lethal ventricular arrhythmias [43,44,45], and even premature cardiac demise [46,47,48,49]. More specifically, AF is associated with a 1.5-time risk of acute myocardial infarction or intellectual decline/dementia, 1.6-time risk of chronic renal disorder, 2.4-time risk of cerebral stroke, 5-time risk of congestive cardiac failure, and 2-time risk of sudden cardiac demise [2]. According to a retrospective investigation of a nationally representative population with the Medicare beneficiaries, after the initial diagnosis of AF in persons ≥65 years, during the first five years the most frequent clinical consequence was death, followed by cardiac failure, stroke, gastrointestinal hemorrhage, and acute myocardial infarction [50]. Additionally, AF is responsible for higher healthcare expenditures [2]. Based on the US data from Optum, subjects with incident AF have annual healthcare disbursements of $63,031, which is $27,896 higher than those without AF [51]. Evidently, AF has given rise to substantial morbidity and mortality as well as huge economic encumbrance [2,52,53,54]. Despite the vital clinical significance, the etiopathogenesis underlying AF remains largely indefinite.
It is generally understood that the etiopathogenesis underpinning AF is exceedingly complicated and multifaceted, and both environmental/non-hereditary pathogenic factors and genetic/inheritable defective components may lead to the occurrence and maintenance of AF [2,55,56,57,58,59]. There is a growing body of non-genetic hazard factors reported to account for AF, such as advancing age, unhealthy/sedentary lifestyle, night shift work, immoderate smoking, excessive alcohol consumption, obstructive sleep apnea, obesity, surgical operations, clonal haematopoiesis, diabetes mellitus, epilepsy, depression, gut microbiota dysbiosis, homocysteine, hyperuricaemia, asthma, pulmonary embolism, hyperthyroidism, bacterial/viral infections, β-thalassemia, chronic kidney disease, essential hypertension, rheumatic/valvular heart disease, coronary heart disease, dilated cardiomyopathy, hypertrophic cardiomyopathy, acoustic/toxicant pollution, and cardiac autonomic nerve system disorder [2,55,56,60,61,62,63,64,65]. However, aggregating genetic investigations have demonstrated that heritable determinants exert crucial effect on the occurrence and sustainment of AF [57,58,59]. At present, no less than 60 AF-causing genes have been reported, of which the overwhelming majority encode sodium ion channel proteins, such as SCN5A (Na+ channel alpha subunit 5) and SCN1B (Na+ channel beta subunit 1); potassium ion channel subunits, such as KCNQ1 (K+ channel subfamily Q member 1) and KCNE2 (K+ channel subfamily E regulatory subunit 2); calcium ion channel proteins, such as CACNB2 (Ca2+ channel auxiliary subunit beta 2) and CACNA2D4 (Ca2+ channel auxiliary subunit alpha2delta 4); gap junction channel proteins/connexins, such as GJA1/Cx43 (gap junction protein alpha 1/connexin 43) and GJA5/Cx40 (gap junction protein alpha 5/connexin 40); transcription factors, such as PITX2 (paired like homeodomain 2) and SHOX2 (short stature homeobox 2); myocardial structural proteins, such as myosin light chain 4 (MYL4) and titin (TTN); and signaling molecules such as atrial natriuretic peptide (ANP) [57,58,59,66,67,68,69,70,71,72,73,74]. In addition, extensive genome- and exome-wide comparison assays between AF patients and controls, such as whole-exome sequencing (WES) analysis, have unveiled genetic variants at >140 chromosomal loci that confer enhanced susceptibility to AF, though the functional effects of these AF-related genetic variants remain largely unclear [59,75]. Nevertheless, due to noteworthy genetic heterogeneity, the hereditary ingredients susceptible to AF in most patients are still obscure.
2. Materials and Methods
2.1. Research Individuals
In the current research program, a four-generation pedigree suffering from idiopathic AF (arbitrarily named as Pedigree AF-001) and another group of 236 cases who were unrelated and suffered idiopathic AF, along with 312 healthy volunteers who were unrelated and had no familial history of AF, were prospectively enlisted from the population of Han race in Shanghai, China. The criteria for the inclusion of AF patients were a definite diagnosis of AF, no known cause of AF, and informed consent. The criteria for the exclusion of AF patients included a definite environmental/genetic cause of AF and no informed consent. Diagnostic ascertainment and clinical classification of AF were conducted as elaborated previously [2]. Phenotypic data, which included personal information on medical history as well as familial history, medical records, physical examination findings, 12-lead electrocardiograms, trans-thoracic echocardiographic parameters, and routine laboratory test reports, were collected from each study subject. A 24-hour ambulatory electrocardiographic monitoring was carried out when indicated. Blood samples were prepared from research participants, which were used to extract genomic DNA utilizing a DNA purification kit (catalogue No. A1620; Promega, USA), following standard protocols. The current case-control investigation was conducted strictly in accordance with the Declaration of Helsinki. Before the enrollment of research participants, the Medical Ethics Committee at Shanghai Fifth People′s Hospital approved the study protocols (ethical approval code: 2022-179; ethical approval date: October 23, 2024). All research participants were of Chinese origin and signed a written informed consent form before the start of clinical investigations.
2.2. Genetic Assay in Study Participants
As described elsewhere [68,73,76,77,78], WES analysis was performed in five AF-affected members and three unaffected/healthy members from Pedigree AF-001. Briefly, 5 µg of genomic DNA from each family member selected for WES analysis were applied to construct a genomic DNA library, from which an exome library was constructed via hybridization capture utilizing the SureSelectXT2 Human All Exon (V6) Kit (catalogue No. 5190-8872; Agilent Technologies, USA). Sequencing of an exome library was implemented using the HiSeq 4000 Genome Analyzer (Illumina, USA), with the HiSeq 3000/4000 SBS Kit (catalogue No. 410-1003; Illumina, USA), according to the manufacturer’s protocols. Raw WES data were treated using Illumina Analysis Pipeline (version 2.6; Illumina, USA) with default settings, and reads were mapped to the human reference genome (GRCh37) with BWA (version 2.10.12). Variant calling was completed by leveraging GATK (version 3.8.1.0), and variant annotation was made by utilizing ANNOVAR (version 2018). Rare deleterious variations linked to AF were subject to Sanger sequencing assays in all the family members from Pedigree AF- 001. Sanger sequencing examination of the entire coding regions, as well as splicing donors/acceptors, of the gene harboring the observed AF-linked variation was implemented in another group of 236 patients with idiopathic AF, along with 312 unrelated healthy volunteers as controls. Besides, the dbSNP (single nucleotide polymorphism database; accessed on 01 September 2025) and gnomAD (version 2.1.1; accessed on 01 September 2025) databases were consulted as described elsewhere [73] to verify the novelty of the discovered AF-linked variation.
2.3. Recombination of Gene-Expressing Vectors
Human heart cDNA was prepared as depicted previously [79,80]. A 2400-bp DNA segment harboring the whole open read frame of the wild-type human sex-determining region (SRY)-related high-mobility-group (HMG) box 5 (SOX5) gene (Nucleotide accession No.: NM_006940.6) was amplified through polymerase chain reaction (PCR) from human heart cDNA with the AccuPrime™ Pfx DNA Polymerase Kit (catalogue No. 12344024; Invitrogen, USA) using the SOX5-specific primers of 5′-CGCGAATTCACTTGACAGGTTCAGTTGGAG-3′ and 5′-CGCCTCGAGTCTTTAAGTCCTAAGGTCAC-3′. The PCR-yielded products and the pcDNA™3.1(+) vector (catalogue No. V79020; Invitrogen, USA) were cut with EcoRI (catalogue No. R0101V; New England Biolabs, USA) and XhoI (catalogue No. R0146V; New England Biolabs, USA), fragmented via 1.6% agarose gel electrophoresis, extracted utilizing the MinElute Gel Extraction Kit (catalogue No. 28604; Qiagen, Germany) and ligated with the T4 DNA ligase (catalogue No. M0202V; New England Biolabs, USA) to produce the wild-type human SOX5-pcDNA™3.1(+) recombinant vector. Using the wild-type human SOX5-pcDNA™3.1(+) vector as a PCR template, the Gln119*-mutant SOX5-pcDNA™3.1(+) vector was created via site-targeted mutagenesis employing a site-directed mutagenesis kit (catalogue No. 210518; Agilent Technologies, USA) and the forward primer of 5′-GAAGAAGGTGGGCGATAGAGTGGCGAGTCCTTG-3′ as well as the reverse primer of 5′-CAAGGACTCGCCACTCTATCGCCCACCTTCTTC-3′. Similarly, the Glu214*-mutant human SOX5-pcDNA™3.1(+) vector was produced by site-targeted mutagenesis with the primers comprising 5′-CTGACCAGCCTCCGATAGCAGCTGTTGGCTGCC-3′ (forward) and 5′-GGCAGCCAACAGCTGCTATCGGAGGCTGGTCAG-3′ (backward). The SHOX2-pcDNA™3.1(+) vector expressing wild-type human SHOX2 was constructed as described elsewhere [81]. The SOX9-pcDNA™3.1(+) vector expressing wild-type human SOX9 (Nucleotide accession No.: NM_000346.4) was constructed like constructing the SOX5-pcDNA™3.1(+) vector, using the primers of 5′-GTAGCTAGCGAAAGCGGAGCTCGAAACTG-3′ and 5′-GTAGCGGCCGCCAAGTGGGTAATGCGCTTGG-3′. Additionally, the GJA1-luciferase (GJA1-luc) and SCN5A-luc vectors expressing Firefly luciferase were constructed as described elsewhere.[73]. All the recombinant vectors were verified by Sanger sequencing analysis.
2.4. Cellular Transfection with Multiple Expression Vectors and Dual-Luciferase Activity Measurement
COS7 and HEK293 cells were cultured as previously narrated [73]. Cells were transiently transfected with multiple expressing vectors utilizing a lipofectamine reagent (catalogue No. 15338100; Invitrogen, USA). Specifically, COS-7 cells were transiently transfected with 400 ng of empty pcDNA™3.1(+) as an external control, or 400 ng of wild-type human SOX5-pcDNA™3.1(+), or 400 ng of Gln119*-mutant human SOX5-pcDNA™3.1(+), or 400 ng of Glu214*-mutant human SOX5-pcDNA™3.1(+), or 200 ng of wild-type human SOX5-pcDNA™3.1(+) + 200 ng of empty pcDNA™3.1(+), or 200 ng of wild-type human SOX5-pcDNA™3.1(+) + 200 ng of Gln119*-mutant human SOX5-pcDNA™3.1(+), or 200 ng of wild-type human SOX5-pcDNA™3.1(+) + 200 ng of Glu214*-mutant human SOX5-pcDNA™3.1(+), together with 1.2 μg of GJA1-luc and 3 ng of pGL4.75 expressing Renilla luciferase (catalogue No. E6931; Promega, USA). To analyze the synergistic transactivation function, HEK293 cells were transiently transfected with 200 ng of each expression vector (empty pcDNA™3.1(+), wild-type human SOX5-pcDNA™3.1(+), wild-type human SHOX2-pcDNA™3.1(+), Gln119*-mutant human SOX5-pcDNA™3.1(+), or Glu214*-mutant human SOX5-pcDNA™3.1(+)) together with 1.0 μg of SCN5A-luc and 2 ng of pGL4.75 (catalogue No. E6931; Promega, USA). Notably, in each cellular transfection, the SOX9-pcDNA™3.1(+) vector (200 ng) was used together, due to its fundamental role for SOX5 function [82,83,84,85]. Cells were collected 48 h after transfection with the above-mentioned expression vectors, and then the activities of dual luciferases were quantitatively assayed using a dual-luciferase assay kit (catalogue No. E2920; Promega, USA) under a fluorescent plate reader (Promega, USA), following the manufacturer's instructions. As depicted elsewhere [73], the activities of the GJA1 and SCN5A promoters were presented as the ratios of Firefly bioluminescence intensities to Renilla bioluminescence intensities. In addition, for every vector used, three independent dual-reporter assays were performed in triplicate, and the means of results from three independent dual-reporter assays were applied to the comparison between two or among no less than three groups.
2.5. Statistical Assessment
Continuous parameters, such as age and promoter activity, are described as mean ± standard deviation (± SD), whereas categorical parameters, such as sex and familial history of AF, are presented as counts/numbers as well as percentages. An unpaired Student’s t-test and a one-way analysis of variance followed by the Tukey–Kramer post-hoc test were utilized for continuous data to evaluate the differences between the two groups and among ≥3 groups, respectively. A Chi-square/χ2 or Fisher’s exact test was utilized for categorical data, as appropriate, to assess the differences between the two groups. A two-tailed p <0.05 indicated significant difference. Statistical analyses were conducted with the aid of SPSS (version 25.0; IBM, USA).
3. Results
3.1. Clinical and Demographic Characteristical Profiles of the Pedigree Members and Other Study Subjects
As portrayed in Figure 1, a four-generation pedigree comprising 28 members with idiopathic AF (arbitrarily termed Pedigree AF-001) was recruited, including 25 living pedigree members.
In Pedigree AF-001, eight members, encompassing four female and four male members, had a definite diagnose of AF in terms of the electrocardiographic findings/medical records. No environmental/acquired risk factors prone to AF were detected in the members from Pedigree AF-001, such as obesity, obstructive sleep apnea, hyperthyroidism, coronary artery disease, essential arterial hypertension, dilated/hypertrophic cardiomyopathy, acute myocarditis, chronic heart failure, cardiac surgery, pulmonary heart disease, chronic kidney disease, and diabetes mellitus. The index patient (member II-6 from Pedigree AF-001), a 66-year-old female individual with 15 years of AF history, was referred to the local hospital due to an acute attack of syncope. One representative electrocardiogram showing AF of the index patient was provided in Figure 2.
The proband (member II-6 in Pedigree AF-001) underwent a successful catheter-based radiofrequency ablation for AF during this hospitalization. Her mother (member I-2 from Pedigree AF-001) had 24 years of AF history and died of an acute attack of cerebral stroke at 69 years of age. The index patient’s elder brother (member II-1 from Pedigree AF-001) had 20 years of AF history and died of stroke at the age of 64 years. The proband’s younger brother (member II-7 in Pedigree AF-001), a 63-year-old male member with 11 years of AF history, underwent a successful radiofrequency ablation of AF aged 58 years. The index case’s elder niece (member III-2 from Pedigree AF-001), a 48-year-old member with four years of AF history, underwent a successful radiofrequency ablation of AF at the age 46 years. The index case’s other relatives affected with AF had a medical history of taking antiarrhythmic drugs but did not undergo surgical/catheter-based therapy for AF until recruitment. The index case’s unaffected relatives, including11 male and nine female individuals, had no history of AF episode, with normal electrocardiograms. The basic clinical and demographic characteristic profiles of the pedigree members suffering AF are narrated in Table 1.
Additionally, all the pedigree members suffering AF also manifested intellectual impairment, developmental delay of language, and diverse facial dysmorphisms including broad nasal bridges, wide mouths, and teeth anomalies. The index case’s elder brother (member II-1 from Pedigree AF-001) had also episodes of seizures.
Additionally, another group of 236 cases affected with idiopathic AF underwent clinical investigation in contrast 312 healthy persons with no familial history of AF, who were enlisted as control subjects. The demographic and baseline clinical characteristic data of this cohort of AF cases along with the control people are described in Table 2.
3.2. Identificaton of Two Novel SOX5 Variations Contributing to AF
WES analysis was completed in six AF members (II-6, II-7, III-2, III-7, III-10 and IV-1) and seven healthy members (II-2, II-3, II-5, II-8, III-1, IV-4 and IV-5) from Pedigree AF-001 (Figure 1), by which only the pathogenic variation of chr12: 23,999,043C>T (GRCh37.p13/GCF_000001405.25/hg19: NC_000012.11), equal to chr12: 24,246,045C>T (GRCh38.p14/GCF_000001405.40/hg38: NC_000012.12) or NM_006940.6: c.355C>T; p.(Gln119*), was discovered to co-segregate with AF, and verified by Sanger sequencing assays to be in co-segregation with AF in the entire family (Pedigree AF-001). The sequencing chromatogram traces illustrating the heterozygous c.355C>T variation in SOX5 along with its wild type as a sequence control are provided in Figure 3.
Moreover, Sanger sequencing examination of the entire coding regions and splicing donors/acceptors of SOX5 was implemented in all the research participants utilizing the primer pairs presented in Table 3, which confirmed that the variation of NM_006940.6: c.355C>T; p.(Gln119*) in SOX5 was shared by all the AF family members but by none of the unaffected family members of Pedigree AF-001. Genetic analysis of Pedigree AF-001 indicated that AF was transmitted in an autosomal-dominant mode.
Additionally, Sanger sequencing examination of the whole coding regions along with splicing junction sites of the SOX5 gene in another group of 236 cases suffering from idiopathic AF unveiled a heterozygous SOX5 variation of NM_006940.6: c.640G>T; p.(Glu214*), residing in the fifth coding exon of SOX5 and resulting in a conversion of glutamic acid codon to stop codon at amino acid position 214 of SOX5, in one male case who was aged 41 years, had no family history of AF, but also suffered intellectual impairmwnt, delayed language development, and mild facial dysmorphisms characterized by broad nasal bridges and teeth abnormalities. This SOX5 variation was not observed in his parents, who had normal electrocardiograms without AF, indicating a de novo mutation. The sequencing chromatogram traces exhibiting the c.640G>T variation in SOX5 together with its wild type as a sequence control are given in Figure 4.
A representative electrocardiogram indicating AF from the case harboring the SOX5 c.640G>T variation was shown in Figure 5
Neither of the idenified two SOX5 nutations responsible for AF was found in the 624 human control chromosomes, or in the databases of gnomAD and dbSNP, confirming the novelty of the two SOX5 nutations.
3.3. Functional failure of Gln119*- or Glu214*-Mutant SOX5 to Transactivate GJA1
As displayed in Figure 6 (A), in cultivated COS-7 cells transfected with multiple expression plasmids, encompassing empty pcDNA™3.1(+) plasmid (‒), wild-type human SOX5-pcDNA™3.1(+) plasmid (SOX5), and Gln119*-mutant human SOX5-pcDNA™3.1(+) plasmid (Gln119*), singly or together, SOX5 and Gln119* induced transactivation of GJA1 by ~9-fold and ~1-fold, respectively (SOX5 vs Gln119*: t = 13.3255; p = 0.0002). When SOX5 and Gln119* were transfected together, the induced transcriptional activation of GJA1 was ~6-fold (SOX5 vs Gln119* + SOX5: t = 6.0364; p = 0.0038). Additionally, similar results were given when multiple comparisons were performed (F = 96.4652, p = 6.045 × 10−8). Specifically, for (‒) vs SOX5, t = 8.3967; p < 0.0001; for (‒) vs Gln119*, t = 0.0367; p = 1.0000; for (‒) vs SOX5 + (‒), t = 4.9753; p < 0.0001; for (‒) vs SOX5 +Gln119*, t = 4.4033; p < 0.0001; for SOX5 vs Gln119*, t = 8.3600; p < 0.0001; for SOX5 vs SOX5 + (‒), t = 3.4213; p = 0.0004; for SOX5 vs SOX5 + Gln119*, t = 3.9933; p = 0.0001; for Gln119* vs SOX5 + (‒), t = 4.9387; p < 0.0001; for Gln119* vs SOX5 + Gln119*, t = 4.3667; p < 0.0001; for SOX5 + (‒) vs SOX5 + Gln119*, t = 0.5720; p = 0.07978. Similarly, as shown in Figure 6 (B), SOX5 and Glu214* transcriptionally activated GJA1 by ~10-fold and ~1-fold, respectively (SOX5 vs Glu214*: t = 12.9871; p = 0.0002). When SOX5 and Glu214* were transfected in combination, the induced transactivation of GJA1 was ~6-fold (SOX5 vs Glu214* + SOX5: t = 6.0837; p = 0.0037). Besides, equal statistical results were generated when multiple comparisons were conducted (F = 97.2192, p = 5.818 × 10−8). Specifically, for (‒) vs SOX5, t = 8.9833; p < 0.0001; for (‒) vs Glu214*, t = 0.0733; p = 0.9999; for (‒) vs SOX5 + (‒), t = 5.4533; p < 0.0001; for (‒) vs SOX5 +Glu214*, t = 4.6233; p < 0.0001; for SOX5 vs Glu214*, t = 8.91; p < 0.0001; for SOX5 vs SOX5 + (‒), t = 3.53; p = 0.0006; for SOX5 vs SOX5 + Glu214*, t = 4.36; p < 0.0001; for Glu214* vs SOX5 + (‒), t = 5.38; p < 0.0001; for Glu214* vs SOX5 + Glu214*, t = 4.55; p < 0.0001; for SOX5 + (‒) vs SOX5 + Glu214*, t = 0.83; p = 0.5779.
3.4. Inability of Gln119*-or Glu214*-Mutant SOX5 to Induce Transactivation of SCN5A singly or Synergistically with SHOX2
As exhibited in Figure 7 (A), in HEK-293 cells cultivated in vitro expressing multiple plasmids, including empty pcDNA™3.1(+) plasmid (‒), wild-type human SHOX2-pcDNA™3.1(+) plasmid (SHOX2), wild-type human SOX5-pcDNA™3.1(+) plasmid (SOX5), and Gln119*-mutant human SOX5-pcDNA™3.1(+) plasmid (Gln119*), separately or in both, SOX5 and Gln119* induced transactivation of SCN5A by ~6-fold and ~1-fold, respectively (SOX5 vs Gln119*: t = 10.0999; p = 0.0005). Together with SHOX2, SOX5 and Gln119* transcriptionally activated SCN5A by ~18-fold and ~4-fold, respectively (SOX5 + SHOX2 vs Gln119* + SHOX2: t = 10.1226; p = 0.0005). In addition, similar results were obtained if multiple comparisons were made (F = 100.0152, p = 2.356 × 10−9). Specifically, for (‒) vs SHOX2, t = 3.3533; p = 0.0310; for (‒) vs SOX5, t = 5.36; p = 0.0009; for (‒) vs Gln119*, t = 0.0633; p = 1.0000; for (‒) vs SOX5 + SHOX2, t = 17.45; p < 0.0001; for (‒) vs Gln119* +SHOX2, t = 2.8233; p = 0.0811; for SHOX2 vs SOX5, t = 2.0067; p = 0.3131; for SHOX2 vs Gln119*, t = 3.29; p = 0.0348; for SHOX2 vs SOX5 + SHOX2, t = 14.0967; p < 0.0001; for SHOX2 vs Gln119 + SHOX2, t = 0.53; p = 0.9909; for SOX5 vs Gln119*, t = 5.2967; p = 0.0010; for SOX5 vs SOX5 + SHOX2, t = 12.09; p < 0.0001; for SOX5 vs Gln119 + SHOX2, t = 2.5367; p = 0.1339; for Gln119* vs SOX5 + SHOX2, t = 17.3867; p < 0.0001; for Gln119* vs Gln119 + SHOX2, t = 2.76; p = 0.0907; for SOX5 + SHOX2 vs Gln119* + SHOX2, t = 14.6267; p < 0.0001. Similarly, as depicted in Figure 7 (B), SOX5 and Glu214* transcriptionally activated SCN5A by ~7-fold and ~1-fold, respectively (SOX5 vs Glu214*: t = 14.1459; p = 0.0001). In the presence of SHOX2, SOX5 and Glu214* transcriptionally activated SCN5A by ~19-fold and ~4-fold, respectively (SOX5 + SHOX2 vs Glu214* + SHOX2: t = 12.3580; p = 0.0002). In addition, similar results were generated if multiple comparisons were carried out (F = 153.8289, p = 1.889 × 10−10). In detail, for (‒) vs SHOX2, t = 3.4833; p = 0.0071; for (‒) vs SOX5, t = 5.46; p = 0.0001; for (‒) vs Glu214*, t = 0.007; p = 1.0000; for (‒) vs SOX5 + SHOX2, t = 18.1133; p < 0.0001; for (‒) vs Glu214* +SHOX2, t = 3.13; p = 0.0154; for SHOX2 vs SOX5, t = 1.9767; p = 0.1804; for SHOX2 vs Glu214*, t = 3.5533; p = 0.0061; for SHOX2 vs SOX5 + SHOX2, t = 14.63; p < 0.0001; for SHOX2 vs Glu214* + SHOX2, t = 0.3533; p = 0.9968; for SOX5 vs Glu214*, t = 5.53; p = 0.0001; for SOX5 vs SOX5 + SHOX2, t = 12.6533; p < 0.0001; for SOX5 vs Glu214*+ SHOX2, t = 2.33; p = 0.0874; for Glu214* vs SOX5 + SHOX2, t = 18.1833; p < 0.0001; for Glu214* vs Glu214* + SHOX2, t = 3.2; p = 0.0132; for SOX5 + SHOX2 vs Glu214* + SHOX2, t = 14.9833; p < 0.0001.
4. Discussion
The SOX family of proteins comprises a highly conserved cluster of transcription factors characterized by harboring the HMG domain composed of three α-helices, which binds the core DNA sequence 5’-AACAAT-3’ in the promoters of downstream genes, regulating the expression levels of target genes [85]. This HMG domain of a SOX protein not merely binds target DNA, but also mediates subcellular trafficking and interactions with transcriptionally cooperative partners/co-factors [85].To date, in vertebrates a group of 20 SOX proteins has been found, which are classified into eight subgroups (from SOXA to SOXH) in terms of the amino acid sequence conservation/identity within the HMG motif as well as the existence of other domains [85]. It has been demonstrated that SOX proteins exert pivotal effect on the embryonic development of most organs and postnatal pathological processes in various tissues derived from the endoderm, mesoderm, and ectoderm, encompassing the cardiovascular system, brain, bone, cartilage, lymphatic system, retina, pancreas, and hematopoietic system [86,87]. In addition, it has been reported that genetically defective SOX proteins contribute to many genetic diseases, so-called ‘SOXopathies’, affecting cardiovascular system, urinary system, central nervous system, muscular system, reproductive system, auditory and ocular systems, as well as skeleton, skin and hair [86,87]. The SOXD subgroup of transcription factors includes SOX13, SOX6 and SOX5, of which SOX5 consists of 763 amino acids, and contains two coiled-coil motifs (amino acids 193-274 and amino acids 448-493) located at the N-terminus and a family-restricted HMG domain (amino acids 555-630) located at the C-terminus [85,88]. The coiled-coil domain functions to regulate SOXD protein dimerization (homo- and hetero-dimerization) and promote preferential binding to the adjoining HMG recognition sites by adding flexibility [85,88]. The SOX5 gene is mapped on human chromosome 12p12.1, which produces multiple transcript isoforms by alternative transcription start site and precursor messenger RNA splicing, including the longest isoform (originally named as L-SOX5; Nucleotide accession No.: NM_006940.6) and the shortest isoform (also termed as S-SOX5; Nucleotide accession No.: NM_178010.4) [85]. The longest isoform of SOX5 encodes a protein with 763 amino acids (encoded by exons 1-15), which is amply expressed in various tissues, encompassing heart and brain, playing a critical role in the development and remodeling of cardiovascular and cerebrovascular systems, predominantly participating in cell proliferation, cell cycle regulation, cellular migration and invasion, cell apoptosis, and inflammatory response [85,89,90,91,92], while the shortest isoform of SOX5 encodes a 377-amino-acid protein (encoded mainly by exons 10-15), which is specifically and highly expressed in the testis, playing a crucial role in the morphogenesis and function of motor cilia in the testes/spermatozoa [93,94,95]. The full-length L-SOX5 is usually referred to as SOX5 and this appellation is consistently adopted in the references, mainly because L-SOX5 is functionally and structurally equal to SOX13 and SOX6, and contains a glutamine-rich region and a leucine zipper, which allows dimerization with other SOX proteins, such as SOX9, to cooperatively activate target genes [95]. In the current research, two new SOX5 mutations linked to AF were discovered, including c.355C>T (p.Gln119*) locating at exon 3 and c.640G>T (p.Glu214*) locating at exon 5, hence were anticipated to produce truncating L-SOX5 proteins without HMG domain along with coiled-coil domain and fail to bind target promoters to transactivate downstream genes but have no effect on S-SOX5. Biological assay demonstrated that both the Gln119*- mutant SOX5 and the Glu214*-mutant SOX5 lost the ability to transactivate the expression of GJA1, an AF-causative gene [96]. Furthermore, both the Gln119*- mutant SOX5 and the Glu214*-mutant SOX5 failed to transactivate the expression of SCN5A, alone or synergistically with SHOX2, and pathogenic variations in both SCN5A and SHOX2 have been discovered to result in AF [81,97,98,99,100]. Therefore, genetically compromised SOX5 predisposes to AF at least in part by lowering the expression levels of its target genes such as SCN5A and GJA1.
In humans, the correlation of genetic variations near the SOX5 gene to AF has been clinically investigated. Olesen and coworkers [101] enlisted 209 patients suffering from AF and 534 control subjects without AF, and a total of 8 SNPs were genotyped in study participants by utilizing TaqMan assays. As a result, three SNPs were discovered to be associated with AF, including rs11047543 near to SOX5, rs2200733 closest to PITX2, and rs3807989 adjacent to CAV1. Even if correction was made for multiple testing, rs11047543 and rs2200733 were both still associated with AF [101]. Pfeufer and colleagues [102] performed a meta-analysis of whole-genome association investigations for the hereditary determinants of electrocardiographic PR intervals and their relation to AF in 28,517 European-descent individuals from seven community-based studies. As a result, nine loci were found to be significantly associated with PR intervals, of which five loci were also significantly associated with AF, including rs11047543 near SOX5 (51 kb 5′ of C12orf67), rs3807989 at intron 2 of CAV1/CAV2, rs11708996 at intron 14 of SCN5A, rs251253 next to NKX2-5 (3 kb 5′ of C5orf41), and rs6800541 at intron 14 of SCN10A [102]. Park and partners [103] genotyped 16 SNPs (including rs11047543, rs2106261, rs6800541, rs13376333, rs2200733, rs3825214, rs10465885, rs3807989, rs853445, rs7193343, rs17042171, rs251253, rs10033464, rs11708996, rs17570669, and rs6843082) in a total of 89 Korean patients with early-onset and drug-refractory AF who experienced catheter-based ablation for AF, and observed that three SNPs, including rs11047543 closest to SOX5 (12p12), rs3825214 neighboring TBX5 (12q24), and rs7193343 adjacent to ZFHX3 (16q22), were associated with the enhanced risk for the recurrence of AF after catheter-based radiofrequency ablation therapy, and the risk-allele number of these three SNPs could independently predict the recurrence of AF. Vogel et al. [104] examined the relationship between eight SNPs (located within or near the genes SOX5, KCNN3, CAV1, PITX2, KCNJ5, ZFHX3, and MYH7) and the risks of AF occurrence and recurrence in 259 AF patients and 108 control persons and revealed that the variation of rs11047543 near to SOX5 conferred a higher risk on the recurrence of AF after treatment with direct current cardioversion. In addition, Seifert et al. [105] explored the correlation between the four SNPs previously implicated with AF and PR interval (rs11047543, rs3807989, rs13376333, and rs2200733) and the electrocardiographic P-wave morphology in 176 cases affected with AF, and found that two SNPs, including rs11047543 next to SOX5 and rs3807989 in the vicinity of CAV1/CAV2, were significantly associated with abnormal P-wave morphology, implying significant effect on atrial conduction properties. Collectively, these observational results suggest that common genetic variations near SOX5 are associated with the emergence and recurrence of AF, though the biological pathway/pathogenic mechanism by which the above-mentioned SNPs lead to the development of AF remains to be experimentally elucidated.
It has been validated that SOX proteins, including SOX5, are involved in the regulation of multiple signaling pathways, and in the canonic WNT pathway, SOX5 functions as a key player to compete with T-cell factors/lymphoid enhancer factors for binding to β-catenin, resulting in the repression of the WNT/β-catenin pathway and hence the reduced expression levels of the WNT/β-catenin target genes [95,106]. The WNT pathway plays a key role in embryogenesis, tissue homeostasis, and a wide variety of pathophysiological processes including the activation of adaptive cardiac remodeling and the increase of cardiac fibrosis [107,108]. Atrial fibrosis has been substantiated to be a hallmark of atrial structural remodeling and electrophysiological dysfunction/heterogeneous conduction, creating a pivotal substrate in favor of the initiation and perpetuation of AF [108,109,110], and ablation of fibrotic atrial areas has been demonstrated to improve the therapeutic results of catheter ablation for AF [111]. Additionally, SOX5 can also promote fibrosis by up-regulating the expression levels of N-fibronectin, cadherin and vimentin [112]. Therefore, SOX5 haploinsufficiency may predispose to AF by increasing the WNT/β-catenin activity, generating an important matrix in favor of the occurrence and maintenance of AF.
The critic roles of SOX5 in cardiac organogenesis and structural remodeling have been shown in animals [92,113,114,115]. In adult Drosophila models, knockdown of Sox102F, a fruit fly ortholog of human SOX5, led to a significant decrease in resting heart rate, ventricular wall velocity and cardiac chamber volume, along with a significant increase in ventricular wall thickness with disrupted myofibril structure and WNT signaling transduction [113]. In mice, knockout of Sox5 led to neonatal lethality, with respiratory distress attributable to anomalous development of lungs, and mild skeletal anomalies, and double knockout of Sox5 and Sox6 resulted in murine embryonic death, with more severe pulmonary and skeletal abnormalities [114,115]. Unfortunately, the early death of Sox5-null mouse models prevented analyzing the effect of Sox5 on adult murine cardiac function [114,115]. In the murine hearts with doxorubicin-induced dilated cardiomyopathy, the expression of SOX5 was increased, the WNT/β-catenin pathway and apoptosis were activated, and inflammation and collagen deposition were also increased, which were consistent with the findings from the hearts of patients with dilated cardiomyopathy [92]. In addition, in the hearts, the action potential elicits Ca2+ entry into cardiac myocytes via L-type Ca2+ channels, while in murine myoblast cells, knockdown of Sox5 led to significant decrease in the maximum charge movement, generated by voltage-gated L-type Ca++ channels [116]. In endothelial cells, SOX5 regulates shear stress-regulated gene expression in a nitric oxide-dependent mode [117], and nitric oxide is a key molecular for endothelial and cardiovascular function and has been associated with AF [118]. Collectively, these investigations indicate that SOX5 may regulate voltage-gated L-type Ca2+ channels, and that genetically defective SOX5 may contribute to AF through altering atrial action potential and WNT signaling transduction as well as atrioventricular conduction.
Previously in humans, dozens of deleterious SOX5 variations, encompassing nonsense, missense and frame-shifting variations, were implicated in the etiopathogenesis of Lamb–Shaffer Syndrome, an uncommon genetic disease with a wide spectrum of clinical manifestations, including intellectual impairment/disability, developmental delay of language, attention deficits, seizures, hypotonia, autism spectrum disorder, hyperactivity, scoliosis, visual problem/strabismus, short stature, abnormal hands/feet, and diverse facial dysmorphisms, such as a bulbous nasal tip, a wide mouth, frontal bossing, deep-set eyes, prominent philtra ridges, and epicanthal folds [85,94,119]. In the current research, two novel SOX5 variations, NM_006940.6: c.355C>T; p.(Gln119*) and NM_006940.6: c.640G>T; p.(Glu214*), were causally linked to AF as a prominent clinical manifestation of Lamb–Shaffer syndrome, therefore expanding the SOX5-related phenotypic spectrum. Given that a larger part of AF occurs paroxysmal or sub-clinical with no apparent symptoms [2], the present investigation suggests that a long-term dynamic electrocardiographic screening of the cases suffering from Lamb–Shaffer syndrome attributed to SOX5 variations is needed for the timely diagnosis of AF.
5. Conclusions
The current research indicates SOX5 as a new gene responsible for AF, which adds more insight to the molecular mechanism underpinning AF, and offers a molecular target for genetic counseling and potential individualized medical management of AF in a subset of cases.
Author Contributions
Methodology and conceptualization, D.-L.Z., Y.-J.X. and Y.-Q.Y.; collecting patients, D.-L.Z., X.-B.Q., N.L., C.-X.Y., Z.-P.K., Y.-J.X. and Y.-Q.Y.; performed the experiment, D.-L.Z., C.-X.Y., Y.-J.X. and Y.-Q.Y.; data curation, D.-L.Z., Y.-J.X. and Y.-Q.Y.; formal analysis, D.-L.Z., X.-B.Q., N.L., Y.-Y.D., C.-X.Y., Z.-P.K., Y.-J.X. and Y.-Q.Y.; validation and visualization, D.-L.Z., Y.-J.X. and Y.-Q.Y.; writing—original draft preparation, D.-L.Z., X.-B.Q., N.L., Y.-Y.D., C.-X.Y., Z.-P.K., Y.-J.X. and Y.-Q.Y.; writing—review and editing, D.-L.Z., Y.-J.X. and Y.-Q.Y.; review the final draft, D.-L.Z., Y.-J.X. and Y.-Q.Y.; project administration, D.-L.Z., Y.-J.X. and Y.-Q.Y.; funding acquisition, Y.-J.X.; supervision, D.-L.Z. and Y.-Q.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Natural Science Foundation of Shanghai (23ZR1450000).
Institutional Review Board Statement
This research was completed in strict abidance by the principles outlined in Declaration of Helsinki and the Medical Ethical Committee of Shanghai Fifth People′s Hospital approved the protocols (ethical approval code: 2022-179; ethical approval date: 23 October 2024).
Informed Consent Statement
An informed consent form was signed by the study subjects.
Data Availability Statement
All the data generated in the present research are included in this manuscript.
Acknowledgments
The authors would like to thank the study participants for devotion to the current research.
Conflicts of Interest
There is no conflict of interest.
References
- Liu, H.; Lu, L.; Xiong, H.; Fan, C.; Fan, L.; Lin, Z.; Zhang, H. A Novel Approach to Dual Feature Selection of Atrial Fibrillation Based on HC-MFS. Diagnostics 2024, 14, 1145. [Google Scholar] [CrossRef] [PubMed]
- Joglar, J.A.; Chung, M.K.; Armbruster, A.L.; Benjamin, E.J.; Chyou, J.Y.; Cronin, E.M.; Deswal, A.; Eckhardt, L.L.; Goldberger, Z.D.; Gopinathannair, R.; et al. 2023 ACC/AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation 2024, 149, e1–e156. [Google Scholar] [CrossRef] [PubMed]
- January, C.T.; Wann, L.S.; Alpert, J.S.; Calkins, H.; Cigarroa, J.E.; Cleveland, J.C., Jr.; Conti, J.B.; Ellinor, P.T.; Ezekowitz, M.D.; Field, M.E.; et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation 2014, 130, e199–e267. [Google Scholar] [CrossRef] [PubMed]
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Lin, J.; Wu, X.Y.; Long, D.Y.; Jiang, C.X.; Sang, C.H.; Tang, R.B.; Li, S.N.; Wang, W.; Guo, X.Y.; Ning, M.; et al. Asymptomatic atrial fibrillation among hospitalized patients: clinical correlates and in-hospital outcomes in Improving Care for Cardiovascular Disease in China-trial Fibrillation. Europace 2023, 25, euad272. [Google Scholar] [CrossRef]
- Abdelhamid, K.; Reissenberger, P.; Piper, D.; Koenig, N.; Hoelz, B.; Schlaepfer, J.; Gysler, S.; McCullough, H.; Ramin-Wright, S.; Gabathuler, A.L.; et al. Fully Automated Photoplethysmography-Based Wearable Atrial Fibrillation Screening in a Hospital Setting. Diagnostics 2025, 15, 1233. [Google Scholar] [CrossRef]
- Turakhia, M.P.; Guo, J.D.; Keshishian, A.; Delinger, R.; Sun, X.; Ferri, M.; Russ, C.; Cato, M.; Yuce, H.; Hlavacek, P. Contemporary prevalence estimates of undiagnosed and diagnosed atrial fibrillation in the United States. Clin. Cardiol. 2023, 46, 484–493. [Google Scholar] [CrossRef]
- Andrade, J.G.; Deyell, M.W.; Bennett, R.; Macle, L. Assessment and management of asymptomatic atrial fibrillation. Heart 2024, 110, 675–682. [Google Scholar] [CrossRef]
- Hulsmans, M.; Schloss, M.J.; Lee, I.H.; Bapat, A.; Iwamoto, Y.; Vinegoni, C.; Paccalet, A.; Yamazoe, M.; Grune, J.; Pabel, S.; et al. Recruited macrophages elicit atrial fibrillation. Science 2023, 381, 231–239. [Google Scholar] [CrossRef]
- Chuang, H.J.; Lin, L.C.; Yu, A.L.; Liu, Y.B.; Lin, L.Y.; Huang, H.C.; Ho, L.T.; Lai, L.P.; Chen, W.J.; Ho, Y.L.; et al. Predicting impaired cardiopulmonary exercise capacity in patients with atrial fibrillation using a simple echocardiographic marker. Heart Rhythm 2024, 21, 1493–1499. [Google Scholar] [CrossRef]
- Beisenbayeva, A.; Bekbossynova, M.; Bakytzhanuly, A.; Aleushinova, U.; Bekmetova, F.; Chinybayeva, A.; Abdrakhmanov, A.; Beyembetova, A. Improvements in Cardiopulmonary Exercise Test Results in Atrial Fibrillation Patients After Radiofrequency Ablation in Kazakhstan. Diagnostics 2024, 14, 2355. [Google Scholar] [CrossRef]
- Foster-Witassek, F.; Aebersold, H.; Aeschbacher, S.; Ammann, P.; Beer, J.H.; Blozik, E.; Bonati, L.H.; Cattaneo, M.; Coslovsky, M.; Felder, S.; et al. Longitudinal Changes in Health-Related Quality of Life in Patients With Atrial Fibrillation. J. Am. Heart Assoc. 2023, 12, e031872. [Google Scholar] [CrossRef] [PubMed]
- Zeitler, E.P.; Li, Y.; Silverstein, A.P.; Russo, A.M.; Poole, J.E.; Daniels, M.R.; Al-Khalidi, H.R.; Lee, K.L.; Bahnson, T.D.; Anstrom, K.J.; et al. Effects of Ablation Versus Drug Therapy on Quality of Life by Sex in Atrial Fibrillation: Results From the CABANA Trial. J. Am. Heart Assoc. 2023, 12, e027871. [Google Scholar] [CrossRef] [PubMed]
- Särnholm, J.; Skúladóttir, H.; Rück, C.; Axelsson, E.; Bonnert, M.; Bragesjö, M.; Venkateshvaran, A.; Ólafsdóttir, E.; Pedersen, S.S.; Ljótsson, B.; et al. Cognitive Behavioral Therapy Improves Quality of Life in Patients With Symptomatic Paroxysmal Atrial Fibrillation. J. Am. Coll. Cardiol. 2023, 82, 46–56. [Google Scholar] [CrossRef]
- Lee, J.M.; Cha, M.J.; Nam, G.B.; Choi, K.J.; Sun, B.J.; Kim, D.H.; Song, J.M.; Kang, D.H.; Song, J.K.; Cho, M.S. Incidence and predictors of left atrial thrombus in patients with atrial fibrillation under anticoagulation therapy. Clin. Res. Cardiol. 2024, 113, 1242–1250. [Google Scholar] [CrossRef]
- Okada, M.; Inoue, K.; Tanaka, N.; Tanaka, K.; Hirao, Y.; Iwakura, K.; Egami, Y.; Masuda, M.; Watanabe, T.; Minamiguchi, H.; et al. Impact of left atrial appendage flow velocity on thrombus resolution and clinical outcomes in patients with atrial fibrillation and silent left atrial thrombi: insights from the LAT study. Europace 2024, 26, euae120. [Google Scholar] [CrossRef]
- Troisi, F.; Guida, P.; Vitulano, N.; Quadrini, F.; Di Monaco, A.; Patti, G.; Grimaldi, M. Atrial Thrombosis Prevalence Before Cardioversion or Catheter Ablation of Atrial Fibrillation: An Updated Systematic Review and Meta-Analysis of Direct Oral Anticoagulants Versus Vitamin K Antagonists. Am. J. Cardiol. 2024, 218, 77–85. [Google Scholar] [CrossRef]
- Gupta, S.; Lutnik, M.; Cacioppo, F.; Lindmayr, T.; Schuetz, N.; Tumnitz, E.; Friedl, L.; Boegl, M.; Schnaubelt, S.; Domanovits, H.; et al. Computed Tomography to Exclude Cardiac Thrombus in Atrial Fibrillation-An 11-Year Experience from an Academic Emergency Department. Diagnostics 2024, 14, 699. [Google Scholar] [CrossRef]
- Gurol, M.E.; Wright, C.B.; Janis, S.; Smith, E.E.; Gokcal, E.; Reddy, V.Y.; Merino, J.G.; Hsu, J.C. Stroke Prevention in Atrial Fibrillation: Our Current Failures and Required Research. Stroke 2024, 55, 214–225. [Google Scholar] [CrossRef]
- Chao, T.F.; Potpara, T.S.; Lip, G.Y.H. Atrial fibrillation: stroke prevention. Lancet Reg. Health Eur. 2024, 37, 100797. [Google Scholar] [CrossRef]
- Goh, B.; Bhaskar, S.M.M. Evaluating Machine Learning Models for Stroke Prognosis and Prediction in Atrial Fibrillation Patients: A Comprehensive Meta-Analysis. Diagnostics 2024, 14, 2391. [Google Scholar] [CrossRef]
- Elsheikh, S.; Hill, A.; Irving, G.; Lip, G.Y.H.; Abdul-Rahim, A.H. Atrial fibrillation and stroke: State-of-the-art and future directions. Curr. Probl. Cardiol. 2024, 49, 102181. [Google Scholar] [CrossRef]
- Certo Pereira, J.; Lima, M.R.; Moscoso Costa, F.; Gomes, D.A.; Maltês, S.; Cunha, G.; Dores, H.; Adragão, P. Stroke in Athletes with Atrial Fibrillation: A Narrative Review. Diagnostics 2024, 15, 9. [Google Scholar] [CrossRef]
- Lip, G.Y.H.; Proietti, M.; Potpara, T.; Mansour, M.; Savelieva, I.; Tse, H.F.; Goette, A.; Camm, A.J.; Blomstrom-Lundqvist, C.; Gupta, D.; et al. Atrial fibrillation and stroke prevention: 25 years of research at EP Europace journal. Europace 2023, 25, euad226. [Google Scholar] [CrossRef]
- Blum, S.; Conen, D. Mechanisms and Clinical Manifestations of Cognitive Decline in Atrial Fibrillation Patients: Potential Implications for Preventing Dementia. Can. J. Cardiol. 2023, 39, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Zelnick, L.R.; An, J.; Harrison, T.N.; Lee, M.S.; Singer, D.E.; Fan, D.; Go, A.S. Incident Atrial Fibrillation and Risk of Dementia in a Diverse, Community-Based Population. J. Am. Heart Assoc. 2023, 12, e028290. [Google Scholar] [CrossRef] [PubMed]
- Kogelschatz, B.; Zenger, B.; Steinberg, B.A.; Ranjan, R.; Jared Bunch, T. Atrial fibrillation and the risk of early-onset dementia and cognitive decline: An updated review. Trends Cardiovasc. Med. 2024, 34, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Venier, S.; Vaxelaire, N.; Jacon, P.; Carabelli, A.; Desbiolles, A.; Garban, F.; Defaye, P. Severe acute kidney injury related to haemolysis after pulsed field ablation for atrial fibrillation. Europace 2023, 26, euad371. [Google Scholar] [CrossRef]
- Jordan, F.; Knecht, S.; Isenegger, C.; Arnet, R.; Krisai, P.; Völlmin, G.; du Fay de Lavallaz, J.; Spreen, D.; Osswald, S.; Sticherling, C.; et al. Acute kidney injury after catheter ablation of atrial fibrillation: Comparison between different energy sources. Heart Rhythm 2024, 21, 1248–1249. [Google Scholar] [CrossRef]
- Odutayo, A.; Wong, C.X.; Hsiao, A.J.; Hopewell, S.; Altman, D.G.; Emdin, C.A. Atrial fibrillation and risks of cardiovascular disease, renal disease, and death: systematic review and meta-analysis. BMJ 2016, 354, i4482. [Google Scholar] [CrossRef]
- Winters, J.; Isaacs, A.; Zeemering, S.; Kawczynski, M.; Maesen, B.; Maessen, J.; Bidar, E.; Boukens, B.; Hermans, B.; van Hunnik, A.; et al. Heart Failure, Female Sex, and Atrial Fibrillation Are the Main Drivers of Human Atrial Cardiomyopathy: Results From the CATCH ME Consortium. J. Am. Heart Assoc. 2023, 12, e031220. [Google Scholar] [CrossRef]
- Masuda, M.; Matsuda, Y.; Uematsu, H.; Sugino, A.; Ooka, H.; Kudo, S.; Fujii, S.; Asai, M.; Okamoto, S.; Ishihara, T.; et al. Prognostic impact of atrial cardiomyopathy: Long-term follow-up of patients with and without low-voltage areas following atrial fibrillation ablation. Heart Rhythm 2024, 21, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Vlachakis, P.K.; Theofilis, P.; Ktenopoulos, N.; Patoulias, D.; Fyntanidou, B.; Antoniadis, A.P.; Fragakis, N. Atrial Cardiomyopathy in Atrial Fibrillation: A Multimodal Diagnostic Framework. Diagnostics 2025, 15, 1207. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Vlachakis, P.K.; Ktenopoulos, N.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Atrial Cardiomyopathy in Atrial Fibrillation: Mechanistic Pathways and Emerging Treatment Concepts. J. Clin. Med. 2025, 14, 3250. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, T.C.; Dahm, C.C.; Preis, S.R.; Lin, H.; Trinquart, L.; Benjamin, E.J.; Kornej, J. The bidirectional association between atrial fibrillation and myocardial infarction. Nat. Rev. Cardiol. 2023, 20, 631–644. [Google Scholar] [CrossRef]
- Popovic, B.; Varlot, J.; Humbertjean, L.; Sellal, J.M.; Pace, N.; Hammache, N.; Fay, R.; Eggenspieler, F.; Metzdorf, P.A.; Camenzind, E. Coronary Embolism Among Patients With ST-Segment-Elevation Myocardial Infarction and Atrial Fibrillation: An Underrecognized But Deadly Association. J. Am. Heart Assoc. 2024, 13, e032199. [Google Scholar] [CrossRef]
- Frederiksen, T.C.; Benjamin, E.J.; Trinquart, L.; Lin, H.; Dahm, C.C.; Christiansen, M.K.; Jensen, H.K.; Preis, S.R.; Kornej, J. Bidirectional Association Between Atrial Fibrillation and Myocardial Infarction, and Relation to Mortality in the Framingham Heart Study. J. Am. Heart Assoc. 2024, 13, e032226. [Google Scholar] [CrossRef]
- Karlsson, E.; Kiviniemi, T.; Halminen, O.; Lehtonen, O.; Teppo, K.; Haukka, J.; Mustonen, P.; Putaala, J.; Linna, M.; Hartikainen, J.; et al. Temporal Relation Between Myocardial Infarction and New-Onset Atrial Fibrillation: Results from a Nationwide Registry Study. Am. J. Cardiol. 2024, 211, 49–56. [Google Scholar] [CrossRef]
- Haller, P.M.; Jarolim, P.; Palazzolo, M.G.; Bellavia, A.; Antman, E.M.; Eikelboom, J.; Granger, C.B.; Harrington, J.; Healey, J.S.; Hijazi, Z.; et al. Heart Failure Risk Assessment Using Biomarkers in Patients With Atrial Fibrillation: Analysis From COMBINE-AF. J. Am. Coll. Cardiol. 2024, 84, 1528–1540. [Google Scholar] [CrossRef]
- Xia, Y.; Jiang, J.; Fan, F.; Pan, Y.; Zhou, J.; Zhang, Y.; Li, J.; Liu, J.; Yang, N.; Hao, Y.; et al. Prevalence, Characteristics, and Treatment Strategy of Different Types of Heart Failure in Patients With Nonvalvular Atrial Fibrillation. J. Am. Heart Assoc. 2024, 13, e033941. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, R.; Yang, M.; Li, W.; Zhang, P.P.; Mo, B.F.; Wang, Q.S.; Chen, M.; Li, Y.G. Combined Radiofrequency Ablation and Left Atrial Appendage Closure in Atrial Fibrillation and Systolic Heart Failure. Diagnostics 2023, 13, 3325. [Google Scholar] [CrossRef] [PubMed]
- Bergonti, M.; Ascione, C.; Marcon, L.; Pambrun, T.; Della Rocca, D.G.; Ferrero, T.G.; Pannone, L.; Kühne, M.; Compagnucci, P.; Bonomi, A.; et al. Left ventricular functional recovery after atrial fibrillation catheter ablation in heart failure: a prediction model. Eur. Heart J. 2023, 44, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Choi, Y.Y.; Han, K.D.; Min, K.; Choi, H.Y.; Shim, J.; Choi, J.I.; Kim, Y.H. Atrial fibrillation is associated with increased risk of lethal ventricular arrhythmias. Sci. Rep. 2021, 11, 18111. [Google Scholar] [CrossRef] [PubMed]
- Fawzy, A.M.; Bisson, A.; Bentounes, S.A.; Bodin, A.; Herbert, J.; Lip, G.Y.H.; Fauchier, L. Ventricular arrhythmias and cardiac arrest in atrial fibrillation patients with pacemakers and implantable cardioverter-defibrillators. Eur. J. Intern. Med. 2023, 115, 70–78. [Google Scholar] [CrossRef]
- Lenhoff, H.; Järnbert-Petersson, H.; Darpo, B.; Tornvall, P.; Frick, M. Mortality and ventricular arrhythmias in patients on d,l-sotalol for rhythm control of atrial fibrillation: A nationwide cohort study. Heart Rhythm 2023, 20, 1473–1480. [Google Scholar] [CrossRef]
- Zuin, M.; Bertini, M.; Vitali, F.; Turakhia, M.; Boriani, G. Heart Failure-Related Death in Subjects With Atrial Fibrillation in the United States, 1999 to 2020. J. Am. Heart Assoc. 2024, 13, e033897. [Google Scholar] [CrossRef]
- Kirchhof P, Haas S, Amarenco P, Turpie AGG, Bach M, Lambelet M, Hess S, Camm AJ. Causes of death in patients with atrial fibrillation anticoagulated with rivaroxaban: a pooled analysis of XANTUS. Europace 2024, 26, euae183. [Google Scholar] [CrossRef]
- Wu, J.; Nadarajah, R.; Nakao, Y.M.; Nakao, K.; Wilkinson, C.; Cowan, J.C.; Camm, A.J.; Gale, C.P. Temporal trends of cause-specific mortality after diagnosis of atrial fibrillation. Eur. Heart J. 2023, 44, 4422–4431. [Google Scholar] [CrossRef]
- Obeid, M.J.; Zhou, J.; Sale, A.J.; Longacre, C.; Zeitler, E.P.; Andrade, J.; Mittal, S.; Piccini, J.P. Early mortality after inpatient versus outpatient catheter ablation in patients with atrial fibrillation. Heart Rhythm 2023, 20, 833–841. [Google Scholar] [CrossRef]
- Piccini, J.P.; Hammill, B.G.; Sinner, M.F.; Hernandez, A.F.; Walkey, A.J.; Benjamin, E.J.; Curtis, L.H.; Heckbert, S.R. Clinical course of atrial fibrillation in older adults: the importance of cardiovascular events beyond stroke. Eur. Heart J. 2014, 35, 250–256. [Google Scholar] [CrossRef]
- Deshmukh A, Iglesias M, Khanna R, Beaulieu T. Healthcare utilization and costs associated with a diagnosis of incident atrial fibrillation. Heart Rhythm O2 2022, 3, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Peigh, G.; Zhou, J.; Rosemas, S.C.; Roberts, A.I.; Longacre, C.; Nayak, T.; Schwab, G.; Soderlund, D.; Passman, R.S. Impact of Atrial Fibrillation Burden on Health Care Costs and Utilization. JACC Clin. Electrophysiol. 2024, 10, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Ngo, L.; Denman, R.; Hay, K.; Kaambwa, B.; Ganesan, A.; Ranasinghe, I. Excess Bed Days and Hospitalization Costs Associated With 30-Day Complications Following Catheter Ablation of Atrial Fibrillation. J. Am. Heart Assoc. 2023, 12, e030236. [Google Scholar] [CrossRef] [PubMed]
- Buja, A.; Rebba, V.; Montecchio, L.; Renzo, G.; Baldo, V.; Cocchio, S.; Ferri, N.; Migliore, F.; Zorzi, A.; Collins, B.; et al. The Cost of Atrial Fibrillation: A Systematic Review. Value Health 2024, 27, 527–541. [Google Scholar] [CrossRef]
- Elliott, A.D.; Middeldorp, M.E.; Van Gelder, I.C.; Albert, C.M.; Sanders, P. Epidemiology and modifiable risk factors for atrial fibrillation. Nat. Rev. Cardiol. 2023, 20, 404–417. [Google Scholar] [CrossRef]
- Lu, Y.; Sun, Y.; Cai, L.; Yu, B.; Wang, Y.; Tan, X.; Wan, H.; Xu, D.; Zhang, J.; Qi, L.; et al. Non-traditional risk factors for atrial fibrillation: epidemiology, mechanisms, and strategies. Eur. Heart J. 2025, 46, 784–804. [Google Scholar] [CrossRef]
- Kany, S.; Jurgens, S.J.; Rämö, J.T.; Christophersen, I.E.; Rienstra, M.; Chung, M.K.; Olesen, M.S.; Ackerman, M.J.; McNally, E.M.; Semsarian, C.; et al. Genetic testing in early-onset atrial fibrillation. Eur. Heart J. 2024, 45, 3111–3123. [Google Scholar] [CrossRef]
- Vinciguerra, M.; Dobrev, D.; Nattel, S. Atrial fibrillation: pathophysiology, genetic and epigenetic mechanisms. Lancet Reg. Health Eur. 2024, 37, 100785. [Google Scholar] [CrossRef]
- Owais, A.; Barney, M.; Ly, O.T.; Brown, G.; Chen, H.; Sridhar, A.; Pavel, A.; Khetani, S.R.; Darbar, D. Genetics and Pharmacogenetics of Atrial Fibrillation: A Mechanistic Perspective. JACC Basic Transl. Sci. 2024, 9, 918–934. [Google Scholar] [CrossRef]
- Ding, M.; Viet, N.N.; Gigante, B.; Lind, V.; Hammar, N.; Modig, K. Elevated Uric Acid Is Associated With New-Onset Atrial Fibrillation: Results From the Swedish AMORIS Cohort. J. Am. Heart Assoc. 2023, 12, e027089. [Google Scholar] [CrossRef]
- Xie, Z.; Liu, C.; Lu, X.; Chen, Z.; Zhang, N.; Wang, X.; Li, X.; Li, Y. Identification and Verification of Biomarkers and Immune Infiltration in Obesity-Related Atrial Fibrillation. Biology 2023, 12, 121. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Choi, E.Y. Multimodality Imaging in Patients with Hypertrophic Cardiomyopathy and Atrial Fibrillation. Diagnostics 2023, 13, 3049. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Jeong, Y.; Kim, Y.L.; Kang, M.; Kang, E.; Ryu, H.; Kim, Y.; Han, S.S.; Ahn, C.; Oh, K.H. Association of Chronic Kidney Disease With Atrial Fibrillation in the General Adult Population: A Nationwide Population-Based Study. J. Am. Heart Assoc. 2023, 12, e028496. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.; Oliveira, M.; Laranjo, S.; Rocha, I. Linking Sleep Disorders to Atrial Fibrillation: Pathways, Risks, and Treatment Implications. Biology 2024, 13, 761. [Google Scholar] [CrossRef]
- Dal Zotto, B.; Barbieri, L.; Tumminello, G.; Saviano, M.; ’ Gentile, D.; Lucreziotti, S.; Frattini, L.; Tarricone, D.; Carugo, S. New Onset Atrial Fibrillation in STEMI Patients: Main Prognostic Factors and Clinical Outcome. Diagnostics 2023, 13, 613. [Google Scholar] [CrossRef]
- Jameson, H.S.; Hanley, A.; Hill, M.C.; Xiao, L.; Ye, J.; Bapat, A.; Ronzier, E.; Hall, A.W.; Hucker, W.J.; Clauss, S.; et al. Loss of the Atrial Fibrillation-Related Gene, Zfhx3, Results in Atrial Dilation and Arrhythmias. Circ. Res. 2023, 133, 313–329. [Google Scholar] [CrossRef]
- O'Reilly, M.; Sommerfeld, L.C.; O'Shea, C.; Broadway-Stringer, S.; Andaleeb, S.; Reyat, J.S.; Kabir, S.N.; Stastny, D.; Malinova, A.; Delbue, D.; et al. Familial atrial fibrillation mutation M1875T-SCN5A increases early sodium current and dampens the effect of flecainide. Europace 2023, 25, 1152–1161. [Google Scholar] [CrossRef]
- Li, N.; Li, Y.J.; Guo, X.J.; Wu, S.H.; Jiang, W.F.; Zhang, D.L.; Wang, K.W.; Li, L.; Sun, Y.M.; Xu, Y.J.; et al. Discovery of TBX20 as a Novel Gene Underlying Atrial Fibrillation. Biology 2023, 12, 1186. [Google Scholar] [CrossRef]
- Chalazan, B.; Freeth, E.; Mohajeri, A.; Ramanathan, K.; Bennett, M.; Walia, J.; Halperin, L.; Roston, T.; Lazarte, J.; Hegele, R.A.; et al. Genetic testing in monogenic early-onset atrial fibrillation. Eur. J. Hum. Genet. 2023, 31, 769–775. [Google Scholar] [CrossRef]
- Vad, O.B.; Angeli, E.; Liss, M.; Ahlberg, G.; Andreasen, L.; Christophersen, I.E.; Hansen, C.C.; Møller, S.; Hellsten, Y.; Haunsoe, S.; et al. Loss of Cardiac Splicing Regulator RBM20 Is Associated With Early-Onset Atrial Fibrillation. JACC Basic Transl. Sci. 2023, 9, 163–180. [Google Scholar] [CrossRef]
- Virk, Z.M.; El-Harasis, M.A.; Yoneda, Z.T.; Anderson, K.C.; Sun, L.; Quintana, J.A.; Murphy, B.S.; Laws, J.L.; Davogustto, G.E.; O'Neill, M.J.; et al. Clinical Characteristics and Outcomes in Patients With Atrial Fibrillation and Pathogenic TTN Variants. JACC Clin. Electrophysiol. 2024, 10, 2445–2457. [Google Scholar] [CrossRef]
- Moreno-Manuel, A.I.; Macías, Á.; Cruz, F.M.; Gutiérrez, L.K.; Martínez, F.; González-Guerra, A.; Martínez Carrascoso, I.; Bermúdez-Jimenez, F.J.; Sánchez-Pérez, P.; Vera-Pedrosa, M.L.; et al. The Kir2.1E299V mutation increases atrial fibrillation vulnerability while protecting the ventricles against arrhythmias in a mouse model of short QT syndrome type 3. Cardiovasc. Res. 2024, 120, 490–505. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.F.; Sun, Y.M.; Qiu, X.B.; Wu, S.H.; Ding, Y.Y.; Li, N.; Yang, C.X.; Xu, Y.J.; Jiang, T.B.; Yang, Y.Q. Identification and Functional Investigation of SOX4 as a Novel Gene Underpinning Familial Atrial Fibrillation. Diagnostics 2024, 14, 2376. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, L.L.; Liu, Z.B.; Chen, C.; Ren, X.; Luo, A.T.; Ma, J.H.; Antzelevitch, C.; Barajas-Martínez, H.; Hu, D. Underlying mechanism of atrial fibrillation-associated Nppa-I137T mutation and cardiac effect of potential drug therapy. Heart Rhythm 2024, 21, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.W.; Chen, J.J.; Wang, C.H.; Chang, S.N.; Chiu, F.C.; Huang, P.S.; Chua, S.K.; Chuang, E.Y.; Tsai, C.T. Identification of a new genetic locus associated with atrial fibrillation in the Taiwanese population by genome-wide and transcriptome-wide association studies. Europace 2025, 27, euaf042. [Google Scholar] [CrossRef]
- Li YJ, Wang J, Ye WG, Liu XY, Li L, Qiu XB, Chen H, Xu YJ, Yang YQ, Bai D, et al. Discovery of GJC1 (Cx45) as a New Gene Underlying Congenital Heart Disease and Arrhythmias. Biology 2023, 12, 346. [Google Scholar] [CrossRef]
- Momoi, M.; Katsumata, Y.; Kunimoto, H.; Inami, T.; Miya, F.; Anzai, A.; Goto, S.; Miura, A.; Shinya, Y.; Hiraide, T.; et al. Clonal Hematopoiesis in Chronic Thromboembolic Pulmonary Hypertension. J. Am. Heart Assoc. 2024, 13, e035498. [Google Scholar] [CrossRef]
- Ilic, N.; Krasic, S.; Maric, N.; Gasic, V.; Krstic, J.; Cvetkovic, D.; Miljkovic, V.; Zec, B.; Maver, A.; Vukomanovic, V.; et al. Noonan Syndrome: Relation of Genotype to Cardiovascular Phenotype-A Multi-Center Retrospective Study. Genes 2024, 15, 1463. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, X.Y.; Yang, C.X.; Zhou, H.M.; Li, Y.J.; Qiu, X.B.; Huang, R.T.; Cen, S.S.; Wang, Y.; Xu, Y.J.; et al. Discovery and functional investigation of BMP4 as a new causative gene for human congenital heart disease. Am. J. Transl. Res. 2024, 16, 2034–2048. [Google Scholar] [CrossRef]
- Ke, Z.P.; Gu, J.N.; Yang, C.X.; Li, X.L.; Zou, S.; Bian, Y.Z.; Xu, Y.J.; Yang, Y.Q. Discovery of ETS1 as a New Gene Predisposing to Dilated Cardiomyopathy. Diagnostics 2025, 15, 2031. [Google Scholar] [CrossRef]
- Li, N.; Wang, Z.S.; Wang, X.H.; Xu, Y.J.; Qiao, Q.; Li, X.M.; Di, R.M.; Guo, X.J.; Li, R.G.; Zhang, M.; et al. A SHOX2 loss-of-function mutation underlying familial atrial fibrillation. Int. J. Med. Sci. 2018, 15, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Li, P.; de Crombrugghe, B. A new long form of Sox5 (L-Sox5), Sox6 and Sox9 are coexpressed in chondrogenesis and cooperatively activate the type II collagen gene. EMBO J. 1998, 17, 5718–5733. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Lefebvre, V. L-Sox5 and Sox6 drive expression of the Aggrecan gene in cartilage by Securing binding of Sox9 to a far-upstream enhancer. Mol. Cell. Biol. 2008, 28, 4999–5013. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Lu, Y.; Li, J.; Gu, J.; Ji, Y.; Shao, Y.; Kong, X.; Sun, W. Interaction of SOX5 with SOX9 promotes warfarin-induced aortic valve interstitial cell calcification by repressing transcriptional activation of LRP6. J. Mol. Cell. Cardiol. 2022, 162, 81–96. [Google Scholar] [CrossRef]
- Wang, P.; Xie, H.; Xiao, X.; Wang, H.; Wang, Y.; Liu, S. Functional characterization of SOX5 variant causing Lamb-Shaffer syndrome and literature review of variants in the SOX5 gene. Orphanet J. Rare Dis. 2025, 20, 300. [Google Scholar] [CrossRef]
- Moreno, C.S. SOX4: The unappreciated oncogene. Semin. Cancer Biol. 2020, 67, 57–64. [Google Scholar] [CrossRef]
- Grippa, M.; Graziano, C. Landscape of Constitutional SOX4 Variation in Human Disorders. Genes 2024, 15, 158. [Google Scholar] [CrossRef]
- Truebestein, L.; Leonard, T.A. Coiled-coils: the long and short of it. BioEssays 2016, 38, 903–916. [Google Scholar] [CrossRef]
- Duff, M.O.; Olson, S.; Wei, X.; Garrett, S.C.; Osman, A.; Bolisetty, M.; Plocik, A.; Celniker, S.E.; Graveley, B.R. Genome-wide identification of zero nucleotide recursive splicing in Drosophila. Nature 2015, 521, 376–379. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics 2014, 13, 397–406. [Google Scholar] [CrossRef]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, B.; Cao, Y.; Chen, W.; Yin, L.; Xu, Y.; Qiu, Z. High expression levels and localization of Sox5 in dilated cardiomyopathy. Mol. Med. Rep. 2020, 22, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Kiselak, E.A.; Shen, X.; Song, J.; Gude, D.R.; Wang, J.; Brody, S.L.; Strauss, J.F. 3rd, Zhang, Z. Transcriptional regulation of an axonemal central apparatus gene, sperm-associated antigen 6, by a SRY-related high mobility group transcription factor, S-SOX5. J. Biol. Chem. 2010, 285, 30496–505. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.Q.; Dong, P.; Li, D.Y.; Hu, C.C.; Li, H.P.; Lu, P.; Pan, X.X.; He, L.L.; Xu, X.; Xu, Q. Clinical characterization of Lamb-Shaffer syndrome: a case report and literature review. BMC Med. Genomics 2023, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Diawara, M.; Martin, L.J. Regulatory mechanisms of SoxD transcription factors and their influences on male fertility. Reprod. Biol. 2023, 23, 100823. [Google Scholar] [CrossRef]
- Thibodeau, I.L.; Xu, J.; Li, Q.; Liu, G.; Lam, K.; Veinot, J.P.; Birnie, D.H.; Jones, D.L.; Krahn, A.D.; Lemery, R.; et al. Paradigm of genetic mosaicism and lone atrial fibrillation: physiological characterization of a connexin 43-deletion mutant identified from atrial tissue. Circulation 2010, 122, 236–244. [Google Scholar] [CrossRef]
- Darbar, D.; Kannankeril, P.J.; Donahue, B.S.; Kucera, G.; Stubblefield, T.; Haines, J.L.; George, A.L., Jr.; Roden, D.M. Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation 2008, 117, 1927–1935. [Google Scholar] [CrossRef]
- Ellinor, P.T.; Nam, E.G.; Shea, M.A.; Milan, D.J.; Ruskin, J.N.; MacRae, C.A. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm 2008, 5, 99–105. [Google Scholar] [CrossRef]
- Hoffmann, S.; Clauss, S.; Berger, I.M.; Weiß, B.; Montalbano, A.; Röth, R.; Bucher, M.; Klier, I.; Wakili, R.; Seitz, H.; et al. Coding and non-coding variants in the SHOX2 gene in patients with early-onset atrial fibrillation. Basic Res. Cardiol. 2016, 111, 36. [Google Scholar] [CrossRef]
- Hoffmann, S.; Paone, C.; Sumer, S.A.; Diebold, S.; Weiss, B.; Roeth, R.; Clauss, S.; Klier, I.; Kääb, S.; Schulz, A.; et al. Functional Characterization of Rare Variants in the SHOX2 Gene Identified in Sinus Node Dysfunction and Atrial Fibrillation. Front. Genet. 2019, 10, 648. [Google Scholar] [CrossRef]
- Olesen, M.S.; Holst, A.G.; Jabbari, J.; Nielsen, J.B.; Christophersen, I.E.; Sajadieh, A.; Haunsø, S.; Svendsen, J.H. Genetic loci on chromosomes 4q25, 7p31, and 12p12 are associated with onset of lone atrial fibrillation before the age of 40 years. Can. J. Cardiol. 2012, 28, 191–195. [Google Scholar] [CrossRef]
- Pfeufer, A.; van Noord, C.; Marciante, K.D.; Arking, D.E.; Larson, M.G.; Smith, A.V.; Tarasov, K.V.; Müller, M.; Sotoodehnia, N.; Sinner, M.F.; et al. Genome-wide association study of PR interval. Nat. Genet. 2010, 42, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Roh, S.Y.; Lee, D.I.; Shim, J.; Choi, J.I.; Park, S.W.; Kim, Y.H. The Effects of Single Nucleotide Polymorphisms in Korean Patients with Early-onset Atrial Fibrillation after Catheter Ablation. J. Korean Med. Sci. 2020, 35, e411. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Rudaka, I.; Rots, D.; Isakova, J.; Kalējs, O.; Vīksne, K.; Gailīte, L. A Higher Polygenic Risk Score Is Associated with a Higher Recurrence Rate of Atrial Fibrillation in Direct Current Cardioversion-Treated Patients. Medicina 2021, 57, 1263. [Google Scholar] [CrossRef] [PubMed]
- Seifert, M.B.; Olesen, M.S.; Christophersen, I.E.; Nielsen, J.B.; Carlson, J.; Holmqvist, F.; Tveit, A.; Haunsø, S.; Svendsen, J.H.; Platonov, P.G. Genetic variants on chromosomes 7p31 and 12p12 are associated with abnormal atrial electrical activation in patients with early-onset lone atrial fibrillation. Ann. Noninvasive Electrocardiol. 2019, 24, e12661. [Google Scholar] [CrossRef]
- Martinez-Morales, P.L.; Quiroga, A.C; Barbas, J.A.; Morales, A.V. SOX5 controls cell cycle progression in neural progenitors by interfering with the WNT-beta-catenin pathway. EMBO Rep. 2010, 11, 466–472. [Google Scholar] [CrossRef]
- Luo, B.; Zheng, R.; Shi, C.; Chen, D.; Jin, X.; Hou, J.; Xu, G.; Hu, B. DACT2 modulates atrial fibrillation through TGF/beta and Wnt signaling pathways. Heliyon 2024, 10, e36050. [Google Scholar] [CrossRef]
- Bai, Y.; Li, R.; Hao, J.F.; Chen, L.W.; Liu, S.T.; Zhang, X.L.; Lip, G.Y.H.; Yang, J.K.; Zou, Y.X.; Wang, H. Accumulated beta-catenin is associated with human atrial fibrosis and atrial fibrillation. J. Transl. Med. 2024, 22, 734. [Google Scholar] [CrossRef]
- Karakasis, P.; Theofilis, P.; Vlachakis, P.K.; Korantzopoulos, P.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Atrial Fibrosis in Atrial Fibrillation: Mechanistic Insights, Diagnostic Challenges, and Emerging Therapeutic Targets. Int. J. Mol. Sci. 2024, 26, 209. [Google Scholar] [CrossRef]
- Pang, Z.; Ren, Y.; Yao, Z. Interactions between atrial fibrosis and inflammation in atrial fibrillation. Front. Cardiovasc. Med. 2025, 12, 1578148. [Google Scholar] [CrossRef]
- Salih, A.; Sutaria, A.; Montaser, Z.; Magar, T.P.; El Ashal, G.; Zaghloul, S.; Tom, A.J.; Ahmad, M.; Creta, A.; Ali, H.; et al. Fibrosis-Guided Ablation in Patients With Atrial Fibrillation: A Meta-Analysis of Randomized Controlled Trials. J. Cardiovasc. Electrophysiol. 2025, 36, 2025–2040. [Google Scholar] [CrossRef]
- Hattori, T.; Coustry, F.; Stephens, S.; Eberspaecher, H.; Takigawa, M.; Yasuda, H.; de Crombrugghe, B. Transcriptional regulation of chondrogenesis by coactivator Tip60 via chromatin association with Sox9 and Sox5. Nucleic Acids Res. 2008, 36, 3011–3024. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ahsen, O.O; Liu, J.J.; Du, C.; McKee, M.L.; Yang, Y; Wasco, W. ; Newton-Cheh, C.H.; O'Donnell, C.J; Fujimoto, J.G.; et al. Silencing of the Drosophila ortholog of SOX5 in heart leads to cardiac dysfunction as detected by optical coherence tomography. Hum. Mol. Genet. 2013, 22, 3798–3806. [Google Scholar] [CrossRef] [PubMed]
- Smits, P.; Li, P.; Mandel, J.; Zhang, Z.; Deng, J.M.; Behringer, R.R.; de Crombrugghe, B.; Lefebvre, V. The transcription factors L-Sox5 and Sox6 are essential for cartilage formation. Dev. Cell 2001, 1, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hersh, C.P.; Silverman, E.K.; Gascon, J.; Bhattacharya, S.; Klanderman, B.J.; Litonjua, A.A.; Lefebvre, V.; Sparrow, D.; Reilly, J.J.; Anderson, W.H.; et al. SOX5 is a candidate gene for chronic obstructive pulmonary disease susceptibility and is necessary for lung development. Am. J. Respir. Crit. Care Med. 2011, 183, 1482–1489. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, Z.M.; Delbono, O. Charge movement and transcription regulation of L-type calcium channel alpha(1S) in skeletal muscle cells. J. Physiol. 2002, 540, 397–409. [Google Scholar] [CrossRef]
- Braam, B.; de Roos, R.; Bluyssen, H.; Kemmeren, P.; Holstege, F.; Joles, J.A.; Koomans, H. Nitric oxide-dependent and nitric oxide-independent transcriptional responses to high shear stress in endothelial cells. Hypertension 2005, 45, 672–680. [Google Scholar] [CrossRef]
- Khan, A.A.; Thomas, G.N.; Lip, G.Y.H.; Shantsila, A. Endothelial function in patients with atrial fibrillation. Ann. Med. 2020, 52, 1–11. [Google Scholar] [CrossRef]
- Tenorio-Castano, J.; Gómez, Á.S.; Coronado, M.; Rodríguez-Martín, P.; Parra, A.; Pascual, P.; Cazalla, M.; Gallego, N.; Arias, P.; Morales, A.V.; et al. Lamb-Shaffer syndrome: 20 Spanish patients and literature review expands the view of neurodevelopmental disorders caused by SOX5 haploinsufficiency. Clin. Genet. 2023, 104, 637–647. [Google Scholar] [CrossRef]
Figure 1.
Pedigree with idiopathic atrial fibrillation. The family was named arbitrarily as Pedigree AF-001. “+” denotes an individual carrying the observed SOX5 variation; “–” signifies a member without the found SOX5 variation.
Figure 1.
Pedigree with idiopathic atrial fibrillation. The family was named arbitrarily as Pedigree AF-001. “+” denotes an individual carrying the observed SOX5 variation; “–” signifies a member without the found SOX5 variation.
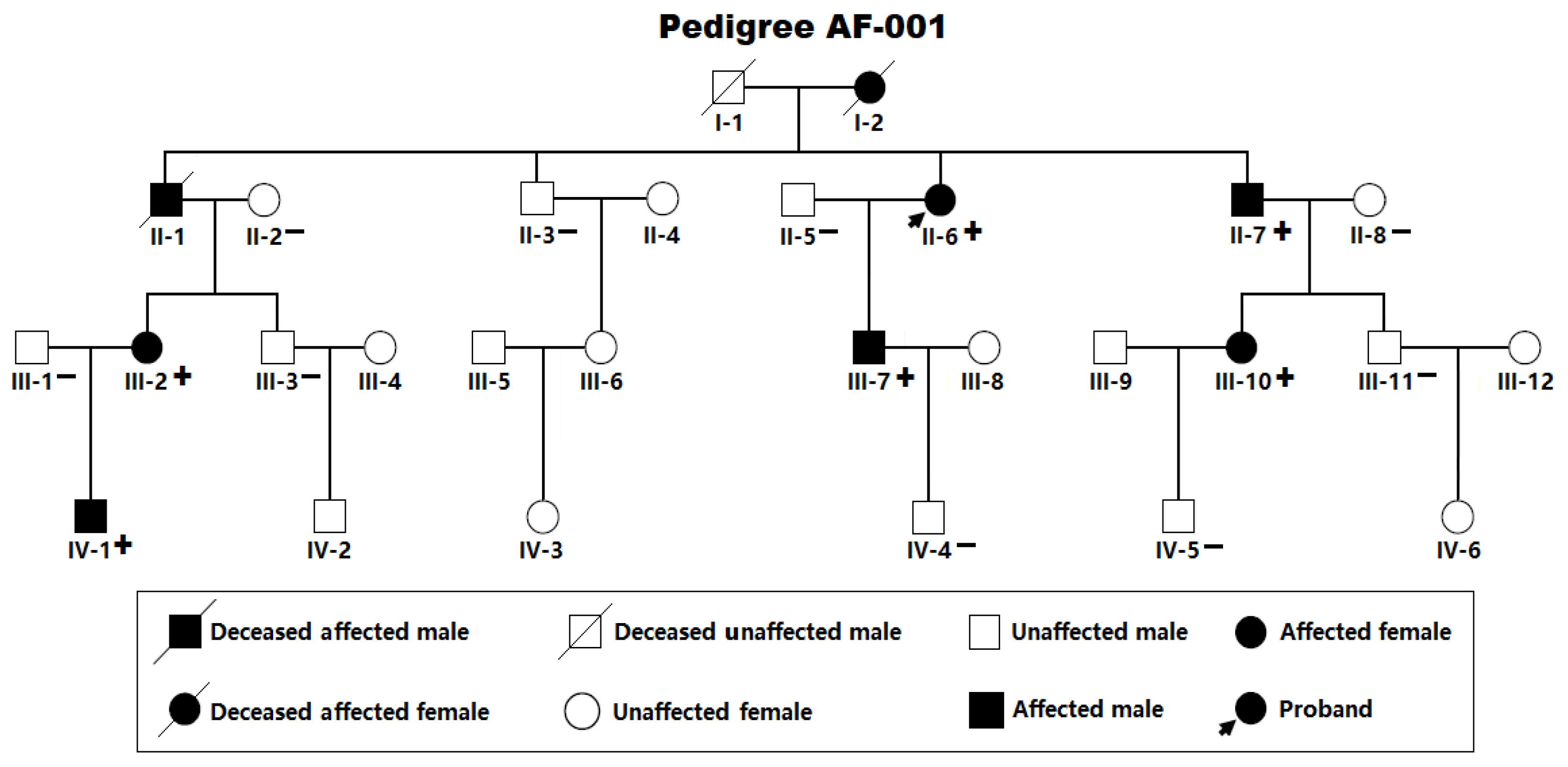
Figure 2.
A representative 12-lead electrocardiogram from the proband (member II-6 in Pedigree AF-001). The standard electrocardiogram documents the occurrence of atrial fibrillation.
Figure 2.
A representative 12-lead electrocardiogram from the proband (member II-6 in Pedigree AF-001). The standard electrocardiogram documents the occurrence of atrial fibrillation.
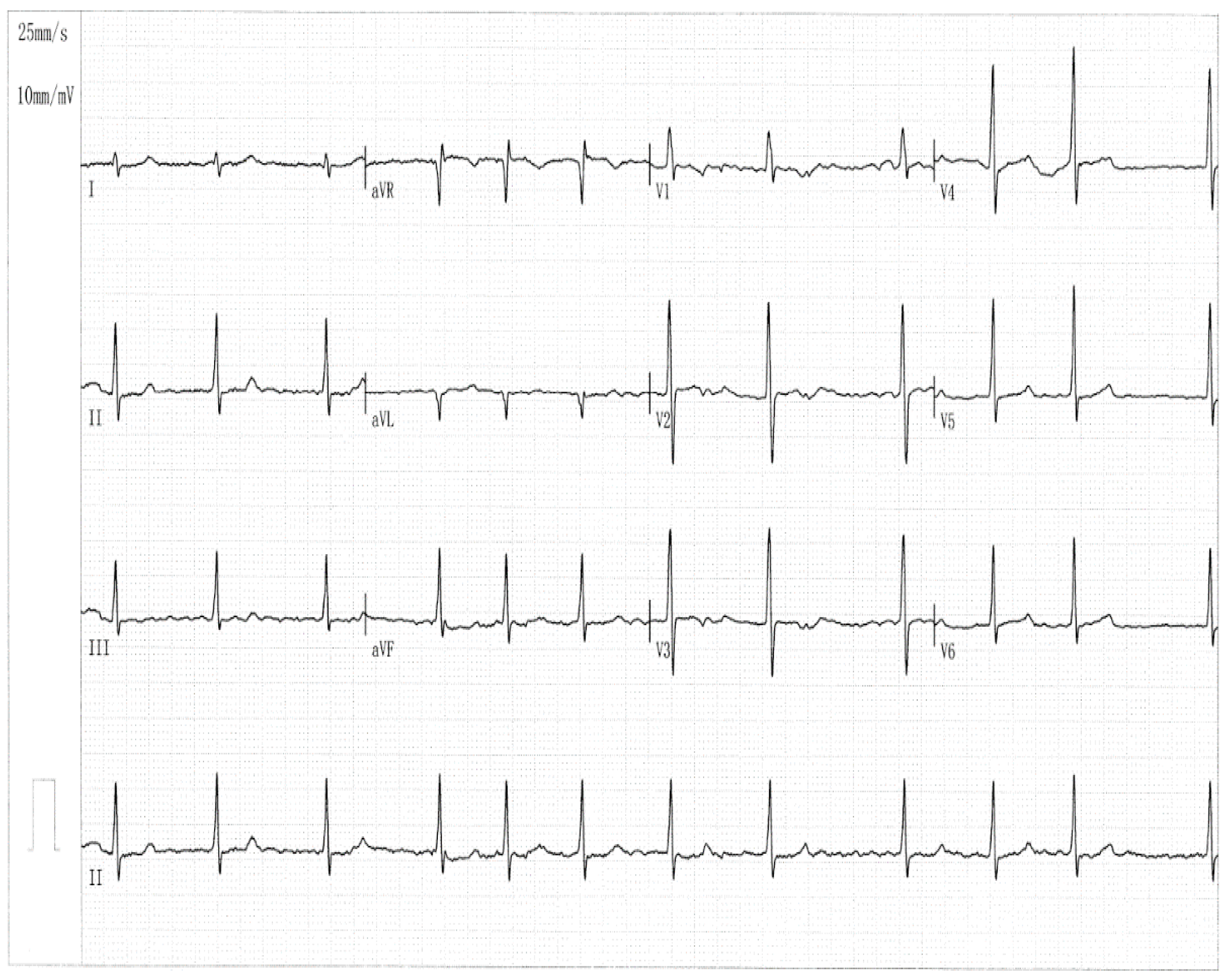
Figure 3.
A novel SOX5 variation accountable for atrial fibrillation. Sequencing electropherograms delineating the heterogeneous SOX5 variation discovered in the proband with atrial fibrillation (Mutant) along with its wild-type control from a healthy person (Wild type). An arrow pinpoints where the variation occurs.
Figure 3.
A novel SOX5 variation accountable for atrial fibrillation. Sequencing electropherograms delineating the heterogeneous SOX5 variation discovered in the proband with atrial fibrillation (Mutant) along with its wild-type control from a healthy person (Wild type). An arrow pinpoints where the variation occurs.
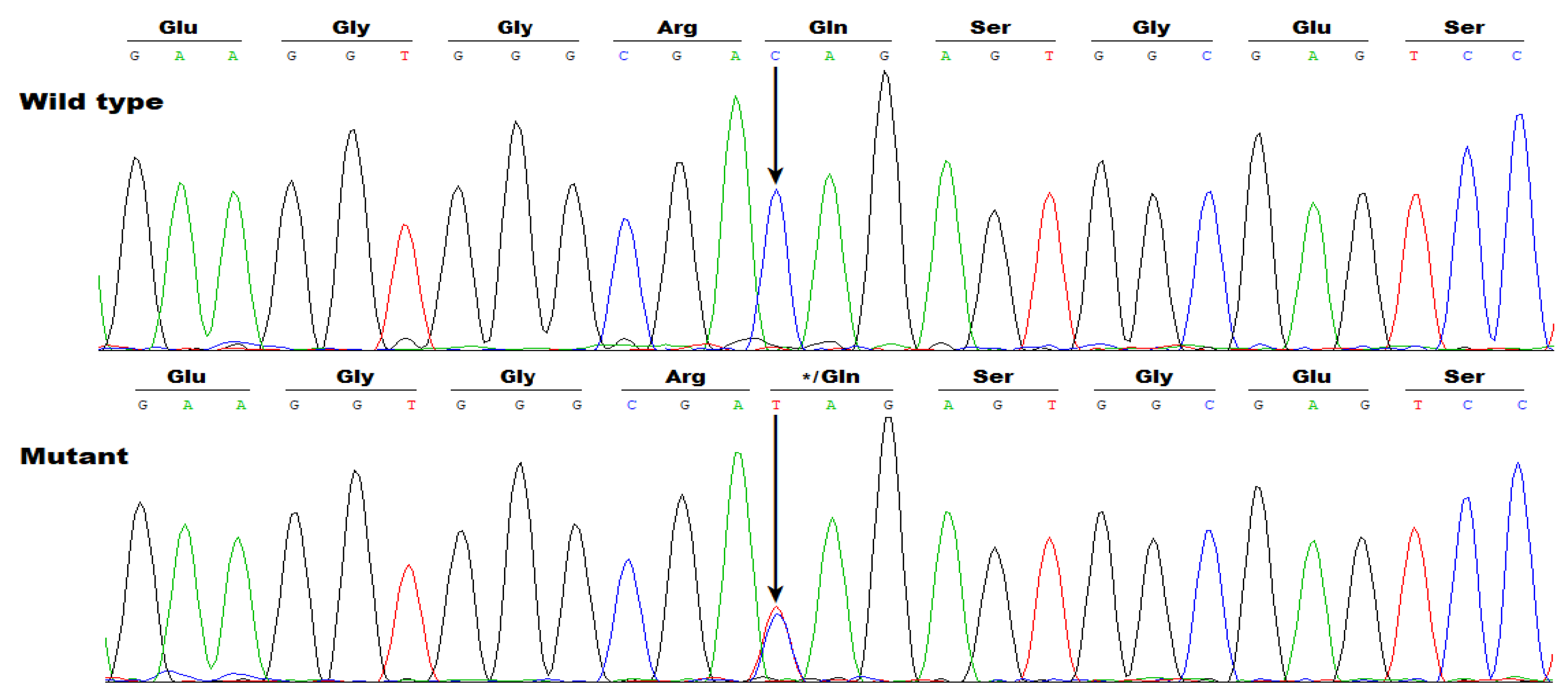
Figure 4.
A de novo SOX5 varitation predispoing to atrial fibrillation. The heterogeneous SOX5 variation (Mutant) was detected in one of 236 patients with atrial fibrillation and its wild type as a sequence control was detected in a healthy subject (Wild type). A vertical arrow orients the nucleotide site where the variation occurs.
Figure 4.
A de novo SOX5 varitation predispoing to atrial fibrillation. The heterogeneous SOX5 variation (Mutant) was detected in one of 236 patients with atrial fibrillation and its wild type as a sequence control was detected in a healthy subject (Wild type). A vertical arrow orients the nucleotide site where the variation occurs.
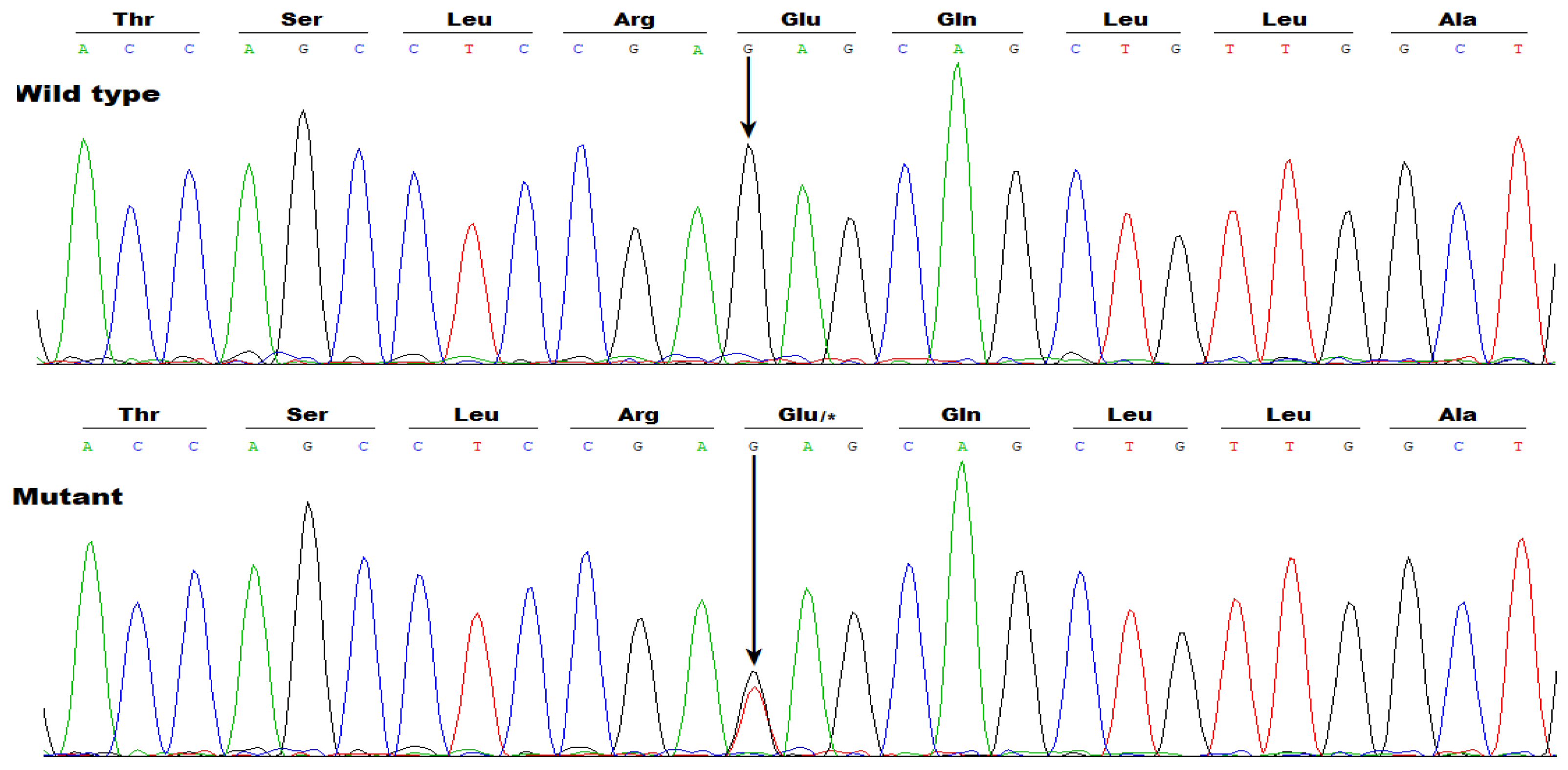
Figure 5.
A representative 12-lead electrocardiogram recorded from the AF case carrying the SOX5 c.640G>T variation. This electrocardiogram manifestates atrial fibrillation.
Figure 5.
A representative 12-lead electrocardiogram recorded from the AF case carrying the SOX5 c.640G>T variation. This electrocardiogram manifestates atrial fibrillation.
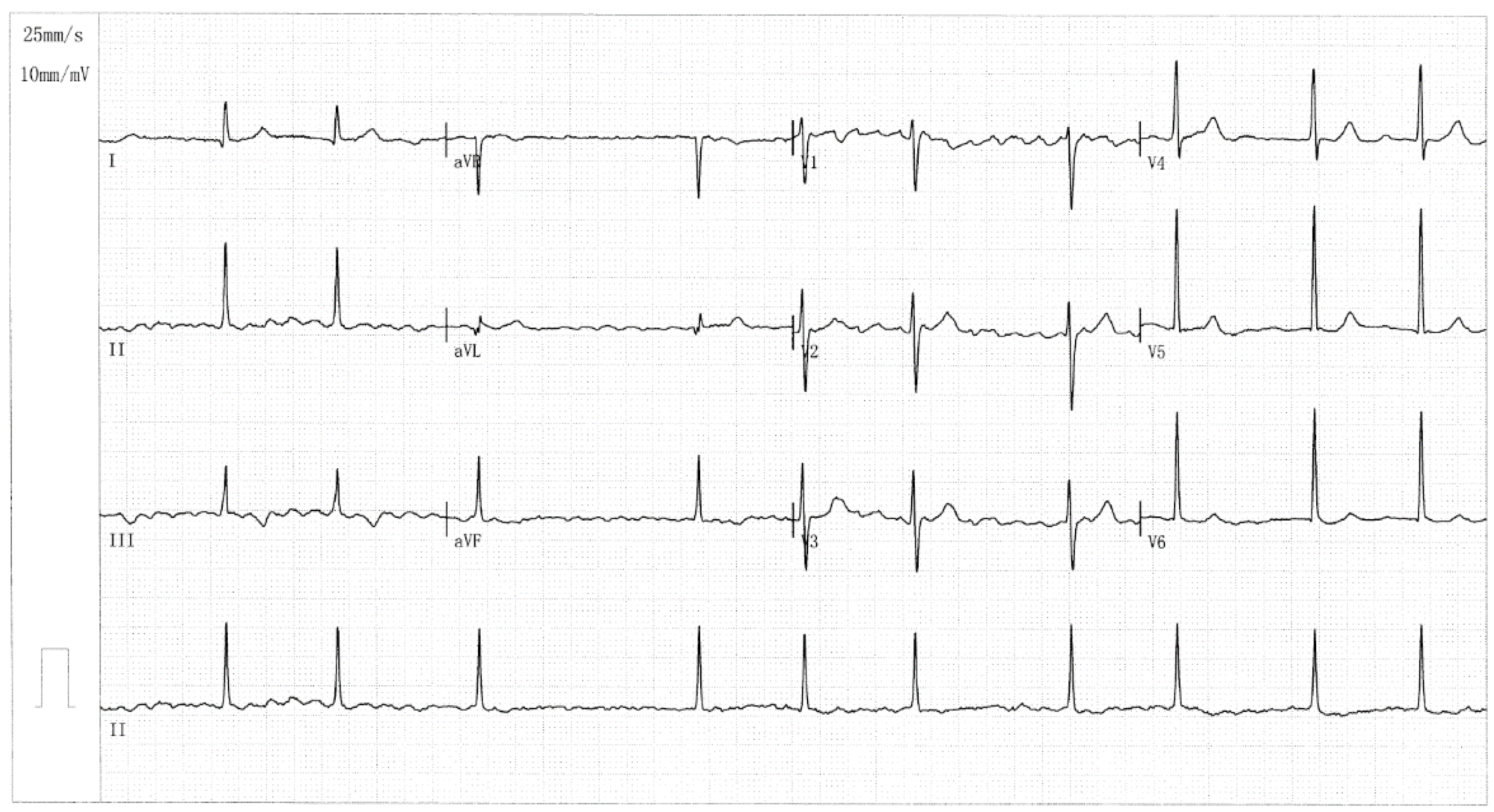
Figure 6.
Failure of Gln119*- or Glu214*-mutant SOX5 to transactivate GJA1. In cultured COS-7 cells in vitro, dual-reporter (Firefly luciferase and Renilla luciferase) gene analysis revealed that both the Gln119*mutant (Figure 6A) and the Glu214* mutant (Figure 6B) failed to transcriptionally activate GJA1. Herein “a” and “c” signify p <0.001, and “c” and “d” denote p <0.005, in comparison with wild-type human SOX5 (400 ng).
Figure 6.
Failure of Gln119*- or Glu214*-mutant SOX5 to transactivate GJA1. In cultured COS-7 cells in vitro, dual-reporter (Firefly luciferase and Renilla luciferase) gene analysis revealed that both the Gln119*mutant (Figure 6A) and the Glu214* mutant (Figure 6B) failed to transcriptionally activate GJA1. Herein “a” and “c” signify p <0.001, and “c” and “d” denote p <0.005, in comparison with wild-type human SOX5 (400 ng).

Figure 7.
Synergistic activation of SCN5A between SOX5 and SHOX2 disrupted by the Gln119*- or Glu214* mutation. In the HEK293 cells maintained in vitro and transfected multiple expression plasmids, dual-luciferase activity measurement demonstrated that the synergistic activation of SCN5A by SOX5 and SHOX2 was disrupted by the Gln119* mutation (Figure 7A) or Glu214* mutation (Figure 7B). Herein “e”, “f”, “g”, and “h” all mean p <0.001, in contrast to the corresponding wild-type counterparts.
Figure 7.
Synergistic activation of SCN5A between SOX5 and SHOX2 disrupted by the Gln119*- or Glu214* mutation. In the HEK293 cells maintained in vitro and transfected multiple expression plasmids, dual-luciferase activity measurement demonstrated that the synergistic activation of SCN5A by SOX5 and SHOX2 was disrupted by the Gln119* mutation (Figure 7A) or Glu214* mutation (Figure 7B). Herein “e”, “f”, “g”, and “h” all mean p <0.001, in contrast to the corresponding wild-type counterparts.
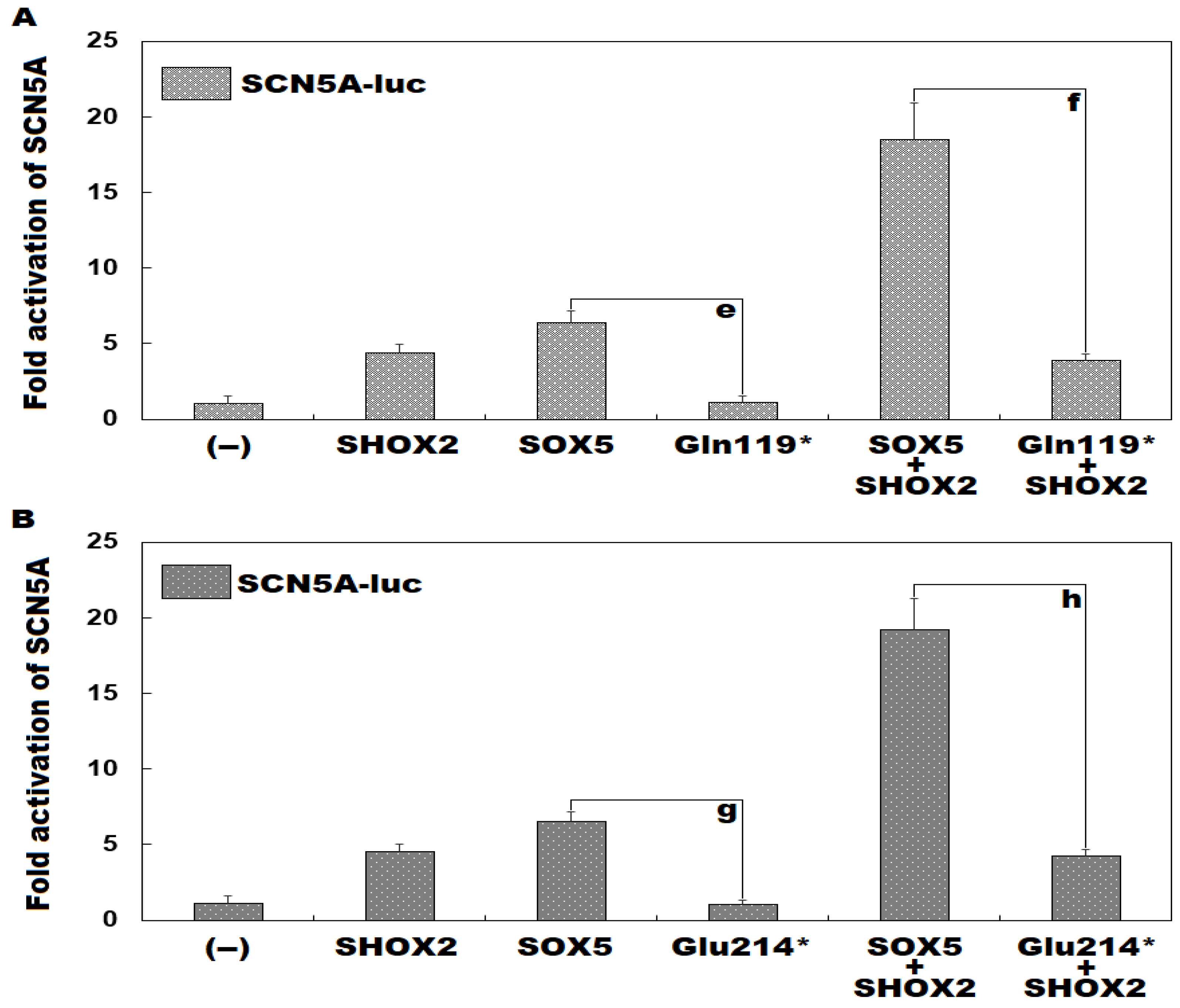

Table 1.
Demographic and baseline clinical characteristics of the living pedigree members affected with atrial fibrillation.
Table 1.
Demographic and baseline clinical characteristics of the living pedigree members affected with atrial fibrillation.
| Individual information | Cardiac manifestation | Electrocardiogram | Echocardiogram | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Identity (Pedigree AF-001) |
Sex | Age at first diagnosis of AF (years) | Age at enrollment (years) | AF (clinical categorizing) | Heart rate (beats/min) | QRS interval (ms) | QTc (ms) |
LAD (mm) | LVEF (%) |
| II-6 II-7 III-2 III-7 III-10 IV-1 |
F M F M F M |
51 47 42 35 40 24 |
66 63 48 42 40 24 |
LSP LSP LSP LSP Persistent Paroxysmal |
69 76 105 83 112 90 |
86 118 92 95 105 81 |
435 507 482 394 416 413 |
39 36 38 33 32 29 |
58 62 60 63 64 66 |
F, female; M, male; AF, atrial fibrillation; QTc, corrected QT interval; LSP, long-standing persistent; LVEF, left ventricular ejection fraction; LAD, left atrial diameter.
Table 2.
Baseline clinical and demographic characteristic data of the group of 236 patients suffering atrial fibrillation along with the 312 control subjects.
Table 2.
Baseline clinical and demographic characteristic data of the group of 236 patients suffering atrial fibrillation along with the 312 control subjects.
| Variable | Case group (n =236) | Control group (n =312) | p-value |
|---|---|---|---|
| Sex (male/female) Age (years) Family history of atrial fibrillation (%) History of cerebral stroke (%) History of implanting a pacemaker (%) Body mass index (kg/m2) Total cholesterol (mmol/L) Fasting blood glucose (mmol/L) Triglyceride (mmol/L) Systolic blood pressure (mmHg) Diastolic blood pressure (mmHg) Resting heart rate (beats/min) Left ventricular ejection fraction (%) Left atrial diameter (mm) |
135/101 54.16 ± 7.39 48 (20.33) 15 (6.36) 12 (5.08) 22.85 ± 3.10 4.15 ± 0.57 4.46 ± 0.62 1.38 ± 0.35 127.84 ± 9.07 84.73 ± 7.46 77.01 ± 8.47 62.25 ± 7.13 37.82 ± 6.52 |
178/134 53.82 ± 8.02 0 (0) 0 (0) 0 (0) 23.06 ± 2.96 4.21 ± 0.61 4.50 ± 0.69 1.40 ± 0.31 128.18 ± 9.41 85.03 ± 9.02 76.88 ± 7.64 62.91 ± 7.22 36.05 ± 6.08 |
0.9716 0.6115 <0.0001* <0.0001* <0.0001* 0.4207 0.2415 0.4832 0.4797 0.6707 0.6785 0.8508 0.2872 0.0011* |
*p <0.05.
Table 3.
Primers to amplify the whole coding exons along with splicing donors/acceptors of the human SOX5 gene.
Table 3.
Primers to amplify the whole coding exons along with splicing donors/acceptors of the human SOX5 gene.
| Coding exon | Forward primer (5´→3´) | Backward primer (5´→3´) | Amplicon (bp) |
|---|---|---|---|
| 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 |
GGTTGTCTAGAGCCTTGCAGC GTTCTGTTGCTACCTGCTTGGC TCAGCTGAATAAGCCATATAACC GAAGTGGGGCTGGGATAGGG TTATTTCCAGCTGGCCCTAGCAT CTGCCGTGGTATCTTAGGCTTC TGGGAAGAAGCATGGAGCATC AAAAGGATGAGGTTTCCGCCT TGTTTCGGGTGCCCATTTCAAG TTTGATGGGAAATGACAGGCTGC GGCCAGACACTACCTATTACCAAGA GGCATACCAAACCCAAACGCC CATTTGCCACCACAAGGCTTATC AGGTACAAAACCACCACCACCT ACATCTAACTATTCACTTACCCACG |
TTTGGTCCGGGCAATCACAAC CTAAGACGCCAGGGGTGAATC CAAGCAGGTGACTATTCCCG GAAGCAGAAGAGGTGAGGGCA TGTTGTGTGCCTAGGACAGTGA TGGTTCCCTGCACCTATCCAG ATGATGCGAGTCCAGAGTCAAGA TTGTTAAGTCGCCTTGCTCCT AGCTGCTGGCATACAATAGACA AAACGGACCTAGGTGGTTCCTC ACAAGCTGGTGGCGTAAAAGG AATGATGAGGTATGAGGTGGCTG ATCCAGGATCCTTCCACAACTGC TGGTAGAGCTAGGAACTTGCAGTG GTGCTTGGCCACTGGTAAGG |
541 555 660 300 656 638 648 678 681 418 500 456 440 592 435 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



