Submitted:
07 October 2025
Posted:
08 October 2025
You are already at the latest version
Abstract
Despite advances in breast cancer treatment, the number of lives lost each year remains high. One of the significant challenges in treating breast cancer is the highly complex and dynamic tumor microenvironment (TME), which often makes tumors resistant to various kinds of therapies. To overcome this, researchers are tuning and tweaking the body's immune cells, especially T cells and subsets, to clear cancerous cell residues more effectively from the patient's body. Recent research shows that long non-coding RNAs (lncRNAs), once considered "junk" DNA, play an essential role in shaping T cells' plasticity in healthy and cancerous tissues. In this review, we presented and discussed our perspectives on how specific lncRNAs control T-cell infiltration into tumors and how they contribute to immune escape. For example, LINC00472, MIAT, HITT, NKILA, and MALAT1 influence how T cells enter tumors, while SNHG1, TINCR, and GATA3-AS1 help cancer cells hide from immune attack. These lncRNAs can shift the balance between regulatory T cells (Tregs) and cytotoxic T cells (CTLs), either slowing tumor growth or helping cancer evade destruction by recruiting Tregs, M2 macrophages, and myeloid-derived suppressor cells (MDSCs). LncRNAs regulate key signalling pathways such as NF-κB, Wnt/β-catenin, PI3K/AKT, and JAK/STAT, often by acting as "sponges" for miRNAs that regulate immune checkpoints like PD-L1. Further, LncRNAs are found to remodel cytokine and chemokine networks, ultimately reshaping the TME. Overexpression of lncRNA, such as TINCR and MIAT, GATA3-AS1, leads to enhanced PD-L1 deubiquitination, thus aiding in tumor immune evasion. Moreover, our in silico analysis suggests that lncRNAs modulate various immune-related signaling pathways in T cells. Together, these insights reveal how lncRNAs could be harnessed to fine-tune T-cell plasticity, paving the way for more personalized immunotherapies and improving outcomes for breast cancer patients.
Keywords:
lncRNA
; T cell
; breast cancer
; Immune Evasion
; T cell infiltration
; and immunotherapy
1. Introduction
The tumor microenvironment (TME) is a complex system that includes a sophisticated network of cancer cells interacting with various stromal components of the host system [1,2,3]. Within the TME, more than twenty types of immune cells exist [4], where T cells, including CD4+ and CD8+ T cells and their subsets, play a significant role in their interactions with cancerous cells, influencing tumor growth and response to immunotherapy [5,6]. The persistent antigen exposure to the tumor microenvironment leads to T-cell exhaustion, progressive dysfunction characterized by diminished effector functions, sustained expression of inhibitory receptors like PD-1(Programmed death 1), CTLA-4 (cytotoxic T lymphocyte associated antigen 4), TIM-3 (T cell immunoglobulin and mucin domain 3), and reduced ability to proliferate [7]. Even though various immunotherapeutic regimens and checkpoint inhibitors have shown promise in restoring diminished T cell activity, their clinical efficacy remains suboptimal, especially in cases of breast cancer. Therefore, it's very important to save the lives of patients suffering from breast cancer, our most loved ones.
In this regard, non-coding RNAs, specifically long non-coding RNAs (lncRNAs) have recently been recognised as crucial molecules in regulating various important cancer hallmarks [8,9,10]. LncRNAs, typically consisting of more than two hundred nucleotides, are often aberrantly expressed in various cancers, including breast cancer [11]. Our previous research has highlighted lncRNA ZFAS1 (Zinc finger NFX1-type containing 1 antisense RNA 1) as a crucial player in the oncogenesis of triple-negative breast cancer (TNBC) [12], wherein we have demonstrated that ZFAS1 acts as a tumor suppressor in TNBC patients, and negatively regulates the STAT3 (Signal transducer and activator of transcription-3) gene expression in TNBC cells. Moreover, in breast cancer, dysregulated lncRNAs have been implicated in tumorigenesis, metastasis, and immune evasion by cancer-associated signaling pathways [11,13]. LncRNAs are also recognized as critical epigenetic regulators that affect gene expression through various mechanisms, such as chromatin remodeling, transcriptional interference, and miRNA and circular RNA sponging [14]. Moreover, many recent studies have also suggested the role of lncRNAs in regulating immune cell dynamics within the TME; thus, influencing T cell activation, infiltration, and differentiation [15,16,17]. In breast cancer, the dysregulation of lncRNAs has been linked to tumor immune evasion [18,19]; however, their specific roles in T cell infiltration and immune escape within the breast TME remain poorly understood.
Very recently, lncRNAs have been found to play an important role in controlling T-cell infiltration in the breast cancer tumor microenvironment. They affect immune surveillance, immune cell recruitment, and pathways that suppress the immune response. LncRNA like NKILA (NF-кB interacting long non-coding RNA) helps tumors escape the immune system by reducing T-cell infiltration and weakening antitumor immunity [20]. Conversely, the downregulation of GATA3-AS1 in triple-negative breast cancer increases tumor infiltration and facilitates cytotoxic T cell-mediated cancer death. Also, lncRNAs affect gene expression and signaling, including the IFN-γ (Interferon gamma) and TGF-β (Transforming growth factor beta) pathways, which are crucial for immune cell movement. They mediate interactions between tumor cells, stromal cells, and immune cells. The detailed role of lncRNAs in regulating T-cell infiltration will be presented in Section 2 of this review.
Immune escape is one of the hallmarks of cancer that allows tumor cells to survive and progress despite immune surveillance. Cancer cells, including breast cancer, use diverse approaches, such as overexpression of immune checkpoints like PD-L1, secretion of immunosuppressive cytokines, induction of regulatory T cells, and metabolic reprogramming, to suppress cytotoxic T-cell activity. Recent research has shown that lncRNAs such as SNHG1, TINCR, and GATA3-AS1 [21,22,23] play a critical role in determining the immune escape from breast cancer, particularly by altering T-cell activity. For example, GATA3-AS1 and TINCR raise PD-L1 expression, which directly inhibits effector T-cell responses, while SNHG1 promotes regulatory T-cell differentiation via the miR-448/IDO axis. Together, they have an impact on immune evasion in breast cancer. A comprehensive mechanistic overview of how lncRNA regulates T-cell dysfunction and immune escape in breast cancer will be discussed (Section 3). A deeper mechanistic understanding could help in uncovering some novel therapeutic strategies to re-activate anti-tumor immunity.
This review highlights the emerging role of lncRNAs in influencing T-cell infiltrations and in immune escape within the breast cancer TME. Herein, we discussed a comprehensive overview of the current understanding of long non-coding RNA-mediated regulation of T cell function. And their potential as a therapeutic target for immunotherapy response. Additionally, we spotlight therapeutic strategies aimed at reversing T cell dysfunction. By elucidating the interaction between lncRNAs and T-cell biology, we aspire to contribute valuable knowledge and novel insights toward overcoming immune resistance in breast cancer and enhancing therapeutic outcomes.
In the subsequent section, we will focus on the emerging role of lncRNAs in T-cell Immune Infiltration (Section 2) and Immune Escape Mechanisms (Section 3) in breast cancer, considering molecular, immunological, and clinical perspectives. We reviewed relevant literature from various sources, including PubMed, Google Scholar, and Embase, from 2010 onward. We discovered a limited amount of information on this topic. Nevertheless, we aim to connect, analyse, and present our perspective to encourage further discussion on this crucial issue. Finally, we used various bioinformatics tools to perform an In Silico analysis (Section 4) of lncRNAs associated with T cell function.
2. Modulatory Functions of lncRNAs in Shaping T-Cell Infiltration Within Breast Cancer
The innate and adaptive immune systems are critical contributors to tumor immunosurveillance and modulation of cancer progression. Immune cells, such as T-cells, natural killer cells, B-cells, or macrophages that travel from the blood circulation and accumulate in the tumor microenvironment, particularly in the tumor stroma and intraepithelial region, are termed tumor-infiltrating immune cells (Figure 1). As key mediators of anti-tumor immune response, the presence and abundance of these immune-infiltrating cells are often associated with therapeutic efficacy and overall patient survival. T cells are identified using specific cell surface markers among these infiltrating cells. CD3+ is a general marker for each T cell, whereas CD4+ and CD8+ T cell markers reflect the distribution of T helper (Th) and cytotoxic lymphocytes (CTL), respectively [24,25]. Cytotoxic T cells aid in anti-tumor activity by secreting perforin and granzyme that induce cell death. T-helper 1 (Th1) promotes the activation of macrophages to target tumor cells by secreting IFN-ɤ (Interferon gamma), IL-2 (Interleukin 2), and TNF-α (Tumor Necrosis Factor alpha), while Th2 (T helper 2) promotes tumor cell proliferation. T cells can differentiate into immunosuppressive T regulatory (Treg) cells in the tumor microenvironment. They promote tumor growth through multiple mechanisms by suppressing the anti-tumor activity of various immune effector cells, including CD4+ T cells, cytotoxic CD8+ T cells, NK cells, and dendritic cells [15].
Long non-coding RNA has been implicated in regulating Treg cells' differentiation and functional activity. Although the concentration of T cells in the breast cancer tumor microenvironment is low, studies have revealed a notable level of T cell infiltration [26]. TIMER (Tumor Immune Estimation Resource) was used to analyze the abundance of tumor-infiltrating immune cells (TIICs). T cell infiltration in tumors is highly variable, not only between different types of cancer but also among tumors of the same type. Compared with Luminal A and B subtypes of breast cancer, HER2+ and triple-negative breast cancer (TNBC) tumors typically show higher levels of T cell infiltration [26]. LncRNA TCL6 expression was strongly associated with increased infiltration of various immune cells, including B cells, CD8+ and CD4+ T cells, neutrophils, and dendritic cells, with all correlations being statistically significant (P<0.001) in breast cancer [27] (Figure 1).
In a recent study, Yang et al. [28] evaluated the immune-related long non-coding RNAs (irlncRNAs) within the tumor microenvironment (TME) of triple-negative breast cancer (TNBC) patients. To classify TNBC tumors based on their immune landscapes, they analysed immune cell infiltration (ICI), PD-L1 expression, and tumor mutational burden (TMB) using bioinformatics tools TCGA-TNBC and datasets GSE33926. The authors revealed that tumors with high ICI showed increased PD-L1 expression and improved overall survival (OS) despite exhibiting lower TMB. In contrast, a higher TMB was linked to better survival outcomes in the low-ICI group, indicating that significant immune infiltration may weaken reliance on TMB for responses to immunotherapy.
Additional immune profiling revealed that high-ICI tumors have many immunostimulatory cells, including CD8+ T cells, activated memory CD4+ T cells, plasma B cells, M1 macrophages, NK cells (Natural killer cells), and dendritic cells. This means the tumor microenvironment is "hot" for the immune system. On the contrary, low-ICI tumors contained additional immunosuppressive cells like M2 macrophages, monocytes, and neutrophils. This distinction shows the intricate relationship between the TME in triple-negative breast cancer, angiogenesis, and resistance to therapy.
To effectively stratify patients into high-risk groups, the authors, by applying LASSO-Cox regression analysis, developed a 13-irlncRNAs (Immune-related long non-coding RNA) prognostic signature. These high-risk group patients exhibited poorer overall survival (p<0.001) and displayed immunosuppressive infiltration with increased plasma B cells, M2 macrophages, and neutrophils, along with diminished PD-L1 expression. A significant association between the expression of irlncRNAs and the infiltration of immune cells like CD8+ T cells, B cells, and macrophages was also demonstrated in this study. Elevated CD8+ T cells and M1 macrophages were associated with improved overall survival (p≈0.04). This suggests that irlncRNAs may play a role in identifying immune-cold, treatment-resistant tumors. Among the 13 irlncRNAs investigated, LINC00472 has been recognized as a tumor suppressor and a predictive marker in breast cancer [29].
The LINC00472 is located on chromosome 6q13, with 2,933 base pairs in length [30]. High expression levels of LINC00472 correlate with ER+ (Estrogen receptor-positive), low-grade breast cancer, and favourable molecular subtypes [31]. Furthermore, experimental cell studies have confirmed that LINC00472 inhibits the phosphorylation of NF-kB (nuclear factor kappa B) by binding to IKKβ (Inhibitor of Nuclear Factor Kappa B Kinase Subunit Beta) in breast cancer [32]. Moreover, chemoresistant breast cancer cells also display increased levels of lncRNA LINC00839, whose overexpression promotes the cancer-related pathway mediated by PI3K/AKT and Myc (myelocytomatosis oncogene) activation. Ultimately, this enhances cell invasion, migration, and proliferation, resulting in worse prognoses for breast cancer [33]. This study provided a detailed and thorough assessment of the tumor microenvironment in TNBC by combining ICI (Immune cell infiltration), PD-L1, TMB (Tumor mutational burden), and irlncRNAs: the ICI score and the irlncRNA signature direct precision immunotherapy and patient stratification in TNBC.
It has been observed that the long non-coding RNA MIAT (Myocardial Infarction Associated Transcript) has aberrant regulation in several kinds of cancers, such as stomach, cervical, lung, liver, and prostate [34]. In these types of cancers, it promotes tumor growth in several ways, particularly by functioning as an miRNA sponge [35]. It is observed that MIAT mainly exists in the nucleus and encompasses about 30 kilobases on chromosome 22q12.1 [36]. Further, it involves significant biological processes involving oncogenesis, immune regulation, and alternative cell transcription [36]. Elevated MIAT levels compared to adjacent tissue are considered a promising non-invasive breast carcinoma diagnostic biomarker [37]. Moreover, upregulated MIAT levels are positively associated with lymph node metastasis and advanced TNM staging, and show strong diagnostic potential, with an AUC (Area under curve) of 0.832, a sensitivity of 75.7%, and a specificity of 85.0 % in ROC (Receiver Operating Characteristic curve) curve analysis [38]. The authors employed bioinformatics analyses further to elucidate MIAT's involvement in the tumor microenvironment, focusing particularly on immune cell infiltration, including T cell subtypes. The study showed that higher MIAT expression in breast tumors is linked to more CD8+ cytotoxic T cells, activating as well as resting CD4+ memory T cells, γδ T cells, and M1-polarized macrophages (Figure 1). In contrast, tumors with high MIAT expression had lower levels of plasma cells, activated NK cells, monocytes, M2 macrophages, and activated mast cells. This unique immune cell profile suggests that MIAT may influence the tumor immune microenvironment by encouraging the infiltration of specific T cell subsets and pro-inflammatory macrophages while reducing immune surveillance and regulatory elements. This may lead to a biased immune response. Previous studies have suggested CD8+ T cells as a prognostically favorable factor of immunotherapy in breast cancer cases [39,40]. The pathway enrichment analysis showed that the genes co-expressed with MIAT significantly participate in several immune processes. These include T cell receptor (TCR) signaling, T helper cell differentiation (Th1, Th2, and Th17), cytokine-cytokine receptor interactions, and primary immunodeficiency.
Moreover, MIAT expression positively correlates with important immune checkpoint molecules, such as PD-1, PD-L1, and CTLA-4, along with other factors that regulate the immune system. This indicates that tumors with high MIAT expression may use immune checkpoint pathways to weaken T-cell responses and avoid being detected by the immune system. Functional validation experiments confirmed these bioinformatic findings. Silencing MIAT in TNBC cell lines significantly reduced cell proliferation, colony formation, and invasiveness. It also triggered apoptosis in both in vitro and xenograft models. Mechanistically, MIAT was shown to act as a competitive endogenous RNA (ceRNA) by sponging miR-150-5p, thereby preventing this miRNA from downregulating CD274, the gene encoding PD-L1 [19] (Figure 2D). miR-150-5p, a miRNA on chromosome 19q13, is highly expressed in TNBC. Its expression levels are associated with tumor grade, patient survival, ethnicity, and regulation of several cancer driver genes [41]. MIAT's distinct impact on tumor growth and immune modulation is supported by its indirect contribution to PD-L1 upregulation, a mechanism that has also been documented for other oncogenic lncRNAs such as HCP5 (Histocompatibility leukocyte antigen complex P5). Together, these results show that MIAT significantly affects the immune microenvironment in breast cancer. It connects to both tumor growth and immune evasion. The study highlights that targeting MIAT could improve anti-tumor immune responses and change the tumor microenvironment, mainly when used with immune checkpoint inhibitors. Additionally, MIAT could serve as a valuable biomarker with strong clinical importance for breast cancer patients.
A similar study by Luan et. al further validated MIAT`s role in breast cancer progression. MIAT functions as ceRNA to miR-155-5p also through its conserved binding sites. This miR-155-5p sponging was found to modulate the expression profile of DUSP7 (dual specificity phosphatase 7). DUSP7 is a diverse family of protein phosphatases that target both phosphotyrosine and phosphoserine/ phosphothreonine within the same substrate. Reduced expression of DUSP7 is often associated with several cancers, suggesting its tumor suppressive roles. In a separate study by Li et. al [42], drug resistance was induced in breast cancer by FOSL1 through DUSP7-mediated dephosphorylation of PEA15 (Proliferation and apoptosis adaptor protein 15). Luan et. al also correlated knockdown of MIAT in MDA-MB-231 and MCF-7 cell lines with decreased epithelial to mesenchymal transitions, increased apoptosis, thereby inhibiting breast cancer migration and invasion [37].
SOX9-AS1 lncRNA is becoming more recognized for its crucial role in cancer progression and immune regulation. It is located on chromosome 17q24.3, close to the SOX9 gene, and spans about 170 kilobases [43]. It is mainly found in the cytoplasm of breast cancer cells [43] and is involved in regulating transcription. It has been significantly overexpressed in several cancers, including TNBC, where it reduces therapy-induced senescence (TIS), favors cell proliferation, and influences immune evasion to exhibit oncogenic activity [44]. Gene enrichment analysis revealed that high expression of SOX9-AS1 in TNBC patients is significantly associated with several biological pathways, like adherens junctions, selenoamino acid metabolism, and the ErbB and Wnt signaling pathways. In cells with TNBC, SOX9-AS1 knockdown impedes cell-cycle progression, causes apoptosis, and minimizes invasive properties, as demonstrated by a study by Xuan Ye et al. [44].
Nevertheless, these effects are mitigated by overexpressing SOX9-AS1, verifying its function in maintaining tumor aggressiveness. Immune infiltration analyses using CIBERSORT showed that lower levels of SOX9-AS1 are linked to more infiltration of naïve B cells, CD8+ T cells, and γδ T cells (Figure 1). This suggests it may suppress the immune response. This suppression seems to relate to the reduction of SASP (senescence-associated secretory phenotype) factors, including IL-6 and IL-8, which are strong chemoattractants. Functional tests confirmed that reducing SOX9-AS1 makes TNBC cells more sensitive to tamoxifen-induced senescence. This is indicated by increased senescence-associated β-galactosidase activity and higher levels of SASP cytokines like IL-1α, IL-1β, IL-6, and IL-8. Mechanistic studies showed that SOX9-AS1 affects the Wnt/β-catenin pathway. When knocked down, it lowers β-catenin levels and alters GSK-3β activity, which promotes SASP expression and enhances senescence. Given that Wnt signaling is known to dampen antigen presentation and T cell activation, SOX9-AS1-mediated Wnt activation may contribute to developing a non-inflamed, immunologically "cold" tumor microenvironment resistant to immune infiltration. Therefore, the SOX9-AS1/Wnt/SASP regulatory axis represents a critical mechanism by which TNBC evades both senescence and immune surveillance, as represented in the Figure 3C. Targeting SOX9-AS1 or its downstream effectors may sensitize TNBC to immunotherapies and senescence-inducing agents, providing a promising therapeutic strategy to convert poorly immunogenic tumors into "hot" tumors more responsive to treatment.
Another lncRNA that garners attention as a critical regulator in immune responses is HITT (HIF-1α Inhibitor at Translation Level). It plays diverse role in tumor suppression and immune modulation. Genomically, its locus lies on chromosome 3p22.1 and with an approximate length of 32 kilobases [45]. Although HITT is transcribed, it does not encode a protein but exerts its functions through interactions with RNA-binding proteins and target mRNAs. It originally emerged as a transcription factor that dampens the translation of HIF-1α, vital to tumor growth and survival in hypoxic environments. Angiogenesis and glycolysis pathways, believed to be crucial to tumor development, are hampered by HITT's inhibition of HIF-1α mRNA translation through binding. A tumor-suppressive function suggests that reduced HITT expression is common in several types of cancers, including colorectal, breast, and cervical cancer [45].
A recent study by Lin et al. [46] demonstrated lncRNA HITT's functional repertoire by exploring its role in immune regulation, specifically within breast cancer's TME. The researchers showed that IFN-γ, a key cytokine involved in immune activation, significantly upregulates HITT expression in MDA-MB-231 cells. They found that upregulation of HITT is IFN-γ time- and dose-dependent, indicating that HITT responds to physiological immune stimuli. Using chromatin immunoprecipitation (ChIP) and promoter-reporter assays, the study identifies E2F1, a transcription factor, as the mediator of this effect. E2F1 directly binds to the HITT promoter following IFN-γ treatment. Silencing E2F1 stops HITT induction and confirms its essential role in regulation. E2F transcription factors control genes that manage DNA replication and cell cycle progression. Uncontrolled cell growth indicates a problem in the E2F pathway in cancer, often due to mutations in the Rb tumor suppressor. Overactive E2F in combination with HITT is a possible therapeutic target because it supports tumor growth, apoptosis resistance, and genomic instability [47].
Functionally, HITT overexpression reduces PD-L1 protein levels without altering PD-L1 mRNA expression, implying that HITT suppresses translation rather than transcription of PD-L1. Conversely, knocking down or knocking out HITT increases PD-L1 protein levels, especially when cells are stimulated with IFN-γ. This translational repression is highly significant given PD-L1's role in immune evasion, where it binds PD-1 on T cells and inhibits their cytotoxic activity, as shown in Figure 2A. Thus, HITT acts as a checkpoint modulator by post-transcriptionally repressing PD-L1 and potentially enhancing anti-tumor immunity. To uncover the mechanistic basis of this repression, Lin et al. investigated whether HITT interacts with the 5′ untranslated region (5′-UTR) of PD-L1 mRNA. Their experiments identified RGS2 (Regulator of G-protein Signaling 2) as a novel binding partner of HITT. Co-immunoprecipitation and RNA pull-down assays confirmed that HITT forms a complex with RGS2 and directly binds the PD-L1 5′-UTR. Mutational analyses of the 5′-UTR revealed that altering nucleotides 1 to 36 disrupts RGS2 binding and abolishes the repression of PD-L1 translation, underscoring the importance of this RNA motif. HITT thus acts as a molecular scaffold, guiding RGS2 to specific RNA structures in PD-L1's 5′-UTR to block its translation initiation.
The immunological relevance of this pathway was validated using co-culture experiments with CD8+ T cells. Cancer cells overexpressing HITT were found to be more susceptible to cytotoxic T cell killing, while HITT-deficient cells were resistant. Importantly, this effect was PD-L1-dependent, neutralizing PD-L1 with antibodies that erased the difference in T cell-mediated killing, confirming the translational control of PD-L1 as the key mechanism. These in vitro results were reconfirmed by in vivo tumor mouse models. Mice with tumors that overexpressed HITT showed slower tumor growth and more T cells entering the tumor. Reintroducing PD-L1 reversed this immune-mediated tumor suppression, affirming that HITT exerts its anti-tumor effects through PD-L1 downregulation.
Lin et al. [46] further analyzed the role of the HITT/RGS2 axis in breast cancer tissue samples. The authors found that the HITT/RGS2 axis may be functionally relevant in patients, as their analysis showed a negative correlation between HITT and PD-L1 levels and a positive correlation between RGS2 expression and low PD-L1 protein. Lower PD-L1 was seen in tumors with high HITT expression, suggesting that immune evasion may be facilitated by tumor downregulation of HITT. This finding supports the translational significance of HITT pathway targeting in cancer treatment. Given that PD-L1 is a central target in immune checkpoint therapy, the ability of HITT to suppress PD-L1 at the translational level introduces a novel therapeutic approach. Unlike conventional methods that use antibodies to block PD-L1 or its receptor PD-1, modulating HITT activity could provide a complementary or synergistic strategy. Therapeutic options may include HITT-mimicking oligonucleotides, gene therapy to overexpress HITT, or modulators that enhance HITT–RGS2 complex formation. These strategies might be especially helpful in overcoming checkpoint blockade therapy resistance. Overall, the authors provide a thorough functional and mechanistic description of HITT as a critical immune response regulator in breast cancer. The study reveals a hitherto unidentified layer of immune checkpoint regulation by clarifying how IFN-γ activates HITT through E2F1 and how HITT inhibits PD-L1 translation via RGS2. The study also makes a strong case for creating HITT-based immunotherapies by confirming HITT's immune-boosting and tumor-suppressive properties in cellular and animal models and connecting these results with clinical data. Future work could expand this paradigm to other cancers and explore combination therapies integrating HITT activation with existing checkpoint inhibitors.
NKILA(NF-κB Interacting Long Non-Coding RNA), an 8.4-kilobase-long cytoplasmic lncRNA, plays a crucial role in cancer immunity [48]. Initially identified for its ability to inhibit the NF-κB signaling pathway in breast cancer cells, NKILA has since been recognized as a key immunoregulatory molecule that affects T cell survival, tumor immune evasion, and the modulation of the immune microenvironment across various cancer types [49].
NKILA directly binds to the NF-κB/IκB complex in the cytoplasm [49]. Maintaining the stability of this complex prevents IκB from being phosphorylated and degraded. As a result, NF-κB is unable to migrate into the nucleus, suppressing the transcription of anti-apoptotic genes. NF-κB plays a key role in T-cell survival, growth, and the production of inflammatory cytokines like IL-2. Therefore, NKILA acts as a negative regulator of NF-κB, shifting the balance toward pro-apoptotic signaling when T cells are activated. This suppression of NF-κB makes T cells, especially cytotoxic CD8⁺ T lymphocytes (CTLs) and type-1 helper T cells (TH1), very vulnerable to activation-induced cell death (AICD). In comparison, regulatory T cells and TH2 cells show relative resistance. Notably, tumor-infiltrating CTLs and TH1 cells in breast and lung cancers demonstrate elevated NKILA expression, which correlates with increased apoptosis and poorer patient survival outcomes.
Mechanistically, the authors illustrate that upon T-cell receptor (TCR) engagement, an influx of intracellular calcium activates calmodulin, facilitating histone acetylation at the NKILA promoter. In cancer, dysregulated calmodulin signaling contributes to tumor progression, metastasis, and therapy resistance by activating oncogenic pathways like PI3K/Akt and MAPK (Mitogen Activated protein kinase) [50]. While histone acetylation at the NKILA promoter mechanism is beneficial in preventing chronic inflammation and autoimmunity, NKILA becomes a disadvantage in cancer, as it induces the premature death of tumor-specific T cells. Its expression is increased by STAT1 and histone acetylation in activated T cells, making NKILA an essential connection between T cell activation and activation-induced cell death (AICD). This process also improves the binding of STAT1 and the transcriptional activation of NKILA. The expression of NKILA leads to a continued inhibition of NF-κB, which usually supports T cell survival. As a result, higher levels of NKILA make tumor-specific T cells more sensitive to apoptosis through Fas/FasL-induced signaling and other pathways that promote cell death during AICD. Authors further support their results by doing electrophoretic mobility shift assays using drugs to block NF-κB, which increased sensitivity to apoptosis.
The authors further demonstrate that cytotoxic T lymphocytes' (CTLs') anti-tumor efficacy and persistence increase when NKILA is silenced. Compared to control groups, NKILA-knockdown CTLs demonstrated enhanced tumor infiltration (Figure 1) and decreased tumor growth in patient-derived breast cancer xenograft models. This illustrates how NKILA modulation in adoptive T-cell immunotherapy has therapeutic potential. To sum up, NKILA is an essential immune-regulatory long non-coding RNA. It facilitates tumor immune evasion by limiting T-cell longevity by suppressing NF-κB. Targeting NKILA also offers a strong way to improve anti-tumor immunity and T-cell persistence in therapeutic settings.
MALAT1 (Metastasis-Associated Lung Adenocarcinoma Transcript 1), also known as NEAT2, is a highly conserved lncRNA [51]. It is located on chromosome 11q13.1 and is about 12.8 kilobases long [52]. This lncRNA is mainly found in nuclear speckles and is key in regulating alternative splicing, gene transcription, and epigenetic modifications [53]. MALAT1 is often overexpressed in various cancers, including lung, breast, colorectal, prostate, and glioblastoma. It is linked to tumor progression, metastasis, and worse prognosis [54]. It promotes cell growth, epithelial-mesenchymal transition (EMT), invasion, and immune evasion. Additionally, it acts as a sponge for tumor-suppressive miRNAs, which affects immune and inflammatory signaling pathways [55].
To investigate the role of lncRNA Malat1 in regulating T cell infiltration in the breast tumor microenvironment, Adewunmi et al. (2023) [56] established syngeneic mouse models using orthotopically implanted T12 and 2208L cell lines. The T12 cell line represents a claudin-low tumor subtype known for high macrophage infiltration, while 2208L is a luminal-like TNBC model with a rich presence of neutrophils. Both cell lines have highly immunosuppressive myeloid environments that support tumor growth, metastasis, and resistance to treatment. In the 2208L model, knocking down Malat1 led to more CD8+ T cells, significantly increasing cytotoxic markers Granzyme B and IFN-γ. There was also a decrease in immunosuppressive CD4+ T cells. In the T12 subtype, although the total number of CD8+ T cells stayed the same, there was a notable increase in Granzyme B+ T cell infiltration. Levels of CD4+ T cells dropped, while Treg populations were unaffected after Malat1 silencing. Overall, analysis of tumor-infiltrating lymphocytes in primary tumors showed that inhibiting Malat1 using Gapmer antisense oligonucleotides (ASOs) created a more immunostimulatory tumor microenvironment, boosting cytotoxic T cell infiltration and division (Figure 1).
Tumor secretory profiles of both cell lines are also altered by Malat1 knockdown. Chemokines like Ccl5 that attract immunosuppressive M2-type macrophages are suppressed in T12 cancer cells. Also, the inflammatory cytokines that attract M1-macrophages, like Cxcl9 and Cxcl10, are decreased, denoting that macrophage repolarization to a more inflammatory state is not supported by Malat1 inhibition. Similarly, reduced secretion of immunosuppressive cytokines/chemokines, including Cxcl1, Gm-csf, TNF-α, Ccl5, and cytokine IL6, was observed in Malat1 ASO-treated 2208L cell lines. All these factors that are known to recruit MDSC (myeloid-derived suppressor cells) and develop an immunosuppressive microenvironment are reduced. By reducing the immunosuppressive activity of tumor-associated macrophages (TAM) and MDSCs on T cells, Malat1 knockdown in the TNBC tumor microenvironment contributes towards a more immunostimulatory landscape, enabling a stronger T cell response towards the tumor.
In a similar exploration of MALAT1's role in T cell modulation within breast cancer, Kumar and colleagues [57] utilized murine breast cancer models and human and mouse datasets. They found that Malat1 is essential for initiating metastatic lesions and their reactivation from dormant states. Mechanistically, among its downstream effects, Malat1 enhances the expression of WNT ligands, thereby reinforcing autocrine signaling loops that support tumor self-renewal. Additionally, it upregulates a specific set of serine protease inhibitors, most notably SERPINB6B [57].
Functionally, SERPINB6B exhibits dual immunomodulatory effects. It inhibits caspase 1 and cathepsin G, thereby preventing the activation and pore formation of gasdermin D, which is the central executor of pyroptosis. This inhibition interrupts a highly inflammatory form of programmed cell death known as pyroptosis, which typically alerts and attracts immune effectors. By impairing pyroptosis, SERPINB6B dampens immunogenic cell death signals, reducing the clearance of early metastatic lesions by cytotoxic T cells.
Genetic or pharmacologic suppression of Malat1 through antisense oligonucleotides (ASOs) effectively disrupts this immune-evasive mechanism. Specifically, Malat1 ASOs downregulate SERPINB6B, lifting the blockade on caspase-1 and cathepsin G. This reinstates gasdermin D-mediated pyroptosis and enhances T cell-mediated tumor lysis. The resulting molecular cascade significantly reduces metastatic colony formation and improves survival outcomes in treated mouse models. Significantly, the metastasis-suppressive effect of Malat1 inhibition is negated when SERPINB6B is knocked out, highlighting SERPINB6B as a crucial mediator of Malat1's pro-metastatic role. SERPINB6B is a serine protease inhibitor (serpin) family member that regulates protease activity and maintains cellular homeostasis. Though its specific role in cancer remains underexplored, aberrant expression of SERPINB6B has been associated with inflammation and immune modulation, suggesting potential involvement in tumor progression, immune evasion, and metastasis [57].
Overall, this study suggests that Malat1 is a master regulator of metastatic emergence by linking tumor-intrinsic signaling with immune suppression and underscoring its potential as a target for anti-metastasis therapies.
SNHG16 (Small Nucleolar RNA Host Gene 16) lncRNA is located on chromosome 17q25.1 [58], and has been identified as an oncogenic regulator in cervical, neuroblastoma, pancreatic, gastric, and breast cancers [59]. It is often overexpressed and associated with adverse clinical outcomes [60]. A pan-cancer meta-analysis indicates that upregulated expression of SNHG16 facilitates proliferation and metastasis while blocking apoptosis by targeting key signaling pathways, such as TGF-β/SMAD, JAK/STAT3, and PI3K/AKT, as well as the Wnt/β-catenin pathway, and others. Additionally, SNHG16 acts as a sponge for several miRNAs, including miR-200a-3p, miR-132-3p, and miR-302a-3p [61].
Recently, Ni et al. (2020) [62] discovered that breast cancer cells create a localized immunosuppressive environment through the exosome-mediated delivery of long SNHG16 lncRNAs. Tumor-infiltrating γδ1 T cells (Vδ1) usually have cytotoxic anti-tumor functions and are plentiful in breast tumors. However, they paradoxically do not eliminate cancer cells. Instead, a specific group of γδ1 T cells expressing CD73, an ectonucleotidase that produces adenosine, a potent immunosuppressant, is found in higher numbers in BC tissues than in normal breast tissue. Furthermore, increased levels of CD73+ γδ1 T cells are present in the peripheral blood of patients, which correlates with tumor size. Functionally, these CD73+ γδ1 T cells strongly suppress the growth of conventional CD4+ and CD8+ T cells, reducing their ability to kill cancer cells, including the release of perforin and granzyme B. Blocking the receptor interactions of CD73 restores T-cell activity and confirms that the immunosuppressive effects of these γδ1 Tregs mainly result from excess adenosine secretion.
To investigate the upregulation of CD73 in γδ1 T cells, the authors performed transwell co-culture experiments involving breast cancer (BC) cells and primary Vδ1 T cells. They found that this effect depended on cancer cell-derived exosomes (TDEs) and the presence of TGF-β1 signaling, but did not require direct contact between the cells. It was determined that exosomes derived from BC cell lines (MCF-7, MDA-MB-231, T-47D) contained significantly higher levels of SNHG16 compared to those from non-malignant breast cells (MCF-10A). Microarray analysis and qRT-PCR validation identified SNHG16 as the crucial lncRNA responsible for the induction of CD73 expression; knocking down SNHG16 in the exosomes abolished their ability to upregulate CD73 in γδ1 T cells.
Mechanistically, SNHG16 acts as a competing endogenous RNA (ceRNA) within γδ1 T cells by sequestering miR-16-5p. This action leads to the derepression of SMAD5, which serves as the downstream effector of the TGF-β1 pathway. The resulting elevation of SMAD5 amplifies TGF-β1 signaling, driving the transcription of CD73. Disrupting this pathway through SNHG16 knockdown, miR-16-5p overexpression, or SMAD5 suppression prevents the induction of CD73. Consequently, the exosomal SNHG16/miR-16-5p/SMAD5 regulatory loop prepares γδ1 T cells for CD73-mediated immunosuppression by leveraging TGF-β1 signaling, Figure 3B. This study identifies CD73+γδ1 T cells as the primary regulatory γδ T-cell population in breast cancer, which plays a significant role in diminishing anti-tumor immunity through the production of adenosine. Notably, exosomes derived from BC cells loaded with SNHG16 induce the phenotypic transformation of γδ1 T cells into immunosuppressive CD73+ regulatory T cells (Tregs). The authors show that TGF-β1, a recognized inducer of CD73, is further enhanced by SNHG16 through the miR-16-5p-SMAD5 signaling pathway. Given the functional implications of γδ1 T cells transitioning from a cytotoxic to a suppressive role, this study reveals the mechanism of immune evasion in BC. It proposes that targeting exosomal communication, SNHG16, or CD73+γδ1 Tregs could open new therapeutic possibilities. The findings highlight the broader impact of exosomal SNHG16 long non-coding RNAs in shaping the tumor microenvironment.
Overall, from the above section, we can conclude that the aforementioned lncRNAs play a vital role in the infiltration of immune cells to the tumor microenvironment, hence controlling the breast cancer tumorigenesis.
3. Immunomodulatory lncRNAs Orchestrating T Cell Dysfunction and Immune Escape
Through the modulation of T cell activity, lncRNA contributes to establishing an immunosuppressive tumor microenvironment, promoting tumor immune evasion. Recruitment of immunosuppressive cells like Treg cells, M2 macrophages, and myeloid-derived suppressor cells (MDSCs) around the tumor leads to inactivation of cytotoxic T lymphocytes (CTLs) by secretion of immunosuppressive chemokines like CCL2, CXCL12 [63]. MDSCs facilitate tumor evasion through multiple mechanisms, such as depletion of arginine and increased production of reactive nitrogen and oxygen species, which impair T cell receptors (TCR). Inhibitory cytokines like transforming growth factor-beta (TGF-ꞵ), interleukin 10 (IL-10) promote differentiation of T regulatory cells, thus inhibiting anti-tumor immunity [64]. In addition to myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs) and tumor-associated neutrophils (TANs) within the tumor microenvironment (TME) show immunosuppressive properties that help tumors grow. These cells can activate immune checkpoint pathways and their related receptors, which promote tumor growth [65,66]. Another way tumors can trigger immunosuppressive responses is by increasing the levels of immunosuppressive molecules and their receptors. CTLA-4 (Cytotoxic T lymphocyte associated protein 4), PD1 Programmed death 1), Lymphocyte activating 3 (LAG3) are the inhibitory receptors on CD4+ T helper cells and CD8+ T cytotoxic cells that interact with specific ligands on tumor cells. These interactions suppress T cell activation, proliferation, and effector functions, resulting in immune escape of tumor cells [67]. CTLA-4 mediates immunosuppression by inhibiting various proliferative pathways, such as the NF-kB and PI3K/Akt pathways [68].
Another long non-coding RNA that has gained recognition as a key regulator of T-cell modulation is SNHG1 [21]. Originating from a host gene linked to small nucleolar RNAs (snoRNAs), SNHG1 is located in the nucleus and cytoplasm, and participates in transcriptional and post-transcriptional regulation. It is on chromosome 11q12.3 and spans approximately 4.6Kb [69,70]. SNHG1 is frequently upregulated in several cancers, including breast, lung, colorectal, liver, and gliomas. Its overexpression is linked to poor clinical outcomes and tumor progression [71]. As an oncogenic lncRNA, SNHG1 promotes proliferation, invasion, metastasis, epithelial-mesenchymal transition (EMT), and resistance to apoptosis by negatively regulating tumor suppressors such as p53 [72]. One of the main mechanisms involves acting as a competitive endogenous RNA (ceRNA) where it binds and sequesters tumor-suppressive miRNAs such as miR-448, miR-195, and miR-145, thereby facilitating oncogenic signaling pathways [21,70,73]. miR-195, miR-448, and miR-14 play a significant role in breast cancer progression, prognosis, and therapy resistance. miR-195 is a tumor suppressor in breast cancer and inhibits cell growth and movement. It also spreads by targeting key oncogenes such as BCL2, CCND1, and FASN [74]. miR-448, on the other hand, acts as a tumor suppressor by blocking genes involved in epithelial-to-mesenchymal transition (EMT), like ZEB1 [75]. It also affects pathways such as PI3K/AKT and Wnt/β-catenin, which reduce metastasis and improve chemosensitivity. miR-14, although not studied as much in humans, has been linked to breast cancer progression based on similarities with Drosophila and findings from comparative studies [76].
Recently, [21] researchers examined SNHG1's role in T cell responses related to breast cancer. The study found that SNHG1 expression was higher in tumor-infiltrating CD4+ T cells (TILs) than in peripheral CD4+ T cells. This increase is linked to lower levels of miR-448 and higher expression of indoleamine-2,3-dioxygenase (IDO), IL-10, and the transcription factor FoxP3. These markers suggest a rise in regulatory T cell (Treg) differentiation. By directly binding to iR-448, SNHG16 inhibits its activity, weakening its tumor suppressor ability. Since miR-448 negatively regulates IDO, IL-10, and FoxP3, its inhibition by SNHG1 results in higher expression of IDO. IDO then breaks down tryptophan into kynurenine, creating an immunosuppressive tumor environment. This change decreases effector T cell activity while promoting Treg expansion. In in vivo murine xenograft models, SNHG1 knockdown reduced tumor growth and lowered IL-10, IDO, and FoxP3, highlighting its role in immune evasion and tumor progression, Figure 3A.
In summary, SNHG1 facilitates immune escape in breast cancer by enhancing Treg cell differentiation through the miR-448/IDO axis, thereby dampening anti-tumor immunity. Future studies warrant investigating whether SNHG1 modulates the infiltration and function of other immune cell subsets within the tumor microenvironment, including CD8+ T cells, dendritic cells, and M2 macrophages.
GATA3-AS1, or GATA3 antisense RNA 1, is a long non-coding RNA in a head-to-head orientation with the GATA3 protein-coding gene on chromosome 10p14 [77]. It spans about 2 kb and has several splice variants, including four main exons. Under normal conditions, GATA3-AS1 is mainly expressed in T-helper 2 (Th2) lymphocytes. It regulates GATA3 transcription by remodeling chromatin [78]. GATA3-AS1 brings histone methyltransferase complexes, like MLL/WDR5, to the shared promoter region of GATA3-AS1 and GATA3. This process helps add H3K4me3 and H3K27ac marks. It also forms DNA: RNA hybrid R-loops that support transcriptional activation [79]. The knockdown of GATA3-AS1 in Th2 cells decreases GATA3 expression and the downstream cytokines IL-5 and IL-13. In the context of cancer, GATA3-AS1 is often aberrantly overexpressed across various tumor types and functions as an oncogenic lncRNA, contributing to tumor growth and immune evasion [23].
Zhang et al. (2020) [23] examined how the lncRNA GATA3-AS1 contributes to aggressive behavior in TNBC through immune evasion. Using bioinformatics tools, the authors found that GATA3-AS1 was significantly overexpressed in both TNBC tissues and cell lines compared to normal breast tissue. This overexpression is linked to larger tumor sizes, lymph node metastasis, advanced TNM staging, and worse overall survival rates. Functional tests performed on TNBC cell lines showed that knocking down GATA3-AS1 with shRNA led to significant reductions in proliferation, colony formation, migration, and invasiveness. Additionally, there was an increase in apoptosis and greater susceptibility to CD8+ T cell-mediated killing. Mechanistically, the study reveals two distinct yet complementary molecular pathways through which GATA3-AS1 promotes oncogenesis and immune evasion. In the first pathway, GATA3-AS1 acts as a competing endogenous RNA (ceRNA), sponging miR-676-3p and thereby relieving the repression on COPS5 (also known as CSN5), a deubiquitinase that specifically stabilizes PD-L1 (Figure 2B). Numerous assays, including RIP, luciferase reporter, and RNA pull-down, confirm the direct interaction between GATA3-AS1 and miR-676-3p. Furthermore, knockdown experiments indicate that decreased levels of GATA3-AS1 lead to lower COPS5 levels, enhanced ubiquitination of PD-L1, and a reduction in its half-life.
They further show that GATA3-AS1 directly interacts with the GATA3 protein, which helps its ubiquitination and eventual degradation. RIP assays confirm this interaction. Treatments with cycloheximide and MG132 (a proteasome inhibitor) reveal that overexpression of GATA3-AS1 speeds up the turnover of GATA3. Since GATA3 is a transcription factor that encourages luminal differentiation and reduces malignancy, its destabilization worsens the malignant phenotype. By stabilizing PD-L1, GATA3-AS1 supports immune escape in TNBC. GATA3-AS1 knockdown leads to increased PD-L1 ubiquitination, decreased protein levels, stronger CD8⁺ T-cell infiltration (Figure 1), and heightened apoptosis, demonstrating restored anti-tumor immunity. This dual mechanism PD-L1 stabilization via miR-676-3p/COPS5 and GATA3 destabilization positions GATA3-AS1 as a central driver of aggressive growth and evasion of host immunity in TNBC. The authors note that these findings highlight GATA3-AS1 as a promising therapeutic target: targeting it could destabilize PD-L1, restore GATA3 tumor-suppressive functions, suppress tumor proliferation, and enhance immunotherapy responses.
This study advances our understanding of lncRNA-mediated cancer progression by shedding light on how lncRNA orchestrates both the tumor microenvironment and malignant behavior. It identifies GATA3-AS1 as a powerful oncogenic driver and immune modulator in TNBC. Further studies are warranted to understand more about the role of GATA3-AS1 in immune evasion.
Tissue differentiation-inducing non-protein coding RNA (TINCR) was first characterized over a decade ago for its role in regulating cellular differentiation and tumor progression. It is situated on chromosome 19p13.3 [8]. The TINCR gene spans approximately 20 kilobases and is transcribed into a long non-coding RNA that lacks significant open reading frames. Functionally, TINCR is highly versatile; it serves as a molecular scaffold for proteins, recruits epigenetic modifiers, and acts as a competing endogenous RNA.
Wang et al. (2023) [22] looked at how lncRNA TINCR weakens the effectiveness of PD-L1 inhibitors in breast cancer. It does this by creating a complex network that involves changes to genes and immune responses. The authors note that PD-L1 blockage often has limited success in breast cancer because tumors can escape the immune system. They focus on TINCR, which is linked to cancer progression, as a possible reason for this immune resistance. Their findings show that TINCR is overexpressed in both breast cancer tissues and cell lines. This overexpression is positively linked to PD-L1 protein levels, suggesting a direct role in avoiding immune attacks. Through functional tests, they show that TINCR encourages breast cancer cell growth, movement, invasion, and tumor development in living organisms. Importantly, silencing TINCR improved the effectiveness of PD-L1 blockage in mouse models. This suggests that TINCR significantly contributes to resistance against immunotherapy.
The study reveals that TINCR regulates PD-L1 primarily at the protein level by inhibiting its ubiquitin-mediated degradation rather than affecting PD-L1 transcript levels. This led the authors to identify the deubiquitinase USP20 as a downstream effector whose expression is promoted by TINCR. The knockdown of USP20 replicates the decline in PD-L1 protein levels, while USP20 overexpression restores these levels in TINCR-deficient cells, thereby confirming a functional relationship. Mechanistically, TINCR acts as a ceRNA in the cytoplasm by sponging miR-199a-5p. This microRNA would otherwise bind to and degrade USP20 mRNA. As a result, the stability of USP20 mRNA improves, which leads to higher levels of USP20 protein and a subsequent decrease in PD-L1 ubiquitination.
TINCR is involved in nuclear epigenetic regulation by recruiting the DNA methyltransferase DNMT1 to the promoter of miR-199a-5p. This recruitment leads to CpG methylation, which causes the transcriptional downregulation of the miRNA and an increase in USP20 expression. These cytoplasmic and nuclear mechanisms significantly raise levels of USP20 and PD-L1 proteins. Further investigation shows that IFN-γ signaling acts as an upstream regulator of TINCR through the phosphorylation and activation of STAT1. After IFN-γ stimulation, STAT1 binds to the TINCR promoter, which boosts TINCR transcription. Chromatin immunoprecipitation (ChIP) and luciferase assays confirm the direct interaction between STAT1 and the TINCR promoter. Moreover, immunohistochemical analysis of tissue specimens also shows a link between STAT1 and TINCR expression.
This study is the first to identify TINCR as a crucial regulator of tumor immunity in breast cancer, broadening its previously established roles beyond tumor progression. The IFN-γ/STAT1→TINCR→USP20→PD-L1 pathway connects inflammatory signaling with immune evasion (Figure 2C), revealing a network in which TINCR fine-tunes the stability of USP20 both epigenetically and post-transcriptionally to increase PD-L1 protein levels. This ultimately dampens T cell-mediated anti-tumor activity and diminishes the efficacy of PD-L1 inhibitors. Experimental results demonstrate that TINCR knockdown significantly enhances tumor sensitivity to PD-L1 blockade in mouse models, suggesting its potential as a therapeutic target. It is clear that TINCR lncRNA functions are significantly exacerbated because it works in two ways: it recruits DNMT1 in the nucleus and acts as a miRNA sponge in the cytoplasm. This suggests that focusing on TINCR could improve the immune system against breast cancer by lowering PD-L1 levels and making it easier for the immune system to reactivate.
Overall, this study discovers an innovative method by which breast cancers can resist immunotherapy and implies that TINCR might serve as a good target for making PD-L1-based treatments operate more effectively.
4. In Silico Analysis of lncRNAs Associated with T Cell Function
4.1. Functional Enrichment Analysis
Functional roles of studied lncRNAs were assessed using Gene Ontology (GO) analysis to explore their target genes and associated products. Pathway annotation using DAVID bioinformatics was carried out, and results were visualized using GraphPad Prism, with the top 10 KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways represented on the Y-axis, while having negative logarithms of false discovery rate (-log10 FDR) on the X-axis, along with gene count (Figure 4). LINC00472 was found to be significantly enriched in transcriptional dysregulation in cancer, hepatocellular carcinoma, HPV infection, PI3/Akt and MAPK signalling pathways [80], and other pathways in cancer, as denoted in Figure 4(A). Downregulation of LINCOO472 is associated with tumor suppression in hepatocellular carcinoma by targeting miR-93-5p/PDCD4 pathway [81]. LncRNA MIAT is associated with chemical carcinogenesis-receptor activation, Human T-cell leukemia virus 1 infection, cell cycle, transcriptional dysregulation, spliceosome, and pathways in cancer, as represented in the Figure 4(B). By regulating the G1/S phase cell cycle transition, higher expression of MIAT contributes to invasion and migration in esophageal cancer [82]. MIAT, through its tandem UACUAAC motif, binds to Splicing factor-1 (SF-1) and alters pre-mRNA splicing [83]. LncRNA SOX9-AS1 was found to be enriched significantly only in four KEGG pathways, including, mRNA surveillance pathway and the spliceosome, Figure 4(C). Top 10 KEGG pathways of NKILA comprise HPV Infection, MAPK signalling pathways, chemical carcinogenesis-receptor activation, cell cycle, altered transcription, HPV infection, pathways of cancer, and Hepatocellular carcinoma (Figure 4D). NKILA, by NF-kB, reduces cell cycle progression, invasion, and metastasis in melanoma cells, and induces apoptosis in them [84]. Figure 4(E) represents significant enrichment of MALAT1 in Ras, PI3-Akt, and MAPK signaling pathways, autophagy, and pathways in cancer. MALAT1 induces autophagy by suppressing miR-204 in gastric cancer, and it serves a pro-tumorigenic role in glioma by modulating the miR-101/STMN1-RAB5A-ATG4D, and increasing cellular proliferation [85]. SNHG16 expression is associated with viral carcinogenesis, HPV infection, cell cycle, spliceosome, HPV, pathways of cancer, and chemical carcinogenesis-receptor activation, shown in Figure 4(F). In bladder cancer and glioma cells, silencing of SNHG16 induces cell cycle arrest at the G1 stage. This was achieved by elevated expression of p21 and downregulation of Cyclin D1 and cyclin B1 [86]. The top 10 significantly enriched KEGG pathways associated with LncRNA SNHG1 are represented in Figure 4(G). This includes cell cycle, HPV infection, Human T-cell leukemia virus 1 infection, disrupted transcriptional activity, and pathways in cancer. Through the increased expression of SNHG1, HPV16E6 promotes angiogenesis in lung cancer [87]. Similarly, enrichment of GATA3-AS1 in various signaling pathways, such as estrogen signaling pathways, transcriptional dysregulation, hepatocellular carcinoma, MAPK, PI3K/Akt, cAMP signaling pathway, and pathways in cancer, is denoted in Figure 4(H). No significant pathway enrichment analysis was found for lncRNAs HITT and TINCR.
4.2. Survival Analysis and Prognostic Value of Studied LncRNAs
Furthermore, to validate the role of lncRNAs in prognosis and overall survival of patients in breast cancer, these previously studied lncRNAs were searched in the breast invasive carcinoma category in the TCGA database. Kaplan-Meier plots are represented in Figure 5(A-H), with lncRNAs MIAT and SNHG16 being statistically significant. Increased expression of MIAT is significantly associated (p-value= 0.0065) with better survival outcomes, suggesting its potential as a promising prognostic biomarker. On the other side, patients with increased SNHG16 expression exhibited poorer survival probabilities when compared to patients with lower SNHG16 expression profiles. With this data being statistically significant (p-value=0.035), SNHG16 could be considered as a negative prognostic marker for invasive breast carcinoma. The data obtained for LINC00472, SOX9-AS1, MALAT1, SNHG1, and GATA3-AS1 were found to be non-significant, with p-values <0.05. However, no data were available to date in the TCGA database for HITT and TINCER lncRNAs in breast invasive carcinoma regarding the overall survival of patients.
Further, analysis of differential expression of these lncRNAs in the context of breast invasive carcinoma was assessed through UALCAN on 29th July, 2025. The expression pattern of the aforementioned lncRNAs in the primary tumor of breast cancer, as compared to normal tissues, is represented in Figure 6. Expression of LINC00472 was found to be downregulated in breast invasive carcinoma with a non-significant p-value of 0.066, denoted in Figure 6(A). Expression profiles of MIAT, NKILA, SNHG1, and GATA3-AS1 are upregulated in breast invasive carcinoma with a statistically significant p-value, with MIAT having a p-value = 2.21 (Figure 6B). NKILA with the p-value < 0.05 (Figure 6D), SNHG1 upregulated with p-value = 1.63 × 10-7, Figure 6(F), and GATA3-AS1 being upregulated in breast cancer with a significant p-value Figure 5(H). No such changes in the expression profile of SNHG16 are observed, Figure 6(G). Conversely, LncRNAs- SOX9-AS1(Figure 6C) and MALAT1, (Figure 6E), are downregulated with significant p-values of 1.50 × 10-14 and 2.25 × 10-3.
4.3. Network Analysis
To explore functional associations of these lncRNAs, by using the RNAInter v2.4 database, a set of target genes that are already reported for each corresponding lncRNA is obtained. RNA-RNA interactions for each of these lncRNAs were explored on this database for the mRNA category and by using Cytoscape, a network was constructed from extracted data, offering a visual representation of interconnected regulatory patterns, Figure 7. Genes like MYC (myelocytomatosis oncogene), SOX2 (Sex determining region Y-box 2), TP53 (Tumor protein 53), BCL6 (B cell lymphoma 6), TARDBP (Transactive Response DNA binding protein), POLR2A (RNA Polymerase II Subunit A) are some of the common target genes of lncRNAs - LINC00472, MIAT, NKILA, SNHG1, SNHG16, and GATA3-AS1 were identified (Figure 7). c-MYC mediates the interaction between breast cancer cells and tumor microenvironment. It facilitates angiogenesis, epithelial to mesenchymal transitions, and cancer-associated fibroblast (CAF) activity. It can further modulate the TAM (tumor-associated macrophage), NK cells, T cells and B cells, along with the expression of immune checkpoint molecules like PD-L1 [88]. Mutant p35 gene promotes tumor aggressiveness and facilitates metastatic behaviour through different molecular routes [89]. BCL6 gene is reported to reduce apoptosis and aid in proliferation, migration, and invasion in breast cancer [90]. Certain genes like BAX (BCL2 Associated X, apoptosis regulator), CDH1 (Cadherin), GATA4 (GATA binding protein 4), ERG (ETS-related gene) and others are also reported targets for specific lncRNAs (Figure 7). CDH1 is a tumor suppressor gene that transcribes into E-cadherin protein, which provides stability to cells by inhibiting cell motility [91]. GATA4 reduces the migration and invasion of breast cancer by decreasing the expression profile of MMP9 (Matrix metalloproteinase 9), which otherwise degrades extracellular matrix and aids in EMT [92]. The network obtained provides a more comprehensive framework for understanding the molecular functions of these lncRNAs within the complex biological systems.
5. Conclusion and Future Outlook
In conclusion, current evidence emphasizes the essential role of lncRNAs as central regulators of the tumor immune landscape, specifically by influencing the T cell dynamics. This review highlights various lncRNAs, such as LINC00472, MIAT, HITT, NKILA, MALAT1, and SNHG16, affecting T cell infiltration. The role of SNHG1, TINCR, and GATA3-AS1 in immune escape is also discussed, providing insights into how lncRNAs facilitate breast cancer immunity, which can be utilised for therapeutic purposes.
By reviewing available literature, we have observed that lncRNA mediates T cell activation, differentiation, exhaustion, and immune checkpoint expression within the tumor microenvironment of breast cancer. By modulating the differentiation and functional aspects of Treg and T cytotoxic cells, lncRNA can inhibit tumor growth or aid in tumor immune evasion by recruiting Treg cells, M2 macrophages, and MDSCs. LncRNA uses key signaling pathways like NF-kB, Wnt/ β-catenin, PI3/AKT, and JAK/ STAT to modulate T-cell plasticity. Interfering with the lncRNA-regulated signaling may improve immunotherapeutic efficacy, better stratification of breast cancer patients, and personalised therapeutic paradigms. LncRNAs exhibit their immunomodulatory effects by various molecular mechanisms, like miRNA decoy activity, transcriptional regulation, and direct interaction with protein complexes involved in immune signaling.
The cytokine and chemokine signaling, which governs T cell activation (e.g., IL-2), T cell differentiation (e.g., IL-12, IL-4), T cell migration (eg, CCL5, CXCL9, CXCL10), and effector functions (IFN-ɣ, TNF-ɑ), has a crucial role in T cell-mediated immune functions. LncRNAs, including SOX9-AS1, NKILA, MALAT1, HITT, and GATA3-AS1, regulate key signaling cascades, driving the changes in cytokine and chemokine production, thus remodelling the tumor microenvironment. By altering the IFN-ɣ signaling, HITT fosters an immunologically active tumor microenvironment. NKILA plays an immunosuppressive role by sensitising T cells to activation cell death. lncRNAs like LINK-A, MALAT1, MIAT, and GATA3 possess strong potential as prognostic and diagnostic breast cancer biomarkers, as their expression profiles are often associated with TNM staging, survival rates, and T cell infiltration rates. Upregulated MIAT, SOX9-AS1, and HITT profiles exhibit delayed tumor growth by increased CD8+ cytotoxic T cells infiltration into tumor sites. Similarly, down regulation of MALAT1, GATA3-AS1 and NKILA promotes CD8+ T cell infiltration, suppressing tumor proliferation and facilitating the immunotherapy response. As a molecular sponge for distinct miRNAs, lncRNAs such as MIAT1 and GATA3-AS1 regulate PDL1 expression through a competitive endogenous RNA (ceRNA) mechanism. Overexpression of lncRNA, such as TINCR and MIAT, GATA3-AS1, and knockdown of HITT is associated with increased PD-L1 protein activity, either by enhanced PD-L1 deubiquitination or by increasing PD-L1 translation, thus aiding in tumor immune evasion.
Based on the current study's findings, to fully understand the roles lncRNAs play in T cell activation and exhaustion completely, more research focusing on the functional validation of lncRNAs using in vivo and single-cell multi-omics technologies is warranted. Clinical evaluation of lncRNA expression profiles is required to establish them as reliable biomarkers for prognosis and patient stratification. Artificial intelligence and machine learning tools can also be utilized to correlate the expression patterns of lncRNAs with T cell function and activation, to potentially improve immunity in breast cancer patients. For therapeutic purposes, lncRNAs that are oncogenic could be targeted, or lncRNAs that are tumor-suppressive could be restored, utilizing promising technologies, like CRISPR, CAR-T cells, and antisense oligonucleotides. Moreover, lncRNA modulation along with immune checkpoint inhibitors may be used to improve personalized immunotherapy for breast cancer. Based on the characteristics of the tumor microenvironment and the levels of immune cells, personalized immunotherapy options, such as immune checkpoint inhibitors or combination therapies, can be provided to improve breast cancer care.
Author Contributions
Conceptualization, A.J.; methodology, A.D.; data curation, A.D., S.S., V.U., and A.J.; writing original draft preparation, A.D., and A.J.; writing and editing, A.D., S.S., V.U., H.P., and A.J.; supervision, A.J.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest to report. All authors have read and agreed to the published version of the manuscript.
Funding Statement
This research received no external funding.
Acknowledgments Statement
We are thankful to DST-PURSE and DST-FIST, Central University of Punjab, for providing us with funds for infrastructure facilities.
Abbreviations
| AICD | Activation-induced cell death |
| ASOs | Antisense oligonucleotides |
| AUC | Area under the curve |
| BAX | BCL2 Associated X, apoptosis regulator |
| BC | Breast cancer |
| BCL6 | B cell lymphoma 6 |
| CAF | Cancer-associated fibroblast |
| CDH1 | Cadherin |
| CeRNA | Competitive endogenous RNA |
| ChIP | Chromatin immunoprecipitation |
| CTLs | Cytotoxic T Lymphocytes |
| CTLA-4 | Cytotoxic T lymphocyte-associated Antigen 4 |
| DNMT1 | DNA methyltransferase |
| DUSP7 | Dual specificity phosphatase 7 |
| EMT | Epithelial to mesenchymal transition |
| ER+ | Estrogen receptor - positive |
| ERG | ETS-related gene |
| GATA4 | GATA binding protein 4 |
| GATA3-AS1 | GATA3 antisense RNA1 |
| HCP5 | Human leukocyte antigen complex P5 |
| HITT | HIF-1α Inhibitor at Translation level |
| ICIs | Immune checkpoint Inhibitors |
| IDO | Indoleamine-2,3-dioxygenase |
| IFN-γ | Interferon- gamma |
| IKKβ | Inhibitor Of Nuclear Factor Kappa B Kinase Subunit Beta |
| IL | Interleukins |
| IL-α | Interleukin alpha |
| irlncRNAα | Immune-related long non-coding RNA |
| JAK | Janus kinase |
| Kb | Kilobases |
| KEGGK | yoto Encyclopedia of Genes and Genomes |
| LAG3 | Lymphocyte activating 3 |
| lncRNA | Long non-coding RNA |
| MALAT1 | Metastasis-associated lung adenocarcinoma transcript1 |
| MAPK | Mitogen-activated protein kinase |
| MDSCs | Myeloid-derived suppressor cells |
| MIAT | Myocardial Infarction Associated Transcript |
| miRNA | Micro RNA |
| MYC | Myelocytomatosis oncogene |
| NK cells | Natural killer cells |
| NKILA | NF-кB interacting long non-coding RNA |
| NF-кB | Nuclear Factor- kappa B |
| OS | Overall survival |
| PEA15 | Proliferation and apoptosis adaptor protein 15 |
| PD1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| POLR2 | ARNA Polymerase II Subunit A |
| TAM | Tumor-associated macrophages |
| TP53 | Tumor protein 53 |
| RGS2 | Regulator of G-protein signalling 2 |
| ROC | Receiver Operating Characteristic curve |
| SASP | Senescence- associated secretory phenotype |
| Serpin | Serine protease inhibitors |
| snoRNA | Small nucleolar RNA |
| SNHG1 | Small nucleolar RNA host gene 1 |
| SNHG16 | Small nucleolar RNA host gene 16 |
| SOX2 | Sex determining region Y-box 2 |
| STAT1 | Signal transducer and activator of transcription 1 |
| STAT3 | Signal transducer and activator of transcription-3 |
| TAM | Tumor-associated macrophages |
| TAN | Tumor-associated neutrophils |
| TARDBP | Transactive response DNA binding protein |
| TCR | T cell receptor |
| TDEs | Tumor-derived exosomes |
| TGF-β | Transforming growth factor-beta |
| Th | T helper |
| TIICs | Tumor-infiltrating immune cells |
| TIMER | Tumor Immune Estimation Resource |
| TIM-3 | T cell immunoglobulin and mucin domain 3 |
| TINCR | Tissue differentiation-inducing non-protein-coding RNA |
| TIS | Therapy-induced senescence |
| TMB | Tumor mutational burden |
| TNBC | Triple-negative breast cancer |
| TNF-α | Tumor Necrosis Factor alpha |
| TNM | Tumor, nodes, and metastasis |
| Treg | T regulatory |
| TME | Tumor microenvironment |
| UTR | Untranslated region |
| ZFAS1 | Zinc finger NFX1-type containing 1 antisense RNA 1 |
References
- Harris MA, Savas P, Virassamy B, O'Malley MMR, Kay J, Mueller SN, et al. Towards targeting the breast cancer immune microenvironment. Nat Rev Cancer 2024, 24, 554–77. [Google Scholar] [CrossRef] [PubMed]
- Anderson NM, Simon MC. The tumor microenvironment. Curr Biol 2020, 30, R921–R5. [Google Scholar] [CrossRef] [PubMed]
- Zhao Y, Shen M, Wu L, Yang H, Yao Y, Yang Q, et al. Stromal cells in the tumor microenvironment: accomplices of tumor progression? Cell Death Dis 2023, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Pansy K, Uhl B, Krstic J, Szmyra M, Fechter K, Santiso A, et al. Immune Regulatory Processes of the Tumor Microenvironment under Malignant Conditions. Int J Mol Sci 2021, 22. [Google Scholar]
- Chen C, Liu X, Chang CY, Wang HY, Wang RF. The Interplay between T Cells and Cancer: The Basis of Immunotherapy. Genes (Basel) 2023, 14. [Google Scholar]
- Ahmed H, Mahmud AR, Siddiquee MF, Shahriar A, Biswas P, Shimul MEK, et al. Role of T cells in cancer immunotherapy: Opportunities and challenges. Cancer Pathog Ther 2023, 1, 116–26. [Google Scholar] [CrossRef]
- Xiang S, Li S, Xu J. Unravelling T cell exhaustion through co-inhibitory receptors and its transformative role in cancer immunotherapy. Clin Transl Med 2025, 15, e70345. [Google Scholar] [CrossRef]
- Sharma U, Barwal TS, Malhotra A, Pant N, Vivek, Dey D, et al. Long non-coding RNA TINCR as potential biomarker and therapeutic target for cancer. Life Sci 2020, 257, 118035. [Google Scholar] [CrossRef]
- Sharma U, Barwal TS, Acharya V, Tamang S, Vasquez KM, Jain A. Cancer Susceptibility Candidate 9 (CASC9): A Novel Targetable Long Noncoding RNA in Cancer Treatment. Transl Oncol 2020, 13, 100774. [Google Scholar] [CrossRef]
- Sharma U, Barwal TS, Acharya V, Singh K, Rana MK, Singh SK, et al. Long Non-Coding RNAs as Strategic Molecules to Augment the Radiation Therapy in Esophageal Squamous Cell Carcinoma. Int J Mol Sci 2020, 21. [Google Scholar]
- Su J, Deng L, Wang YD. Roles and Mechanisms of Long Non-Coding RNAs in Breast Cancer. Int J Mol Sci 2022, 24. [Google Scholar]
- Sharma U, Barwal TS, Khandelwal A, Malhotra A, Rana MK, Singh Rana AP, et al. LncRNA ZFAS1 inhibits triple-negative breast cancer by targeting STAT3. Biochimie 2021, 182, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Zhao S, Zhang X, Chen S, Zhang S. Long noncoding RNAs: fine-tuners hidden in the cancer signaling network. Cell Death Discov 2021, 7, 283. [Google Scholar] [CrossRef]
- Zhang X, Wang W, Zhu W, Dong J, Cheng Y, Yin Z, et al. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int J Mol Sci 2019, 20. [Google Scholar]
- Guo Y, Xie Y, Luo Y. The Role of Long Non-Coding RNAs in the Tumor Immune Microenvironment. Front Immunol 2022, 13, 851004. [Google Scholar] [CrossRef]
- Plasek LM, Valadkhan S. lncRNAs in T lymphocytes: RNA regulation at the heart of the immune response. Am J Physiol Cell Physiol 2021, 320, C415–C27. [Google Scholar]
- Erber J, Herndler-Brandstetter D. Regulation of T cell differentiation and function by long noncoding RNAs in homeostasis and cancer. Front Immunol 2023, 14, 1181499. [Google Scholar] [CrossRef]
- Zhan DT, Xian HC. Exploring the regulatory role of lncRNA in cancer immunity. Front Oncol 2023, 13, 1191913. [Google Scholar] [CrossRef]
- Fonseca-Montano MA, Vazquez-Santillan KI, Hidalgo-Miranda A. The current advances of lncRNAs in breast cancer immunobiology research. Front Immunol 2023, 14, 1194300. [Google Scholar] [CrossRef]
- Huang D, Chen J, Yang L, Ouyang Q, Li J, Lao L, et al. NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death. Nat Immunol 2018, 19, 1112–25. [Google Scholar] [CrossRef]
- Pei X, Wang X, Li H. LncRNA SNHG1 regulates the differentiation of Treg cells and affects the immune escape of breast cancer via regulating miR-448/IDO. Int J Biol Macromol. 2018, 118, 24–30. [Google Scholar] [CrossRef]
- Wang Q, Li G, Ma X, Liu L, Liu J, Yin Y, et al. LncRNA TINCR impairs the efficacy of immunotherapy against breast cancer by recruiting DNMT1 and downregulating MiR-199a-5p via the STAT1-TINCR-USP20-PD-L1 axis. Cell Death Dis 2023, 14, 76. [Google Scholar] [CrossRef]
- Zhang M, Wang N, Song P, Fu Y, Ren Y, Li Z, et al. LncRNA GATA3-AS1 facilitates tumour progression and immune escape in triple-negative breast cancer through destabilization of GATA3 but stabilization of PD-L1. Cell Prolif 2020, 53, e12855. [Google Scholar] [CrossRef] [PubMed]
- Zou R, Gu R, Yu X, Hu Y, Yu J, Xue X, et al. Characteristics of Infiltrating Immune Cells and a Predictive Immune Model for Cervical Cancer. J Cancer 2021, 12, 3501–14. [Google Scholar]
- Rathore AS, Kumar S, Konwar R, Makker A, Negi MP, Goel MM. CD3+, CD4+ & CD8+ tumour infiltrating lymphocytes (TILs) are predictors of favourable survival outcome in infiltrating ductal carcinoma of breast. Indian J Med Res 2014, 140, 361–9. [Google Scholar]
- Yang W, Liu S, Mao M, Gong Y, Li X, Lei T, et al. T-cell infiltration and its regulatory mechanisms in cancers: insights at single-cell resolution. J Exp Clin Cancer Res 2024, 43, 38. [Google Scholar] [CrossRef]
- Zhang Y, Li Z, Chen M, Chen H, Zhong Q, Liang L, et al. lncRNA TCL6 correlates with immune cell infiltration and indicates worse survival in breast cancer. Breast Cancer 2020, 27, 573–85. [Google Scholar] [CrossRef]
- Yang W, Qiu Z, Zhang J, Zhi X, Yang L, Qiu M, et al. Correlation Between Immune Cell Infiltration and PD-L1 Expression and Immune-Related lncRNA Determination in Triple-Negative Breast Cancer. Front Genet 2022, 13, 878658. [Google Scholar] [CrossRef]
- Shao G, Fan X, Zhang P, Liu X, Huang L, Ji S. Methylation-dependent MCM6 repression induced by LINC00472 inhibits triple-negative breast cancer metastasis by disturbing the MEK/ERK signaling pathway. Aging (Albany NY) 2021, 13, 4962–75. [Google Scholar]
- Shen Y, Katsaros D, Loo LW, Hernandez BY, Chong C, Canuto EM, et al. Prognostic and predictive values of long non-coding RNA LINC00472 in breast cancer. Oncotarget 2015, 6, 8579–92. [Google Scholar] [CrossRef] [PubMed]
- Shen Y, Wang Z, Loo LW, Ni Y, Jia W, Fei P, et al. LINC00472 expression is regulated by promoter methylation and associated with disease-free survival in patients with grade 2 breast cancer. Breast Cancer Res Treat 2015, 154, 473–82. [Google Scholar] [CrossRef]
- Wang Z, Katsaros D, Biglia N, Shen Y, Loo L, Yu X, et al. ERalpha upregulates the expression of long non-coding RNA LINC00472 which suppresses the phosphorylation of NF-kappaB in breast cancer. Breast Cancer Res Treat 2019, 175, 353–68. [Google Scholar] [CrossRef]
- Chen Q, Shen H, Zhu X, Liu Y, Yang H, Chen H, et al. A nuclear lncRNA Linc00839 as a Myc target to promote breast cancer chemoresistance via PI3K/AKT signaling pathway. Cancer Sci 2020, 111, 3279–91. [Google Scholar] [CrossRef]
- Fu S, Wang Y, Li H, Chen L, Liu Q. Regulatory Networks of LncRNA MALAT-1 in Cancer. Cancer Manag Res 2020, 12, 10181–98. [Google Scholar] [CrossRef]
- Yan C, Jin Y. Silencing of long noncoding RNA MIAT inhibits the viability and proliferation of breast cancer cells by promoting miR-378a-5p expression. Open Med (Wars) 2023, 18, 20230676. [Google Scholar] [CrossRef] [PubMed]
- Zeinelabdeen Y, Abaza T, Yasser MB, Elemam NM, Youness RA. MIAT LncRNA: A multifunctional key player in non-oncological pathological conditions. Noncoding RNA Res 2024, 9, 447–62. [Google Scholar] [CrossRef] [PubMed]
- Luan T, Zhang X, Wang S, Song Y, Zhou S, Lin J, et al. Long non-coding RNA MIAT promotes breast cancer progression and functions as ceRNA to regulate DUSP7 expression by sponging miR-155-5p. Oncotarget 2017, 8, 76153–64. [Google Scholar] [CrossRef] [PubMed]
- Ye T, Feng J, Cui M, Yang J, Wan X, Xie D, et al. LncRNA MIAT Services as a Noninvasive Biomarker for Diagnosis and Correlated with Immune Infiltrates in Breast Cancer. Int J Womens Health 2021, 13, 991–1004. [Google Scholar] [CrossRef]
- Ibrahim EM, Al-Foheidi ME, Al-Mansour MM, Kazkaz GA. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: a meta-analysis. Breast Cancer Res Treat 2014, 148, 467–76. [Google Scholar] [CrossRef]
- Sconocchia G, Eppenberger S, Spagnoli GC, Tornillo L, Droeser R, Caratelli S, et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology 2014, 3, e952197. [Google Scholar] [CrossRef]
- Sugita BM, Rodriguez Y, Fonseca AS, Nunes Souza E, Kallakury B, Cavalli IJ, et al. MiR-150-5p Overexpression in Triple-Negative Breast Cancer Contributes to the In Vitro Aggressiveness of This Breast Cancer Subtype. Cancers (Basel) 2022, 14. [Google Scholar]
- Li L, Wang N, Xiong Y, Guo G, Zhu M, Gu Y. Transcription Factor FOSL1 Enhances Drug Resistance of Breast Cancer through DUSP7-Mediated Dephosphorylation of PEA15. Mol Cancer Res 2022, 20, 515–26. [Google Scholar] [CrossRef]
- Cisneros-Villanueva M, Fonseca-Montano MA, Rios-Romero M, Lopez-Camarillo C, Jimenez-Morales S, Langley E, et al. LncRNA SOX9-AS1 triggers a transcriptional program involved in lipid metabolic reprogramming, cell migration and invasion in triple-negative breast cancer. Sci Rep 2024, 14, 1483. [Google Scholar] [CrossRef]
- Ye X, Cen Y, Li Q, Zhang YP, Li Q, Li J. Immunosuppressive SOX9-AS1 Resists Triple-Negative Breast Cancer Senescence Via Regulating Wnt Signalling Pathway. J Cell Mol Med 2024, 28, e70208. [Google Scholar] [CrossRef]
- Wang X, Li L, Zhao K, Lin Q, Li H, Xue X, et al. A novel LncRNA HITT forms a regulatory loop with HIF-1alpha to modulate angiogenesis and tumor growth. Cell Death Differ 2020, 27, 1431–46. [Google Scholar] [CrossRef] [PubMed]
- Lin Q, Liu T, Wang X, Hou G, Xiang Z, Zhang W, et al. Long noncoding RNA HITT coordinates with RGS2 to inhibit PD-L1 translation in T cell immunity. J Clin Invest 2023, 133. [Google Scholar]
- Nakajima R, Zhao L, Zhou Y, Shirasawa M, Uchida A, Murakawa H, et al. Deregulated E2F Activity as a Cancer-Cell Specific Therapeutic Tool. Genes (Basel) 2023, 14. [Google Scholar]
- Zhang X, Li Y, Huan C, Hou Y, Liu R, Shi H, et al. LncRNA NKILA inhibits HBV replication by repressing NF-kappaB signalling activation. Virol Sin 2024, 39, 44–55. [Google Scholar] [CrossRef]
- Zhang T, Ma C, Zhang Z, Zhang H, Hu H. NF-kappaB signaling in inflammation and cancer. MedComm (2020) 2021, 2, 618–53. [Google Scholar]
- Bahar ME, Kim HJ, Kim DR. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct Target Ther 2023, 8, 455. [Google Scholar] [CrossRef]
- Meseure D, Vacher S, Lallemand F, Alsibai KD, Hatem R, Chemlali W, et al. Prognostic value of a newly identified MALAT1 alternatively spliced transcript in breast cancer. Br J Cancer 2016, 114, 1395–404. [Google Scholar] [CrossRef]
- Arun G, Aggarwal D, Spector DL. MALAT1 Long Non-Coding RNA: Functional Implications. Noncoding RNA 2020, 6. [Google Scholar]
- Tsyganov MM, Ibragimova MK. MALAT1 Long Non-coding RNA and Its Role in Breast Carcinogenesis. Acta Naturae 2023, 15, 32–41. [Google Scholar] [CrossRef]
- Li ZX, Zhu QN, Zhang HB, Hu Y, Wang G, Zhu YS. MALAT1: a potential biomarker in cancer. Cancer Manag Res 2018, 10, 6757–68. [Google Scholar] [CrossRef]
- Mazarei M, Shahabi Rabori V, Ghasemi N, Salehi M, Rayatpisheh N, Jahangiri N, et al. LncRNA MALAT1 signaling pathway and clinical applications in overcome on cancers metastasis. Clin Exp Med 2023, 23, 4457–72. [Google Scholar] [CrossRef]
- Adewunmi O, Shen Y, Zhang XH, Rosen JM. Targeted Inhibition of lncRNA Malat1 Alters the Tumor Immune Microenvironment in Preclinical Syngeneic Mouse Models of Triple-Negative Breast Cancer. Cancer Immunol Res 2023, 11, 1462–79. [Google Scholar] [CrossRef]
- Kumar D, Gurrapu S, Wang Y, Bae SY, Pandey PR, Chen H, et al. LncRNA Malat1 suppresses pyroptosis and T cell-mediated killing of incipient metastatic cells. Nat Cancer 2024, 5, 262–82. [Google Scholar] [CrossRef]
- Wang X, Hu K, Chao Y, Wang L. LncRNA SNHG16 promotes proliferation, migration and invasion of osteosarcoma cells by targeting miR-1301/BCL9 axis. Biomed Pharmacother 2019, 114, 108798. [Google Scholar]
- Gong CY, Tang R, Nan W, Zhou KS, Zhang HH. Role of SNHG16 in human cancer. Clin Chim Acta 2020, 503, 175–80. [Google Scholar] [CrossRef]
- Yu Y, Chen F, Yang Y, Jin Y, Shi J, Han S, et al. lncRNA SNHG16 is associated with proliferation and poor prognosis of pediatric neuroblastoma. Int J Oncol 2019, 55, 93–102. [Google Scholar]
- Ghafouri-Fard S, Khoshbakht T, Taheri M, Shojaei S. A Review on the Role of Small Nucleolar RNA Host Gene 6 Long Non-coding RNAs in the Carcinogenic Processes. Front Cell Dev Biol 2021, 9, 741684. [Google Scholar]
- Ni C, Fang QQ, Chen WZ, Jiang JX, Jiang Z, Ye J, et al. Breast cancer-derived exosomes transmit lncRNA SNHG16 to induce CD73+gammadelta1 Treg cells. Signal Transduct Target Ther 2020, 5, 41. [Google Scholar] [CrossRef]
- Cheng JN, Yuan YX, Zhu B, Jia Q. Myeloid-Derived Suppressor Cells: A Multifaceted Accomplice in Tumor Progression. Front Cell Dev Biol 2021, 9, 740827. [Google Scholar]
- Markowitz J, Wesolowski R, Papenfuss T, Brooks TR, Carson WE, 3rd. Myeloid-derived suppressor cells in breast cancer. Breast Cancer Res Treat 2013, 140, 13–21. [Google Scholar] [CrossRef]
- He S, Zheng L, Qi C. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and their targeting in cancer therapy. Mol Cancer 2025, 24, 5. [Google Scholar] [CrossRef]
- Munir MT, Kay MK, Kang MH, Rahman MM, Al-Harrasi A, Choudhury M, et al. Tumor-Associated Macrophages as Multifaceted Regulators of Breast Tumor Growth. Int J Mol Sci 2021, 22. [Google Scholar]
- Chang L, Li J, Ding J, Lian Y, Huangfu C, Wang K. Roles of long noncoding RNAs on tumor immune escape by regulating immune cells differentiation and function. Am J Cancer Res 2021, 11, 2369–85. [Google Scholar]
- Tang S, Ning Q, Yang L, Mo Z, Tang S. Mechanisms of immune escape in the cancer immune cycle. Int Immunopharmacol 2020, 86, 106700. [Google Scholar] [CrossRef]
- Zhang L, Sheng M, Cao H, Zhang L, Shao W. Decoding the role of long non-coding RNAs in periodontitis: A comprehensive review. Biomed Pharmacother 2023, 166, 115357. [Google Scholar]
- Tian T, Qiu R, Qiu X. SNHG1 promotes cell proliferation by acting as a sponge of miR-145 in colorectal cancer. Oncotarget 2018, 9, 2128–39. [Google Scholar] [CrossRef]
- Huang L, Jiang X, Wang Z, Zhong X, Tai S, Cui Y. Small nucleolar RNA host gene 1: A new biomarker and therapeutic target for cancers. Pathol Res Pract 2018, 214, 1247–52. [Google Scholar] [CrossRef]
- Saadh MJ, Hamid JA, Malathi H, Kazmi SW, Omar TM, Sharma A, et al. SNHG family lncRNAs: Key players in the breast cancer progression and immune cell's modulation. Exp Cell Res 2025, 447, 114531. [Google Scholar] [CrossRef]
- Zeng H, Zhou S, Cai W, Kang M, Zhang P. LncRNA SNHG1: role in tumorigenesis of multiple human cancers. Cancer Cell Int 2023, 23, 198. [Google Scholar] [CrossRef]
- McAnena P, Tanriverdi K, Curran C, Gilligan K, Freedman JE, Brown JAL, et al. Circulating microRNAs miR-331 and miR-195 differentiate local luminal a from metastatic breast cancer. BMC Cancer 2019, 19, 436. [Google Scholar]
- Ma P, Ni K, Ke J, Zhang W, Feng Y, Mao Q. miR-448 inhibits the epithelial-mesenchymal transition in breast cancer cells by directly targeting the E-cadherin repressor ZEB1/2. Exp Biol Med (Maywood) 2018, 243, 473–80. [CrossRef]
- Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nat Rev Drug Discov 2010, 9, 775–89. [Google Scholar] [CrossRef]
- Figueiredo JC, Hsu L, Hutter CM, Lin Y, Campbell PT, Baron JA, et al. Genome-wide diet-gene interaction analyses for risk of colorectal cancer. PLoS Genet 2014, 10, e1004228. [Google Scholar]
- Keshavarz F, Mokhtari MJ, Poursadeghfard M. Increased level of GATA3-AS1 long non-coding RNA is correlated with the upregulation of GATA3 and IL-4 genes in multiple sclerosis patients. Mol Biol Rep 2024, 51, 874. [Google Scholar]
- Gibbons HR, Shaginurova G, Kim LC, Chapman N, Spurlock CF, 3rd, Aune TM. Divergent lncRNA GATA3-AS1 Regulates GATA3 Transcription in T-Helper 2 Cells. Front Immunol 2018, 9, 2512. [Google Scholar] [CrossRef]
- Ghafouri-Fard S, Askari A, Hussen BM, Rasul MF, Taheri M, Ayatollahi SA. A review on the role of LINC00472 in malignant and non-malignant disorders. Pathol Res Pract 2023, 247, 154549. [Google Scholar] [CrossRef]
- Chen C, Zheng Q, Kang W, Yu C. Long non-coding RNA LINC00472 suppresses hepatocellular carcinoma cell proliferation, migration and invasion through miR-93-5p/PDCD4 pathway. Clin Res Hepatol Gastroenterol 2019, 43, 436–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang W, Chen Q, Lei C. lncRNA MIAT promotes cell invasion and migration in esophageal cancer. Exp Ther Med 2020, 19, 3267–74. [Google Scholar]
- Tsuiji H, Yoshimoto R, Hasegawa Y, Furuno M, Yoshida M, Nakagawa S. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 2011, 16, 479–90. [Google Scholar] [CrossRef]
- Bian D, Gao C, Bao K, Song G. The long non-coding RNA NKILA inhibits the invasion-metastasis cascade of malignant melanoma via the regulation of NF-kB. Am J Cancer Res 2017, 7, 28–40. [Google Scholar]
- Fu Z, Luo W, Wang J, Peng T, Sun G, Shi J, et al. Malat1 activates autophagy and promotes cell proliferation by sponging miR-101 and upregulating STMN1, RAB5A and ATG4D expression in glioma. Biochem Biophys Res Commun 2017, 492, 480–6. [Google Scholar] [CrossRef]
- Xiao Y, Xiao T, Ou W, Wu Z, Wu J, Tang J, et al. LncRNA SNHG16 as a potential biomarker and therapeutic target in human cancers. Biomark Res 2020, 8, 41. [Google Scholar]
- Nie Z, Zhang K, Li Z, Bing X, Jin S, Li M. Human papillomavirus 16 E6 promotes angiogenesis of lung cancer via SNHG1. Cell Biochem Biophys 2023, 81, 325–36. [CrossRef]
- Gao FY, Li XT, Xu K, Wang RT, Guan XX. c-MYC mediates the crosstalk between breast cancer cells and tumor microenvironment. Cell Commun Signal 2023, 21, 28. [Google Scholar] [CrossRef]
- Marvalim C, Datta A, Lee SC. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics 2023, 13, 1421–42. [Google Scholar] [CrossRef] [PubMed]
- Wu Q, Liu X, Yan H, He YH, Ye S, Cheng XW, et al. B-cell lymphoma 6 protein stimulates oncogenicity of human breast cancer cells. BMC Cancer 2014, 14, 418. [Google Scholar] [CrossRef]
- Shenoy, S. CDH1 (E-Cadherin) Mutation and Gastric Cancer: Genetics, Molecular Mechanisms and Guidelines for Management. Cancer Manag Res 2019, 11, 10477–86. [Google Scholar] [CrossRef] [PubMed]
- Yang Y, Song S, Li S, Kang J, Li Y, Zhao N, et al. GATA4 regulates the transcription of MMP9 to suppress the invasion and migration of breast cancer cells via HDAC1-mediated p65 deacetylation. Cell Death Dis 2024, 15, 289. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
LncRNAs affecting immune cell infiltration in breast cancer tumor microenvironment. Downregulation or upregulation of specific lncRNAs alters immune cell recruitment, shaping anti-tumor cytotoxic responses or promoting tumor progression.
Figure 1.
LncRNAs affecting immune cell infiltration in breast cancer tumor microenvironment. Downregulation or upregulation of specific lncRNAs alters immune cell recruitment, shaping anti-tumor cytotoxic responses or promoting tumor progression.
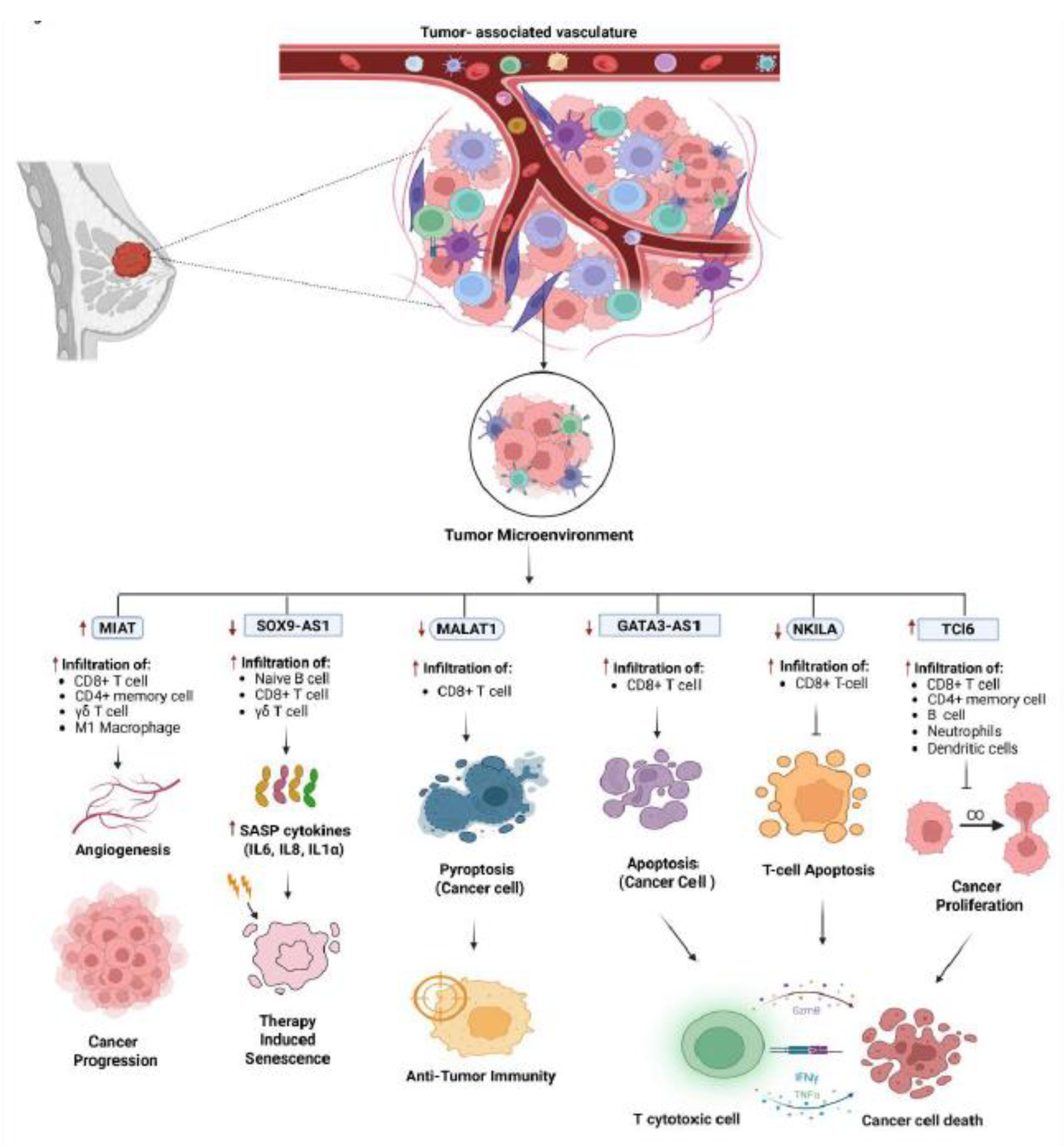
Figure 2.
Various LncRNAs facilitate immune escape by impairing T-cell cytotoxicity via upregulating PD-L1 expression on tumor cells. [Note: ↑- Upregulation, ↓- Downregulation] (Created with Biorender.com).
Figure 2.
Various LncRNAs facilitate immune escape by impairing T-cell cytotoxicity via upregulating PD-L1 expression on tumor cells. [Note: ↑- Upregulation, ↓- Downregulation] (Created with Biorender.com).
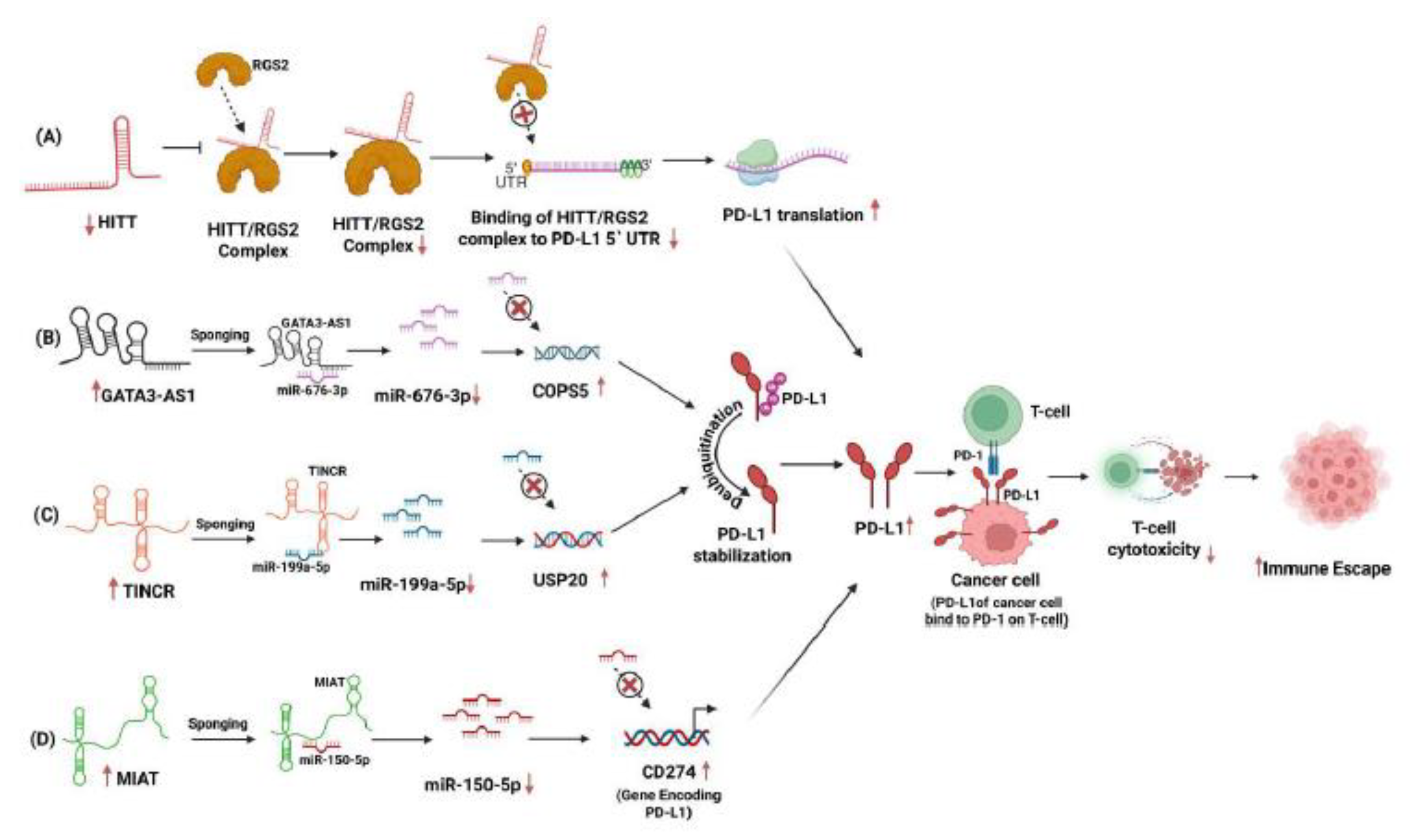
Figure 3.
Different pathways engaged by LncRNAs in modulating immune escape in breast cancer. [Note: ↑- Upregulation, ↓- Downregulation] (Created with Biorender.com).
Figure 3.
Different pathways engaged by LncRNAs in modulating immune escape in breast cancer. [Note: ↑- Upregulation, ↓- Downregulation] (Created with Biorender.com).
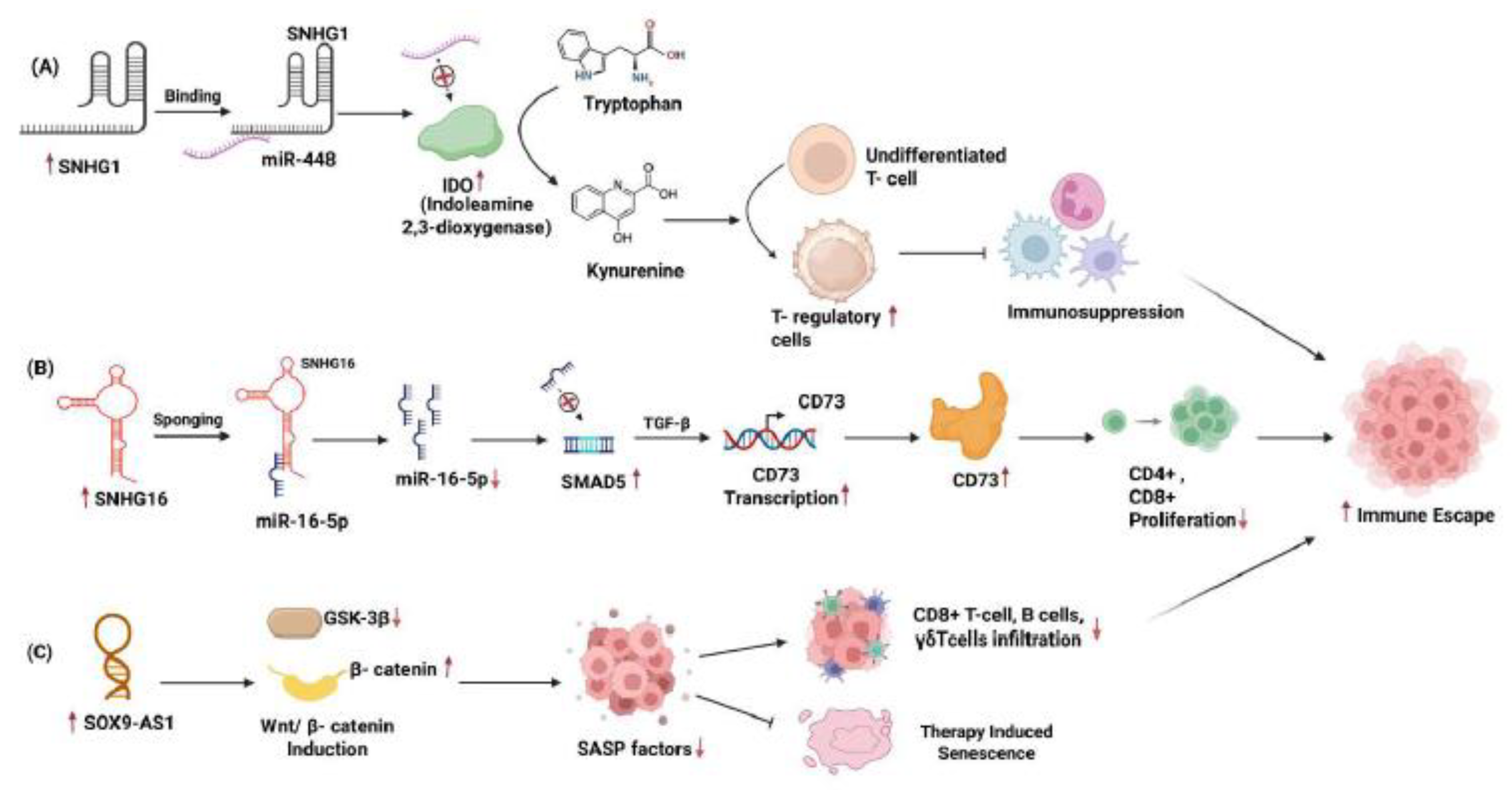
Figure 4.
Gene enrichment analysis of the top 10 significantly enriched KEGG pathways related to lncRNAs and their coexpressed genes. (A) KEGG pathways for LINC00472 and its coexpressed genes. (B) MIAT (C) SOX9-AS1 (D) NKILA (E) MALAT1 (F) SNHG16 (G) SNHG1 (H) GATA3-AS1. The horizontal line indicates gene count, and colors in the index represent -log 10 values of FDR. Only 4 KEGG pathways are associated with SOX9-AS1 lncRNA, and no data were found for HITT and TINCR.
Figure 4.
Gene enrichment analysis of the top 10 significantly enriched KEGG pathways related to lncRNAs and their coexpressed genes. (A) KEGG pathways for LINC00472 and its coexpressed genes. (B) MIAT (C) SOX9-AS1 (D) NKILA (E) MALAT1 (F) SNHG16 (G) SNHG1 (H) GATA3-AS1. The horizontal line indicates gene count, and colors in the index represent -log 10 values of FDR. Only 4 KEGG pathways are associated with SOX9-AS1 lncRNA, and no data were found for HITT and TINCR.
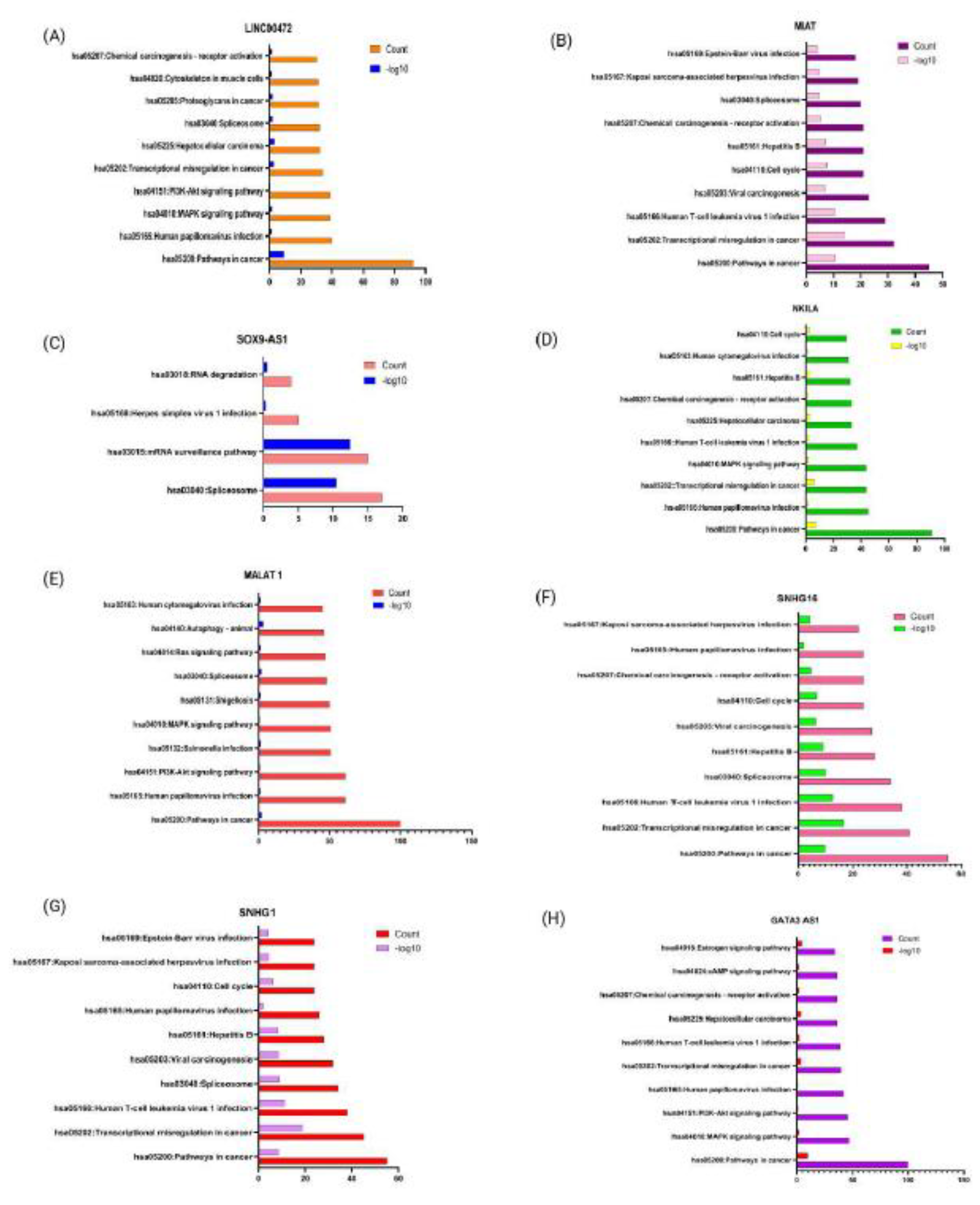
Figure 5.
Plots of Kaplan-Meier estimate the overall survival of a group of patients retrieved from the TCGA database, accessed on 29 July 2025. Survival probability of invasive breast cancer patients is represented against the time of survival associated with various lncRNAs. (A) LINC00472 (B) MIAT (C) SOX9-AS1 (D) NKILA (E) MALAT1 (F) SNHG16 (G) SNHG1 (H) GATA3-AS1.
Figure 5.
Plots of Kaplan-Meier estimate the overall survival of a group of patients retrieved from the TCGA database, accessed on 29 July 2025. Survival probability of invasive breast cancer patients is represented against the time of survival associated with various lncRNAs. (A) LINC00472 (B) MIAT (C) SOX9-AS1 (D) NKILA (E) MALAT1 (F) SNHG16 (G) SNHG1 (H) GATA3-AS1.
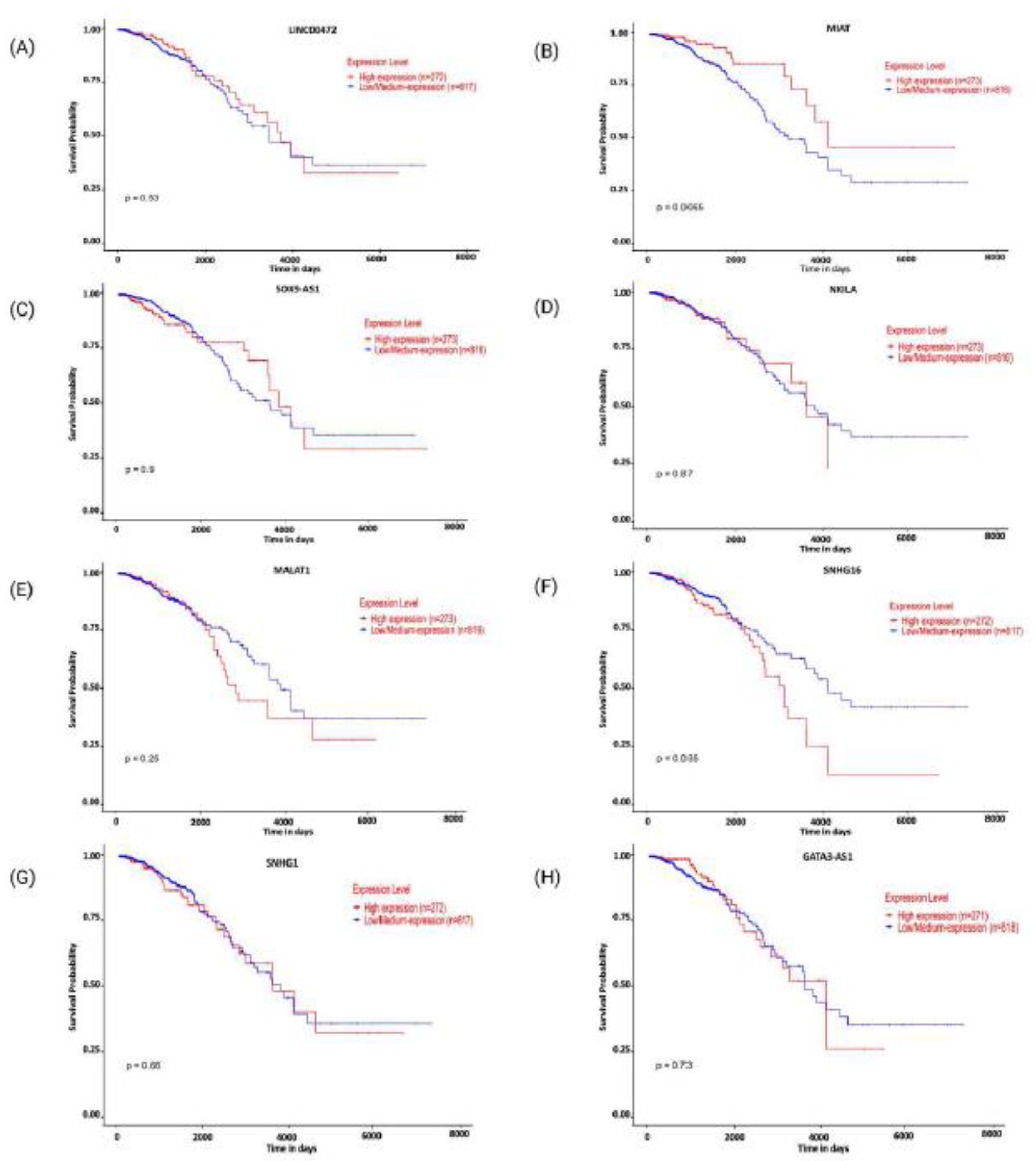
Figure 6.
Differential expression of various lncRNAs in invasive breast carcinoma in the ENCORI database on 29 July 2025. (A) LINC00472 (B) MIAT (C) SOX9-AS1 (D) NKILA (E) MALAT1 (F) SNHG1 (G) SNHG16 (H) GATA3-AS1. No data found for HITT and TINCR. Expression of MIAT, NKILA, SNHG1, SNHG16, and GATA3-AS1 is upregulated in invasive breast carcinoma. Expression of LINC00472, SOX9-AS1, and MALAT1 is downregulated in invasive breast carcinoma, but the literature suggests their upregulation in TNBC.
Figure 6.
Differential expression of various lncRNAs in invasive breast carcinoma in the ENCORI database on 29 July 2025. (A) LINC00472 (B) MIAT (C) SOX9-AS1 (D) NKILA (E) MALAT1 (F) SNHG1 (G) SNHG16 (H) GATA3-AS1. No data found for HITT and TINCR. Expression of MIAT, NKILA, SNHG1, SNHG16, and GATA3-AS1 is upregulated in invasive breast carcinoma. Expression of LINC00472, SOX9-AS1, and MALAT1 is downregulated in invasive breast carcinoma, but the literature suggests their upregulation in TNBC.
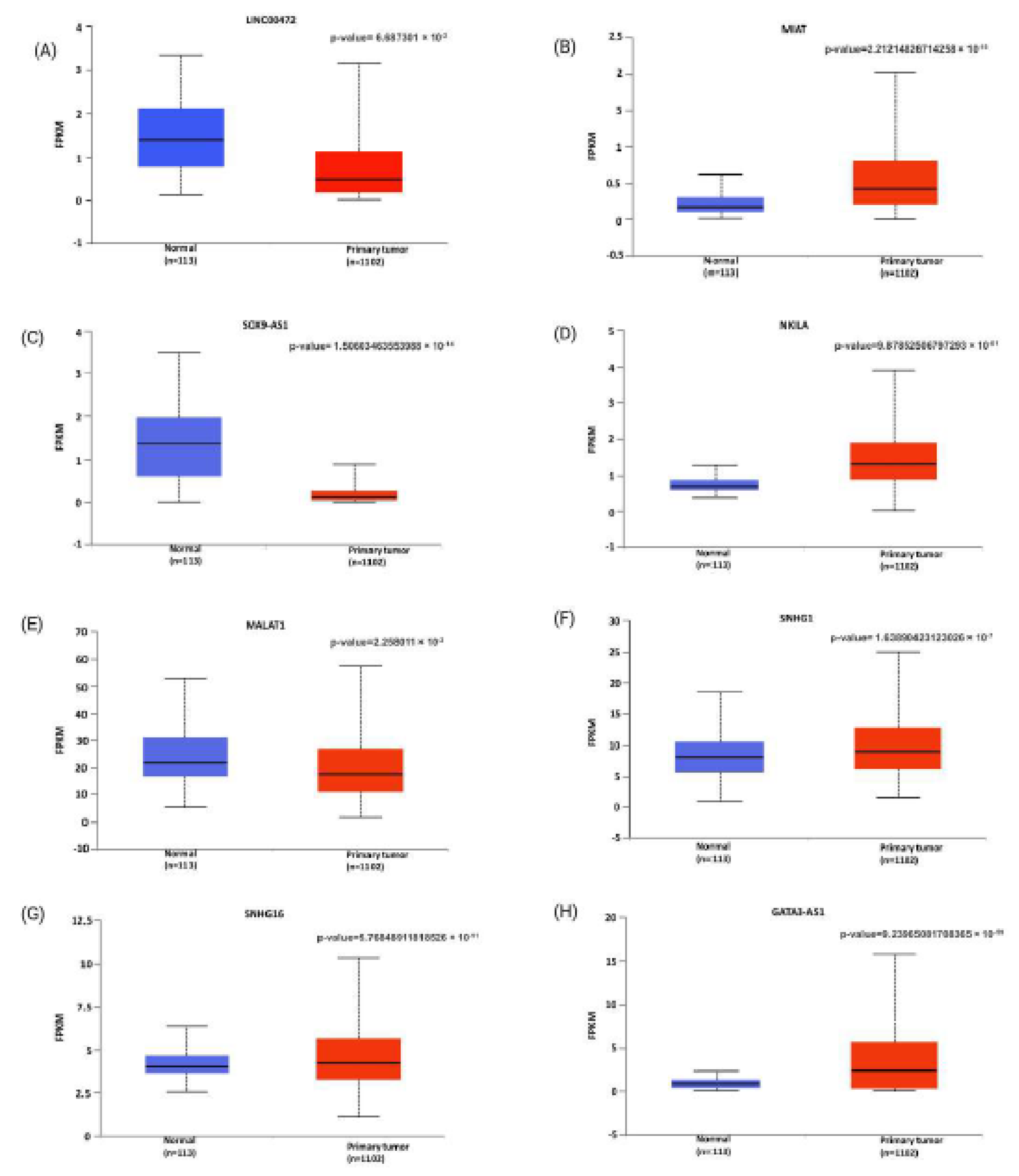
Figure 7.
Network representing various lncRNAs and their overlapping and some distinct target genes. Red boxes represent lncRNAs, target genes in green boxes, and lines denote interactions or regulatory connections (Created with Cytoscape).
Figure 7.
Network representing various lncRNAs and their overlapping and some distinct target genes. Red boxes represent lncRNAs, target genes in green boxes, and lines denote interactions or regulatory connections (Created with Cytoscape).
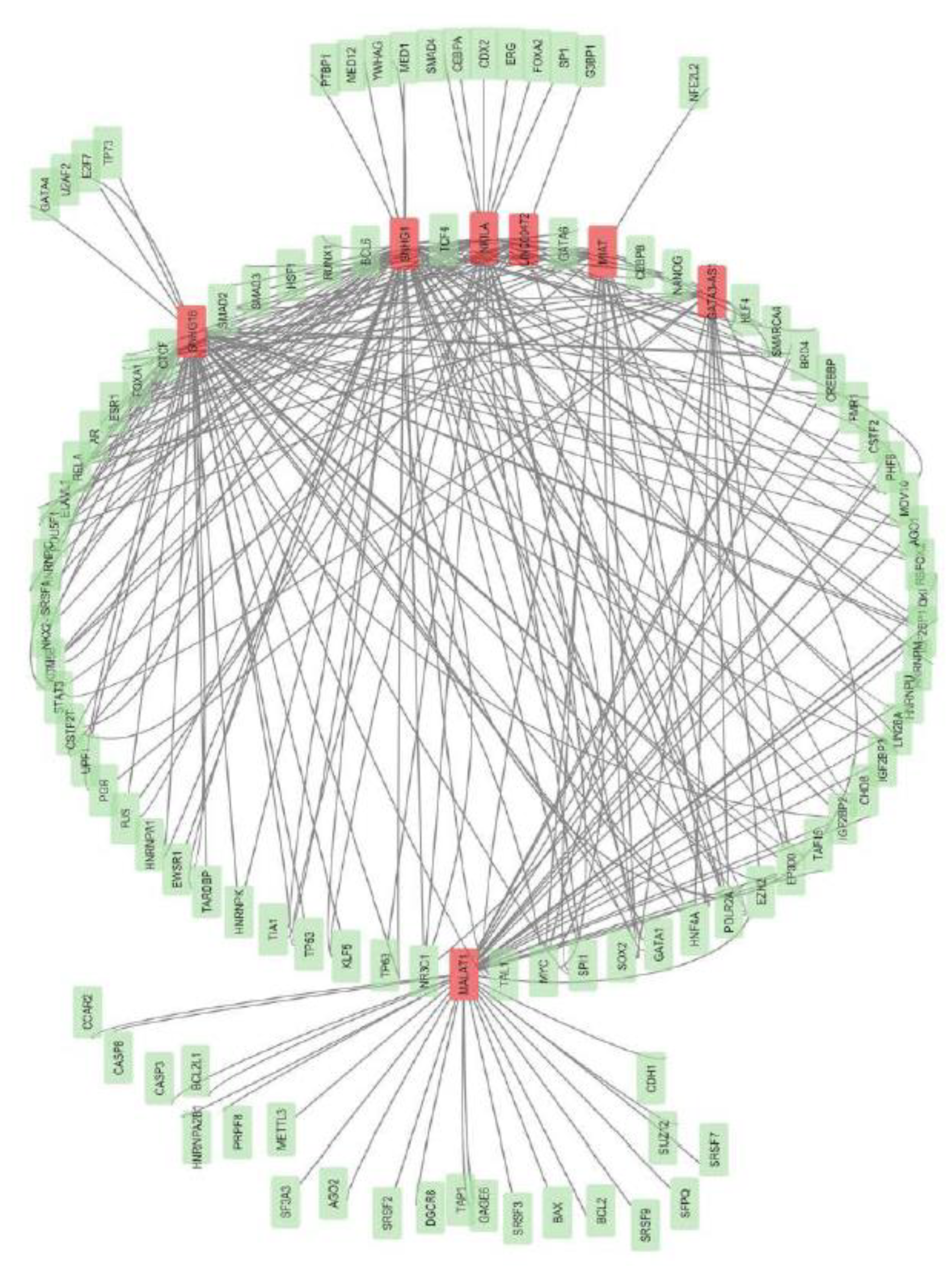
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



