Submitted:
07 October 2025
Posted:
08 October 2025
You are already at the latest version
Abstract
Anticancer strategies targeting the DNA damage response are largely centered on a number of false hypotheses. For example, engaging apoptosis in solid tumors is universally assumed to represent a tumor suppression response. But what is “apoptosis,” really? Time-lapse microscopy and other single-cell assays have revealed that engaging apoptosis in solid tumor cells is accompanied by anastasis, the homeostatic process of cell recovery from late stages of apoptosis, even after the formation of apoptotic bodies. Furthermore, apoptotic cells secrete a variety of prosurvival factors that contribute to overall tumor repopulation. Not surprisingly, numerous clinical studies reported since the 1990s have demonstrated that increased apoptosis in solid tumors is associated with cancer aggressiveness rather than representing a favorable clinical outcome. Another major false hypothesis pertains to the role of wild-type p53 in regulating apoptosis. Several recent articles addressing the challenges that have been encountered in implementing p53-based cancer therapies assume that p53 is pro-apoptotic. This assumption, which has become an almost indisputable fact, is shocking given that by mid-2000s it was already well established that p53 serves to inhibit apoptosis through upregulating ~40 anti-apoptotic proteins. The complexity of cancer cell response to therapeutic agents is discussed herein with a focus on the significance of p53-p21WAF1 signaling in suppressing the apoptosis-anastasis tumor repopulation pathway.
Keywords:
p53
; p21
; WIP1
; apoptosis
; anastasis
; senescence
; therapy resistance
; polyploid giant cancer cells
; PGCCs
1. Introduction
Sarabjot Pabla has recently published an online (LinkedIn) article entitled “Contrarian Thinking in Bioinformatics: Unlocking Breakthroughs by Challenging Assumptions” in which he states that some of the major advances in science have not come from following the obvious path, but from asking whether the current way of doing things is exactly what’s holding us back [1]. “Contrarian logic doesn’t mean rejecting consensus for its own sake. It means re-examining defaults, finding blind spots, and testing counterintuitive ideas that lead to better answers,” stated Pabla, a clinical and research bioinformatics expert [1].
The current review presents “contrarian thinking” based on solid preclinical and clinical data regarding regulated (or programmed) cell death and p53 function (Figure 1). I prefer to use “false hypothesis” rather than “contrarian thinking” for highly simplistic and outdated (1990s) assumptions that have derailed cancer research for decades and, unfortunately, continue to do so. Some of these false hypotheses have been discussed [2,3,4].
Over the past three decades, apoptosis and other modes of regulated cell death, together with the transcription regulators p53 and p21WAF1 (p21), have been among the most extensively studied and highly reviewed fields in the context of cancer progression and therapy. Despite this, the following three fundamental questions still remain: (i) What is apoptosis? (ii) How is apoptosis influenced by p53 signaling? And (iii) What are the reasons for repeated failures in implementing novel anticancer strategies? The intention of the current article is to shed some light on these questions.
Figure 1.
A highly simplified graphic summary and the main take home messages of the studies reviewed herein.
Figure 1.
A highly simplified graphic summary and the main take home messages of the studies reviewed herein.
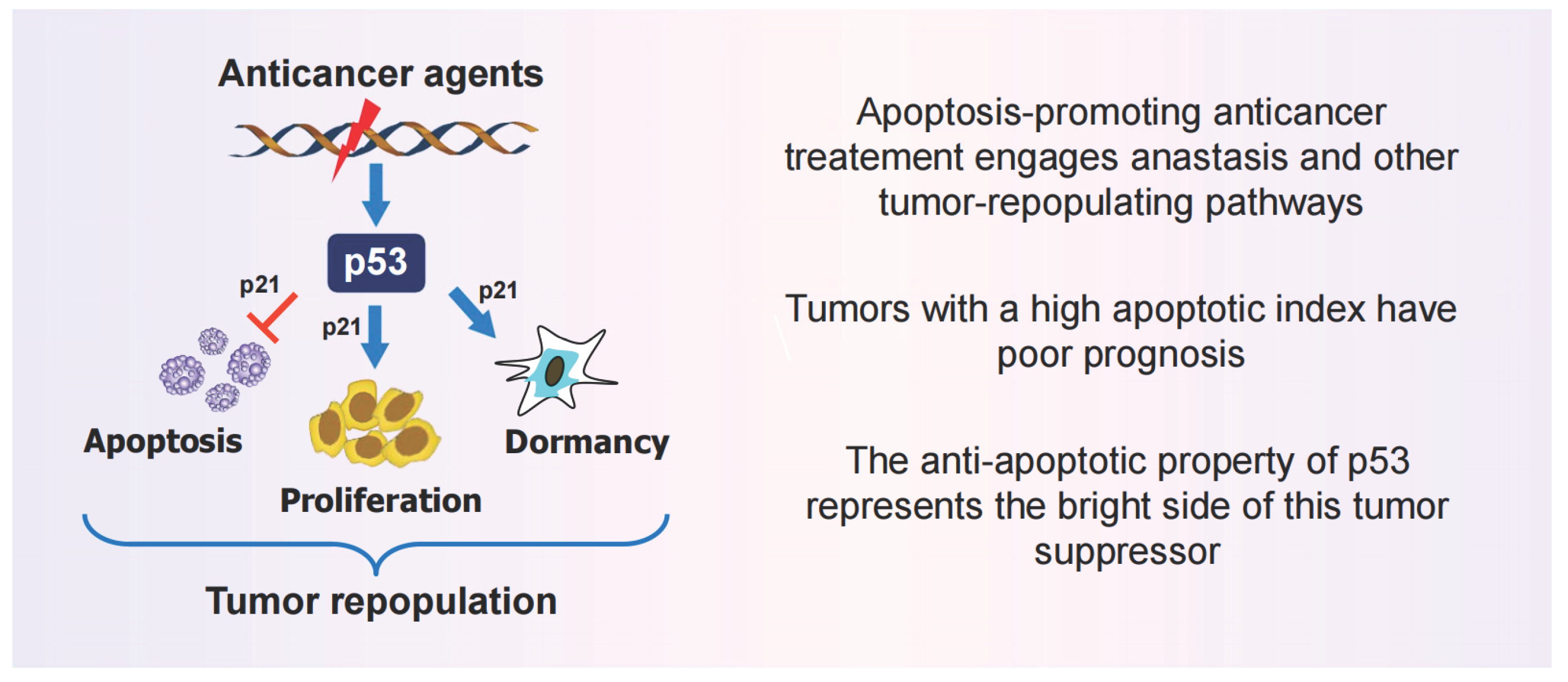
Please note that the discoveries highlighted herein are made with cell types (e.g., solid tumor cells) that predominantly undergo dormancy (active sleep) under stressful conditions. The situation might be quite different for other cell types such as lymphocytes and thymocytes that are programmed to be eliminated via apoptosis during negative selection or in response to stress.
2. Is Apoptosis a Tumor Suppression Mechanism?
2.1. Precision Oncology Targeting Apoptosis: Reality or False Promises?
“Precision oncology is inspirational. What doctor or patient would not want to harness genetics to tailor a therapy to an individual? But traveling back in a time machine is also inspirational. Who would not want to wind back the clock to remove their cancer before it spreads? In both cases, however, as of 2016, the proposal is neither feasible, cost-effective nor assured of future success. Yet in only one of these cases does the rhetoric so far outpace the reality that we risk fooling even ourselves” [5].
These remarks were made by Vinay Prasad in a Perspective article entitled “The precision-oncology illusion” that was published in Nature a decade ago [5]. While numerous authors have argued that precision oncology is not an illusion, a handful of other authors have highlighted compelling preclinical and clinical data that strongly support Prasad’s conclusion and have referred to personalized/precision oncology as “failed medicine” or (empty) promises that remain to be fulfilled [6,7,8,9,10,11,12,13,14,15,16,17,18].
There is no doubt that a small fraction of cancer patients do respond exceptionally well to radiotherapy, chemotherapy, and other mainstream treatments [19]. For the majority of cancer patients, however, particularly for patients with metastatic disease, traditional or targeted (“precision”) anticancer treatment, which is designed to eradicate solid tumors, has proven to cause more harm than benefit. In fact, as pointed out by , Frank Arguello, the life expectancy of patients with esophageal cancers, for example, has not improved significantly over the span of a century (reviewed in [2]). This is perhaps not surprising, given that a presumed friend (apoptosis) has turned out to be the worst enemy in cancer therapy, fueling the oncogenic process, rather than promoting cancer cell demise (“suicide”) [20,21,22,23,24,25,26,27,28,29,30,31,32] (also see below).
2.2. The Apoptosis-Anastasis Tumor-Repopulating Pathway
The process known as apoptosis has two components (Figure 2): The canonical component (traditionally referred to as “apoptosis”) accompanied by anastasis. The former involves the activation of initiator caspases, mitochondrial outer membrane permeabilization (MOMP), release of cytochrome c and other apoptogenic factors from the mitochondria into the cytoplasm, activation of apoptotic proteases (executioner caspases), nuclear fragmentation and formation of apoptotic bodies [33,34,35]. This is followed by anastasis, the natural phenomenon by which cells return from late stages of apoptosis and other forms of regulated cell death [23,24,28,29,30,31,32]. Thus, the formation of apoptotic bodies, which is traditionally labeled as “apoptosis” (presumably implying cell demise) is not the end of the apoptosis-anastasis journey.
Cancer cells undergoing anastasis exhibit increased invasiveness, metastatic potential, and therapy resistance when compared to non-anastatic (bulk) cancer cells [32]. The cell adhesion protein cadherin 12 (CDH12) [36], cIAP2/NFκB [37], and p38 MAPK signaling [38] are implicated in anastasis-driven tumor angiogenesis and metastasis.
The cell surface expression of CD24 has been recently reported to be preferentially enriched in anastatic cancer (melanoma) cells that exhibit tumorigenic properties [39,40]. According to Vasileva et al. [39], even CD24-positive cancer cells that display various cell “death” indicators are able to recover and form large colonies under 3D culture conditions. These indicators included trypan blue staining (a marker of transient loss of cell membrane integrity), annexin V staining (a marker of phosphatidylserine externalization), nuclear fragmentation, and cell detachment from the culture surface.
2.3. Other Apoptosis-Related Tumor-Repopulating Pathways
In addition to anastasis, various other pro-survival pathways are associated with cancer cells undergoing apoptosis. These include phoenix rising [41,42], nuclear expulsion [43], senescence reversal [44], and the blebbishield emergency program (observed in cancer stem cells) [45,46]. (For details, please see our recent reviews [2,3,4].)
2.4. Increased Apoptosis in Solid Tumors is linked to an Unfavorable Clinical Outcome
Clinical studies reported since 1996 [47] have established that increased apoptosis in solid tumors is associated with cancer aggressiveness and poor patient outcomes (e.g., [48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64]). Some of these studies involved a large cohort of cancer patients. For example, the meta-analysis reported by Yang et al. in 2018 [54] was performed with 3091 breast cancer cases.
2.5. Take Home Messages
Collectively, these pre-clinical and clinical observations challenge the popular hypothesis that apoptosis might be a tumor suppression mechanism. They also underscore the danger of relying on molecular, biochemical, and morphological manifestations of apoptosis as a marker of cancer cell death, and call for revisiting thousands of articles that have used the terms “apoptosis” and “death” interchangeably.
3. Apoptotic Cancer Cells Promote Tumor Diversity and Heterogeneity
In response to moderate levels of stress, such as clinically relevant chemotherapy exposure, virtually all apoptotic cancer cells are known to undergo anastasis when determined in tissue culture studies (see, e.g., [4] and the video in [65]). The situation might be quite different in the tumor microenvironment, where cancer cell fate is influenced not only by the interplay between different cell types, but also by a myriad of molecules released from dying cells (Figure 3). Thus, to what extent apoptotic cancer cells that display the “eat me” signals (e.g., phosphatidylserine exposure) will be eliminated by the immune system remains unknown.
Irrespective of what proportion of apoptotic cancer cells will be destroyed, such cells are known to “sacrifice themselves at the altar of heterogeneity” via “treacherous apoptosis” [22]. This phenomenon refers to the presence of densely populated caspase 3-positive cells within an individual tumor (apoptotic cell islands) that fuel the proliferation and survival of cancerous and non-cancerous cells nearby, thus creating a diverse tumor population [66].
Figure 3.
Left: Complex heterogeneity within a solid tumor (adapted from Fu et a;. [67]). A region enriched with apoptotic cells is marked. Such “apoptotic cell islands” stimulate the proliferation of surrounding cancerous cells, which is in part mediated by oncogenic caspase 3 [41,42]. Right: Examples of cancer cell types that can promote tumor repopulation post therapy. A subset of cancer cells increase their p21 protein levels in response to treatment which is “just right” to enable them to temporarily halt their cell cycle, repair their genome and resume proliferation [68]. Anastasis refers to the natural process of cell recovery from late stages of regulated cell death [23,24]. Dying cells (e.g., through apoptosis) release a panel of pro-survival factors, including prostaglandin E2 (PGE2) via the phoenix rising pathway [20,25]. Polyploid/multinucleated giant cancer cells (PGCCs), senescent cancer cells, and therapy-tolerant cancer cells are three cell subgroups that enter a state of transient dormancy (active sleep) post-therapy [3,69]. Cancer stem cells undergoing apoptosis resist destruction (phagocytosis) by fusing their apoptotic belbs to form a blebbishield [26,45].
Figure 3.
Left: Complex heterogeneity within a solid tumor (adapted from Fu et a;. [67]). A region enriched with apoptotic cells is marked. Such “apoptotic cell islands” stimulate the proliferation of surrounding cancerous cells, which is in part mediated by oncogenic caspase 3 [41,42]. Right: Examples of cancer cell types that can promote tumor repopulation post therapy. A subset of cancer cells increase their p21 protein levels in response to treatment which is “just right” to enable them to temporarily halt their cell cycle, repair their genome and resume proliferation [68]. Anastasis refers to the natural process of cell recovery from late stages of regulated cell death [23,24]. Dying cells (e.g., through apoptosis) release a panel of pro-survival factors, including prostaglandin E2 (PGE2) via the phoenix rising pathway [20,25]. Polyploid/multinucleated giant cancer cells (PGCCs), senescent cancer cells, and therapy-tolerant cancer cells are three cell subgroups that enter a state of transient dormancy (active sleep) post-therapy [3,69]. Cancer stem cells undergoing apoptosis resist destruction (phagocytosis) by fusing their apoptotic belbs to form a blebbishield [26,45].
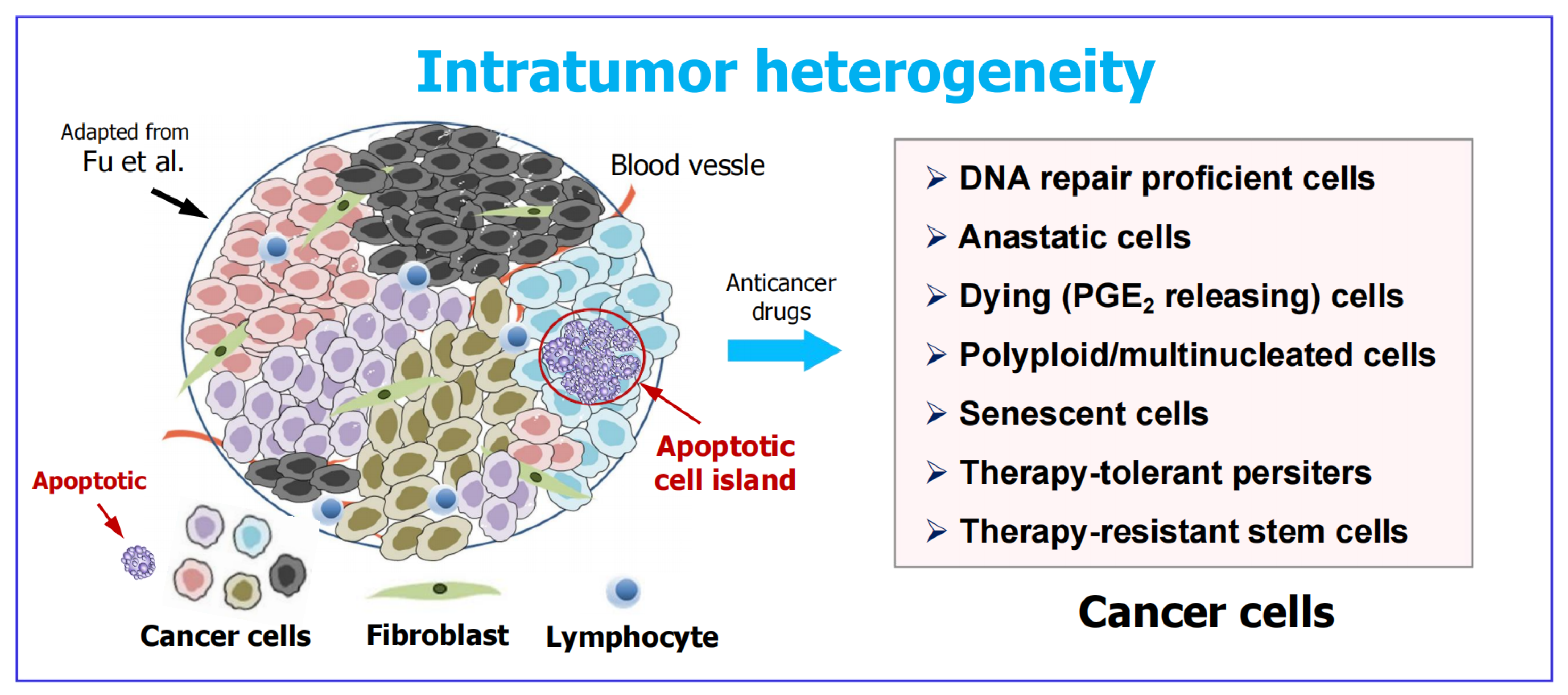
At first glance, it would appear that targeting (inhibiting) treacherous apoptosis together with signaling pathways associated with anastasis might improve the outcome of cancer therapy. This possibility is unlikely to be tenable given that a number of apoptosis-unrelated responses are known to contribute to intratumor heterogeneity [67,68,69,70,71]. These include cancer cell dormancy (a potential characteristic or mechanism underlying minimal residual disease) [70], as well as extrinsic factors such as angiogenesis, hypoxia, oxidative stress and acidosis [71].
4. Intratumor Heterogeneity: A Well Established (Yet Widely Overlooked) Obstacle in Cancer Therapy
The impact of tumor heterogeneity on implementing the various branches of precision oncology (e.g., strategies targeting p53, p21, DNA-damage response, regulated cell death, etc.) is becoming increasingly appreciated (e.g., [72,73,74,75,76,77,78,79,80,81,82]). But this knowledge is not new!
The discovery of cancer stem cells over two decades ago underscored the significance of cellular heterogeneity within a given tumor in terms of therapy resistance and disease recurrence (reviewed in [83]). By mid 2000s, a handful of pioneering cancer biologists who had relied on single-cell studies demonstrated that cancer cells with a highly enlarged nucleus or multiple nuclei (manifestations of mitotic “catastrophe” or “death”) give rise to progeny with stem cell-like properties. In 2001, for example, Erenpreisa and Cragg published a review entitled “Mitotic Catastrophe: A Mechanism of Survival...” in which they concluded that “the features of mitotic death do not simply represent aberrations of dying cells but are indicative of a switch to amitotic modes of cell survival that may provide additional mechanisms of genotoxic resistance” [84].
Cancer cells with extensive nuclear abnormalities (polyploidy, multinucleation, micronucleation) are now referred to as polyploid giant cancer cells (PGCCs) [85,86,87], and have emerged as the root causes of therapy resistance and relapse based on numerous preclinical and clinical studies (reviewed in [69,88]). Like cancer stem cells, PGCCs represent only a small proportion of cells within a solid tumor, thus contributing to intratumor heterogeneity.
In addition to PGCCs, cancer cell dormancy can also represent one or more of the following responses depending on the type of anticancer agent administered and the genetic background of cells (reviewed in [4]): therapy-induced premature senescence (which is often associated with a highly enlarged morphology due to extensive cytoplasmic mass), and the development of drug-tolerant persister cancer cells, radiation-tolerant persister cancer cells, and quiescent cancer cells. Each of these responses is reversible and can lead to the emergence of tenaciously proliferating cancers. It is feasible to assume that more than one these dormancy states can occur in different subsets of cancer cells within a tumor.
For those who are interested in further reading, the aforementioned recent reviews [72,73,74,75,76,77,78,79,80,81,82] have provided a wide range of overview of the cellular, molecular and clinical heterogeneity in the context of cancer progression, therapy resistance, and recurrence of metastatic disease. It is noteworthy that these reviews do not point out the contribution of therapy-induced responses (anastasis, treacherous apoptosis, PGCCs, cell fusion, etc.) to intratumor heterogeneity discussed herein and in our previous publications (e.g., [2,3,4]). This underscores the tremendous multifactorial nature of tumor heterogeneity. (Perhaps the reader might think that this is a rather wishy-washy explanation. I totally agree. How can a therapy-related article by reputable authors disregard the dark sides of apoptosis, PGCCs, senescence, etc.?)
Several questions arise when considering all these therapy-resistance responses, that can underlie tumor diversity, including the following two:
- Is cancer cell dormancy a greater threat in managing solid tumors or treacherous apoptosis (encompassing anastasis)? Probably the former is a bigger fish to fry based on reasons discussed previously [3,89,90]. For example, judging from tissue culture studies, clinically relevant anticancer exposure (radiation, drugs) triggers cancer cell dormancy but rarely engages regulated cell death [3,89,90,91]. This observation gives credence to the emerging trend of deintensification in cytotoxic cancer therapy [92], which would be expected to minimize the occurrence of side effects as well as regulated cell death and other tumor-repopulating events [4].
- Given that intratumor heterogeneity was well established over two decades ago [83], why did it take so long for most cancer research community members to appreciate its impact on resistance and relapse? Who knows! Perhaps “in the quest for the next cancer cure, few researchers bother to look back at the graveyard of failed medicines to figure out what went wrong” [93].
5. What Are the Reasons for Repeated Failures in Treating Solid Tumor Malignancies?
5.1. Most Preclinical Anticancer Studies Generate Clinically Irrelevant Information
The main objective of the various Special Issues of MDPI publications that I have Guest Edited in recent years has been to provide a comprehensive update on the growing complexity of cellular and molecular responses to DNA-damaging anticancer agents in human solid tumors and tumor-derived cell lines (see, e.g., [94]). Most articles published in these collections focused on therapy resistance reflecting genome chaos (e.g., polyploidy), regulated cell death (apoptosis), atavistic reprogramming (unicellular-like stress-resistant traits in cancer), and cell fusion [2,94,95].
The impetus behind leading these Special Issues, as well as writing the current review, has been the following grim reality: These various therapy-resistant and tumor-repopulating responses, as revealed by single-cell analysis, are either overlooked or scored as “death” in ubiquitously used preclinical radiosensitivity and chemosensitivity assays, including those listed in Figure 4 (left).
The danger of relying on these so-called “down” assays (decreased viability, colony forming ability, protein levels, etc.) for measuring cancer cell death has been discussed by us [3,69,95] and others [23,96,97,98]. The conventional in vitro colony formation assay, for example, which is considered the gold standard for measuring cancer cell radiosensitivity and chemosensitivity, determines the ability of a test agent to covert proliferating cancer cells to dormant, tumor-repopulating cells, and not dead cells [95]. In fact, the observation that cancer cells (HeLa) that lose their colony-forming ability in response to stress (exposure to ionizing radiation) remain viable and secrete proliferation-stimulating factors dates back to 1950s (for details, see [69]). (The image shown in Figure 4 is reproduced from the seminal study reported by Puck and Marcus in 1956 [99].)
With respect to high content multiwell plate assays, as pointed out by Eastman [96], drug treatment conditions typically used to obtain IC50 values (50% inhibitory concentration) are clinically irrelevant (also see Appendix A). The same caveats also pertain to assessing cancer cell apoptosis, which is often performed under clinically-irrelevant conditions (continuous treatment with high drug concentrations [3,89,96]).
5.2. The Consequences of Dishonesty in Data Reporting
We have recently discussed the growing complexity of tumor heterogeneity in terms of therapy resistance in various articles, including a review entitled “What are the reasons for continuing failures in cancer therapy? Are misleading/inappropriate preclinical assays to be blamed? Might some modern therapies cause more harm than benefit? [2]. An important, albeit disturbing topic that we covered in these articles relates to the consequences of dishonesty in data reporting, with numerous publications in major journals containing massaged or falsified results (see, e.g., section 4 in [2], and subsection 4.3 in [69]).
Thanks to the artificial intelligence technology, a significant number of such publications have been (and continue to be) retracted. For example, based on PubMed searches, only in 2025, at least TWENTY p53-related articles and over SIXTY apoptosis-related articles have been retracted (I stopped counting!).
A decade ago, a blog on “retraction watch” was published which highlighted five major cancer therapy-related publications from a reputable laboratory that were retracted [100]. In that blog, someone (Todd) raised the following profound question: has anyone, or organization, “started to audit meta-analyses, systematic reviews, practice guidelines, etc—to determine the impact of these retractions?”
We have a similar concern regarding thousands of p53/cancer-related articles in which “apoptosis” is used as another word for death. Like the retracted papers, how are these highly biased articles going to impact “meta-analysis, systematic reviews, practice guidelines, etc?” As stated by Dr. Otis Brawley (previous chief medical officer at the American Cancer Society), the consequences of such sloppiness in biomedical research “are real—and they can be deadly. Patients and their families have bought into treatments that either don’t work, cost a fortune or cause life-threatening side effects” [101].
It is for such reasons that our group has committed to writing articles and leading Special Issues in order to highlight false hypotheses that have derailed cancer research for decades. These include the biological output of p53 signaling discussed below.
6. Activation of Wild-Type p53 Signaling Following Clinically Relevant Anticancer Treatment Serves to Suppress (“Treacherous”) Apoptosis
There is a common trend in most reviews on the biological outputs of p53/p21 signaling (e.g., [102,103,104,105,106]). These articles typically provide comprehensive discussion on, e.g., different modes of cell “death” (e.g., apoptosis; cellular senescence; autophagy, ferroptosis), cell metabolism, and the immune system, and the roles played by p53 signaling in regulating these responses. Some reports also discuss the significance of p53 dynamics and cell fate decisions following treatment with ionizing radiation, chemotherapeutic drugs, and small molecule p53 activators such as nutlins. Although the growing complexity of cancer cell response to genotoxic stress has been generally appreciated, these reviews typically focus on the two-armed model of cell fate outcome in response to DNA damage, which was highlighted by Lane in 1992 [107]. Namely, p53-dependent cell cycle arrest and apoptosis (e.g., [103]). In this model, p21 is considered to be merely an activator of cell cycle checkpoints.
There are four fundamental issues with this canonical model. First, the biological output of p53-p21 signaling is context dependent (see below). Second, unlike the conventional wisdom [108,109], apoptosis and senescence are not permanent cell fates. In fact, as recently pointed out by Kandouz [110], it is still uncertain as to what constitutes cancer cell death. Third, clinically relevant anticancer exposure rarely (if at all) engages apoptosis in p53 wild-type solid tumor cells [3,89]. Forth, the landscape of p21 functions has expanded far beyond its classical role as a regulator of cell cycle progression ([111]; also see Figure 5).
6.1. Impact of p53 on Apoptosis Under Non-Physiological Versus Clinically-Relevant Conditions
The pro-apoptotic property of wild-type p53, which is often regarded as “indisputable fact” (similar to apoptosis being equal to death), needs to be put into context as follows:
- ➢
- ➢
- Strong p53 activation (above the apoptotic threshold) is observed under non-physiological conditions, such as cell exposure to very high doses of genotoxic agents (cisplatin; UVC) that induce bulky (transcription-blocking) DNA lesions [115].
- ➢
- Under these conditions, bulky lesions prevent transcriptional activation of MDM2 and other p53 negative regulators (e.g., p21, WIP1), resulting in a strong accumulation of p53 protein that triggers apoptosis presumably via its proline-rich region [115].
- ➢
- ➢
In short, activation of p53 signaling under physiological (clinically relevant) conditions appears to function as a strong barrier (“molecular brick wall” [90]) that protects against apoptosis, rather than engaging it. The antiapoptotic property of p53 was originally suggested to reflect its “dark” side [117], but it turns out that preventing treacherous apoptosis (which fuels the oncogenic fire [25]) represents the “bright” side of this important tumor suppressor.
6.2. The “Goldilocks Zone” for Cancer Cell Proliferation Following Clinically Relevant Chemotherapy Exposure
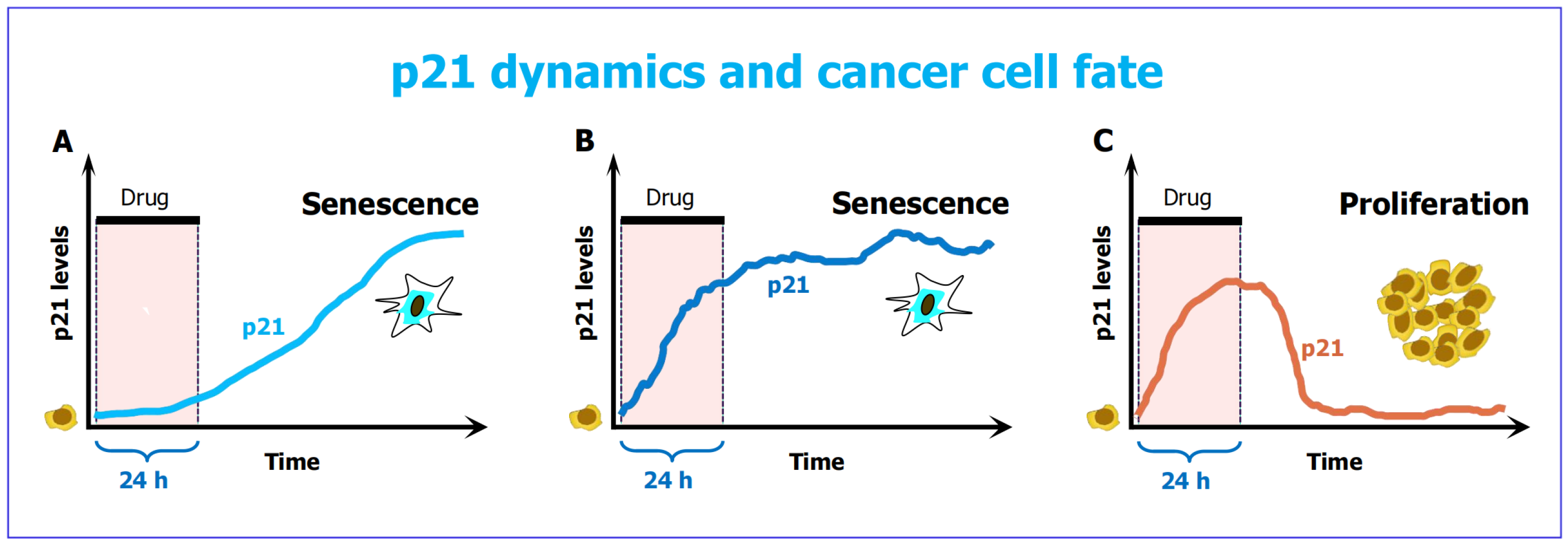
Time lapse microscopy has revealed that early p21 dynamics predict and shape cellular fate [91]: cancer cells with either low or very high levels of early p21, following chemotherapy exposure, are fated toward premature senescence (Figure 6, panels A and B), whereas cells with intermediate amounts of early p21 exhibit transient cell cycle arrest, decrease their p21 levels and resume proliferation (Figure 6C]. The latter scenario has been termed the p21 “Goldilocks zone” for proliferation. Different p21 dynamics and cell fate outcomes in “DNA repair proficient cancer cells” (noted in Figure 3) contributes to intratumor heterogeneity.
The study was reported by Hsu et al. [91] in 2019 (also see [118,119]). The experiments were performed with A549 (p53 wild-type) lung carcinoma cells that were treated with 50 nM doxorubicin for 1 day, and then incubated with fresh medium (without drug) for 4 days. The authors stressed that this treatment condition, which is known to be clinically relevant, did not engage regulated cell death.
Another pro-survival property of p21 pertains to reversibility of senescence-associated proliferation arrest, giving rise to highly metastatic progeny. This observation was first reported by Igor Roninson’s group over 25 years ago [120,121,122]. The authors concluded that, in solid tumor cells, the “re-entry into cell cycle after high-level induction of p21 may serve as a major cause of genetic destabilization that contributes to carcinogenesis and tumor progression” [122]. More recently, other groups have demonstrated that the reversal of senescence can be accelerated following treatment with apoptosis-triggering drugs (e.g., camptothecin; ABT-737) or ectopic expression of caspase 3 [123].
The prosurvival properties of p21 has led to the notion that perhaps selective inhibition of p21 function in cancer cells might result in a favorable patient outcome. This possibility turned out to be untenable. Single cell studies have demonstrated that loss of p21 (or p53) in cancer cells is permissive for the development of PGCCs, that are known to underlie resistance, metastasis and relapse (reviewed in [124]).
These discoveries need to be taken into consideration when designing therapeutic strategies targeting the p53-p21 pathway.
7. Conclusions
Preclinical anticancer studies are designed on the premise that therapy-induced apoptosis and cell proliferation arrest (dormancy) are permanent fates, ultimately leading to cancer cell demise. Accordingly, an enormous effort has been devoted to developing therapeutic strategies centered on apoptosis and durable proliferation arrest (senescence), mediated by wild-type p53 and its downstream effector p21, respectively.
Although apoptosis and senescence are scored as “death” in multiwell plate cell viability and other ubiquitously used preclinical radiosensitivity and chemosensitivity assays, single-cell studies have demonstrated that these responses are reversible, resulting in the emergence of more aggressive cancers (reviewed in, e.g., [4,23,32]). Furthermore, apoptotic cancer cells are known to promote the reversal of proliferation arrest in cancer cells undergoing senescence [123].
7.1. Who Would Disregard the Treacherous Side of Apoptosis in Treating Solid Tumors?
The dark side of apoptosis in cancer therapy is highlighted in only a handful of articles (perhaps in no more than a dozen, when excluding publications by our own group). Thus, the majority of publications (articles, reviews, editorials, online blogs) that discuss the challenges and opportunities in implementing precision oncology continue to propose novel apoptosis-stimulating anticancer strategies. The reasons for this serious oversight remain unknown.
Some authors, however, have started to discuss the need for a paradigm shift in the study of cell death in general, and specifically oncology, based on a wealth of preclinical observations (i.e., even after overlooking the aforementioned clinical reports) (e.g., [28,29,30,31]). These preclinical studies argue against the hypothesis proposed over 20 years ago, which is still widely cited (e.g., by the Nomenclature Committee on Cell Death) [35]. Namely, in mammalian cells, “the activation of executioner caspases occurs after the cells are committed to die” [35]. Now we know that there is no points of no return in apoptosis and probably other regulated cell death pathways [29,30]. Thus, “a paradigm shift in the study of cell death is currently occurring” [30].
7.2. Call For Contrarian Logic In Cancer Research
I trust that the discoveries highlighted herein and previously [2,3,4] are sufficient to put the following three fundamental questions into perspective:
- ➢
- What is apoptosis? Is it an irreversible mode of cell death based on cell “viability” and other misleading preclinical assays? Or engaging apoptosis in solid tumors represents a treacherous, tumor repopulating outcome? (I think it is the latter.)
- ➢
- Is “evading apoptosis” a hallmark of cancer, contributing to tumor progression and therapy resistance, as hypothesized by Hanahan and Weinberg over two decades ago [125]? Or, like normal cells, cancer cells simply employ the homeostatic process of anastasis to survive after engaging regulated cell death? (I think it is the latter.)
- ➢
- Is deregulated anastasis a hallmark of cancer? The availability of anastasis markers such as cell surface CD24 expression will hopefully lead to addressing this and other outstanding questions in cancer progression and therapy.
Understanding and counteracting different layers of tumor heterogeneity is paramount for an improved management of cancer in general, and the metastatic disease in particular [74]. The integration of novel molecular diagnostic technologies aided by machine learning tools offers a promising avenue in this regard [74]. These machine learning tools need to take into consideration the various layers of intratumor heterogeneity discussed herein and previously [2,69,95], which includes non-mutational/non-genetic events such as cell fusion, giving rise to PGCCs that generate tumor-repopulating progeny via amitosis, depolyploidization, and other mechanisms.
The outstanding contributions of pioneering cancer biologists in the complex fields of polyploidy and genome chaos has been recently presented in a commentary entitled “Amitotic Cell Division, Malignancy, and Resistance to Anticancer Agents” that we recently published as a tribute to Drs. Kirsten Walen and Rengaswami Rajaraman [126].
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflicts of interest.
Appendix A
Figure 1.
A. Misinformation that can be perpetrated by the misguided use of cell “viability” or “cytotoxicity” assays (reproduced from Eastman’s review article published in 2017 [96]).
Figure 1.
A. Misinformation that can be perpetrated by the misguided use of cell “viability” or “cytotoxicity” assays (reproduced from Eastman’s review article published in 2017 [96]).

References
- Pabla, S. Contrarian Thinking in Bioinformatics: Unlocking Breakthroughs by Challenging Assumptions. 2025. Available online: https://www.linkedin.com/pulse/contrarian-thinking-bioinformatics-unlocking-sarabjot-pabla-y693e (accessed on 5 October 2025).
- Mirzayans, R.; Murray, D. What are the reasons for continuing failures in cancer therapy? Are misleading/inappropriate preclinical assays to be blamed? Might some modern therapies cause more harm than benefit? Int. J. Mol. Sci. 2022, 23, 13217. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R. Changing the landscape of solid tumor therapy from apoptosis-promoting to apoptosis-inhibiting strategies. Curr. Issues Mol. Biol. 2024, 46, 5379–5396. [Google Scholar] [CrossRef]
- Mirzayans, R. Anastasis and other apoptosis-related prosurvival pathways call for a paradigm shift in oncology: Significance of deintensification in treating solid tumors. Int. J. Mol. Sci. 2025, 26, 1881. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef]
- Maeda, H.; Khatami, M. Analyses of repeated failures in cancer therapy for solid tumors: Poor tumor-selective drug delivery, low therapeutic efficacy and unsustainable costs. Clin. Transl. Med. 2018, 7, 11. [Google Scholar] [CrossRef]
- Joyner, M.J.; Paneth, N. Promises, promises, and precision medicine. J. Clin. Invest. 2019, 129, 946–948. [Google Scholar] [CrossRef]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Pich, O.; Bailey, C.; Watkins, T.B.K.; Zaccaria, S.; Jamal-Hanjani, M.; Swanton, C. The translational challenges of precision oncology. Cancer Cell 2022, 40, 458–478. [Google Scholar] [CrossRef]
- Heng, J.; Heng, H.H. Genome chaos, information creation, and cancer emergence: Searching for new frameworks on the 50th anniversary of the “war on cancer”. Genes 2022, 13, 101. [Google Scholar] [CrossRef]
- Lohse, S. Mapping uncertainty in precision medicine: A systematic scoping review. J. Eval. Clin. Pract. 2023, 29, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T. Journeys to failure that litter the path to developing new cancer therapeutics. JAMA Netw. Open 2023, 6, e2324949. [Google Scholar] [CrossRef] [PubMed]
- Suehnholz, S.P.; Nissan, M.H.; Zhang, H.; Kundra, R.; Nandakumar, S.; Lu, C.; Carrero, S.; Dhaneshwar, A.; Fernandez, N.; Xu, B.W.; et al. Quantifying the expanding landscape of clinical actionability for patients with cancer. Cancer Discov. 2024, 14, 49–65. [Google Scholar] [CrossRef]
- Kailen, W.G. Preclinical cancer target validation: How not to be wrong. NIH Wednesday Afternoon Lectures (WELS) series, January 24, 2018 [https://videocast.nih.gov/watch=27066] (accessed on 5 October 2025).
- Belluz, J. Most cancer drugs fail in testing. This might be a big reason why. Science—VOX blog (2019) https://www.vox.com/2019/9/16/20864066/cancer-studies-fail (acceaassed on 5 October 2025).
- Horgan, J. The cancer industry: hype vs. reality. Cancer medicine generates enormous revenues but marginal benefits for patients (2020) https://www.scientificamerican.com/blog/cross-check/the-cancer-industry-hype-vs-reality/ (accessed on 5 October 2025).
- Horgan, J. The cancer industry: Hype versus reality (2023) https://johnhorgan.org/cross-check/the-cancer-industry-hype-versus-reality (accessed on 5 October 2025).
- Azra, R. The First Cell: And the Human Costs of Pursuing Cancer to the Last. New York, Basic Books, 2019.
- Conley, B.A.; Staudt, L.; Takebe, N.; Wheeler, D.A.; Wang, L.; Cardenas, M.F.; Korchina, V.; Zenklusen, J.C.; McShane, L.M.; Tricoli, J.V.; et al. The Exceptional Responders initiative: Feasibility of a National Cancer Institute pilot study. J. Natl. Cancer Inst. 2021, 113, 27–37. [Google Scholar] [CrossRef]
- Ichim, G.; Tait, S.W.G. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.V. The dark side of apoptosis. In Molecular Mechanisms of Tumor Cell Resistance to Chemotherapy, Resistance to Targeted Anti-Cancer Therapeutics 1; Bonavida, B., Ed.; Springer: New York, NY, USA, 2013; pp. 245–258.
- Dhanasekaran, R. Treacherous apoptosis—Cancer cells sacrifice themselves at the altar of heterogeneity. Hepatology 2022, 76, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return journey from cell death. Cancers 2021, 13, 3671. [Google Scholar] [CrossRef]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef]
- Castillo Ferrer, C.; Berthenet, K.; Ichim, G. Apoptosis—Fueling the oncogenic fire. FEBS J. 2021, 288, 4445–4463. [Google Scholar] [CrossRef]
- Jinesh, G.G. Exposing the deadly dark side of apoptotic cancer stem cells. Oncoscience 2017, 4, 124–125. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef]
- Kalkavan, H.; Rühl, S.; Shaw, J.J.P.; Green, D.R. Non-lethal outcomes of engaging regulated cell death pathways in cancer. Nat. Cancer 2023, 4, 795–806. [Google Scholar] [CrossRef]
- Nano, M.; Montell, D.J. Apoptotic signaling: Beyond cell death. Semin Cell Dev. Biol. 2024, 156, 22–34. [Google Scholar] [CrossRef]
- Green, D.R. Cell death: Revisiting the roads to ruin. Dev. Cell 2024, 59, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Not dead yet: Cell death and survival in cancer and normal physiology. Front. Cell Death 2024, 3, 147734. [Google Scholar] [CrossRef]
- Tang, H.M.; Tang. H.L. Unravelling the pathological roles of anastasis in cancer recurrence. Open Biol. 2025, 15, 240270.
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Kin, T.; Beck, W.T. Impact of complex apoptotic signaling pathways on cancer cell sensitivity to therapy. Cancers 2024, 16, 984. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Pietrocola, F.; Guilbaud, E.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostini, M.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023, 30, 1097–1154. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Liu, X.; Liu, M.; Sun, L.; Pan, X.; Hu, H.; Jiang, B.; Zou, Y.; Liu, Q.; et al. Chemotherapy-induced executioner caspase activation increases breast cancer malignancy through epigenetic de-repression of CDH12. Oncogenesis 2023, 12, 34. [Google Scholar] [CrossRef]
- Wang, R.; Wang, Y.; Liu, X.; Liu, M.; Sun, L.; Pan, X.; Hu, H; Jiang, B.; Zou, Y.; Liu, et al. Anastasis enhances metastasis and chemoresistance of colorectal cancer cells through upregulating cIAP2/NFκB signaling. Cell Death Dis. 2023, 14, 388.
- Sun, L.; Yao, C.; Li, X.; Wang, Y.; Wang, R.; Wang, M.; Liu, Q.; Montell, D.J.; Shao, C.; Gong, Y.; Sun, G. Anastasis confers ovarian cancer cells increased malignancy through elevated p38 MAPK activation. Cell Death Differ. 2023, 30, 809–824. [Google Scholar] [CrossRef]
- Vasileva, M.H.; Bennemann, A.; Zachmann, K.; Schön, M.P.; Frank, J.; Ulaganathan, V.K. CD24 flags anastasis in melanoma cells. Apoptosis 2025, 30, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ulaganathan, V.K. CD24: A Cell Surface Marker for Anastasis in Melanoma. 2024. Available online: https://communities.springernature.com/posts/cd24-a-cell-surface-marker-for-anastasis-in-melanoma (accessed on 5 October 2025).
- Corsi, F.; Capradossi, F.; Pelliccia, A.; Briganti, S.; Bruni, E.; Traversa, E.; Torino, F.; Reichle, A.; Ghibelli, L. Apoptosis as driver of therapy-induced cancer repopulation and acquired cell-resistance (CRAC): A simple in vitro model of Phoenix Rising in prostate cancer. Int. J. Mol. Sci. 2022, 23, 1152. [Google Scholar] [CrossRef] [PubMed]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef] [PubMed]
- Park, W.Y.; Gray, J.M.; Holewinski, R.J.; Andresson, T.; So, J.S.; Carmona-Rivera, C.; Hollander, M.C.; Yang, H.H.; Lee, M.; Kaplan, M.J.; et al. Apoptosis-induced nuclear expulsion in tumor cells drives S100a4-mediated metastatic outgrowth through the RAGE pathway. Nat. Cancer 2023, 4, 419–435. [Google Scholar] [CrossRef]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Jinesh, G.G.; Brohl, A.S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 2022, 7, 296. [Google Scholar] [CrossRef]
- Ergün, S.; Aslan, S.; Demir, D.; Kayaoğlu, S.; Saydam, M.; Keleş, Y.; Kolcuoğlu, D.; Hekim, N.T.; Güneş, S. Beyond death: Unmasking the intricacies of apoptosis escape. Mol. Diagn. Ther. 2024, 28, 403–423. [Google Scholar] [CrossRef]
- Tatebe, S.; Ishida, M.; Kasagi, N.; Tsujitani, S.; Kaibara, N.; Ito, H. Apoptosis occurs more frequently in metastatic foci than in primary lesions of human colorectal carcinomas: Analysis by terminal-deoxynucleotidyl-transferase-mediated dUTP-biotin nick end labeling. Int. J. Cancer 1996, 65, 173–177. [Google Scholar] [CrossRef]
- Huang, Q.; Li, S.; Cheng, I.; Deng, M.; He, X.; Wang, Z.; Yang, C.H.; Zhao, X.Y.; Huang, J. High expression of anti-apoptotic protein Bcl-2 is a good prognostic factor in colorectal cancer: Result of a meta-analysis. World J. Gastroenterol. 2017, 23, 5018–5033. [Google Scholar] [CrossRef]
- Flanagan, L.; Meyer, M.; Fay, J.; Curry, S.; Bacon, O.; Duessmann, H.; John, K.; Boland, K.C.; McNamara, D.A.; Kay, E.W.; et al. Low levels of Caspase-3 predict favourable response to 5FU-based chemotherapy in advanced colorectal cancer: Caspase-3 inhibition as a therapeutic approach. Cell Death Dis. 2016, 7, e2087. [Google Scholar] [CrossRef]
- Alcaide, J.; Funez, R.; Rueda, A.; Perez-Ruiz, E.; Pereda, T.; Rodrigo, I.; Covenas, R.; Munoz, M.; Redondo, M. The role and prognostic value of apoptosis in colorectal carcinoma. BMC Clin. Pathol. 2013, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.; Morrison, I.; Heriot, A.G.; Bartlett, J.B.; Finlayson, C.; Dalgleish, A.G.; Kumar, D. The correlation between colorectal cancer rates of proliferation and apoptosis and systemic cytokine levels; plus their influence upon survival. Br. J. Cancer 2006, 94, 1412–1419. [Google Scholar] [CrossRef]
- Hilska, M.; Collan, Y.U.; Laine, V.J.O.; Kössi, J.; Hirsimäki, P.; Laato, M.; Roberts, P.J. The significance of tumor markers for proliferation and apoptosis in predicting survival in colorectal cancer. Dis. Colon. Rectum. 2005, 48, 2197–2208. [Google Scholar] [CrossRef] [PubMed]
- Bendardaf, R.; Ristamaki, R.; Kujari, H.; Laine, J.; Lamlum, H.; Collan, Y.; Pyrhonen, S. Apoptotic index and bcl-2 expression as prognostic factors in colorectal carcinoma. Oncology 2003, 64, 435–442. [Google Scholar] [CrossRef]
- Yang, X.; Zhong, D.N.; Qin, H.; Wu, P.R.; Wei, K.L.; Chen, G.; He, R.Q.; Zhong, J.C. Caspase-3 over-expression is associated with poor overall survival and clinicopathological parameters in breast cancer: A meta-analysis of 3091 cases. Oncotarget 2018, 9, 8629–8641. [Google Scholar] [CrossRef]
- Lindner, A.U.; Lucantoni, F.; Varešlija, D.; Resler, A.; Murphy, B.M.; Gallagher, W.M.; Hill, A.D.K.; Young, L.S.; Prehn, J.H.M. Low cleaved caspase-7 levels indicate unfavourable outcome across all breast cancers. Mol. Med. 2018, 96, 1025–1037. [Google Scholar] [CrossRef]
- Pu, X.; Storr, S.J.; Zhang, Y.; Rakha, E.A.; Green, A.R.; Ellis, I.O.; Martin, S.G. Caspase-3 and caspase-8 expression in breast cancer: Caspase-3 is associated with survival. Apoptosis 2017, 22, 357–368. [Google Scholar] [CrossRef]
- Koshida, Y.; Saegusa, M.; Okayasu, I. Apoptosis, cell proliferation and expression of Bcl-2 and Bax in gastric carcinomas: Immunohistochemical and clinicopathological study. Br. J. Cancer 1997, 75, 367–373. [Google Scholar] [CrossRef]
- Beer, T.W.; Carr, N.J.; Whittaker, M.A.; Pullinger, N. Mitotic and in situ end-labeling apoptotic indices as prognostic markers in malignant mesothelioma. Ann. Diagn. Pathol. 2000, 4, 143–148. [Google Scholar] [CrossRef]
- Kahlos, K.; Soini, Y.; Paakko, P.; Saily, M.; Linnainmaa, K.; Kinnula, V.L. Proliferation, apoptosis, and manganese superoxide dismutase in malignant mesothelioma. Int. J. Cancer 2000, 88, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Mangili, F.; Cigala, C.; Arrigoni, G.; Rovere, E.; Gattuso, C.; Santambrogio, G.; Garancini, P. Cell loss and proliferation in non-small cell lung carcinoma: Correlation with histological subtype. Eur. J. Histochem. 1998, 42, 287–295. [Google Scholar]
- Tormanen, U.; Eerola, A.K.; Rainio, P.; Vahakangas, K.; Soini, Y.; Sormunen, R.; Bloigu, R.; Lehto, V.P.; Paakko, P. Enhanced apoptosis predicts shortened survival in non-small cell lung carcinoma. Cancer Res. 1995, 55, 5595–5602. [Google Scholar]
- Meggiato, T.; Calabrese, F.; Valente, M.; Favaretto, E.; Baliello, E.; Del Favero, G. Spontaneous apoptosis and proliferation in human pancreatic cancer. Pancreas 2000, 20, 117–122. [Google Scholar] [CrossRef]
- Magistrelli, P.; Coppola, R.; Tonini, G.; Vincenzi, B.; Santini, D.; Borzomati, D.; Vecchio, F.; Valeri, S.; Castri, F.; Antinori, A. Apoptotic index or a combination of Bax/Bcl-2 expression correlate with survival after resection of pancreatic adenocarcinoma. J. Cell Biochem. 2006, 97, 98–108. [Google Scholar] [CrossRef]
- Naresh, K.N.; Lakshminarayanan, K.; Pai, S.A.; Borges, A.M. Apoptosis index is a predictor of metastatic phenotype in patients with early stage squamous carcinoma of the tongue: A hypothesis to support this paradoxical association. Cancer 2001, 91, 578–584. [Google Scholar] [CrossRef]
- Montell, D.J. Cellular Survival by Anastasis. 2024. Available online: https://denisemontell.mcdb.ucsb.edu/research/cellular-survival-anastasis (accessed on 5 October 2025).
- Khatib, S.A.; Ma, L.; Dang, H.; Forgues, M.; Chung, J.-Y.; Ylaya, K.; Hewitt, S.M.; Chaisaingmongkol, J.; Rucchirawat, M.; Wang, X.W. Single-cell biology uncovers apoptotic cell death and its spatial organization as a potential modifier of tumor diversity in HCC. Hepatology 2022, 76, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.C.; Liang, S.B.; Luo, M.; Wang, X.P. Intratumoral heterogeneity and drug resistance in cancer. Cancer Cell Int. 2025, 25, 103. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hemann, M.T. A dynamic view of chemotherapy effectiveness. Nature 2019, 572, 321–322. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and treatment resistance of solid tumors with a focus on polyploid/senescent giant cancer cells (PGCCs). Int. J. Mol. Sci. 2023, 24, 11534. [Google Scholar] [CrossRef] [PubMed]
- Min, H.Y.; Lee, H.Y. Cellular dormancy in cancer: mechanisms and potential targeting strategies. Cancer Res. Treat. 2023, 55, 720–736. [Google Scholar] [CrossRef]
- Sun, X.X.; Yu, Q. Intra-tumor heterogeneity of cancer cells and its implications for cancer treatment. Acta Pharmacol. Sin. 2015, 36, 1219–1227. [Google Scholar] [CrossRef]
- Rouault, C.D.; Charafe-Jauffret, E.; C.; Ginestier. The interplay of DNA damage, epigenetics and tumour heterogeneity in driving cancer cell fitness. Nat. Commun. 2025, 16, 8733. [CrossRef]
- Liu, Y.; Su, Z.; Tavana, O.; Gu, W. Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell. 2024, 42, 946–967. [Google Scholar] [CrossRef]
- MacDonald, W.J.; Purcell, C.; Pinho-Schwermann, M.; Stubbs, N.M.; Srinivasan, P.R.; El-Deiry, W.S. Heterogeneity in cancer. Cancers 2025, 17, 441. [Google Scholar] [CrossRef]
- Maleki, E.H.; Bahrami, A.R.; Matin, M.M. Cancer cell cycle heterogeneity as a critical determinant of therapeutic resistance. Genes Dis. 2023, 11, 189–204. [Google Scholar] [CrossRef]
- Zhu, H.; Tian, Y.; Chen, H.; Y.; Qian, J. Li.; Niu, D.; Zhao, W.; Wu, Y.; Zhang, X.; Tang, T.; et al. Targeting DNA damage response pathways in tumor drug resistance: Mechanisms, clinical implications, and future directions. Drug Resist. Updat. 2025, 83, 101287. [CrossRef] [PubMed]
- Oh, M.S.; Abascal, J.; Rennels, A.K.; Salehi-Rad, R.; Dubinett, S.M.; Liu, B. Tumor heterogeneity and the immune response in non-small cell lung cancer: Emerging insights and implications for immunotherapy. Cancers 2025, 17, 1027. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Miao, K.; Sun, H.; Deng, C.X. Tumor heterogeneity reshapes the tumor microenvironment to influence drug resistance. Int. J. Biol. Sci. 2022, 18, 3019–3033. [Google Scholar] [CrossRef] [PubMed]
- Proietto, M.; Crippa, M.; Damiani, C.; Pasquale, V.; Sacco, E.; Vanoni, M.; Gilardi, M. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front. Oncol. 2023, 13, 1164535. [Google Scholar] [CrossRef]
- Tomasik B, Garbicz F, Braun M, Bieńkowski M and Jassem J. Heterogeneity in precision oncology. Camb. Prism. Precis. Med. 2024, 2, e2.
- Tonello, S.; Rolla, R.; Tillio, P.A.; Sainaghi, P.P.; Colangelo, D. Microenvironment and tumor heterogeneity as pharmacological targets in precision oncology. Pharmaceuticals 2025, 18, 915. [Google Scholar] [CrossRef]
- Clinton, T.N.; Chen, Z.; Wise, H.; Lenis, A.T.; Chavan, S.; Donoghue, M.T.A.; Almassi, N.; Chu, C.E.; Dason, S. Genomic heterogeneity as a barrier to precision oncology in urothelial cancer. Cell Rep. 2022, 41, 111859. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Pharmacological modulation of p53 function in cancer therapy. Curr. Signal. Transduct. Ther. 2008, 3, 183–194. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Cragg, M.S. Mitotic death: a mechanism of survival? A review. Cancer Cell Int. 2001, 1, 1–7. [Google Scholar] [CrossRef]
- Chen, J.; Niu, N.; Zhang, J.; Qi, L.; Shen, W.; Donkena, K.V.; Feng, Z.; Liu, J. Polyploid giant cancer cells (PGCCs): The evil roots of cancer. Curr. Cancer Drug Targets 2019, 19, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Erenpreisa, J.; Sikora, E. Polyploid giant cancer cells: An emerging new field of cancer biology. Semin. Cancer Biol. 2022, 81, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2022, 81, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Trabzonlu, L.; Pienta, K.J.; Trock, B.J.; De Marzo, A.M.; Amend. S.R. Presence of cells in the polyaneuploid cancer cell (PACC) state predicts the risk of recurrence in prostate cancer. Prostate 2023, 83, 277–285. [CrossRef]
- Murray, D.; Mirzayans, R. Cellular responses to platinum-based anticancer drugs and UVC: Role of p53 and implications for cancer therapy. Int. J. Mol. Sci. 2020, 21, 5766. [Google Scholar] [CrossRef]
- Mirzayans, R. When Therapy-induced cancer cell apoptosis fuels tumor relapse. Onco. 2024, 4, 37–45. [Google Scholar] [CrossRef]
- Hsu, C.H.; Altschuler, S.J.; Wu, L.F. Patterns of early p21 dynamics determine proliferation-senescence cell fate after chemotherapy. Cell 2019, 178, 361–373. [Google Scholar] [CrossRef]
- Soon, J.A.; Franchini, F.; IJzermanm, N.J.; McArthur, G.A. Leveraging the potential for deintensification in cancer care. Nat. Cancer 2024, 5, 1597–1599. [Google Scholar] [CrossRef]
- Belluz, J. Most cancer drugs fail in testing. This might be a big reason why. Science—VOX blog (2019) https://www.vox.com/2019/9/16/20864066/cancer-studies-fail (accessed on 5 October 2025).
- Mirzayans, R. The cellular response to DNA damage: From DNA repair to polyploidy and beyond. Int. J. Mol. Sci. 2023, 24, 6852. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854–8866. [Google Scholar] [CrossRef] [PubMed]
- Forgie, B.N.; Prakash, R.; Goyeneche, A.A.; Telleria, C.M. Vitality, viability, long-term clonogenic survival, cytotoxicity, cytostasis and lethality: What do they mean when testing new investigational oncology drugs? Discov. Oncol. 2024, 15, 5. [Google Scholar] [CrossRef] [PubMed]
- Nicoletto, R.E.; Ofner, C.M. Cytotoxic mechanisms of doxorubicin at clinically relevant concentrations in breast cancer cells. Cancer Chemother. Pharmacol. 2022, 89, 285–311. [Google Scholar] [CrossRef]
- Puck, T.T.; Marcus, P.I. Action of X-rays on mammalian cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef]
- Retraction Watch: Tracking retractions as a window into the scientific process. Available online: https://retractionwatch.com/2015/07/06/cancer-research-retraction-is-fifth-for-robert-weinberg-fourth-for-his-former-student/ (accessed on 5 October 2025).
- Szabo, L. Cancer treatment hype gives false hope to many patients. Kaiser Health News (2017) https://www.usatoday.com/story/news/2017/04/27/cancer-treatment-hype-gives-false-hope-many-patients/100972794/ (last accessed on 5 October 2025).
- Hernández Borrero, L.J.; El-Deiry, WS. Tumor suppressor p53: Biology, signaling pathways, and therapeutic targeting. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188556. [Google Scholar] [CrossRef]
- Lees, A.; Sessler, T.; McDade, S. Dying to survive—The p53 paradox. Cancers 2021, 13, 3257. [Google Scholar] [CrossRef]
- Zhang, S.; Carlsen, L.; Hernandez Borrero, L.; Seyhan, A.A.; Tian, X.; El-Deiry, W.S. Advanced strategies for therapeutic targeting of wild-type and mutant p53 in cancer. Biomolecules 2022, 12, 548. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, J.; Long, Y.; Maimaitijiang, A.; Su, Z.; Li, W.; Li, J. Unraveling the guardian: p53’s multifaceted role in the DNA damage response and tumor treatment strategies. Int. J. Mol. Sci. 2024, 25, 12928. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The cell-cycle arrest and apoptotic functions of p53 in tumor initiation and progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Levine, A.J. Targeting the p53 protein for cancer therapies: The translational impact of p53 research. Cancer Res. 2022, 82, 362–364. [Google Scholar] [CrossRef]
- Oren, M.; Prives, C. p53: A tale of complexity and context. Cell 2024, 187, 1569–1573. [Google Scholar] [CrossRef]
- Kandouz, M. Cell death, by any other name. Cells 2024, 13, 325. [Google Scholar] [CrossRef]
- Murray, D.; Mirzayans, R.; McBride, W.H. Defenses against pro-oxidant forces—Maintenance of cellular and genomic integrity and longevity. Radiat. Res. 2018, 190, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Barley, R.D.C.; Enns, L.; Paterson, M.C.; Mirzayans, R. Aberrant p21WAF1-dependent growth arrest as the possible mechanism of abnormal resistance to ultraviolet light cytotoxicity in Li-Fraumeni syndrome fibroblast strains heterozygous for TP53 mutations. Oncogene 1998, 17, 533–543. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Q.; Gong, X.; Yem, F.; Liou, Y.C. Dose-dependent mutual regulation between Wip1 and p53 following UVC irradiation. Int. J. Biochem. Cell Biol. 2011, 43, 535–544. [Google Scholar] [CrossRef]
- Kracikova, M.; G. ; Akiri, George, A.; Sachidanandam, R.; Aaronson, S.A. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death and Differ. 2013, 20, 576–588. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Kumar, P.; Murray, D. Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: Impact on chemotherapy outcome. Int. J. Mol. Sci. 2017, 18, 928. [Google Scholar] [CrossRef]
- Jänicke, R.U.; Sohn, D.; Schulze-Osthoff, K. The dark side of a tumor suppressor: Anti-apoptotic p53. Cell Death Differ. 2008, 15, 959–976. [Google Scholar] [CrossRef]
- Ashraf, H.M.; Moser, J.; Spencer, S.L. Senescence Evasion in Chemotherapy: A Sweet Spot for p21. Cell 2019, 178, 267–269. [Google Scholar] [CrossRef]
- Liu, Y.; Hemann, M.T. A dynamic view of chemotherapy effectiveness. Nature 2019, 572, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Broude, E.V.; Dokmanovic, M.; Zhu, H.; Ruth, A.; Xuan, Y.; Kandel, E.S.; Lausch. E.; Christov, K.; Roninson, I.B. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999, 59, 3761–3767.
- Chang, B.D.; Xuan, Y.; Broude, E.V.; Zhu, H.; Schott, B.; Fang, J.; Roninson, I.B. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene 1999, 18, 4808–4818. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.D.; Broude, E.V.; J. Fang.; T.V. Kalinichenko.; R. Abdryashitov.; J.C. Poole.; I.B. Roninson. p21Waf1/Cip1/Sdi1-induced growth arrest is associated with depletion of mitosis-control proteins and leads to abnormal mitosis and endoreduplication in recovering cells. Oncogene 2000, 19, 2165–2170. [Google Scholar] [CrossRef]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability assessment following anticancer treatment requires single-cell visualization. Cancers 2018, 10, 255. [Google Scholar] [CrossRef]
- Hanahan, D. , Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Amitotic cell division, malignancy, and resistance to anticancer agents: A tribute to Drs. Walen and Rajaraman. Cancers 2024, 16, 3106. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Early (canonical) and late (pro-survival) events after triggering apoptosis in solid tumors. For details, see text and [4]. The illustration of the canonical component is adapted from Kim et al. [34]).
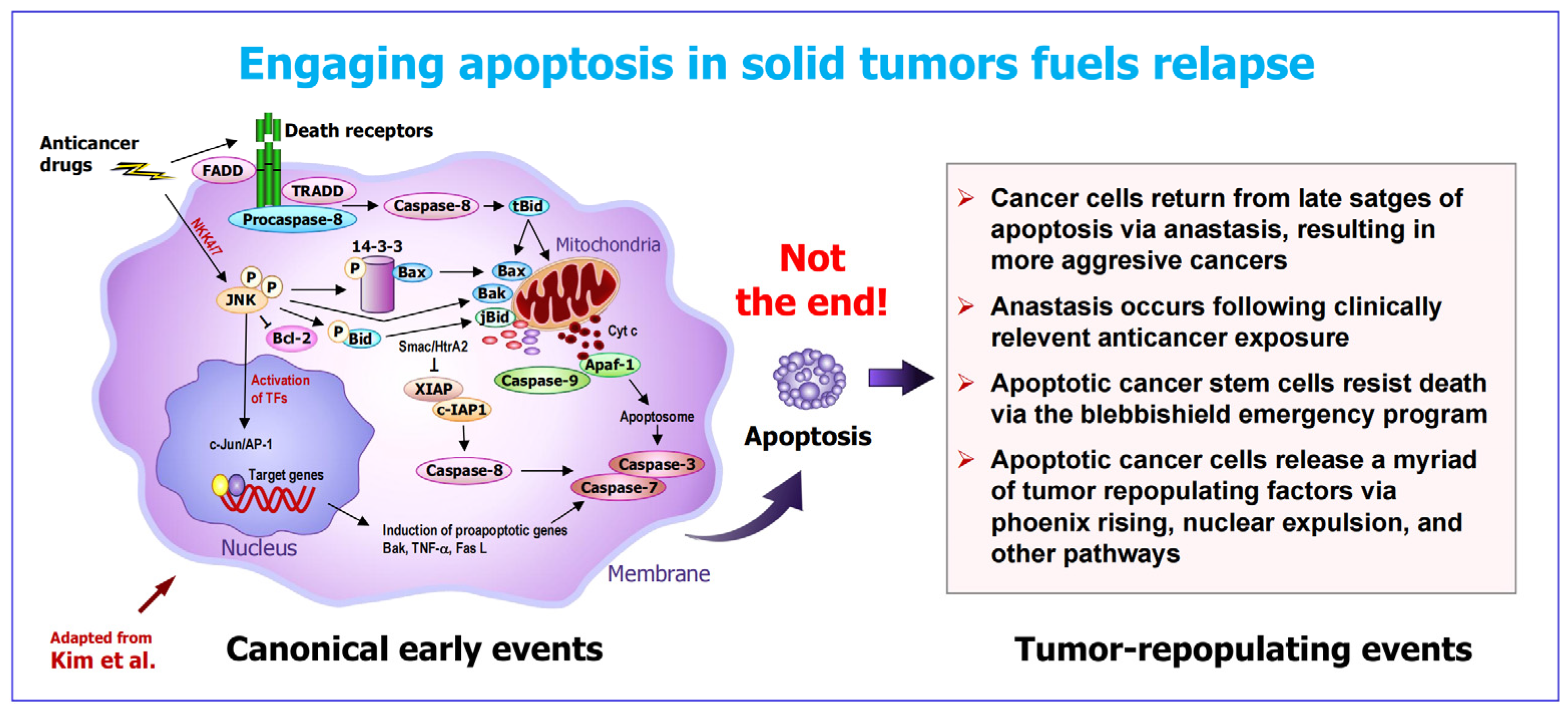
Figure 4.
Left: Preclinical assays that are widely used in anticancer drug discovery studies. These so-called “down” assays measure averaged responses of a large number of cells (i.e., overlook intratumor heterogeneity). Old-fashioned microscopy (right), advanced time-lapse microscopy and other single-cell assays are required in order to reveal and study the complexity and heterogeneity that exists within a tumor. The image, showing a remarkable heterogeneity within HeLa cell cultures in response to radiation exposure, was reported 70 years ago (for details, see [69]). Small sized cells, a macroscopic colony containing small sized cells, and polyploid giant cells are marked.
Figure 4.
Left: Preclinical assays that are widely used in anticancer drug discovery studies. These so-called “down” assays measure averaged responses of a large number of cells (i.e., overlook intratumor heterogeneity). Old-fashioned microscopy (right), advanced time-lapse microscopy and other single-cell assays are required in order to reveal and study the complexity and heterogeneity that exists within a tumor. The image, showing a remarkable heterogeneity within HeLa cell cultures in response to radiation exposure, was reported 70 years ago (for details, see [69]). Small sized cells, a macroscopic colony containing small sized cells, and polyploid giant cells are marked.
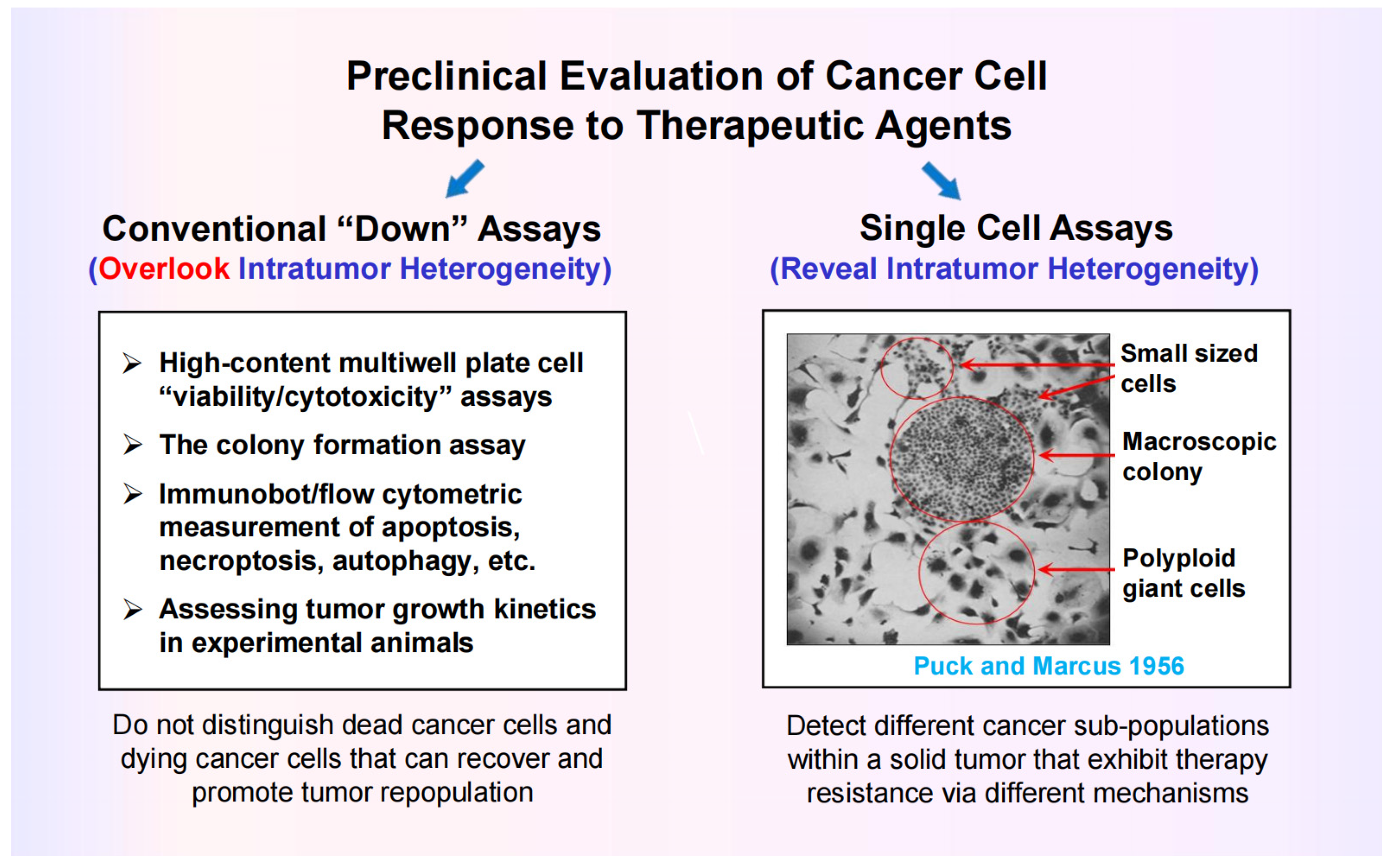
Figure 5.
A partial schematic of the DNA damage response network illustrating the importance of negative regulation of p53 by p21, WIP1 (wild-type p53-induced phosphatase 1), and other p53 targets (e.g., MDM2, DNAJB9; not shown) in suppressing regulated cell death. Arrows indicate stimulation and T-shaped lines indicate inhibition. Multiple functions of p21 in the DNA damage response network are indicated.
Figure 5.
A partial schematic of the DNA damage response network illustrating the importance of negative regulation of p53 by p21, WIP1 (wild-type p53-induced phosphatase 1), and other p53 targets (e.g., MDM2, DNAJB9; not shown) in suppressing regulated cell death. Arrows indicate stimulation and T-shaped lines indicate inhibition. Multiple functions of p21 in the DNA damage response network are indicated.
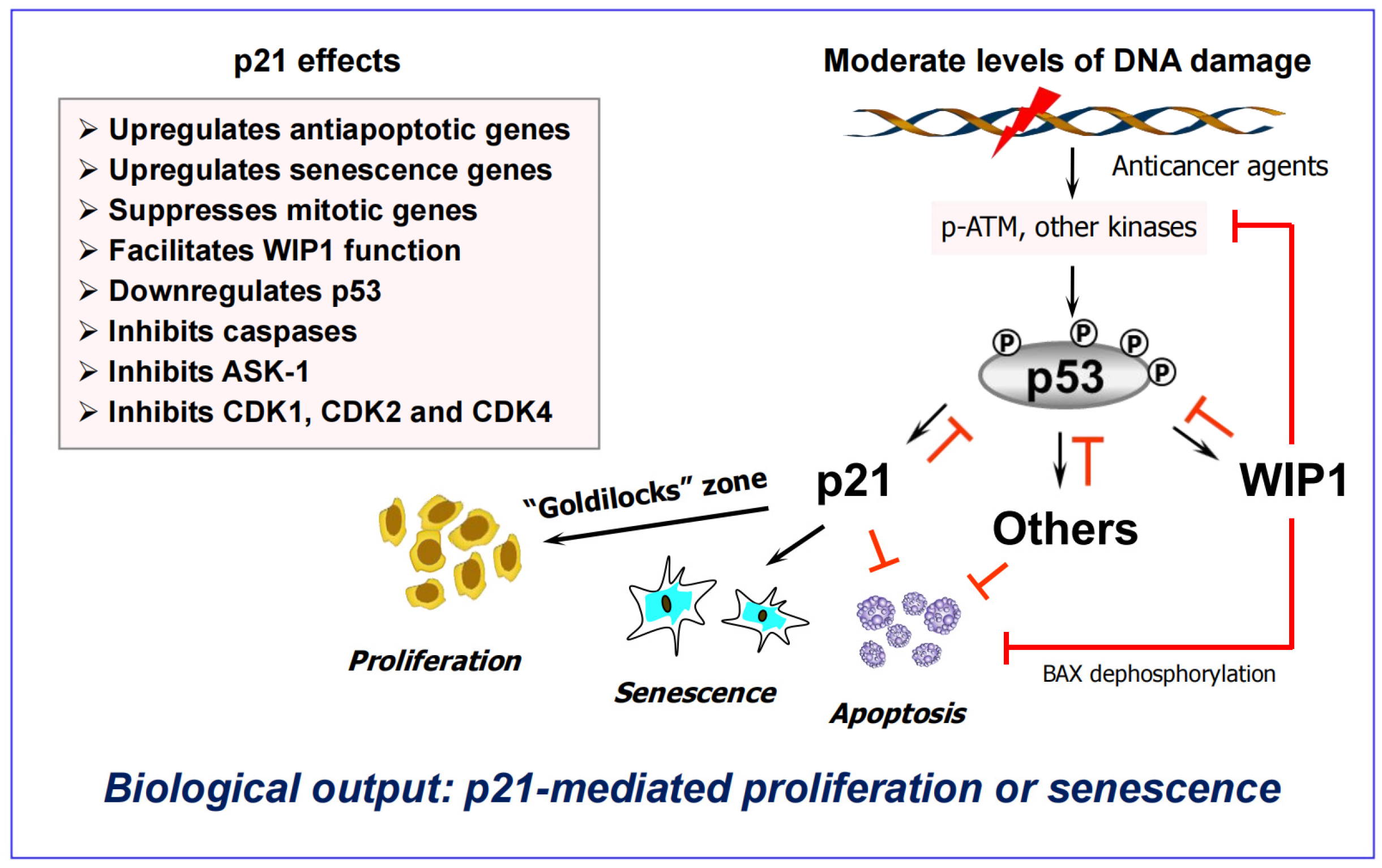
Figure 6.
The influence of p21 dynamics on cancer cell fate following a 24- h treatment with the chemotherapeutic drug doxorubicin, reported by Hsu et al. [91]. Cells with either low levels of p21 (A) or very high levels of p21 (B) following treatment undergo senescence. On the other hand, cells that their p21 to a “just right” level post-treatment activate cell cycle checkpoints (to facilitate DNA repair) and resume proliferation. The latter scenario is called the p21 “Goldilocks zone” for proliferation [91].
Figure 6.
The influence of p21 dynamics on cancer cell fate following a 24- h treatment with the chemotherapeutic drug doxorubicin, reported by Hsu et al. [91]. Cells with either low levels of p21 (A) or very high levels of p21 (B) following treatment undergo senescence. On the other hand, cells that their p21 to a “just right” level post-treatment activate cell cycle checkpoints (to facilitate DNA repair) and resume proliferation. The latter scenario is called the p21 “Goldilocks zone” for proliferation [91].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



