Submitted:
03 October 2025
Posted:
06 October 2025
You are already at the latest version
Abstract
Multilocus pathogenic variation—when multiple genetic disorders coexist in a single individual—remains rare but is increasingly recognized in the era of genomic medicine. Reporting such cases is essential to improve diagnostic accuracy, refine clinical management, and inform genetic counseling. We describe a pediatric case with three distinct genetic diagnoses contributing to an atypical and complex phenotype. The patient was born to consanguineous parents, presented with neurological symptoms, gastrointestinal dysfunction, endocrine abnormalities, and dysmorphic features. Trio-based exome sequencing identified three independent findings: cerebrotendinous xanthomatosis (CTX) due to a homozygous pathogenic variant in the cytochrome P450 family 27 subfamily A member 1 (CYP27A1) gene, Klinefelter syndrome (47,XXY), and an incidental heterozygous pathogenic variant in the breast cancer 2 susceptibility gene (BRCA2), associated with hereditary cancer predisposition. The overlapping manifestations of CTX and Klinefelter syndrome produced a non-classical presentation that delayed diagnosis. The BRCA2 variant, though unrelated to presenting symptoms, has major implications for future cancer surveillance and family risk assessment. This case underscores the utility of exome sequencing in elucidating complex phenotypes, particularly in consanguineous populations, and highlights the need for multidisciplinary care and tailored guidelines for patients with multiple genetic diagnoses, including the management of incidental findings with actionable implications.
Keywords:
Xanthomatosis
; Cerebrotendinous
; Klinefelter syndrome
; BRCA2 Protein
; whole exome sequencing
; genetic diseases
; inborn
; genetic counseling
1. Introduction
Advancements in massive parallel sequencing technologies, particularly whole exome sequencing (WES), have significantly increased the detection of multiple independent genetic findings in a single individual—a phenomenon referred to as multigene or multilocus diagnoses [1]. Far from anecdotal, the estimated prevalence of such findings ranges from 4% to 7% in clinical genomic sequencing studies, with higher rates reported among patients with neurological disorders, consanguinity, atypical phenotypes, multisystem involvement, or protracted diagnostic journeys [2,3]. The identification of multiple pathogenic variants provides a more comprehensive framework for understanding complex clinical presentations, enhances diagnostic accuracy, and enables the implementation of personalized therapeutic and preventive strategies.
The recognition of dual or even triple genetic diagnoses is becoming increasingly relevant in clinical genetics, particularly when each variant carries distinct etiologic, prognostic, therapeutic, or familial implications [4]. This diagnostic complexity aligns with the principles of precision medicine, wherein integrated genomic data allow clinicians to manage patients not only according to the primary disease but also in consideration of coexisting genetic contributions that may modulate phenotypic expression, disease progression, or treatment response. Furthermore, acknowledging the coexistence of multiple genetic conditions can help avoid diagnostic anchoring, shorten the diagnostic timeline, and improve reproductive counseling and risk stratification for family members [5].
Cerebrotendinous xanthomatosis (CTX, MIM: 606530) is a rare autosomal recessive lipid storage disorder caused by biallelic mutations in the cytochrome P450 family 27 subfamily A member 1 (CYP27A1) gene, which encodes sterol 27-hydroxylase. Enzymatic deficiency leads to impaired bile acid synthesis and accumulation of cholestanol and cholesterol in the brain, tendons, and other tissues. CTX typically manifests in childhood with chronic diarrhea, juvenile cataracts, epilepsy, neuropsychiatric disturbances, hyporeflexia, and progressive cognitive decline [6,7].
Klinefelter syndrome (47, XXY), a common sex chromosome aneuploidy occurring in approximately 1 in 600 males, presents with a broad phenotypic spectrum that may include hypogonadism, gynecomastia, pubertal delay, infertility, micrognathia, and neurodevelopmental challenges. Despite being a well-characterized condition, its clinical variability often contributes to delayed or missed diagnoses, particularly in pediatric populations [8,9].
In contrast, pathogenic variants in the breast cancer 2 susceptibility gene (BRCA2) are more commonly associated with hereditary breast and ovarian cancer syndromes in adults. However, such variants may be incidentally identified in pediatric patients undergoing WES for unrelated indications. The presence of a heterozygous BRCA2 mutation confers increased lifetime risks for male breast cancer, prostate cancer, pancreatic cancer, melanoma, and central nervous system (CNS) tumors, thus warranting early cancer surveillance strategies and genetic counseling for the patient and at-risk relatives [10].
In this report, we present the case of a 14-year-old male with a complex constellation of neurological, gastrointestinal, dysmorphic, and endocrine manifestations, in whom three clinically relevant and genetically distinct diagnoses were identified: a homozygous pathogenic variant in CYP27A1 (CTX), a 47, XXY karyotype (Klinefelter syndrome), and a heterozygous pathogenic variant in BRCA2. Each finding contributes uniquely to the patient’s phenotype, clinical course, and long-term management, illustrating the challenges and value of comprehensive genomic analysis in complex pediatric presentations.
2. Case Description
A 14-year-old male patient, born in Venezuela and currently residing in Cali, Colombia, was referred to the medical genetics service due to early-onset seizures (most recently occurring two months prior to evaluation)), mild cognitive impairment, gynecomastia, facial dysmorphisms and intermittent chronic diarrhea. The family history was notable for parental consanguinity, as the patient’s parents are first cousins on the paternal side. Additionally, consanguinity was documented in the previous generation, with a consanguineous union among the patient’s great-grandparents The patient’s mother, a 40-year-old woman with no relevant medical history, reported an uneventful pregnancy and a term cesarean delivery due to fetal macrosomia.
Since the age of 3, the patient has experienced nocturnal focal motor seizures involving the right hemibody, which have been refractory to multiple antiepileptic drugs, including oxcarbazepine, valproic acid, and levetiracetam. According to his mother, cognitive and language neurodevelopment was age-appropriate until approximately 6 years of age, at which point academic challenges emerged, accompanied by a progressive decline in attention, academic performance, and behavioral regulation. Psychometric assessment reported an Intelligence Quotient (IQ) of 70, consistent with mild intellectual disability. A six-hour electroencephalogram telemetry showed bursts of generalized sharp waves, and brain Magnetic Resonance Imaging (MRI) revealed a subcortical malacic lesion in the right basal frontal region, interpreted as a chronic structural abnormality with potential epileptogenic significance
The mother also reported intermittent chronic diarrhea since the age of 4, with variable frequency, no seasonal pattern or dietary triggers, characterized by Bristol type 6 stools without mucus, blood, or steatorrhea. A gastroenterology consultation suggested a possible genetic etiology. Basic metabolic screening revealed elevated total cholesterol (220 mg/dL) and low-density lipoprotein (LDL) cholesterol (140 mg/dL).
On physical examination, the patient measured 159 cm in height, weighed 54 kg, and had a head circumference of 57 cm, all within normal percentiles. However, several facial dysmorphisms were noted: synophrys, webbed neck, epicanthal folds, left palpebral ptosis, dorsal telangiectasias, asymmetric auricles, micrognathia, bifid uvula, a café-au-lait spot on the trunk, as well as mild pectus excavatum, cavus foot, and bilateral gynecomastia. This combination of morphological and neurological findings raised suspicion of a multisystemic genetic disorder (Figure 1).
Given the phenotypic complexity, the prolonged diagnostic odyssey, and the history of parental consanguinity, whole-exome sequencing in trio and karyotyping with G-banding were performed. The karyotype revealed a 47, XXY pattern, consistent with Klinefelter syndrome, which explained gynecomastia, body morphology, and part of the observed neurocognitive profile (Figure 2).
Molecular analysis of the exome identified a pathogenic homozygous variant in the CYP27A1 gene (c.1183C>T; p.Arg395Cys), which was confirmed in heterozygosity in both parents. This finding is consistent with a diagnosis of cerebrotendinous xanthomatosis (CTX), a rare autosomal recessive lipid storage disorder characterized by abnormal cholestanol accumulation and progressive neurological, gastrointestinal, and systemic involvement. Additionally, a heterozygous pathogenic variant in BRCA2 (c.4936_4939del; p.Glu1646Glnfs*23), inherited from his mother, was also identified. This frameshift variant is associated with increased lifetime risk for several malignancies, including male breast cancer, prostate cancer, and pancreatic cancer and highlights the need for structured genetic counseling and long-term surveillance strategies for the proband and at-risk family members.
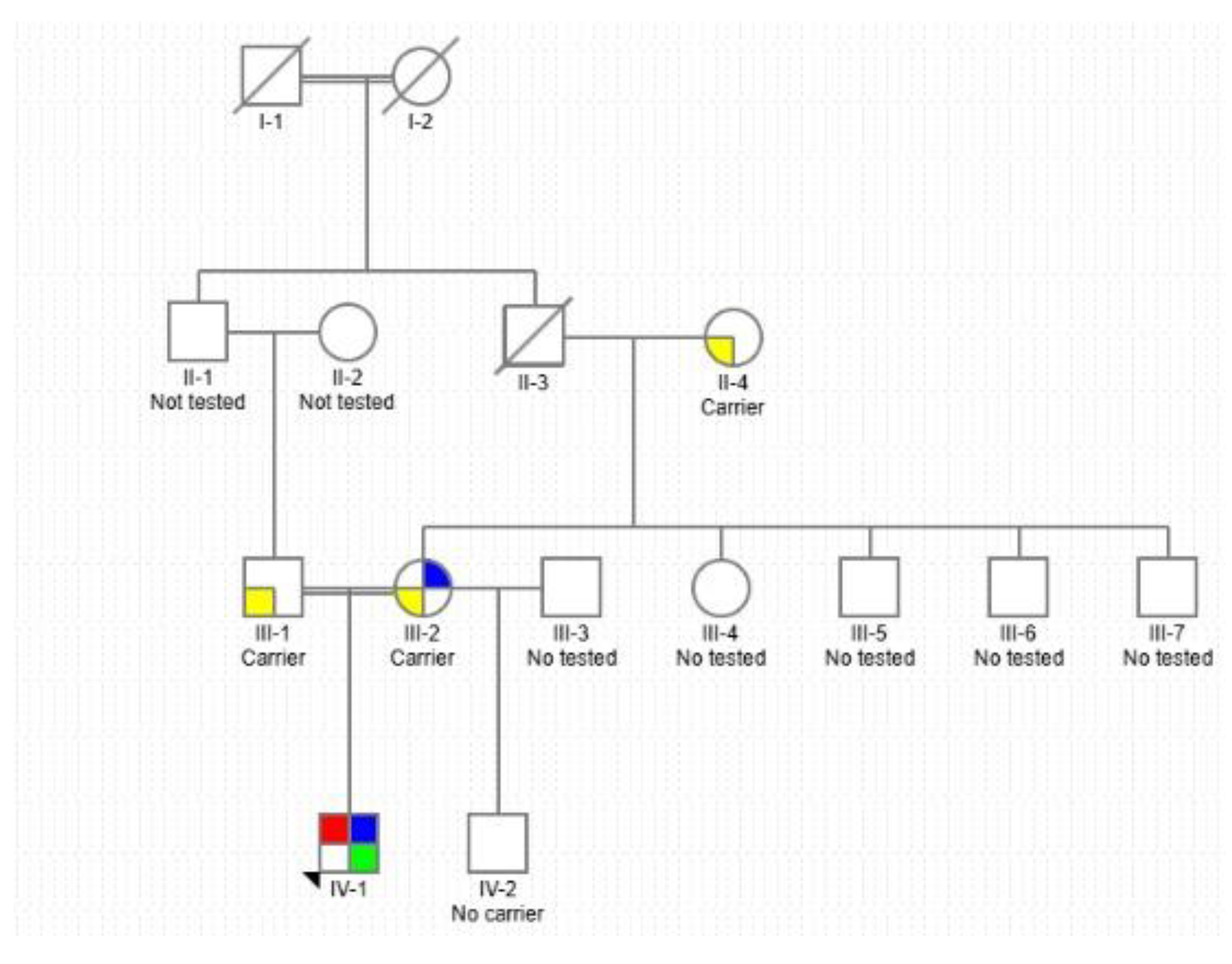
Given the documented consanguinity and the autosomal recessive inheritance pattern of CTX, a three-generation pedigree was constructed. The pedigree illustrates the paternal consanguinity, the segregation pattern of the homozygous CYP27A1 pathogenic variant, and the inheritance of the BRCA2 mutation, which is associated with increased familial cancer risk (Figure 3).
3. Discussion and Conclusions
This case illustrates the diagnostic and clinical challenges associated with multilocus pathogenic variation in pediatric patients with complex, multisystemic presentations. The simultaneous identification of cerebrotendinous xanthomatosis (CTX), Klinefelter syndrome (47, XXY), and a heterozygous pathogenic variant in BRCA2 underscores the increasing relevance of considering multilocus diagnoses in individuals with consanguinity, atypical evolution, or phenotypic expansion [11].
The systematic implementation of high-throughput sequencing technologies, such as whole exome sequencing (WES), has shifted the diagnostic paradigm by enabling the detection of multiple causative variants in a single individual. Current studies estimate that 4% to 7% of patients undergoing genomic testing harbor more than one molecular diagnosis, which may result in blended phenotypes or overlapping symptomatology [11]. This phenomenon, referred to as “genetic collision,” requires a comprehensive, multidisciplinary diagnostic framework.
Comparable reports in the literature support this emerging complexity. Posey et al. [12], for example, found that 31.6% of patients with presumed phenotypic expansion harbored multiple pathogenic variants when reassessed by exome sequencing. Other published cases have identified up to four concurrent diagnoses in a single patient [13,14], while dual molecular diagnoses—including rare combinations such as Angelman syndrome with Krabbe disease—have been reported in children with atypical disease progression [15]. These findings underscore the growing need to adopt multigenic models in clinical reasoning.
For the patient in this report, clinical management required a domain-specific approach. CTX is a treatable metabolic disorder; administration of chenodeoxycholic acid can restore bile acid homeostasis and prevent progression of neurological symptoms [16]. Management of Klinefelter syndrome includes testosterone replacement therapy to support pubertal development, improve bone density, and address neurobehavioral outcomes [17]. Additionally, the BRCA2 variant, although not responsible for the presenting symptoms, mandates high-risk cancer surveillance and cascade testing in accordance with National Comprehensive Cancer Network (NCCN) guidelines [18].
The concurrent identification of three unrelated genetic conditions in a single individual is extraordinarily rare. Assuming statistical independence, the joint prevalence of CTX (0.0003%) [20], Klinefelter syndrome (0.17%) [21], and BRCA2 pathogenic variants (0.25%) [22] yields an estimated probability of approximately 1.275 × 10⁻¹¹—or roughly one in 78 billion births—emphasizing the uniqueness of this case.
Importantly, the BRCA2 variant was detected as an incidental finding. Its disclosure is aligned with current American College of Medical Genetics and Genomics (ACMG) guidelines, which, in version 3.2 of their secondary findings list, include BRCA2 among 81 genes whose pathogenic variants must be reported due to their actionable nature and potential to significantly reduce morbidity through early intervention [23]. Beyond its clinical novelty, this case calls for the development of specific care protocols for individuals with multilocus diagnoses. These include integrated follow-up frameworks, criteria for therapeutic prioritization when multiple conditions co-occur, and expansion of genetic counseling models to address both treatable and predictive conditions simultaneously. As the field of genomic medicine evolves, there is a growing need to incorporate multigenic reasoning into clinical training and decision-making pathways [15,19].
This case exemplifies the increasing complexity introduced by multilocus genetic diagnoses in pediatric medicine. The co-occurrence of a treatable metabolic disorder, a syndromic aneuploidy, and a cancer predisposition variant in the same patient necessitates an integrated, multidisciplinary approach. The case underscores the clinical utility of exome sequencing, the importance of early diagnosis in treatable conditions, and the ethical imperative to manage incidental findings appropriately. It also highlights the need for the development of evidence-based guidelines for the diagnosis, counseling, and longitudinal care of patients with multiple molecular diagnoses.
Author Contributions
All authors contributed equally to the preparation, writing and proofreading of this manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study was approved by the ethics committee #2022.1919 of the Hospital Universitario Fundación Valle del Lili.
Informed Consent Statement
The consent was signed by the legal representative of the minor and an informed consent form was requested.
Data Availability Statement
Information is available upon request from the correspondence author.
Acknowledgments
We thank the Genomic Medicine Laboratory of Universidad Icesi for their support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mitani T, Isikay S, Gezdirici A, et al. High prevalence of multilocus pathogenic variation in neurodevelopmental disorders in the Turkish population. The American Journal of Human Genetics 2021; 108: 1981–2005. [CrossRef]
- Posey JE, Harel T, Liu P, et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. New England Journal of Medicine 2017; 376: 21–31. [CrossRef]
- Herman I, Jolly A, Du H, et al. Quantitative dissection of multilocus pathogenic variation in an Egyptian infant with severe neurodevelopmental disorder resulting from multiple molecular diagnoses. Am J Med Genet A 2022; 188: 735–750. [CrossRef]
- Espada-Musitu D, Manero-Azua Á, Vado Y, et al. Genetic counselling in the era of next generation sequencing. Anales de Pediatría (English Edition) 2025; 102: 503712. [CrossRef]
- Narayanan DL, Udyawar D, Kaur P, et al. Multilocus disease-causing genomic variations for Mendelian disorders: role of systematic phenotyping and implications on genetic counselling. European Journal of Human Genetics 2021; 29: 1774–1780. [CrossRef]
- Nóbrega PR, Bernardes AM, Ribeiro RM, et al. Cerebrotendinous Xanthomatosis: A practice review of pathophysiology, diagnosis, and treatment. Front Neurol; 13. Epub ahead of print 23 December 2022. [CrossRef]
- Islam M, Hoggard N, Hadjivassiliou M. Cerebrotendinous Xanthomatosis: diversity of presentation and refining treatment with chenodeoxycholic acid. Cerebellum Ataxias 2021; 8: 5. [CrossRef]
- Blackburn J, Ramakrishnan A, Graham C, et al. Klinefelter Syndrome: A Review. Clin Endocrinol (Oxf) 2025; 102: 565–573. [CrossRef]
- Butler G, Srirangalingam U, Faithfull J, et al. Klinefelter syndrome: going beyond the diagnosis. Arch Dis Child 2023; 108: 166–171. [CrossRef]
- Li S, Silvestri V, Leslie G, et al. Cancer Risks Associated With BRCA1 and BRCA2 Pathogenic Variants. Journal of Clinical Oncology 2022; 40: 1529–1541. [CrossRef]
- Baldridge D, Heeley J, Vineyard M, et al. The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genetics in Medicine 2017; 19: 1040–1048. [CrossRef]
- Karaca E, Posey JE, Coban Akdemir Z, et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genetics in Medicine 2018; 20: 1528–1537. [CrossRef]
- Bozkurt-Yozgatli T, Pehlivan D, Gibbs RA, et al. Multilocus pathogenic variants contribute to intrafamilial clinical heterogeneity: a retrospective study of sibling pairs with neurodevelopmental disorders. BMC Med Genomics 2024; 17: 85. [CrossRef]
- Narayanan DL, Udyawar D, Kaur P, et al. Multilocus disease-causing genomic variations for Mendelian disorders: role of systematic phenotyping and implications on genetic counselling. European Journal of Human Genetics 2021; 29: 1774–1780. [CrossRef]
- Liu Y, Ma X, Chen Z, et al. Dual rare genetic diseases in five pediatric patients: insights from next-generation diagnostic methods. Orphanet J Rare Dis 2024; 19: 159. [CrossRef]
- Berginer VM, Salen G, Shefer S. Long-Term Treatment of Cerebrotendinous Xanthomatosis with Chenodeoxycholic Acid. New England Journal of Medicine 1984; 311: 1649–1652. [CrossRef]
- Groth KA, Skakkebæk A, Høst C, et al. Klinefelter Syndrome—A Clinical Update. J Clin Endocrinol Metab 2013; 98: 20–30.
- Gupta S, Weiss JM, Burke CA, et al. NCCN Guidelines Version 4.2024 Genetic/Familial High-Risk Assessment: Colorectal, Endometrial, and Gastric Continue. https://www.nccn.org/home/ (2025).
- Dumbuya JS, Zeng C, Deng L, et al. The impact of rare diseases on the quality of life in paediatric patients: current status. Front Public Health; 13. Epub ahead of print 24 March 2025. [CrossRef]
- Pramparo T, Steiner RD, Rodems S, et al. Allelic prevalence and geographic distribution of cerebrotendinous xanthomatosis. Orphanet J Rare Dis 2023; 18: 13. [CrossRef]
- Ridder LO, Berglund A, Stochholm K, et al. Morbidity, mortality, and socioeconomics in Klinefelter syndrome and 47,XYY syndrome: a comparative review. Endocr Connect; 12. Epub ahead of print 24 March 2023. [CrossRef]
- Matta BP, Gomes R, Mattos D, et al. Familial history and prevalence of BRCA1, BRCA2 and TP53 pathogenic variants in HBOC Brazilian patients from a public healthcare service. Sci Rep 2022; 12: 18629. [CrossRef]
- Miller DT, Lee K, Abul-Husn NS, et al. ACMG SF v3.2 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine 2023; 25: 100866. [CrossRef]
Figure 1.
Dysmorphic features and physical findings observed in the patient.
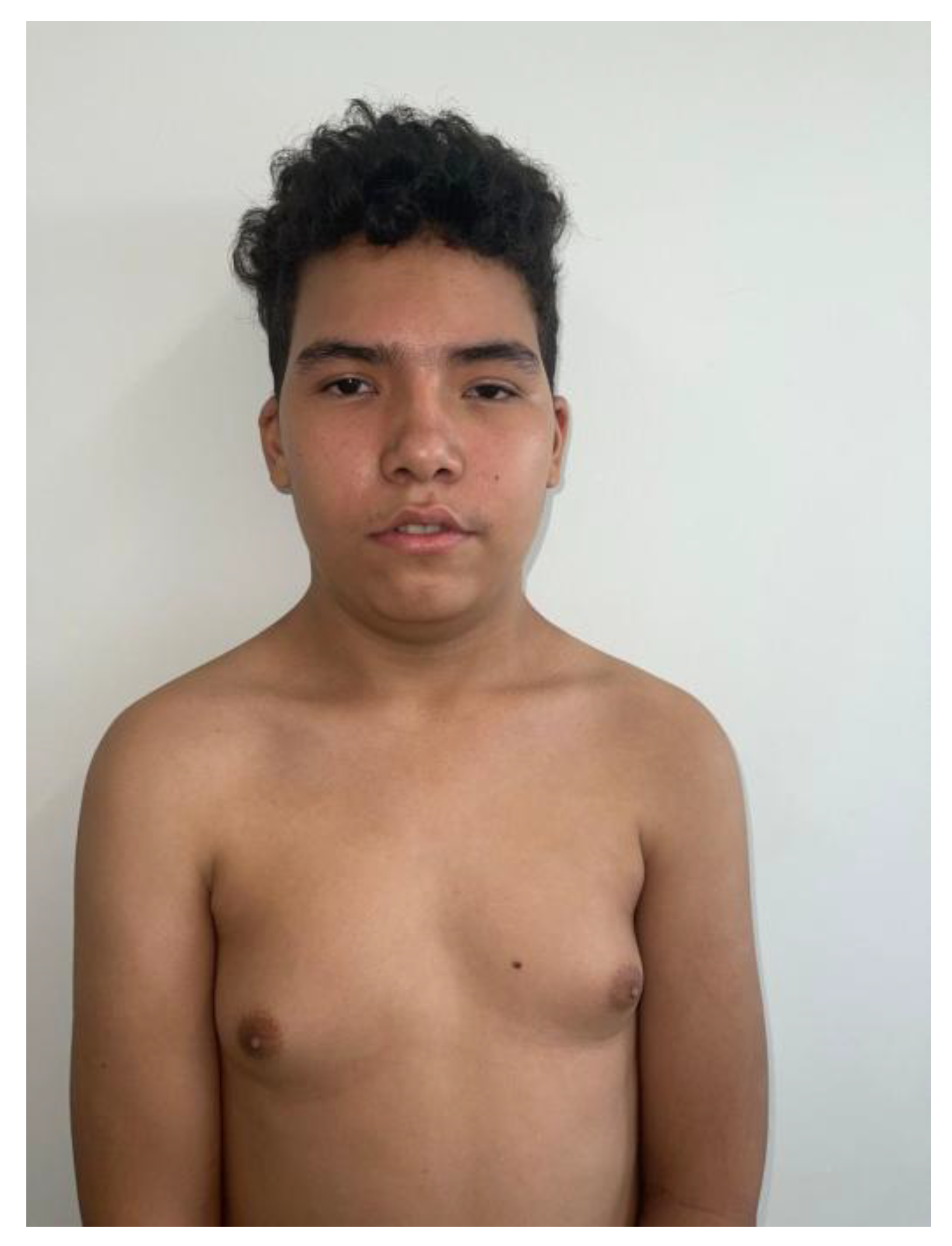
Figure 2.
The Karyotype of the patient shows a 47, XXY chromosomal pattern.
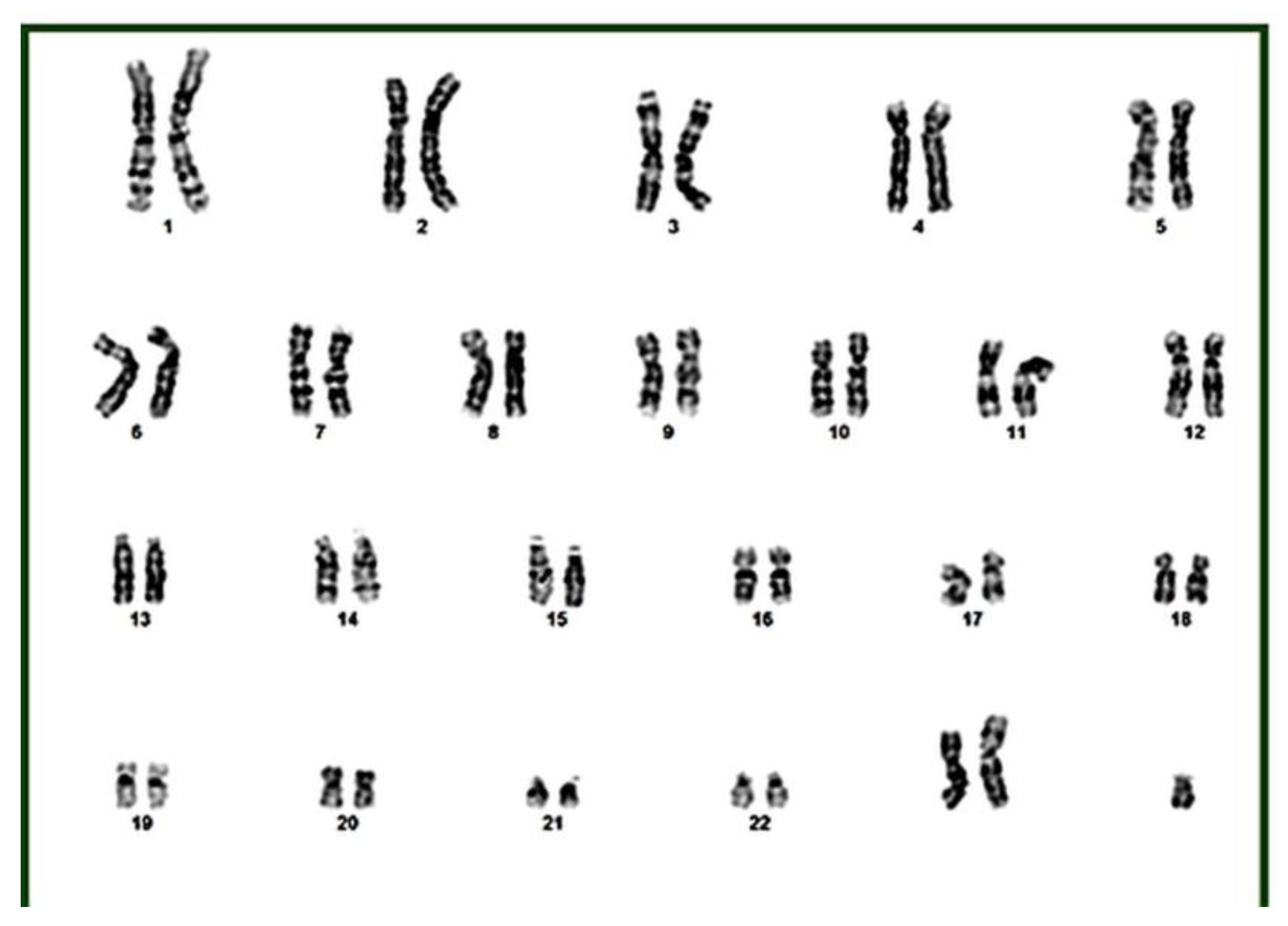
Figure 3.
Pedigree showing segregation of genetic variants across four generations. Squares represent males, circles females, and slashed symbols indicate deceased individuals. The proband (IV-1), marked by an arrow, is homozygous for a pathogenic CYP27A1 variant (red) and also carries variants associated with Klinefelter syndrome (blue) and BRCA2 (green). Yellow indicates heterozygous carrier status. Individuals labeled “Carrier” are confirmed carriers; “No carrier” tested negative; “Not tested” indicates no genetic testing performed.
Figure 3.
Pedigree showing segregation of genetic variants across four generations. Squares represent males, circles females, and slashed symbols indicate deceased individuals. The proband (IV-1), marked by an arrow, is homozygous for a pathogenic CYP27A1 variant (red) and also carries variants associated with Klinefelter syndrome (blue) and BRCA2 (green). Yellow indicates heterozygous carrier status. Individuals labeled “Carrier” are confirmed carriers; “No carrier” tested negative; “Not tested” indicates no genetic testing performed.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



