
Submitted:
30 September 2025
Posted:
01 October 2025
You are already at the latest version
Abstract
Bacteriophage receptor-binding proteins are often attached to the tail via a conserved N-terminal adapter/anchor domain, presumed to function independently from the distal receptor-binding/catalytic domain. Using synthetic phage technology, we demonstrated that the N-terminal domain in Przondovirus phages KP192 and KP195 substantially modulates the receptor-binding and hydrolytic activities of their type A tailspikes. A bioinformatics analysis of related proteins revealed a high correlation between the N-terminal domain and the distal receptor-binding region. Furthermore, it was shown that an imperfect structural fit between the N-terminal domain and the adjacent tail proteins (gatekeeper and nozzle proteins) can reduce virion assembly efficiency, thereby impairing phage fitness. These results underscore the importance of selecting an appropriate N-terminal domain of receptor-binding proteins when engineering bacteriophages with altered host specificity.
Keywords:
bacteriophage
; Klebsiella
; tailspike depolymerase
; receptor-binding protein
; anchor domain
; adapter domain
; synthetic biology
; genome assembly
; transformation-associated recombination cloning
1. Introduction
The escalating prevalence of multidrug-resistant (MDR) and extensively drug-resistant (XDR) bacteria represents a significant global health threat. These pathogens, resistant to most conventional antibiotics, are directly linked to prolonged illnesses, elevated mortality rates, and untreatable infections, requiring the urgent development of novel therapeutic strategies. Bacteriophages (phages), the natural enemies of bacteria, represent a promising alternative for the effective control of bacterial infections [1,2]. Due to their distinct mechanisms of action, antibiotics and phages can complement each other to overcome bacterial resistance [3,4,5].
Advances in genetic engineering and synthetic biology have enabled the design of synthetic phages with tailored properties based on natural phage scaffolds (reviewed in [6,7,8,9,10,11]). A particularly inspiring application of phage engineering is the reprogramming of host specificity by exchanging receptor-binding proteins (RBPs) between phages. This strategy has been successfully used to redirect phage specificity both within and across bacterial genera [12,13,14].
Phage RBPs often exhibit a modular architecture: a conserved N-terminal adapter domain anchoring the RBP to the phage tail and a highly variable C-terminal domain that determines receptor specificity [15,16]. This modularity is believed to facilitate rapid phage adaptation to new hosts through horizontal gene transfer and domain swapping [13,17].
Many podoviruses utilize a T7gp17-like adapter domain (Pfam PF03906) to attach RBPs to the tail [15,16,18,19]. In particular, tailspike proteins (tsp) of Klebsiella-infecting phages belonging to the Przondovirus and Drulisvirus genera contain this domain [20,21]. In addition to the type A tailspikes, which are directly attached to the tail using the T7gp17-like adapter domain, phages of these genera can also contain auxiliary type B tailspikes that are attached to the type A tailspikes via a T4gp10-like branching domain [15,20,22]. Although the T7gp17-like adapter domain is structurally conserved across many phages, its amino acid sequence is highly divergent. This sequence variation, even among phages of the same genus, can prevent the incorporation of heterologous RBPs into a recipient phage’s tail, complicating the engineering of phages with an altered host range [12]. A potential solution is the construction of chimeric RBPs, in which the N-terminal adapter domain from the recipient phage is fused to the receptor-binding domain from a donor phage, thereby ensuring proper tail assembly [13].
In this study, we applied this approach to reprogram the host specificity of two Przondovirus phages, KP192 and KP195, targeting Klebsiella strains of different capsular types. While first-generation synthetic phages with switched specificity were previously constructed through whole RBP gene replacement [14], we hypothesized that preserving the native N-terminal domain of the recipient phage’s type A tailspikes would enhance tail assembly efficiency. We therefore engineered second-generation phages featuring chimeric type A tailspikes with the specificity-determining C-terminal part of the donor phage’s tailspikes and the N-terminal domain of the recipient phage’s tailspikes.
The obtained results indicated that the N-terminal domain of the type A tailspikes serves not merely as a structural adapter but also modulates the receptor-binding and hydrolytic activities of the tailspikes. Consequently, its replacement can impair phage fitness. These findings underscore the importance of choosing an appropriate N-terminal domain of receptor-binding proteins when engineering bacteriophages with altered host specificity.
2. Results
2.1. Experimental Design and Construction of Synthetic Phages
To study the effect of the T7gp17-like adapter domain of type A tailspike proteins on phage properties, phages KP192 and KP195, belonging to the Przondovirus genus, were used. Tailspike proteins of phages KP192 and KP195 share >95% identity with those of Klebsiella phages Kp9 and SH-Kp 152410, respectively [23,24]. The KP192 virion contains six copies of homotrimeric KL111-specific type A tailspikes (tspA192) integrated into the phage tail, and an additional six copies of homotrimeric K2-specific type B tailspikes (tspB192), attached to the T4gp10-like branching domain of the type A tailspikes (Figure 1A, Table 1) [14,20,25]. Thus, this phage infects Klebsiella with K2 and KL111 capsular types. The KP195 virion contains only K64-specific type A tailspikes (tspA195) embedded directly into the tail (Figure 1A, Table 1), and therefore infects Klebsiella with a K64-type capsule. The sequences of the N-terminal domains (the first 149 amino acid residues) of the tspA192 and tspA195 proteins differ significantly (68% identity), although the AlphaFold3-generated models have a similar fold, which is typical for other T7gp17-like NTDs (PDB ID: 7BOZ, 8DSP, 7EY9, and 7Y1C) (Figure 1B,C). Some of the amino acid substitutions are located at the interface formed by the NTD of the type A tailspike and the nozzle and gatekeeper proteins that form the phage tail (Figure 1D). Differences in these regions can reduce the efficiency of incorporation of type A spikes of the KP192 phage into the tail of the KP195 phage and vice versa, thereby reducing the replication efficiency.
To investigate this effect, synthetic phages similar to the previously described KP192_tspA195 and KP195_tspAB192 [14], but featuring chimeric type A tailspikes, were constructed. The N-terminal domain (the first 149 amino acid residues) in such chimeric spikes corresponded to the tail of the recipient phage, and the receptor-binding domains were transferred from the RBP of the donor phage and provided the required capsular specificity (Figure 1A, Table 1). Following the previously established naming scheme, these phages were designated KP195_tspN195AB192 and KP192_tspN192A195, where the first part indicates the genomic scaffold, followed by an underscore and the name of the transplanted gene(s). It is worth noting that phages KP195_tspN195AB192 and KP192_tspN192A195 differed from the corresponding phages KP195_tspAB192 and KP192_tspA195 only in the region encoding the first 149 amino acid residues of the tailspike A proteins, and were otherwise identical. Therefore, a comparison of the properties of these phages allowed us to determine the effect of differences in the N-terminal domain of the type A tailspikes on phage properties.
Synthetic phage genomes were assembled via transformation-associated recombination (TAR) cloning in yeast (Figure 2A) [26,27,28,29]. Each genome was split into nine overlapping fragments (3–6 kbp) in relatively conserved regions. These fragments were PCR-amplified and combined with a part of the yeast centromeric plasmid pRSII415, following the previously described approach [12,13,14,30]. The detailed scheme of the assembly is shown in Figure 2B. To ensure that the mutation rate was acceptable and did not interfere with the formation of viable virions, the genomes of control synthetic phages KP192ctrl and KP195ctrl, identical to the parental phages KP192 and KP195, were assembled similarly. Following yeast transformation, individual colonies were screened using PCR. Yeast plasmid DNAs containing synthetic phage genomes were isolated from the positive clones after yeast cultivation and used for phage genome “rebooting”. Following phage amplification, the correct assembly of the synthetic phage genomes was confirmed using PCR verification and sequencing. The properties of the wild-type and the synthetic phages are summarized in Table 1.
2.2. “Rebooting” of the Synthetic Klebsiella Phage Genomes
Since K. pneumoniae strains often demonstrate low electrocompetence [31], synthetic phages were produced by transformation of E. coli cells as an intermediate host, similarly to the method described previously [12,13,14]. E. coli cell extracts containing phage particles were used to infect K. pneumoniae strains with a suitable K-type. The K. pneumoniae strain CEMTC-2274 (hereinafter AKL111) with the capsular type KL111 was chosen for propagation of KP195_tspN195AB192 and KP192ctrl phages, since both had KL111-specific tailspikes. The K. pneumoniae strain CEMTC-2337 (hereinafter EK64) was used for K64-specific phages KP192_tspN192A195 and KP195ctrl for similar reasons.
The genomes of the control synthetic phages, KP192ctrl and KP195ctrl, were successfully “rebooted”, resulting in the formation of numerous plaques on a Klebsiella lawn (Figure 2C). The shape and size of plaques formed by the corresponding control and wild-type phages were the same. This indicated a high efficiency of assembly and “rebooting” of the genomes of synthetic Klebsiella phages using E. coli as an intermediate host. However, phages KP195_tspN195AB192 and KP192_tspN192A195 could not be “rebooted” using strains AKL111 and EK64, respectively. It has been previously shown that replacing the phage KP192 genomic scaffold with the phage KP195 genomic scaffold (and vice versa) resulted in a decrease in the replication efficiency of chimeric phages KP195_tspAB192 and KP192_tspA195 on strains AKL111 and EK64, respectively, compared to wild-type phages KP192 and KP195 [14]. Therefore, strains CEMTC-2291 (hereinafter BK2) and CEMTC-11039 (hereinafter HK64), on which phages KP195_tspAB192 and KP192_tspA195 replicated more efficiently [14], were also used to “reboot” the genomes of phages KP195_tspN195AB192 and KP192_tspN192A195. Finally, the genomes of phages KP195_tspN195AB192 and KP192_tspN192A195 were successfully “rebooted” (Figure 2C), resulting in the formation of phage particles. The phages were eluted from plaques and used in further experiments.
2.3. N-Terminal Domains of Type A Tailspikes Alter Phage Replication Efficiency via Different Mechanisms
To test whether the N-terminal domain sequence can impact phage properties, the infectious characteristics of the KP195_tspN195AB192 and KP192_tspN192A195 phages were compared to those of the KP195_tspAB192 and KP192_tspA195 synthetic phages, described previously [14] (Figure 1A). The efficiency of plating (EOP) and the efficiency of planktonic cell lysis were studied using Klebsiella strains with suitable K-types. In order to correctly compare different phages, equalization of the phage particle concentration in the phage suspensions was performed using protein electrophoresis followed by densitometry, as described and validated previously [14].
The infectious properties of phages KP195_tspAB192 and KP195_tspN195AB192 were studied on strains AKL111, BK2, and two additional K2 strains CEMTC-2573 and CEMTC-3533 (hereinafter CK2 and DK2, respectively). It was found that infectious titers differed by ≤ 1.5 orders of magnitude on K2 strains, and the appearance of the plaques was the same (Figure 3A, B). However, a significant difference was observed on the AKL111 strain with the KL111-type capsule: phage KP195_tspN195AB192 formed very small plaques, and the infectious titer of the phage sample was 4 orders of magnitude lower than that of phage KP195_tspAB192 (Figure 3A, B). The adsorption efficiency of the KP195_tspN195AB192 phage also depended on the K-type of the Klebsiella strain (Figure 3C). On the BK2 strain, both synthetic phages KP195_tspAB192 and KP195_tspN195AB192, as well as the parental phage KP192, were adsorbed with the same efficiency. However, on the AKL111 strain, the adsorption efficiency of the KP195_tspN195AB192 phage was significantly lower. In addition, the capsule hydrolytic (depolymerase) activity of UV-inactivated phages was studied. No differences were found on the K2-type strains, but on the KL111-type strain, the inactivated KP195_tspN195AB192 phage showed weaker activity (Figure 3D). These results show a clear pattern: the KP195_tspAB192 and KP195_tspN195AB192 phages differed only in the NTD of the KL111-specific type A tailspikes, and the most striking differences in the properties of these phages were observed exactly on the KL111-type strain.
The efficiency of plating for the second pair of phages (KP192_tspA195 and KP192_tspN192A195) could not be compared quantitatively: it was not possible to obtain a suspension of the KP192_tspN192A195 phage with a concentration high enough to determine the concentration of phage particles by electrophoresis. This was due to the fact that the replication efficiency of the KP192_tspN192A195 phage was low: this phage formed small turbid plaques even on those strains on which the KP192_tspA195 phage formed large plaques (strains CEMTC-11039 and CEMTC-11041 (hereinafter IK64) (Figure 3E). In addition, the KP192_tspN192A195 phage did not cause lysis of planktonic cultures of these strains, while the KP192_tspA195 phage effectively lysed them. However, the differences in the properties of these phages clearly indicated that the chimeric tailspike protein tspN192A195 reduced the reproduction efficiency of the KP192_tspN192A195 phage compared to that of the KP192_tspA195 phage. This is similar to the reduction in the reproduction efficiency of phage KP195_tspN195AB192 compared to that of phage KP195_tspAB192 on strain AKL111 due to the chimeric tailspike tspN195A192.
Next, the efficiency of lysis of planktonic cultures of strains AKL111 and BK2 by phages KP195_tspAB192 and KP195_tspN195AB192 was studied. It was found that KP195_tspN195AB192, unlike KP195_tspAB192, did not cause lysis of the AKL111 culture (Figure 3F). This agreed well with the lower efficiency of plating for KP195_tspN195AB192 on this strain. However, KP195_tspN195AB192 resulted in significantly more effective lysis of the BK2 culture (Figure 3F). To study this phenomenon, one-step growth curves were measured, allowing us to determine the burst size and lag time for KP195_tspAB192 and KP195_tspN195AB192. It turned out that the burst size values for these phages substantially differed when using the BK2 strain: one cycle of KP195_tspN195AB192 infection resulted in approximately 80 phages/cell, while for KP195_tspAB192 the value was only 16 phages/cell, i.e., 5 times lower (Figure 3G). Apparently, some degree of mismatch between the N192 domain of the tspA192 protein and the rest of the tail proteins (the nozzle and gatekeeper proteins inherited from the KP195 phage) of KP195_tspAB192 led to a decrease in the efficiency of assembly of mature phage particles. This led to a decrease in the burst size and the efficiency of lysis of the BK2 strain culture by KP195_tspAB192 compared to those of KP195_tspN195AB192.
2.4. Sequence Conservation of N-Terminal Domains of tspA192 and tspA195 Tailspikes
To determine whether there is a correlation between the sequence of the enzymatic part of type A tailspikes and the sequence of the NTD of these proteins, a bioinformatics search for tailspike proteins similar to tspA192 and tspA195 was performed using sequences from the GenBank database. All 18 proteins closely related to tspA192 had a highly conserved NTD sequence similar to that of the N192 domain (Table 2). All 84 proteins closely related to tspA195 also had a conserved NTD sequence similar to that of the N195 domain (Table 2). Meanwhile, the mean distance between the N192-like group and the N195-like group was 0.403, indicating that these two groups differed substantially. In addition, a phylogenetic analysis of the N-terminal part of the studied sequences formed the same groups as an analysis of their enzymatic part (Figure 4). Thus, a clear correlation was demonstrated between the sequences of the enzymatic part of type A tailspikes and the sequences of their NTD in natural phages. No genes were found that encode a hybrid tsp protein containing an NTD from one group and enzymatic/receptor-binding domains from another group. This apparently reflects the fact that such hybrid tsp proteins reduced the efficiency of phage reproduction and were eliminated during evolution.
3. Discussion
Engineering of synthetic phages with altered specificity requires correct docking of RBPs of a donor phage with the virion of a recipient phage. Several options are possible: 1) to transfer the entire RBP gene, including the part encoding the adapter domain; 2) to assemble the gene of a chimeric RBP containing the adapter domain from the recipient phage; and 3) to transfer all the genes encoding the tail proteins (i.e., the RBP(s), the gatekeeper and the nozzle proteins) from the donor phage. The first option is suitable only for closely related phages, since only in this case the N-terminal adapter domain of the RBP of one phage can seamlessly dock with the tail of another phage. The second option ensures that the chimeric RBP can integrate into the tail due to the presence of the appropriate adapter domain. Therefore, it allows transplantation of RBP between distant phages, including phages of different morphological types. However, the transferred RBP does not always function properly in the context of a "foreign" tail [12,13]. The third option is sometimes the only working solution, for example, when constructing Klebsiella-specific phages based on a genomic scaffold of phage T7 [12]. However, this option is only applicable to phages with the same tail morphology and requires smooth docking of the transferred tail of one phage with the portal of another phage.
In this study, we compared the first two approaches using phages belonging to the Przondovirus genus and infecting K. pneumoniae. Surprisingly, the first approach (replacement of the entire gene encoding a type A tailspike) turned out to be more effective and robust, despite a mismatch between the N-terminal domains of the type A tailspike and the rest of the tail proteins. The efficiency of replication of the KP195_tspAB192 and KP192_tspA195 phages using Klebsiella strains with the capsular types KL111 or K64, respectively, was substantially higher compared to that of the second-generation phages KP195_tspN195AB192 and KP192_tspN192A195 containing chimeric tspA proteins. Since the genomes of the first- and second-generation phages differed only in the region encoding the N-terminal domain of the type A tailspike, it can be concluded that either the N-terminal domain itself or the corresponding region of the genome significantly influenced the efficiency of phage reproduction.
Several mechanisms can be proposed to explain the observed results. One of the most obvious reasons is the misfolding of the type A spikes, resulting in the formation of phages with defective tails. However, since type B spikes are attached to the phage tail only through the T4gp10-like branching domain of the type A spikes [13,15,20,25], misfolding or the absence of a significant number of type A spikes would lead to more radical differences in the properties (efficiency of plating, size and transparency of plaques, adsorption rate, depolymerase activity of UV-inactivated phages) of the KP195_tspN195AB192 and KP195_tspAB192 phages on K2 strains, comparable to the differences on the AKL111 strain. This was not observed.
Another hypothesis is that the N-terminal domain of the type A tailspikes is directly involved in the binding (and, possibly, hydrolysis) of the capsular polysaccharide, thereby enhancing the depolymerase activity of the enzymatic/binding domains of the type A tailspikes. This hypothesis is supported by the fact that the adsorption efficiency of the KP195_tspN195AB192 phage on the cells of the AKL111 strain, as well as the depolymerase activity of the UV-inactivated KP195_tspN195AB192 phage against this strain, were reduced compared to those of the KP195_tspAB192 phage. Apparently, the N195 domain located in the chimeric type A spikes of the KP195_tspN195AB192 phage was unable to enhance the binding of these spikes to the capsule of the AKL111 strain. This hypothesis is also supported by the fact that the gatekeeper and nozzle proteins of the phage tail can exhibit polysaccharide-hydrolytic activity [32,33,34,35], although this activity is auxiliary for these proteins. Our study provides the first evidence that the NTD of the phage tailspikes, generally considered as an adapter domain, can modulate receptor-binding and degrading activity. It is possible that this additional receptor-binding site proposed above is formed only upon contact of the N-terminal domain of the tailspikes with other tail proteins (gatekeeper and nozzle) and therefore cannot be detected by studying individual tailspike proteins.
In addition, the effect of partial steric hindrance between the N-terminal domain of the type A tailspike and the remaining tail proteins was investigated. It was shown that an imperfect match between these proteins resulted in a 5-fold reduction in the number of mature virions of the KP195_tspAB192 phage in the progeny compared to that of the KP195_tspN195AB192 phage, presumably due to a decrease in the efficiency of phage tail assembly. This resulted in weaker lytic activity of KP195_tspAB192 when using the BK2 strain compared to that of KP195_tspN195AB192. Given that such an effect could compromise the therapeutic potential of synthetic phages with altered host specificity, it must be carefully considered in their design.
Our findings contrast with a previous study that reported no functional difference between native and chimeric type A tailspikes of related Przondovirus phages [13]. This discrepancy may be explained by two factors: first, the prior study may not have quantitatively compared replication efficiencies; second, the sequence divergence between the NTDs in that study (11%) was considerably lower than the 32% divergence in our KP192/KP195 system, which could make functional differences more pronounced.
It is believed that the adapter/anchor domains and receptor-binding domains of RBPs of bacteriophages, including those of the Przondovirus genus, are independent modules with clearly distinct functions [13,17]. It is also believed that phages can exchange receptor-binding domains of these proteins via horizontal transfer of the corresponding parts of the genes [13,17]. Our results demonstrated that the T7gp17-like N-terminal domains of phage RBPs can not only function as an adapter, but also enhance the receptor-binding and hydrolytic activities of the tailspikes. Therefore, combining different N-terminal domains with the same enzymatic/binding part of an RBP can lead to significant changes in the efficiency of receptor binding and hydrolysis.
A bioinformatics analysis of tspA192-like and tspA195-like tailspikes also indicated a high correlation between the sequence of the NTD of the type A spikes and the sequence of the enzymatic/binding part of these proteins in natural phages. This apparently reflects the fact that the functions of these domains are interconnected, and unfavorable combinations reduced phage fitness and were eliminated during evolution.
Thus, the study demonstrated that the sequence of the N-terminal domain of the type A tailspikes of the Przondovirus phages can exert a complex influence on the infectious properties of the phage, affecting both virion assembly and receptor-binding efficiency. To improve the efficiency of the synthetic phage design and the predictability of their properties, the above factors should be taken into account when choosing an appropriate strategy for the transfer of receptor-binding proteins between phages.
4. Materials and Methods
4.1. Phages, Bacterial, and Yeast Strains
The wild-type Klebsiella phages KP192 and KP195 (GenBank accession numbers NC_047968 and NC_047970, respectively) were obtained from the Collection of Extremophilic Microorganisms and Type Cultures (CEMTC) at the Institute of Chemical Biology and Fundamental Medicine, Siberian Branch of the Russian Academy of Sciences (ICBFM SB RAS), Novosibirsk. Two synthetic phages, KP195_tspAB192 and KP192_tspA195, were used from a previous study [14]. Saccharomyces cerevisiae strain BY4741 (ATCC 4040002) was employed for transformation-associated recombination (TAR) cloning. Escherichia coli TOP10 (Thermo Fisher Scientific, Waltham, MA, USA) was used for the “rebooting” of phage genomes. The following Klebsiella pneumoniae strains were used for phage amplification and characterization: KL111-type strain CEMTC-2274 (designated AKL111); K2-type strains CEMTC-2291 (BK2), CEMTC-2573 (CK2), and CEMTC-3533 (DK2); and K64-type strains CEMTC-2337 (EK64), CEMTC-11039 (HK64), and CEMTC-11041 (IK64). The K-locus types of these strains were previously determined either by wzi gene sequencing (GenBank accession numbers: MN371474, MN371475, MN371483, MN371512, and MN371476) or by wzy allele-specific PCR [14,36].
4.2. Culturing Conditions
Bacterial cultures were grown at 37 °C on Lysogeny Broth (LB) agar plates or in liquid LB medium with shaking at 180 rpm. S. cerevisiae suspension cultures were incubated at 27–30 °C with shaking at 180 rpm in either rich YPD medium (1% yeast extract, 2% peptone, 2% dextrose) or synthetic selective YNB-Leu medium, composed of 0.67% Yeast Nitrogen Base with ammonium sulfate (BD Biosciences), 2% dextrose, and 0.069% Complete Supplement Mixture lacking leucine (CSM-Leu; MP Biomedicals, Santa Ana, CA, USA).
4.3. Preparation of DNA Fragments for Assembly of Phage Genomes
Overlapping fragments of the phage genomes were amplified by PCR with Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific, Waltham, MA, USA) using the primers listed in Tables S1 and S2 and following the manufacturer’s protocol. A small aliquot (0.1–0.2 μL) of a phage suspension at a titer of 108–1010 PFU/mL was used as a template. The vector fragment was amplified from the yeast centromeric plasmid pRSII-415 (Addgene plasmid #35454) using the primers pRSII415_192/5_genome_dir and pRSII415_192/5_genome_rev (Table S1). The resulting vector fragment was used to assemble both KP192- and KP195-based phage genomes. All the fragments were purified with the GeneJET Gel Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA), and DNA concentration values were determined using a NanoDrop One spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
4.4. Phage Genome Assembly in Yeast
Transformation-associated recombination (TAR) cloning in yeast was used for phage genome assembly [26,29,37]. Competent cells of the S. cerevisiae strain BY4741 were prepared according to previously described methods [38,39]. For the transformation, approximately 300 ng of each appropriate phage genome fragment (Table S3) and 300 ng of the vector backbone fragment were combined with 240 μL of 50% PEG-3350, 36 μL of 1 M lithium acetate, and 25 μL of denatured salmon sperm DNA (2 mg/mL). The final reaction volume was adjusted to 360 μL. Subsequently, approximately 108 freshly prepared competent yeast cells were added to the mixture. The cell suspension was heat-shocked at 42 °C for 30–60 min. Following incubation, the cells were pelleted by centrifugation at 12,000× g for 30 seconds and resuspended in 200 μL of sterile water. The transformed yeast cells were plated on YNB-Leu agar plates and incubated at room temperature for 3–4 days to allow for colony formation.
4.5. Isolation of a Yeast Centromeric Plasmid Harboring the Phage Genome
Individual yeast colonies were inoculated separately into YNB-Leu medium and grown at 30 °C with shaking (180 rpm) until the optical density OD600 reached 6–9. Total DNA, including the centromeric plasmid pRSII-415 harboring the phage genome, was extracted from the yeast cells using a previously described method [12,13].
4.6. Phage Genome “Rebooting”
Phage genomes were rebooted using E. coli as an initial phage propagation host, as previously described [12,13]. Briefly, an aliquot of the total yeast DNA preparation, containing the cloned phage genome, was used to transform electrocompetent E. coli TOP10 cells (Thermo Fisher Scientific, Waltham, MA, USA) via electroporation. Following electroporation, the cells were recovered in 1 mL of SOC medium and were grown at 37 °C for 3 h with shaking. To induce phage release, the cells were lysed by the addition of 50 μL of chloroform, followed by vigorous vortexing and centrifugation at 12,000× g for 1 min. The resulting supernatant was mixed with 0.5 mL of an exponentially growing culture of the appropriate K. pneumoniae strain and 4 mL of molten top agar (0.8% w/v). The mixture was poured onto LB agar plates. Phage plaques were observed following incubation at 37 °C for 3–16 h.
4.7. Verification of Genome Assembly Accuracy
Four pairs of phage-specific primers (Table S4) were used in PCR to verify that the synthetic phage samples contained chimeric genomes (the genomic scaffold from one phage and the transferred tailspike gene from another phage), as previously described [14].
A 700 bp region, starting at nucleotide position 33,024 and covering a junction between the genes encoding the internal virion protein D and the type A tailspike (including the entire N-terminal domain-encoding sequence), was verified in the genomes of phages KP192_tspA195 and KP192_tspN192A195. The region was amplified by PCR using primers pt7_ivpD192/5_seq_dir and pt8_tsp195_seq_v2_rev (Table S4) and sequenced by the Sanger method with a BigDye Terminator v3.1 kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions.
Complete genome sequencing of phages KP195_tspAB192 and KP195_tspN195AB192 was performed as described previously [40]. Briefly, phage genomic DNA was fragmented using a Covaris Ultrasonicator (Covaris, Woburn, MA, USA), and DNA libraries were constructed using the NEB Next Ultra II DNA Library Prep Kit for Illumina (both from New England BioLab, Ipswich, MA, USA). Paired-end sequencing was performed on an Illumina MiSeq sequencer using a v.2 reagent kit (2 × 250 base reads) (Illumina Inc., San Diego, CA, USA). The phage genomes were assembled de novo using SPAdes Genome Assembler v. 3.15.4 [41].
4.8. Phage Propagation and Purification
Phages were propagated by infecting 50 mL of an exponentially growing culture (OD600 = 0.4–0.7) of the appropriate K. pneumoniae host strain at a multiplicity of infection (MOI, i.e., the ratio of phage to bacterium, calculated based on the infectious titer of the phage sample) of 0.01 (1 PFU of phage per 100 cells). The following phage-host pairs were used: KP192ctrl and KP195_tspAB192 with strain AKL111; KP195_tspN195AB192 with strain BK2; KP195crtl with strain EK64; and KP192_tspA195 with strain HK64. Infected cultures were incubated with shaking at 37 °C until complete lysis was observed. Cellular debris was removed by centrifugation, and phages were purified from the supernatant by precipitation with polyethylene glycol 6000 (PEG-6000), as described previously [42,43]. The phage pellet was resuspended in 800 μL of SM buffer (10 mM NaCl, 10 mM MgCl₂, 50 mM Tris-HCl, pH 7.5, 0.05% NaN3) and stored at 4 °C.
4.9. Determination of Infectious Titer
Two types of phage titers were used in this study: the infectious titer (specific for a particular Klebsiella strain) and the pseudo-physical titer (independent of Klebsiella strain), introduced previously [14]. The infectious titer (PFU/mL) of phage samples was determined by the double-agar overlay plaque assay. An appropriate indicator Klebsiella strain was grown in LB medium at 37 °C with shaking (180 rpm) to mid-exponential phase (OD600 = 0.5–0.6). A 0.5 mL aliquot of the bacterial culture was mixed with 4 mL of molten soft agar (0.8%) and overlaid onto an LB agar plate. Ten-fold serial dilutions (10-1 to 10-8) of each phage sample were prepared in LB medium. Aliquots (6 µL) of each dilution were applied onto the prepared bacterial lawns. Plates were incubated overnight at 37 °C, and plaques were counted to calculate the titer. All assays were performed in triplicate. Data analysis was performed using Microsoft Excel 2010.
4.10. Equalization of Phage Particle Concentration in Phage Samples
To enable a direct and fair comparison between phages with different efficiencies of plating, the concentrations of phage virions in phage stocks were normalized (or equalized) using protein electrophoresis followed by densitometry, as validated previously [14]. A pseudo-physical phage titer (titerPP), measured in “protein concentration-linked units” per milliliter (PCLU/mL), was used to describe phage particle concentration. The infectious titer of phage KP192 determined on the bacterial lawn formed by an exponentially growing culture of strain AKL111 was chosen as a reference for determining the PCLU units. Thus, if a sample of the KP192 phage had an infectious titer of 1011 PFU/mL when analyzed using strain AKL111, then it was considered to contain exactly 1011 PCLU in 1 mL.
To determine the pseudo-physical titer of a test phage, two-fold serial dilutions of the sample were analyzed by SDS-PAGE together with an aliquot of the reference sample containing 3 × 108 PCLU of purified phage KP192. Following electrophoresis, the gels (12% w/v) were stained using Coomassie G-250 (Figure S1). The band intensities corresponding to the major capsid protein (MCP; ~37 kDa) were quantified using Image-Lab 6.0 software (Bio-Rad). The dilution of the test sample whose MCP band intensity most closely matched that of the reference sample was used to calculate the titerPP using the following equation:
where ODtest and ODreference are the densitometry values of the major capsid protein bands for the test and reference samples, respectively, Vtest is the volume of the test sample aliquot analyzed by SDS-PAGE, and DF is the dilution factor of the test sample. The estimated error rate for this method is 20–30%, which is acceptable for the purposes of this study.
titerPP = ODtest × DF × (3 × 108 PCLU)/(ODreference × Vtest)
4.11. Determination of Phage Adsorption Efficiency
A test strain of K. pneumoniae, used for phage adsorption, and an indicator strain of K. pneumoniae, needed to count plaques formed by unbound phage particles, were used in each adsorption experiment. Strain AKL111 was used as the indicator strain for phage KP192, while strain BK2 was used as the indicator for the KP195_tspAB192 and KP195_tspN195AB192 phages since these phage-strain combinations provided efficient formation of large plaques. When used for phage adsorption, K. pneumoniae strains AKL111 and BK2 were cultivated at 37 °C until the OD600 reached 0.2. Bacterial cultures used as indicator strains (i.e., for plaque formation) were grown under the same conditions to the OD600 of 0.4–0.6. An aliquot of the test phage containing 5 × 106 PCLU in 10 μL was mixed with 100 μL of the adsorption strain suspension (107 CFU) or with 100 μL of LB medium (control experiment). Following incubation at 37 °C for 7 min, all samples were centrifuged at 12,000× g for 30 s to settle down the cells and adsorbed phages.
A 50 μL aliquot of the supernatant containing unadsorbed phages was mixed with 350 μL of PBS and 20 μL of chloroform. Following centrifugation at 12,000× g for 1 min, the supernatants were serially ten-fold diluted in LB medium. Then, 100 μL of each dilution was mixed with 500 μL of the indicator strain culture and 3.5 mL of molten top agar (0.8%), and the mixture was overlaid onto an LB agar plate. After incubation at 37 °C for 3–16 h, phage plaques were counted and the adsorption efficiency was calculated according to the following equation:
where Ncontrol is the plaque count from the control sample (phage mixed with LB), and Nfree is the plaque count on the experimental plate. The experiments were performed in triplicate, and the mean values and standard deviations (SD) were calculated.
adsorption efficiency (%) = [1 – (Nfree / Ncontrol) ] × 100%
4.12. Determination of Depolymerase Activity of UV-Inactivated Phages
Twenty microliters of phage suspensions (1010–1011 PCLU/mL) were irradiated with hard ultraviolet (UV) light under a low-pressure mercury-vapor discharge lamp for 90 min to inactivate the phages. To assay depolymerase activity, bacterial lawns were prepared by mixing 100 μL of an overnight culture of K. pneumoniae with 3.5 mL of molten top agar (0.8%) and overlaying the mixture onto LB agar plates. Serial ten-fold dilutions of the UV-inactivated phages in LB medium were applied to the top layer of agar (6 μL per spot). The plates were incubated at 37 °C for 3–5 h and examined for the formation of translucent zones indicating depolymerase activity.
4.13. Bacterial Killing Assay
An appropriate K. pneumoniae strain was cultivated at 37 °C with shaking at 180 rpm until the OD600 reached 0.5. A test phage aliquot containing 108 PCLU was added to 5 mL of the bacterial culture containing approximately 109 CFU (MOI = 0.1). After a 30-min adsorption period at 37 °C without shaking, the culture was incubated with shaking (180 rpm) at 37°C. Starting from the moment of mixing the cells with the phage, 100 μL aliquots were collected, and the bacterial titer in each aliquot was determined by plating ten-fold serial dilutions onto LB agar plates. The experiments were performed in triplicate for each Klebsiella strain.
4.14. One-Step Growth Assay
The BK2 strain of K. pneumoniae was cultivated at 37 °C with shaking until the OD600 reached 0.4–0.6. Phages were mixed with the test strain culture at an MOI of 0.02 (1 PCLU per 50 CFU). Following incubation at 37 °C for 5 min, the cells were centrifuged at 8,000× g for 2 min to remove unbound phages. The cell pellet was suspended in fresh LB medium, and a fraction of the suspended cells was transferred to an incubation flask containing 10 mL of LB medium. The volume of the fraction was chosen to achieve a suitable concentration of infected cells for accurate plaque counting. To determine the number of infected cells that were able to undergo lysis, a 100 μL aliquot of the cell suspension from the incubation flask was immediately mixed with 500 μL of an exponential-phase BK2 culture (OD600 = 0.4–0.6) and 3.5 mL of molten top agar, and the mixture was overlaid onto an LB agar plate (“infected cells count” plate). Aliquots of the cell suspension were collected from the incubation flask every 5 min for 70 min and kept on ice. Following centrifugation at 12,000× g for 1 min at 4 °C, the aliquots were analyzed in the same way as the aliquot used for the “infected cells count” plate. After incubation at 37 °C for 3–16 h, phage plaques were counted and the burst size was calculated according to the following equation:
where Nprogeny is the number of plaques formed on a plate corresponding to a plateau in the growth curve, DF is the dilution factor of the aliquot assayed, and Ninf. cells is the plaque count from the "infected cells count" plate.
Burst size = Nprogeny × DF / Ninf. cells
4.15. Protein Structure Modeling and Visualization
4.16. Bioinformatics Analysis
Sequences of the C-terminal part (excluding the 149 N-terminal aa residues) of the tspA192 or tspA195 proteins were used to perform a BLAST (blastp) search using the NCBI GenBank non-redundant protein sequences (nr) database. Sequences with ≤75% identity and/or ≤80% query coverage were removed. Each of the resulting sequences was split into N-terminal (the first 149 aa residues) and C-terminal regions. Phylogenetic analysis was conducted using MEGA v.7.0.26 [46]. Neighbor-joining trees with 500 bootstrap replicates were constructed separately for the N-terminal domain (NTD) and C-terminal (Cterm) sequence groups. The genetic distances between the NTDs of tspA192-like and tspA195-like proteins were estimated using the Dayhoff model, calculating the overall mean distance within each group and between the groups.
4.17. Quantification and Statistical Analysis
Data are presented as the mean ± standard deviation (SD). Statistical analyses were performed using GraphPad Prism 8.0.1 (GraphPad Software, USA). Where applicable, statistical significance was assessed by one-way analysis of variance (ANOVA) followed by Tukey’s test for multiple comparisons.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, I.K.B., E.E.M., and N.V.T.; investigation, I.K.B., E.E.M., A.V.M., M.I.K.; resources, N.V.T. and I.K.B.; writing—original draft preparation, I.K.B. and N.V.T.; writing—review and editing, I.K.B., N.V.T., and V.V.M.; visualization, I.K.B. and E.E.M.; supervision, N.V.T.; project administration, I.K.B.; funding acquisition, I.K.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 24-24-00553.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated in this study are available from the corresponding authors upon reasonable request.
Acknowledgments
The authors would like to thank Yuliya N. Kozlova from the Institute of Chemical Biology and Fundamental Medicine of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia, for initial characterization and providing phages KP192 and KP195. The authors would also like to thank Roman B. Gorodnichev from the Lopukhin Federal Research and Clinical Center of Physical-Chemical Medicine of the Federal Medical Biological Agency, Moscow, Russia, for providing some of the Klebsiella strains used in the study.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| CEMTC | Collection of Extremophilic Microorganisms and Type Cultures |
| CFU | Colony forming unit |
| EOP | Efficiency of plating |
| MOI | Multiplicity of infection |
| NTD | N-terminal domain |
| OD600 | Optical density at 600 nm |
| PCLU | Protein concentration-linked unit |
| PFU | Plaque-forming unit |
| pLDDT | Predicted local distance difference test |
| RBP | Receptor-binding protein |
| RMSD | Root mean square deviation |
| TAR | Transformation-associated recombination cloning |
| titerPP | Pseudo-physical phage titer |
| tsp | Tailspike protein |
References
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage Therapy: From Biological Mechanisms to Future Directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef]
- Cui, L.; Watanabe, S.; Miyanaga, K.; Kiga, K.; Sasahara, T.; Aiba, Y.; Tan, X.-E.; Veeranarayanan, S.; Thitiananpakorn, K.; Nguyen, H.M.; et al. A Comprehensive Review on Phage Therapy and Phage-Based Drug Development. Antibiotics 2024, 13, 870. [Google Scholar] [CrossRef]
- Segall, A.M.; Roach, D.R.; Strathdee, S.A. Stronger Together? Perspectives on Phage-Antibiotic Synergy in Clinical Applications of Phage Therapy. Curr. Opin. Microbiol. 2019, 51, 46–50. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Międzybrodzki, R.; Drulis-Kawa, Z.; Cater, K.; Knežević, P.; Winogradow, C.; Amaro, K.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Rękas, J.; et al. Bacteriophages and Antibiotic Interactions in Clinical Practice: What We Have Learned so Far. J. Biomed. Sci. 2022, 29, 23. [Google Scholar] [CrossRef]
- Supina, B.S.I.; Dennis, J.J. The Current Landscape of Phage–Antibiotic Synergistic (PAS) Interactions. Antibiotics 2025, 14, 545. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming Bacteriophage Host Range: Design Principles and Strategies for Engineering Receptor Binding Proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Lenneman, B.R.; Fernbach, J.; Loessner, M.J.; Lu, T.K.; Kilcher, S. Enhancing Phage Therapy through Synthetic Biology and Genome Engineering. Curr. Opin. Biotechnol. 2021, 68, 151–159. [Google Scholar] [CrossRef]
- Łobocka, M.; Dąbrowska, K.; Górski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 2021, 35, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Costa, A.R.; Van Beljouw, S.P.B.; Fineran, P.C.; Brouns, S.J.J. Approaches for Bacteriophage Genome Engineering. Trends Biotechnol. 2023, 41, 669–685. [Google Scholar] [CrossRef]
- Meile, S.; Du, J.; Staubli, S.; Grossmann, S.; Koliwer-Brandl, H.; Piffaretti, P.; Leitner, L.; Matter, C.I.; Baggenstos, J.; Hunold, L.; et al. Engineered Reporter Phages for Detection of Escherichia Coli, Enterococcus, and Klebsiella in Urine. Nat. Commun. 2023, 14, 4336. [Google Scholar] [CrossRef]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef]
- Latka, A.; Lemire, S.; Grimon, D.; Dams, D.; Maciejewska, B.; Lu, T.; Drulis-Kawa, Z.; Briers, Y. Engineering the Modular Receptor-Binding Proteins of Klebsiella Phages Switches Their Capsule Serotype Specificity. mBio 2021, 12, e00455–21. [Google Scholar] [CrossRef]
- Baykov, I.K.; Kurchenko, O.M.; Mikhaylova, E.E.; Miroshnikova, A.V.; Morozova, V.V.; Khlebnikova, M.I.; Tikunov, A.Yu.; Kozlova, Y.N.; Tikunova, N.V. Replacement of the Genomic Scaffold Improves the Replication Efficiency of Synthetic Klebsiella Phages. Int. J. Mol. Sci. 2025, 26, 6824. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef]
- Evseev, P.V.; Sukhova, A.S.; Tkachenko, N.A.; Skryabin, Y.P.; Popova, A.V. Lytic Capsule-Specific Acinetobacter Bacteriophages Encoding Polysaccharide-Degrading Enzymes. Viruses 2024, 16, 771. [Google Scholar] [CrossRef] [PubMed]
- Pas, C.; Latka, A.; Fieseler, L.; Briers, Y. Phage Tailspike Modularity and Horizontal Gene Transfer Reveals Specificity towards E. Coli O-Antigen Serogroups. Virol. J. 2023, 20, 174. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xiao, H.; Wang, X.; Song, S.; Han, Z.; Li, X.; Yang, F.; Wang, L.; Song, J.; Liu, H.; et al. Structural Changes of a Bacteriophage upon DNA Packaging and Maturation. Protein Cell 2020, 11, 374–379. [Google Scholar] [CrossRef]
- Chen, W.; Xiao, H.; Wang, L.; Wang, X.; Tan, Z.; Han, Z.; Li, X.; Yang, F.; Liu, Z.; Song, J.; et al. Structural Changes in Bacteriophage T7 upon Receptor-Induced Genome Ejection. Proc. Natl. Acad. Sci. 2021, 118, e2102003118. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef]
- Cheetham, M.J.; Huo, Y.; Stroyakovski, M.; Cheng, L.; Wan, D.; Dell, A.; Santini, J.M. Specificity and Diversity of Klebsiella Pneumoniae Phage-Encoded Capsule Depolymerases. Essays Biochem. 2024, 68, 661–677. [Google Scholar] [CrossRef]
- Prokhorov, N.S.; Riccio, C.; Zdorovenko, E.L.; Shneider, M.M.; Browning, C.; Knirel, Y.A.; Leiman, P.G.; Letarov, A.V. Function of Bacteriophage G7C Esterase Tailspike in Host Cell Adsorption. Mol. Microbiol. 2017, 105, 385–398. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, Z.; Tao, X.; Shi, X.; Lin, P.; Liao, D.; Ma, C.; Cai, X.; Lin, W.; Jiang, X.; et al. Structural and Functional Basis of Bacteriophage K64-ORF41 Depolymerase for Capsular Polysaccharide Degradation of Klebsiella Pneumoniae K64. Int. J. Biol. Macromol. 2024, 265, 130917. [Google Scholar] [CrossRef]
- Huang, L.; Huang, X.; Zhao, T.; Zhang, J.; Xiang, Y. Isolation and Characterization of Three Lytic Podo-Bacteriophages with Two Receptor Recognition Modules against Multidrug-Resistant Klebsiella Pneumoniae 2024.
- Napolitano, V.; Privitera, M.; Drulis-Kawa, Z.; Marasco, D.; Fallarini, S.; Berisio, R.; Squeglia, F. Structural and Functional Features of Klebsiella Pneumoniae Capsular Degradation by the Phage Depolymerase KP32gp38: Implications for Vaccination against K. Pneumoniae. Int. J. Antimicrob. Agents 2025, 66, 107596. [Google Scholar] [CrossRef]
- Larionov, V.; Kouprina, N.; Graves, J.; Resnick, M.A. Highly Selective Isolation of Human DNAs from Rodent–Human Hybrid Cells as Circular Yeast Artificial Chromosomes by Transformation-Associated Recombination Cloning. Proc. Natl. Acad. Sci. 1996, 93, 13925–13930. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete Chemical Synthesis, Assembly, and Cloning of a Mycoplasma Genitalium Genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A Fully Decompressed Synthetic Bacteriophage øX174 Genome Assembled and Archived in Yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef]
- Costa, A.R.; Azeredo, J.; Pires, D.P. Synthetic Biology to Engineer Bacteriophage Genomes. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2024; Vol. 2734, pp. 261–277. ISBN 978-1-0716-3522-3. [Google Scholar]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J. Designing P. Aeruginosa Synthetic Phages with Reduced Genomes. Sci. Rep. 2021, 11, 2164. [Google Scholar] [CrossRef]
- Fournet-Fayard, S.; Joly, B.; Forestier, C. Transformation of Wild Type Klebsiella Pneumoniae with Plasmid DNA by Electroporation. J. Microbiol. Methods 1995, 24, 49–54. [Google Scholar] [CrossRef]
- Pyra, A.; Brzozowska, E.; Pawlik, K.; Gamian, A.; Dauter, M.; Dauter, Z. Tail Tubular Protein A: A Dual-Function Tail Protein of Klebsiella Pneumoniae Bacteriophage KP32. Sci. Rep. 2017, 7, 2223. [Google Scholar] [CrossRef]
- Pyra, A.; Filik, K.; Szermer-Olearnik, B.; Czarny, A.; Brzozowska, E. New Insights on the Feature and Function of Tail Tubular Protein B and Tail Fiber Protein of the Lytic Bacteriophage φYeO3-12 Specific for Yersinia Enterocolitica Serotype O:3. Molecules 2020, 25, 4392. [Google Scholar] [CrossRef]
- Brzozowska, E.; Pyra, A.; Pawlik, K.; Janik, M.; Górska, S.; Urbańska, N.; Drulis-Kawa, Z.; Gamian, A. Hydrolytic Activity Determination of Tail Tubular Protein A of Klebsiella Pneumoniae Bacteriophages towards Saccharide Substrates. Sci. Rep. 2017, 7, 18048. [Google Scholar] [CrossRef]
- Brzozowska, E.; Pyra, A.; Pawlik, K.; Górska, S.; Gamian, A. The Antibiofilm Activity of Dual-Function Tail Tubular Protein B from KP32 Phage 2018.
- Morozova, V.; Babkin, I.; Kozlova, Y.; Baykov, I.; Bokovaya, O.; Tikunov, A.; Ushakova, T.; Bardasheva, A.; Ryabchikova, E.; Zelentsova, E.; et al. Isolation and Characterization of a Novel Klebsiella Pneumoniae N4-like Bacteriophage KP8. Viruses 2019, 11, 1115. [Google Scholar] [CrossRef]
- Baykov, I.; Kurchenko, O.; Mikhaylova, E.; Morozova, V.V.; Tikunova, N.V. Robust and Reproducible Protocol for Phage Genome “Rebooting” Using Transformation-Associated Recombination (TAR) Cloning into Yeast Centromeric Plasmid. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2024; ISBN 978-1-0716-3522-3. [Google Scholar]
- Gietz, D.; Jean, A.St.; Woods, R.A.; Schiestl, R.H. Improved Method for High Efficiency Transformation of Intact Yeast Cells. Nucleic Acids Res. 1992, 20, 1425–1425. [Google Scholar] [CrossRef]
- Gietz, R.D. Yeast Transformation by the LiAc/SS Carrier DNA/PEG Method. In Yeast Protocols; Xiao, W., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Vol. 1163, pp. 33–44. ISBN 978-1-4939-0798-4. [Google Scholar]
- Morozova, V.V.; Yakubovskij, V.I.; Baykov, I.K.; Kozlova, Y.N.; Tikunov, A.Yu.; Babkin, I.V.; Bardasheva, A.V.; Zhirakovskaya, E.V.; Tikunova, N.V. StenM_174: A Novel Podophage That Infects a Wide Range of Stenotrophomonas Spp. and Suggests a New Subfamily in the Family Autographiviridae. Viruses 2023, 16, 18. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinforma. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Chapter 2. Bacteriophage λ and Its Vectors. In Molecular cloning: a laboratory manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Vol. 1, pp. 2.25–2.106. ISBN 978-0-87969-577-4. [Google Scholar]
- Carroll-Portillo, A.; Coffman, C.N.; Varga, M.G.; Alcock, J.; Singh, S.B.; Lin, H.C. Standard Bacteriophage Purification Procedures Cause Loss in Numbers and Activity. Viruses 2021, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Differences between bacteriophages KP192 and KP195 and the derived synthetic phages. (A) Schematic representation of the wild-type and synthetic phages. The capsular specificity of the synthetic phages is indicated in parentheses. “N192” and “N195” denote the N-terminal adapter domains of the tspA192 and tspA195 proteins, respectively. (B) Alignment of the N-terminal domain sequences of tspA192 and tspA195; colored bars below the alignment correspond to the domains shown in panel C. (C) Ribbon representation of the N-terminal domain of a tailspike protein homotrimer (based on the Kp9 phage tail structure, PDB ID: 7Y1C). (D) The interface between the N-terminal domain of a tailspike homotrimer (colored same as in C) and a ring formed by the gatekeeper protein (two interacting subunits out of twelve are colored in pink and violet, others are shown as a beige surface). One out of six subunits of the nozzle protein is shown as a sky-blue ribbon. Contact-forming residues that differ between the N192 and N195 domains are shown in red. The six-fold symmetry axis of the phage tail is shown as a dashed orange line. The images were prepared using UCSF Chimera 1.13.1.
Figure 1.
Differences between bacteriophages KP192 and KP195 and the derived synthetic phages. (A) Schematic representation of the wild-type and synthetic phages. The capsular specificity of the synthetic phages is indicated in parentheses. “N192” and “N195” denote the N-terminal adapter domains of the tspA192 and tspA195 proteins, respectively. (B) Alignment of the N-terminal domain sequences of tspA192 and tspA195; colored bars below the alignment correspond to the domains shown in panel C. (C) Ribbon representation of the N-terminal domain of a tailspike protein homotrimer (based on the Kp9 phage tail structure, PDB ID: 7Y1C). (D) The interface between the N-terminal domain of a tailspike homotrimer (colored same as in C) and a ring formed by the gatekeeper protein (two interacting subunits out of twelve are colored in pink and violet, others are shown as a beige surface). One out of six subunits of the nozzle protein is shown as a sky-blue ribbon. Contact-forming residues that differ between the N192 and N195 domains are shown in red. The six-fold symmetry axis of the phage tail is shown as a dashed orange line. The images were prepared using UCSF Chimera 1.13.1.
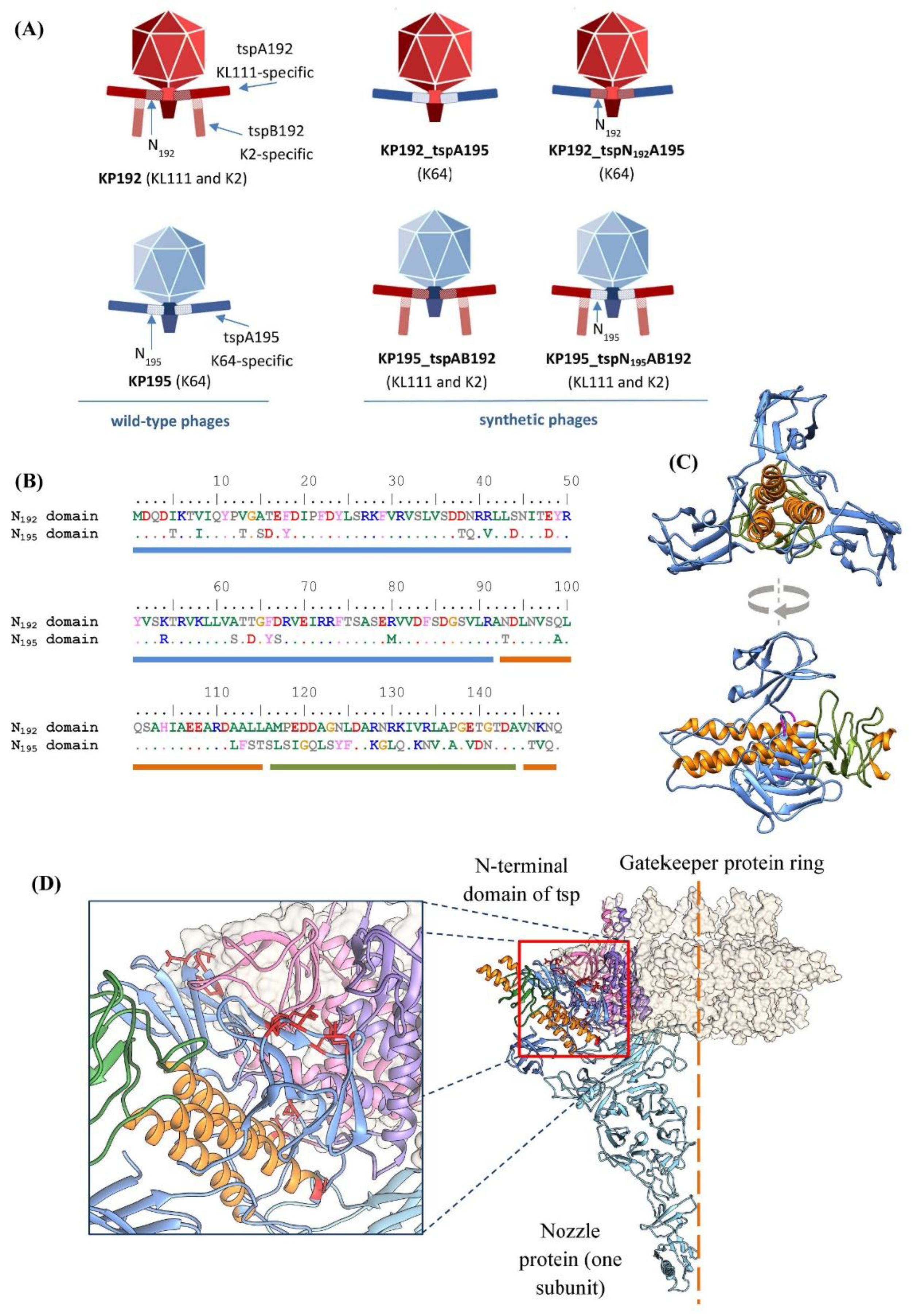
Figure 2.
Assembly and “rebooting” of the synthetic phage genomes. (A) An outline of phage genome assembly in yeast and “rebooting” of phage genomes. “YCp”—yeast centromeric plasmid; “tsp gene”—a DNA fragment containing the gene(s) encoding tailspike protein(s). The red fragment represents an assembled phage genome integrated into a yeast plasmid. (B) Detailed diagram of the assembly of synthetic phage genomes (see also Tables S2–S4). Primers are indicated by arrows. Regions of overlapping DNA fragments are shown as colored squares. Regions of overlap with the yeast centromeric plasmid are marked with blue diamonds. Parts 1 to 9 of the genome are designated “pt1”–“pt9”. The dashed lines indicate regions that differed between phages within the same scaffold group. (C) “Rebooting” of synthetic phage genomes. Plates containing a Klebsiella lawn and phage plaques are shown. “tspN195AB192” and “tspN192A195” denote KP195_tspN195AB192 and KP192_tspN192A195.
Figure 2.
Assembly and “rebooting” of the synthetic phage genomes. (A) An outline of phage genome assembly in yeast and “rebooting” of phage genomes. “YCp”—yeast centromeric plasmid; “tsp gene”—a DNA fragment containing the gene(s) encoding tailspike protein(s). The red fragment represents an assembled phage genome integrated into a yeast plasmid. (B) Detailed diagram of the assembly of synthetic phage genomes (see also Tables S2–S4). Primers are indicated by arrows. Regions of overlapping DNA fragments are shown as colored squares. Regions of overlap with the yeast centromeric plasmid are marked with blue diamonds. Parts 1 to 9 of the genome are designated “pt1”–“pt9”. The dashed lines indicate regions that differed between phages within the same scaffold group. (C) “Rebooting” of synthetic phage genomes. Plates containing a Klebsiella lawn and phage plaques are shown. “tspN195AB192” and “tspN192A195” denote KP195_tspN195AB192 and KP192_tspN192A195.
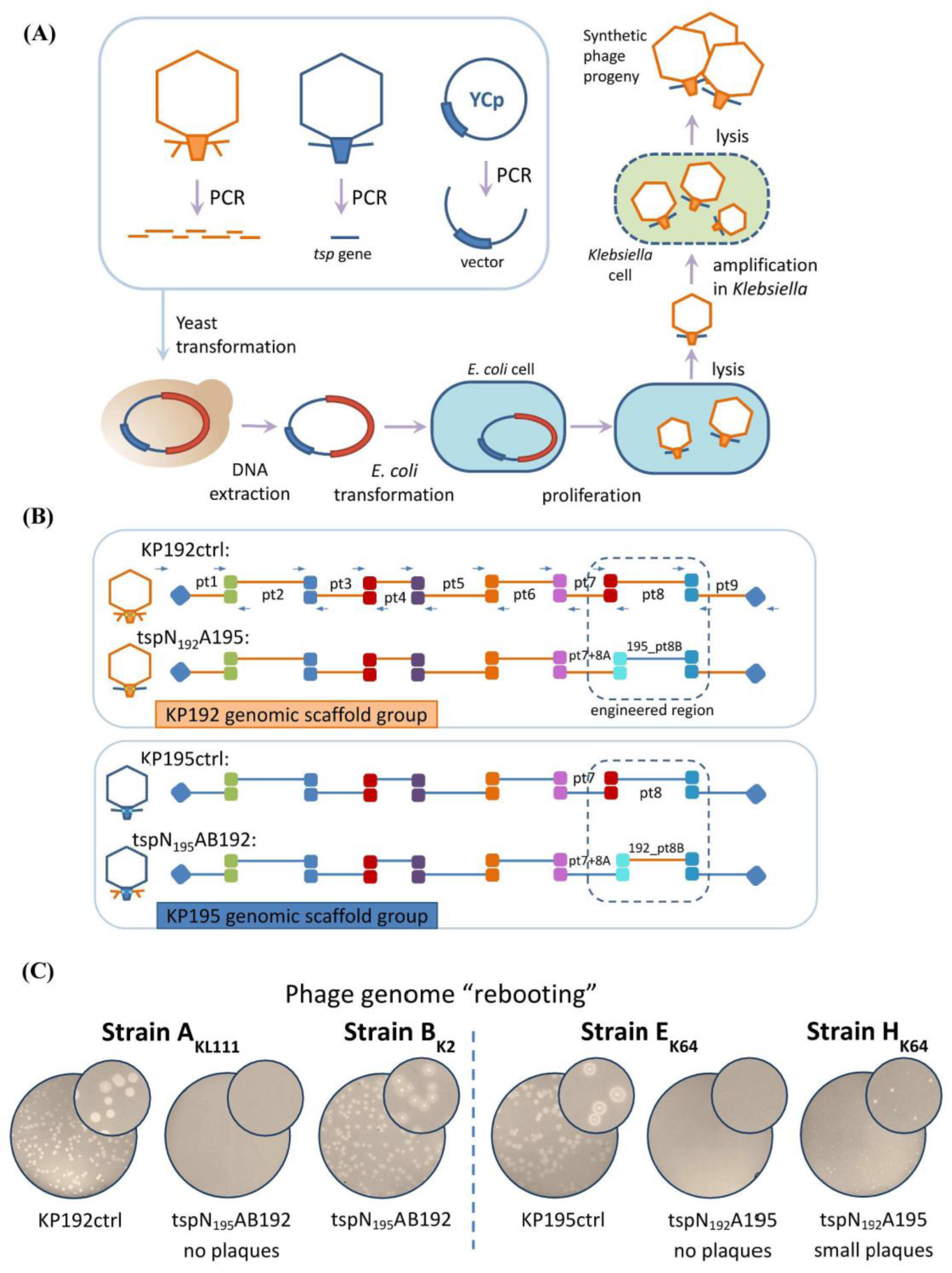
Figure 3.
Differences in the genomic scaffolds of synthetic phages affect their replication efficiency. (A) Infectious titers of KP195_tspAB192 and KP195_tspN195AB192 phage samples. The phage particle concentration in the samples was the same (4.6 × 1011 PCLU/mL). Data from n = 3 independent experiments are represented as the mean ± SD. Statistical significance of log-transformed infectious titer values was determined using a one-way ANOVA with Tukey’s multiple comparison test (** p < 0.01, *** p < 0.001). (B) Plaque morphology. Numbers represent the sample dilution factor, e.g., “–8” indicates a 1:108 dilution of the phage stock. The phage particle concentration in the samples was the same. The size of the area shown is 12 × 12 mm. (C) Adsorption efficiency. The fraction (%) of adsorbed phages following a 7-minute adsorption. Test strains are indicated. Indicator strains are listed in the Materials and Methods section. Data from n = 4 independent experiments are represented as the mean ± SD. Statistical significance was determined using a one-way ANOVA with Tukey’s multiple comparisons test (*** p < 0.001, **** p < 0.0001). (D) Spot test of the depolymerase activity of UV-inactivated phage samples, “tspAB” and “chAB” denote phages KP195_tspAB192 and KP195_tspN195AB192. Dilutions of the phage stocks are indicated on the left. The phage particle concentration in the stocks was the same (4.6 × 1011 PCLU/mL). (E) Plaque morphology for the second pair of phages. The size of the area shown is 12 × 12 mm. (F) Bacteria killing (lytic) curves; 1 PCLU of phage per 10 cells was used for infection. Data from n = 3 independent experiments are represented as the mean ± SD. (G) One-step growth curves. Data from n = 3 independent experiments are represented as the mean ± SD.
Figure 3.
Differences in the genomic scaffolds of synthetic phages affect their replication efficiency. (A) Infectious titers of KP195_tspAB192 and KP195_tspN195AB192 phage samples. The phage particle concentration in the samples was the same (4.6 × 1011 PCLU/mL). Data from n = 3 independent experiments are represented as the mean ± SD. Statistical significance of log-transformed infectious titer values was determined using a one-way ANOVA with Tukey’s multiple comparison test (** p < 0.01, *** p < 0.001). (B) Plaque morphology. Numbers represent the sample dilution factor, e.g., “–8” indicates a 1:108 dilution of the phage stock. The phage particle concentration in the samples was the same. The size of the area shown is 12 × 12 mm. (C) Adsorption efficiency. The fraction (%) of adsorbed phages following a 7-minute adsorption. Test strains are indicated. Indicator strains are listed in the Materials and Methods section. Data from n = 4 independent experiments are represented as the mean ± SD. Statistical significance was determined using a one-way ANOVA with Tukey’s multiple comparisons test (*** p < 0.001, **** p < 0.0001). (D) Spot test of the depolymerase activity of UV-inactivated phage samples, “tspAB” and “chAB” denote phages KP195_tspAB192 and KP195_tspN195AB192. Dilutions of the phage stocks are indicated on the left. The phage particle concentration in the stocks was the same (4.6 × 1011 PCLU/mL). (E) Plaque morphology for the second pair of phages. The size of the area shown is 12 × 12 mm. (F) Bacteria killing (lytic) curves; 1 PCLU of phage per 10 cells was used for infection. Data from n = 3 independent experiments are represented as the mean ± SD. (G) One-step growth curves. Data from n = 3 independent experiments are represented as the mean ± SD.
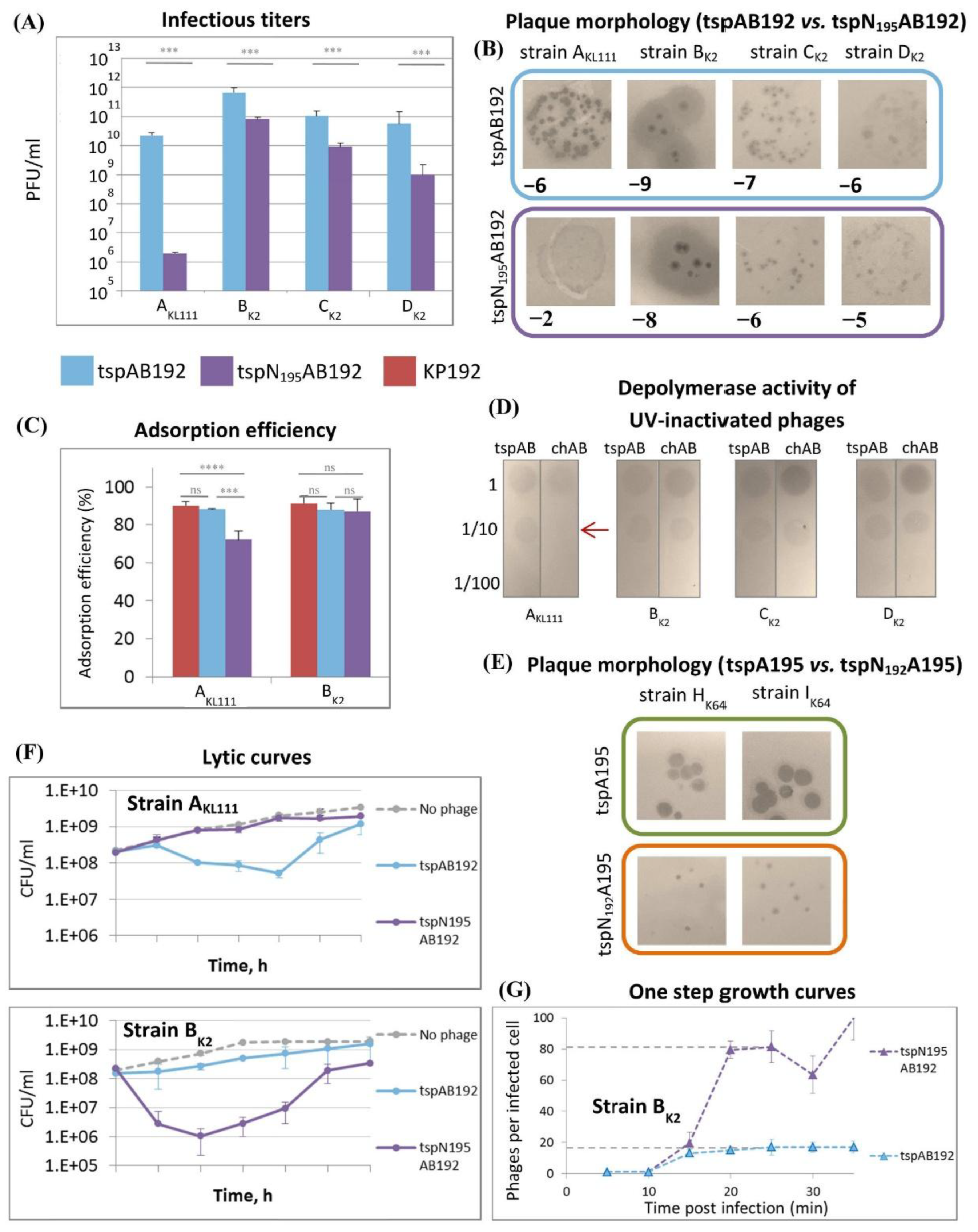
Figure 4.
Phylogenetic trees constructed from the sequences of the enzymatic part (the entire tailspike protein except for the first 149 amino acid residues) or the N-terminal domain of tspA192-like and tspA195-like tailspikes. The numbers indicate the bootstrap values.
Figure 4.
Phylogenetic trees constructed from the sequences of the enzymatic part (the entire tailspike protein except for the first 149 amino acid residues) or the N-terminal domain of tspA192-like and tspA195-like tailspikes. The numbers indicate the bootstrap values.


Table 1.
Summary of the properties of the wild-type and synthetic phages.
| Phages with tspA192 and tspB192 tailspikes | Phages with tspA195 tailspikes | |||||
|---|---|---|---|---|---|---|
| Name | KP192 / KP192ctrl |
KP195_ tspAB192 |
KP195_ tspN195AB192 |
KP195 / KP195ctrl |
KP192_ tspA195 |
KP192_ tspN192A195 |
| Type | wild-type1 | synthetic | synthetic | wild-type1 | synthetic | synthetic |
| Pictogram |  |
 |
 |
 |
 |
 |
| Capsular specificity | KL111 and K2 | KL111 and K2 | KL111 and K2 | K64 | K64 | K64 |
| Genomic scaffold |
KP192 | KP195 | KP195 | KP195 | KP192 | KP192 |
| N-terminal domain of the type A tailspikes | N192 | N192 | N195 | N195 | N195 | N192 |
| NTD of the type A tailspikes is native to the gatekeeper and nozzle proteins | Yes | No | Yes | Yes | No | Yes |
1 Phages KP192 and KP195 were wild-type, while KP192ctrl and KP195ctrl were their synthetic analogs.
Table 2.
Sequence conservation of the N-terminal domains of tspA192-like and tspA195-like tailspikes.
Table 2.
Sequence conservation of the N-terminal domains of tspA192-like and tspA195-like tailspikes.
| tailspike group | Number of sequences | Capsular specificity | Amino acid identity of NTD (within group) |
Mean distance (within group) | Amino acid identity of NTD (between groups) |
Mean distance (between groups) |
|---|---|---|---|---|---|---|
| tspA192-like proteins | 19 | KL111 | ≥ 95% | 0.018 | ≤ 70% | 0.403 |
| tspA195-like proteins | 85 | K64 | ≥ 93% | 0.026 | ≤ 70% | 0.403 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



